

Abstract
Sickle cell disease (SCD) is a monogenetic disorder caused by a mutation in the [H9252]-globin gene HBB leading to polymerization of red blood cells causing damage to cell membranes, increasing its rigidity and intravascular hemolysis. Multiple lines of evidence suggest that SCD can be viewed as pan-vasculopathy associated with multiple mechanisms but driven by hemoglobin S polymerization. Here we review the pathophysiology, clinical manifestations and management strategies for cerebrovascular disease, pulmonary hypertension and renal disease associated with SCD. These “vascular phenotypes” reflect the systemic nature of the complications of SCD and are a major threat to the well-being of patients with the disorder.
Keywords: Sickle cell disease, cerebral vasculopathy, pulmonary hypertension, renal disease, nitric oxide
1. Introduction
Sickle cell disease (SCD) is a hematological hereditary condition characterized by structurally abnormal hemoglobin (HbS) that polymerizes to “sickle” red blood cells under unfavorable conditions such as hypoxia and stress. Historically, it has been traced back to a Ghanaian family who suffered from signs and symptoms suggestive of SCD in the year 1670 [1]. It was, however, not until 1910 when Dr. James B. Herrick brought attention to a peculiar case of a dental student who had what is now known as SCD [2].
It is estimated that 5% of the world’s population carry genetic imprints responsible for hemoglobinopathies (primarily sickle cell and thalassemia) [3]. More specifically, 100,000 Americans are thought to have SCD [4, 5] thus making it the most common hemoglobinopathy in the United States and likely worldwide. The “sickling” process does not have benign consequences and leads to various disease processes including vascular and non vascular complications. These can lead to significant impact on morbidity and mortality in patients with SCD. In fact, the median survival of adults with SCD was 42 in men and 48 in women in the 1990s [6]. There has been some interval improvement in survival with more recent data suggesting that the median survival has increased to 6th or 7th decade in life but continues to be consistently lower than contemporary adults without SCD [7–9].
SCD is due to a point mutation (substitution from glutamic acid to valine) in the beta-globin chain exclusively found on hemoglobin. In homozygous state, this leads to polymerization of hemoglobin causing damage to red blood cell membranes and increasing its rigidity [10–12]. This rigidity can have two distinct effects as these deformed erythrocytes circulate throughout the body and can offer explanation to the spectrum of sickle cell complications. One effect is an increase in gelation and an obstructive adhesion of sickled cells which leads to hypoxic-ischemic-reperfusion injury to various organs. Phenotypically, it conforms to patients who suffer from vaso-occlusive pain crisis, acute chest syndrome and osteonecrosis. The other spectrum is recurrent hemolysis which leads to endothelial dysfunction and proliferative vasculopathy. This sub phenotype appears to be more common in patients with pulmonary hypertension, priapism, leg ulceration and stroke [13–16]. The latter is likely the main driver of vascular complications seen in SCD although both processes are not mutually exclusive (Fig. 1). The focus of this review will encompass the vascular complications of SCD which include cardiopulmonary vasculopathy, cerebral vasculopathy, and renal vasculopathy.
Fig. 1.
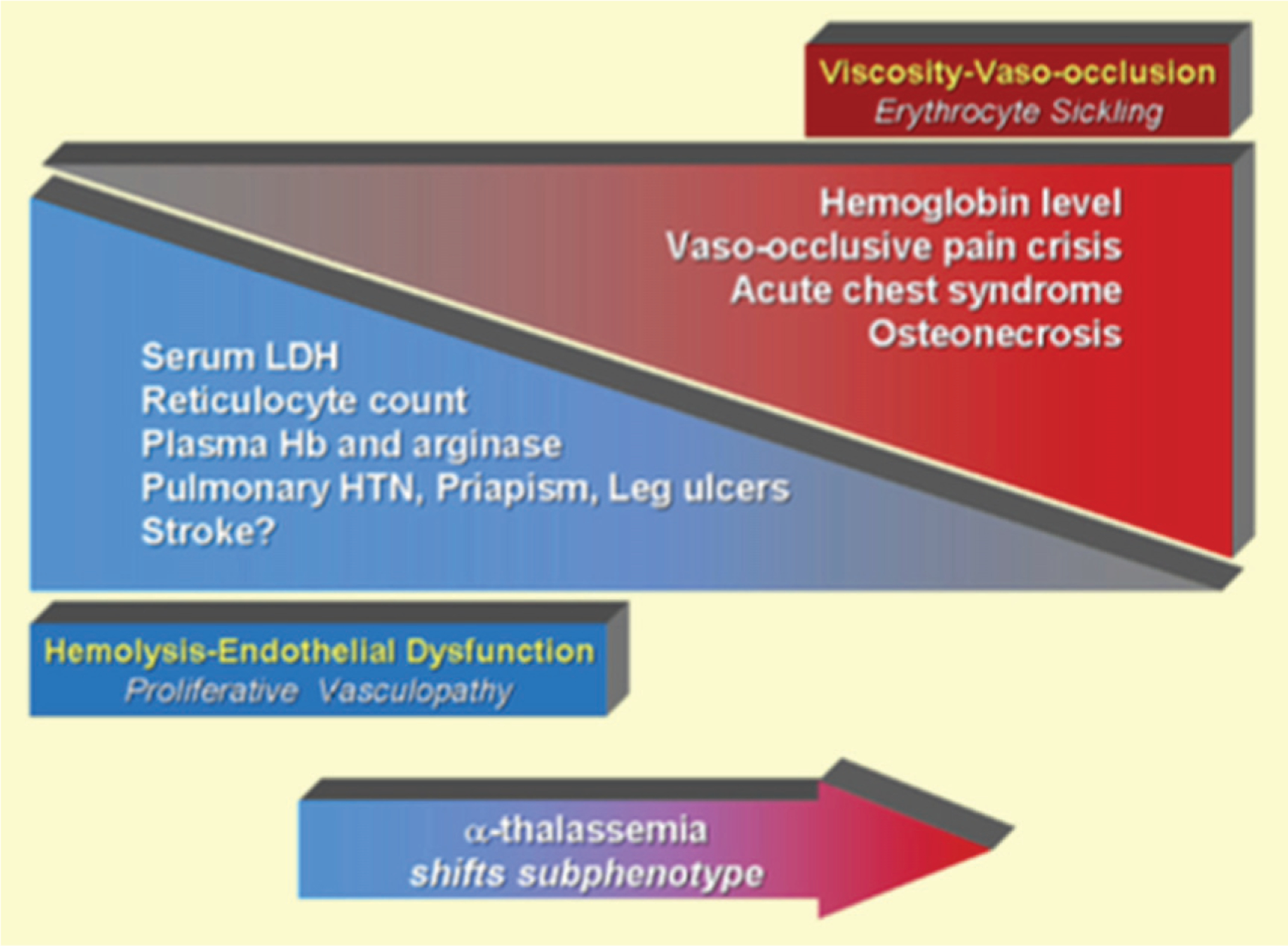
Model of overlapping subphenotypes of sickle cell disease. Published data suggest that patients with sickle cell disease with higher hemoglobin levels have a higher frequency of viscosity-vaso-occlusive complications closely related to polymerization of sickle hemoglobin, resulting in erythrocyte sickling and adhesion. Such complications include vaso-occlusive pain crisis, acute chest syndrome, and osteonecrosis. In contrast, a distinct set of hemolysis-endothelial dysfunction complications involving a proliferative vasculopathy and dysregulated vasomotor function, including leg ulcers, priapism, pulmonary hypertension, and possibly non-hemorrhagic stroke, is associated with low hemoglobin levels, and high levels of hemolytic markers such as reticulocyte counts, serum lactate dehydrogenase, plasma hemoglobin and arginase, producing a state of impaired nitric oxide bioavailability. The spectrum of prevalence and severity of each of these subphenotypes overlap with each other. 16Adapted from Kato et al. Blood Rev. 2007 January; 21(1): 37–47.
2. Pathophysiology
The fundamental role of the endothelium in various physiologic processes cannot be understated. In SCD, the vascular endothelium is markedly disrupted and dysregulated and is the converging cascade of the complex pathophysiologic processes linked to sickle cell vasculopathy [17]. Central to this endothelial dysfunction is the biodeficiency of nitric oxide (NO). NO is an endogenous vasodilator synthesized by endothelial cells by converting L-arginine to citrulline and nitric oxide by endothelial NO synthase (eNOS). It is responsible for one-fourth of the resting blood flow and inhibition of which leads to increase in systemic blood pressure and decrease in regional blood flow [18–20]. NO does so by regulating basal vascular tone and inhibiting transcriptional factors implicated in endothelial adhesion and homeostatic activation [21–24]. The half-life of NO in the vasculature is very short because of rapid reaction with red blood cell hemoglobin to form methemoglobin and nitrate [25]. Yet, it is able to exert its vasodilatory effects on vascular smooth muscle because of the compartmentalization of hemoglobin within the erythrocytes. This compartmentalization minimizes scavenging of NO by intracellular hemoglobin [26]. However, intravascular hemolysis and resultant release of free hemoglobin potentiates the scavenging of NO leading to regional biodeficiency of NO. This reaction is so fast and irreversible that cell-free plasma hemoglobin levels of only 6–10 microM are sufficient to inhibit all NO signaling and produce vasoconstriction [27]. The release of heme from the hemoglobin molecule in plasma leads to activation of TLR4 and inflammasome pathways [28]. Additionally, hemolysis releases erythrocyte arginase-1 which metabolizes arginine into ornithine thus reducing the substrate needed for NO synthesis and decreasing its overall bioavailability. The redirection of arginine to ornithine by arginase-1 has downstream effects in the formation of polyamines and proline, which are involved in collagen synthesis and linked to proliferative vasculopathy of pulmonary hypertension and cardiovascular disease [29–32]. The free hemoglobin also increases vasoconstrictor tone and amplifies NO resistance by inhibiting NO-dependent vasodilators [33]. Other contributors of increased NO resistance include NO synthase inhibitor such as asymmetric dimethylarginine. Interestingly, plasma levels of asymmetric dimethylarginine are 3-fold high in patients with SCD compared to healthy controls [34]. In fact, elevated levels of asymmetric dimethylarginine have also been linked to increased hemolysis, pulmonary hypertension and death [35]. Patients with SCD also have lower levels of apolipoprotein A-I which has been associated with abnormal endothelial function [36].
One other piece of this complex pathophysiologic picture is the role of reactive oxygen species (ROS) in generating NO resistance. Xanthine oxidase, NADPH oxidase and uncoupled endothelial NO synthase (eNOS) have been postulated to be sources of superoxide and hydrogen peroxide in SCD [37–39]. In particular, eNOS becomes dysfunctional “or uncoupled” in the setting of low levels of L-arginine or co-factor tetrahydrobiopterin (BH4) and starts production of superoxide instead of NO [40]. The ROS in turn can react with NO to form oxygen radicals such as peroxynitrite (ONOO-) further depleting NO bioavailability [41]. The downstream effect of chronic NO depletion leads to vasoconstriction, increased adhesion and platelet activation. There is endothelial activation which results in up regulation of potent vasoconstrictors such as endothelin −1. Adhesion molecules such as VCAM −1 are up regulated, platelet expression of intracellular arginase is increased and procoagulant factors are increased, all of which correlate with the degree of intravascular hemolysis [42–45].
Based on these observations, SCD can be viewed as “pan-vasculopathy” associated with multiple mechanisms but driven by hemoglobin S polymerization. The “vascular phenotypes” described in the next sections reflect the systemic nature of the complications of SCD.
3. Cerebral vasculopathy
There are many systemic consequences of SCD with cerebral vasculopathy such as stroke and chronic cerebral ischemia arguably being one of the most debilitating. Stroke was described in autopsy of children with SCD 13 years after Dr. Herrick’s original description [46]. Despite the numerous advances made in treatment, SCD remains one of the most common causes of stroke in children. The risk is highest in the first decade though, due to the protective effects of fetal hemoglobin, it is quite uncommon in the first two years of life. Strokes are clinically evident in 11% of patients with SCD before the age of 20 and up to 24% by the age of 45. The occurrences of various stroke phenotypes differ with age with the ischemic subtype being more common up to the age of 20. However, during the third decade, for unclear reasons, the hemorrhagic variant becomes the dominant subtype [47]. Additionally, 10% to 30% of SCD patients may have silent strokes that exhibit radiologic findings consistent with diffuse white matter disease [48, 49]. Other manifestations include Moyamoya (Japanese term for hazy puff of smoke) which is defined as a chronic, occlusive cerebrovascular disease involving bilateral stenosis or occlusion of the terminal portion of the internal carotid arteries and/or the proximal portions of the anterior cerebral arteries and middle cerebral arteries [50]. The smoky angiographic appearance is the origin of its name [51]. It has been associated with increased risk of recurrence of cerebrovascular events (CVAs) and thus suggests a poor prognosis [52].
The risks associated with stroke also vary with each subtype in SCD. Risk factors for silent strokes include lower hemoglobin levels, higher baseline systolic blood pressure and male sex [53]. Clinically evident ischemic strokes are associated with prior transient ischemic attacks, acute chest syndrome, and nocturnal hypoxemia [54]. Hemorrhagic variants have been linked to lower steady state hemoglobin and higher steady state leukocyte counts. Clinical features of CVA in SCD do not vary as compared to non-SCD control except that it is to be suspected in a younger cohort. However, in the event of silent infarcts, the most common presentation can be subtle cognitive deficits [55]. Therefore, high index of suspicion should be maintained at all times when evaluating patients with SCD especially in the pediatric age group. Unfortunately, often the clinical presentation represents the end point of a complex process that leads to devastating consequences.
A conscious effort was made to develop screening parameters to prevent cerebrovascular complications in children with SCD. In the 1990s, transcranial doppler (TCD) was explored as a potential imaging modality that could be used for the purposes of screening [56]. Subsequent work from the same group showed that children with “abnormal” TCD velocities, defined as greater than 200 cm/s in the large cerebral vessels, had an associated 40% stroke risk within 3 years [57]. Furthermore, certain predisposing conditions can impact the development of abnormal TCD values in kids with SCD. The absence of alpha thalassemia, presence of G6PD (Glucose-6 phosphate dehydrogenase) deficiency, lower hemoglobin and increased LDH levels are associated with aberrant TCD values [58]. The use of TCD has been proven to have excellent sensitivity but lacks sufficient specificity which compromises positive predictive value [59]. However, a landmark study, the Stroke Prevention Trial in Sickle Cell Anemia (STOP I) successfully incorporated an “abnormal” TCD value as a criteria to transfuse children 2 to 16 years of age with SCD or sickle cell thalassemia with no prior history of stroke to maintain hemoglobin S (HbS) <30% and showed an absolute risk reduction of stroke from 30% to 3% over 30 months [60]. However, replicating the same results in adults has been challenging due to the fact the adults with SCD have lower doppler velocities and therefore the criteria used in children may not be predictive of determining individuals who are at greatest risk of developing stroke [61]. Based on these convoluted intricacies, consensus guidelines recommend annual screening with TCDs in children from age 2 to 16 but do not recommend such measures in adults with SCD [62]. Also, it is recommended to institute chronic transfusion therapy in children with elevated TCD parameters.
Receiving regular transfusions is not a benign phenomenon and is associated with some adverse effects which include alloimmunization, high ferritin levels and transfusion hemosiderosis. This was evident in the STOP I cohort where sixteen percent of transfused patients had treatment discontinued because of alloimmunization and ferritin levels rose by 1500% over 24 months in the transfusion group. This has led to exploration of non-transfusion modalities. Hydroxyurea emerged as a potential therapeutic option based on its ability to decrease the incidence of acute chest syndrome and painful crises [63]. Initial studies showed encouraging results in a small Belgian cohort [64] and more recently the TWiTCH (TCD With Transfusions Changing to Hydroxyurea) trial confirmed its non-inferiority when compared to chronic blood transfusions [65]. Perhaps, its role will expand to an acceptable alternative particularly if patients develop adverse effects to chronic transfusion therapy or do not have access to blood transfusions.
Management of ischemic stroke in SCD is divided in acute and long term management. Diagnostic imaging studies seem to favor the use of MRI/MRV if it can be obtained within 60 minutes of presentation and can be paired with sequences to detect cerebral hemorrhage [66]. Acute management is not much different than the standard care of patients with ischemic stroke with two additional specific goals, i.e. reducing sickle cell hemoglobin levels to <30% of the total hemoglobin and raising the total hemoglobin to a level of 10 g/dL. Both goals are concurrently achieved with administration of blood transfusions, either simple or exchange transfusions. Thrombolysis in SCD remains controversial because of the inherent risk of hemorrhagic conversion and increased incidence of hemorrhagic CVA in SCD compared to general population.
Long term management is arguably more complex with a common theme of secondary prevention. Roughly two-thirds of sickle cell patients who suffer from a cerebrovascular event have recurrence of such events [67]. Studies incorporating chronic blood transfusions with a goal of keeping sickle cell hemoglobin to less than 30% have consistently shown a reduction in recurrent strokes to less than 15% [68, 69]. This has been further validated with patients with silent cerebral infarcts (SCI). In the SIT (Silent cerebral Infarct Transfusion) trial, children with SCIs assigned to transfusion arm had statistically significant reduction in development of clinically evident strokes or new or enlarged silent cerebral infarcts (6% vs 14%) [70]. In fact, cessation of such transfusion yields adverse outcomes with high risk of recurrent cerebrovascular events [71, 72]. Unlike its emergence in primary prevention, results of hydroxyurea as an alternative have been discouraging with the Stroke with Transfusions Changing to Hydroxyurea (SWiTCH) trial reporting a negative outcome in the hydroxyurea group. The trial was stopped early with the reporting of 7 cerebrovascular events in the hydroxyurea group when compared to none in the group received blood transfusions [73]. Interestingly, bone marrow transplantation seems to retard the progression of cerebral vasculopathy with subsequent reduction in strokes [74–76]. However further studies are needed to further elucidate the beneficial effects of bone marrow transplant in this subpopulation.
4. Cardiopulmonary vasculopathy
As scientific advances are being made to improve the survival of the sickle cell population, newer challenges have emerged that were previously under-recognized. Perhaps and arguably, the most challenging of these complications is the development of pulmonary hypertension. Pulmonary hypertension is defined as mean pulmonary artery pressure ≥25 mm Hg at rest as determined by right heart catheterization [77]. Clinically, pulmonary hypertension is divided into 5 categories (Table 1) but hemodynamically it can be divided into two subtypes. If the pulmonary capillary wedge pressure (PCWP) or left ventricular end-diastolic pressure (LVEDP) at right heart catheterization is ≤15 mm Hg, it is termed as precapillary pulmonary hypertension and if the PCWP or LVEDP is >15mm Hg, it is terms as postcapillary pulmonary hypertension.
Table 1.
Updated classification of pulmonary hypertension
| 1. Pulmonary arterial hypertension |
| 1.1 Hdiopathic PAH |
| 1.2 Heritable PAH |
| 1.2.1 BMPR2 |
| 1.2.2 ALK-1, ENG, SMAD9, CAV1, KCNK3 |
| 1.2.3 Unknown |
| 1.3 Drug and toxin induced |
| 1.4 Associated with: |
| 1.4.1 Connective tissue disease |
| 1.4.2 HIV infection |
| 1.4.3 Portal hypertension |
| 1.4.4 Congenital heart diseases |
| 1.4.5 Schistosomiasis |
| 1’ Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis |
| 1” Persistent pulmonary hypertension of the newborn (PPHN) |
| 2. Pulmonary hypertension due to left heart disease |
| 2.1 Left ventricular systolic dysfunction |
| 2.2 Left ventricular diastolic dysfunction |
| 2.3 Valvular disease |
| 2.4 Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies |
| 3. Pulmonary hypertension due to lung diseases and/or hypoxia |
| 3.1 Chronic obstructive pulmonary disease |
| 3.2 Interstitial lung disease |
| 3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern |
| 3.4 Sleep-disordered breathing |
| 3.5 Alveolar hypoventilation disorders |
| 3.6 Chronic exposure to high altitude |
| 3.7 Developmental lung diseases |
| 4. Chronic thromboembolic pulmonary hypertension (CTEPH) |
| 5. Pulmonary hypertension with unclear multifactorial mechanisms |
| 5.1 Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy |
| 5.2 Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis |
| 5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders |
| 5.4 Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH |
[83]Adapted from Simmonneau JACC 2013;62(25 Suppl):D34–41.
Recent studies have shown that a minimum prevalence of pulmonary hypertension in adults from sickle cell disease ranges from 6–11%. This may be an underestimation due to certain exclusion criteria in some of the studies. It has also been linked to increasing morbidity and mortality [78–80]. The mortality in patients with SCD and pulmonary hypertension ranged from 12.5% to 37.5% when compared to SCD patients without pulmonary hypertension where mortality ranged from 0.8% to 17% at varying intervals of follow up. In an earlier smaller study, of patients undergoing right heart catheterization, 11 of 20 (55.0%) patients with pulmonary hypertension died over up to 9 years of follow up compared with 3 of 14 (21.4%) without pulmonary hypertension [81]. This clearly signifies the importance of prompt and appropriate diagnosis.
The development of pulmonary hypertension is SCD is multifactorial resulting from the influence of chronic hemolysis, as detailed earlier, increased risk of thromboembolism, hypoxia driven gene expression and/or left ventricular dysfunction. Interestingly, pulmonary hypertension in SCD can be precapillary, postcapillary or a combination of both in a single patient [82]. This has led to its reclassification as World Health Organization (WHO) PH group 5 [83, 84]. It has long been known that patients with SCD have an inherent risk of developing venous thromboembolism [85]. Hemolysis and chronic NO depletion have been linked to increased platelet activation and contributes to a hypercoagulable state. The development of acute pulmonary embolism can progress to complications such as chronic thromboembolic pulmonary hypertension (CTEPH). Previously considered very rare (0.1–0.5%) [86], more recent studies have suggested it to be in the range of 0.4–8.8% [87]. Therefore, thromboembolism has to be considered a contributor to the development pulmonary hypertension in SCD. This was outlined in one study where scintigraphic evidence suggestive of CTEPH occurred in approximately 12% of SCD patients with pulmonary hypertension [88]. Autopsies series confirm these findings including the presence of micro thrombi and distal thrombi [89–91]. Additionally, postmortem diagnoses of pulmonary hypertension and its association with thromboembolism has been described in another autopsy series [92].
Hypoxia is a potent vasoconstrictor of the pulmonary vasculature [93] and influences various processes that can lead to the development of pulmonary hypertension [94]. In SCD due to chronic anemia, there is an upregulation of erythropoietin which, in turn, increases the levels of circulating hypoxia inducible factor (HIF)-1[H9251] [95]. HIF-1[H9251] activation has been linked to the development of various forms of pulmonary hypertension through changes in mitochondrial redox signaling, fission, and numbers, and contributes to the development of a proliferative, apoptosis-resistant phenotype in pulmonary vascular cells [96–98]. Altered gene expression in SCD has been described which leads to increased levels of HIF-1 and HIF-2 at normoxia [99]. Additionally, MAPK8, a gene involved in promoting apoptosis, which is normally downregulated in response to hypoxia, has decreased expression in normoxic condition in patients with SCD. This resistance to apoptosis coupled with abnormal proliferation of pulmonary vascular smooth muscle is distinguished feature of pulmonary arterial hypertension. [100].
Roughly half of the patients with pulmonary hypertension in SCD have post capillary PH. High cardiac output resulting from chronic anemia is primarily driven by increase in stroke volume and less by increase in heart rate. This leads to significant dilation of the left ventricle [101]. The response to this dilation is the development of eccentric hypertrophy in which wall thickening is increased and myofibers are elongated [102]. Over time, with increase in hypertrophy and left ventricular mass, diastolic dysfunction ensues. In fact, studies using standard Doppler parameters have shown that diastolic dysfunction is even found in children with SCD [103, 104]. In adults, this diastolic dysfunction is known to be an independent risk factor for decreased exercise tolerance [105] and increased mortality with a risk ratio of 4.8 [106].
There are various non-invasive tools that can be used to screen SCD patients for development for pulmonary hypertension. Doppler echocardiography, and NT-pro BNP are the most commonly used indices to gauge to possibility of developing pulmonary hypertension. Yet, both are imperfect tests which may have acceptable sensitivities but lack overall accuracy. However, they provide context for further testing which, even though is invasive, is the gold standard for diagnosing pulmonary hypertension i.e. right heart catheterization. Doppler echocardiography identifies important cardiac structures and their respective abnormalities but also estimates pulmonary artery systolic pressures via tricuspid regurgitation velocity (TRV). Using the Bernoulli equation, the tricuspid regurgitation velocity (TRV) can estimate right ventricular and pulmonary artery systolic pressures (PASP ≈ 4*TRV2) after adding an estimate of the central venous or right atrial pressure. There is a positive correlation of TRV and echocardiography-estimated systolic pulmonary artery pressure with systolic pulmonary pressure measured at right heart catheterization in patients with SCD [107]. Pulmonary hypertension is considered to be unlikely if the TRV is ≤2.8 m/sec and there are no other echocardiographic changes suggestive of pulmonary hypertension such as enlargement of right-sided chambers and right ventricular systolic dysfunction. The diagnosis is considered to be possible if the TRV is 2.9–3.4 m/sec and to be likely if it is ≥3.4 m/sec [108]. This has been validated in various studies using the above criteria. In one study, a TRV of at least 2.5 m/second identified PH with a sensitivity and specificity of 78 and 19%, respectively. The sensitivity decreased but the specificity increased to 67 and 81%, respectively, when a TRV of at least 2.88 m/second was used instead [109]. Serum NT-pro BNP level has also been evaluated as a non-invasive test that can be used to detect PH. However, most studies validating its use have marked limitations as the reference standard for PH used in those studies have been elevated TRV as opposed to direct hemodynamic measurements [110, 111]. Nonetheless, it appears to have value as a screening tool because the use of right heart catheterization for primary screening may not be acceptable to many patients due to costs and invasiveness.
Interestingly, there is data supporting the correlation of elevated TRV with mortality in SCD. Gladwin and colleagues [105] showed that mortality risk among patients with a TRV of at least 2.5 m/second was 10 times greater than that observed among patients with a TRV less than 2.5 m/second. Further analysis demonstrated that the degree of TRV elevation correlated with mortality; among patients with a TRV of 2.5–2.9 m/second and patients with a TRV of at least 3.0 m/second, the risk ratio for mortality was 4.4 and 10.6, respectively [112]. Further studies have reported similar findings [103, 113]. Similarly, NT-pro BNP has also shown to correlate with mortality in SCD [108]. This was evident in a study of 330 adults recruited as part of the Cooperative Study of Sickle Cell Disease, in which an NT-pro-BNP level of at least 160 pg/ml was associated with a relative risk of death of 6.24 compared with those with lower levels [114]. On the basis of these studies, consensus guidelines advocate for the use of non-invasive testing for risk stratification, particularly doppler echocardiography, every 1 to 3 years in patients with diagnosed pulmonary hypertension and SCD [115].
As hemolysis appears to play a central role in the development pulmonary hypertension, logically, interventions that can improve anemia can lead to a favorable response. Hydroxyurea is lucrative option due to its ability to decrease hemolysis. Furthermore, extrapolated data from the MSH trial seems to favor the use of hydroxyurea in this cohort. Moreover, case-series targeting decreasing hemolysis with hydroxyurea has shown to improve pulmonary artery systolic parameters in SCD [116]. Therefore, in SCD patients with pulmonary hypertension who have increased risk of mortality, consideration should be made to start treatment with hydroxyurea. For patients with SCD who either are not responsive to hydroxyurea or not suitable candidates for hydroxyurea, chronic transfusion therapy has been suggested but there is no direct evidence to support its use. Advocacy of its use is based on extrapolated data from the STOP-1 cohort. Use of targeted pulmonary vasodilator therapy remains controversial. This is based primarily on the negative outcome of the Walk-PHaSST trial [117] where patients in the treatment group with sildenafil resulted in an increase in serious adverse events, primarily hospitalization for pain. However, for select patients who have catheterization confirmed marked elevation of PVR (≥2 Wood units), normal PAWP (≤15 mm Hg), and related symptoms, trial of either a prostacyclin agonist or an endothelin receptor antagonist can be considered [113].
Table 2 summarizes our suggested approach to the evaluation and management of patients with sickle and pulmonary hypertension.
Table 2.
Suggested approach to screening, diagnosis and management of pulmonary hypertension in SCD
|
[82]Adapted from Gordeuk et al. Blood 2016; 127(7): 820–828
5. Renal vasculopathy
The kidneys are the recipients of the entire cardiac output and therefore present as targets for vascular complications of SCD (Fig. 2). Thus renal dysfunction is one of the most common presentations of sickle cell vasculopathy. It is estimated that renal dysfunction in SCD ranges from 5 to 18% of the total population of SCD patients [118]. The median age of developing kidney disease in SCD ranges from 23.1 years in one series [119] to 37 years in a more recent study [120]. Clinical manifestations can vary from mild to severe dysfunction and the severity has been linked to increased mortality [121]. In one study, 25% of the patients die within 12 months of starting dialysis [122]. Thus, it is of paramount importance to recognize the development of renal vasculopathy in SCD as appropriate management can prevent progression of kidney disease and impact survival.
Fig. 2.
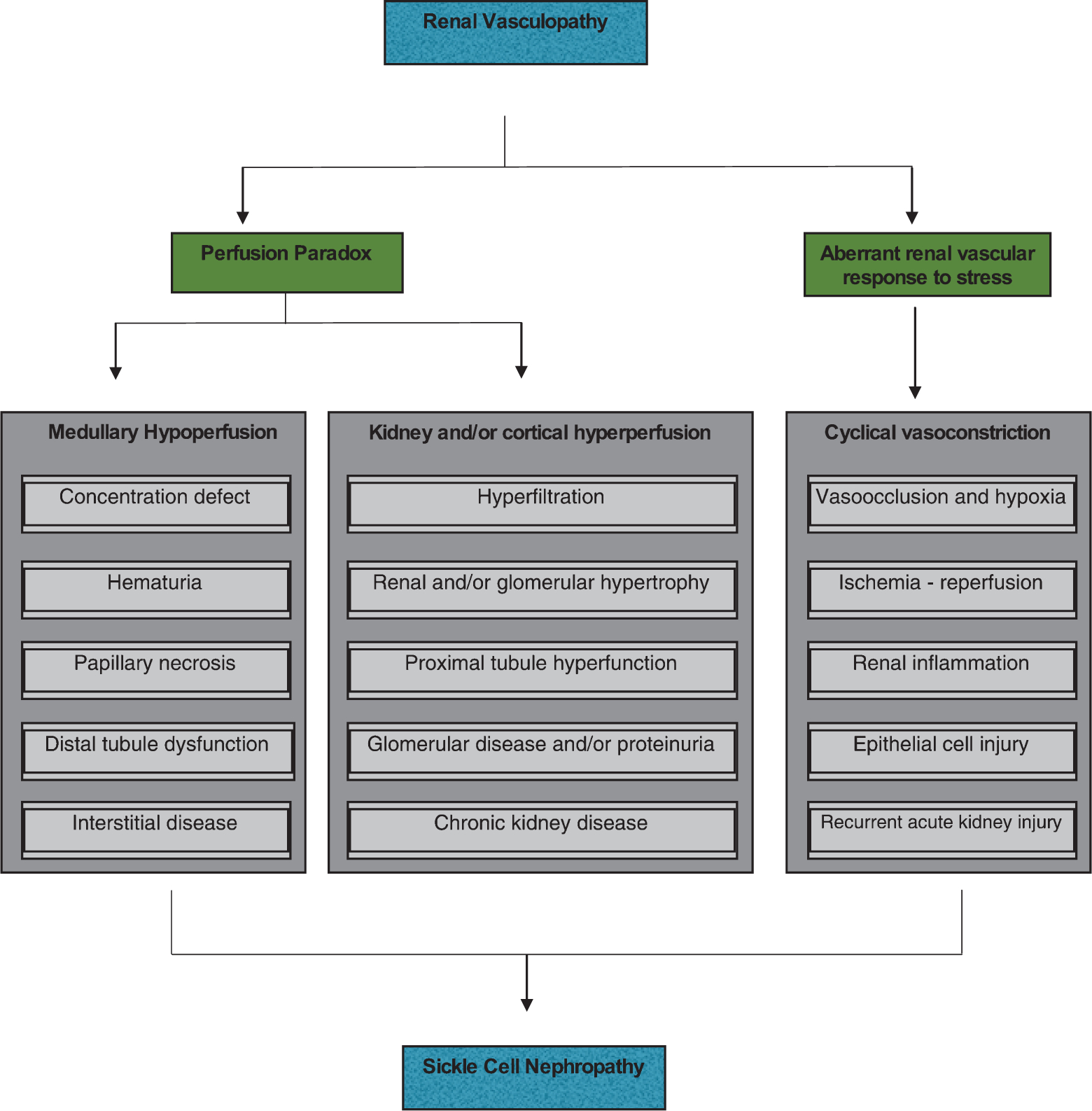
Salient pathogenic processes in the development of sickle cell nephropathy. Sickle cell nephropathy largely reflects an underlying functional vasculopathy. This vasculopathy leads to a perfusion paradox, wherein medullary hypoperfusion occurs in conjunction with kidney and/or cortical hyperperfusion. The renal vasculopathy also leads to aberrant renal vascular responses to stress that occur systemically or in distant organs and tissues. This response is characterized by enhanced renal vasoconstriction and resultant vasoocclusion. Recurrent cycles of ischemia and ischemia–reperfusion injury thus occur, thereby leading to subclinical and clinical acute kidney injury. These processes summate in the initiation and progression of sickle cell nephropathy. [131]Adapted from Nath et al. Nat Rev Nephrol. 2015;11 : 161–171.
Apart from the general aberrant vascular response to stress seen in various other organs and described above, the most characteristic finding suggestive of renal vasculopathy in SCD is alteration in renal hemodynamics. Hyperperfusion, which occurs systemically, also occurs regionally; blood flow is increased in the forearm, pedal cutaneous area, brain, and kidney [123]. Yet, the hypoxic, acidotic, and hyperosmolar environment of the inner medulla promote sickling of red blood cells with resultant impairment in renal medullary blood flow thus leading to microcirculatory regional defects. The combination of the hyperperfused whole kidney and the microcirculatory regional defects in the renal medulla leads to a “perfusion paradox” in SCD. This combined medullary hypoperfusion with cortical hyperperfusion and cyclical vasoconstriction leads to various renal manifestations of SCD.
Increased blood flow in SCD naturally results in increased filtration across the glomerulus thus leading to increased GFR [124]. This hyperfiltration has been observed in infancy and has been linked to the development of increased renal growth [125]. The leading hypothesis revolves around increased delivery of salt and water to the proximal tubule via hyperfiltration, which increases tubular reabsorption of sodium and water to restore glomerulotubular balance. As this is a highly aerobic metabolic process, it potentially stimulates mitochondrial and adaptive cellular responses which can lead to proximal tubular hyperfunction and renal enlargement [126]. Additionally, adaptive cellular responses can lead to cytokine and growth factor elevations which have positive effect on renal growth [127]. However, these processes over time can lead to cortical and glomerular capillary proliferation, interstitial edema, glomerular basement membrane thickening, glomerulosclerosis and fibrosis similar to histology seen in focal segmental gloumerulosclerosis (FSGS) [128–132]. This is thought to be primarily driven difference in the rate of glomerular enlargement when compared to relatively slower podocyte hypertrophy thus leading to areas of the glomerular basement membrane without podocyte foot processes. These denuded areas adhere to the parietal epithelium, thereby predisposing to FSGS. Additionally, the influx of cytokines promoting renal inflammation can lead to podocyte injury [133].
Glomerular disease inherently manifests as proteinuria which has been described as an age dependent phenomenon in SCD. It is estimated that about 30% patients with SCD develop proteinuria [134]. The presence of proteinuria is also associated with progressive CKD [135]. This occurs by the process described above i.e. continuous hyperfiltration causing gradual glomerular injury, increased glomerular growth causing damage to the endothelium leading to loss of podocyte integrity, and endothelial dysfunction from recurrent hemolysis [136].
Concomitantly, but contrary to hyperperfusion is the medullary hypoperfusion which manifests with simultaneous but distinguished clinical features. The high oxygen demand in the renal medulla is related to solute resorption which can lead to relatively low regional oxygen tension. This promotes sickling and congestion in the vasa recta, and resultant ischemia, impair both solute reabsorption by the ascending limb of the long loops of Henle and the capacity of the vasa recta to serve as countercurrent exchangers [137, 138]. These impairments lead to a decrease in solute accrual and retention in the medullary interstitium, thereby reducing interstitial osmolality. Over time, this leads to medullary fibrosis and clinically exhibits as the inability of concentrate urine (or hyposthenuria). This impairment has been described in infancy [139] and can lead to polyuria and eventual dehydration thus increasing the risk of precipitating a sickle cell crisis. Congestion of medullary vessels can also cause rupture and extravasation of red blood cells into the tubular lumen thus precipitating a very common clinical presentation in SCD i.e. hematuria. Sometimes necrosis of the renal papilla from severe ischemia in the vasa recta can also present as hematuria [140]. Lastly, an additional effect of medullary hypoperfusion is the distal nephron dysfunction leading to impaired urine acidification and potassium excretion.
Despite a wide array of renal manifestations, there are no specific treatments for each manifestation. Most of the management is based on general management of SCD patients e.g. prevention of vaso-occlusive crisis as well as management related to chronic kidney disease e.g. optimal blood pressure control and dietary modifications. One relatively specific treatment is the use of ACE inhibitors if proteinuria develops. In a short trial with ACE inhibitor therapy in small number of patients with mild sickle cell nephropathy, there was a reduction in proteinuria by 57%, but returned to high levels after treatment withdrawal [141]. Similar results were reproduced in smaller studies [142, 143]. The combined use of an ACE inhibitor with hydroxyurea has been suggested to prevent the progression of microalbuminuria to frank proteinuria [144]. However, this combination therapy should be tested prospectively in patients with sickle nephropathy. Renal transplantation is a viable option in SCD related ESRD. In fact, no difference in one-year cadaveric renal graft survival rates has been observed between patients with SCD and the general transplant population [145]. However, the three-year cadaveric graft survival rate was slightly lower in patients with sickle cell nephropathy (48% versus 60%; p = 0.055), and the adjusted three-year risk of graft loss was significantly greater (p = 0.003) thus signifying the importance of continued close follow up in this population.
6. Conclusion
Sickle cell disease therefore should not be only viewed by its hematological roots but rather should be understood as a systemic pathological process with global complications that are often life threatening. A major unifying feature of the disease, however, seems to arise from endothelial dysfunction and its downstream effects which have varied phenotypic presentations. Therefore, ongoing and future research should also focus on the endothelium so that targeted therapies can reduce the risk of vascular complications of sickle cell disease.
References
- [1].Konotey-Ahulu FID. Effect of environment on sickle cell disease in West Africa: Epidemiologic and clinical considerations Chapter 3: Sickle Cell Disease, Diagnosis, Management, Education and Research. Abramson H, Bertles JF, Wethers DL, eds. CV Mosby Co, St. Louis: 1973;20. [Google Scholar]
- [2].Herrick JB. Peculiar elongated and sickle shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. 1910;6:517. [PMC free article] [PubMed] [Google Scholar]
- [3].Sickle-cell disease and other haemoglobin disorders. WHO Fact sheet; 2011. [Google Scholar]
- [4].Data and Statistics. CDC; 2016. [Google Scholar]
- [5].Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4S):S512–21. [DOI] [PubMed] [Google Scholar]
- [6].Platt O, Brambilla D, Rosse W. Mortality in sickle cell disease–Life expectancy and risk factors for early death. NEJM. 1994;330:1639–44. [DOI] [PubMed] [Google Scholar]
- [7].Elmariah H, Garrett ME, De Castro LM, Jonassaint JC, Ataga KI, Eckman JR, Ashley-Koch AE, Telen MJ. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89(5):530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gardner K, Douiri A, Drasar E, Allman M, Mwirigi, Awogbade M, Thein SL. Survival in adults with sickle cell disease in a high-income setting. Blood. 2016;128(10):1436–8. [DOI] [PubMed] [Google Scholar]
- [9].Pleasants S Epidemiology: A moving target. Nature. 2014;515(7526):S2–3. [DOI] [PubMed] [Google Scholar]
- [10].Bunn HF, Forget BG, Ranney HM. Hemoglobinopathies. Major Probl Intern Med. 1997;12:1–291. [PubMed] [Google Scholar]
- [11].Hebbel RP, Schwartz RS, Mohandas N. The adhesive sickle erythrocyte: Cause and consequence of abnormal interactions with endothelium, monocytes/macrophages and model membranes. Clinics in Haematology. 1985;14:141–61. [PubMed] [Google Scholar]
- [12].Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: Pathophysiological implications. Proc Natl Acad Sci USA. 1989;86:3356–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ballas SK. Sickle cell anemia with few painful crises is characterized by decreased red cell deformability and increased number of dense cells. American Journal of Hematology. 1991;36:122–30. [DOI] [PubMed] [Google Scholar]
- [14].Schnog JJ, Lard LR, Rojer RA, van der Dijs FP, Muskiet FA, Duits AJ. New concepts in assessing sickle cell disease severity. American Journal of Hematology. 1998;58:61–6. [DOI] [PubMed] [Google Scholar]
- [15].Alexander N, Higgs D, Dover G, Serjeant GR. Are there clinical phenotypes of homozygous sickle cell disease? Br J Haematol. 2004;126:606–11. [DOI] [PubMed] [Google Scholar]
- [16].Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Reviews. 2007;21:37–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Morris CR. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematology Am Soc Hematol Educ Program. 2008;177–85. [DOI] [PubMed] [Google Scholar]
- [18].Vargas HM, Cuevas JM, Ignarro LJ, Chaudhuri G. Comparison of the inhibitory potencies of N(G)-methyl-, N(G)-nitro-and N(G)-amino- L-arginine on EDRF function in the rat: Evidence for continuous basal EDRF release. J Pharmacol Exp Ther. 1991;257:1208–15. [PubMed] [Google Scholar]
- [19].Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci U S A. 1991;86:3375–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aisaka K, Gross SS, Griffith OW, Levi R. NG-methylarginine, an inhibitor of endothelium-derived nitric oxide synthesis, is a potent pressor agent in the guinea pig: Does nitric oxide regulate blood pressure in vivo? Biochem Biophys Res Commun. 1989;160:881–6. [DOI] [PubMed] [Google Scholar]
- [21].De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. Journal of Clinical Investigation. 1995;96:60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Panza JA, Casino PR, Kilcoyne CM, Quyyumi AA. Role of endothelium-derived nitric oxide in the abnormal endothelium-dependent vascular relaxation of patients with essential hypertension. Circulation. 1993;87:1468–74. [DOI] [PubMed] [Google Scholar]
- [23].Spiecker M, Darius H, Kaboth K, Hubner F, Liao JK. Differential regulation of endothelial cell adhesion molecule expression by nitric oxide donors and antioxidants. J Leukoc Biol. 1998;63:732–9. [PubMed] [Google Scholar]
- [24].Dambisya YM, Lee TL. A thromboelastography study on the in vitro effects of L-arginine and L-NG-nitro arginine methyl ester on human whole blood coagulation and fibrinolysis. Blood Coagul Fibrinolysis. 1996;7:678–83. [DOI] [PubMed] [Google Scholar]
- [25].Doherty DH, Doyle MP, Curry SR, Vali RJ, Fattor TJ, Olson JS, Lemon DD. Rate of reaction with nitric oxide determines the hypertensive effect of cell-free hemoglobin. Nature Biotechnology. 1998;16:672–6. [DOI] [PubMed] [Google Scholar]
- [26].Schechter AN, Gladwin MT. Hemoglobin and the paracrine and endocrine functions of nitric oxide. NEJM. 2003;348:1483–5. [DOI] [PubMed] [Google Scholar]
- [27].Donadee C, Raat NJ, Kanias T. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282(28):20221–9. [DOI] [PubMed] [Google Scholar]
- [29].Schnog JJ, Jager EH, van der Dijs FP, Duits AJ, Moshage H, Muskiet FD, Muskiet FA. Evidence for a metabolic shift of arginine metabolism in sickle cell disease. Ann Hematol. 2004;83:371–5. [DOI] [PubMed] [Google Scholar]
- [30].Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, S V. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension and mortality in sickle cell disease. JAMA. 2005;294(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Omodeo-Sale F, Cortelezzi L, Vommaro Z, Scaccabarozzi D, Dondorp AM. Dysregulation of L-arginine metabolism and bioavailability associated to free plasma heme. Am J Physiol Cell Physiol. 2010;299(1):C148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tang WHW, Wang Z, Cho L, Brennan DM, Hanzen SL. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J Am Coll Cardiol. 2009;53(22): 2061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon III RO, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–9. [DOI] [PubMed] [Google Scholar]
- [34].Landburg PP, Teerlink T, Muskiet FA, Duits AJ, Schnog JJ. Plasma concentrations of asymmetric dimethylarginine, an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell patients but do not increase further during painful crisis. American Journal of Hematology. 2008;83:577–9. [DOI] [PubMed] [Google Scholar]
- [35].Kato GJ, Wang Z, Machado RF, Blackwelder WC, Taylor JGT, Hazen SL. Endogenous nitric oxide synthase inhibitors in sickle cell disease: Abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br J Haematol. 2009;145(4):506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yuditskaya S, Tumblin A, Hoehn GT, Wang G, Drake SK, Xu X, Ying S, Chi AH, Remaley AT, Shen RF, Munson PJ, Suffredini AF, Kato GJ. Proteomic identification of altered apolipoprotein patterns in pulmonary hypertension and vasculopathy of sickle cell disease. Blood. 2009;113:1122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci USA. 2001;98:15215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Griendling KK, Sorescu D, Ushio-Fukai M. NADPH oxidase: Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. [DOI] [PubMed] [Google Scholar]
- [39].Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: Structure, function and inhibition. Biochem J. 2001;357:593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moncada S, Higgs EA. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995;9: 1319–30. [PubMed] [Google Scholar]
- [41].Wood KC, Hsu LL, Gladwin MT. Sickle cell disease vasculopathy: A state of nitric oxide resistance. Free Radical Biology & Medicine. 2008;44:1506–28. [DOI] [PubMed] [Google Scholar]
- [42].Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. JAMA. 2005;293:1653–62. [DOI] [PubMed] [Google Scholar]
- [43].Raghavachari N, Xu X, Harris A, Villagra J, Logun C, Barb J, Solomon MA, Suffedini AF, Danner RL, Kato G, Munson PJ, Morris SM Jr, Gladwin MT. Amplified expression profiling of platelet transcriptome reveals changes in arginine metabolic pathways in patients with sickle cell disease. Circulation. 2007;115(2);1551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ataga KI, Moore CG, Hillery CA, Jones S, Whinna HC, Strayhorn D, Sohier C, Hinderliter A, Parise LV, Orringer EP. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica. 2008;93:20–6. [DOI] [PubMed] [Google Scholar]
- [46].Sickle cell anemia. Report of two cases in children, with necropsy in one case. By Sydenstricked VP, Mulherin WA and Houseal RW. The AJDC archives; 1923. [PubMed] [Google Scholar]
- [47].Ohene-Frempong K, Weiner SJ, Sleeper LA. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood. 1998;91(1):288–94. [PubMed] [Google Scholar]
- [48].Pavlakis SG, Bello J, Prohovnik I. Brain infarction in sickle cell anemia: Magnetic resonance imaging correlates. Ann Neurol. 1988;23(2):125–130. [DOI] [PubMed] [Google Scholar]
- [49].Moser FG, Miller ST, Bello JA. The spectrum of brain MR abnormalities in sickle-cell disease: A report from the Cooperative Study of Sickle Cell Disease. Am J Neuroradiol. 1996;17(5):965–72. [PMC free article] [PubMed] [Google Scholar]
- [50].Burke GM, Burke AM, Sherma AK. Moyamoya disease: A summary. Neurosurg Focus. 2009;26(4):E11. [DOI] [PubMed] [Google Scholar]
- [51].Suzuki J, Takaku A, et al. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20(3):288. [DOI] [PubMed] [Google Scholar]
- [52].Hulbert ML, McKinstry RC, Lacey JL, et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood. 2011;117(3):772–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].DeBaun MR, Sarnaik SA, Rodeghier MJ, et al. 2012. Associated risk factors for silent cerebral infarcts in sickle cell anemia: Low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood. 119(16):3684–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Webb J, Kwiatkowski JL. Stroke in patients with sickle cell disease. Expert Rev Hematol. 2013;6:301–16. [DOI] [PubMed] [Google Scholar]
- [55].Armstrong FD, Thompson RJ Jr, Wang W. Cognitive functioning and brain magnetic resonance imaging in children with sickle cell disease: Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Pediatrics. 1996;97(6):864–70. [PubMed] [Google Scholar]
- [56].Adams R, Mckie V, Nichols F, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. NEJM. 1992;326(9):605–10. [DOI] [PubMed] [Google Scholar]
- [57].Adams RJ, McKie VC, Carl EM, et al. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann Neurol. 1997;42(5):699–704. [DOI] [PubMed] [Google Scholar]
- [58].Bernaudin F, Verlhac S, Chevret S, Torres M, Coic L, Arnaud C, Kamdem A, Hau I, Grazia Neonato M, Delacourt C. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood. 2008;112(10):4314–7. [DOI] [PubMed] [Google Scholar]
- [59].Seibert JJ, Glasier CM, Kirby RS, et al. Transcranial Doppler, MRA, and MRI as a screening examination for cerebrovascular disease in patients with sickle cell anemia: An 8-year study. Pediatr Radiol. 1998;28(3):138–42. [DOI] [PubMed] [Google Scholar]
- [60].Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. NEJM. 1998;339(1):5–11. [DOI] [PubMed] [Google Scholar]
- [61].Valadi N, Silva GS, Bowman LS, et al. Transcranial Doppler ultrasonography in adults with sickle cell disease. Neurology. 2006;67(4):572–4. [DOI] [PubMed] [Google Scholar]
- [62].Yawn BP, et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–48. [DOI] [PubMed] [Google Scholar]
- [63].Charache S, Terrin ML, Moore RD. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia: Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. NEJM. 1995;332(20):1317–22. [DOI] [PubMed] [Google Scholar]
- [64].Gulbis B, Haberman D, Dufour D. Hydroxyurea for sickle cell disease in children and for prevention of cerebrovascular events: The Belgian experience. Blood. 2005;105(7):2685–90. [DOI] [PubMed] [Google Scholar]
- [65].Ware RE, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia—TCD With Transfusions Changing to Hydroxyurea (TWiTCH): A multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387(10019):661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kassim AA, Galadanci NA, Pruthi S, DeBraun MR. How I treat and manage strokes in sickle cell disease. Blood. 28;125(22):3401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65(3):461–71. [DOI] [PubMed] [Google Scholar]
- [68].Pegelow CH, Adams RJ, McKie V. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126(6):896–9. [DOI] [PubMed] [Google Scholar]
- [69].Russell MO, Goldberg HI, Hodson A, et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood. 1984;63(1):162–9. [PubMed] [Google Scholar]
- [70].DeBraun MR, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. NEJM. 2014;371:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. NEJM. 2005;353(26):2769–78. [DOI] [PubMed] [Google Scholar]
- [72].Wang WC, Kovnar EH, Tonkin IL, et al. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. J Pediatr. 1991;118(3):377–82. [DOI] [PubMed] [Google Scholar]
- [73].Ware RE, Helms RW, et al. , Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood. 2012;119(17):3925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. NEJM. 2009;361(24):2309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Vermylen C, Cornu G, Ferster A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: The first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998;22(1):1–6. [DOI] [PubMed] [Google Scholar]
- [77].Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42–D50. [DOI] [PubMed] [Google Scholar]
- [78].Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. NEJM. 2011;365(1):44–53. [DOI] [PubMed] [Google Scholar]
- [79].Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterization in sickle cell disease. Eur Respir J. 2012;39(1):112–18. [DOI] [PubMed] [Google Scholar]
- [80].Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA. 2012;307(12):1254–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: Cardiac catheterization results and survival. Blood. 2003;101(4):1257–61. [DOI] [PubMed] [Google Scholar]
- [82].Gordeuk VR, Castro OL, Machado RF, et al. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood. 2016;127(7):820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Simonneau G, Gatzoulis MA, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–41. [DOI] [PubMed] [Google Scholar]
- [84].Hayes MM, Amar V, et al. , Pulmonary Hypertension in Sickle Cell Disease. Ann Am Thorac Soc. 2014;11(9):1488–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lim MY, Ataga KI, Key NS. Hemostatic abnormalities in sickle cell disease. Curr Opin Hematol. 2013;20(5):472–7. [DOI] [PubMed] [Google Scholar]
- [86].Fedullo PF, Auger WR, Kerr KM, et al. Chronic thromboembolic pulmonary hypertension. NEJM. 2001;345:1465–72. [DOI] [PubMed] [Google Scholar]
- [87].Yang S, et al. Incidence and risk factors of chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. J Thorac Dis. 2015;7(11):1927–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Anthi A, Machado RF, Jison ML, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med. 2007;175(12):1272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Graham JK, Mosunjac M, Hanzlick RL, Mosunjac M. Sickle cell lung disease and sudden death: A retrospective/prospective study of 21 autopsy cases and literature review. Am J Forensic Med Pathol. 2007;28(2):168–72. [DOI] [PubMed] [Google Scholar]
- [90].Manci EA, Culberson DE, Yang YM, et al. Investigators of the Cooperative Study of Sickle Cell Disease. Causes of death in sickle cell disease: An autopsy study. Br J Haematol. 2003;123(2):359–65. [DOI] [PubMed] [Google Scholar]
- [91].Haque AK, Gokhale S, Rampy BA, Adegboyega P, Duarte A, Saldana MJ. Pulmonary hypertension in sickle cell hemoglobinopathy: A clinicopathologic study of 20 cases. Hum Pathol. 2002;33(10):1037–43. [DOI] [PubMed] [Google Scholar]
- [92].Darbari DS, Kple-Faget P, Kwagyan J, Rana S, Gordeuk VR, Castro O. Circumstances of death in adult sickle cell disease patients. Am J Hematol. 2006;81(11):858–63. [DOI] [PubMed] [Google Scholar]
- [93].Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Shimoda LA, Semenza GL. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. 2011;183(2):152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fijalkowska I, Xu W, Comhair SA, et al. Hypoxia inducible-factor1a regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176(3):1130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1a-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation. 2006;113(22):2630–41. [DOI] [PubMed] [Google Scholar]
- [98].Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation. 2010;121(18):2045–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhang X, Zhang W, Ma SF, et al. Hypoxic response contributes to altered gene expression and precapillary pulmonary hypertension in patients with sickle cell disease. Circulation. 2014;129(16):1650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Malenfant S, Neyron AS, Paulin R, et al. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ. 2013;3(2):278–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Varat MA, Adolph RJ, Fowler NO. Cardiovascular effects of anemia. Am Heart J. 1972;83:415–26. [DOI] [PubMed] [Google Scholar]
- [102].Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Johnson MC, Kirkham FJ, Redline S, Rosen CL, Yan Y, Roberts I, Gruenwald J, Marek J, Debaun MR. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood. 2010;116(1):16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hankins JS, McCarville MB, Hillenbrand CM, et al. Ventricular diastolic dysfunction in sickle cell anemia is common but not associated with myocardial iron deposition. Pediatr Blood Cancer. 2010;55(3):495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Sachdev V, Kato GJ, Gibbs JS, et al. Walk- PHASST Investigators. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation. 2011;124(13):1452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Sachdev V, Machado RF, Shizukuda Y, Gladwin MT, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49:472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. NEJM. 2004;350:886–95. [DOI] [PubMed] [Google Scholar]
- [108].Galie N, Hoeper MM, Humbert M, et al. Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC); European Respiratory Society (ERS); International Society of Heart and Lung Transplantation (ISHLT). Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6):1219–63. [DOI] [PubMed] [Google Scholar]
- [109].Fitzgerald M, Fagan K, Herbert DE, Al Ali M, Mugal M, Haynes J Jr. Misclassification of pulmonary hypertension in adults with sickle hemoglobinopathies using Doppler echocardiography. South Med J. 2012;105:300–5. [DOI] [PubMed] [Google Scholar]
- [110].Machado RF, Anthi A, Steinberg MH, Bonds D, Sachdev V, Kato GJ, Taveira-DaSilva AM, Ballas SK, Blackwelder W, Xu X. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. 2006;296: 310–8. [DOI] [PubMed] [Google Scholar]
- [111].Voskaridou E, Tsetsos G, Tsoutsias A, Spyropoulou E, Christoulas D, Terpos E. Pulmonary hypertension in patients with sickle cell/b thalassemia: Incidence and correlation with serum N-terminal pro-brain natriuretic peptide concentrations. Haematologica. 2007;92:738–43. [DOI] [PubMed] [Google Scholar]
- [112].Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. NEJM. 2008;359:2254–65. [DOI] [PubMed] [Google Scholar]
- [113].De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A, Telen MJ. Pulmonary hypertension associated with sickle cell disease: Clinical and laboratory endpoints and disease outcomes. Am J Hematol. 2008;83:19–25. [DOI] [PubMed] [Google Scholar]
- [114].Machado RF, Hildesheim M, Mendelsohn L, Remaley AT, Kato GJ, Gladwin MT. NT-pro brain natriuretic peptide levels and the risk of death in the Cooperative Study of Sickle Cell Disease. Br J Haematol. 2011;154:512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Klings ES, Machado RF, et al. An official american thoracic society clinical practice guideline: Diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med. 2014;189(6):727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Olnes M, Chi A, Haney C, May R, Minniti C, Taylor J, Kato GJ. Improvement in hemolysis and pulmonary arterial systolic pressure in adult patients with sickle cell disease during treatment with hydroxyurea. Am J Hematol. 2009;84:530–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, Gibbs JS, Little JA, Schraufnagel DE, Krishnamurti L, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118(4):855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Scheinman JI. Sickle cell nephropathy In: Pediatric Nephrology, edited by Holliday M, Barratt TM, Avner ED, Baltimore, Williams & Wilkins, 1994, pp. 908–19. [Google Scholar]
- [119].Powars DR, Elliot-Mills DD, Chan L, Hiti AL, Opas LM, Johnson C. Chronic renal failure in sickle cell disease: Risk factors, clinical course, and mortality. Ann Intern Med. 1991;115:614–20. [DOI] [PubMed] [Google Scholar]
- [120].Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: A 4-decade observational study of 1056 patients. Medicine (Baltimore). 2005;84:363–76. [DOI] [PubMed] [Google Scholar]
- [121].Platt RS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. NEJM. 1994;330:1639–44. [DOI] [PubMed] [Google Scholar]
- [122].McClellan AC, Luthi JC, Lynch JR, Soucie JM, Kulkarni R, Guasch A, Huff ED, Gilbertson D, McClellan WM, DeBaun MR. High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159:360–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Nath KA, Katusic ZS, Gladwin MT. The perfusion paradox and vascular instability in sickle cell disease. Microcirculation. 2004;11:179–93. [DOI] [PubMed] [Google Scholar]
- [124].Hatch FE Jr, Azar SH, Ainsworth TE, Nardo JM, Culbertson JW. Renal circulatory studies in young adults with sickle cell anemia. J Lab Clin Med. 1970;76:632–40. [PubMed] [Google Scholar]
- [125].Ware RE, Rees RC, Sarnaik SA, Iyer RV, Alveraz OA, Casella JF, Shulkin BL, Shalaby-Rana E, Strife CF, Miller JH, Lane PA, Wang WC, Miller ST. Renal function in infants with sickle cell anemia: Baseline data from the BABY HUG trial. J Pediatr. 2010;156:66–70.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Fine L The biology of renal hypertrophy. Kidney Int. 1986;29:619–34. [DOI] [PubMed] [Google Scholar]
- [127].Brittain JE, Parise LV. Cytokines and plasma factors in sickle cell disease. Curr Opin Hematol. 2007;14:438–43. [DOI] [PubMed] [Google Scholar]
- [128].Walker BR, Alexander F, Birdsall TR, Warren RL. Glomerular lesions in sickle cell nephropathy. JAMA. 1971;215: 437–40. [PubMed] [Google Scholar]
- [129].Elfenbein IB, Patchefsky A, Schwartz W, Weinstein AG. Pathology of the glomerulus in sickle cell anemia with and without nephrotic syndrome. Am J Pathol. 1974;77:357–74. [PMC free article] [PubMed] [Google Scholar]
- [130].Bhathena DB, Sondheimer JH. The glomerulopathy of homozygous sickle hemoglobin (SS) disease: Morphology and pathogenesis. J Am Soc Nephrol. 1991;1:1241–52. [DOI] [PubMed] [Google Scholar]
- [131].Nasr SH, Markowitz GS, Sentman RL, D’Agati VD. Sickle cell disease, nephrotic syndrome, and renal failure. Kidney Int. 2006;69:1276–80. [DOI] [PubMed] [Google Scholar]
- [132].Maigne G, Ferlicot S, Galacteros F, Belefant X, Ulinski T, Niaudet P, Ronco P, Godeau B, Durrbach A, Sahali S, Lang P, Lambotte O, Audard V. Glomerular lesions in patients with sickle cell disease. Medicine (Baltimore). 2010;89:18–27. [DOI] [PubMed] [Google Scholar]
- [133].Nath KA, Hebbel RP. Nat Rev Nephrol. 2015;11(3):161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Falk RJ, Jennette JC. Sickle cell nephropathy. Adv Nephrol. 1994;23:133–47. [PubMed] [Google Scholar]
- [135].Gosmanova EO, Zaidi S, Wan JY, Adams-Graves PE. Prevalence and progression of chronic kidney disease in adult patients with sickle cell disease. J Investig Med. 2014;62:804–7. [DOI] [PubMed] [Google Scholar]
- [136].Nath KA, Katusic ZS. Vasculature and kidney complications in sickle cell disease. J Am Soc Nephrol. 2012;23:781–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].de Jong PE, Statius van Eps LW. Sickle cell nephropathy: New insights into its pathophysiology. Kidney Int. 1985;27:711–7. [DOI] [PubMed] [Google Scholar]
- [138].Buckalew VM Jr, Someren A. Renal manifestations of sickle cell disease. Arch Intern Med. 1994;133:660–9. [PubMed] [Google Scholar]
- [139].Wesson DE. The initiation and progression of sickle cell nephropathy. Kidney Int. 2002;61:2277–86. [DOI] [PubMed] [Google Scholar]
- [140].Scheinman JI. Sickle cell disease and the kidney. Nat Clin Pract Nephrol. 2009;5:78–88. [DOI] [PubMed] [Google Scholar]
- [141].Falk RJ, Scheinman J, Phillips G, Orringer E, Johnson A, Jennette C. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. NEJM. 1992;326:910–5. [DOI] [PubMed] [Google Scholar]
- [142].Aoki RY, Saad ST. Enalapril reduces the albuminuria of patients with sickle cell disease. Am J Med 1995;98(5):432–35. [DOI] [PubMed] [Google Scholar]
- [143].Foucan L, Bourhis V, Bangou J, et al. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. Am J Med. 1998;104(4):339–42. [DOI] [PubMed] [Google Scholar]
- [144].Fitzhugh CD, Wigfall DR, Ware RE. Enalapril and hydroxyurea therapy for children with sickle nephropathy. Pediatr Blood Cancer. 2005;45(7):982–5. [DOI] [PubMed] [Google Scholar]
- [145].Ojo AO, Govaerts TC, Schmouder RL. Renal transplantation in end-stage sickle cell nephropathy. Transplantation. 1999;67(2):291–95. [DOI] [PubMed] [Google Scholar]