

SUMMARY
The immunosuppressive tumor microenvironment (TME) is a major barrier to immunotherapy. Within solid tumors, why monocytes preferentially differentiate into immunosuppressive tumor-associated macrophages (TAMs) rather than immunostimulatory dendritic cells (DCs) remains unclear. Using multiple murine sarcoma models, we find that the TME induces tumor cells to produce retinoic acid (RA), which polarizes intratumoral monocyte differentiation toward TAMs and away from DCs via suppression of DC-promoting transcription factor Irf4. Genetic inhibition of RA production in tumor cells or pharmacologic inhibition of RA signaling within TME increases stimulatory monocyte-derived cells, enhances T cell-dependent anti-tumor immunity, and synergizes with immune checkpoint blockade. Furthermore, an RA-responsive gene signature in human monocytes correlates with an immunosuppressive TME in multiple human tumors. RA has been considered as an anti-cancer agent, whereas our work demonstrates its tumorigenic capability via myeloid-mediated immune suppression and provides proof of concept for targeting this pathway for tumor immunotherapy.
Graphical Abstract

In Brief
Tumor cells evade immune responses by producing high levels of retinoic acid, which directs intratumoral monocytes to differentiate into immunosuppressive macrophages rather than immunostimulatory dendritic cells.
INTRODUCTION
Tumor immunity involves the capture and processing of tumor antigens by antigen presenting cells (APCs), migration of APCs to draining lymph nodes to prime T cells, and migration of primed T cells to the tumor where they exert cytotoxic anti-tumor effects (Chen and Mellman, 2013). APCs are important at multiple steps; dendritic cells (DCs) process tumor antigens to prime anti-tumor T cells, DCs and immunostimulatory tumor-associated macrophages (TAMs) support T cell function, while immunosuppressive TAMs counteract T cell function (Chen and Mellman, 2013). The TME promotes suppressive TAMs and inhibits DCs, impeding the generation and maintenance of functional anti-tumor T cells (Veglia and Gabrilovich, 2017). Hence, identifying pathways that polarize APC distribution in the TME is critical for developing new approaches for cancer immunotherapy.
Macrophages originate from embryonic precursors or from circulating monocytes (Haldar and Murphy, 2014). Likewise, DCs differentiate from bone marrow-derived DC-precursors (conventional DCs or cDCs) or from circulating monocytes (monocyte-derived DCs or moDCs) (Mildner and Jung, 2014). While cDCs are critical for antitumor immune responses, the role of moDCs in tumor immunity is less clear (Veglia and Gabrilovich, 2017). Circulating monocytes are vastly more abundant than cDC precursors and under inflammatory conditions can differentiate into moDCs (Merad et al., 2013). Within solid tumors, however, monocytes preferentially generate immunosuppressive TAMs (Richards et al., 2013). The molecular basis for this is largely unknown (Salmon et al., 2019).
Retinoic acid (RA) has established roles in development, including in the functional specification of peritoneal macrophages (Gundra et al., 2017; Okabe and Medzhitov, 2014). Despite diverse functions in immunity, RA is generally thought to promote tolerance via multiple immune cell types (Erkelens and Mebius, 2017). While some studies have reported that RA promotes DC differentiation, others have reached the opposite conclusion (Jin et al., 2010; Mohty et al., 2003). Similarly, although RA can induce Foxp3+ T regulatory cells and Arg1 producing anti-inflammatory macrophages, it has also been reported to promote anti-tumor Th1 or Th17 T cell responses and dampen myeloid-derived suppressor cell function (Bhatt et al., 2014; Bhattacharya et al., 2016; Hill et al., 2008; Nefedova et al., 2007; Vellozo et al., 2017). Factors contributing to these conflicting results include the exclusive reliance on cell surface markers to represent cellular identity and function, the use of inconsistent or supra-physiological doses of RA in experimentation, and the presence of additional environmental factors that modulate RA’s effects (Duester, 2017). Further, the impact of RA on tumor-associated APCs remains unclear.
RA is generated from vitamin A-derivative retinol through a series of enzyme-catalyzed steps, the rate limiting step being the conversion of retinaldehyde to RA via retinaldehyde dehydrogenases (Raldh1, Raldh2, or Raldh3) (Duester, 2008). Raldh enzymes are highly expressed in many human tumors (Khoury et al., 2012; Li et al., 2010, 2014; Marcato et al., 2015; Wei et al., 2015; Zhang et al., 2013). Indeed, given RA’s role as a pro-differentiation agent during development, the effect of RA to promote tumor cell differentiation and suppress tumor growth has been studied for decades, albeit with limited clinical translation outside of promyelocytic leukemia (Tang and Gudas, 2011). However, solid tumors represent a complex niche comprising of many different cell types other than malignant cells (Binnewies et al., 2018). Hence, we wanted to explore whether RA could act on immune cells in the TME to promote immune suppression and facilitate tumor growth.
Here, we uncover an important role of RA in regulating intratumoral APC distribution and anti-tumor immune responses in sarcomas. We find that TME-associated factors such as interleukin(IL)-13 induce RA production in sarcoma cells. Tumor-derived RA blocks monocyte differentiation into DCs within the TME, instead generating immunosuppressive TAMs. Blocking RA production in tumor cells or inhibiting RA signaling within the TME increases the frequency of immunostimulatory APCs and engenders anti-tumor immune responses, which show strong synergy with immune checkpoint blockade. Finally, using RA-induced gene signatures in human monocytes, we computed an “RA response score” to identify human tumors wherein this pathway may underlie tumor immune evasion. Hence, the decades-old paradigm of RA as an anti-tumor agent is context-dependent as our findings clearly show a pro-tumor effect of RA mediated via RA’s impact on anti-tumor immune responses.
RESULTS
TME Promotes Monocyte Differentiation into Immunosuppressive TAMs
Mononuclear phagocytes (MPs) have been extensively examined in many common tumors, but sparsely in sarcoma (Broz and Krummel, 2015; Broz et al., 2014; Ehnman and Larsson, 2015; Laoui et al., 2011). Sarcomas have over 60 subtypes, but based on their underlying genetic aberrations can be broadly divided into: (1) translocations generating fusion oncogenes, (2) mutations in tumor suppressors or oncogenes, and (3) genomic instability without a consistent mutation (Taylor et al., 2011). We utilize a representative mouse model for each of these genetic categories: synovial sarcoma (SS) (driven by SYT-SSX fusion oncogene), undifferentiated pleomorphic sarcoma (UPS) (driven by loss of p53 and activation of KRAS), and fibrosarcoma (FS) (syngeneic transplant of sarcoma cell lines derived from methylcolanthrene induced murine fibrosarcomas) (Haldar et al., 2007; Kirsch et al., 2007; Schreiber and Podack, 2009).
In all three sarcoma models, monocytes and TAMs comprised the majority of intratumoral leukocytes (Figures 1A, S1A, and S1C). In contrast, CD11b+ DCs were rare and CD103+ DCs exceedingly rare (Figures 1B, S1B, and S1C). To assess the impact of the different MP subsets on T cell proliferation, we isolated monocytes, TAMs, or DCs from tumors and co-cultured them with αCD3/28-stimulated splenic T cells (Figure 1C). DC isolation was greatly facilitated by use of the Zbtb46GFP mouse that allowed clear distinction of DCs from TAMs (Satpathy et al., 2012). Both TAMs and monocytes suppressed T cell proliferation, with TAMs displaying greater suppression (~2–3×) (Figures 1D, S1D, and S1E). In contrast, DCs stimulated T cell proliferation (Figure 1E). Hence, the abundant TAMs were the most immunosuppressive MP cells in the sarcoma TME.
Figure 1. TME Promotes Differentiation of Monocytes into Immunosuppressive Macrophages.

(A and B) Flow cytometry (FCM) showing monocytes (CD45+CD11b+Ly6C+) and TAMs (CD45+CD11b+F4/80+Ly6C−) (A), and DCs (CD45+ZBTB46+) (B) in FS tumors implanted into Zbtb46GFP hosts.
(C) T cell suppression assay (D and E) with tumor MPs (gating shown in A and B).
(D) T cell proliferation with tumor monocytes or TAMs (n = 5 tumors, pooled). Shown are frequency and absolute number of proliferated CD4 and CD8 T cells. Representative of three independent experiments.
(E) Frequency and absolute number of proliferated T cells following co-culture with DCs or TAMs (n = 3 tumors, pooled). Representative of two independent experiments.
(F) Intratumoral monocyte transplantation experiment.
(G and H) FCM plots of tdT+ cells (derived from Lyz2Cre: Rosa26tdT: Zbtb46GFP transplanted monocytes) in FS tumors at 3 days or 7 days post-monocyte transplant
(G). Frequency of F4/80+ or Zbtb46-GFP+ cells within the tdT+ fraction at indicated time points (H). Data aggregated from three independent experiments.
(I) Ex vivo intratumoral monocyte differentiation experiment.
(J) FCM plots (left) and frequencies (right) of DCs and macrophages after 3 days in culture. Representative of three independent experiments. Each dot represents an individual mouse.
*p < 0.05, **p < 0.01, ***p < 0.001. Two-tailed t test. (D) and (E) were analyzed with one-way ANOVA with Tukey’s post hoc test. All error bars represent SEM. Events shown in FCM plots are pregated on live singlets unless otherwise specified and numbers represent percentage of cells within indicated gates.
See also Figure S1.
Studies in other solid tumors have shown monocytes to be a major source of TAMs (Richards et al., 2013). To identify monocyte progenies in sarcoma, we performed monocyte lineage-tracing by generating FS tumors (syngeneic transplant) in LysM-Cre: Rosa26tdT hosts (Abram et al., 2014). Monocytes generated the majority of TAMs but few DCs (Figure S1F). These findings were corroborated in a monocyte-deficient mouse model of UPS generated by breeding the UPS model to Ccr2−/− mice (Figures S1G and S1H).
Monocyte subsets differ in their ability to generate DCs versus macrophages (Briseño et al., 2016; Menezes et al., 2016; Olingy et al., 2019). Hence, skewed monocyte differentiation within TME may reflect selective recruitment of monocyte subsets that are primed for macrophage differentiation. Alternatively, factors in the TME may drive macrophage differentiation from “uncommitted” monocytes. To address this, we injected LysMCre: Rosa26tdT: Zbtb46GFP bone marrow monocytes into FS tumors and analyzed their differentiation over time (Figure 1F). The vast majority of transplanted monocytes (tdT+ cells) differentiated into TAMs but not DCs (Figures 1G and 1H). Next, we asked whether intratumoral monocytes retain the ability to differentiate into DCs by isolating tumor monocytes and culturing them ex vivo with DC promoting cytokines GM-CSF and IL-4 (Figures 1I and S1I). In this setting, the vast majority of monocytes differentiated into DCs but not macrophages (Figure 1J). Further, we were able to detect the major monocyte subsets within sarcoma TME (Figure S1J). Taken together, these findings suggest that TME-associated factors promote monocyte differentiation into suppressive TAMs but not DCs.
IL-13 Promotes Tumor Cell RA Production
To identify TME factors driving monocyte differentiation into TAMs, we performed microarray-based gene expression profiling of TAMs isolated from our murine sarcoma models. Compared to tissue-resident macrophages (Immgen), TAMs expressed higher cellular retinoic acid binding proteins (CRABPs) (Figures 2A, S2A, and S2B). This was independently validated via qRT-PCR on monocytes, macrophages, and DCs isolated from tumors and normal tissues (Figure S2C). Gene expression profiling of bulk tumor also revealed higher expression of Raldh enzymes (that catalyze the rate-limiting step of RA production) compared to surrounding normal muscle (Figure 2B). To directly quantify RA, we performed liquid chromatography/mass spectrometry for the major bioactive isoform of RA, all-trans retinoic acid (ATRA), and found elevated ATRA in all three mouse sarcoma models compared to normal mesenchymal tissues (Figure 2C).
Figure 2. TME Induces Tumor Cells to Produce High Levels of RA.
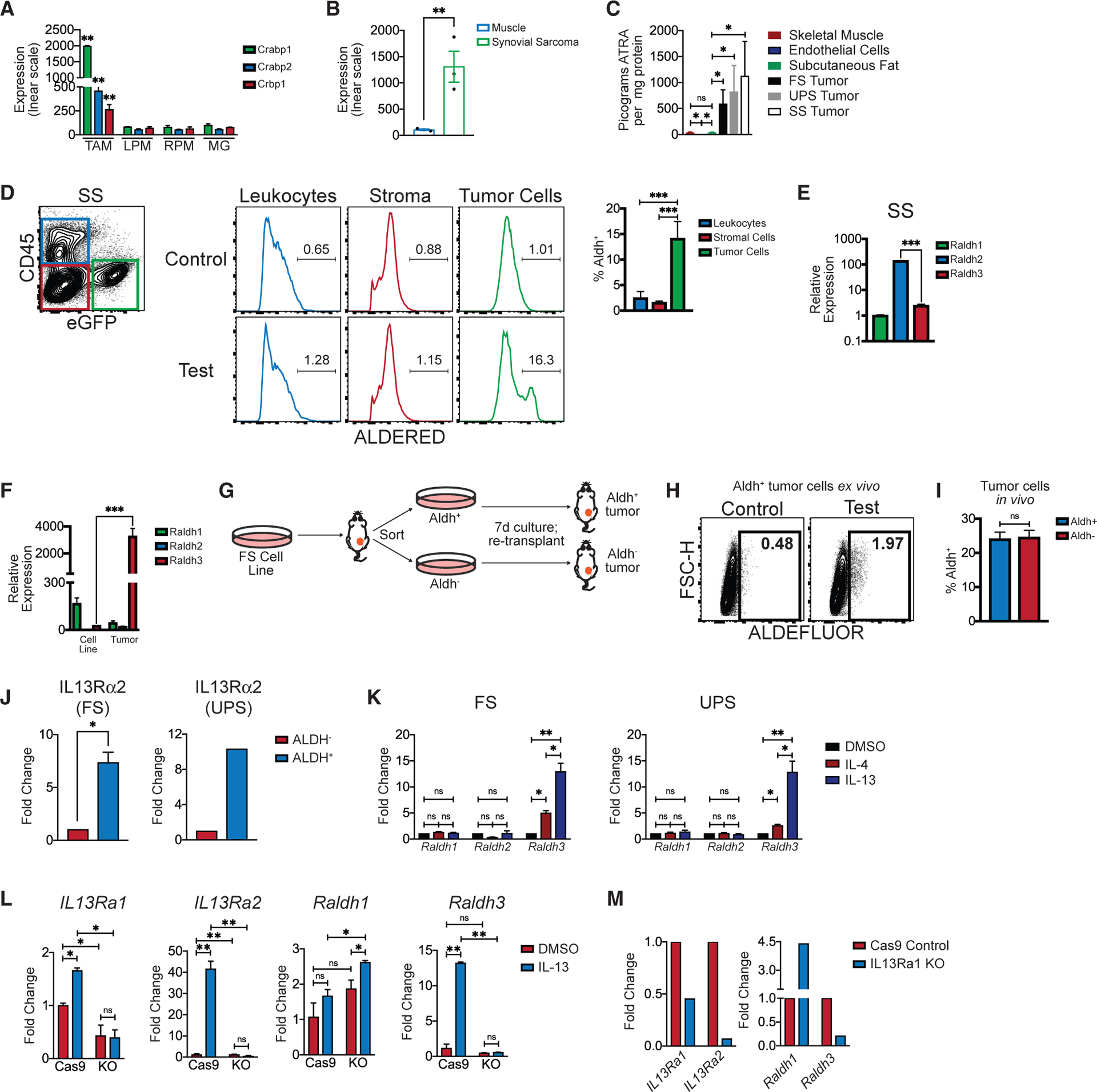
(A) Microarray (Affymetrix, mouse gene 1.0ST) analyses of CD45+ F4/80+ CD11c− TAMs sorted from autochthonous UPS. Shown is the expression (y axis, linear scale) of cellular retinoic acid binding proteins (CRABPs) in TAMs compared to selected tissue resident macrophages (expression data from ImmGen.org). LPM, large peritoneal macrophage; RPM, red pulp macrophage; MG, microglia.
(B) Microarray (Affymetrix mouse gene 1.0ST)-based expression of Aldh1a2 (Raldh2) in autochthonous SS compared to surrounding skeletal muscle. Each dot represents an individual mouse tumor (n = 3 per group).
(C) Liquid chromatography/mass spectrometry for all-trans retinoic acid (ATRA) was performed on frozen subcutaneous adipose tissue (n = 3), primary lung microvascular endothelial cells (n = 3), FS tumors (n = 10), UPS tumors (n = 5), or SS tumors (n = 5).
(D) ALDERED assay on mouse SS. FCM show eGFP+ tumors cells, CD45+ leukocytes and eGFP− CD45− stromal cells. Representative histograms of ALDERED fluorescence in the aforementioned populations (right). “Control” (top) show ALDERED with Aldh inhibitor (DEAB) while “test” (bottom) show the same without the inhibitor, which distinguishes fluorescence via Aldh activity from background. Bar graph shows frequency of Aldh+ cells within indicated populations (n = 6 tumors).
(E) Aldh+ or Aldh− cells were sorted from mouse SS (n = 3) tumors and the expression of Raldh isoforms quantified by qPCR (normalized to Hprt). Representative of two independent experiments.
(F) Raldh expression in FS tumor cells cultured in vitro compared to FS tumors in vivo (n = 4 FS tumors, qPCR normalized to Hprt expression). Representative of three independent experiments.
(G) Experimental outline for (H) and (I).
(H) ALDEFLUOR assay on cultured Aldh+ tumor cells showing loss of Aldh activity in vitro.
(I) Tumors generated from re-transplanted Aldh+ or Aldh− tumor cells (as described in G) were harvested and assayed by ALDEFLUOR. Frequency of ALDEFLUOR+ cells is shown (n = 5 tumors per group). Representative of two independent experiments.
(J) ALDH+ or ALDH–tumor cells were sorted from FS or UPS flank tumors and the expression ofIL13Ra2 was measured by qPCR (n = 3 FS tumors individually sorted; n = 5 UPS tumors pooled and sorted).
(K) FS or UPS cells were treated in vitro with recombinant IL-4 (20 ng/mL), IL-13 (20 ng/mL), or DMSO and the relative expression of Raldh1 and Raldh3 measured.
(L) Cas9 Control or IL13Ra1 KO UPS cells were treated in vitro with recombinant IL-13 (20 ng/mL) or DMSO. Relative expression ofIl13ra1, Il13ra2, Raldh1, and Raldh3 quantified by qPCR.
(M) C57BL/6 mice were transplanted (s.c.) with Cas9 Control or IL13Ra1 KO UPS cell lines. Tumors were harvested 11 days post-implantation, tumor cells were fluorescence-activated cell sorted (FACS), and relative expression of Il13ra1, Il13ra2, Raldh1, and Raldh3 in sorted tumor cells is shown (qPCR). All expression normalized to Hprt.
*p < 0.05, **p < 0.01, ***p < 0.001. Two-tailed t test. (A), (C)–(E), (K), and (L) were analyzed with one-way ANOVA with Tukey’s post hoc test. All error bars represent SEM. Events shown in FCM plots are pregated on live singlets unless otherwise specified and numbers represent percentage of cells within indicated gates. See also Figure S2.
To identify the cellular source of RA in TME, we performed a fluorescence-based assay detecting enzymatic activity of aldehyde dehydrogenases (Aldh), a larger family of enzymes that includes Raldh (Ginestier et al., 2009). RA production was largely restricted to tumor cells in all of our murine models (Figures 2D, S2D, and S2E). To assess whether Aldh fluorescence is truly indicative of Raldh activity, we performed gene expression profiling on sorted Aldh+ and Aldh− tumor cells. Raldh transcripts were the only members of the Aldh enzyme family that were upregulated (data not shown). Further, Raldh isoforms were differentially expressed between sarcoma subtypes, although all three isoforms have redundant catalytic functions (Kumar et al., 2012) in the production of RA (Figures 2E and S2F). These data demonstrate that tumor cells produce the vast majority of RA in sarcoma TME.
Only a subset of tumor cells exhibited RA production and FS tumor cells massively upregulated Raldh in vivo compared to in vitro, suggesting that RA production is induced by the TME (Figure 2F). To further address this, we isolated Aldh+ (RA-producing) and Aldh− (RA-negative) tumor cells from FS tumors, cultured the cells separately for 7 days, and re-transplanted them each into syngeneic recipients (Figure 2G). RA-producing tumor cells ceased RA production in vitro (Figure 2H), and RA-negative cells regained the capacity to produce RA in vivo (Figure 2I), demonstrating that TME induces tumor cells to produce RA.
To identify TME factors that may induce RA production, we revisited the microarray data from Aldh+ and Aldh− tumor cells. IL-13 receptor alpha 2 (IL13Ra2), which is known to be strongly induced by IL-13 signaling (Suzuki et al., 2015), was significantly higher in Aldh+ cells (confirmed by qPCR in sorted ALDH+ versus ALDH− tumor cells) (Figure 2J). IL-13 (and to a lesser extent IL-4) treatment induced Raldh expression in murine sarcoma cells in vitro (Figure 2K). To abrogate IL-13 signaling, we generated IL13Ra1 knockout sarcoma cell lines using CRISPR-Cas9.IL13Rα1 KO tumor cells did not upregulate IL13Rα2 or Raldh3 upon exposure to IL-13 in vitro (Figure 2L). Correspondingly, transplanted IL13Ra1 KO tumor cells showed reduced Raldh3 (Figure 2M). Finally, we identified T cells as the primary source of IL-13 in TME, while the expression of IL-13 receptors was largely restricted to myeloid and tumor cells (Figure S2G). Taken together, these findings suggest that T cell-derived IL-13 can induce RA production by tumor cells.
RA Promotes Macrophage and Suppresses DC Differentiation from Monocytes
The results above show high levels of RA in sarcomas and evidence of RA exposure in TAMs. Hence, we explored whether RA might impact monocyte differentiation in sarcoma TME. We utilized a well-established assay of in vitro monocyte differentiation with GM-CSF and IL-4 that yields a mixture of macrophages and DCs (Helft et al., 2015). Addition of RA at the onset of culture impaired DC differentiation of both murine and human monocytes (Figures 3A and 3B). RA doses were based on published literature (Duester, 2017) and our finding that plasma RA ranges between ~5–20 nM. The effects of RA were recapitulated by RAR agonist CH55 and blocked by pan-RAR inverse agonist BMS493, suggesting that RA acts on monocytes via the RAR signaling (Figure 3C). Conversely, RA promoted macrophage differentiation from both mouse and human monocytes (Figures 3D and 3E). These effects were even more striking when IL-13 was used instead of IL-4 (Figures S3A and S3B). A brief bioinformatics-based analysis of human tumors suggested that the levels of IL-13 may be higher than IL-4 in many solid tumors, including sarcomas (Figure S3C) (Suzuki et al., 2015). Hence, IL-13 likely plays an important role in MP cell differentiation and function in the TME.
Figure 3. RA Promotes Immunosuppressive Macrophage and Inhibits DC Differentiation from Monocytes In Vitro.

(A and B) Zbtb46GFP bone marrow monocytes (A) or human monocytes from normal donors (B) were cultured with GM-CSF, IL-4, and RA (100 nM for mice and 20 nM for human) or DMSO. FCM plots and cumulative frequencies of DCs are shown. Representative of five independent experiments.
(C) Frequency of CD11c+ CD1a+ cells generated from human monocytes cultured with GM-CSF and IL-4 with either DMSO, RA (20 nM), CH55 (RAR agonist; 10 nM), BMS493 (pan-RAR inverse agonist; 1 mM), or RA + BMS493. Representative of two independent experiments.
(D) FCM plots and frequencies of F4/80+ cells generated from mouse monocytes cultured with GM-CSF and IL-4 with DMSO or RA. Representative of five independent experiments.
(E) Frequency of CD68+ cells in human monocytes cultured with GM-CSF and IL-4 with DMSO or RA. Representative of three independent experiments.
(F and G) Ly6C+ Zbtb46-GFP− monocytes from FS tumors were cultured with GM-CSF and IL-4. DMSO or RA (100 nM) was added at the onset of culture. FCM plots and frequencies of macrophages (G) and DCs (bar graph, F) are shown (n = 3 FS tumors, representative of three independent experiments).
(H and I) BM monocytes were cultured with GM-CSF and IL-4 (H) or with M-CSF (I). RA (100 nM) or DMSO was added at day 0. After 3 days, differentiated APCs were washed, and co-cultured for 3 days with CFSE-labeled aCD3/28 stimulated splenic T cells. Shown are histograms and frequency of proliferated T cells. Representative of four independent experiments.
(J) Human monocytes from normal donors were cultured with M-CSF. After 5 days, differentiated macrophages were washed and co-cultured for 3 days with CFSE-labeled aCD3/28 stimulated T cells (from different human donor). RA or DMSO was added to the co-culture at day 0. Shown is frequency of proliferated T cells. Representative of two independent experiments.
(K and L) Mouse (C57BL/6J) BM monocytes (K) or human monocytes from normal donors (L) were cultured with GM-CSF and IL-4 with DMSO or RA. Cells were harvested 1 day later, RNA was extracted, and microarray (Affymetrix Mouse Gene 2.0ST) analysis was performed. Shown are fold changes of selected macrophage and DC signature genes.
(M) Mouse BM monocytes were cultured with GM-CSF and IL-4 with either RA, CH55 (RAR agonist), BMS493, or DMSO. After 3 days, RNA was extracted and expression of Irf4 and Zbtb46 measured by qPCR. Representative of two independent experiments.
(N) Human monocytes from normal donors were cultured with GM-CSF and IL-4. After 5 days, RNA was extracted and expression of Irf4 and Zbtb46 measured by qPCR. Representative of two independent experiments.
(O) Mouse BM monocytes from LysMCre: Irf4fl/fl or LysMCre: Irf4+/+ mice and cultured with GM-CSF and IL-4. RA or DMSO was added at Day 0. After 3 days, differentiated APCs were washed and co-cultured for 3 days with CFSE-labeled αCD3/28 stimulated splenic T cells. Shown is frequency of proliferated T cells. Data aggregated from three independent experiments.
(P) Human monocytes were transfected with control plasmid (pMax-GFP from Lonza) or IRF4 (IRF4-IRES2-eGFP (GeneCopoeia) using Human Monocyte Nucleofector Kit (Lonza). 2 × 106 cells were transfected with 1 mg plasmid. Expression of IRF4 and ZBTB46 is shown. Data aggregated from two independent experiments. Normalized to Hprt.
*p < 0.05, **p < 0.01, ***p < 0.001. Two-tailed t test. (C), (H)–(J), (M)–(P) were analyzed with one-way ANOVA with Tukey’s post hoc test. All error bars represent SEM. Events shown in FCM plots are pregated on live singlets unless otherwise specified and numbers represent percentage of cells within indicated gates. See also Figure S3.
To examine whether RA impacts monocyte differentiation within tumors, we isolated tDT+ GFP− intratumoral monocytes from FS tumors in LysMCre: Rosa26tdT: Zbtb46GFP mice and cultured them with GM-CSF and IL-4 with or without RA. Consistent with results above, RA inhibited DC and promoted macrophage differentiation from tumor monocytes (Figures 3F, 3G, and S3D). Notably, these effects of RA on monocyte differentiation were significantly augmented in the presence of tumor-conditioned media (Figure S3E).
Finally, we tested aforementioned effects of RA in the context of human cancer. Although most tumors tested produced very little RA in vitro, only inducing its production in vivo as described above, we found that hepatocellular carcinoma SNU398 expressed high levels of RALDH1 in vitro (Figure S3F). Thus, we co-cultured human monocytes with SNU398 in the presence or absence of BMS493. Consistent with our observations above, DC differentiation was suppressed in the presence of SNU398. Strikingly, BMS493 completely rescued this effect, suggesting that tumor-derived RA was largely responsible for the skewed monocyte differentiation (Figure S3G). Together, these results demonstrate that RA signaling inhibits tumor monocyte differentiation into DC and promotes differentiation into macrophage.
RA Regulates Key Transcription Factors to Promote Monocyte Differentiation into Immunosuppressive Macrophages
As TAMs are generally immunosuppressive, we examined RA’s impact on the immunosuppressive capacity of monocyte-derived APCs. GM-CSF and IL-4 differentiated APCs with aCD3/28 stimulated splenic T cells and measured T cell proliferation and activation. As expected, the addition of RA during monocyte differentiation enhanced APC suppressive function (Figures 3H and S3H). The findings were reproduced in monocyte differentiation systems involving GM-CSF alone or with tumor condition media (Figures S3I–S3K). RA also endowed additional suppressive capability on differentiated macrophages generated by culturing monocytes with M-CSF (Figures 3I and S3L). This finding was recapitulated in human macrophage T cell co-cultures (Figure 3J). Of note, the addition of RA to T cells in the absence of macrophages did not significantly impact T cell proliferation (Figure S3M), suggesting that RA- induced T cell suppression in co-cultures were largely mediated via macrophages. Together, these results demonstrated that RA polarizes monocyte differentiation toward suppressive APCs, and additionally induces suppressive functions on mature macrophages.
RA regulates gene expression by binding to retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers (Duester, 2008). Microarray-based gene expression profiling of mouse or human monocytes treated with RA revealed that RA induced key genes associated with macrophage differentiation while downregulating genes associated with moDC differentiation (Figures 3K and 3L). Notably, RA downregulated transcription factor Irf4, which regulates monocyte differentiation into DCs (Briseño et al., 2016; Lehtonen et al., 2005). We further validated this via qPCR and confirmed that this effect was dependent on RAR signaling (Figures 3M and 3N). Similar to RA-treated wild-type (WT) APCs, Irf4-deficient APCs (LysMCre: Irf4fl/fl) demonstrated heightened T cell suppression (Figure 3O). Importantly, the addition of RA to Irf4-deficient monocytes did not further enhance suppressive capacity of these APCs (Figure 3O). We also performed the complementary experiment of overexpressing Irf4 in human monocytes, which rescued RA-mediated suppression of DC differentiation (Figure 3P). Taken together, these findings suggest that RA impacts monocyte differentiation by regulating key transcription factors such as Irf4.
Reducing Tumor RA Production Enhances Intratumoral Stimulatory APCs
Our data thus far show high levels of RA production by tumor cells and the ability of RA to generate immunosuppressive TAMs from monocytes. Hence, we next examined the impact of reducing tumor cell-derived RA on intratumoral APCs in vivo. Using CRISPR-Cas9, we generated a Raldh3 KO FS cell line, given that Raldh3 was specifically upregulated in RA producing FS cells in vivo. Deletion of Raldh3 resulted in compensatory increase in Raldh1 expression (Figure S4A). Thus, we generated a Raldh1/3 double KO (DKO) cell line, which showed >80% reduction in Raldh1 and Raldh3 transcripts (Figure S4B), modestly reduced Aldh enzymatic activity in vivo (Figure S4C), and significantly reduced ATRA levels compared to control tumors (Figure S4D). DKO tumors showed reduced TAMs and increased CD11b+ DCs, while CD103+ DCs remained largely unchanged (Figures 4A, 4B, and S4E). Notably, a significantly higher fraction of F4/80+ TAMs in DKO tumors were CD11c+ MHCII+ and expressed higher levels of activation markers CD40, CD86, and tumor necrosis factor alpha (TNF-α) (Figures 4C, S4F, and S4G). We further characterized RA-driven changes in intratumoral leukocytes by using single-cell RNA-sequencing (scRNA-seq), comparing tumor-infiltrating CD45+ leukocytes in DKO to Cas9 control tumors (Figure 4D). The largest differences were observed in the myeloid compartment, with DKO tumors harboring reduced TAMs with immunosuppressive markers (TAM 1), increased TAMs with immunostimulatory markers (TAM 2), and increased DCs (Figures 4E–4G). Intriguingly, the majority of DCs expressed Ccr2, suggesting a monocyte origin (Figure 4E). Consistent with reduction in immunosuppressive TAMs by scRNA-seq, we found that F4/80+ TAMs from DKO tumors were significantly less suppressive to T cell proliferation and activation ex vivo (Figures 4H, S4H, and S4I). Of note, the distribution of blood neutrophils and monocytes were similar in mice harboring control or DKO tumors, suggesting that the effects of decreasing tumor cell RA production on immune responses are mostly confined to the TME (Figures 4I and S4J).
Figure 4. Decreasing Tumor RA Enhances Intratumoral Stimulatory APCs.
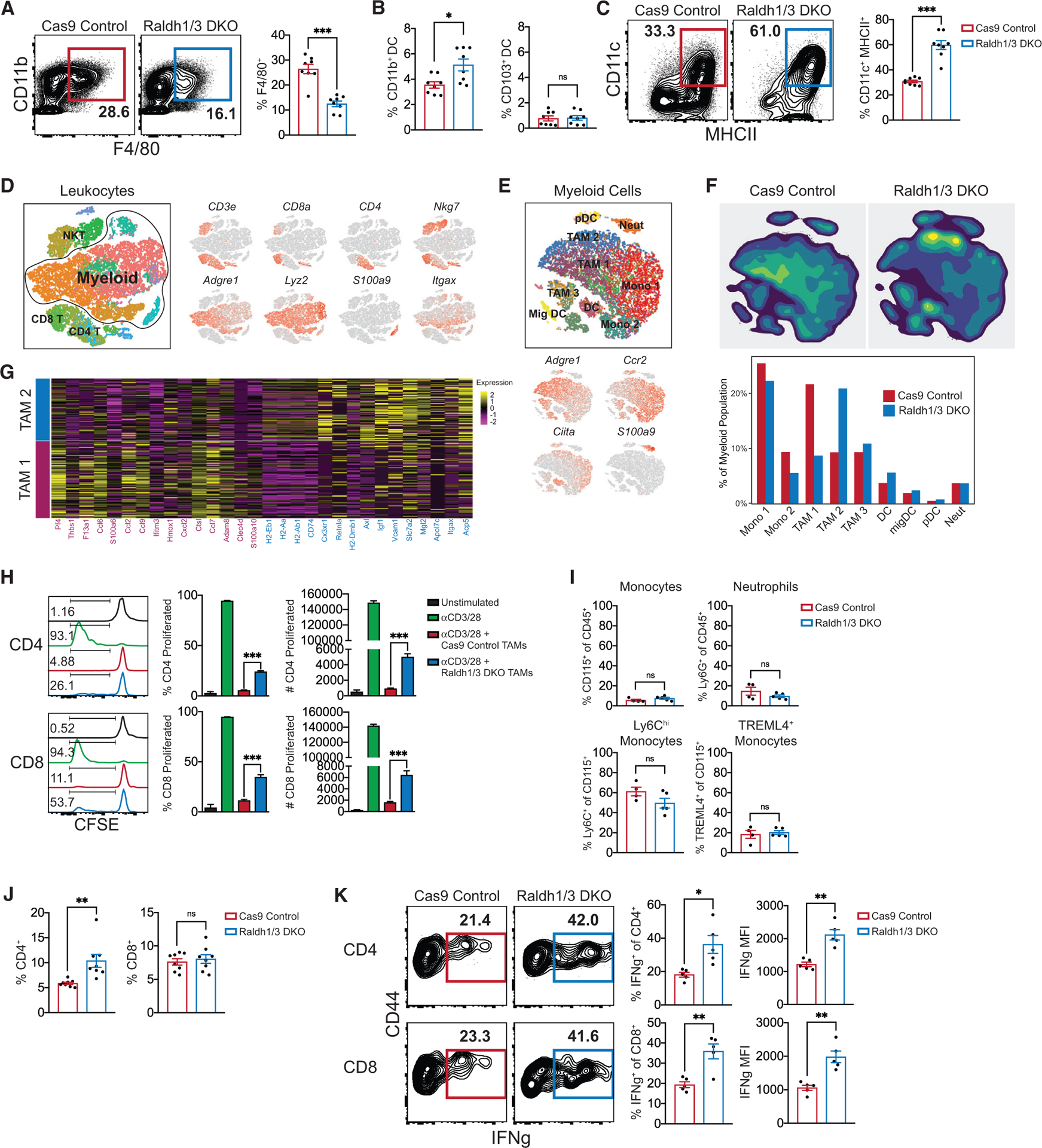
(A) Frequency of CD11b+ F4/80+ TAMs in Raldh1/3 DKO or Cas9 control FS tumors (n = 8 tumors).
(B) Frequency of CD11b+ or CD103+ DCs in Raldh1/3 DKO or Cas9 control (n = 8 tumors).
(C) Frequency of TAMs expressing both CD11c and MHCII (pregated on CD11b+ F4/80+) in Raldh1/3 DKO or Cas9 control FS tumors (n = 8 tumors per group).
(D) CD45+ leukocytes from Raldh1/3 DKO or Cas9 control FS tumors were profiled by scRNA-seq (n = 4 tumors per group). Shown are merged t-distributed stochastic neighbor embedding (tSNE) plots of identified immune populations (left) and selected marker gene expression (right).
(E) Merged tSNE plot of reclustered myeloid populations (top) and selected marker gene expression (bottom).
(F) Density plots (top) and relative frequencies (bottom) of myeloid clusters in Raldh1/3 DKO or Cas9 Control tumors.
(G) Heatmap of top 15 differentially expressed genes in TAM 1 compared to TAM 2 myeloid populations.
(H) T cell suppression assay using CD11b+ F4/80+ TAMs sorted from Raldh1/3 DKO or Cas9 control tumors. Sorted TAMs were co-cultured with CFSE-labeledαCD3/28 stimulated splenic T cells obtained from a non-tumor-bearing host. Representative histograms, frequencies, and absolute numbers of proliferated T cells are shown. Representative of two independent experiments.
(I) Monocyte and neutrophil frequency in peripheral blood of C57BL/6 hosts bearing Raldh1/3 DKO or Cas9 control tumors (n = 5 mice per group). Representative of two independent experiments.
(J) CD4+ or CD8+ T cell frequencies within CD45+ leukocytes in Raldh1/3 DKO or Cas9 control tumors (n = 8 tumors per group). Representative of three independent experiments.
(K) IFNγ production in T cells from Raldh1/3 DKO or Cas9 control tumors. Intratumoral T cells were incubated with GolgiStop and stimulated for 4 h with PMA/ionomycin. Shown are representative contour plots (left) and frequencies (right) of IFNγ+ CD4 or CD8 T cells (n = 5 tumors per group). Representative of two independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001. Two-tailed t test. (H) was analyzed with one-way ANOVA with Tukey’s post hoc test. All error bars represent SEM. Events shown in FCM plots are pregated on live singlets unless otherwise specified and numbers represent percentage of cells within indicated gates.
See also Figure S4.
Additional immunophenotyping revealed increased CD4+ T cells in DKO tumors (Figures 4J and S4K). Importantly, both CD4+ and CD8+ T cells in DKO tumors produced more interferon gamma (IFNγ) compared to those from control tumors (Figure 4K). Concomitant with a more stimulatory myeloid and T cell compartment, DKO FS demonstrated markedly decreased tumor volume and weight, as well as extended survival (Figures 5A, 5B, S5A, and S5B). The effects of Raldh1/3 DKO on immune cells and tumor size were also consistent in an intramuscular orthotropic transplant-based model, suggesting that the impact of RA deficiency is independent of the anatomical location of the tumor (Figures S5C–S5F). To confirm the role of RA in the observed effects with DKO tumors, we overexpressed Raldh2 in the Raldh1/3 DKO cells to restore RA production (Figure S5G). Over- expression of Raldh2 restored tumor growth in DKO FS (Figure 5C). Correspondingly, Raldh2 overexpression reversed the stimulatory myeloid compartment of DKO tumors (Figure 5D). Together, these findings show that reducing tumor cell RA production engenders stimulatory APCs, augments intratumoral T cell responses, and inhibits tumor progression.
Figure 5. Decreasing Tumor RA Augments Anti-tumor T Cell Responses and Synergizes with PD-1 Blockade.
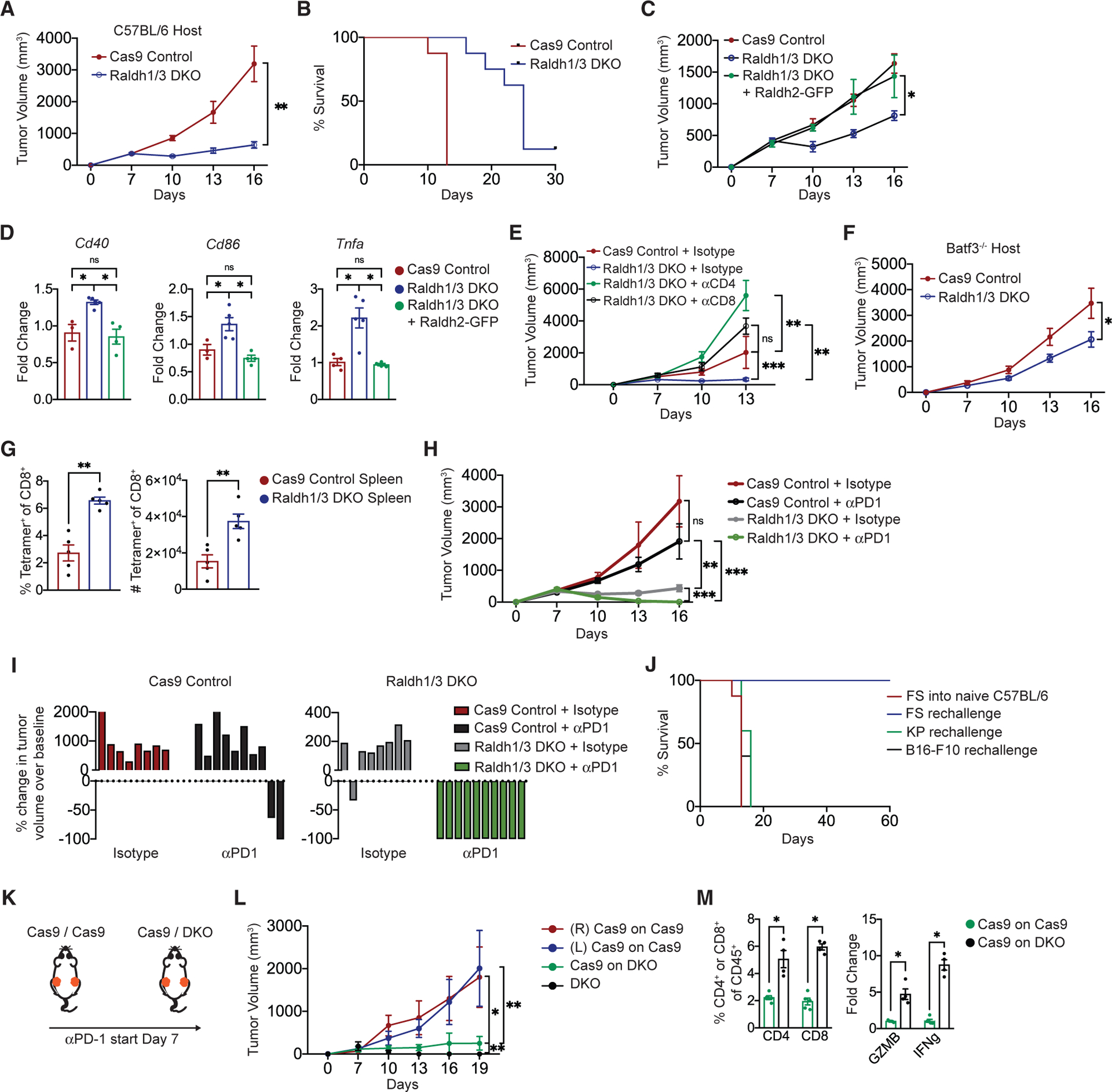
(A) Volume of Raldh1/3 DKO or Cas9 control FS tumors implanted s.c. in C57BL/6 mice measured every 3 days starting 7 days post-implantation (n = 8 tumors per group, representative of three independent experiments).
(B) Survival of mice bearing Raldh1/3 DKO or Cas9 control FS tumors implanted s.c. in C57BL/6 mice (n = 12 tumors per group, data aggregated from three independent experiments).
(C) Volume of Raldh1/3 DKO overexpressing Raldh2-GFP (Raldh rescue), Raldh1/3 DKO, or Cas9 control FS tumors implanted s.c. in Lyz2Cre:Rosa26-LSLCas9-IRES-GFP mice. These mice were used as hosts to minimize potential immune responses against Cas9 and GFP. Tumor volume measured every 3 days starting at 7 days post-implantation (n = 10 tumors per group, representative of two independent experiments).
(D) Relative expression of Cd40, Cd86, and Tnfa by qPCR in indicated FS tumors (n = 5 tumors per group, representative of two independent experiments).
(E) Tumor volume following CD4+ or CD8+ T cell depletion. aCD4, aCD8, or isotype control antibody was administered to 3 days before s.c. implantation of Raldh1/3 DKO or Cas9 control FS tumors. Thereafter, antibodies were administered intraperitoneally (i.p.) every 3 days (n = 5 tumors per group).
(F) Growth of Raldh1/3 DKO or Cas9 control FS tumors implanted s.c. in in Batf3−/− hosts (n = 7 mice per group).
(G) Raldh1/3 DKO or Cas9 control FS tumor cell lines expressing cytoplasmic OVA-ZsGreen were injected s.c. into C57BL/6 hosts. Shown are frequency and number of H-2kb/SINFEKL tetramer-positive splenic CD8+ T cells at 11 days post-tumor implantation.
(H and I) αPD1 or isotype control antibody was administered to C57BL/6 mice starting 7 days post-implantation of Raldh1/3 DKO or Cas9 control tumors. Three doses (200 mg i.p.) were given at days 7, 10, and 13. Shown are tumor growth curves (H) and waterfall plots (I) of change in tumor volume after 12 days of therapy.
(J) Parental FS, KP sarcoma, or B16-F10 melanoma tumor cell lines were implanted s.c. into mice that previously rejected (60 days tumor-free) Raldh1/3 DKO tumors upon αPD1 therapy or into naive C57BL/6 mice as control (n = 10 FS, n = 5 KP, n = 5 B16-F10). Shown is the survival curve for indicated groups.
(K) Experimental outline: C57BL/6 mice received either Cas9 control tumors on both sides or Cas9 control on one side and Raldh1/3 DKO tumor on the other. αPD-1 was administered (200 mg i.p. on days 7, 10, and 13).
(L) Tumor volume measured every 3 days starting at 7 days post-implantation (n = 5 tumors per group).
(M) Frequencies of CD4+ or CD8+ T cells in Cas9 tumors contralateral to Cas9 tumors or Cas9 tumors contralateral to Raldh1/3 DKO tumors (left). Relative expression of Gzmb and Ifnγ in bulk tumors (right). Normalized to Hprt expression.
*p < 0.05, **p < 0.01, ***p < 0.001. Two-tailed t test. (D) was analyzed with one-way ANOVA with Tukey’s post hoc test. (A)–(C), (E), (F), (H), (J), and (L) were analyzed with linear mixed-effects modeling with Tukey’s HSD post-test or Kaplan-Meier with log-rank test. All error bars represent SEM.
See also Figure S5.
We next asked whether reduced growth of DKO tumors reflects tumor cell autonomous effects or anti-tumor immune responses. DKO cells did not demonstrate a significant proliferation defect in vitro (Figure S5H). Depletion of either CD4+ or CD8+ T cells allowed for rapid growth of DKO tumors in vivo, demonstrating a role of T cells (Figure 5E). Additionally, DKO tumors grew significantly faster in Batf3 KO mice (deficient in cDC1s) compared to WT controls, suggesting that anti-tumor immune responses upon RA reduction required the presence of cDC1s that cross-present tumor antigens to prime CD8+ T cells (Figures 5F and S5I).
Next, we examined the tumor-specificity of T cell responses induced by RA-deficiency by expressing the model antigen ovalbumin (OVA) in Cas9 control and DKO FS cells. We found a higher frequency and number of OVA (hence tumor)-specific splenic CD8+ T cells in mice harboring DKO-OVA tumors (Figure 5G). Consistent with published data, parental (control) FS were poorly responsive to αPD1 monotherapy (Figure 5H) (Gubin et al., 2018). Strikingly, administration of αPD1 in established DKO tumors lead to significant tumor regression (Figures 5H and 5I). Mice that experienced complete regression of DKO tumors rejected re-challenge with parental FS but not unrelated tumor cells (Figure 5J). Finally, reducing RA levels in the primary tumor (Raldh1/3 DKO) enhanced the sensitivity to αPD-1 in a distant (contralateral) Cas9 Control tumor, providing additional evidence of enhanced anti-tumor T cell immunity upon tumor RA reduction (Figures 5K–5M). Together, these results show that reducing tumor RA production engenders a strong anti-tumor T cell response that synergizes with checkpoint blockade to control murine sarcoma.
Inhibiting RAR Signaling in TME Enhances Anti-tumor Immune Responses
Although pharmacologic approaches to specifically inhibit Raldh enzymes are not available, potent antagonists of RAR signaling exist (Chiba et al., 2016). We first asked whether RAR signaling blockade could recapitulate the aforementioned effects of reducing tumor RA production. As RAR antagonism has pleiotropic effects on myelopoesis and peripheral immunity, we restricted RA-signaling blockade to the TME via intratumoral administration of BMS493. The results were similar to those observed in RA-deficient DKO tumors, including decreased frequency of TAMs, increased activation markers on APCs, and increased frequency of T cells (Figures 6A–6C). Strikingly, intratumoral BMS493 robustly synergized with systemic αPD1 therapy in FS (Figures 6D and S6A). Of interest, Raldh1 and Raldh3 were significantly upregulated in tumors treated with BMS493, suggesting a potential resistance mechanism whereby tumors upregulate RA production in response to RA signaling blockade (Figure S6B). However, administration of BMS493 to DKO tumors (in which this resistance mechanism cannot operate) did not show superior response compared to control, suggesting the existence of other resistance mechanisms (Figure S6C).
Figure 6. Intratumoral RAR Signaling Inhibition Engenders Stimulatory APCs and Synergizes with PD-1 Blockade.
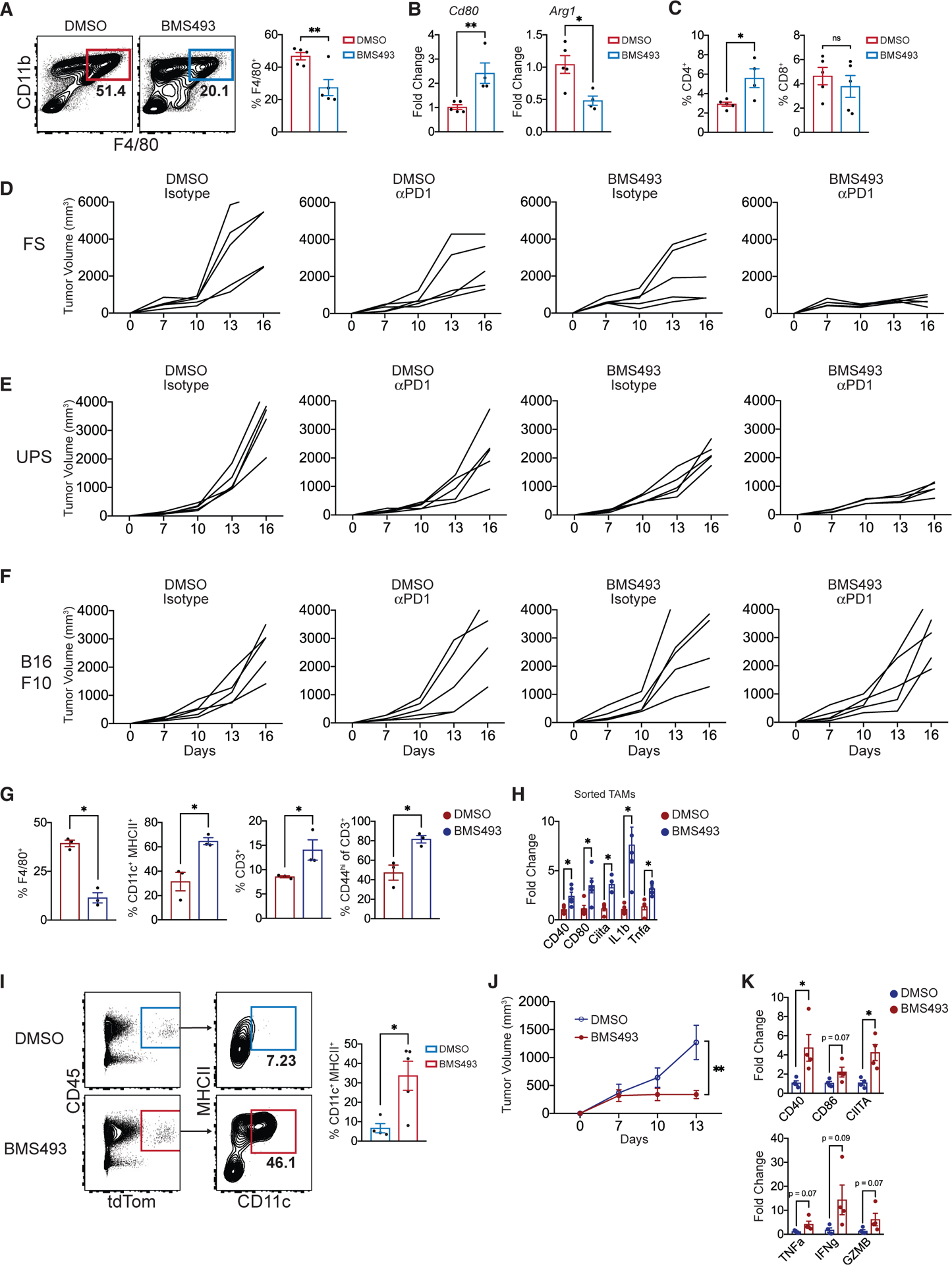
(A) Frequency of CD11b+ F4/80+ TAMs in FS tumors treated with BMS493 or DMSO. Three doses (200 mg, intratumorally) at days 7, 10, and 13 (n = 5 tumors per group, harvested 15 days post-transplant).
(B) Cd80 and Arg1 expression (qPCR) in FS tumors treated with intratumoral BMS493 or DMSO (n = 5 tumors per group).
(C) Frequencies of CD4+ or CD8+ T cells within CD45+ leukocytes in FS tumors treated with intratumoral BMS493 or DMSO (n = 5 tumors per group).
(D–F) Individual growth curves of FS (D), UPS (E), and B16-F10 melanoma (F) tumors treated with αPD1 (or isotype control) in combination with intratumoralBMS493 (or DMSO). BMS493 (200 mg intratumorally) and/or αPD1 (200 mg i.p.) were administered at days 7, 10, and 13 (n = 5 tumors per group).
(G) Mice with established SS tumors (autochthonous model) were treated with DMSO or BMS493 i.p. (3 doses of 200 mg at days 1, 3, and 5; mice euthanized on day 7). Shown are frequencies of specified myeloid and lymphoid populations in SS tumors (n = 3 per group).
(H) Relative expression of Cd40, Cd80, Ciita, Il1b, and Tnfa in sorted TAMs from DMSO- or BMS493-treated SS tumors (n = 3 per group).
(I) Monocytes from bone marrow of Lyz2Cre: Rosa26tdT mice were treated with either DMSO or BMS493 (1 mM) for 1 h and washed twice in PBS. Subsequently, 5 × 105 monocytes were injected directly into FS flank tumors (three injections; 7 days, 9 days, and 11 days post- transplant). FCM plots of tdT+ cells (derived from transplanted monocytes) and frequencies of CD11c+ MHCII+ within the tdT+ fraction at 13 days post-tumor implantation.
(J) Growth of FS tumors injected with DMSO- or BMS493-treated monocytes. Tumor volume was measured every 3days starting at 7 days post-implantation (n = 4 tumors per group, representative of three independent experiments).
(K) Relative expression of Cd40, Cd86, Ciita, Tnfa, Ifng, and Gzmb in bulk FS tumors transplanted with either DMSO- or BMS493-treated monocytes. Normalized to Hprt expression.
*p < 0.05, **p < 0.01, ***p < 0.001. Two-tailed t test. (J) was analyzed with linear mixed-effects modeling with Tukey’s HSD post-test. All error bars represent SEM. Events shown in FCM plots are pregated on live singlets unless otherwise specified and numbers represent percentage of cells within indicated gates.
See also Figure S6.
We also tested intratumoral RAR inhibition on transplanted UPS cells, finding similar synergy between RAR blockade and αPD1 therapy (Figures 6E and S6D). In contrast to our findings in FS and UPS, RAR blockade with or without αPD1 did not show any effect in B16-F10 melanoma, a tumor where we found little to no RA production (Figures 6F and S6E). The aforementioned effects of RAR signaling inhibitors raise exciting translational possibilities in RA-rich tumors but were shown in syngeneic transplant-based systems with intratumoral injections. Hence, we tested the effects of systemic delivery (intraperitoneal) of BMS493 in autochthonous mouse models of SS and UPS. Similar to transplantation-based models, BMS493 increased stimulatory myeloid cells and T cells within both types of tumors (Figures 6G, 6H, S6F, and S6G).
Thus far, we show that reducing RA in TME promotes monocyte differentiation into immunostimulatory APCs and enhances anti-tumor T cell responses. To further examine the link between these two observations, we devised a monocyte-transplant experiment whereby bone marrow monocytes from LysMCre: Rosa26tdT mice were treated ex vivo with DMSO (control) or BMS493 (irreversible RAR blockade), washed, and transplanted into syngeneic FS tumors. The fate of the transplanted monocytes and their impact on tumor growth were monitored. In contrast to control monocytes, a significant fraction of BMS493-treated monocytes differentiated into CD11c+ MHCII+ DCs in the TME (Figure 6I). Concomitantly, tumors transplanted with BMS493-treated monocytes grew at a significantly slower rate and displayed an immunostimulatory TME with evidence of activated APCs and T cells (Figures 6J and 6K).
Taken together, these findings provide proof-of-concept for targeting RA-RAR signaling in solid tumor immunotherapy.
Human Tumors Display Evidence of RA-Mediated Immunosuppression
RA producing enzymes are known to be elevated in many human cancers (including breast, lung, colon, prostate) and have been associated with poor treatment response and worse survival (Khoury et al., 2012; Li et al., 2010, 2014; Marcato et al., 2015; Wei et al., 2015; Zhang et al., 2013). We found that human UPS expressed higher levels of RALDH3 compared to non-malignant skeletal muscle (Figure 7A). Consistent with murine UPS, ALDEFLUOR analysis of human UPS showed that a subset of CD45− cells produced the majority of RA (Figure 7B). CD45− cells of human SS also expressed significantly higher levels of RALDH2 and RALDH3 compared to CD45+ cells (Figure 7C). Further, analysis of The Cancer Genome Atlas (TCGA) mRNA data revealed that RALDH isoforms are expressed in many types of cancers with RALDH1 and RALDH3 generally being more highly expressed than RALDH2 (Figure S7A). Expression of RALDH1 or RALDH3 was negatively correlated with survival in multiple types of human cancer (Figure S7B). In sarcoma, sub categorization by Raldh expression was not predictive of poor clinical outcome, but interpretation of these analyses is significantly hampered by small sample sizes and heterogeneity in sarcoma clinical datasets and will require additional investigation (Figure S7C).
Figure 7. Monocyte RA-Responsive Gene Signature Correlates with an Immunosuppressive TME in Human Cancer.
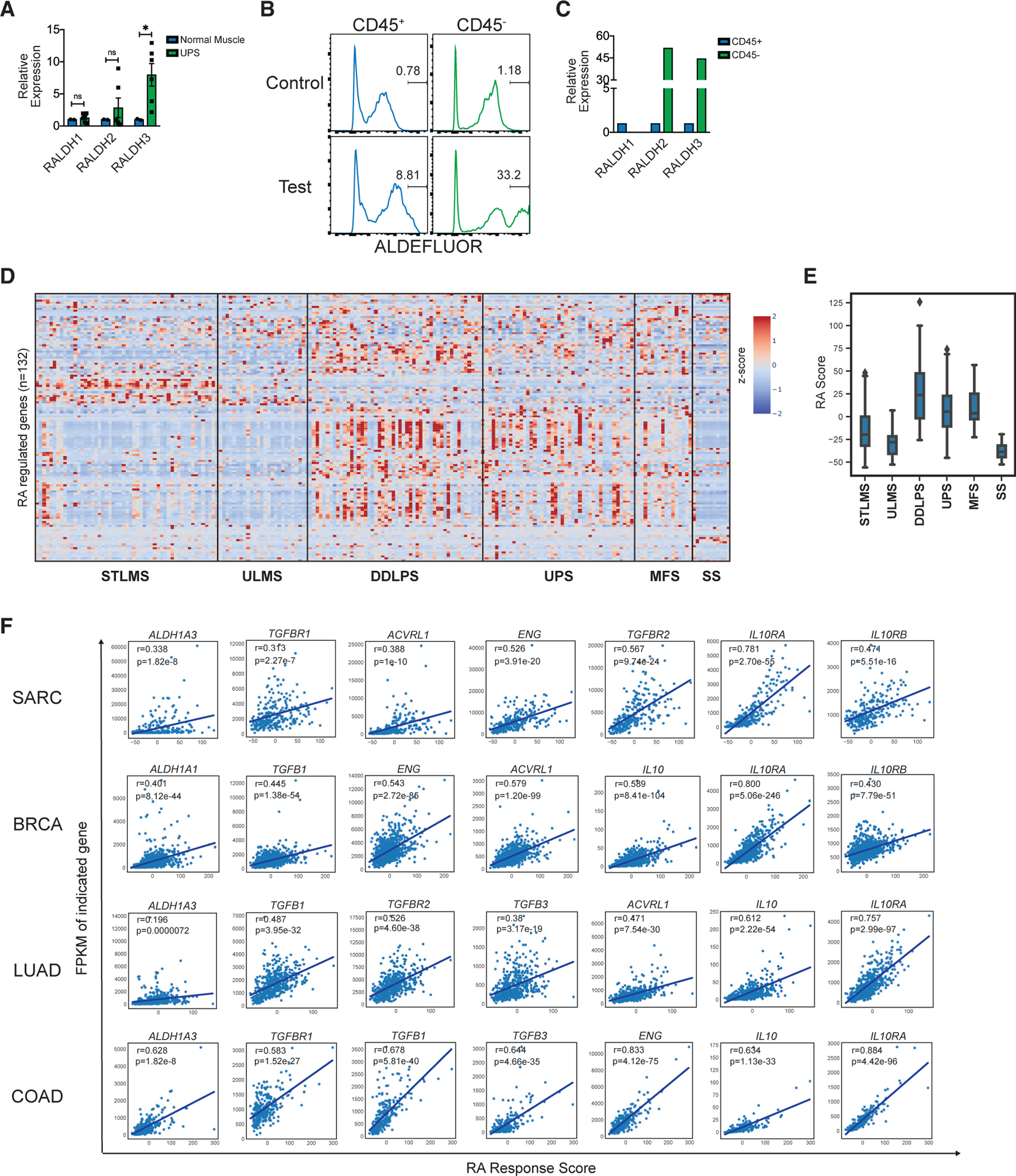
(A) RALDH1, RALDH2, and RALDH3 expression (qPCR) in human UPS compared to human skeletal muscle. Each dot represents tissue sampled from a different location of tumor (two tissue samples from n = 3 tumors).
(B) ALDEFLUOR assay on human UPS. Shown are histograms of “control” and “test” samples pregated on CD45+ (left) or CD45− cells (right). Representative of 2 human UPS.
(C) Relative expression of RALDH1, RALDH2, and RALDH3 (qPCR) in sorted CD45+ compared to CD45− cells from human synovial sarcoma (SS). Representative of 2 human SS. Normalized to Hprt.
(D) Heatmap of Z scores of human monocyte RA regulated genes (n = 132; y axis) for each tumor sample (n = 259; x axis) in TCGA SARC (sarcoma) dataset. SARC dataset was further subcategorized into soft tissue leiomyosarcoma (STLMS), uterine leiomyosarcoma (ULMS), dedifferentiated liposarcoma (DDLPS), undifferentiated pleomorphic sarcoma (UPS), myxofibrosarcoma (MFS), and synovial sarcoma (SS). Human monocyte RA regulated gene list was obtained by analyzing microarray data of human monocytes cultured with GM-CSF and IL-4 treated with RA (20 nM) versus DMSO. The names of individual genes on the y axis have been listed in Figure S7. Additional details in Figure S7 and STAR Methods.
(E) Boxplots of “RA response score” for samples in SARC TCGA dataset (grouped as described in D). “RA response score” for each tumor sample was calculated by summing over the Z score for all RA-regulated genes.
(F) “RA response score” plotted against fragments per kilobase of exon model per million reads mapped (FPKM) values for each tumor sample for the indicated genes. Pearson’s correlation analysis to determine r and p values. BRCA, breast invasive carcinoma; LUAD, lung adenocarcinoma; COAD, colon adenocarcinoma. The individual p value for each correlation is shown within each plot.
*p < 0.05. Two-tailed t test. All error bars represent SEM. Events shown in FCM plots are pregated on live singlets unless otherwise specified and numbers represent percentage of cells within indicated gates.
See also Figure S7.
To identify human tumors where high levels of RA may drive myeloid mediated immunosuppression, we generated an unbiased list of RA-regulated genes from our microarray-based RA-treated human monocyte gene expression profile (Figure S7D). We queried for the expression of these genes in the TCGA mRNA database and determined an “RA response score” for individual tumor samples. In TCGA sarcoma dataset, we found that the RA score clustered uniquely within certain sarcoma subtypes, with DDLPS, UPS, and MFS having higher average RA scores compared to STLMS, ULMS, and SS (Figures 7D and 7E). The RA score was significantly predictive of survival in ULMS (Figure S7E). Further, the RA score (compared to a “random score”) was enriched in a group of samples across all cancer types in TCGA, suggesting that a subset of human tumors may experience heightened RA signaling (Figure S7F). The RA score was significantly correlated with RALDH3 expression, providing transcriptional evidence that this score is likely reflective of higher tumor RA (Figure 7F). Notably, the RA score was significantly correlated with multiple members of transforming growth factor β (TGF-β) and IL-10 signaling pathways, which are known to drive immunosuppression in cancer (Figure 7F). This correlation was not restricted to sarcoma and included several common tumor types (Figure 7F). Hence, similar to murine sarcomas, RA may drive immunosuppression in many distinct human solid tumors.
In summary, our study identifies an RA-dependent immune evasion pathway in solid tumors, provides proof of concept for targeting this pathway for solid tumor immunotherapy, and describes a method to potentially identify human tumors in which this pathway is active and hence amenable to RA-targeted therapeutic approaches.
DISCUSSION
The immunosuppressive TME prevents spontaneous and treatment-induced anti-tumor immune responses. Myeloid cells are key drivers of immunosuppression and the molecular basis of immunosuppressive myeloid cell function in TME has begun to be elucidated. Some examples include PI3Kγ signaling driving anti-inflammatory function of TAMs, lipid accumulation driving tumor DC dysfunction, and prostaglandin E2 driving suppressive neutrophil functions (Cubillos-Ruiz et al., 2015; Kaneda et al., 2016; Veglia et al., 2019). Here, we describe an RA-dependent mechanism affecting myeloid differentiation that promotes an immunosuppressive TME and tumor immune escape. Previous work has demonstrated the importance of Mafb and Irf4 in controlling monocyte fate commitment into macrophages and DCs, respectively (Goudot et al., 2017). Our work suggests that tumors target this transcriptional circuitry to increase TAMs and reduce DCs. Hence, repolarizing the immune TME from a suppressive to stimulatory milieu may benefit from targeting both dysfunction and differentiation of myeloid cells in the TME.
RA is a well-studied signaling molecule and a powerful morphogen with important roles in the development and function of various cells. Solid tumors represent a complex niche comprising of many different cell types besides malignant cells. Hence, the overall impact of RA signaling on solid tumors is likely a sum total of its effects on the constituent cells of the TME. RA has been shown to have “pro-differentiation” effects on several types of tumor cells and hence can be considered anti-tumor in this context. Indeed, this has been the dominant theme in the field of RA cancer research. In contrast, we show here that RA drives anti-inflammatory effects in immune cells and can be considered pro-tumor in this context. Therefore, targeting RA signaling for tumor therapy requires a clear understanding of its role in specific tumor types as well as selective targeting of the appropriate cell type within TME. We have attempted to identify tumor types where the RA-dependent myeloid pathway may play a role in immune evasion by computing a monocyte “RA response score.” The overarching intent is to identify appropriate patient populations for potential clinical trials based on RA-targeting for tumor immunotherapy.
DCs can originate from HSC-derived DC precursors (cDCs) or from monocytes (moDCs). In this study, we primarily focused on the effects of RA on moDCs, given that monocytes are vastly more abundant than cDC precursors and continuously infiltrate solid tumors. Nonetheless, previous studies have examined the role of RA on splenic and intestinal cDC development, finding that RA can actually promote the development of cDC2s (Klebanoff et al., 2013). Although outside the scope of the current manuscript, we also observed that RA did not suppress cDC development in FLT3 culture systems, in contrast to its effects on moDC development (not shown). This suggests that the effects of RA on DCs may depend on ontogeny.
Previous studies have shown links between Th2 cytokine milieu and immunosuppressive TME, although the underlying pathways are not fully understood (Fridman et al., 2012; McCormick and Heller, 2015; Suzuki et al., 2015; Zou, 2005). RA induction by IL-13 described here provides one mechanistic basis for this observation. Notably, both IL-4 and IL-13 play a role in the differentiation of monocytes into DCs and M2-polarized macrophages. Induction of RA via these cytokines blocks DC differentiation while allowing M2 macrophages to develop from monocytes, representing an interesting pathway by which tumor cells sense and respond to “immune pressure.” However, additional factors likely regulate RA production in solid tumors. It is also unclear whether tumor cells are the only RA-producing cells within the TME of all tumor types. Given the pro-differentiation effects of RA, other non-malignant cells of TME, such as fibroblasts, may be the primary source of RA in some tumors. We anticipate future work to clarify these issues, which will help develop RA-targeted approaches for tumor immunotherapy.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for additional information about the manuscript or for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Malay Haldar (mhaldar@pennmedicine.upenn.edu). All unique reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Tumor models
Two GEMMs were used in these studies: Rosa26syt-ssx: Catnblox(e3) mice (generated as previously described in (Barrott et al., 2015) and KrasG12D: Trp53flox mice (a generous gift from Dr. David Kirsch) (Kirsch et al., 2007). Oncogenesis was initiated by local injection of Cre protein (TAT-Cre, Millipore) in the hind limb musculature to minimize immune response to virus. Syngeneic fibrosarcoma flank tumors were established in C57BL/6 mice purchased from Jackson Laboratories.
Mononuclear phagocyte models
Zbtb46GFP mice were a generous gift from Dr. Kenneth Murphy (Satpathy et al., 2012). Ccr2−/− mice were obtained from Jackson Laboratories and bred to KrasG12D: Trp53flox mice. LysMCre mice were obtained from Jackson Laboratories and bred to 1) Rosa26LSL-tdT 2) Irf4flox or 3) Rosa26Cas9-eGFP mice. Batf3−/− mice were obtained from Jackson Laboratories and/or bred in house for experimental use.
Sex (both male and female) and aged matched mice between 6–8 weeks were used for these studies. Mice were bred and maintained in specific pathogen free facilities at the University of Pennsylvania. All animal procedures were conducted according to National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Tumor cells
C57BL/6 syngeneic fibrosarcoma cell line was a generous gift from Dr. Robert Schrieber at Washington University in St. Louis and was used for experimentation as previously described (Gubin et al., 2018). KrasG12D: Trp53flox sarcoma cell line was a generous gift from Dr. Sandra Ryeom and B16-F10 melanoma cell line was a generous gift from Dr. Andy Minn. Tumor cell lines were cultured in DMEM with 10% FBS, 1% Pen/Strep and 2mM glutamine. Low passage (< P15) cell lines were used for in vitro and in vivo experimentation. All cells were confirmed to be negative for mycoplasma contamination as assessed by MycoAlert Mycoplasma Detection Kit (Lonza).
Human samples
Human undifferentiated pleomorphic sarcoma, synovial sarcoma, or non-malignant muscle samples were obtained from surgically resected tumors from patients (de-identified) undergoing therapeutic surgical resection in accordance with protocol approved by the Institutional Review Board at the University of Pennsylvania. Single cell suspensions from human tumor samples were generated using methods described in sections below. Similarly, ALDEFLUOR assay, cell sorting and qPCR analyses were performed as described below.
METHOD DETAILS
“RA response score” computation and TCGA analysis
We performed differential expression analysis, using DESeq2, on microarray data of human monocytes treated with RA or DMSO. Genes up or downregulated by at least three-fold (with false discovery rate q-value < 0.05) were collected and assigned to the RA response gene signature that contained a total of 146 genes. For analysis of TCGA mRNA datasets, only genes that were expressed with a FPKM value > 0.5 across all patient samples in a cancer group were retained. This reduced the total number of genes in the RA response gene signature to n = 132 for the SARC, BRCA, LUAD, and COAD datasets.
From the TCGA mRNA expression data, we computed the FPKM z-score for the n = 132 RA response signature genes and hierarchically ranked them as shown in Figure 7D. The RA response score for every patient sample, in a given cancer, for a given gene set, was taken to be the z-score summed over all genes. In order to evaluate the heterogeneity in the RA response score, we also computed a random response score employing a randomly generated gene set (n = 132 genes) as described above. To identify the genes in a specific cancer that may be regulated by the RA response score, we performed a genome-wide correlation between the patient RA response score and the corresponding FPKM values and quantified the degree of correlation using a linear regression model.
Additionally, Kaplan-Meier analyses based Raldh mRNA substratification in selected TCGA datasets was performed using UALCAN (Chandrashekar et al., 2017). High expression indicates the top quartile, while low expression indicates the bottom quartile.
Microarray analyses
Microarray services were provided by the UPENN Molecular Profiling Facility, including quality control tests of the total RNA samples by agilent bioanalyzer and nanodrop spectrophotometry. All protocols were conducted as described in the Affymetrix WT Pico Reagent Kit Manual and the Affymetrix GeneChip Expression Analysis Technical Manual. Gene expression data were normalized and values modeled using ArrayStar4 (DNASTAR). The data is available for download at the Gene Expression Omnibus (GEO) through accession number GSE144612 (reference series for all microarray experiments associated with this manuscript).
Implantation of tumor cells, tumor growth measurements and survival analyses
Cultured tumor cells were detached using 0.05% trypsin (GIBCO), washed once with DMEM media and once with 1x PBS, and counted in preparation for implantation. Tumor cells were propagated in vitro for two passages prior to implantation and injected cells were greater than 90% viable. 1 × 106 tumor cells were implanted subcutaneously (s.c.) into shaved flanks of recipient mice. Tumor dimensions were measured using a caliper starting at Day 7 and every three days thereafter; volume was calculated by using formula (ab2)π/2, where a is the longest measurement and b is the shortest. Tumor volumes of 1000mm3 were used as endpoints for survival analyses. Waterfall plots were generated by comparing tumor volume at the start of treatment to tumor volume 11d later. For re-challenge experiments with tumor cell lines, tumor cells were injected into mice cured of primary tumors for > 60 days.
Flow cytometry of murine samples
Tumors were harvested and minced at indicated time points post-implantation for analysis. Single cell suspensions were generated by digestion with collagenase B and DNase I for 45 minutes at 37°C and filtration through 70uM cell strainer. Mouse blood was collected in EDTA tubes and RBCs were lysed using ACK lysing buffer. Samples were incubated for 5 minutes at room temperature with anti-mouse CD16/32 Fc Block, and subsequently stained on ice with primary-fluorophore conjugated antibodies for identification of cell populations by FACS. 7AAD (BioLegend) was used for dead cell discrimination. Flow cytometry was performed on an LSR II Flow Cytometer (BD Biosciences) and analyzed using FlowJo software (Treestar). List of antibodies can be found in table above.
T cell IFNγ ex vivo assay
Single cell suspensions generated from mouse tumors were incubated for 4 hours at 37°C with PMA (50ng/mL; Sigma), Ionomycin (750ng/mL; Sigma) and GolgiStop (5ug/mL; BD). Intracellular staining was performed using a fixation/permeabilization kit (eBiosciences) according to manufacturer’s instructions.
In vitro tumor cell proliferation assay
After tumor cells were propagated for two weeks in vitro, 5 × 104 of indicated tumor cell lines were plated in triplicate. Viable and nonviable cell numbers were counted each day for five days.
ALDEFLUOR assay
The ALDEFLUOR assay (STEMCELL Technologies) was performed according to manufacturer’s instructions to identify cells with Aldh activity. In brief, single cell suspensions generated from indicated tumors were incubated with a fluorescently tagged substrate of the Aldh enzyme; the fluorescent product accumulates in cells proportional to their Aldh activity. DEAB, a potent inhibitor of Aldh, was used as a negative control for each sample. Cells were subsequently stained with surface antibodies and fluorescence was quantified by flow cytometry. Gates for Aldh+ cells were drawn relative to baseline fluorescence as determined by DEAB negative control. To avoid spectral overlap, ALDERED assay (STEMCELL Technologies) was performed in lieu of ALDEFLUOR assay for samples with endogenous GFP fluorescence.
Cell sorting
Cells were sorted on MoFlo Astrios or FACS Jazz at the Children’s Hospital of Philadelphia (CHOP) Flow Cytometry Core Laboratory. For measurement of gene expression in sorted cells, RNA was isolated from sorted cell pellets and qPCR performed as described elsewhere. For T cell suppression assays, tumor APCs were sorted into complete RPMI media and T cell suppression assay performed as described elsewhere.
LC-MS for ATRA
All-trans retinoic acid was extracted from snap frozen mouse tumors or normal muscle as described (Kane et al., 2008). Quantification of ATRA was performed by the Children’s Hospital of Philadelphia Metabolomics Core using liquid chromatography tandem mass spectrometry (LC-MS).
Bone marrow monocyte isolation
Monocytes were isolated from bone marrow of indicated mice using the Mouse BM Monocyte Isolation Kit (Miltenyi Biotec) according to manufacturer’s instructions. Viability and purity of negative selection monocyte isolation was assessed by flow cytometry to be > 90%.
Intratumoral monocyte transfer
Monocytes were isolated (as described above) from mouse bone marrow of LysMCre: Rosa26tdT: Zbtb46GFP mice. Subsequently, 5 × 105 monocytes were resuspended in 50uL 1x PBS and injected directly into established FS flank tumors at 7 days post implantation. Tumors were harvested at specified time points and analyzed by flow cytometry to track tdT+ monocytic progeny.
In vitro and ex vivo mouse and human monocyte differentiation assays
Mouse monocytes (isolated from bone marrow or sorted from LysMCre: Rosa26tdT: Zbtb46GFP tumors) were cultured with GM-CSF (20ng/mL) & IL-4 (20ng/mL), GM-CSF & IL-13 (20ng/mL), GM-CSF alone, or M-CSF alone (20ng/mL). Normal donor human monocytes (obtained the Human Immunology Core at the University of Pennsylvania were cultured with GM-CSF (50ng/mL) & IL-4 (50ng/mL), or M-CSF (50ng/mL). Murine or human cytokines were purchased from PeproTech. RA (100nM for mouse and 20nM for human monocytes) or DMSO was added at specified time points for indicated differentiation assays. Cellular identity and function of differentiated monocytes was assessed by a combination of unbiased transcriptional analyses, flow cytometry based protein analyses, and functional T cell suppression assays.
Mouse and human T cell suppression assays
Mouse splenic T cells were isolated from a non-tumor bearing C57BL/6 mouse using Pan T Cell Isolation Kit (Miltenyi Biotec). Viability and purity of negative selection T cell isolation was assessed by flow cytometry to be > 95%. Normal donor human T cells were obtained from the Human Immunology Core at the University of Pennsylvania. 8 3 104 mouse or human T cells were labeled with CFSE and cultured for 3 days at 37°C with 2uL of αCD3/28 beads (Dynabeads Mouse or Human T-Activator CD3/28, GIBCO) along with 30U recombinant human IL-2. Mononuclear phagocytes generated under various in vitro differentiation conditions (or sorted from mouse tumors) were co-cultured with stimulated T cells. T cell proliferation and activation was measured by flow cytometry to quantify mononuclear phagocyte suppressive ability.
Depletion of CD4+ or CD8+ T cells in vivo
200ug of clone GK1.5 (CD4+ T cell depletion), clone 2.43 (CD8+ T cell depletion), or clone LTF-2 (isotype control) antibody was administered i.p. starting three days prior to tumor implantation and repeated every three days until mouse sacrifice. All antibodies were purchased from BioXCell. CD4+ and CD8+ depletion was confirmed in peripheral blood and within tumors by flow cytometry.
In vivo reagents
200ug of αPD1 monoclonal blocking antibody (clone RMP 1–14, BioXCell) or isotype control antibody (clone 2A3, BioXCell) was administered i.p. at Days 7, 10 and 13 post tumor implantation. 200ug of BMS493 (Torcis) was administered intratumorally at Days 7, 10 and 13 post tumor implantation. BMS493 was dissolved in 10uL DMSO and diluted to a final volume of 50uL in 1x PBS for intratumoral injection. As a vehicle control, 10uL DMSO in a final volume of 50uL was injected intratumorally.
RNA isolation and qPCR analysis for gene expression
Total RNA was isolated using GenElute Mammalian Total RNA Miniprep Kit (Sigma). Reverse transcription was performed using High Capacity RNA to cDNA Kit (Life Technologies). qRT-PCR was performed using ViiA7 Real-Time PCR machine and TaqMan probes used for gene specific amplification (purchased from ThermoFisher Scientific) are listed below. Arg1 (Mm00475988_m1), Hprt (Mm03024075_m1), Tnfa (Mm00443258_m1), Il1b (Mm00434228_m1), Cd40 (Mm00441891_m1), Cd80 (Mm00441891_m1), Cd86 (Mm00444543_m1), Raldh1 (Mm00657317_m1), Raldh2 (Mm00501306_m1), Raldh3 (Mm00474049_m1), Irf4 (Mm00516431_m1), Zbtb46 (Mm00511327_m1), Raldh1 (Hs00946916_m1), Raldh2 (Hs00180254_m1), Raldh3 (Hs00167476_m1), Irf4 (Hs01056533_m1), Zbtb46 (Hs01008168_m1), Hprt (Cat #4333768).
CRISPR mediated gene deletion in tumor cells
LentiCRISPRv2 vector was a gift from Feng Zhang (Addgene plasmid #52961). In brief, the vector was transfected into 293T cells using polyethylenimine (PEI) along with lentivirus packaging plasmids. Lentivirus supernatant was collected 48 hours later and passed through a 40uM filter. Subsequently, tumor cells were transduced and selected on 3ug/mL of puromycin for two weeks. Clones were generated using single cell sorting and knockout efficiency determined by genomic sequencing and gene-specific qPCR analysis. CRISPR sequences were identified using mouse Geckov2 library (Feng Zhang): Raldh1: TAA-ATC-CGA-CAA-GTA-TGC-AT; Raldh3: TAC-TTA-CAG-CCA-GGA-TCG-CT; IL13Ra1: GAG-ACG-CTC-AAA-TTC-GTC-AC. In vivo knockout efficiency of Raldh1/3 DKO cell line was determined by qPCR for Raldh1 and Raldh3 gene expression in bulk tumor tissue, by ALDEFLUOR assay measuring functional enzymatic activity on tumor single cell suspension, and by liquid chromatography / mass spectrometry for all-trans retinoic acid in tumor tissue.
Overexpression of target genes in tumor cells
Raldh2-GFP ORF expression clone was purchased from GeneCopoeia and transfected into Cas9 Control or Raldh1/3 DKO FS cell lines using electroporation and nucleofection (Amaxa Nucleofector II, Lonza). After three days, cell lines were sorted based on GFP expression using a BD FACS Jazz instrument and subsequently cultured for two weeks in vitro prior to s.c. implantation. In vivo confirmation of overexpression was determined by qPCR for Raldh2 and maintenance of GFP expression in bulk tumor tissue.
OVA-encoding plasmid was a kind gift from Dr. Robert Vonderheide at UPenn. Raldh1/3 DKO or Cas9 Control FS tumor cell lines were transfected with plasmid encoding cytoplasmic OVA-ZsGreen. 3d later, cells were FACsorted for ZsGreen to ensure equal expression of OVA-ZsGreen between cell lines.
Single cell sequencing preparation
Cas9 Control or Raldh1/3 DKO tumors (n = 4 per group) were harvested on Day 11 (as described elsewhere) and CD45+ live cells were FACS sorted. Subsequently, 10x Genomics Controller and the v3 Library and Gel Bead kit (10x Genomics) were used to obtain single-cell emulsions. 10× 3′v3 kit protocol was followed as described to generate RNA sequencing libraries. The generated libraries were sequenced using an Illumina NovaSeq SP. The data are available for download at the Gene Expression Omnibus (GEO) through accession number GSE144507.
Single cell RNA sequencing analysis
Downstream analysis of the single-cell RNA-sequenced samples was performed using Seurat (v. 3.1.0) in R (v. 3.6.0). Genes expressed in less than three cells were removed. Cells that expressed less than 500 genes or had over 14% mitochondrial content were filtered out. The four samples were merged and counts for all genes were log2 normalized and scaled (NormalizeData and ScaleData). Principal components (PCs) were determined using the 2000 most variable genes (FindVariableFeatures). Subsequently, the top 50 PCs were used for graph-based cluster identification and dimensionality reduction by t-distributed stochastic neighbor embedding (t-SNE). Myeloid clusters were subsetted out using Adgre1, Lyz2, S100a9, Itgax and negative for lymphoid markers and re-analyzed as described above. Genes separating TAM_1 and TAM_2 clusters were determined using the default Wilcoxon Rank Sum test in the FindMarkers function.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was calculated between two groups by student’s unpaired t test. One-way ANOVA with Tukey’s HSD post- test was used to calculate statistical significance between multiple groups. Significance for survival was calculated by Kaplan-Meier with long-rank analysis. Analyses were performed using GraphPad Prism 8. Error bars represent SEM and p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001).
DATA AND CODE AVAILABILITY
The datasets generated in this study are available for download at the Gene Expression Omnibus (GEO) through accession numbers GSE144612 (microarray dataset) and GSE144507 (scRNaseq dataset).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD11b | Invitrogen | 25-0112-82 |
| Anti-mouse CD115 | BioLegend | 135531 |
| Anti-mouse CD64 | BD Biosciences | 558539 |
| Anti-mouse CD103 | BioLegend | 121406 |
| Anti-mouse Ly6G | BioLegend | 127633 |
| Anti-mouse CD11c | BioLegend | 117324 |
| Anti-mouse CD45 | BioLegend | 103138 |
| Anti-mouse MHCII | BioLegend | 107635 |
| Anti-mouse F4/80 | Invitrogen | MF48005 |
| Anti-mouse TREML4 | BioLegend | 143304 |
| Anti-mouse Ly6C | BioLegend | 128026 |
| Anti-mouse CD24 | BioLegend | 101814 |
| Anti-mouse CD44 | BioLegend | 103043 |
| Anti-mouse CD4 | BioLegend | 100437 |
| Anti-mouse CD8a | BioLegend | 100758 |
| Anti-mouse CD3e | BioLegend | 100351 |
| Anti-mouse IFNg | BioLegend | 505809 |
| Anti-human CD1a | BD Biosciences | 563938 |
| Anti-human CD11c | BD Biosciences | 340714 |
| Anti-human CD68 | eBioscience | 14-0688-82 |
| Anti-human CD45 | BioLegend | 304058 |
| Bacterial and Viral Strains | ||
| Stbl3 competent E. coli | ThermoFisher Scientific | C737303 |
| Critical Commercial Assays | ||
| MycoAlert Mycoplasma Detection Kit | Lonza | LT07–118 |
| Intracellular Fixation and Permeabilization Buffer Set | eBioScience | 88-8824-00 |
| ALDEFLUOR Assay | STEMCELLTechnologies | 01700 |
| AldeRed Assay | Sigma Aldrich | SCR150 |
| Monocyte Isolation Kit (BM), mouse | Miltenyi Biotec | 130-100-629 |
| Pan T cell Isolation Kit II, mouse | Miltenyi Biotec | 130-095-130 |
| CellTrace CFSE Cell Proliferation Kit | ThermoFisher Scientific | C34554 |
| Dynabeads Mouse T-Activator CD3/CD28 | ThermoFisher Scientific | 11452D |
| Dynabeads Human T-Activator CD3/CD28 | ThermoFisher Scientific | 11131D |
| GenElute Mammalian Total RNA Miniprep Kit | Sigma Aldrich | RTN-70 |
| Human Monocyte Nucleofector Kit | Lonza | VPA-1007 |
| Critical Reagents | ||
| TAT-CRE Recombinase | Millipore | SCR508 |
| Collagenase B | Sigma Aldrich | 11088815001 |
| DNase I | Sigma Aldrich | D4527 |
| RBC Lysis Buffer 10X | BioLegend | 420301 |
| 7AAD Viability Staining Solution | BioLegend | 420403 |
| PMA | Sigma Aldrich | P1585 |
| Ionomycin | Sigma Aldrich | I9657 |
| GolgiStop | BD Biosciences | 554724 |
| Recombinant Murine GM-CSF | PeproTech | 315–03 |
| Recombinant Murine M-CSF | PeproTech | 315–02 |
| Recombinant Murine IL-4 | PeproTech | 214–14 |
| Recombinant Murine IL-13 | PeproTech | 210–13 |
| Recombinant Human GM-CSF | PeproTech | 300–03 |
| Recombinant Human M-CSF | PeproTech | 300–25 |
| Recombinant Human IL-4 | PeproTech | 200–04 |
| Recombinant Human IL-2 | PeproTech | 200–02 |
| Retinoic Acid | Sigma Aldrich | R2625 |
| InVivoMAb anti-mouse CD4 Clone GK1.5 | BioXCell | BE0003-1 |
| InVivoMab anti-mouse CD8a Clone 2.43 | BioXCell | BE0061 |
| InVivoMab rat IgG2b isotype control Clone LTF-2 | BioXCell | BE0090 |
| InVivoMab anti-mouse PD-1 Clone RMP1–14 | BioXCell | BE0146 |
| InVivoMab rat IgG2a isotype control Clone 2A3 | BioXCell | BE0089 |
| BMS493 | Tocris | 3509 |
| LentiCRISPRv2 vector | Addgene | 52961 |
| Puromycin | InvivoGen | ANT-PR-1 |
| Raldh2-GFP ORF expression clone | GeneCopoeia | EX-Mm21038-M61 |
| Software and Algorithms | ||
| CRISPR Design | Broad Institute | http://zlab.bio/guide-design-resources |
| FlowJo | Treestar | https://flowjo.com |
| R language | R Core Team, 2019 The R Project for Statistical Computing | http://www.r-project.org |
| Seurat v. 3.1.0 | Satija Lab | https://satijalab.org/seurat |
| Graphpad Prism 8 | GraphPad | https://graphpad.com |
| Deposited Data | ||
| Microarray data | This study | GSE144612 |
| scRNaseq data | This study | GSE144507 |
| Experimental Models: Tumor Cells | ||
| C57BL/6 Fibrosarcoma | Robert Schrieber | N/A |
| KrasG12D: Trp53flox Sarcoma | Sandra Ryeom | N/A |
| B-16 F10 Melanoma | Andy Minn | N/A |
| Experimental Models: Mouse Strains | ||
| Rosa26syt-ssx: Catnblox(e3) | Barrott et al., 2015 | N/A |
| KrasG12D: Trp53flox | Kirsch et al., 2007 | N/A |
| KrasG12D: Trp53flox: Ccr2−/− | Generated | N/A |
| LysMCre: Rosa26LSL-tdT: Zbtb46GFP | Generated | N/A |
| LysMCre: Irf4flox | Generated | N/A |
| LysMCre: Rosa26Cas9-eGFP | Generated | N/A |
| Batf3−/− | Jackson Laboratories | 013755 |
| C57BL/6 | Jackson Laboratories | 000664 |
Highlights.
The tumor microenvironment induces tumor cells to produce retinoic acid
Retinoic acid skews monocyte differentiation toward macrophage rather than DC
Blocking retinoic acid production enhances anti-tumor T cell immunity
Pharmacological blockade of retinoic acid signaling synergizes with anti-PD-1 therapy
ACKNOWLEDGMENTS
We thank Drs. Brian Keith and Ivan Maillard for their scientific input and Dr. Michael Gonzalez for help with scRNA-seq. We thank Y. Daikhin, O. Horyn, and Ilanna Nissim for performing the ATRA measurements in the Metabolomics Core Facility, The Children’s Hospital of Philadelphia (CHOP). We also thank FCM cores at UPenn and CHOP, CAG Core at CHOP, and HIC core at UPenn. Thisworkwas supported by the NCI-R37CA234027(M.H.), Burroughs Wellcome CAMS (M.H.), the Concern Foundation (M.H.), the Slay Sarcoma Research Initiative (M.H.), the Sarcoma Program at UPenn (M.H.), and NIH F30CA236464 (S.D.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrott JJ, Illum BE, Jin H, Zhu J-F, Mosbruger T, Monument MJ, Smith-Fry K, Cable MG, Wang Y, Grossmann AH, et al. (2015). β-catenin stabilization enhances SS18-SSX2-driven synovial sarcomagenesis and blocks the mesenchymal to epithelial transition. Oncotarget 6, 22758–22766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Qin J, Bennett C, Qian S, Fung JJ, Hamilton TA, and Lu L (2014). All-trans retinoic acid induces arginase-1 and inducible nitric oxide synthase-producing dendritic cells with T cell inhibitory function. J. Immunol 192, 5098–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, Krois CR, Kenkel JA, Pham TD, Carmi Y, et al. (2016). Normalizing Microbiota-Induced Retinoic Acid Deficiency Stimulates Protective CD8(+) T Cell-Mediated Immunity in Colorectal Cancer. Immunity 45, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briseño CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, Durai V, Grajales-Reyes GE, Iwata A, Bagadia P, et al. (2016). Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte Derived Dendritic Cells. Cell Rep. 15, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, and Krummel MF (2015). The emerging understanding of myeloid cells as partners and targets in tumor rejection. Cancer Immunol. Res 3, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, and Varambally S (2017). UAL- CAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 19, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. [DOI] [PubMed] [Google Scholar]
- Chiba T, Skrypnyk NI, Skvarca LB, Penchev R, Zhang KX, Rochon ER, Fall JL, Paueksakon P, Yang H, Alford CE, et al. (2016). Retinoic Acid Signaling Coordinates Macrophage-Dependent Injury and Repair after AKI. J. Am. Soc. Nephrol 27, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. (2015). ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duester G (2008). Retinoic acid synthesis and signaling during early organogenesis. Cell 134, 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duester G (2017). Retinoic acid’s reproducible future. Science 358, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehnman M, and Larsson O (2015). Microenvironmental Targets in Sarcoma. Front. Oncol 5, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkelens MN, and Mebius RE (2017). Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol. 38, 168–180. [DOI] [PubMed] [Google Scholar]
- Fridman WH, Pagès F, Sautès-Fridman C, and Galon J (2012). The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306. [DOI] [PubMed] [Google Scholar]
- Ginestier C, Wicinski J, Cervera N, Monville F, Finetti P, Bertucci F, Wi-cha MS, Birnbaum D, and Charafe-Jauffret E (2009). Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 8, 3297–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudot C, Coillard A, Villani A-C, Gueguen P, Cros A, Sarkizova S, Tang-Huau T-L, Bohec M, Baulande S, Hacohen N, et al. (2017). Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages. Immunity 47, 582–596. [DOI] [PubMed] [Google Scholar]
- Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. (2018). High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 175, 1014–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundra UM, Girgis NM, Gonzalez MA, San Tang M, Van Der Zande HJP, Lin J-D, Ouimet M, Ma LJ, Poles J, Vozhilla N, et al. (2017). Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat. Immunol 18, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, and Murphy KM (2014). Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev 262, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, Hancock JD, Coffin CM, Lessnick SL, and Capecchi MR (2007). A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell 11, 375–388. [DOI] [PubMed] [Google Scholar]
- Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, and Reis e Sousa C (2015). GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity 42, 1197–1211. [DOI] [PubMed] [Google Scholar]
- Hill JA, Hall JA, Sun C-M, Cai Q, Ghyselinck N, Chambon P, Belkaid Y, Mathis D, and Benoist C (2008). Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 29, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C-J, Hong CY, Takei M, Chung S-Y, Park J-S, Pham T-NN, Choi S-J-N, Nam J-H, Chung I-J, Kim H-J, and Lee JJ (2010). All-trans retinoic acid inhibits the differentiation, maturation, and function of human monocyte-derived dendritic cells. Leuk. Res 34, 513–520. [DOI] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Wang C, and Napoli JL (2008). Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem 80, 1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, et al. (2016). PI3Kg is a molecular switch that controls immune suppression. Nature 539, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury T, Ademuyiwa FO, Chandrasekhar R, Jabbour M, Deleo A, Ferrone S, Wang Y, and Wang X (2012). Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Mod. Pathol 25, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, et al. (2007). A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat. Med 13, 992–997. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, Sukumar M, Muranski P, Scott CD, Hall JA, et al. (2013). Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J. Exp. Med 210, 1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Sandell LL, Trainor PA, Koentgen F, and Duester G (2012). Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta 1821, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoui D, Van Overmeire E, Movahedi K, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, and Van Ginderachter JA (2011). Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology 216, 1192–1202. [DOI] [PubMed] [Google Scholar]
- Lehtonen A, Veckman V, Nikula T, Lahesmaa R, Kinnunen L, Matikainen S, and Julkunen I (2005). Differential expression of IFN regulatory factor 4 gene in human monocyte-derived dendritic cells and macrophages. J. Immunol 175, 6570–6579. [DOI] [PubMed] [Google Scholar]
- Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, Stass SA, and Jiang F (2010). ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Invest 90, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XS, Xu Q, Fu XY, and Luo WS (2014). ALDH1A1 overexpression is associated with the progression and prognosis in gastric cancer. BMC Cancer 14, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcato P, Dean CA, Liu R-Z, Coyle KM, Bydoun M, Wallace M, Clements D, Turner C, Mathenge EG, Gujar SA, et al. (2015). Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol. Oncol 9, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick SM, and Heller NM (2015). Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 75, 38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, Patel R, Gautier EL, Hugues S, Longhi MP, et al. (2016). The Heterogeneity of Ly6Chi Monocytes Controls Their Differentiation into iNOS+ Macrophages or Monocyte-Derived Dendritic Cells. Immunity 45, 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, and Mortha A (2013). The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, and Jung S (2014). Development and function of dendritic cell subsets. Immunity 40, 642–656. [DOI] [PubMed] [Google Scholar]
- Mohty M, Morbelli S, Isnardon D, Sainty D, Arnoulet C, Gaugler B, and Olive D (2003). All-trans retinoic acid skews monocyte differentiation into interleukin-12-secreting dendritic-like cells. Br. J. Haematol 122, 829–836. [DOI] [PubMed] [Google Scholar]
- Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, and Gabrilovich DI (2007). Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 67, 11021–11028. [DOI] [PubMed] [Google Scholar]
- Okabe Y, and Medzhitov R (2014). Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157, 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olingy CE, Dinh HQ, and Hedrick CC (2019). Monocyte heterogeneity and functions in cancer. J. Leukoc. Biol 106, 309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DM, Hettinger J, and Feuerer M (2013). Monocytes and macrophages in cancer: development and functions. Cancer Microenviron. 6, 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Remark R, Gnjatic S, and Merad M (2019). Host tissue determinants of tumour immunity. Nat. Rev. Cancer 19, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM (2012). Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med 209, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber TH, and Podack ER (2009). A critical analysis of the tumour immunosurveillance controversy for 3-MCA-induced sarcomas. Br. J. Cancer 101, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Leland P, Joshi BH, and Puri RK (2015). Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine 75, 79–88. [DOI] [PubMed] [Google Scholar]
- Tang X-H, and Gudas LJ (2011). Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol 6, 345–364. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, and Ladanyi M (2011). Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 11, 541–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, and Gabrilovich DI (2017). Dendritic cells in cancer: the role revisited. Curr. Opin. Immunol 45, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. (2019). Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 569, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellozo NS, Pereira-Marques ST, Cabral-Piccin MP, Filardy AA, Ribeiro-Gomes FL, Rigoni TS, DosReis GA, and Lopes MF (2017). All-Trans Retinoic Acid Promotes an M1- to M2-Phenotype Shift and Inhibits Macrophage-Mediated Immunity to Leishmania major. Front. Immunol 8, 1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Peng J-J, Gao H, Zhang T, Tan Y, and Hu Y-H (2015). ALDH1 Expression and the Prognosis of Lung Cancer: A Systematic Review and Meta-Analysis. Heart Lung Circ 24, 780–788. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yan W, You G, Bao Z, Wang Y, Liu Y, You Y, and Jiang T (2013). Genome-wide DNA methylation profiling identifies ALDH1A3 promoter methylation as a prognostic predictor in G-CIMP-primary glioblastoma. Cancer Lett. 328, 120–125. [DOI] [PubMed] [Google Scholar]
- Zou W (2005). Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 5, 263–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated in this study are available for download at the Gene Expression Omnibus (GEO) through accession numbers GSE144612 (microarray dataset) and GSE144507 (scRNaseq dataset).