

Abstract
Pediatric cancer is a leading cause of death in children and adolescents. Improvements in pediatric cancer treatment that include the alleviation of long-term side effects require a deeper understanding of the genetic, epigenetic and developmental factors driving these cancers. Here, we review how the unique attributes of the zebrafish model system in embryology, imaging and scalability have been used to identify new mechanisms of tumor initiation, progression and relapse and for drug discovery. We focus on zebrafish models of leukemias, neural tumors and sarcomas - the most common and difficult childhood cancers to treat.
Keywords: Zebrafish, Pediatric, Cancer, Leukemia, Sarcoma, Brain Tumors
Childhood and Adolescent Cancer
Pediatric cancer is one of the leading causes of death in children and adolescents (ages 0 to 19) and is mainly comprised of leukemias, cancers of the nervous system, and sarcomas [1]. Recent genomic profiling efforts to better understand the etiology of this group of diseases have enabled the stratification of tumor types based on molecular signatures and have led to the identification of potential genetic drivers and cooperating molecular events that underlie the development of different pediatric cancers. Interestingly, most pediatric malignancies are mutationally quiet and are thought to be driven by a single driver gene, a fusion oncoprotein or structural/copy number alterations [2,3]. In contrast, adult tumors frequently exhibit high mutational burdens, likely due to a longer period of mutational acquisition under selective pressure [2,3]. Despite these differences, however, pediatric cancer treatments are still largely modeled after treatments designed for the adult version of the disease and can cause debilitating, long-term side effects when administered to children. The development of robust pre-clinical pediatric cancer models that accurately recapitulate these diseases will ultimately be necessary for the design of more precise, targeted therapies to improve outcomes for pediatric cancer patients. Here, we discuss how the zebrafish model has advanced the pediatric cancer field as a preclinical model for gene and drug discovery.
Zebrafish in Cancer Research
The zebrafish (Danio rerio) was established as a developmental biology model because it is uniquely amenable to forward genetics, embryology and imaging, allowing the discovery of new mechanisms controlling embryogenesis, neurogenesis and organ formation. These features also make zebrafish an attractive model to study cancer because: (1) optically-transparent zebrafish embryos and adults enables direct observation of tumor cell behavior in vivo, (2) rapid production of zebrafish animals provide a highly scalable platform for genetic and drug screening, and (3) significant conservation of cancer signaling pathways between fish and human allow identification of new molecular mechanisms of tumorigenesis.
Over fifty genetically-engineered zebrafish models of human cancer have been established that closely resemble their human counterparts at the histological and/or genomic levels [4,5]. Zebrafish cancer models have accelerated the discovery of new mechanisms driving human cancers and identified new drugs for clinical trials [6]. A number of recent reviews detailing the experimental approaches for generating different leukemia [7], sarcoma [8], neuroblastoma [9] and germ-cell tumor [10] models are published elsewhere. Here, we focus on how genetically engineered zebrafish lines that specifically model pediatric and adolescent cancers, which we define as zebrafish tumors that arise within the first 90 days of life, have informed pediatric cancer research and therapeutics (see Figure 1 (Key Figure) and Table 1).
Figure 1. Key Figure. Frequency of Pediatric Cancers and Corresponding Anatomical Location in Zebrafish.
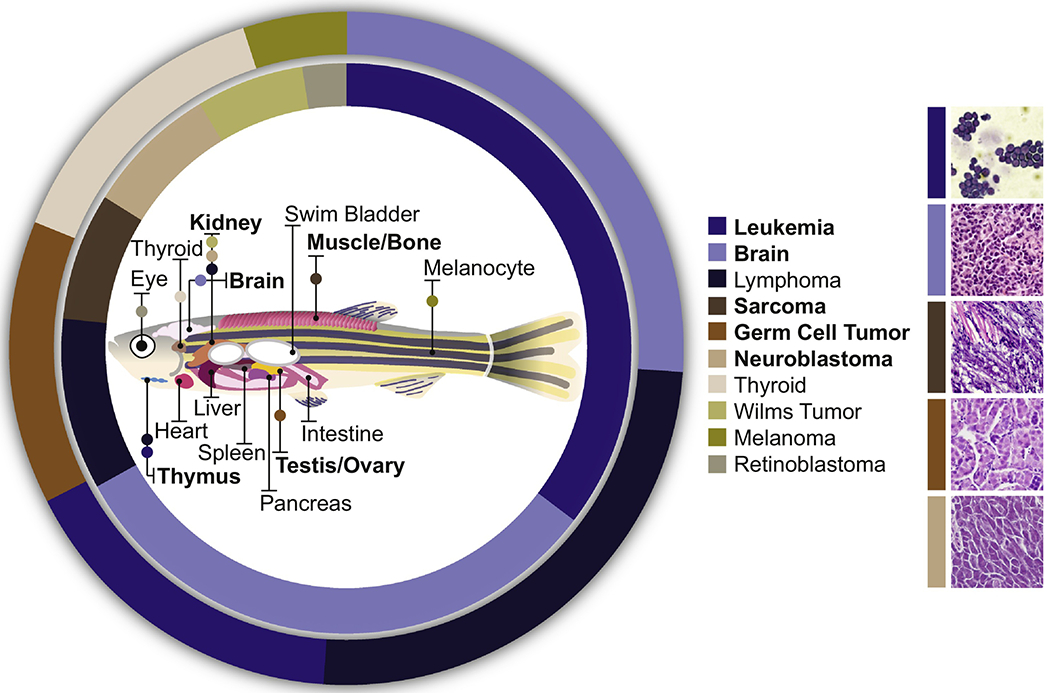
The frequency of childhood (inner circle) and adolescent (outer circle) cancers (as reported in [11,36]) with corresponding anatomical tumor location in zebrafish is illustrated. Zebrafish models have been developed for pediatric leukemia, brain tumors, sarcomas, germ-cell tumors and neuroblastoma (bolded). Representative histology of these zebrafish models is shown on the right. Histology images from: [26] (T-ALL), [39] (CNS-PNET), [62] (ERMS), [71] (Germ-Cell Tumor), and [50] (NB).
Table 1:
Models of pediatric cancer in zebrafish
| Cancer | Driver Oncogene | Model | Oncogene Description | Reference |
|---|---|---|---|---|
| T-ALL | Myc | Tg(rag2:EGFP-mMyc) | Amplification of Myc | [15] |
| Myc | Tg(rag2-loxP-dsRED2-loxP-EGFP-mMyc) | Cre inducible Myc amplification | [16] | |
| Myc | Tg(rag2:LDL-EGFP-mMyc); Tg(Hsp70:Cre) | Heat-shock-inducible Myc amplification | [17] | |
| MYC | Tg(rag2:hMYC-ER) | 4-hydroxytamoxifen inducible MYC amplification | [18] | |
| NOTCH | Tg(rag2:NICD-EGFP) | NOTCH1 intracellular domain amplification | [20] | |
| ARID5B | Tg(rag2:ARID5B) | ARID5B amplification | [23] | |
| B-ALL/T-ALL | MYC | Tg(rag2:hMYC) | Mixed-lineage ALL with MYC amplification | [30] |
| B-ALL | TEL(ETV6)-AML1(RUNX1) | Tg(XEF:EGFP-TEL-AML1) or Tg(ZBA:EGFP-TEL-AML1) | TEL-AML1 fusion | [29] |
| AML | AML1-ETO | Tg(hsp70:AML1-ETO) | AML1-ETO fusion | [32] |
| FLT3 | Tg(CMV:FLT3-ITD) or Tg(CMV:FLT3-TKD) | FLT3-ITD (internal tandem duplication) or FLT3-TKD (tyrosine kinase inhibitor resistant mutation D835Y) | [33] | |
| CNS-PNET | nf1a/b | nf1a−/−;nf1b−/− | Loss of nf1a/b | [41] |
| rb1 | rb1−/− | Loss of rb1 | [42,43] | |
| NRAS | Tg(sox10:mCherry-NRASWT) or Tg(sox10:mCherry-NRASQ61R) | Amplification of wild-type or activated NRAS | [39] | |
| NB | MYCN | Tg(dβh:EGFP-MYCN) | Amplification of EGFP-MYCN | [48] |
| MYCN | Tg(dβh:MYCN;dβh:EGFP) | Amplification of MYCN | [49] | |
| cMYC | Tg(dβh:cMYC;dβh:EGFP) | Amplification of cMYC | [53] | |
| ERMS | KRAS | Tg(rag2:KRASG12D) | Activating KRAS mutation | [60] |
| KRAS | Tg(cdh15:KRASG12D) | Activating KRAS mutation | [61] | |
| ARMS | PAX3-FOXO1 | Tg(CMV-GFP-PAX3-FOXO1) | PAX3-FOXO1 fusion | [69] |
Leukemia
Acute lymphoblastic leukemia (ALL) arises in T- and B-lymphoblasts in the bone marrow and is the most commonly diagnosed cancer in children [11]. Current 5-year survival rates for these children are upwards of 90% with typical treatments involving chemotherapy and allogeneic bone marrow transplantation for high-risk or recurrent cases [11]. Although survival for these patients is high, there are many long-term detrimental side effects to the current treatments [11]. For these reasons, current efforts are focused on reducing treatment toxicity and developing targeted therapies for high-risk patients and those with recurrent disease.
T-Cell Acute Lymphoblastic Leukemia
T-Cell ALL (T-ALL) accounts for 15% of ALL cases that arise in children [12]. T-ALL forms in the thymus from immature thymocytes that have acquired genetic or epigenetic changes and migrated to the bone marrow, peripheral blood and lymph nodes. Genetic alterations in T-ALL patients range from translocations, gene fusions, chromosomal gains and deletions, and epigenetic abnormalities [12,13]. Unlike most other pediatric cancers that differ significantly from adult cancers in mutational landscape, both pediatric and adult T-ALLs exhibit frequent alterations in the NOTCH, PI3K-AKT, JAK-STAT, and RAS pathways [14]. Early zebrafish models of T-ALL recapitulated mouse and human studies and rapidly identified novel mechanisms of tumor progression and survival in this disease.
Zebrafish Models of Myc-driven T-ALL
The first genetically-engineered zebrafish model of cancer was classified as T-ALL, in which the recombination activating gene 2 (rag2) promoter was used to drive expression of the mouse proto-oncogene Myc in T- and B-lymphocytes [15]. In the Tg(rag2:mMyc) transgenic model, mosaic expression of Myc induces T-ALL in 6% of animals, with a mean latency of 44 days [15]. Conditional germline transgenic models of Myc-driven T-ALL have now been established that use one of three inducible approaches: 1) injecting Cre mRNA into Tg(rag2-loxP-dsRED2-loxP-EGFP-mMyc) embryos [16], 2) crossing Tg(rag2:LDL-EGFP-mMyc) and Tg(hsp70:Cre) fish [17], and 3) treating Tg(rag2:hMYC-ER) fish with 4-hydroxytamoxifen [18]. As detailed below, the increased reproducibility and tumor penetrance in these refined models (80-100% tumor penetrance by 35 days of life) have allowed the identification of new genes and/or genetic pathways that promote or repress Myc-driven T-ALL initiation, progression and survival as well as the discovery of drugs that inhibit tumorigenesis in vivo.
Zebrafish Models of Notch-driven T-ALL
The NOTCH signaling pathway is aberrantly activated in over 60% of T-ALL cases [13] and can promote T-ALL tumorigenesis through both MYC-dependent and -independent pathways [19,20]. Moreover, NOTCH1 and Myc collaborate during T-ALL pathogenesis in zebrafish since rag2-mediated expression of both Myc and the constitutively active intracellular domain of NOTCH1 (NICD) enhanced T-ALL progression compared to expression of either Myc or NICD alone [20]. Interestingly, enhanced Notch signaling did not increase the number of leukemia-propagating cells, indicating that NOTCH controls pre-malignant cell expansion, while Myc primarily drives clonal cell growth and survival [20]. Notch pathway mutations are also associated with activation of the Hedgehog pathway in ~16% of relapsed pediatric T-ALL cases [21]. Loss of ptch1, a negative regulator of the Hedgehog pathway, accelerated the onset of notch1-induced T-ALL in zebrafish, suggesting that activation of the Hedgehog signaling pathway cooperates with NOTCH1 to drive T-cell transformation [21]. These zebrafish models highlight the therapeutic potential for targeting multiple developmental pathways during T-ALL disease progression.
Identifying New Genetic Mechanisms in T-ALL Tumorigenesis
Zebrafish have been used to identify new oncogenic co-factors that drive T-ALL pathogenesis by different mechanisms. For example, the thymocyte selection-associated high mobility group box factor TOX was found to synergize with Myc, as well as NICD, to promote T-ALL in zebrafish by inducing genomic instability through impaired non-homologous-end-joining DNA repair [22]. Elevated expression of the ARID family of genes resulted in T-ALL tumors that had elevated myc levels, suggesting that ARID5B contributes to the overexpression of Myc during T-ALL tumorigenesis [23]. Aberrant lymphocyte-specific expression of JDP2, a gene that encodes a bZIP protein whose expression correlates with poor overall survival in pediatric T-ALL cases [24], was sufficient to drive T-ALL in over 50% of zebrafish at 40 weeks post-fertilization (wpf) and caused a higher tumor penetrance when co-expressed with Myc [24]. These studies therefore revealed new mechanisms that could potentially be targeted to disable MYC-driven T-ALL tumors.
Identifying New Therapeutics for T-ALL using Zebrafish
Pediatric leukemia patients have a greater-than-90% chance of survival. Yet, current treatment regimens cause systemic toxicities, and a subset of patients eventually relapse. To identify novel T-ALL therapies to potentially address this problem, a small molecule screen of 4,880 FDA-approved compounds was performed using the zebrafish Tg(rag2:MYC-ER) model of T-ALL [25]. Perphenazine, a phenothiazine antipsychotic, was found to reduce thymic fluorescence in embryo assays, overall tumor burden in zebrafish with established MYC-induced T-ALL, and growth of human T-ALL cells in a murine xenograft model [25]. In Myc;Akt-driven T-ALLs, the combination of the glucocorticoid dexamethasone and an AKT inhibitor (MK2206) was highly effective against leukemia-propagating cells [26]. Using serial transplantation, MK2206 was also shown to overcome dexamethasone resistance, a finding recapitulated in human T-ALL cells [26]. MK2206 entered a phase I clinical trial for refractory childhood malignancies and was well tolerated at tested doses, but only a limited benefit has been observed so far [27]. Interestingly, expression of jdp2 was found to confer dexamethasone resistance in zebrafish Myc-driven T-ALL models [24]. The zebrafish data suggests that stratification of patients with activation of the AKT pathway and/or elevated JDP2 could be useful for predicting responses to therapy involving glucocorticoids.
Zebrafish Models of B-Cell Acute Lymphoblastic Leukemia
B-cell ALL (B-ALL) comprises 85% of all childhood ALL cases. While B-ALL and T-ALL are morphologically indistinguishable [12], each disease is characterized by distinct molecular subtypes. Similar genetic and chromosomal abnormalities exist in both adult and pediatric B-ALL, including fusion genes such as BCR-ABL1 and ETV6-RUNX, MLL-rearrangements, hyperploidy and hypoploidy, albeit at different frequencies [28]. The first model of B-ALL in zebrafish ubiquitously expressed TEL-AML1 (ETV6-RUNX) using the xenopus elongation factor 1α and the zebrafish β-actin promoters to generate B-ALL tumors with 3% penetrance [29]. In addition, it was recently found that a subset of animals from the Tg(rag2:hMYC) line also develop B-ALL tumors with distinct gene expression profiles from the T-ALL tumors [30], indicating that this transgenic line actually models mixed-lineage ALL. These studies emphasize the need to incorporate modern comparative onco-genomics methods to verify the exact type and/or sub-type of human tumor that the zebrafish lines are modeling.
Zebrafish Models of Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is defined by an accumulation of immature myeloid cells that comprises at least 20% of the patient’s bone marrow. The overall 5-year survival rate for AML is 64%, but rates differ significantly depending on the molecular subtype [11]. Unlike other liquid malignancies, some pediatric AML patients have strikingly different somatic variations than those observed in adult AML. Thus, childhood AML is uniquely typified by gene fusion events such as AML1-ETO (also called RUNX1-RUNX1T1), NUP98-NSD1, and KMT2A-MLLT3 [31]. Expression of AMLl-ETO driven by the hsp70 promoter [32], and FLT3-ITD or FLT3-TKD driven by the CMV promoter [33], led to myeloid cell expansion in the early zebrafish embryo. Hematopoietic defects in AML1-ETO embryos could be reversed using the COX2 inhibitor Nimesulide to block a β-catenin-mediated increase in myelopoiesis [34]. Additionally, myeloid expansion in FLT3-ITD, but not FLT3-TKD, animals could be abrogated by treatment with a tyrosine kinase inhibitor (AC220), suggesting the existence of different therapeutic opportunities for seemingly similar genetic aberrations [33]. These studies illustrate the utility of the zebrafish embryo for understanding basic mechanisms of oncogenic fusion genes in hematopoiesis and for the discovery of new therapies for pediatric leukemia.
Brain and CNS Tumors
Brain and central nervous system (CNS) tumors are the leading cause of pediatric-cancer-related deaths [35]. Approximately 75% of pediatric brain tumors are malignant and have a collective 5-year survival rate of 78% [36]. Moreover, surgical debulking of the tumor, radiation and general chemotherapy cause long-term complications on a developing brain [11]. Fortunately, significant progress in the genomic characterization of pediatric brain tumors has identified specific brain tumor entities with distinct molecular profiles, allowing for more accurate classification of tumors with recurrent genomic features. The challenge now is to generate cell- and animal-based models of the new brain tumor entities for gene and drug discovery.
Zebrafish Models of Pediatric Brain Cancer
Models of Central Nervous System Primitive Neural Ectodermal Tumors
Central nervous system primitive neural ectodermal tumors (CNS-PNETs) typically arise in the cerebrum and account for 3-5% of pediatric brain tumors [37,38]. One subgroup of CNS-PNET, called CNS NB-FOXR2, expresses transcription factors critical for oligodendrocyte precursor cells (OPCs) and activation of the RAS/MAPK pathway [39]. To model this subgroup, we activated NRAS signaling in sox10-expressing OPCs [39], which initiated brain tumor formation by 6 wpf, showing OPCs are a cell of origin for these tumors and can be transformed by activated RAS/MAPK signaling. Histological and cross-species genomic analysis showed that the zebrafish brain tumors were significantly more similar to the human CNS NB-FOXR2 tumors than any other CNS-PNET subtypes or even normal zebrafish brain. Strikingly, the zebrafish CNS NB-FOXR2 tumors were selectively sensitive to MEK inhibitor (AZD6244) treatment, showing dramatic reductions in tumor burden and an increase in overall survival [39]. AZD6244 is currently in clinical trials for treatment of pediatric brain tumors and could be effectively repurposed for the treatment of CNS NB-FOXR2 patients [40]. These studies show how zebrafish can be used to rapidly test the cellular origin and pathway activation predictions arising from recent genomic data that now exists for pediatric brain tumor entities, including rare subgroups, for the development of targeted therapies.
The zebrafish appears to be an excellent animal model for testing genetic drivers of OPC-derived pediatric tumors. For example, loss of the neurofibromatosis-1a/b (nf1a/b) or retinoblastoma-1 (rb1) tumor suppressor genes in zebrafish, often with p53 deficiency, generates brain tumors that resemble oligodendrogliomas [41] or embryonal tumors similar to CNS-PNETs [42,43]. Comparative genomic analysis between the Tg(sox10:NRAS) model [39] and the rb1-deficient models [43] showed a significant overlap in gene expression changes. These studies show that OPC-derived pediatric tumors acquire genetic or epigenetic changes that culminate in activation of the E2F family of transcription factors, and suggest that such tumors will be sensitive to inhibitors of E2F, such as HLM006474 [44], regardless of the initial oncogenic drivers.
Neuroblastoma
Neuroblastoma (NB) is a solid tumor that arises from the neural-crest-derived peripheral sympathetic nervous system. It is the most common non-cranial solid tumor in infants and often presents in the adrenal glands [45,46]. Treatments for NB patients typically include surgical resection, chemotherapy, radiation and bone marrow transplantation [47]. Currently, there is a need to better understand the mechanisms underlying high-risk NB, as these patients only have a 50% five-year survival rate [46]. The zebrafish system has been extremely valuable for identifying new oncogenic drivers, cooperating genetic mutations and developmental transcriptional networks that promote high-risk NB, as well as new treatment paradigms.
Zebrafish Models of Neuroblastoma
Amplification of MYCN is observed in ~20% of NBs and is associated with a poor prognosis [46]. Zebrafish models of NB are driven by targeted expression of the human MYCN gene to the developing peripheral sympathetic nervous system using the dopamine-β-hydroxylase (dβh) promoter [9]. Zebrafish models of NB with both low (17%) and high (70%) penetrance have been developed in order to detect cooperating mutations that either increase or decrease tumor burden, respectively [48,49]. The low-penetrance NB model Tg(dβh:EGFP-MYCN) expresses an EGFP-MYCN fusion protein, while the highly penetrant Tg(dβh:EGFP;dβh:MYCN) model involves the co-integration, but not fusion, of the EGFP and MYCN genes [48,49]. In both models, the zebrafish NB tumors histologically resemble human NB and arise predominately in the inter-renal gland, the zebrafish equivalent of the human adrenal medulla.
The low-penetrance Tg(dβh:EGFP-MYCN) NB model has been useful for the identification of a number of genes that cooperate with MYCN to accelerate NB tumorigenesis. For example, patients with MYCN amplification frequently present with modifications in the RAS signaling pathway, including mutations in ALK, NF1, and PTPN11 [9]. Co-expression of MYCN with ALKF1174L [48], PTPN11E69K [50], or loss of nf1 [51] reduced the latency to tumor onset and increased the rate of tumor penetrance. These studies show that activation of the RAS pathway is an important event in generating aggressive NB in vivo and that RAS pathway inhibitors may be effective in slowing or preventing disease progression in MYCN-amplified NB. Importantly, ALK inhibitors are currently being assessed for clinical efficacy in high-risk NB [52].
High-risk NB patients frequently exhibit large genomic rearrangements that include chromosomal gains and losses [45]. As such, the zebrafish NB model has been used to screen for potential malignancy-associated genes that reside in regions of chromosomal imbalance. Candidate genes located at chromosome 1q, which represents a typical chromosomal gain in a subset of high-risk NB cases, were tested for their ability to modify the penetrance of the Tg(dβh:EGFP-MYCN) NB model, and the digestive organ expansion factor (DEF) gene was identified as a cooperating driver of NB in vivo [49]. In addition, def haploinsufficiency significantly reduced tumor growth in the highly penetrant Tg(dβh:EGFP;dβh:MYCN) strain as well as human NB cell lines by inducing apoptosis [49]. Thus, DEF could represent a new therapeutic target for NB patients with chromosome lq gains.
Almost 50% of high-risk NB cases do not have MYCN amplification, so other genetic or epigenetic events likely drive these highly malignant cancers. For example, a recent study showed cMYC, a homolog of MYCN, was sufficient to drive aggressive NB in zebrafish [53]. This was the first animal model generated to support data from human cells and patient samples showing that 10% of MYCN-independent high-risk NBs have aberrant expression of cMYC mRNA and protein levels due to focal amplification of distant enhancers or enhancer hijacking via 8q24 translocations [53]. Thus, high-risk NB patients can now be stratified based on expression of MYCN versus cMYC protein to eventually support the administration of precision therapy.
A significant clinical challenge in NB is the treatment of metastasis, which is often present at the time of NB diagnosis. Genome-wide association studies identified the LIM-only domain gene (LMO1) as strongly associated with high-grade, metastatic NB tumors [54]. Interestingly, co-expression of LMO1 with MYCN led to only mild alterations in tumor penetrance and onset but significantly increased the development of NB metastases [55]. LMO1 overexpression also enhanced the migration capacity of human NB cells and upregulated genes involved in extracellular matrix remodeling [55]. These studies have paved the way for the rapid identification of other genetic events and/or drugs that impact NB metastasis in vivo [50,51,56].
Sarcoma
Sarcomas of the soft tissue and bone represent 7% of all childhood cancers [11]. The most common sarcoma to arise in children and adolescents is rhabdomyosarcoma. It forms from undifferentiated muscle cells and typically arises in the head/neck, genitourinary tract or extremities [11]. There are two major histological subtypes of rhabdomyosarcoma, embryonal (ERMS) and alveolar (ARMS). ERMS accounts for 75% of all RMS cases and typically features loss of heterozygosity at the 11p15.5 locus or point mutations in MYOD1, FGFR4, as well as in any one of the genes encoding the major RAS GTPases [11,57]. In contrast, 80% of ARMS patients exhibit overexpression of the PAX3-FOXO1 or PAX7-FOXO1 fusion gene [58]. RMS therapies, which are limited to local resection, radiation, and multi-agent chemotherapy, have remained largely unchanged for the last 50 years [11], and despite advances in next generation sequencing and molecular typing of patients, the 5-year survival rate remains at 82% for ERMS and 65% for ARMS [59]. As described below, zebrafish RMS models have significantly contributed to our understanding of the etiology of these pediatric cancers.
Zebrafish Models of Fusion-negative Rhabdomyosarcoma
The most studied model of zebrafish ERMS was generated using a rag2 promoter to drive the expression of a constitutively active KRAS (KRASG12D) gene [60]. In this model, KRASG12D is aberrantly expressed in muscle satellite cells (due to aberrant activation of the rag2 promoter fragment in non-lymphoid cells) in a mosaic fashion [60]. Nearly 50% of these fish develop tumors rapidly (10-80 days-post-fertilization (dpf)) [60]. RNA in situ hybridization studies using clinical markers of human RMS as well as gene set enrichment analysis (GSEA), confirmed that the genomic landscape of zebrafish tumors closely resembles human ERMS [60]. These studies showed that muscle satellite cells represent at least one cell of origin for ERMS [61].
Tumor recurrence is a significant clinical challenge for the effective treatment of ERMS. The zebrafish ERMS model has been used to identify potential genes that influence the behavior of tumor-propagating cells (TPCs) during ERMS initiation. The Tg(rag2:KRASG12D) ERMS driver was co-expressed with genes previously demonstrated to be important in muscle regeneration and stem-cell self-renewal, including rag2-ICN1 (zebrafish intracellular activated NOTCH1) [62], rag2-Vangl2 [63], or mylpfa-myf5 [64]. These studies showed that Notchl activation inhibited the muscle differentiation factor MEF2C and thereby caused the dedifferentiation of ERMS cells into self-renewing myf5-positive TPCs [62]. Similarly, activation of Vangl2, which controls the non-canonical Wnt/planar-cell-polarity pathway, increased TPC numbers through regulation of the downstream RhoA GTPase [63]. Importantly, the genetic mechanisms controlling TPCs in ERMS were shown to be conserved in human cells using mouse RMS xenografts as well as in vitro sphere-formation assays. Thus, the zebrafish model of ERMS has been instrumental in the discovery of new, conserved mechanisms of tumor recurrence and ERMS self-renewal that can now be explored in pre-clinical settings to improve survival for children with recurrent or metastatic disease.
The zebrafish Tg(rag2:KRASG12D) model has been used as a pre-clinical model to test the efficacy of drugs that inhibit KRAS signaling during ERMS tumor growth [65]. Pharmacological inactivation of downstream KRAS effectors, such as MEK (PD98059) and S6K1 (TPCK), acted synergistically suggesting that dual inhibition of the MAPK and AKT pathway could be an effective strategy to target KRAS-driven ERMS [65]. Furthermore, in both zebrafish and human RMS cells, inhibition of the RAS and mTOR pathways inhibited translation initiation, providing a mechanistic basis for the growth-inhibiting effects of these drugs in ERMS [65]. Similar studies have identified a VEGFA inhibitor (Cediranib) that decreased ERMS tumor growth by three-fold and reduced microvessel density, suggesting that VEGFA signaling may promote ERMS tumor progression through angiogenesis [66]. Additionally, inhibitors of GSK3 (6-bromoindiubin-3’-oxime) and HDAC (trichostatin A or vorinostat) blocked the growth of ERMS tumors [67] and promoted cellular differentiation [68]. Importantly, all of these studies utilized human RMS-cell-based models to show conservation of drug effects on cell growth/proliferation, differentiation and migration, supporting the likelihood that these drugs will also demonstrate efficacy in the clinic.
Zebrafish Models of Fusion-positive Sarcomas
A zebrafish model of ARMS was recently developed by ubiquitously expressing the human PAX3-FOXO1 fusion gene using the CMV promoter in a p53-deficient genetic background [69]. Thus, in contrast to the ERMS model, the cell of origin is unknown in ARMS. Nonetheless, these studies showed that the PAX3-FOXO1 human fusion oncogene is sufficient to transform zebrafish cells into either ARMS tumors or CNS-PNETs with distinct histological features [69]. PAX3-FOXO1 was found to cause aberrant expression of the transcription factor hes3 in muscle cells, which blocks the expression of muscle differentiation markers [69]. Consistently, expression of human HES3 in zebrafish and muscle progenitors inhibits myogenesis, and increased HES3 expression in human RMS patients signifies a poor prognosis [69]. This zebrafish model has therefore provided important insight into the conserved genetic mechanisms through which the PAX3-FOXO1 fusion gene mediates tumorigenesis. Indeed, an important advance from studies on fusion-positive sarcomas is the ability to use the zebrafish system to rapidly test the transforming potential of fusion oncoproteins without prior knowledge of the cell of origin.
Concluding Remarks
Cancer in children is a relatively rare occurrence compared to adult cancers. As such, many pediatric tumor entities have no associated cell-line- or animal-based model system with which to define the mechanisms of disease pathogenesis or test potential therapeutics. Preclinical drug studies traditionally require costly and time-consuming rodent models to assess the efficacy of novel therapies, and the smaller size of the pediatric cancer population often discourages the development of new therapies by industry due to limited financial returns. Thus, in our opinion, the most important benefit of the zebrafish system to pediatric cancer research is the ability to develop rare tumor subtypes in a timely and affordable manner for preclinical drug discovery, as described above for leukemias, brain tumors, neuroblastoma and sarcomas. In addition, the unique imaging attributes of the zebrafish represent a powerful tool for directly monitoring tumor cell behaviors during invasion and metastasis, as well as response to drug treatments. Such attributes are equally important for studying adult cancers and have been recently reviewed elsewhere [70]. These features and a comparison of the different advantages of zebrafish and murine models for pediatric cancer research is described in Figure 2.
Figure 2: Comparison of Zebrafish and Mouse Models of Pediatric Cancer.
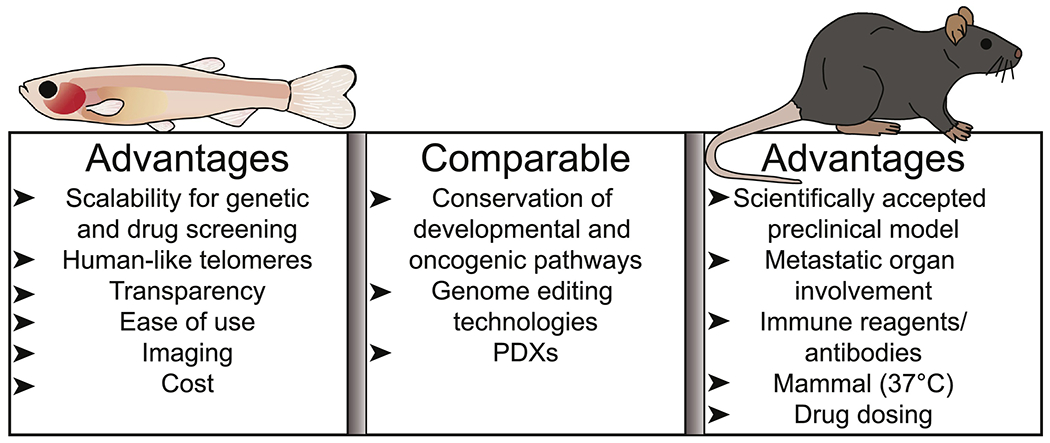
Zebrafish and mouse models of pediatric cancer have complementary attributes for identifying new mechanisms driving childhood tumors. Both model systems have conserved developmental and oncogenic pathways, can be utilized for PDXs, and have genome-editing technologies available. Zebrafish have unique advantages in scalability, cost and imaging. In contrast, mice have conserved physiological features (body temperature and organ systems) and more accurate drug delivery, dosing and metabolism.
There are still a number of outstanding challenges that need to be addressed by the zebrafish pediatric cancer community (see Outstanding Questions). Refinement of some of the existing zebrafish models using specific promoter elements, or CRISPR/Cas9-mediated knock-in alleles, promises to advance the field considerably. New models of major classes of pediatric tumors, including medulloblastoma, retinoblastoma, Wilms tumor, thyroid cancer and cancer susceptibility syndromes (Box 1) could also be established in zebrafish with the help of recent pan-cancer genomic profiling and the use of cell-of-origin specific promoters. Ultimately, the complementary use of zebrafish pediatric cancer models and experiments in human cell lines or zebrafish larval and adult PDX models (Box 2) may enable us to bypass more time-consuming and expensive animal models, thereby allowing for the identification of new therapeutics on an accelerated time-scale. The field of zebrafish pediatric cancer research therefore promises to address many of the current needs for childhood cancers, particularly with respect to treating rare tumor types using precision-medicine-based approaches.
Box: Outstanding Questions.
How can we refine the zebrafish cancer models to better analyze mechanisms of tumor recurrence, metastasis, and chemoresistance in zebrafish?
How do we generate a “humanized” zebrafish model system to perform cancer immunology studies in zebrafish models?
How do we effectively model cancer susceptibility syndromes in zebrafish?
What is the best way to model structural/copy number variants in zebrafish that represent major drivers of pediatric cancers?
How can we streamline the application of small molecule inhibitors identified in zebrafish cancer models to the clinic?
Box 1: Cancer Susceptibility Syndromes.
Approximately 10% of childhood and adolescent cancers are caused by mutations in cancer-predisposing genes, such as P53, NF1, RB1 and APC [72]. Common cancer susceptibility disorders include neurofibromatosis, Noonan syndrome, Costello syndrome, retinoblastoma, Down syndrome, Gorlin syndrome and Li-Fraumeni syndrome [73]. Some mutations give rise to specific kinds of tumors (e.g. RB1 mutations and retinoblastoma), while others predispose patients to multiple different cancer types (e.g. the P53 mutations associated with Li-Fraumeni syndrome). Cancer predisposition syndromes are often associated with a diagnosis of multiple cancers, the childhood onset of a typically adult-specific cancer, co-occurring congenital abnormalities, and the development of certain tumors [73]. Yet, between knowledge of the initial driver mutations and the ability to diagnose the disorders there remains a considerable gap in our mechanistic understanding of how pediatric cancer susceptibility syndromes underlie the development of cancer. Previous efforts to model Costello syndrome in zebrafish using a Tol2-mediated gene trap to introduce constitutively active HRASV12-GFP [74] led to the development of some features associated with Costello syndrome patients, such as shorter body length, craniofacial defects and a propensity to develop RMS, albeit at a low frequency [74]. In addition, a model of Li-Fraumeni syndrome was generated with a p53-deletion mutant that spontaneously develops angiosarcoma, germ-cell tumors, leukemia, and malignant peripheral nerve sheath tumors (MPNSTs) [75]. However, the field ultimately awaits additional genomic studies and the development of additional animal models for other cancer predisposition syndromes before further progress can be made towards the discovery of effective treatments.
Box 2: Xenotransplantation Models of Pediatric Cancers.
Patient-derived xenografts (PDXs) are an important tool for evaluating new anticancer therapies and integrating precision medicine through the transplantation of primary patient tumors. Immunodeficient mice, in which engraftment requires 2 to 8 months before treatments can be assessed, have historically been necessary for these studies [76]. Zebrafish larvae have also been used for generating tumor xenograft models, as the immune system does not fully mature until 4 weeks of age [70]. Embryonic and larval transplantation has enabled high-throughput drug screening and direct visualization of tumor cell behaviors in a short 10-day window. However, the adaptive immune system and lower temperatures required for larval growth (28°C – 35°C) eventually kill the human cells. An exciting advance in the field is the generation of the optically clear prkdc−/−;il2rga−/− zebrafish strain that lacks T-, B-, and natural killer cells and grows at 37°C, which has enabled robust long-term engraftment of human PDXs with the same growth, proliferation and survival kinetics observed in tumor-matched immune-compromised mouse PDX models [77]. This model also enables the use of photo-conversion, in-vivo assessment of single-cell dynamics, and cost-effective large-scale chemical screening coupled with long-term engraftment [77]. Since these studies can be performed within a 28-day period, they allow for real-time therapy assessment, an important feature that is currently unavailable for the development of precision medicine. Yet, immunocompromised models are still not ideal as many cancer patients retain functional immune systems prior to therapy. The development of humanized PDX models, however, in which the animal is engrafted with both human hematopoietic stem cells and PDXs, enables both the assessment of tumor dynamics in the context of a partial immune response and the testing of relevant anticancer immunotherapies [76]. This rapidly advancing field is expected to be feasible in zebrafish in the near future by co-transplanting human immune progenitor cells with patient tumors and generating transgenic zebrafish strains optimized for growth of human cells, including expression of human growth factors (e.g., IL3 and GM-CSF).
Box: Highlights.
Zebrafish develop tumors that are histologically and genetically similar to human tumors.
Zebrafish enable the rapid identification of molecular drivers of tumor development and can be used to model cancers with unknown cells of origin using heat-shock and β-actin promoters.
Zebrafish models are amenable to high-throughput drug screening through larval drug submersion approaches as well as transplantation of primary patient tumors into immunocompromised lines.
Tumor-cell dynamics can be visualized in vivo throughout the lifetime of the animal by coupling oncogenes to fluorescent markers.
Acknowledgements
We would like to thank Cicely Jette Stewart for professional editorial assistance. This work was supported by grants from the National Institute of Health (R01NS106527, P30 CA042014) (R.A.S.) and T32 DK007115 (M.J.C.) as well as the Huntsman Cancer Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cunningham R et al. (2018) The major causes of death in children and adolescents in the United States. N Engl J Med. 379, 2468–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grobner SN et al. (2018) The landscape of genomic alterations across childhood cancers. Nature. 555, 321–327. [DOI] [PubMed] [Google Scholar]
- 3.Ma X et al. (2018) Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 555, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yen J et al. (2014) Zebrafish models of cancer: progress and future challenges. Curr Opin Genet Dev. 24, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White R et al. (2013) Zebrafish cancer: the state of the art and the path forward. Nat Rev Cancer. 13, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Letrado P et al. (2018) Zebrafish: speeding up the cancer drug discovery process. Cancer Res. 78, 6048–6058. [DOI] [PubMed] [Google Scholar]
- 7.He S et al. (2017) Zebrafish models of leukemia In Methods in Cell Biology. (Detrich HW, Westerfield M, and Zon LI, eds), pp. 563–592, Elsevier. [Google Scholar]
- 8.Hayes MN and Langenau DM (2017) Discovering novel oncogenic pathways and new therapies using zebrafish models of sarcoma In Methods in Cell Biology. (Detrich HW, Westerfield M, and Zon LI, eds), pp. 525–561, Elsevier. [DOI] [PubMed] [Google Scholar]
- 9.Casey MJ and Stewart RA (2017) Zebrafish as a model to study neuroblastoma development. Cell Tissue Res. 372, 223–232. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez A and Amatruda JF (2016) Zebrafish germ cell tumors. Adv Exp Med Biol. 916, 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward E et al. (2014) Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 64, 83–103. [DOI] [PubMed] [Google Scholar]
- 12.Karrman K and Johansson B (2017) Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 56, 89–116. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y et al. (2017) The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat genet. 49, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girardi T et al. (2017) The genetics and molecular biology of T-ALL. Blood. 129, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langenau DM et al. (2003) Myc-induced T cell leukemia in transgenic zebrafish. Science. 299, 887–890. [DOI] [PubMed] [Google Scholar]
- 16.Langenau DM et al. (2005) Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. PNAS. 102, 6068–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng H et al. (2007) Heat-shock induction of T-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br J Haematol. 138, 169–175. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez A et al. (2011) Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J Exp Med. 208, 1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Martin M and Ferrando A (2017) The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. 129, 1124–1133. [DOI] [PubMed] [Google Scholar]
- 20.Blackburn JS et al. (2012) Notch signaling expands a pre-malignant pool of T-cell acute lymphoblastic leukemia clones without affecting leukemia-propagating cell frequency. Leukemia. 26, 2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns MA et al. (2018) Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 32, 2126–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lobbardi R et al. (2017) TOX regulates growth, DNA repair, and genomic instability in T-cell acute lymphoblastic leukemia. Cancer Discov. 7, 1336–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leong WZ et al. (2017) ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 31, 2343–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansour MR et al. (2018) JDP2: An oncogenic bZIP transcription factor in T cell acute lymphoblastic leukemia. J Exp Med. 215, 1929–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutierrez A et al. (2014) Phenothiazines induces PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest. 124, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackburn JS et al. (2014) Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 25, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fouladi M et al. (2014) A phase I trial of MK-2206 in children with refractory malignancies: a children’s oncology group study. Pediatr Blood Cancer. 61, 1246–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lilljebjorn H and Fioretos T (2017) New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood. 130, 1395–1401. [DOI] [PubMed] [Google Scholar]
- 29.Sabaawy HE et al. (2006) TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. PNAS. 103, 15166–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borga C et al. (2019) Simultaneous B and T cell acute lymphoblastic leukemias in zebrafish driven by transgenic MYC: implications for oncogenesis and lymphopoiesis. Leukemia. 33, 333–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolouri H et al. (2018) The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 24, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeh JJ et al. (2008) AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development. 135, 401–410. [DOI] [PubMed] [Google Scholar]
- 33.He B et al. (2014) Functions of flt3 in zebrafish hematopoiesis and its relevance to human acute myeloid leukemia. Blood. 123, 2518–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh JJ et al. (2009) Discovering chemical modifiers of oncogene-regulated hematopoietic differentiation. Nat Chem Biol. 5, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curtin SC et al. (2016) Declines in cancer death rates among children and adolescents in the United States, 1999–2014. NCHS Data Brief. 257, 1–8. [PubMed] [Google Scholar]
- 36.Siegel RL et al. (2018) Cancer statistics, 2018. CA Cancer J Clin. 68, 7–30. [DOI] [PubMed] [Google Scholar]
- 37.Picard D et al. (2012) Integrative genomic analyses identify LIN28 and OLIG2 as markers of survival and metastatic potential in childhood central nervous system primitive neuro-ectodermal brain tumours. Lancet Oncol. 13, 838–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sturm D et al. (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 164, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Modzelewska K et al. (2016) MEK inhibitors reverse growth of embryonal brain tumors derived from oligoneural precursor cells. Cell Rep. 17, 1255–1264. [DOI] [PubMed] [Google Scholar]
- 40.Banerjee A et al. (2017) A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a pediatric brain tumor consortium (PBTC) study. Neuro Oncol. 19, 1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin J et al. (2012) Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis Model Mech. 5, 881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solin SL et al. (2015) Rapid tumor induction in zebrafish by TALEN-mediated somatic inactivation of the retinoblastoma1 tumor suppressor rb1. Sci Rep. DOI: 10.1038/srep13745 (http://www.nature.com) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schultz L et al. (2018) Epigenetic regulators Rbbp4 and Hdac1 are overexpressed in a zebrafish model of RB1 embryonal brain tumor, and are required for neural progenitor survival and proliferation. Dis Model Mech. DOI: 10.1242/dmm.034124 (http://www.dmm.biologists.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Y et al. (2008) A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 68, 6292–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matthay KK et al. (2016) Neuroblastoma. Nat Rev Dis Primers. DOI: 10.1038/nrdp.2016.78 (http://www.nature.com) [DOI] [PubMed] [Google Scholar]
- 46.Whittle S et al. (2017) Overview and recent advances in the treatment of neuroblastoma. Expert Rev Anticancer Ther. 17, 369–386. [DOI] [PubMed] [Google Scholar]
- 47.Cheung NV and Dyer MA (2013) Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu S et al. (2012) Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 21, 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao T et al. (2017) The pre-rRNA processing factor DEF is rate limiting for the pathogenesis of MYCN-driven neuroblastoma. Oncogene. 36, 3852–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X et al. (2017) Critical role for GAB2 in neuroblastoma pathogenesis through the promotion of SHP2/MYCN cooperation. Cell Rep. 18, 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He S et al. (2016) Synergy between loss of NF1 and overexpression of MYCN in neuroblastoma is mediated by the GAP-related domain. eLIFE. DOI: 10.7554/eLife.14713 (http://www.elifesciences.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trigg RM and Turner SD (2018) ALK in neuroblastoma: biological and therapeutic implications. Cancers (Basel). DOI: 10.3390/cancers10040113 (http://www.mdpi.com) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimmerman MW et al. (2018) MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 8, 320–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang K et al. (2011) Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 469, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu S et al. (2017) LMOl synergizes with MYCN to promote neuroblastoma initiation and metastasis. Cancer Cell. 32, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carter DR et al. (2015) Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci Transl Med. DOI: 10.1126/scitranslmed.aabl803 (http://www.stm.sciencemag.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shern JF et al. (2015) Pediatric rhabdomyosarcoma. Crit Rev Oncog. 20, 227–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malempati S and Hawkins DS (2012) Rhabdomyosarcoma: review of the children’s oncology group (COG) soft-tissue sarcoma committee experience and rationale for current COG studies. Pediatr Blood Cancer. 59, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meza JL et al. (2006) Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The children’s oncology group. J Clin Oncol. 24, 3844–3851. [DOI] [PubMed] [Google Scholar]
- 60.Langenau DM et al. (2007) Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 21, 1382–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Storer NY et al. (2013) Zebrafish rhabdomyosarcoma reflects the developmental stage of oncogene expression during myogenesis. Development. 140, 3040–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ignatius MS et al. (2017) The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep. 19, 2304–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hayes MN et al. (2018) Vangl2/RhoA signaling pathway regulates stem cell self-renewal programs and growth in rhabdomyosarcoma. Cell Stem Cell. 22, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tenente IM et al. (2017) Myogenic regulatory transcription factors regulator growth in rhabdomyosarcoma. eLIFE. DOI: 10.7554/eLife.19214 (http://www.elifesciences.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le X et al. (2013) A novel chemical screening strategy in zebrafish identifies common pathways in embryogenesis and rhabdomyosarcoma development. Development. 140, 2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen EY et al. (2013) Cross-species array comparative genomic hybridization identifies novel oncogenic events in zebrafish and human embryonal rhabdomyosarcoma. PLoS Genet. DOI: 10.1371/journal.pgen.1003727 (http://www.journals.plos.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen EY et al. (2014) Glycogen synthase kinase 3 inhibitors induce the canonical WNT/P-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. PNAS. 111, 5349–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vleeshouwer-Neumann T et al. (2015) Histone deacetylase inhibitors antagonize distinct pathways to suppress tumorigenesis of embryonal rhabdomyosarcoma. PLoS One. DOI: 10.1371/journal.pone.0144320 (http://www.journals.plos.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kendall GC et al. (2018) PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLIFE. DOI: 10.7554/eLife,33800 (http://www.elifesciences.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Osmani N and Goetz JG (2019) Multiscale imaging of metastasis in zebrafish. Trends Cancer. 5, 766–778. [DOI] [PubMed] [Google Scholar]
- 71.Neumann JC et al. (2011) Zebrafish models of germ cell tumor. Methods Cell Biol. 105, 3–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J et al. (2015) Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 373, 2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ripperger T et al. (2017) Childhood cancer predisposition syndromes - A concise review and recommendations by the cancer predisposition working group of the society for pediatric oncology and hematology. Am J Med Genet. 173, 1017–1037. [DOI] [PubMed] [Google Scholar]
- 74.Santoriello C et al. (2009) Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis Model Mech. 2, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ignatius M et al. (2018) tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. eLIFE. DOI: 10.7554/eLife.37202 (http://www.elifesciences.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jung J et al. (2018) The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat. 50, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan C et al. (2019) Visualizing engrafted human cancer and therapy responses in immunodeficient zebrafish. Cell. 177, 1903–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]