

Abstract
PURPOSE
Response to programmed cell death protein 1 (PD-1) blockade is often conceptualized as resulting from reinvigoration of tumor-infiltrating lymphocytes. However, recruited antitumor immunity from the periphery may also be an important contributor to response. A detailed assessment of the response dynamics of individual metastasis could provide insight to the systemic and local features that mediate response and resistance to immunotherapy.
MATERIALS AND METHODS
Patients with metastatic non–small-cell lung cancer (NSCLC) or mismatch repair deficiency (MMRD) carcinoma treated with PD-1 monotherapy were evaluated independently. Absolute and percent change of each target lesion were quantified at each computed tomography scan using RECIST. Patterns of progression were predefined as systemic or mixed and were correlated with clinical outcomes.
RESULTS
A total of 761 individual lesions from 214 patients with NSCLC and 290 lesions from 78 patients with MMRD carcinoma were examined. Individual target lesion responses aligned with best overall response of each patient (85% NSCLC and 93% MMRD lesions responded in patients with partial response/complete response). In responding patients, timing of response was uniform (73% NSCLC and 76% MMRD lesions responded synchronously), and deeper responses were associated with prolonged progression-free survival and overall survival. By contrast, at progression, mixed progression was common (45% of NSCLC and 53% of MMRD) and associated with improved survival compared with those who experienced systemic progression (NSCLC hazard ratio [HR], 0.58; P = .001; MMRD HR, 0.40; P = .07). Organ sites had differential responses, with lymph node and liver metastasis among the most and least responsive, respectively.
CONCLUSION
Temporal-spatial patterns of response across individual metastases tend to be uniform, favoring the role of peripheral, clonally directed antitumor immunity as a key mediator of response to PD-1 blockade. In contrast, progression is more heterogeneous, potentially revealing the clinical importance of local features and intertumoral heterogeneity.
INTRODUCTION
Monoclonal antibodies that inhibit the programmed cell death protein 1 (PD-1) or its ligand, programmed cell death ligand 1 (PD-L1), have revolutionized cancer therapeutics.1-3 Inhibition of the PD-1/PD-L1 signaling pathway stimulates T-cell–mediated immunity, leading to potent antitumor activity and durable responses in a variety of cancers.4
Clinically meaningful antitumor immune response appears to be dependent on a complex interplay between peripheral immunity and the local tumor microenvironment. Although the mechanism of action of PD-1 blockade is often conceptualized as reinvigoration of T cells from within the tumor microenvironment, recent studies demonstrating terminal dysfunction of tumor-infiltrating lymphocytes (TILs)5-7 highlight the possibility that immigration of peripheral T cells is also necessary. Still, the impact of tumor inflammation,8,9 metastatic organ microenvironment,10,11 and tumor clonal heterogeneity12,13 on response to T cell checkpoint blockade all emphasize that local, tumor-specific features contribute to effectiveness of immune responses. In short, the interplay between the peripheral and tumoral immune compartments to define the nature of response and progression to PD-1 blockade is not well understood.
We hypothesized that an examination of the patterns of response to PD-1 blockade at the level of individual metastases—in contrast to the more typical summation within a given patient—could provide insight to the impact of systemic and local features on the antitumor immune response. To do so, we systematically assessed, at both individual metastatic site-specific and patient-specific levels, the dynamics of response and patterns of progression to PD-1 blockade in two independent cohorts of patients with metastatic cancers.
MATERIALS AND METHODS
Patients
After institutional review board approval, two cohorts of patients were examined: patients with metastatic non–small-cell lung cancer (NSCLC) treated with anti–PD-1 monotherapy (n = 214) and patients with metastatic carcinoma with mismatch repair deficiency (MMRD) treated with pembrolizumab (n = 78).14 All patients received at least one dose of PD-1 blockade and were evaluable for response.
Radiologic Assessments and Predefined Patterns of Response and Resistance
Computed tomography (CT) scans of individual patients during PD-1 treatment were reviewed by trained radiologists, and response was determined by RECIST v1.1. The absolute size and percent change of overall tumor burden and within individual metastases were quantified using unidimensional measurements (millimeters) of target lesions. Nontarget lesions were captured and followed qualitatively. The appearance of new lesions was captured at the time of onset.
In all patients, irrespective of their best overall response by RECIST, the pattern of progression was determined using predefined characteristics at the time that any target and/or nontarget lesion met the criteria for progression of disease (PD) and/or when a new lesion was present, whichever occurred first (Table 1). The patterns of progression were defined as:
TABLE 1.
Definition of Patterns of Progression

Systemic progression: PD in two or more lesions (including target, nontarget, or new lesion) in patients with three or fewer target lesions or PD in three or more lesions (including target, nontarget, or new lesion) in patients with four or more target lesions.
Mixed progression: PD in only one lesion (including target, nontarget, new lesion) in patients with three or fewer target lesions; or PD in two or fewer lesions (including target, nontarget, new lesion) in patients with four or more target lesions.
Timing of radiologic assessments was not prespecified in the NSCLC cohort; the median time of the first and second CT scans were 1.9 and 4.0 months, respectively, after starting treatment. Timing of CT scans in the MMRD cohort were prespecified as described.3
Statistical Approach
Longitudinal changes in tumor size were quantified and captured as percent change. Progression-free survival (PFS; defined as start of PD-1 therapy until RECIST-defined progression or death, or, if no progression, were censored at the data lock) and overall survival (OS; defined as start of PD-1 therapy until death, or, if no death, were censored at the data lock) among groups were depicted using the Kaplan-Meier method and compared using log-rank (Mantel-Cox) test. Two-way comparisons of continuous variables were performed using the Mann-Whitney test, and three-way comparisons were performed using the Kruskal-Wallis test. Two-way comparisons of categorical variables were performed using Fisher’s exact test. Correlation analyses were performed using Spearman correlation test. All statistical tests were two sided, and P < .05 was set as the level of significance.
RESULTS
Patients
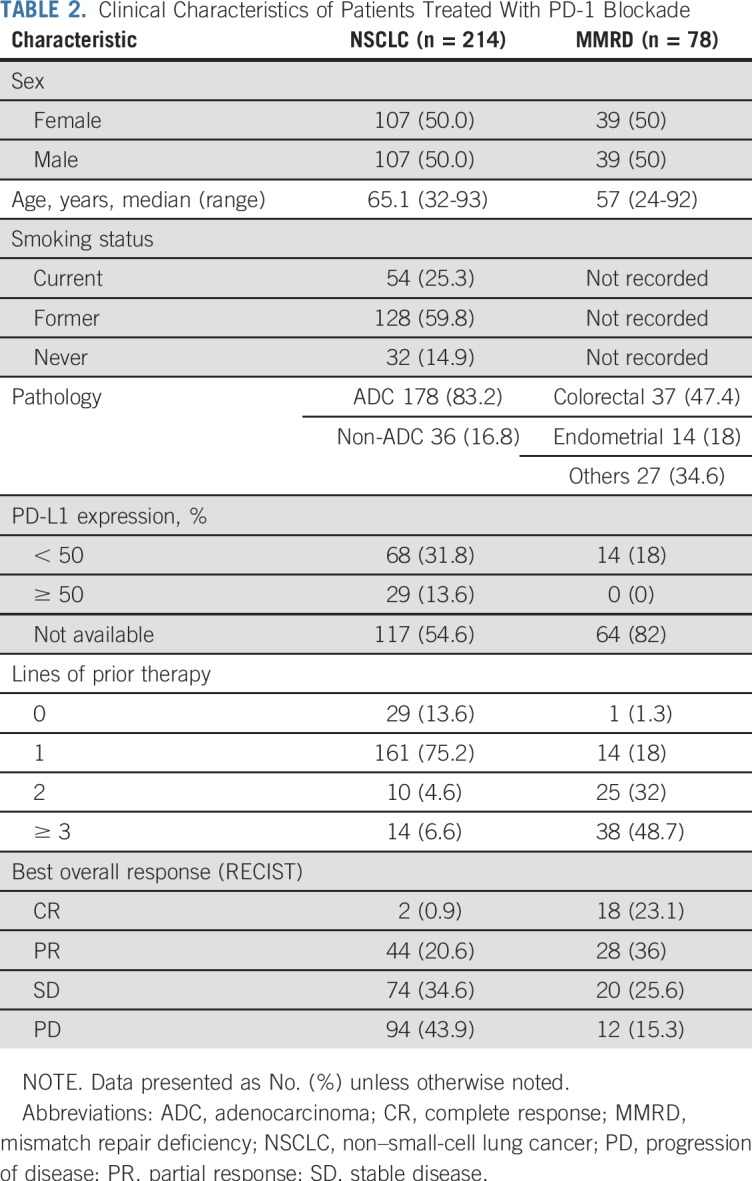
In the NSCLC cohort, 214 patients with stage IV NSCLC were included, with a total of 761 baseline lesions (501 target, 260 nontarget) identified on imaging. The MMRD cohort consisted of 78 patients with metastatic MMRD carcinoma, with a total of 290 baseline lesions identified on imaging (187 target and 103 nontarget; Table 2; Data Supplement [online only]). The overall response rates were 21.5% in the NSCLC cohort and 59.1% in the MMRD cohort.
TABLE 2.
Clinical Characteristics of Patients Treated With PD-1 Blockade
Temporal-Spatial Patterns of Response
We first evaluated the change in size of each target lesion over time in patients with NSCLC. We found that individual target lesion responses aligned with the RECIST-defined best overall response (BOR) of each patient (Figs 1A-1C). In NSCLC, among patients who achieved complete response or partial response (CR/PR; n = 46), 91 of 107 (85%) of individual target lesions also had objective response (> 30% reduction; Data Supplement). Patients with stable disease (SD) or PD as BOR also aligned in their individual target lesion response, although to a lesser extent. Among patients with SD (n = 74), 118 of 159 (74%) individual target lesions had SD, and among patients with PD (n = 94), 130 of 235 (55%) individual target lesions had PD (> 20% growth; Data Supplement). A similar alignment of individual target lesion responses relative to BOR was seen in patients with metastatic MMRD carcinoma (Figs 1D-1F; Data Supplement).
FIG 1.

Patterns of response in patients treated with programmed cell death protein 1 (PD-1) blockade. (A-F) Spider plot of tumor burden changes of target lesions during the course of treatment with PD-1 blockade. Tumor burden was considered 0 at baseline, and longitudinal follow-up (x-axis) was captured in percentage of change (y-axis). (A) Target lesions (n = 107) from patients with non–small-cell lung cancer (NSCLC) who achieved complete response (CR) or partial response (PR) as their best overall response (BOR) per RECIST (n = 46). (B) Target lesions (n = 159) from patients with NSCLC and stable disease (SD; n = 74). (C) Target lesions (n = 235) from patients with NSCLC and progression of the disease (PD; n = 94). (D) Target lesions (n = 101) from patients with mismatch repair deficiency (MMRD) carcinoma with CR/PR (n = 46). (E) Target lesions (n = 57) from patients with MMRD carcinoma and SD (n = 20). (F) Target lesions (n = 29) from patients with MMRD carcinoma and PD (n = 12). (G-H) Violin plots of the timing of response of individual target lesions in patients that achieve CR or PR per RECIST. The first time at least one target lesion responded (> 30% response) was considered 0; then other target lesions were assessed for relative timing of initial response on the same or subsequent scans. (G) Time to achieve response in target lesions from patients who achieved CR or PR with NSCLC (n = 107). (H) Time to achieve response in target lesions from patients who achieved CR or PR with MMRD carcinomas (n = 101).
Of note, pseudoprogression, defined as tumor growth of ≥ 20% followed by partial or complete response by RECIST, occurred in only one patient in the NSCLC cohort (Data Supplement). Two additional patients with NSCLC had isolated pseudoprogression in individual target lesions (Data Supplement). In the MMRD cohort, no pseudoprogression was observed, either in terms of overall response or in individual lesions.
We analyzed the synchrony of response in the lesions from patients who achieved CR or PR by RECIST (107 lesions in the NSCLC cohort and 101 lesions in the MMRD carcinoma cohort). We found that 82 of the 107 lesions (76%) in the NSCLC cohort achieved initial response (≥ 30%) synchronously with other target lesions at the same CT scan, within a given patient (Fig 1G). In addition, 90% and 95% of lesions responded by 3 and 6 months, respectively, from the first responding lesion in a given patient. In responders with MMRD carcinoma, 74 of 101 responding lesions (73%) achieved initial partial response at the same CT scan within a given patient; 81% and 93% lesions responded within 3 or 6 months, respectively, of the first responding lesion (Fig 1H).
We also evaluated whether PD-L1 expression, which may be a proxy for the degree of intratumoral cytotoxic immune infiltration,15,16 influenced the patterns of response in the NSCLC cohort (available in 28 out of 46 responders). There were no differences in the patterns of response, synchrony, timing, or clinical outcomes (PFS and OS) between patients with high (≥ 50% PD-L1, n = 15) and low (< 50% PD-L1, n = 13) expression of PD-L1 (Data Supplement). In the MMRD cohort, direct quantification of CD8+ TILs was available in a subset of patients (n = 14). There were no differences in outcomes between patients with low (< 200 tumor CD8+ cells/mm2, n = 7) and high (> 200 tumor CD8+ cells/mm2, n = 7) TILs (Data Supplement). Similarly, the patterns and synchrony of responses were unchanged regardless of TIL status (Data Supplement).
Depth of Response and Baseline Tumor Burden
We examined if there was a correlation between overall depth of response and survival. Among responders in both the NSCLC and MMRD cohorts, patients with depth of response equal or greater than the median response in each cohort (−51% in NSCLC, −72% in MMRD) had improved PFS compared with those responders with depth of response lower than the median response (NSCLC cohort: PFS hazard ratio [HR], 0.39; 95% CI, 0.13 to 1.15; P = .03; MMRD cohort: PFS HR, 0.08; 95% CI, 0.01 to 0.5; P = .008; Figs 2A and 2B). NSCLC responders with depth of response equal or greater than the median response also had a significantly prolonged OS when compared with those responders with depth of response lower than the median response (OS HR, 0.22; 95% CI, 0.05 to 0.93; P = .01; Fig 2C). In MMRD, all responders were alive at time of follow-up, and therefore there were no differences between groups (Fig 2D).
FIG 2.

Depth of response and association with clinical outcomes. (A-D) Patients with complete response (CR) or partial response (PR) by RECIST in the non–small-cell lung cancer (NSCLC) cohort (n = 46) and the mismatch repair deficiency (MMRD) cohort (n = 46) were divided into two groups on the basis of the median best overall response (BOR; NSCLC cohort, ≤ v ≥ −51%; and MMRD cohort, ≤ v ≥ −72%, respectively). (A) Progression-free survival (PFS) in the NSCLC cohort, and (B) MMRD cohort comparing these two groups. (C) Overall survival (OS) in the NSCLC, and (D) MMRD cohort comparing these two groups.
The impact of baseline tumor burden on response was also assessed. There was no difference in the baseline tumor burden among RECIST-defined responders (CR/PR) versus nonresponders (SD or PD) among patients with NSCLC or MMRD (P = .83 and P = .09, respectively, Kruskal-Wallis test; Data Supplement). In the NSCLC cohort, there was also no correlation between tumor burden and percent best response (Spearman rho, 0.0003; P = .9). In the MMRD cohort, there was a weak positive correlation between responders (CR/PR) and tumor burden (Spearman rho, 0.37; P = .008). Furthermore, the patterns, timing, synchrony, and overall survival in responders in both cohorts were unaffected by high versus low tumor burden (Data Supplement).
Patterns of Progression
We next analyzed the patterns of progression in all patients (irrespective of their initial BOR) and the association with clinical outcomes. Patterns of progression were predefined to assess heterogeneity in the pattern of progression (Table 1). In patients with NSCLC, mixed pattern of progression was common (78 of 214 [36.4% of all patients treated, 45% of those who progressed at some point during treatment]); systemic progression pattern occurred in 94 patients (43.9% and 55%, respectively), and 42 (19.6%) did not experience progression (Fig 3A). Patients with mixed progression had improved PFS (per RECIST) and OS when compared with patients with systemic progression (PFS HR, 0.45; 95% CI, 0.32 to 0.61; P < .0001; OS HR, 0.58; 95% CI, 0.41 to 0.82; P = .001; Figs 3B and 3C).
FIG 3.

Patterns of progression of disease (PD) during programmed cell death protein 1 (PD-1) blockade. (A-C) Patterns of progression in patients with non–small-cell lung cancer (NSCLC) on the basis of prespecified criteria. (A) Proportion of patients who experienced systemic or mixed progression. Median time (months) and range of progression are described. (B) Kaplan-Meier plots of progression-free survival (PFS), and (C) overall survival (OS) in patients with systemic (n = 94) or mixed progression patterns (n = 78) in the NSCLC cohort. (D-F) Patterns of progression in patients with mismatch repair deficiency (MMRD) carcinoma on the basis of prespecified criteria. (D) Proportion of patients who experienced systemic or mixed progression. (E) Kaplan-Meier plots of PFS, and (F) OS in patients with systemic (n = 14) or mixed progression patterns (n = 16) in the MMRD cohort.
In patients with MMRD carcinoma, most patients did not experience progression (61.5%), but among those who did experience progression, mixed progression pattern was again common (20.5% of all patients treated, 53% of those who experience progression), whereas systemic progression occurred in 17.9% and 47%, respectively (Fig 3D). Similar to the NSCLC cohort, patients with MMRD with mixed progression had improved PFS and OS when compared with patients who experienced systemic progression (PFS HR, 0.28; 95% CI, 0.11 to 0.72; P = .0009; OS HR, 0.40; 95% CI, 0.13 to 1.21; P = .07; Figs 3E and 3F).
Impact of Site of Metastasis on Response and Resistance
Last, we examined whether organ site of the metastasis was associated with response to PD-1 blockade. In the NSCLC cohort, individual metastases in lymph nodes tended to have the best responses; lung, adrenal, and pleura had intermediate responses, and bone and liver had the least responses (Fig 4A). Lymph node lesions had significantly better responses when compared with lung, pleural, and liver lesions (P = .011, P = .002, and P = .003, respectively). In the MMRD cohort, most lesions responded regardless of site of disease, and there were no significant differences among metastatic sites; numerically, lymph nodes and GI tract lesions were more responsive to PD-1 blockade, whereas responses were less substantial in liver metastases (Fig 4B). When considering the appearance of new lesions (n = 125 in NSCLC; n = 37 in MMRD), the liver, lung, and lymph nodes were the most common sites of new metastases in both cohorts (Data Supplement).
FIG 4.

Site of disease and response to programmed cell death protein 1 (PD-1) blockade. Distribution of best response (%) of individual target lesions to PD-1 blockade grouped by organ of metastasis in patients with (A) non–small-cell lung cancer (NSCLC), and (B) mismatch repair deficiency (MMRD) carcinoma. Response by RECIST criteria, median and range of the percentage of response, and interquartile range of best responses are depicted. Comparisons among responses: *P < .05; †P < .01. CR, complete response; PD, progression of disease; PR, partial response; SD, stable disease.
DISCUSSION
We investigated whether an assessment of individual metastases dynamics in two independent cohorts could contribute to a better understanding of how response and progression to PD-1 blockade occurs. We found in patients with NSCLC and MMRD cancers, individual target lesion responses closely aligned with the best overall response of each patient. In particular, when response occurred, it tended to be uniform across metastases and occur synchronously. By contrast, progression tended to occur more heterogeneously across metastases, with differential survival seen in patients with mixed versus systemic progression.
We infer that the largely uniform dynamics of response within a given patient suggest that response to PD-1 blockade is a result of systemic, clonally directed antitumor immunity that is recruited from the periphery to invade tumors (Fig 5). It seems more challenging to achieve such temporal-spatial uniformity solely through coordinated reinvigoration of local infiltrating lymphocytes. In addition, differential PD-L1 expression or TIL quantity among responders did not influence the patterns of response, which may have been expected if reinvigorated TILs were the dominant effector of response. This proposed model emphasizing the importance of recruited rather than reinvigorated immune responses aligns with emerging preclinical17 and clinical data.18,19 For example, a recent analysis using single-cell RNA sequencing of paired biopsies of several patients with melanoma treated with immune checkpoint blockade found that a cluster of T cells characterized by increased memory, early activation, and survival were enriched in responders and found distinctly in on-treatment, rather than pretreatment, samples.20 In addition, another study using paired single-cell RNA and T-cell receptor sequencing to track individual T-cell clones in response to PD-1 blockade found that on-treatment expanded intratumoral T-cell clones were new rather than preexisting in pretreatment tissue.21 Overall, these single-cell data favor that T-cell clones involved in response to immune checkpoint blockade are largely recruited from outside of the tumor microenvironment. Other studies have also incorporated analysis of peripheral immune cell populations22-24 as potential predictors of response to PD-1 blockade. We believe our report similarly encourages further pursuit of biomarkers of immunotherapy response by profiling the circulating immune landscape.
FIG 5.

Hypothesized model of patterns of response and progression to programmed cell death protein 1 (PD-1) blockade. PD-1 blockade is proposed to achieve benefit largely from a systemic, clonal-directed antitumor immune response, which is recruited from the periphery to infiltrate tumors (lavender arrows), consistent with the uniform spatiotemporal pattern of response across metastases seen within a given patient (top right panel). In contrast, patterns of progression can be more mixed, which may reflect the heterogenous and organ-specific properties of the local tumor microenvironment (red dashed circles) that can block or restrain the systemic immune response to PD-1 blockade, and produce mixed progression across metastases within a given patient (bottom right panel).
Of course, even if not the dominant source, intratumoral T cells still undoubtedly contribute to the process of response to PD-1 blockade. Tumor PD-L1 expression and gene expression signatures of intratumoral cytotoxic lymphocytes unequivocally associate with whether response occurs.16,25,26 In addition, data are emerging to refine the biology of responsive infiltrating T-cell populations. A recent study, for example, demonstrated that intratumoral exhausted CD8+ T cells are heterogeneous, and a subtype, termed progenitor exhausted CD8+ T cells, distinctly retain responsiveness to immune checkpoint blockade in a murine model of melanoma.27 Future mechanistic studies are critical to dissect the contributions and interactions of tumor-specific T cells derived from both the peripheral and intratumoral compartments. Ultimately, these findings will have important therapeutic implications for rationally guiding synergistic combinations and guiding diagnostic and biomarker discovery efforts.
Beyond the originating site of effective antitumor immunity, the systemic pattern of clinical response also appears to align with the importance of clonal neoantigens as a primary target of response to PD-1 blockade.13 If response was achieved through targeting of subclonal antigens, we would have expected more substantial spatiotemporal heterogeneity of response. Potentially relatedly, we did not, in contrast to other studies,28 identify a correlation between tumor burden and response to PD-1 blockade. As intratumoral heterogeneity of neoantigens has been demonstrated to associate with response to PD-1 blockade,13 we conclude that tumor size is not proportional to heterogeneity. Therefore, tumor heterogeneity is not simply a byproduct of tumor growth but rather mediated by distinct characteristics of a tumor and selective immune pressure.
In contrast to the uniformity of response dynamics, progression is characterized by two different patterns—mixed and systemic—which are associated with distinct survival outcomes. We speculate that progression might be more affected by local intratumoral features and intertumor heterogeneity (Fig 5). The frequency of and survival difference in those patients with mixed progression suggest that selective immune pressure against individual metastasis can modulate the relative immunogenicity of a metastasis at a local level. Consistent with these results, other groups have demonstrated robust pharmacodynamic peripheral immune changes in response to PD-1 blockade even among nonresponding patients.23,29 Overall, our data suggest that progression may not be simply a lack of antitumor immunity but rather its own active process governed by a confluence of features that define the ultimate outcome.
Another local feature that may dictate the heterogeneous radiographic response is the site of metastasis. Lymph nodes were among the most responsive in both NSCLC and MMRD, which may be explained by the local populations of T cells and the role of tumor-draining lymph nodes in priming and conditioning antigen-specific responses to PD-1 blockade.30,31 Similar to previous data,11 liver metastases had among the poorest responses to PD-1 blockade and were the most common site of appearance of new lesions. The liver appears to have distinct immune tolerance affecting local32,33 and systemic34-36 immunity. These trends of differential response depending on the location of metastasis aligns conceptually with recent data demonstrating organ-specific “immunostats,” that is, varying thresholds for immune activation within each organ.37 Additional work is needed to explain the mechanistic basis for how local organ sites affect the immunophenotype and immunogenicity of metastases.
Our study has important limitations. We performed a largely retrospective analysis using moderate-sized cohorts. Still, both independent cohorts demonstrated similar results in nearly each analysis, adding confidence to the conclusions. In addition, our predefined “systemic” and “mixed” progression criteria were applied for the first time in this study. Additional studies in larger cohorts are needed to corroborate and refine these concepts of patterns of progression. Interestingly, these categories differentiated phenotypes of progression with significant differences in mortality, suggesting they have clinical relevance.
In conclusion, our study characterized the individual lesion-level dynamics of response and progression to PD-1 blockade in two independent tumor types. We found that individual metastases tend to be consistent and align with the overall response of each patient and, by contrast, patterns of progression are more heterogeneous. Although our study lacks mechanistic evidence, it provides clinical evidence to support the inference of peripherally recruited, clonally directed tumor immunity as a key mechanism of action of immune checkpoint blockade and that progression may be more affected by intertumor heterogeneity and specific features of the local tumor microenvironment.
Footnotes
Supported by Memorial Sloan Kettering Cancer Center Support Grant/Core Grant No. P30 CA008748 and the Druckenmiller Center for Lung Cancer Research at Memorial Sloan Kettering Cancer Center; M.D.H. is a Damon Runyon Clinical Investigator supported in part by the Damon Runyon Cancer Research Foundation Grant No. CI-98-18 and is a member of the Parker Institute for Cancer Immunotherapy.
AUTHOR CONTRIBUTIONS
Conception and design: Juan C. Osorio, Luis A. Diaz Jr, Matthew D. Hellmann
Financial support: Matthew D. Hellmann
Provision of study material or patients: Dung T. Le, Luis A. Diaz Jr, Matthew D. Hellmann
Collection and assembly of data: Juan C. Osorio, Dung T. Le, Jennifer N. Durham, Andrew J. Plodkowski, Darragh F. Halpenny, Michelle S. Ginsberg, Peter Sawan, Helena A. Yu, Azadeh Namakydoust, Jamie E. Chaft, Gregory J. Riely, Hira Rizvi, Luis A. Diaz Jr, Matthew D. Hellmann
Data analysis and interpretation: Juan C. Osorio, Kathryn C. Arbour, Dung T. Le, Andrew J. Plodkowski, Joseph G. Crompton, Helena A. Yu, Barzin Y. Nabet, Jamie E. Chaft, Hira Rizvi, Luis A. Diaz Jr, Matthew D. Hellmann
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Lesion-Level Response Dynamics to Programmed Cell Death Protein (PD-1) Blockade
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Kathryn C. Arbour
Consulting or Advisory Role: AstraZeneca
Dung T. Le
Honoraria: Merck
Consulting or Advisory Role: Merck, Bristol-Myers Squibb
Research Funding: Merck, Bristol-Myers Squibb, Aduro BioTech, Curegenix, Medivir
Patents, Royalties, Other Intellectual Property: Inventor of technology, “Microsatellite Instability as a Pharmacogenomic Marker of Therapeutic Response to Immune Checkpoint Inhibition”
Jennifer N. Durham
Employment: AstraZeneca/MedImmune (I)
Stock and Other Ownership Interests: AstraZeneca/MedImmune (I)
Helena A. Yu
Consulting or Advisory Role: AstraZeneca, Eli Lilly
Research Funding: Clovis Oncology (Inst), AstraZeneca (Inst), Astellas Pharma (Inst), Eli Lilly (Inst), Novartis (Inst), Pfizer (Inst), Daiichi Sankyo (Inst)
Travel, Accommodations, Expenses: Eli Lilly
Other Relationship: Astellas Pharma
Azadeh Namakydoust
Consulting or Advisory Role: Bayer
Travel, Accommodations, Expenses: Bayer
Barzin Y. Nabet
Patents, Royalties, Other Intellectual Property: I have a patent pending related to immunomodulatory nucleic acids (Inst)
Jamie E. Chaft
Consulting or Advisory Role: Genentech/Roche, AstraZeneca/MedImmune, Merck, Bristol-Myers Squibb
Research Funding: Genentech/Roche (Inst), Bristol-Myers Squibb (Inst), AstraZeneca/MedImmune (Inst), Merck (Inst)
Gregory J. Riely
Research Funding: Novartis (Inst), Roche/Genentech (Inst), GlaxoSmithKline (Inst), Pfizer (Inst), Infinity Pharmaceuticals (Inst), Mirati Therapeutics (Inst), Merck (Inst), Takeda (Inst)
Patents, Royalties, Other Intellectual Property: Patent application submitted covering pulsatile use of erlotinib to treat or prevent brain metastases (Inst)
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Other Relationship: Pfizer, Roche/Genentech, Takeda
Luis A. Diaz
Leadership: Personal Genome Diagnostics, Jounce Therapeutics
Stock and Other Ownership Interests: PapGene, Personal Genome Diagnostics, Jounce Therapeutics, Zydecom, Thrive Detect, NeoPhore, Amgen (I)
Consulting or Advisory Role: Merck, Personal Genome Diagnostics, Cell Design Labs, Lyndra, Caris Life Sciences, Genocea Biosciences, Zydecom, NeoPhore
Research Funding: Merck (Inst)
Patents, Royalties, Other Intellectual Property: US-2010041048-A1: Circulating Mutant DNA to Assess Tumor Dynamics; US-2015344970-A1: Personalized Tumor Biomarkers; WO-2010118016-A2: Digital quantification of DNA methylation; US-2005202465-A1: Thymidylate synthase gene and metastasis; US-2014227271-A1: Somatic mutations in ATRX in brain cancer; WO-2012094401-A2: Genes frequently altered in pancreatic neuroendocrine tumors; US-2013323167-A1: Detecting and treating solid tumors through selective disruption of tumor vasculature; EP-2912468-B1: Papanicolaou test for ovarian and endometrial cancers; US-9976184-B2: Mutations in pancreatic neoplasms; US-2017267760-A1: Checkpoint Blockade and Microsatellite Instability; US-2018171413-A1: Head and neck squamous cell carcinoma assays; US-2018086832-A1: HLA-restricted epitopes encoded by somatically mutated genes; US-2018258490-A1: Assaying ovarian cyst fluid; US-2016208340-A1: TERT Promoter Mutations in Urothelial Neoplasia; US-2015252415-A1: Arid1b and neuroblastoma; WO-2018071796-A2: Compositions and methods for identifying functional anti-tumor T cell responses; EP-3322824-A1: Detection of tumor-derived DNA in cerebrospinal fluid; US-2016273049-A1: Systems and methods for analyzing nucleic acid (Inst); US-2018135044-A1: Non-unique barcodes in a genotyping assay (Inst); US-2017016075-A1: Neoantigen analysis (Inst)
Travel, Accommodations, Expenses: Merck
Matthew D. Hellmann
Stock and Other Ownership Interests: Shattuck Labs, Immunai
Honoraria: AstraZeneca, Bristol-Myers Squibb
Consulting or Advisory Role: Bristol-Myers Squibb, Merck, Genentech, AstraZeneca/MedImmune, Nektar, Syndax, Mirati Therapeutics, Shattuck Labs, Immunai, Blueprint Medicines
Research Funding: Bristol-Myers Squibb (Inst)
Patents, Royalties, Other Intellectual Property: A patent has been filed by Memorial Sloan Kettering (PCT/US2015/062208) for the use of tumor mutation burden for prediction of immunotherapy efficacy, and which is licensed to Personal Genome Diagnostics
Travel, Accommodations, Expenses: AstraZeneca, Bristol-Myers Squibb, Eli Lilly
No other potential conflicts of interest were reported.
REFERENCES
- 1.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaz LA, Jr, Le DT. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;373:1979. doi: 10.1056/NEJMc1510353. [DOI] [PubMed] [Google Scholar]
- 4.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Philip M, Fairchild L, Sun L, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghoneim HE, Fan Y, Moustaki A, et al: De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170:142-157.e19, 2017. [DOI] [PMC free article] [PubMed]
- 8.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 9.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calne RY, Sells RA, Pena JR, et al. Induction of immunological tolerance by porcine liver allografts. Nature. 1969;223:472–476. doi: 10.1038/223472a0. [DOI] [PubMed] [Google Scholar]
- 11.Tumeh PC, Hellmann MD, Hamid O, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res. 2017;5:417–424. doi: 10.1158/2326-6066.CIR-16-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hellmann MD, Nathanson T, Rizvi H, et al: Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33:843-852.e4, 2018. [DOI] [PMC free article] [PubMed]
- 13.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. doi: 10.1126/science.aar3593. Cristescu R, Mogg R, Ayers M, et al: Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362:eaar3593, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spitzer MH, Carmi Y, Reticker-Flynn NE, et al: Systemic immunity is required for effective cancer immunotherapy. Cell 168:487-502.e15, 2017. [DOI] [PMC free article] [PubMed]
- 18.Sade-Feldman M, Yizhak K, Bjorgaard SL, et al. Defining T Cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J, Ji Z, El Asmar M, et al: Peripheral T cell dynamics in resectable NSCLC patients treated with neoadjuvant PD-1 blockade. Presented at the Society for Immunotherapy of Cancer (SITC) Conference, Washington DC, November 11, 2018. [Google Scholar]
- 20. Sade-Feldman M, Yizhak K, Bjorgaard SL, et al: Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175:998-1013.e20, 2018. [DOI] [PMC free article] [PubMed]
- 21.Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang AC, Orlowski RJ, Xu X, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25:454–461. doi: 10.1038/s41591-019-0357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang AC, Postow MA, Orlowski RJ, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. doi: 10.1038/nm.4466. Krieg C, Nowicka M, Guglietta S, et al: High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med 24:144-153, 2018 [Erratum: Nat Med 24:1773-1775, 2018] [DOI] [PubMed] [Google Scholar]
- 25.Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 26. Spranger S, Dai D, Horton B, et al: Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31:711-723.e4, 2017. [DOI] [PMC free article] [PubMed]
- 27.Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. doi: 10.1158/1078-0432.CCR-17-2386. Joseph RW, Elassaiss-Schaap J, Kefford R, et al: Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res 24:4960-4967, 2018 [Erratum: Clin Cancer Res 24:6098, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Callahan MK, Horak CE, Curran MA, et al: Peripheral and tumor immune correlates in patients with advanced melanoma treated with combination nivolumab (anti-PD-1, BMS-936558, ONO-4538) and ipilimumab. J Clin Oncol 31, 2013 (no 15_suppl; abstr 3003) [Google Scholar]
- 30.Thomas SN, Rutkowski JM, Pasquier M, et al. Impaired humoral immunity and tolerance in K14-VEGFR-3-Ig mice that lack dermal lymphatic drainage. J Immunol. 2012;189:2181–2190. doi: 10.4049/jimmunol.1103545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stachura J, Wachowska M, Kilarski WW, et al. The dual role of tumor lymphatic vessels in dissemination of metastases and immune response development. OncoImmunology. 2016;5:e1182278. doi: 10.1080/2162402X.2016.1182278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bamboat ZM, Stableford JA, Plitas G, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol. 2009;182:1901–1911. doi: 10.4049/jimmunol.0803404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knolle PA, Schmitt E, Jin S, et al. Induction of cytokine production in naive CD4(+) T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology. 1999;116:1428–1440. doi: 10.1016/s0016-5085(99)70508-1. [DOI] [PubMed] [Google Scholar]
- 34.Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006. doi: 10.1038/ni.2691. [DOI] [PubMed] [Google Scholar]
- 35.Limmer A, Ohl J, Wingender G, et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur J Immunol. 2005;35:2970–2981. doi: 10.1002/eji.200526034. [DOI] [PubMed] [Google Scholar]
- 36.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 37.Pao W, Ooi CH, Birzele F, et al. Tissue-specific immunoregulation: A call for better understanding of the “immunostat” in the context of cancer. Cancer Discov. 2018;8:395–402. doi: 10.1158/2159-8290.CD-17-1320. [DOI] [PubMed] [Google Scholar]