

Abstract
Accumulating evidence suggests that sporadic Parkinson's disease has a long prodromal period during which several non‐motor features develop, in particular, impairment of olfaction, vagal dysfunction and sleep disorder. Early sites of Lewy pathology are the olfactory bulb and enteric plexus of the stomach. We propose that a neurotropic pathogen, probably viral, enters the brain via two routes: (i) nasal, with anterograde progression into the temporal lobe; and (ii) gastric, secondary to swallowing of nasal secretions in saliva. These secretions might contain a neurotropic pathogen that, after penetration of the epithelial lining, could enter axons of the Meissner's plexus and, via transsynaptic transmission, reach the preganglionic parasympathetic motor neurones of the vagus nerve. This would allow retrograde transport into the medulla and, from here, into the pons and midbrain until the substantia nigra is reached and typical aspects of disease commence. Evidence for this theory from the perspective of olfactory and autonomic dysfunction is reviewed, and the possible routes of pathogenic invasion are considered. It is concluded that the most parsimonious explanation for the initial events of sporadic Parkinson's disease is pathogenic access to the brain through the stomach and nose – hence the term ‘dual‐hit’.
Keywords: aetiology, autonomic regulation, olfaction, Parkinson's disease
Introduction
Sporadic Parkinson's disease (PD) is the most frequent degenerative disorder of the human nervous system after Alzheimer's disease. It is not known to occur spontaneously in other vertebrates and does not directly affect other organs apart from the nervous system. Motor dysfunction (hypokinesia, postural imbalance, cogwheel rigidity, resting tremor) indicates the presence of disease, but also can appear in the guise of ‘Parkinsonism’ in other disorders that are associated with a significant reduction of dopamine in the central nervous system (CNS). Parkinsonism may develop as a sequel to intoxication, trauma, vascular disease and infections. There are genetically based familial forms and degenerative or sporadic forms, the last of which include the tauopathy progressive supranuclear palsy (PSP) and synucleinopathies such as multiple system atrophy (MSA) and Lewy body disease. Lewy body disease has been further subdivided into pure autonomic failure, sporadic PD and dementia with Lewy bodies (DLB) [1, 2, 3, 4, 5]. This review will refer mainly to the sporadic form of PD.
The pathological process that underlies sporadic PD is linked to the development of α‐synuclein‐containing inclusion bodies in the form of Lewy bodies (LBs) in perikarya and Lewy neurites (LNs) in neuronal processes [6, 7, 8]. Of the diverse neuronal types within the human nervous system, only a few develop pathological inclusions, and this selective involvement is reflected in the regional pattern of the pathology. Vulnerable cells are distributed throughout the peripheral, enteric and central portions of the nervous system (ENS/CNS) [9, 10, 11, 12]. All of the susceptible cells are projection neurones that generate a long and thin axon, which is unmyelinated, or poorly myelinated [13]. Despite their greater prevalence with advancing age, PD‐associated inclusion bodies do not occur consistently in all aged non‐symptomatic cases and, as such, they are pathological rather than protective or neutral age changes. The presence of these pathognomonic inclusions is a prerequisite for the post mortem diagnosis of sporadic PD [14, 15].
These disease‐related inclusions occur in symptomatic PD patients as well as in individuals who did not manifest any of the characteristic motor symptoms in life. Thus, the pathological process has a pre(motor)‐symptomatic and a symptomatic phase [16, 17, 18]. The term ‘presymptomatic phase’ implies that even when only a few LNs/LBs are detectable in non‐symptomatic cases, such ‘incidental’ inclusion bodies represent incipient PD or the harbinger of the symptomatic disease phase [14, 19].
It has been postulated that sporadic PD might be a primary disorder of olfaction, given that smell loss is an early event in the course of this disorder [20]. This theory was derived from sources based on psychology, physiology, anatomy and pathology. Additional studies not only have corroborated the initial involvement of anterior olfactory structures, but also have pointed to an early involvement of the enteric nerve cell plexuses as well as of the dorsal motor nucleus of the vagus and the intermediate reticular zone in the lower brainstem [21, 22, 23, 24, 25]. To be plausible, any theory attempting to explain sporadic PD must incorporate these extra‐nigral sites, which consistently become affected in the course of the disorder [9]. Furthermore, any speculation regarding the cause and beginnings of PD must take into account the involvement of olfactory and autonomic systems that generally develop prior to the onset of the classical somatomotor symptoms [26, 27, 28]. This article will review the evidence for such involvement and summarize several hypotheses developed on the basis of these findings.
Evidence for PD‐related olfactory dysfunction
Psychophysical tests
The first case–control study that demonstrated smell abnormality in PD was conducted by Ansari and Johnson [29] in 22 clinically diagnosed PD patients. A subsequent larger study used detection threshold tests to amyl acetate in 78 subjects and 40 controls [30]. Thresholds were reduced, but no correlation was found with age, gender or treatment with levodopa. Unlike the first study, there was no association with disease duration. The next sizeable investigations using the University of Pennsylvania Smell Identification Test (UPSIT) showed that age‐matched olfactory dysfunction did not relate to odour type, was independent of disease duration, and did not correspond with motor function, tremor or cognition [31, 32]. The authors also demonstrated that the deficit was of the same magnitude in both nostrils, and not influenced by anti‐Parkinsonian medication. A comparable survey was undertaken using UPSIT in 155 cognitively normal, depression‐free PD patients, and 156 age‐matched controls [33]. UPSIT scores for PD patients were dramatically lower than those for controls. There was no correlation between disease duration and UPSIT score (r = 0.07). Impairment of smell sense has also been documented in PD patients using Sniffin Sticks [34, 35]. Although the psychophysical evidence provides overwhelming support for olfactory involvement in PD, it does not completely eliminate the possibility of confounding factors from cognitive dysfunction, nor can it be determined whether the smell defect is initially peripheral or central [36, 37, 38, 39].
Neurophysiological tests
A further measure of smell sense is the olfactory event‐related response (OERP) pioneered by Kobal and Plattig [40], which has the advantage of minimizing the potential effect of cognitive dysfunction. An initial examination compared OERP recording of 73 PD patients with that of 47 controls of similar age and gender [33]. In 36 patients (49%), responses were either absent or unsatisfactory for technical reasons. Analysis of the 37 with a measurable trace showed that, for hydrogen sulphide (H2S), a highly significant latency difference existed between diagnostic groups. Similar results were obtained in 31 patients with clinically labelled PD tested by OERP to vanillin and H2S [41]. Prolonged latencies were seen in these individuals whether they were taking anti‐Parkinson medication or not.
The above‐cited evidence from psychophysical and neurophysiological sources gives virtually unassailable support to the presence of olfactory dysfunction in established PD, a feature that occurs more frequently [33] than tremor (80% vs. 70%).
Olfactory dysfunction in presymptomatic phases of PD
If the previous observations about early olfactory involvement are correct, this should be reflected by tests inindividuals at risk for future disease or by prospective studies of those with prior olfactory impairment [42]. Montgomery and colleagues [43, 44] implemented a test battery comprising motor function, olfaction (UPSIT) and mood for PD first‐degree relatives. There were significant differences in sons and daughters, particularly where the affected parent was the father. This work has been criticized [45] because there may have been self‐selection in allegedly unaffected relatives – who may have had undisclosed motor complaints resulting in the unusually high 2‐year positive prediction rate (40 out of 59 subjects). Ponsen et al. [45] evaluated prospectively 78 asymptomatic first‐degree relatives of non‐familial PD patients by olfactory tests and Dopamine Transporter Scan (DATScan). Forty were hyposmic at baseline and, when reviewed 2 years later, four had abnormal DATScans and showed clinical signs of PD. In the remaining 36 hyposmics, who displayed no sign of PD, the rate of decline of dopamine transporter binding was higher than in normosmic relatives. Others [46] tested 30 patients with unexplained and isolated smell impairment to determine whether any might be in the premotor phase of PD. Apart from detailed olfactory testing, subjects were evaluated by DATScan and transcranial sonography of the substantia nigra. Eleven displayed increased (that is, abnormal) echogenicity on transcranial sonography characteristic of PD. Ten subjects volunteered for DATScan and, of them, five had abnormal scans and an additional two were borderline, suggesting that they might be in a presymptomatic phase of PD. Two of the five scan‐positive patients have now developed clinically confirmed PD (Hummel, pers. comm., 2006).
The first long‐term, community‐based prospective study has been published, in connection with these relations [47]. The authors used the cross‐cultural Brief Smell Identification Test (BSIT) [48] in 263 healthy Japanese‐American men aged 71–95 years who participated in the Honolulu‐Asia Ageing Study. After 7‐year follow‐up, 19 men developed PD at an average latency of 2.7 years from baseline assessment. Adjustment for multiple confounders gave relative odds for PD in the lowest tertile of BSIT score of 4.3 (95% CI 1.1–16.1; P = 0.02), thus indicating a moderate predictive power of olfactory testing. In the same cohort, those who later died underwent brain autopsy to detect the presence of brainstem LBs [49]. Of 163 autopsied men without clinical PD or dementia, 17 were found to have incidental LBs. Those who scored in the lowest tertile of the BSIT were significantly more likely to have LBs at autopsy. Potentially conflicting findings were obtained in a study of 70 male twin pairs discordant for PD [50], 62 of whom agreed to undergo olfactory tests. At baseline, the authors confirmed impaired UPSIT scores in the affected twins, but not in their brothers who were rated normal. After a mean of 7.3 years, 28 brothers were still alive and, of them, 19 were retested using the 12 odour BSIT, whereby 2 had developed PD. Neither had impaired UPSIT at baseline, but the average decline in UPSIT percentile score in both was greater than the remaining 17, who had not developed the disease. It is suggested that smell testing may not be a reliable predictor. The dropout rate was unusually high and smell assessment was by different methods, that is, initially by UPSIT (40‐item) then by BSIT (12‐item). On the first occasion, the test was unsupervised; moreover, a 7.3‐year interval may have been too short.
Further considerations regarding olfactory dysfunction
Despite such evidence, theories regarding olfactory dysfunction as a characteristic symptom of PD have had difficulty gaining acceptance, because 10–20% with a clinical diagnosis of PD have normal smell identification. In some PD patients with normal UPSIT scores, the OERP was absent or delayed [33], which implies that the olfactory deficit may be more frequent than indicated by UPSIT. Pathology‐based studies indicate that the diagnostic error rate for PD is 10% or higher, and that olfactory identification is sometimes abnormal in other diseases showing Parkinsonism [51]. Mistaken diagnosis of PD occurs most often in MSA, PSP, vascular Parkinsonism and, occasionally in individuals with essential tremor. All these disorders may be characterized by either normal or slight impairment of olfaction. It is not yet known whether persons with normal UPSIT score are the very patients who have received an incorrect diagnosis, but prospective studies would resolve this issue.
Evidence for PD‐related autonomic dysfunction
An understanding of the applied anatomy of the vagus is necessary in view of its early and severe pathological involvement. Three major components of the vagus can be distinguished in the medulla oblongata: (i) the ambiguus nucleus, a motor nucleus that innervates muscles of the palate, pharynx, larynx and heart; (ii) the dorsal motor nucleus, which harbours the preganglionic visceromotor neurones that control the postganglionic parasympathetic nerve cells in internal organs of the chest and abdomen; and (iii) the complex of nuclei accompanying the solitary tract. The parvocellular nuclei of the latter complex receive taste information and an abundance of visceral sensations. The dorsal motor nucleus is damaged severely and early on in PD, the solitary tract nuclei exhibit little change until later stages, and the nucleus ambiguus remains intact [21] (Figure 1, right).
Figure 1.
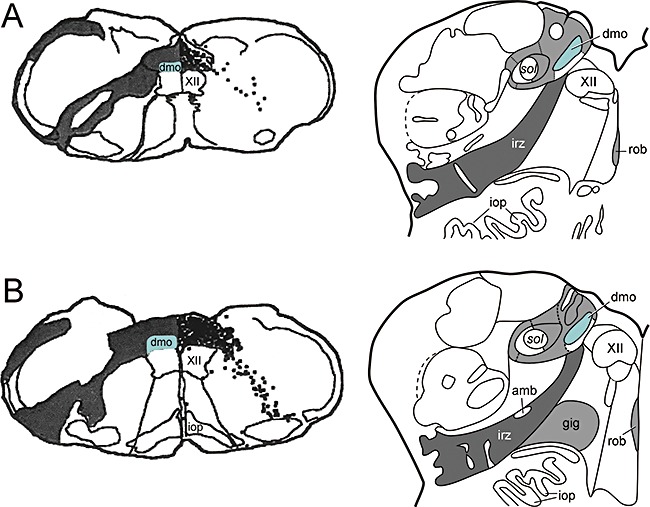
(A–B, left hand side) to illustrate the mode of spread of herpes simplex virus following injection into peripheral portions of the vagus nerve in the rat medulla oblongata, adapted and reproduced in part with permission from Blessing et al.[154]. (A–B, right hand side) to show the topographical distribution pattern of α‐synuclein pathology in the human medulla in stages 1 and 2 of sporadic Parkinson's disease, adapted and reproduced in part with permission from Del Tredici et al.[21]. amb, ambiguus nucleus; dmo, dorsal motor nucleus of the vagus (marked in green); gig, gigantocellular reticular nucleus, iop, inferior olivary nucleus, principal subnucleus; irz, intermediate reticular zone; rob, nucleus raphes obscurus; sol, solitary tract; XII, motor nucleus of the hypoglossal nerve.
Gastrointestinal disorder
The gastrointestinal tract is supplied by two major nerves: the vagus, which is for excitatory parasympathetic control, and sympathetic nerves, which are inhibitory. There is abundant evidence of pathological involvement in both systems, as detailed below, and clinically features of this pathology become increasingly obvious [52, 53, 54, 55]. Dysphagia is a common feature sometimes demonstrable in asymptomatic individuals [56]. In one survey of 17 autopsy‐confirmed cases of PD, swallowing difficulties appeared after an average period of 10 years from onset of initial extrapyramidal symptoms [57]. A variety of mechanisms account for swallowing difficulty, with evidence of dysfunction at either oropharyngeal or oesophageal level. Although dysphagia may relate to mechanical problems (e.g. Zenker's diverticulum) or dry mouth, the majority can be explained on the basis of vagal dysfunction.
Reduction of appetite and weight loss are common features that typically affect more advanced cases, and this may also relate to impairment of smell and taste [58], or to poor diet, increased energy expenditure or depression [59]. Although many patients drool saliva, the consensus view is that the volume of saliva produced is either unchanged or decreased, although it may be increased by levodopa preparations [60]. Gastric dysfunction is likewise a recognized feature of established PD [61, 62]. These investigators reported frequent bloating or nausea suggesting delayed gastric emptying ,and this phenomenon has indeed been confirmed by measurement of gastric emptying time in patients whether they are receiving levodopa or not [63]. While little is known about small intestinal motility in PD, there are several studies relating to colonic disorder.
If ENS involvement and disease of both the parasympathetic and sympathetic systems is an early event in PD, there should be relevant symptoms in the premotor phase. There is presently only one investigation in support of this, namely the prospective study of bowel habit in elderly patients who took part in the Honolulu Heart Program [64]. This comprised 6790 men without extrapyramidal disease at enrolment, followed up for 24 years, of whom 96 developed PD. The adjusted risk of PD in those with less than one bowel movement per day, compared with those with one or more per day, was increased almost threefold (OR 2.7; 95% CI 1.3, 5.5), implying that constipation is a harbinger of PD [27]. Prevalence estimates of constipation in PD have been inconsistent because of variable definitions of constipation, but a conservative estimate would be at least 20% [65]. Colonic transit time is raised from control values of around 20 h to 44–130 h [66, 67], probably worsening as the disease advances.
Cardiac disorder
In established PD, several investigators have found reduced cardiac uptake of the noradrenaline analogue, metaiodobenzylguanidine (MIBG) [68, 69, 70, 71, 72]. This procedure evaluates postganglionic noradrenergic cardiac sympathetic function. Heart rate variability may be abnormal in PD and is thought to reflect involvement of both sympathetic and parasympathetic pre‐ and postganglionic neurones [73]. Incidental LBs have been found in the cervical sympathetic ganglia and cardiac plexus [74]. In another study, it was clearly shown that tyrosine hydroxylase‐immunoreactive nerve fibres in the heart had almost entirely disappeared in patients with PD (and DLB), whereas they were well preserved in all those with PSP and pure Alzheimer's disease [75]. Only one patient with PD displayed abnormalities in the sympathetic ganglia, which indicates that cardiac sympathetic denervation precedes neuronal loss there. Autonomic failure presaging a Parkinsonian syndrome has been shown in two patients [76], but, to date, there have been no long‐term prospective epidemiological studies of cardiac function. One investigation showed a significant correlation between MIBG uptake and olfactory disorder in 26 patients with PD (but not MSA), thereby confirming the close association of these two modalities [77].
Sleep disorder
A variety of sleep disorders are recognized in established PD [78], of which the most studied are excessive daytime sleepiness and rapid eye movement sleep behaviour disorder [79]. The pathophysiology is not understood fully but most likely reflects cellular changes found in the Parkinson neuropathological stages 1 and 2, involving the reticular formation, coeruleus/subcoeruleus complex, pedunculopontine nucleus and hypothalamus [80]. In the prospective Honolulu‐Asia Ageing Study [81], 43 of 3078 men aged 71–93 years developed PD over a 7‐ to 10‐year period, and those reporting excessive daytime sleepiness were approximately three times more likely to develop PD (OR 3.3; 95% CI 1.4, 7.0). According to one source [82], 11/29 subjects with unexplained REM sleep behaviour disorder (RBD) subsequently developed Parkinsonism, complementing the finding of decreased striatal transporter uptake in RBD by others [83]. In other studies of olfaction and sleep [84, 85, 86], many RBD patients had significantly impaired smell function, once more implying that olfaction and RBD are early features. A difficulty with most of these studies, however, is the lack of post mortem confirmation. The investigation by Iranzo and co‐workers [87] is of particular interest in this regard. Apart from corroborating the suspected status of RBD as a precursor of PD, they document the time interval between onset of RBD and clinically manifest sporadic PD. In seven patients, the latent period was on average 12 years (range 3–17 years). Given that RBD is a sign of PD stage 2 pathology, this estimate provides a lower time limit for the presymptomatic phase and suggests that the earliest evidence of PD pathology (stage 1) would be 15–20 years before the onset of typical clinical manifestations.
The pathological process associated with sporadic PD
Braak and colleagues [22] performed detailed pathoanatomical analyses of 41 cases of PD by α‐synuclein immunostaining. A similar approach was taken in 69 subjects who had no PD‐associated somatomotor symptoms in life but displayed LNs and/or LBs (incidental cases). A third group consisted of 58 age‐ and gender‐matched cases without LNs or LBs and no history of neurological or psychiatric illness. The results of the cross‐sectional study led the authors to propose that the PD‐related pathological process in the CNS progresses, apparently without remission, through presymptomatic and symptomatic disease phases [22]. A subsequent study identified LNs and LBs in enteric nerve cell plexuses in both presymptomatic and clinically diagnosed PD cases [88] (Figure 2). This led to the suggestion that the pathological process begins at two sites simultaneously, that is, in the olfactory bulb/anterior olfactory nucleus and within enteric nerve cell plexuses. Following damage of these predilection sites, the preganglionic parasympathetic projection neurones of the dorsal motor nucleus of the vagus (Figure 3) and, shortly thereafter, the post‐ and preganglionic sympathetic projection neurones in the coeliac ganglion and intermediolateral nucleus of the spinal cord may become drawn into the disease process [89]. Next to show the lesions are superordinate supraspinal centres such as the coeruleus/subcoeruleus complex, magnocellular portions of the reticular formation, and posterior raphe nuclei (1, 4, right). Additional CNS regions might then follow successively: central subnucleus of the amygdala [90, 91, 92, 93], pars compacta of the substantia nigra, and magnocellular nuclei of the basal forebrain [23]. In other words, all of the vulnerable sites do not become involved at the same time but, rather, in a predictable topographic sequence. Within the brain, the disease process displays a stereotypical caudal‐rostral advance from the lower brainstem through basal portions of the mid‐ and forebrain, finally reaching the cerebral cortex (Figure 4A ). A distinctive lesional pattern, including neuronal loss, emerges. With one exception [94], subsequent studies have confirmed, for the most part, the results of this pathoanatomical study [95, 96, 97, 98], and inter‐rater reliability of the pathological samples was high [99].
Figure 2.
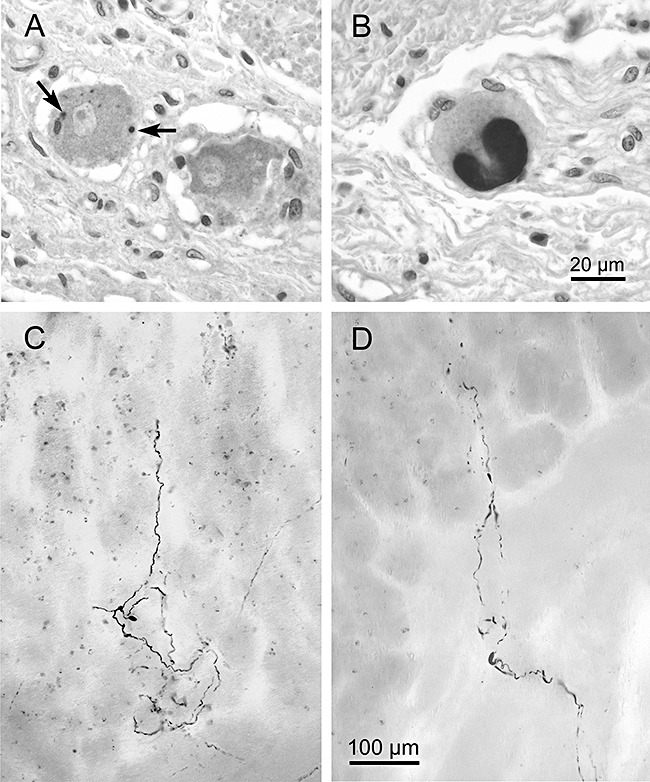
Alpha‐synuclein immunoreactions (Syn‐1: 1:2 000, Transduction Laboratories) in 6‐μm paraffin sections showing Lewy bodies (A, arrows and B) in the Auerbach plexus of the human oesophagus. Scale bar in (B) is valid for (A). Reproduced in part with permission from Braak et al. [24]. Aggregated axonal α‐synuclein inclusions (Lewy neurites) in the gastric Meissner plexus (C,D). Notice how the terminal ramifications of abnormally altered axons extend through the lamina propria of the gastric mucosa and run parallel to the gastric glands (D). Syn‐1 immunoreactions in 150‐μm cryosections. Scale bar in (D) also applies to (C). Adapted and reproduced with permission from Braak et al. [88].
Figure 3.
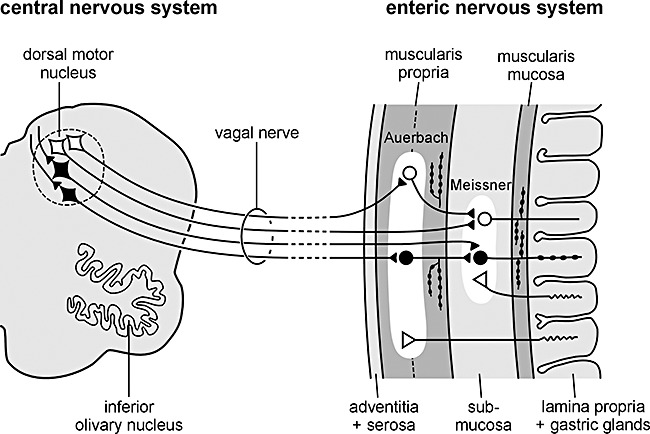
Simplified diagram showing the vagal interconnections between the enteric nervous system and medulla oblongata. A neurotropic agent capable of passing through the mucosal epithelial barrier of the stomach could enter terminal axons of postganglionic VIPergic neurones (black, rounded cell somata) in the submucosal Meissner plexus and, via retrograde axonal and transneuronal transport (black, rounded cell somata in the Auerbach plexus), reach the preganglionic cholinergic neurones (black, diamond‐shaped cell somata) of the dorsal motor nucleus of the vagus in the lower brainstem. Reproduced with permission from Braak et al. [88].
Figure 4.
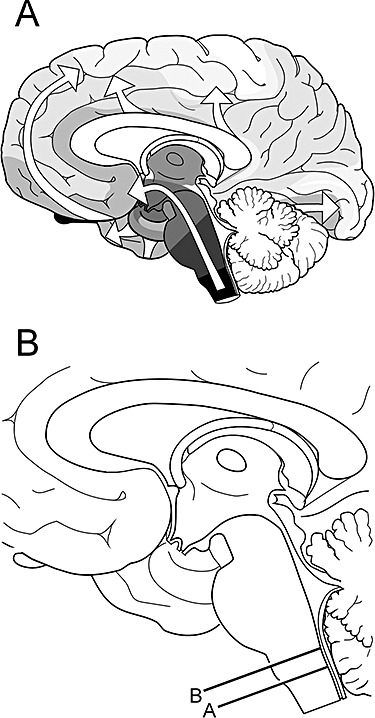
Two schematic drawings. (A) The essentially caudal‐rostral expansion (white arrows) of the Lewy body pathology in the brain after it attains a foothold in the dorsal motor nucleus of the vagus in the medulla oblongata (here, in black). The other consistently involved induction site in sporadic Parkinson's disease is the olfactory bulb (black). Reproduced with permission from Braak et al. [22]. (B) Median view of a mediosagittal section through the human brain: lines A and B indicate the respective planes of the frontal sections through the medulla oblongata depicted in Figure 1. Adapted and reproduced in part with permission from Del Tredici et al. [21].
Pathological changes in olfactory structures
PD‐associated α‐synuclein‐containing inclusion bodies as yet have not been found in the olfactory epithelium of autopsied PD patients [100]. Nasal biopsy specimens from seven patients with symptomatic PD [101] were compared with four anosmic controls using antibodies against olfactory marker protein (OMP), neurotubulin, protein gene product 9.5 (PGP 9.5) and mRNA for OMP. Irregular areas of olfactory epithelium were positive for PGP 9.5 and neurotubulin but mostly negative for OMP, although mRNA for OMP was found in the olfactory cleft and respiratory mucosa. In this small series, there was no clear difference between PD and anosmic controls; however, sections were not examined for presence of LNs/LBs, and it could be argued that those with anosmia may have been in the presymptomatic phase of PD. Daniel and Hawkes [102] examined olfactory bulbs and tracts in eight controls as well as eight patients with a clinical and pathological diagnosis of PD taken from the United Kingdom Parkinson's Disease Brain Bank. All PD cases contained LBs, which were most numerous in the anterior olfactory nucleus but also were found in mitral cells, the first projection neurones to receive input from the bipolar neurones in the olfactory epithelium. It was subsequently shown that loss of neurones in the anterior olfactory nucleus correlated with disease duration [103]. One report [104] suggested that expression of tyrosine hydroxylase in the olfactory bulb is increased 100‐fold, and that the consequent excess of dopamine might explain the hyposmia that develops in PD. Braak and colleagues [22] confirmed the presence of PD‐related lesions in mitral cells and tufted neurones of the olfactory bulb and in projection neurones of the anterior olfactory nucleus, which is dispersed throughout the olfactory tract. A tightly woven network of LNs rapidly develops within the anterior olfactory nucleus. From there, the pathology tends to spread slowly into more remote olfactory sites (olfactory tubercle, piriform and periamygdalear cortex, entorhinal cortex of the ambient gyrus) [105] without advancing into non‐olfactory cortical areas [8, 22, 24].
Pathological changes in the ENS
It is known that ENS lesions occur in symptomatic cases of PD [106, 107, 108, 109, 110]. As in the brain, only a few of the many neuronal types within the ENS [111, 112, 113] are prone to develop PD‐associated lesions (Figure 2A,B ). Once again, the vulnerable cells apparently are projection neurones with a long and unmyelinated axon [13]. The inhibitory nitrergic vasoactive intestinal polypeptide neurones have been shown to develop LNs and LBs [109, 110]. Axons containing α‐synuclein from the submucous plexus have been seen to protrude through the muscle layer of the mucosa, extending widely and ramifying within the mucosal lamina propria [88] (2, 3). Axons of affected ENS cells contain thread‐like or spindle‐shaped LNs of varying size. In the mucosal lamina propria, the unmyelinated axons are only micrometers away from the body's innermost environment, their only protection being a single layer of epithelial cells (2, 3). Such involvement of the enteric nerve cell plexuses may represent a particularly early event – if not the earliest vagal‐associated event – because LNs were observed both in clinically diagnosed cases and in non‐symptomatic individuals with PD‐related brain lesions limited to the lower brainstem [88]. In summary, the studies indicate that early involvement of the ENS with widespread and thinly distributed lesions throughout the walls of the upper gastrointestinal tract is accompanied by mild CNS lesions. It remains to be seen whether ENS lesions also occur in the absence of lesions in the brain.
Pathological changes in preganglionic parasympathetic projection neurones
The very first brainstem lesions appear in the dorsal motor nucleus of the vagus [21]. The preganglionic parasympathetic projection neurones there generate long and thin unmyelinated axons [114, 115]. Pathological α‐synuclein aggregates are detectable in both proximal intramedullary and peripheral portions of these axons. Other nuclei in the dorsal vagal area, including the nucleus gelatinosus, area postrema, and the nuclei surrounding the solitary tract, are minimally affected or uninvolved. The catecholaminergic melanoneurones in this area and those in the intermediate reticular zone [116] remain intact, at least initially [21]. These neurones do not project to the periphery but to higher levels of the brain [117]. The multipolar motorneurones of the ambiguus nucleus have thickly myelinated axons and remain free of LNs/LBs.
Pathological changes in post‐ and preganglionic sympathetic projection neurones
PD‐associated pathological changes in sympathetic pathways (intermediolateral column of the spinal cord, coeliac ganglion and other peripheral sympathetic ganglia) have been studied and, to some extent, characterized already [74, 118]. Recently, such lesions also have been shown in presymptomatic patients dying of non‐neurological causes, thereby confirming the early involvement of the autonomic system [89, 119, 120]. All cases examined also displayed PD‐related involvement of the lower brainstem, which, in turn suggests that the spinal cord abnormalities within the sympathetic nervous system may occur after the parasympathetic preganglionic projection neurones of the vagus have become affected.
Is PD caused by a neurotropic pathogen?
It is unlikely that the lesions in the olfactory system and in other predilection sites within the ENS and CNS develop independently of each other. A more plausible explanation is that a common pathologic insult, a hitherto unknown neurotropic factor or pathogenic substance, induces the disease and triggers the sequential involvement of vulnerable regions. Anatomical analysis reveals a continuous chain of long‐axoned and sparsely myelinated projection neurones that interconnect not only the olfactory epithelium but also the ENS with the brain (Figure 3) and, within the brain, all of the vulnerable regions [23]. After uptake, such a neurotropic pathogen might utilize these pathways and progress within the nervous system by way of axonal transport and trans‐synaptic transmission [121, 122, 123, 124]
Neuroactive substances, including neurotropic viruses, unconventional pathogens with prion‐like properties, or slow neurotoxins, usually are taken up at synapses, where they are frequently controlled by receptor‐mediated endocytosis. The substances are transported to the cell body via the axon [125, 126, 127, 128]. Neurotropic viruses that pass from the surroundings into axons of susceptible nerve cells can be prevented from doing so by the existence of a myelin sheath, which functions as a physical barrier against virus penetration [129]. Thus, the absence of a myelin sheath around axons of the first neurones in the potential chain of vulnerable projection neurones may facilitate entrance and damage by viruses or other pathogens [125, 127, 130, 131, 132]. In addition, most of the neuronal types located within the CNS are protected against uptake of substances from the extracellular milieu beyond the CNS by the blood–brain barrier. Only axons of nerve cells that project from the olfactory epithelium into the CNS and those that project from the CNS into the periphery, such as the preganglionic parasympathetic and sympathetic fibres, lack such a protective barrier. Accordingly, an intravenous injection of horseradish peroxidase (HRP) results in retrograde labelling of the dorsal motor vagal nucleus [133].
Despite enormous gaps in the present state of knowledge, the main line of reasoning that favours a ‘dual‐hit’ PD‐related process (with olfactory and enteric means of access) is that both are in close and constant contact to the (potentially hostile) outer environment. From the enteric plexuses, the prospective pathogen may gain access retrogradely to the CNS via parasympathetic pathways (vagus; Figure 3) and, thereafter, through post‐ and preganglionic sympathetic fibres. Once in the CNS, the disease process could ascend from spinal cord and lower brainstem through vulnerable portions of the basal mid‐ and forebrain until it reaches the cerebral cortex (Figure 4A ). From the olfactory epithelium, on the other hand, the pathogen would follow an anterograde route, to reach medial amygdala, olfactory tubercle, as well as piriform and periamygdalear cortex.
Possible role of viral invasion
The possibility that PD might be of viral aetiology was formally addressed by Elizan and Casals [134]. The inability to transfer PD to primates and the lack of viral antibodies in the then newly described Guam PD‐dementia individuals, who were usually anosmic [135], argued against a viral aetiology for PD, although the theory was not entirely discounted. Influenza virus has been associated with PD, as discussed in detail by Takahashi and Yamada [136]. Despite many negative studies that searched for direct evidence of influenza A in PD [137, 138, 139, 140, 141], nearly all pointed out that influenza A behaves as a persistent virus possibly capable of initiating autoimmunity. On the basis of human and experimental models, these authors proposed that the virus shows a predilection for the substantia nigra, cerebellum and hippocampus, and may be responsible for the formation of LBs.
Parkinsonism is seen on rare occasion during or after infection with herpes simplex encephalitis [142], and a link between chronic herpes simplex type 1 encephalitis, PD and tic doloureux has been suggested [143]. There is no evidence of antibodies to herpes simplex virus (HSV) type 1 and 2 when compared with measles or cytomegalovirus antibodies in the serum and spinal fluid of PD subjects [144, 145]. Similar findings were documented by Elizan and Casals [134]. One unconfirmed report suggests an association between PD and coronavirus infection on the basis of enzyme‐linked immunosorbent assay in cerebrospinal fluid in 20 patients [146].
There is recent evidence that diseases of intermediate type hypersensitivity (asthma, allergic rhinitis, seasonal rhinitis) may be associated with PD [147]. Retrospectively, the authors reviewed medical records in 196 cases of PD and 196 healthy controls and found a significant, approximately twofold increase of prior intermediate‐type sensitivity disorder in general, but particularly for allergic rhinitis (OR 2.9; 95% CI 1.3, 6.4; P < 0.01). There was a trend towards protection against PD in those who had used anti‐inflammatory drugs. It is suggested that patients with PD might initiate an inflammatory response that could be directed towards the CNS. There is some evidence of impaired smell sense in those with allergic rhinitis [148], raising the possibility that allergic rhinitis might facilitate entry of a pathogen from the nose into the olfactory bulb and tract.
Altered protein handling may be a factor in the PD‐associated pathogenic process. Many viruses, e.g. Epstein‐Barr virus, encode proteins that exploit the ubiquitin‐proteasome system to regulate virus latency and allow the persistence of infected cells in immunocompetent hosts [149, 150].
There is longstanding evidence that neurotropic virus can enter the brain via the nasal route. It was shown that HSV type 1 placed intranasally in 6‐week‐old mice was detectable in the trigeminal root entry zone and olfactory bulbs 4 days later [151]. In some mice, virus which had entered the olfactory bulb, spread via axons as far as the temporal lobe, hippocampus and cingulate cortex. Another study in the rat showed that HRP applied intranasally can be transported to the bulb, anterior olfactory nucleus, as well as to cholinergic neurones of the diagonal band, serotonergic raphe neurones, and noradrenergic cells of the locus coeruleus [152]. It is not widely appreciated that direct connections exist between primary olfactory areas and the substantia nigra. For example, application of HRP into the olfactory tubercle results in anterograde labelling of the ventral tegmental area, the pars reticulata of the substantia nigra, and anterior olfactory nucleus. As anticipated, there was retrograde labelling in the olfactory bulb and anterior olfactory nucleus [153].
Blessing and colleagues [154] studied connections of the vagus using HSV type 1 in the rat (Figure 1, left). Live HSV‐1 was injected into the cervical vagus, and its distribution was examined using polyclonal antiserum. On day 2, virus was detected in glial cells of the area postrema, in nuclei of the solitary tract, the vagal dorsal motor nucleus, spinal trigeminal nucleus, and the great raphe nucleus, whereas the hypoglossal nucleus was spared, as in PD. On day 3, there was rostral spread to involve the locus coeruleus, parvocellular reticular nucleus and periaqueductal grey, but not the substantia nigra. HSV‐1‐positive neurones were seen in an oblique area corresponding to the human intermediate reticular zone [114]. This route of infection is shown in Figure 1 (left) and should be compared with the similar distribution of PD‐associated brainstem pathology (Figure 1, right). There are clear differences between this animal model and the pathology in humans. For instance, in contrast to the situation in PD, the trigeminal nerve in the rodents is involved and the substantia nigra is spared. Nevertheless, the rostral migration of HSV‐1 has remarkable similarity to the suggested progression of the α‐synuclein pathology in PD (Figure 1) and reaffirms speculation that a neurotropic agent may utilize the vagus as a means of entering the medulla [22, 23]. The experiments of Blessing and co‐workers [154] are of additional interest in that they show that the first site of attack is in glial cells, which raises the possibility that residual virus in these cells may be responsible for the slow progression of PD.
Transvagal spread in animal models has been reported for other neurotropic enteroviruses, reovirus, pseudorabies virus and haemagglutinating encephalomyelitis virus [132]. The same group was unable to introduce CNS infection in mice with influenza A virus through non‐autonomic routes, such as the anterior chamber of the eye, the brachial plexus, knee joint, sciatic nerve and hindlimb footpad. Conversely, pseudorabies virus spreads though both somatic and autonomic nerves. A strain of Hong Kong influenza virus inoculated intranasally in mice reached the CNS through afferent fibres of the olfactory, vagal, trigeminal and sympathetic nerves following replication in the respiratory mucosa [155]. Intranasally inoculated avian influenza virus in mice gave rise to lesions in lung and brainstem [156]. The main areas involved were the nuclei of the solitary tract and the trigeminal ganglia. Although haematogenous CNS spread is well documented in many viral strains, the findings of these experiments indicate that influenza viruses have preferential routes of access to the CNS along the vagus and olfactory nerves following replication in the lungs, but the pattern of brainstem involvement is variable; that is, the trigeminal nerve is sometimes involved, at other times spared. These observations have some parallels with the pattern of proposed CNS invasion in PD. Although no virus mirrors exactly the pathological profile seen in PD, it is possible that a particular viral strain might display a predilection for the olfactory and vagal routes of entry.
Discussion
We propose that an unknown neurotropic pathogen initiating the pathological process underlying sporadic PD adopts a two‐pronged attack on the nervous system: anterogradely, via olfactory pathways; and retrogradely, via enteric plexuses and preganglionic vagal fibres. If the pathogen is viral, then brain entry via the nasal route or uptake through the gastrointestinal tract has ample precedent, as described above. A nasal infection would have direct access to the olfactory nerve; infected saliva or mucous could be swallowed and reach the upper digestive tract to infect axons of Meissner's plexus and, after transneuronal passage, ascend retrogradely in preganglionic parasympathetic fibres of the vagus nerve to the lower brainstem. Direct access to the medulla via the viscerosensory fibres of the vagus in the pharynx or via the trigeminal nerve, while anatomically appealing, is not compatible with virtual sparing of the solitary tract and trigeminal nuclei during the entire course of the disorder.
Our observations relate to the commonest, sporadic variety of PD. As mentioned earlier, not all neuropathologists are in accord with the Frankfurt PD classification [94, 95], the main objection being (in a few cases) the absence of medullary or pontine Lewy pathology where the substantia nigra was affected. Another study used the Frankfurt staging procedure to group an autopsy cohort into: preclinical (stages 1 and 2); early (stages 3 and 4, 35% with clinical PD); and late (stages 5 and 6, 86% with clinical PD) cases [157]. Preclinical compared with early or late‐stage cases should progressively be more elderly at the time of sampling, but this feature was not observed. Despite this, the proposed sequential progression of Lewy pathology, and the associated clinical features that have been demonstrated in many prospective epidemiological studies [47, 49, 64, 81], give strong support to the staging concept.
The enigma of PD is to explain why a condition that probably begins in anterior olfactory structures and portions of enteric nerve cell plexuses, has a presymptomatic period of several years, possibly decades, during which time the disease‐related destruction inexorably advances retrogradely along fibre tracts in the brainstem and anterogradely along olfactory pathways in a predictable sequence to destroy the substantia nigra and then initiate a disease that runs a further course of approximately 15 additional years. What the clinician observes is the latter phase of a chronic illness, about which most patients are unaware or relatively untroubled. In contrast to most virally induced infection, the intraneuronal lesions appear to progress with remarkable slowness; yet, at the same time, they display only minimal interindividual deviation from the predictable topographical distribution [23, 88].
Acknowledgements
The authors wish to thank Ms Inge Szász‐Jacobi for expert assistance with the graphics. This work was made possible in part by funding from the Deutsche Forschungsgemeinschaft (DFG) and Hilde‐Ulrichs Foundation (Florstadt‐Staden, Germany).
References
- 1. Hague K, Lento P, Morgello S, Caro S, Kaufmann H. The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol 1997; 94: 92–6 [DOI] [PubMed] [Google Scholar]
- 2. Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson's disease. Arch Neurol 1999; 56: 33–9 [DOI] [PubMed] [Google Scholar]
- 3. Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord 2003; 18 (Suppl. 6): 2–12 [DOI] [PubMed] [Google Scholar]
- 4. Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I. SIC Task force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18: 467–86 [DOI] [PubMed] [Google Scholar]
- 5. Geser F, Wenning GK, Poewe W, McKeith I. How to diagnose dementia with Lewy bodies: state of the art. Mov Disord 2005; 20 (Suppl. 12): 11–20 [DOI] [PubMed] [Google Scholar]
- 6. Lowe J. Lewy bodies In Neurodegenerative Diseases. Ed. Calne DP. Philadelphia, PA: Saunders, 1994; 51–69 [Google Scholar]
- 7. Duda JE, Lee VMY, Trojanowski JQ. Neuropathology of synuclein aggregates: new insights into mechanism of neurodegenerative diseases. J Neurosci Res 2000; 61: 121–7 [DOI] [PubMed] [Google Scholar]
- 8. Del Tredici K, Braak H. Idiopathic Parkinson's disease: staging an α‐synucleinopathy with a predictable pathoanatomy In Molecular Mechanisms in Parkinson's Disease. Eds Kahle P, Haass C. Georgetown, TX: Landes Bioscience, 2004; 1–32 [Google Scholar]
- 9. Jellinger K. Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol 1991; 14: 153–97 [DOI] [PubMed] [Google Scholar]
- 10. Takahashi H, Wakabayashi K. The cellular pathology of Parkinson's disease. Neuropathology 2001; 21: 315–22 [DOI] [PubMed] [Google Scholar]
- 11. Takahashi H, Wakabayashi K. Controversy: is Parkinson's disease a single disease entity? Yes. Parkinsonism Relat Disord 2005; 11: 31–7 [DOI] [PubMed] [Google Scholar]
- 12. Braak H, Del Tredici K. Preclinical and clinical stages of intracerebral inclusion body pathology in idiopathic Parkinson's disease In Parkinson's Disease: Progress in Research. Ed. Willow. JM. Hauppauge, NY: Nova Science, 2005; 1–49 [Google Scholar]
- 13. Braak H, Del Tredici K. Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging 2004; 25: 19–23 [DOI] [PubMed] [Google Scholar]
- 14. Thal DR, Del Tredici K, Braak H. Neurodegeneration in normal brain aging and disease. Sci Aging Knowledge Environ 2004; 23: 1–13 [DOI] [PubMed] [Google Scholar]
- 15. Mikolaenko I, Pletnikova O, Kawas CH, O'Brien R, Resnick SM, Crain B, Troncosco JC. Alpha‐synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA). J Neuropathol Exp Neurol 2005; 64: 156–62 [DOI] [PubMed] [Google Scholar]
- 16. Foley P, Riederer P. Pathogenesis and preclinical course of Parkinson's disease. J Neural Transm Suppl 1999; 56: 31–74 [DOI] [PubMed] [Google Scholar]
- 17. Wolters EC, Francot C, Bergmans P, Winogrodzka A, Booij J, Berendse HW, Stoof JC. Preclinical premotor; Parkinson's disease. J Neurol 2000; 247 (Suppl. 2): 103–9 [PubMed] [Google Scholar]
- 18. Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson's disease: conceptual aspects. J Neural Transm 2004; 111: 201–16 [DOI] [PubMed] [Google Scholar]
- 19. Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 1988; 51: 745–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hawkes CH, Shephard BC, Daniel SE. Is Parkinson's disease a primary olfactory disorder? Q J Med 1999; 92: 473–80 [DOI] [PubMed] [Google Scholar]
- 21. Del Tredici K, Rüb U, de Vos RAI, Bohl JRE, Braak H. Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 2002; 61: 413–26 [DOI] [PubMed] [Google Scholar]
- 22. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24: 197–211 [DOI] [PubMed] [Google Scholar]
- 23. Braak H, Rüb U, Del Tredici K. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 2003; 110: 517–36 [DOI] [PubMed] [Google Scholar]
- 24. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res 2004; 318: 121–34 [DOI] [PubMed] [Google Scholar]
- 25. Braak H, Müller CM, Rüb U, Ackermann H, Bratzke H, de Vos RAI, Del Tredici K. Pathology associated with sporadic Parkinson's disease – where does it end? J Neural Transm Suppl 2006; 70: 89–97 [DOI] [PubMed] [Google Scholar]
- 26. Lang AE, Obeso JA. Challenges in Parkinson's disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol 2004; 3: 309–16 [DOI] [PubMed] [Google Scholar]
- 27. Ahlskog JE. Challenging conventional wisdom: the etiologic role of dopamine oxidative stress in Parkinson's disease. Mov Disord 2005; 20: 271–82 [DOI] [PubMed] [Google Scholar]
- 28. Langston JW. The Parkinson's complex: Parkinsonism is just the tip of the iceberg. Ann Neurol 2006; 59: 591–6 [DOI] [PubMed] [Google Scholar]
- 29. Ansari KA, Johnson A. Olfactory function in patients with Parkinson's disease. J Chronic Dis 1975; 28: 493–7 [DOI] [PubMed] [Google Scholar]
- 30. Quinn NP, Rossor MN, Marsden CD. Olfactory threshold in Parkinson's disease. J Neurol Neurosurg Psychiatry 1987; 50: 88–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 1988; 38: 1237–44 [DOI] [PubMed] [Google Scholar]
- 32. Doty RL, Stern MB, Pfeiffer C, Gollomp SM, Hurtig HI. Bilateral olfactory dysfunction in early stage treated and untreated idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 1992; 55: 138–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hawkes CH, Shephard BC, Daniel SE. Olfactory dysfunction in Parkinson's disease. J Neurol Neurosurg Psychiatry 1997; 62: 436–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hummel T, Sekinger B, Wolf SR, Pauli E, Kobal G. ‘Sniffin sticks’: olfactory performance assessed by the combined testing of odor identification, odor discrimination and olfactory threshold. Chem Senses 1997; 22: 39–52 [DOI] [PubMed] [Google Scholar]
- 35. Daum RF, Sekinger B, Kobal G, Lang CJ. Olfactory testing with ‘sniffin’ sticks' for clinical diagnosis of Parkinson's disease. Nervenarzt 2000; 71: 643–50 [DOI] [PubMed] [Google Scholar]
- 36. Liberini P, Parola S, Spano PF, Antonini I. Olfaction in Parkinson's disease: methods of assessment and clinical relevance. J Neurol 2000; 247: 88–96 [DOI] [PubMed] [Google Scholar]
- 37. Hawkes CH. Olfaction in neurodegenerative disorder. Mov Disord 2003; 18: 364–72 [DOI] [PubMed] [Google Scholar]
- 38. Doty RL. Olfaction. Annu Rev Psychol 2001; 52: 423–52 [DOI] [PubMed] [Google Scholar]
- 39. Katzenschlager R, Lees AJ. Olfaction and Parkinson's syndromes: its role in differential diagnosis. Curr Opin Neurol 2004; 17: 417–23 [DOI] [PubMed] [Google Scholar]
- 40. Kobal G, Plattig KH. Objective olfactometry: methodological annotations for recording olfactory EEG‐responses from the awake human. EEG EMG Z Elektroenzephalogr Elektromyogr Verwandte Geb 1978; 9: 135–45 [PubMed] [Google Scholar]
- 41. Barz S, Hummel T, Pauli E, Majer M, Lang CJ, Kobal G. Chemosensory event‐related potentials in response to trigeminal and olfactory stimulation in idiopathic Parkinson's disease. Neurology 1997; 49: 1424–31 [DOI] [PubMed] [Google Scholar]
- 42. Tissingh G, Berendse HW, Bergmanns P, De Waard R, Drukarch B, Stoof JC, Wolters EC. Loss of olfaction in de novo and treated Parkinson's disease: possible implication for early diagnosis. Mov Disord 2001; 16: 41–6 [DOI] [PubMed] [Google Scholar]
- 43. Montgomery EB Jr, Baker KB, Lyons K, Koller WC. Abnormal performance on the PD test battery by asymptomatic first‐degree relatives. Neurology 1999; 52: 757–62 [DOI] [PubMed] [Google Scholar]
- 44. Montgomery EB Jr, Lyons K, Koller WC. Early detection of probable idiopathic Parkinson's disease: II. A prospective application of a diagnostic test battery. Mov Disord 2000; 15: 474–8 [DOI] [PubMed] [Google Scholar]
- 45. Ponsen MM, Stoffers D, Booij J, van Eck‐Smit BL, Wolters EC, Berendse HW. Idiopathic hyposmia as a preclinical sign of Parkinson's disease. Ann Neurol 2004; 56: 173–81 [DOI] [PubMed] [Google Scholar]
- 46. Sommer U, Hummel T, Cormann K, Mueller A, Frasnelli J, Kropp J, Reichmann H. Detection of presymptomatic Parkinson's disease. combining smell tests transcranial sonography and SPECT. Mov Disord 2004; 19: 1196–202 [DOI] [PubMed] [Google Scholar]
- 47. Ross W, Petrovitch H, Abbott RD, Tanner CM, Popper Masaki K. Association of olfactory dysfunction with risk of future Parkinson's disease. Mov Disord 2005; 20 (Suppl. 10): 439 [DOI] [PubMed] [Google Scholar]
- 48. Doty RL, Marcus A, Lee WW. Development of the 12‐item cross‐cultural smell identification test CC‐SIT. Laryngoscope 1996; 106: 353–6 [DOI] [PubMed] [Google Scholar]
- 49. Ross GW, Abbott RD, Petrovitch H, Tanner CM, Davis DG, Nelson J, Markesbery WR, Hardman J, Masaki K, Launer L, White LR. Association of olfactory dysfunction with incidental Lewy bodies. Mov Disord 2006; 21: 2062–7 [DOI] [PubMed] [Google Scholar]
- 50. Marras C, Goldman S, Smith A, Barney P, Aston D, Comyns K, Korell M, Langston JW, Ross GW, Tanner CM. Smell identification ability in twin pairs discordant for Parkinson's disease. Mov Disord 2005; 20: 687–93 [DOI] [PubMed] [Google Scholar]
- 51. Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology 2001; 57: 1497–9 [DOI] [PubMed] [Google Scholar]
- 52. Jost WH. Autonomic dysfunctions in idiopathic Parkinson's disease. J Neurol 2003; 250 (Suppl. 1): 28–30 [DOI] [PubMed] [Google Scholar]
- 53. Micieli G, Tosi P, Marcheselli S, Cavallini A. Autonomic dysfunction in Parkinson's disease. Neurol Sci 2003; 24: 32–4 [DOI] [PubMed] [Google Scholar]
- 54. Visser M, Marinus J, Stiggelbout AM, van Hilten J. Assessment of autonomic dysfunction in Parkinson's disease: the SCOPA‐AUT. Mov Disord 2004; 19: 1306–12 [DOI] [PubMed] [Google Scholar]
- 55. Goetze O, Wieczorek J, Mueller T, Przuntek H, Schmidt WE, Woitalla D. Impaired gastric emptying of a solid test meal in patients with Parkinson's disease using 13C‐sodium octanoate breadth test. Neurosci Lett 2005; 375: 170–3 [DOI] [PubMed] [Google Scholar]
- 56. Bine JE, Frank EM, McDade HL. Dysphagia and dementia in subjects with Parkinson's disease. Dysphagia 1995; 10: 160–4 [DOI] [PubMed] [Google Scholar]
- 57. Muller J, Wenning GK, Verny M, McKee A, Chaudhuri KR, Jellinger K. Progression of dysarthria and dysphagia in postmortem‐confirmed parkinsonian disorders. Arch Neurol 2001; 58: 259–64 [DOI] [PubMed] [Google Scholar]
- 58. Hawkes CH. Smell and Taste Complaints. Boston, MA: Butterworth‐Heinemann/Elsevier, 2002. [Google Scholar]
- 59. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease. Lancet Neurol 2003; 2: 107–16 [DOI] [PubMed] [Google Scholar]
- 60. Tumilasci OR, Cersosimo MG, Belforte JE, Micheli FE, Benarroch EE, Pazo JH. Quantitative study of salivary secretion in Parkinson's disease. Mov Disord 2006; 21: 660–7 [DOI] [PubMed] [Google Scholar]
- 61. Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology 1992; 42: 726–32 [DOI] [PubMed] [Google Scholar]
- 62. Hardoff R, Sula M, Tamir A, Soil A, Front A, Badarna S. Gastric emptying time and gastric motility in patients with Parkinson's disease. Mov Disord 2001; 16: 1041–7 [DOI] [PubMed] [Google Scholar]
- 63. Sulla M, Hardoff R, Giladi N. Gastric emptying time and gastric motility in patients with untreated Parkinson's disease. Mov Disord 1996; 11: 1041–7 [DOI] [PubMed] [Google Scholar]
- 64. Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS, Ross GW. Frequency of bowel movements and the future risk of Parkinson's disease. Neurology 2001; 57: 456–62 [DOI] [PubMed] [Google Scholar]
- 65. Siddiqui MF, Rast S, Lynn MJ, Auchus AP, Pfeiffer RF. Autonomic dysfunction in Parkinson's disease: a comprehensive symptom survey. Parkinsonism Relat Disord 2002; 8: 277–84 [DOI] [PubMed] [Google Scholar]
- 66. Jost WH, Jung G, Schimrigk K. Colonic transit time in non–idiopathic Parkinson's syndrome. Eur Neurol 1994; 34: 329–31 [DOI] [PubMed] [Google Scholar]
- 67. Jost WH, Schrank B. Defecatory disorders in de novo Parkinsonians – colonic transit and electromyogram of the external anal sphincter. Wien Klin Wochenschr 1998; 110: 535–7 [PubMed] [Google Scholar]
- 68. Iwasa K, Nakajima K, Yoshikawa H, Tada A, Taki J, Takamori M. Decreased myocardial 123I‐MIBG uptake in Parkinson's disease. Acta Neurol Scand 1998; 97: 303–6 [DOI] [PubMed] [Google Scholar]
- 69. Satoh A, Serita T, Seto M, Tomita I, Satoh H, Iwanaga K, Takashima H, Tsujihata M. Loss of 123I‐MIBG uptake by the heart in Parkinson's disease: assessment of cardiac sympathetic denervation and diagnostic value. J Nucl Med 1999; 40: 371–5 [PubMed] [Google Scholar]
- 70. Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson's disease. Ann Intern Med 2000; 133: 338–47 [DOI] [PubMed] [Google Scholar]
- 71. Takatsu H, Nishida H, Matsuo H, Watanabe S, Nagashima K, Wada H, Noda T, Nishigaki K, Fujiwara H. Cardiac sympathetic denervation from the early stage of Parkinson's disease: clinical and experimental studies with radiolabeled MIBG. J Nucl Med 2000; 41: 71–7 [PubMed] [Google Scholar]
- 72. Taki J, Yoshita M, Yamada M, Tonami N. Significance of 123I‐MIBG scintigraphy as a pathophysiological indicator in the assessment of Parkinson's disease and related disorders: it can be a specific marker for Lewy body disease. Ann Nucl Med 2004; 18: 453–61 [DOI] [PubMed] [Google Scholar]
- 73. Haapaniemi TH, Pursiainen V, Korpelainen JT, Huikuri HV, Sotaniemi KA, Myllyla VV. Ambulatory ECG and analysis of heart rate variability in Parkinson's disease. J Neurol Neurosurg Psychiatry 2001; 70: 305–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Iwanaga K, Wakabayashi K, Yoshimoto M, Tomita I, Satoh H, Takashima H. Lewy body‐type degeneration in cardiac plexus in Parkinson's and incidental Lewy body diseases. Neurology 1999; 52: 1269–71 [DOI] [PubMed] [Google Scholar]
- 75. Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol (Berl) 2005; 109: 583–8 [DOI] [PubMed] [Google Scholar]
- 76. Kaufmann H, Nahm K, Purohit D, Wolfe D. Autonomic failure as the initial manifestation of Parkinson's disease and dementia with Lewy bodies. Neurology 2004; 63: 1093–5 [DOI] [PubMed] [Google Scholar]
- 77. Lee PH, Yeo SH, Kim HJ, Youm HY. Correlation between cardiac 123I‐MIBG and odor identification in patients with Parkinson's disease and multiple system atrophy. Mov Disord 2006; 21: 1975–7 [DOI] [PubMed] [Google Scholar]
- 78. Adler CH, Thorpy MJ. Sleep issues in Parkinson's disease. Neurology 2005; 64 (Suppl. 3): 12–20 [DOI] [PubMed] [Google Scholar]
- 79. Schenck CH, Bundlie SR, Mahowald MW. REM behavior disorder (RBD), delayed emergence of parkinsonism and/or dementia in 65% of older men initially diagnosed with idiopathic RBD, and an analysis of the minimum & maximum tonic and/or phasic electromyographic abnormalities found during REM sleep. Sleep 2003; 26 (Suppl.): 316 [Abs 0794.M; A316] [Google Scholar]
- 80. Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman‐Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007; doi: 10.1093/brain/awm056 [DOI] [PubMed] [Google Scholar]
- 81. Abbott RD, Ross GW, White LR, Tanner CM, Masaki KH, Nelson JS, Curb JD, Petrovitch H. Excessive daytime sleepiness and subsequent development of Parkinson disease. Neurology 2005; 65: 1442–6 [DOI] [PubMed] [Google Scholar]
- 82. Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology 1996; 46: 388–93 [DOI] [PubMed] [Google Scholar]
- 83. Eisensehr I, Linke R, Tatsch K, Kharraz B, Gildehaus JF, Wetter CT, Trenkwalder C, Schwarz J, Noachtar S. Increased muscle activity during rapid eye movement sleep correlates with decrease of striatal presynaptic dopamine transporters. IPT and IBZM SPECT imaging in subclinical and clinically manifest idiopathic REM sleep behavior disorder, Parkinson's disease, and controls. Sleep 2003; 26: 507–12 [DOI] [PubMed] [Google Scholar]
- 84. Stiasny‐Kolster K, Doerr Y, Möller JC, Hoffken H, Behr TM, Oertel WH, Mayer G. Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for α‐synucleinopathy demonstrated by dopamine transporter FP‐CIT‐SPECT. Brain 2005; 128: 126–37 [DOI] [PubMed] [Google Scholar]
- 85. Postuma RB, Lang AE, Massicotte‐Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurology 2006; 66: 845–51 [DOI] [PubMed] [Google Scholar]
- 86. Fantini ML, Postuma RB, Montplaisir J, Ferini‐Strambi L. Olfactory deficit in idiopathic rapid eye movements sleep behavior disorder. Brain Res Bull 2006; 70: 386–90 [DOI] [PubMed] [Google Scholar]
- 87. Iranzo A, Santamaria J, Rye DB, Valldeoriola F, Marti MJ, Munoz E. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology 2005; 65: 247–52 [DOI] [PubMed] [Google Scholar]
- 88. Braak H, de Vos RAI, Bohl J, Del Tredici K. Gastric alpha‐synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease‐related brain pathology. Neurosci Lett 2006; 396: 67–72 [DOI] [PubMed] [Google Scholar]
- 89. Braak H, Sastre M, Bohl JRE, de Vos RAI, Del Tredici K. Parkinson's disease: Lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre‐ and postganglionic neurons. Acta Neuropathol 2007; 113: 421–9 [DOI] [PubMed] [Google Scholar]
- 90. Braak H, Braak E, Yilmazer D, de Vos RA, Jansen ENH, Bohl J, Jellinger K. Amygdala pathology in Parkinson's disease. Acta Neuropathol 1994; 88: 493–500 [DOI] [PubMed] [Google Scholar]
- 91. Bohus B, Koolhaas JM, Luiten PGM, Korte SM, Roozendaal B, Wiersma A. The neurobiology of the central nucleus of the amygdala in relation to neuroendocrine and autonomic outflow. Prog Brain Res 1996; 107: 447–60 [DOI] [PubMed] [Google Scholar]
- 92. Liubashima O, Jolkkonen E, Pitkänen A. Projections from the central nucleus of the amygdala to the gastric related area of the dorsal vagal complex: a Phaseolus vulgaris leucoagglutinin study in rat. Neurosci Lett 2000; 291: 85–8 [DOI] [PubMed] [Google Scholar]
- 93. Zhang X, Cui J, Tan Z, Jiang C, Fogel R. The central nucleus of the amygdala modulates gut‐related neurons in the dorsal vagal complex in rats. J Physiol 2003; 553: 1005–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I. α‐Synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 2005; 57: 82–91 [DOI] [PubMed] [Google Scholar]
- 95. Jellinger KA. Lewy body‐related alpha‐synucleinopathy in the aged human brain. J Neural Transm 2004; 111: 1219–35 [DOI] [PubMed] [Google Scholar]
- 96. Neumann M, Müller V, Kretzschmar HA, Haass C, Kahle PJ. Regional distribution of proteinase‐K‐resistent α‐synuclein correlates with Lewy body disease stage. Neuropathol Exp Neurol 2004; 63: 1225–35 [DOI] [PubMed] [Google Scholar]
- 97. Saito Y, Ruberu NN, Sawabe M, Arai T, Kazama H, Hosoi T, Yamanouchi H, Murayami S. Lewy body‐related alpha‐synucleinopathy in aging. J Neuropathol Exp Neurol 2004; 63: 742–9 [DOI] [PubMed] [Google Scholar]
- 98. Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amgydala Lewy bodies: a distinct form of alpha‐synucleinopathy. J Neuropathol Exp Neurol 2006; 65: 685–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Müller CM, de Vos RAI, Maurage CA, Thal DR, Tolnay M, Braak H. Staging of sporadic Parkinson disease‐related alpha‐synuclein pathology: inter‐ and intra‐rater reliability. J Neuropathol Exp Neurol 2005; 64: 623–8 [DOI] [PubMed] [Google Scholar]
- 100. Duda JE, Shah U, Arnold SE, Lee VM, Trojanowski JQ. The expression of alpha‐, beta‐, and gamma‐synucleins in olfactory mucosa from patients with and without neurodegenerative diseases. Exp Neurol 1999; 160: 515–22 [DOI] [PubMed] [Google Scholar]
- 101. Witt M, Gudziol V, Haehner A, Reichmann H, Hummel T. Nasal mucosa in patients with Parkinson's disease. Chem Senses 2006; 31: 479–93 (Abstract 106) [Google Scholar]
- 102. Daniel SE, Hawkes CH. Preliminary diagnosis of Parkinson's disease by olfactory bulb pathology. Lancet 1992; 340: 186 [DOI] [PubMed] [Google Scholar]
- 103. Pearce RK, Hawkes CH, Daniel SE. The anterior olfactory nucleus in Parkinson's disease. Mov Disord 1995; 10: 283–7 [DOI] [PubMed] [Google Scholar]
- 104. Huisman E, Uylings HB, Hoogland PV. A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson's disease. Mov Disord 2004; 19: 687–92 [DOI] [PubMed] [Google Scholar]
- 105. Price JL. Olfaction In The Human Nervous System, 2nd edn Eds Paxinos G, Mai. JM. San Diego, CA: Elsevier Academic Press, 2004; 1198–212 [Google Scholar]
- 106. Jager W, den Hartog WA, Bethlem J. The distribution of Lewy bodies in the central and autonomic nervous system in idiopathic paralysis agitans. J Neurol Neurosurg Psychiatry 1960; 23: 283–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Qualman SJ, Haupt HM, Yang P, Hamilton SR. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson's disease. Gastroenterol 1984; 87: 848–56 [PubMed] [Google Scholar]
- 108. Wakabayashi K, Takahashi H. Neuropathology of autonomic nervous system in Parkinson's disease. Eur Neurol 1997b; 38 (Suppl. 2): 2–7 [DOI] [PubMed] [Google Scholar]
- 109. Wakabayashi K, Takahashi H, Ohama E, Ikuta F. Parkinson's disease: an immunohistochemical study of Lewy body‐containing neurons in the enteric nervous system. Acta Neuropathol 1990; 79: 581–3 [DOI] [PubMed] [Google Scholar]
- 110. Wakabayashi K, Takahashi H, Ohama E, Takeda S, Ikuta F. Lewy bodies in the visceral autonomic nervous system in Parkinson's disease. Adv Neurol 1993; 60: 609–12 [PubMed] [Google Scholar]
- 111. Costa M, Brookes SJH, Hennig GW. Anatomy and physiology of the enteric nervous system. Gut 2000; 47 (Suppl. 4): iv15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Furness JB. Types of neurons in the enteric nervous system. J Auton Nerv Syst 2000; 81: 87–96 [DOI] [PubMed] [Google Scholar]
- 113. Anlauf M, Schäfer MKH, Eiden L, Weihe E. Chemical coding of the human gastrointestinal nervous system: cholinergic, VIPergic, and catecholaminergic phenotypes. J Comp Neurol 2003; 459: 90–111 [DOI] [PubMed] [Google Scholar]
- 114. Huang XF, Törk I, Paxinos G. Dorsal motor nucleus of the vagus nerve: a cyto‐ and chemoarchitectonic study in the human. J Comp Neurol 1993; 330: 158–82 [DOI] [PubMed] [Google Scholar]
- 115. Hopkins DA, Bieger D, de Vente J, Steinbusch HWM. Vagal efferent projections: viscerotopy, neurochemistry and effects of vagotomy. Prog Brain Res 1996; 107: 79–96 [DOI] [PubMed] [Google Scholar]
- 116. Huang XF, Paxinos G. Human intermediate reticular zone: a cyto‐ and chemoarchitectonic study. J Comp Neurol 1995, 2004; 360: 571–88 [DOI] [PubMed] [Google Scholar]
- 117. Blessing WW. Lower brain stem regulation of visceral, cardiovascular, and respiratory function In The Human Nervous System, 2nd edn Eds Paxinos G, Mai JK. San Diego, CA: Elsevier Academic Press, 2004; 464–78 [Google Scholar]
- 118. Wakabayashi K, Takahashi H. The intermediolateral nucleus and Clarke's column in Parkinson's disease. Acta Neuropathol 1997a; 94: 287–9 [DOI] [PubMed] [Google Scholar]
- 119. Bloch A, Probst A, Bissig H, Adams H, Tolnay M. α‐Synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol 2006; 12: 284–95 [DOI] [PubMed] [Google Scholar]
- 120. Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF. Alpha‐synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology 2006; 66: 1100–2 [DOI] [PubMed] [Google Scholar]
- 121. Pearson RCA, Esiri MM, Hiorns RW, Wilcock GK, Powell TPS. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer's disease. Proc Natl Acad Sci USA 1985; 82: 4531–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Saper CB, Wainer BH, German DC. Axonal and transneuronal transport in the transmission of neurological disease: potential role in system degenerations, including Alzheimer's disease. Neuroscience 1987; 23: 389–98 [DOI] [PubMed] [Google Scholar]
- 123. Kristensson K. Sorting signals and targeting of infectious agents through axons: an annotation to the 100 years' birth of the name ‘axon’. Brain Res Bull 1996; 41: 327–33 [DOI] [PubMed] [Google Scholar]
- 124. Pearson RCA. Cortical connections and the pathology of Alzheimer's disease. Neurodegeneration 1996; 5: 429–34 [DOI] [PubMed] [Google Scholar]
- 125. Card JP. Exploring brain circuitry with neurotropic viruses: new horizons in neuroanatomy. Anat Rec 1998; 253: 176–85 [DOI] [PubMed] [Google Scholar]
- 126. Mufson EJ, Kroin JS, Sendera TJ, Sobreviela T. Distribution and retrograde transport of trophic factors in the central nervous system: functional implications for the treatment of neurodegenerative diseases. Prog Neurobiol 1999; 57: 451–84 [DOI] [PubMed] [Google Scholar]
- 127. Rinaman L, Levitt P, Card JP. Progressive postnatal assembly of limbic‐autonomic circuits revealed by central transneuronal transport of pseudorabies virus. J Neurosci 2000; 20: 2731–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nicotera P. A route for prion neuroinvasion. Neuron 2001; 31: 345–8 [DOI] [PubMed] [Google Scholar]
- 129. Hill TJ. Ocular pathogenicity of herpes simplex virus. Curr Eye Res 1987; 6: 1–7 [DOI] [PubMed] [Google Scholar]
- 130. Morrison LA, Sidman RL, Fields BN. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci USA 1991; 88: 3852–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Enquist LW, Card JP. Recent advances in the use of neurotropic viruses for circuit analysis. Curr Opin Neurobiol 2003; 13: 603–6 [DOI] [PubMed] [Google Scholar]
- 132. Matsuda K, Park CH, Synden Y, Kimura T, Ochiai K, Kida H. The vagus nerve is one route of transneural invasion for intranasally inoculated influenza A virus in mice. Vet Pathol 2004; 41: 101–7 [DOI] [PubMed] [Google Scholar]
- 133. Broadwell RD, Brightman MV. Entry of peroxidase into neurons of the central and peripheral nervous systems from extracerebral and cerebral blood. J Comp Neurol 1976; 166: 257–84 [DOI] [PubMed] [Google Scholar]
- 134. Elizan TS, Casals J. The viral hypothesis in parkinsonism. J Neural Transm Suppl 1983; 19: 75–88 [PubMed] [Google Scholar]
- 135. Ahlskog JE, Waring SC, Petersen RC, Esteban‐Santillan C, Craig UK, O'Brien PC, Plevak MF, Kurland LT. Olfactory dysfunction in Guamanian ALS, parkinsonism, and dementia. Neurology 1998; 51: 1672–7 [DOI] [PubMed] [Google Scholar]
- 136. Takahashi M, Yamada T. A possible role of influenza A virus infection for Parkinson's disease. Adv Neurol 2001; 86: 91–104 [PubMed] [Google Scholar]
- 137. Gamboa ET, Wolf A, Yahr MD, Harter DH, Duffy PE, Barden H, Hsu KC. Influenza virus antigen in postencephalitic parkinsonism brain. Arch Neurol 1974; 31: 228–32 [DOI] [PubMed] [Google Scholar]
- 138. Marttila RJ, Halonen P, Rinne UK. Influenza virus antibodies in Parkinsonism. Comparison of postencephalic and idiopathic Parkinson patients and matched controls. Arch Neurol 1977; 34: 99–100 [DOI] [PubMed] [Google Scholar]
- 139. Elizan TS, Madden DL, Noble GR, Herrmann KL, Gardner J, Schwartz J, Smith H, Sever JL, Yahr MD. Viral antibodies in serum and CSF of parkinsonian patients and controls. Arch Neurol 1979; 36: 529–34 [DOI] [PubMed] [Google Scholar]
- 140. Schwartz J, Elizan TS. Search for viral particles and virus‐specific products in idiopathic Parkinson disease brain material. Ann Neurol 1979; 6: 261–3 [DOI] [PubMed] [Google Scholar]
- 141. Wetmur JG, Schwartz J, Elizan TS. Nucleic acid homology studies of viral nucleic acids in idiopathic Parkinson's disease. Arch Neurol 1979; 36: 462–4 [DOI] [PubMed] [Google Scholar]
- 142. Solbrig MV, Nashef L. Acute parkinsonism in suspected herpes simplex encephalitis. Mov Disord 1993; 8: 233–4 [DOI] [PubMed] [Google Scholar]
- 143. Howard JS. Tic douloureux, Parkinson's disease and the herpes connection. Integr Physiol Behav Sci 1997; 32: 257–64 [DOI] [PubMed] [Google Scholar]
- 144. Marttila RJ, Rinne UK, Halonen P, Madden DL, Sever JL. Herpesviruses and Parkinsonism. Herpes simplex virus types 1 and 2, and cytomegalovirus antibodies in serum and CSF. Arch Neurol 1981; 38: 19–21 [DOI] [PubMed] [Google Scholar]
- 145. Marttila RJ, Rinne UK, Tilikainen A. Virus antibodies in Parkinson's disease. J Neurol Sci 1982; 54: 227–38 [DOI] [PubMed] [Google Scholar]
- 146. Fazzini E, Fleming J, Fahn S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson's disease. Mov Disord 1992; 7: 153–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bower JH, Maraganore DM, Peterson BJ, Ahlskog JE, Rocca WA. Immunologic diseases, anti‐inflammatory drugs, and Parkinson disease: a case‐control study. Neurology 2006; 67: 494–6 [DOI] [PubMed] [Google Scholar]
- 148. Klimek L. Sense of smell in allergic rhinitis. Pneumologie 1998; 52: 196–202 [PubMed] [Google Scholar]
- 149. Masucci MG. Epstein‐Barr virus oncogenesis and the ubiquitin‐proteasome system. Oncogene 2004; 23: 2107–15 [DOI] [PubMed] [Google Scholar]
- 150. Shackelford J, Pagano JS. Targeting of host‐cell ubiquitin pathways by viruses. Essays Biochem 2005; 41: 139–56 [DOI] [PubMed] [Google Scholar]
- 151. Tomlinson AH, Esiri MM. Herpes simplex encephalitis: immuno‐histological demonstration of spread of virus via olfactory pathways in mice. J Neurol Sci 1983; 60: 473–84 [DOI] [PubMed] [Google Scholar]
- 152. Shipley MT. Transport of molecules from nose to brain: transneuronal anterograde and retrograde labelling in the rat olfactory system by wheatgerm agglutin‐horseradish peroxidase applied to the nasal epithelium. Brain Res Bull 1985; 15: 129–42 [DOI] [PubMed] [Google Scholar]
- 153. Newman R, Winans SS. An experimental study of the ventral striatum of the golden hamster. II. Neuronal connections of the olfactory tubercle. J Comp Neurol 1980; 191: 193–212 [DOI] [PubMed] [Google Scholar]
- 154. Blessing WW, Li YW, Wesselingh SL. Transneuronal transport of herpes simplex virus from the cervical vagus to brain neurons with axonal inputs to central vagal sensory nuclei in the rat. Neuroscience 1991; 42: 261–74 [DOI] [PubMed] [Google Scholar]
- 155. Park CH, Ishinaka M, Takada A, Kida H, Kimura T, Ochiai K, Umimura T. The invasion routes of neurovirulent A/Hong Kong/483/97, H5N1; influenza virus into the central nervous system after respiratory infection in mice. Arch Virol 2002; 147: 1425–36 [DOI] [PubMed] [Google Scholar]
- 156. Shinya K, Shimada A, Ito T, Otsuki K, Morita T, Tanaka H, Takada A, Kida H, Umemura T. Avian influenza virus intranasally inoculated infects the central nervous system of mice through the general visceral afferent nerve. Arch Virol 2000; 145: 187–95 [DOI] [PubMed] [Google Scholar]
- 157. Halliday G, Del Tredici K, Braak H. Critical appraisal of the Braak staging of brain. pathology related to sporadic Parkinson's disease. J Neural Transm (Suppl) 2006; 70: 99–103 [DOI] [PubMed] [Google Scholar]