

Summary
Host genetic factors play a fundamental role in regulating humoral immunity to viral infection, including influenza A virus (IAV). Here, we utilize the Collaborative Cross (CC), a mouse genetic reference population, to study genetic regulation of variation in antibody response following IAV infection. CC mice show significant heritable variation in the magnitude, kinetics, and composition of IAV-specific antibody response. We map 23 genetic loci associated with this variation. Analysis of a subset of these loci finds that they broadly affect the antibody response to IAV as well as other viruses. Candidate genes are identified based on predicted variant consequences and haplotype-specific expression patterns, and several show overlap with genes identified in human mapping studies. These findings demonstrate that the host antibody response to IAV infection is under complex genetic control and highlight the utility of the CC in modeling and identifying genetic factors with translational relevance to human health and disease.
Keywords: Collaborative Cross, humoral immunity, influenza, influenza virus, antibody, host genetics, genetic architecture, genetic mapping, genetic reference population, complex trait
Graphical Abstract
Highlights
-
•
Humoral response to influenza A virus varies across genetically diverse mice
-
•
Distinct genetic loci are important for different aspects of the humoral response
-
•
Loci that regulate antibody to influenza are also important for other pathogens
-
•
Comparison across datasets informs rational selection of candidate genes
Noll et al. use the Collaborative Cross, a mouse genetic reference population, to map genetic loci associated with variation in the humoral response to influenza virus infection. Cross-dataset comparison shows that mapped loci are important for antibody response to multiple pathogens, and candidate genes with likely translational relevance are identified.
Introduction
The humoral immune response protects against pathogens through mechanisms such as antibody-mediated neutralization, opsonization, antibody-dependent cellular cytotoxicity, and initiation of the classical complement pathway (Forthal, 2014). The quality and/or magnitude of the antibody response is an important correlate of protection against many pathogens, including viruses such as influenza A virus (IAV) and severe acute respiratory syndrome coronavirus (SARS-CoV) (Couch et al., 2013, Subbarao et al., 2004). IAV is a particularly large public health burden, with seasonal IAV strains incurring significant morbidity and mortality annually (estimated 50 million cases, 1 million hospitalizations, and 80 thousand deaths in the 2017-2018 season) (Centers for Disease Control and Prevention, 2018). Though vaccination is an important component of influenza control, existing influenza vaccines often exhibit low efficacy. While this is partially due to antigenic mismatch between vaccines and circulating influenza strains (Lewnard and Cobey, 2018), numerous studies have shown that some individuals fail to mount a sufficient protective antibody response upon vaccination (Wiedermann et al., 2016).
Multiple factors contribute to variation in an individual’s ability to mount protective antigen-specific antibody responses following infection or vaccination. These factors include age, underlying disease states, and prior exposure history to related pathogens or vaccines (Lewnard and Cobey, 2018). Despite some dissent (Brodin et al., 2015), multiple studies across different pathogens and vaccines indicate that antibody responses have a strong heritable component, with many estimates of ∼50% (Kruskall et al., 1992, Linnik and Egli, 2016, Ovsyannikova et al., 2012). In the case of the measles vaccine, the antibody response was nearly 90% heritable, suggesting that genetics are a predominant factor contributing to interindividual variation in vaccine response (Tan et al., 2001). Despite this strong evidence for the role of genetic variation in modulating antibody response, the genetic factors regulating antibody response to IAV are unknown. Complicating this type of analysis is the fact that antibody response is the downstream result of a complex immunological process involving multiple tissues, cell types, and cell signaling pathways. Genes underlying variation in antibody response may be involved in any stage of the response, from general regulatory aspects of B cell development to more IAV-specific effects such as innate immune sensing. An understanding of naturally polymorphic genes involved in the antibody response, how they function, and whether their effects are broad or pathogen specific could lead to improved vaccine design, such as by utilization of adjuvants that specifically target relevant pathways and boost antibody response.
Despite the importance of understanding genetic regulation of pathogen-specific immune responses, identifying and studying genetic determinants of host antibody responses or other aspects of immunity is challenging in humans. Demographic and environmental factors such as viral dose and prior immune history affect immune responses to infection or vaccination (Lewnard and Cobey, 2018) and confound analyses. Furthermore, a lack of replicates in outbred humans, as well as difficulties accessing relevant tissues, presents challenges for phenotyping and mechanistic validation. Historically, mouse models have been used to overcome many of the logistical, experimental, and ethical issues that confound human studies. The genetic tractability of the mouse genome, including the ready availability of many gene-specific knockouts, and the ability to easily generate new knockouts, has provided key insights into the host pathways that control both development of the immune system and the quality and durability of vaccine-induced immunity (Li et al., 2008, Vidal et al., 2008). Increasingly, it is appreciated that standard laboratory mouse strains do not recapitulate the genetic diversity observed in the outbred human population (Saul et al., 2019), and this lack of diversity has limited the utility of the mouse in identifying polymorphic genes and pathways that contribute to the variation in the adaptive immune responses observed in humans.
The Collaborative Cross (CC), a multi-parental mouse genetic reference population, was developed to serve as a representative mammalian model of population-wide genetic diversity while still providing the experimental tractability of the reproducible inbred mouse (Churchill et al., 2004). CC strains have fixed and known genomes, facilitating genetic mapping to identify quantitative trait loci (QTLs) driving phenotypes of interest (Keele et al., 2018). Studies across multiple pathogens have highlighted the utility of the CC for studying infectious disease outcomes; CC mice show disease phenotypes as diverse as their genetics, facilitating discovery of novel and extreme phenotypes as well as in QTL mapping (reviewed in Noll et al., 2019). Furthermore, use of the CC enables analysis of correlations between mapped QTL and additional phenotypes across studies, extending the ability to interpret the breadth of phenotypic effect of mapped loci beyond the context in which they were originally mapped. Relevant to this study, CC mice display a wide range of phenotypes in response to challenge with IAV (Ferris et al., 2013, Kollmus et al., 2018, Maurizio et al., 2017) as well as variation in antibody glycosylation (Krištić et al., 2018).
To investigate the impact of host genetic variation on the humoral immune response to IAV, we evaluated IAV-specific antibody responses over a broad infection time course across a large set of F1 crosses between CC strains (CC-F1s). We observed significant variation in antibody response to IAV across these CC-F1s, including variation in the magnitude and kinetics of virus-specific immunoglobulin M (IgM) and IgG, as well as IgG subtype composition, and found these responses were highly heritable. We identified multiple genetic loci associated with variation in specific antibody subtypes at and across different time points post-infection. We highlight the most significant and interpretable loci, show that they have robust effects on antibody responses to other viruses, and identify candidate genes for each.
Results
Antibody Response to IAV Varies across Genetically Diverse Mice
To study the role of genetic factors in driving the magnitude, kinetics, and composition of the antibody response to IAV, we measured IAV hemagglutinin (HA)-specific antibody levels (henceforth referred to as IAV-specific antibody) in a large population of F1 crosses between CC strains (N = 116). This population provided a reproducible population of heterozygous animals to mimic the genetic heterozygosity of the human population (see Data S1 for full list of the F1s with complete strain names including laboratory codes). Our analysis focused on HA-specific antibody responses, as the bulk of the anti-IAV antibody response is directed against the HA protein, and HA-specific antibody responses are most correlated with protective neutralization (Johansson et al., 1987). Female mice from 116 CC-F1s were infected intranasally with IAV A/CA/04/09 (H1N1) and sacrificed at multiple time points post-infection (3 mice per CC-F1 at each time point for a total of ∼1,350 mice). IAV-specific antibody was measured from animals sacrificed at days 7, 10, 15, and 45 post-infection to capture both early and late humoral immune responses. IgM, IgG1, IgG2a+IgG2c (combined detection of subtypes that segregate across mouse strains (Zhang et al., 2012b)), IgG2b, IgG3, and total IgG were quantified from each animal (a total of ∼8,000 antibody measurements across the 116 CC-F1s and 4 time points) to capture differential dynamics of individual antibody subtypes and isotypes across our infection time course.
Overall, IAV-specific antibody kinetics largely matched expected patterns. On average, IgM levels peaked early and then declined, while IgG levels increased throughout our time course (Figure 1 A). However, as shown in Figure 1C, individual F1s varied in overall levels and kinetics of the IAV-specific antibody response, with some F1s displaying responses that were much more rapid and/or greater in magnitude than others (e.g., CC018xCC009 versus CC036xCC051). A number of F1s showed abnormal IgM responses, with later peaks (e.g., at day 15, CC012xCC038 and CC032xCC017) or increasing through day 45 (e.g., CC011xCC032; Figure 1D). Other F1s were particularly notable outliers more generally across isotypes. For example, excluding its’ IgG1 levels, CC003xCC062 showed some of the highest magnitude antibody responses at day 45 across isotypes (Figure S1).
Figure 1.

The CC-F1 Population Exhibits Broad Between-Strain Variation in the Magnitude, Kinetics, and Composition of IAV-Specific Antibody Responses
(A) The CC-F1 population generally exhibited an overall pattern of antibody responses that is consistent with canonical antibody maturation (e.g., IgM peaking early and then waning, concurrent with a continual expansion of IgG isotypes). Bar heights represent mean raw area under the curve (AUC) values across all CC-F1s.
(B) The correlation structure of antibody isotypes and subtype responses in the F1s over time was evaluated to determine the relationship between the development of various antibody types across the population.
(C and D) Representative examples of variation in the magnitude and kinetics of IgG (C) and IgM (D), with some exceptionally notable outliers for IgM. Each point represents an individual mouse, and bars represent mean values for F1s. (D). This variation was independent of Mx1 haplotype (for both panel sets red = Mx1 −/−, blue = Mx1 +/−).
See also Figures S1–S3.
We next investigated the relationships between antibody subtypes across the time points. Correlations between individual subtypes at different time points were largely driven by temporal kinetics, aside from IgM and some IgG1 responses that cluster independently (Figure 1B). Interestingly, antibody composition on days 7 and 15 clustered most similarly, followed by day 45 and lastly day 10. This clustering suggests that day 10 may represent a unique inflection point in the antibody response across the CC population, as opposed to a natural continuation of the responses between days 7 and 15.
We also evaluated whether virus-induced disease correlated with the antibody response. Similar to the variation in antibody response (Figure 1), CC-F1s exhibited a wide range of disease outcomes following IAV infection, ranging from asymptomatic to severe weight loss and mortality (Figure S2). We have previously shown that these differences in disease susceptibility can largely be attributed to variation in the IAV resistance gene Mx1 (Maurizio et al., 2017), although other smaller effect loci also contribute to variation in IAV susceptibility (Ferris et al., 2013). Given the role of Mx1 in regulating both disease susceptibility and viral load, we assessed whether Mx1 haplotype correlated with the antibody response. Protective Mx1 haplotypes (associated with decreased viral load and weight loss) showed some relationship with decreased antibody response throughout infection, with the strongest relationship seen early in infection (days 7 and 10) and with IgG1 and IgG3 (Table S1). However, these relationships waned over time and were not sufficient to fully explain the variation in antibody responses we observed throughout our study. In order to further clarify the role of Mx1 and viral load in influencing antibody responses, we measured viral load by genome copy assay at day 2 post-infection for a subset of CC-F1s selected for either high or low IgG responses at day 10 post-infection. We observed no trend between viral load and IgG levels on any day post-infection (Figure S3). We also assessed whether the antibody response was associated with IAV-induced disease as measured by maximal weight loss, with control for Mx1 haplotype. We observed correlations between maximal weight loss and the IgG1 response across time points, in addition to with early IgM and late total IgG (Table S2), with increased antibody levels associated with enhanced weight loss. These observations suggest that there is a relationship between antibody response and the severity of virus-induced disease that is independent of Mx1-mediated effects.
Genetic Mapping Identifies Loci Associated with Variation in Virus-Specific Antibody Response
IAV-specific antibody responses were significantly influenced by genetic factors across isotypes/subtypes and time points, with heritability estimates ranging from 26%–72% (median 44%; Table S1). This roughly aligns with estimates reported in human studies of antibody responses (Kimman et al., 2007). Antibody responses were least heritable at day 7 (39% average across isotypes) and most heritable at day 45 (57% average across isotypes), suggesting that the early antibody response is more driven by stochastic factors, while genetic factors play a progressively more important role later in the antibody response. The exception to this trend was IgM, which was most heritable at day 7 (56%), fitting with the classical kinetic profile of the IgM response.
Given the high heritability of these antibody phenotypes, we performed genetic mapping to identify QTLs (genomic regions in which genotypes correlate with a phenotype of interest) associated with variation in antibody response. We performed genetic mapping on both total amount of antibody isotypes/subtypes at single time points (i.e., days 7, 10, 15, and 45 individually) in addition to the kinetics (slope) between adjacent time points (i.e., between days 7 and 10, 10 and 15, and 15 and 45) to identify genetic factors contributing to variation in the overall magnitude, composition, and kinetics of the antibody response. Significant (genome-wide p < 0.05) and suggestive (genome-wide p < 0.1) QTLs are summarized in Table 1 (see also Table S3 for additional suggestive QTLs where p < 0.2). In total, we mapped 2 significant QTLs and 21 suggestive QTLs for antibody response to influenza (Ari1–Ari23) (Figure 2 ).
Table 1.
Significant and Suggestive (p < 0.1) QTLs
| Name | Day | Phenotype | Chr | p Value | Start (Mb) | Max (Mb) | End (Mb) | Haplotype Effects |
|---|---|---|---|---|---|---|---|---|
| Ari1 | 7 | IgG2a+IgG2c | 17 | 7.65E-02 | 47 | 52.6 | 54.4 | low: NOD, WSB |
| Ari2 | 10 | IgG3 | 11 | 2.80E-02 | 69.1 | 71.7 | 72.6 | low: WSB |
| Ari3 | 15 | IgM | 8 | 8.15E-02 | 108.7 | 109.4 | 113.1 | high: WSB |
| Ari4 | 15–45 | IgG2b | 5 | 3.95E-02 | 36.8 | 38.7 | 45.3 | high: B6, NOD, NZO, PWK |
| low: AJ, 129, CAST, WSB | ||||||||
| Ari5 | 15 | IgG1 | 16 | 8.00E-02 | 40 | 40.6 | 44.7 | high: CAST, PWK |
| low: AJ | ||||||||
| Ari6 | 15 | IgG2b | 7 | 9.65E-02 | 109.1 | 114.3 | 115.5 | high: NZO |
| Ari7 | 45 | IgG3 | 9 | 7.35E-02 | 7.8 | 13.5 | 22.3 | high: 129, NZO, CAST |
| low: AJ, B6, NOD, PWK | ||||||||
| Ari8 | 15–45 | TotalG | 15 | 9.45E-02 | 51.6 | 53.2 | 55 | high: PWK |
| low: CAST |
See also Table S3.
Figure 2.

Multiple Loci Drive Antibody Responses to IAV
QTL mapping allowed us to identify 23 loci contributing to the antibody response composition, magnitude, and kinetics. We summarize these loci (Ari1–Ari23) in a chromosomal ideogram showing their positions, as well as the antibody type and the time point for which they were mapped on their corresponding genomic loci.
The majority of QTLs were mapped for individual subtypes at given time points (16 QTLs) compared with responses between time points (7 QTLs). At individual time points, the number of QTLs mapped tracked with heritability, progressively increasing from earlier to later time points (2 at day 7, 4 at day 10, 4 at day 15, and 6 at day 45) (Table S4). For kinetic measurements, the majority of QTLs were mapped between days 15 and 45 (1 between days 7 and 10, 1 between days 10 and 15, and 5 between days 15 and 45). IgM, the least abundant isotype, was mapped least frequently (1 QTL), while IgG2a+IgG2c, the most abundant subtype, was mapped most frequently (6 QTLs). The rest of the subtypes (IgG1, IgG2b, and IgG3) and total IgG were each mapped 4 times (Table S5). We identified evidence for complex architecture of specific responses (same phenotype, multiple QTLs), as illustrated by Ari1 and Ari8, which were both mapped for IgG2a+IgG2c at day 7 (Tables 1 and S3). While QTLs were largely mapped across many chromosomes, we also found evidence for pleiotropy (same QTL, multiple phenotypes), as evidenced by the overlap between Ari14-16 on chromosome 5. Importantly, none of the QTL mapped to the Mx1 locus, which further supports the idea that, while Mx1 alleles may have some relationship with antibody responses (Table S1), the genetic regulation of the IAV-specific antibody response is predominantly independent of Mx1.
We prioritized QTLs for follow-up candidate gene analysis based on their genome-wide significance as well as clarity of haplotype effects. From the 8 QTLs with p < 0.1, we focused on 4 QTLs, Ari1–Ari4 (Figure 3 ). Ari1 (chromosome 17 [chr17]: 46.1–54.5 Mb, p = 0.0765) was mapped for IgG2a+IgG2c at day 7 post-infection and accounts for 4.5% of the total phenotypic variation (15.2% of heritable variation), driven by a low response from F1s with WSB/EiJ and NOD/ShiLtJ haplotypes at the locus. Ari2 (chr11: 69.1–72.6 Mb, p = 0.028) was mapped for IgG3 at day 10 post-infection and accounts for 9.8% of the total phenotypic variation (30.7% of heritable variation), driven by a low response from F1s with a WSB/EiJ haplotype at the locus. Ari3 (chr8: 108.7–113.1 Mb, p = 0.0815) was mapped for IgM at day 15 post-infection and accounts for 5.8% of the total phenotypic variation (14.8% of the heritable variation), driven by a high response from F1s with a WSB/EiJ haplotype at the locus. Finally, Ari4 (chr5: 37.0–46.1 Mb, p = 0.0395) was mapped for change in IgG2b between days 15 and 45, with less change between time points in F1s carrying 129S1/SvImJ, A/J, CAST/EiJ, and WSB/EiJ haplotypes at the locus.
Figure 3.

Representative Ari QTLs Associated with Variation in IAV-Specific Antibody Responses
(A–C) show Ari1, D–F show Ari2, G–I show Ari3, and J–L show Ari4.
(A, D, G, and J) Phenotypic distributions for the traits mapped to Ari1–Ari4, respectively. Each plot is independently ordered by the CC-F1 means for that trait (black points) and also shows the individual mice (purple points). The exception is (J), which only shows mean values, as the ratios of antibody response between time points were calculated using the mean value for each F1 at the relevant individual time points.
(B, E, H, and K) show the associated QTL LOD plots (significance score across the genome) for Ari1–Ari4. Significance thresholds are shown in red (genome-wide p = 0.05), blue (p = 0.1), and green (p = 0.2). Following identification of QTLs, we determined the causal haplotypes driving these responses (C, F, I, and L). Each plot is zoomed in to the relevant QTL loci on the x axis. The lower black line shows the LOD score for that region, and the colored lines display the estimated effect of each of the 8 CC founder haplotypes (A/J = yellow, C57BL/6J = gray, 129S1/SvImJ = pink, NOD/ShiLtJ = dark blue, NZO/HlLtJ = light blue, CAST/EiJ = green, PWK/PhJ = red, and WSB/EiJ = purple). Causal haplotype groups are determined based on direction and distance from mean effect and the largest split distance between lines (e.g., in I, the WSB line is furthest away from all others).
Antibody response loci may be specifically associated with the mapped phenotype of interest (e.g., IAV-specific IgG3) or reflect a broader effect on immune system development or response. Therefore, we interrogated the extent to which Ari1–Ari4 haplotypes further correlated with other aspects of IAV-induced pathology or immune responses in our CC-F1 population. We reduced the number of states being analyzed from the 8 independent founder haplotypes used in our QTL mapping analysis to the 2 variant haplotype groups (e.g., high or low response) at the locus, thereby enhancing power to detect less strong phenotypic associations. None of the Ari loci showed correlations with gross IAV-induced disease as measured by weight loss, with control for Mx1 haplotype (Table S4).
The Ari loci showed numerous significant correlations with additional IAV-specific antibody isotypes and time points. Ari1 (IgG2a+IgG2c, day 7) showed broad correlations across multiple isotypes at days 7, 10, and 15. Additionally, the association between Ari1 and IgG1 persisted through day 45 (Table S1). This suggests that Ari1 plays a role in the early antibody response that carries over through the mid-stage response, with a unique persistent effect on IgG1. Ari2 (IgG3, day 10) was broadly correlated with the antibody response at day 7 but showed the strongest correlations at day 10 and was also associated with total IgG through days 15 and 45 (Figure 4 ), suggesting that Ari2 broadly impacts the overall early antibody response with some prolonged effect. Ari3 (IgM, day 15) also broadly correlated with the antibody response at day 7, including a strong correlation with total IgG. Ari3 was correlated with IgG2a+IgG2c and total IgG day 15 post-infection, suggesting that Ari3 is a driver in the early-to-mid stage antibody response for both IgG and IgM (Table S1). When tested across individual time points, Ari4 (IgG2b, ratio between days 15 and 45) showed associations with multiple isotypes, including IgG2b, at day 15, but not day 45 (Table S1), indicating that the association between Ari4 and temporal changes in IgG2b levels are predominantly driven by effects on antibody isotype levels on day 15.
Figure 4.

Ari2 Shows Broad Effects on IAV-Specific Antibody Responses in the CC-F1 Population
We assessed the impact that a WSB/EiJ haplotype (x axis; 1 = one WSB haplotype; 2 = two non-WSB haplotypes) have on other IAV antibody responses in this study (y axis: CC-F1 mean levels of transformed AUC levels for given isotypes/time points). Points represent mean values for CC-F1s, with ~3 mice per CC-F1. Annotations refer to transformations applied to datasets (∗p < 0.1, ∗∗p < 0.05, ∗∗∗p < 0.01).
See also Table S1.
To further assess the role of Ari1–Ari4 in virus-specific antibody responses, we tested to see if Ari1–Ari4 haplotypes were associated with antibody responses to other viruses. For this analysis, we took advantage of existing datasets from related but independent studies with SARS-CoV, a respiratory virus, and chikungunya virus (CHIKV), an arbovirus. In the SARS-CoV study, mice from a similar panel of CC-F1s were infected with SARS-CoV, and antibody response to the spike (S) protein was measured at days 7, 15, and 29 post-infection. An additional measurement was taken 4 days following a secondary challenge (“rechallenge”), which was administered at day 28 post-primary infection. Ari1 haplotypes were not associated with SARS-CoV-specific antibody responses (Table S6). In contrast, Ari2 haplotype showed broad associations with antibody specific to SARS-CoV at days 7 and 29, and with IgM at all time points post-primary and secondary challenge (Table S6). Ari3 haplotype showed only a few correlations with SARS-CoV antibody, which were not indicative of trends either across isotypes or time points (Table S6). Ari4 showed the strongest correlations with the SARS-CoV-specific antibody response, most potently at day 7 but lasting through days 15 and 29 (Table S6). In the CHIKV study, mice from a panel of 64 inbred CC strains were infected with CHIKV. Virus-specific IgM, IgG, and neutralizing antibody were measured at day 7 post-infection. While Ari1, Ari2, and Ari4 did not correlate with the CHIKV-specific antibody response (IgM, IgG, and neutralization titer) in CC-RI mice at day 7 post-infection, Ari3 haplotypes showed strong associations with the CHIKV antibody response (Table S6). Interestingly, these associations were in the opposite direction as observed for IAV (i.e., WSB/EiJ haplotype associated with decreased antibody response). Overall, these findings suggest that Ari1 is uniquely associated with variation in IAV-specific antibody responses, while Ari2–Ari4 show broader effects on virus-specific antibody responses. Ari2 and Ari4 are associated with variation in SARS-CoV-specific antibody responses, while Ari3 was also found to be strongly associated with variation in the CHIKV-specific antibody response.
Candidate Gene Analysis
To identify candidate genes at each of the loci, we applied a series of criteria to filter through the variants (Figure 5 ). We first conducted association testing as implemented in the R package DOQTL (Gatti et al., 2014), comparing the pattern of single-nucleotide polymorphisms (SNPs) at each marker within the QTL to the observed haplotype effects at the corresponding location, and filtering out variants below the LOD threshold. Next, candidate SNPs were categorized based on the classification of the affected genetic element (e.g., regulatory region, protein-coding gene, or non-coding RNA). While variation in regulatory regions, microRNAs (miRNAs), long non-coding RNAs (lncRNAs), or other genetic elements may drive QTLs, we focused on SNPs in protein-coding genes for our analysis, as they have more interpretable and better defined functional consequences. The protein-coding genes at each locus were next evaluated based on whether the gene exhibited baseline expression in the lung or immune tissue (i.e., spleen or thymus) utilizing ENCODE data (Yue et al., 2014).
Figure 5.
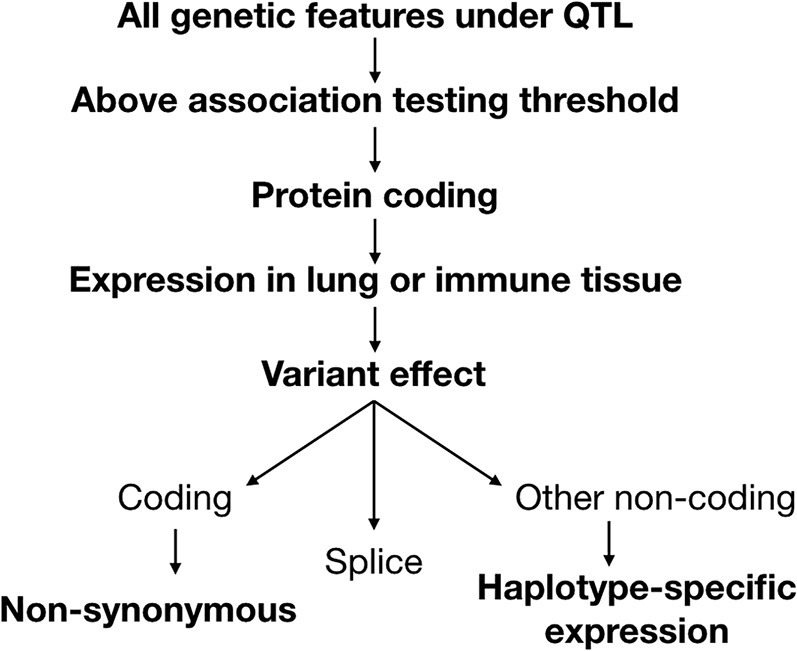
Pipeline to Narrow Down Candidate Genes under Ari1–Ari4
Variants under each QTL were considered if they were above the association testing threshold and were contained in a protein-coding gene. Protein-coding genes were then evaluated based on whether they were expressed in the lung or immune tissue. Using a separate transcriptional dataset from 11 CC strains infected with H3N2 influenza, IAV-specific and haplotype-specific differential expression was evaluated. Genes were considered if they had non-synonymous coding variants or splice region variants or showed haplotype-specific expression.
We next evaluated candidate genes based on the predicted consequences of their variants, which were determined using the Ensembl Variant Effect Predictor (McLaren et al., 2010). Genes with non-synonymous coding variants and splice variants were included as candidates. To assess the relevance of non-coding variants, which were determined most likely to have a role in differential expression, we leveraged transcriptional datasets from lungs and peripheral blood of 11 CC strains, which were either mock-infected or infected with IAV H3N2 (days 3 and 5 post-infection for lung; days 3, 5, and 8 for blood). We applied this data to assess if genes with non-coding variants were differentially expressed between haplotype groups (see examples in Figure S4). By applying these criteria, we identified a set of high-priority candidate genes (Table 2 ).
Table 2.
High-Priority Candidate Genes Identified under Ari Loci based on Evaluation Criteria
| QTL | Gene | Coding Variant | Splice Variant | UTR Variant | Other Non-coding Variant | Lung or Immune Tissue Expression | IAV-Induced Differential Expression |
Haplotype-Specific Expression |
Criteria Met | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lung | Blood | Lung | Blood | ||||||||
| Ari1 | Unc5cl | NA | NA | NA | yes | yes | no | yes | no | yes | non-coding |
| Ari2 | Mis12 | NA | NA | 3′, 5′ | yes | yes | yes | no | yes | yes | non-coding |
| Ari2 | Nlrp1b | Mis | Yes | NA | yes | yes | yes | yes | yes | no | coding, non-coding |
| Ari2 | Nup88 | NA | NA | NA | yes | yes | yes | yes | yes | no | non-coding |
| Ari2 | Rpain | Mis | NA | NA | yes | yes | no | yes | yes | no | non-coding |
| Ari2 | Spns3 | Syn | NA | NA | yes | yes | yes | no | yes | no | non-coding |
| Ari2 | Wscd1 | Mis | NA | NA | yes | yes | yes | no | yes | no | coding, non-coding |
| Ari3 | Bcar1 | Mis | NA | NA | yes | yes | yes | yes | NA | NA | coding |
| Ari3 | Hydin | Mis | NA | NA | yes | yes | yes | no | NA | NA | coding |
| Ari3 | Kars | Syn | yes | 3′, 5′ | yes | yes | yes | yes | NA | NA | splice |
| Ari3 | Mlkl | NA | yes | NA | yes | yes | yes | yes | NA | NA | splice |
| Ari3 | Pkd1l3 | Non | NA | NA | yes | yes | no | yes | NA | NA | coding |
| Ari3 | Zfhx3 | Mis | NA | NA | yes | yes | yes | yes | NA | NA | coding |
| Ari4 | Cc2d2a | NA | NA | NA | yes | yes | yes | no | yes | no | non-coding |
| Ari4 | Ldb2 | NA | NA | NA | yes | yes | yes | no | yes | no | non-coding |
| Ari4 | Prom1 | NA | NA | NA | yes | yes | yes | yes | yes | no | non-coding |
| Ari4 | Tapt1 | NA | NA | 3′ | yes | yes | no | yes | yes | no | non-coding |
High priority candidate genes were identified based on evaluation criteria illustrated in Figure 5. (UTR, untranslated region; Mis, missense; Non, nonsense; Syn, synonymous). See also Figures S4 and S5 and Table S7.
Ari1 contains 139 genetic elements (e.g., protein-coding genes, large intergenic noncoding RNAs [lincRNAs], retained introns, pseudogenes) (25,732 SNPs), which was narrowed to 15 genetic elements (100 SNPs) above the association threshold (LOD > 3.2) (Table S7), and then to six protein-coding genes (46 SNPs), five of which were expressed in relevant tissues. All of these candidate genes contained non-coding variants, while none of the SNPs resulted in non-synonymous coding changes or splice variants. Analysis of haplotype-specific expression found while none of the candidates showed haplotype-specific expression in the lungs, one gene, unc-5 family C-terminal like (Unc5cl or Zud), showed haplotype-specific expression in the peripheral blood, making Unc5cl the lead candidate gene under the locus.
Ari2 contains 99 genetic elements (11,236 SNPs), which was narrowed to 40 genetic elements (286 SNPs) above the association threshold (LOD > 4) (Table S7) and then to 20 protein-coding genes (166 SNPs), 18 of which were expressed in relevant tissues. One of these genes (NOD-like receptors, pyrin domain containing 1B [Nlrp1b]) had non-synonymous variants and splice variants and showed haplotype-specific expression in the lung. Two genes had both non-synonymous variants and haplotype-specific expression in the lung (WSC domain containing 1 [Wscd1] and RPA-interacting protein [Rpain]). Three additional genes under Ari2 with non-coding variants showed haplotype-specific expression in the lung (spinster homolog 3 [Spns3], MIS12 kinetochore complex component [Mis12], and nucleoporin 88 [Nup88]).
Ari3 contains 82 genetic elements (14,707 SNPs), which was narrowed to 57 genetic elements (607 SNPs) above the association threshold (LOD > 2.85) (Table S7) and then to 40 protein-coding genes (565 SNPs), 30 of which were expressed in relevant tissues. Under Ari3, there were four genes with non-synonymous coding variants (breast cancer anti-estrogen resistance protein 1 [Bcar1], HYDIN axonemal central pair apparatus protein [Hydin], polycystic kidney disease 1 like 3 [Pkd1l3], and zinc-finger homeobox 3 [Zfhx3]). There were two additional genes with splice variants (lysyl-tRNA synthetase [Kars] and mixed lineage kinase domain-like [Mlkl]). We were not able to evaluate Ari3 non-coding candidates based on haplotype-specific expression due to a lack of representation of the WSB/EiJ haplotype at Ari3 in the strains included in the transcriptional dataset.
Ari4 contains 22 genetic elements (4,899 SNPs), which was narrowed to 16 genetic elements (68 SNPs) above the association threshold (LOD > 0.597) (Table S7) and then to six protein-coding genes (51 SNPs), all of which were expressed in relevant tissues. Under Ari4, 4 genes with non-coding variants showed haplotype-specific expression in the lung (coiled-coil and C2 domain containing 2A [Cc2da2], LIM-domain-binding protein 2 [Ldb2], Prominin 1 [Prom1], and transmembrane anterior posterior transformation 1 [Tapt1]).
Discussion
The humoral immune response is an important component of protection against viruses such as IAV. However, studying the role of the genetic factors that contribute to regulation of the antibody response is challenging in humans due to confounding demographic and environmental variables, as well as the relatively late stage in infection when the humoral response occurs. To study the role of host genetics in driving the antibody response to IAV, we used a diverse panel of CC-F1s and found that CC mice exhibit high levels of variation in susceptibility to disease as well as the generation and maintenance of specific antibody isotypes and subtypes. In concordance with human studies (Kruskall et al., 1992, Linnik and Egli, 2016, Ovsyannikova et al., 2012), we found strong evidence for genetic control of these traits (median heritability, 44%) and were able to map multiple loci associated with antibody responses.
In addition to genetic mapping, the CC provides a platform for phenotypic discovery and model development. Unique combinations of alleles present in CC mice may present as novel phenotypes, which can provide an opportunity to study interesting disease features and/or more closely model human disease (Rasmussen et al., 2014, Rogala et al., 2014). We identified multiple CC-F1s with outlier antibody response phenotypes in our investigation, including multiple F1s with persistent IAV-specific IgM responses. Importantly, those F1s with persistent IgM responses were not defective in IAV-specific IgG responses, and some had protective Mx1 alleles, suggesting that the sustained IgM response was not due to failure in class switching or inability to control viral replication. Notably, two highlighted CC-F1s with persistent IgM responses shared the parental strain CC032, suggesting that genetic contributions from CC032 (independent of Ari3, for which CC032 has the low-response haplotype) may be involved in longer lasting IgM responses. Such outlier strains can be used to model these aberrant responses and/or included in future targeted mapping studies (e.g., F2 crosses).
All antibody response measures were driven to varying degrees by genetically variable (e.g., heritable) effects; however, not all phenotypes were mapped to QTL. This issue of “missing heritability” (the inability to identify all the genetic factors that contribute to heritable variation) is prevalent across genetic mapping studies. Missing heritability can be explained by numerous factors, including lack of sufficient power or informative lines to detect variants of small effect, phenotypic heterogeneity (i.e., same phenotype, different cause), complex genetic architecture, and blindness to genetic features such as copy number variants or epigenetic modifications (Manolio et al., 2009). More targeted studies, such as 2-strain intercrosses, allow for a more complete discovery of phenotypic modulation with greater power to identify more complex genetic interactions such as epistasis (Gralinski et al., 2017, Rogala et al., 2014). Nonetheless, we had sufficient power to identify major loci with effects across our CC-F1 population.
We further investigated our mapped QTLs to identify broader relationships with additional phenotypes to further assess the breadth of effect of each QTL. When testing for further associations with Ari1–Ari4 haplotypes across antibody datasets (IAV, SARS-CoV, and CHIKV), rather than focusing on any individual association, we were interested in robust and persistent trends in correlations across isotypes and/or time points. This is critical due to the differing heritability and potential genetic architecture across isotypes and time points. Therefore, while we did not utilize false discovery rate or multiple-test corrections in assessing these relationships, these trends are indicative of important relationships that merit further study. Furthermore, consideration of these trends, or lack thereof, can provide insight into possible mechanisms by which the genes driving QTL impact the antibody response. Ari1 showed broad effects on the early antibody response to IAV only and was not associated with antibody response to SARS-CoV or CHIKV, suggesting that it may be involved with specific responses to IAV such as innate immune recognition and response or antigen presentation. Ari2 showed broad early effects with IAV as well as SARS-CoV-specific responses, suggesting that the underlying gene is involved in early stages of infection or immune response for respiratory pathogens. Ari3 also showed broad early effects with IAV and a strong correlation with antibody response to CHIKV, though the haplotype effects were in opposing directions for these two different viruses. Ari4 was most relevant for day 15 antibody response to IAV and also showed strong associations with antibody response to SARS-CoV, suggesting that it may be widely relevant for the development of the humoral response. These broad associations highlight the utility of the CC as a resource to extend analyses into related datasets to gain further understanding of the role of loci across phenotypes while illustrating the need for future work to define the specific mechanisms by which each of these loci affect antiviral antibody response.
In addition to impacting the scope of pathogens for which a QTL may play a role, the mechanisms by which these loci mediate antibody response to primary H1N1 infection is also relevant for their potential to affect antibody response to other strains of IAV or even IAV vaccination. QTLs that are driven by genes involved in early innate response in the lung are more likely to be specific to primary infection and may also be virus-strain specific (Ryan et al., 2018). Alternatively, QTLs that appear to be more generally involved in the later humoral response, such as Ari4, may be more likely to have a broader effect that is relevant for other IAV strains and vaccination.
Ultimately, the goal of genetic mapping studies is to identify the causal genes and variants driving phenotypic differences. Insights into possible mechanisms gained from analyzing broad associations across an array of phenotypes can help guide selection of candidate genes, along with other considerations such as gene classification, predicted consequence of mutations, expression in relevant tissues, and any known relevance in infection and/or immunity. We leveraged a transcriptional dataset from lungs and blood of CC strains infected with IAV H3N2, which allowed us to assess haplotype-specific expression of candidate genes in the context of the relevant tissue systems. These results were able to help us to narrow our focus on smaller sets of genes (as shown in Figure 5). However, it is possible that due to disease kinetics, the tissue complexity, and our above-discussed description of strain-differences, this analysis is overly restrictive.
Under Ari1, we identified one candidate gene, Unc5cl (Zud), which showed flu-induced and haplotype-specific expression in the peripheral blood, but not the lungs. While the function of Unc5cl is largely unknown, it contains a death domain and has been shown to be involved in the regulation of nuclear factor κB (NF-κB) (Heinz et al., 2012, Zhang et al., 2004). To our knowledge, a role of Unc5cl in host response to viral infection has not been evaluated, and this gene presents a clear candidate for additional analysis.
Under Ari2, we identified 6 candidate genes (Mis12, Nup88, Nlrp1b, Rpain, Spns3, and Wscd1). There is no documented role in immunity for Wscd1 or Mis12. Rpain (also known as RIP and HRB) has been shown to play an important role in the nuclear export of viral ribonucleoproteins (vRNPs) during IAV replication via interaction with the IAV nuclear export protein NS2. Spns3 is highly expressed in the spleen and thymus, and its paralog, Spsn2, has been shown to be important for lymphocyte development and humoral immune response (Nijnik et al., 2012). Nup88 has been shown to be important for MX2 inhibition of HIV (Dicks et al., 2018), and its Drosophila paralog is involved in innate immune activation (Uv et al., 2000). Nlrp1b is an inflammasome gene, of which different genetic variants have been shown to play a role in susceptibility to anthrax lethal toxin (Moayeri et al., 2010), although it has not been previously linked to viral infection.
Under Ari3, we identified 6 candidates genes with non-synonymous or splice region variants (Bcar1, Hydin, Kars, Mlkl, Pkd1l3, and Zfhx3). Bcar1 and Zfhx3 have no established role in immunity or viral infection. Hydin function is important for ciliary projections and motility, which could affect influenza tropism (Lechtreck et al., 2008, Smith et al., 2019). Kars (also known as LysRS) encodes a tRNA synthetase, which plays a fundamental role in protein translation in addition to acting as a signaling molecule that responds to immunological stimulation (Yannay-Cohen et al., 2009). Mlkl encodes a protein that plays a critical role in necroptosis, and its activity is enhanced by interaction with the NS1 protein of IAV (Gaba et al., 2019). Pkd1l3 encodes a calcium channel component and is largely conserved across classical inbred strains but has a unique nonsense mutation in the WSB allele (which drives the QTL effect).
We were unable evaluate haplotype-specific expression for genes under Ari3 due to a lack of haplotype representation in our transcriptional dataset. However, there are several genes with non-coding variants under the interval that could be considered additional candidates based on precedence in the literature. DEAD box polypeptide 19b (Ddx19b) inhibits type I interferon (IFN) and is required for IAV replication via a role in nuclear export of vRNPs (Diot et al., 2016, Zhang et al., 2019). Dihydroorotate dehydrogenase (Dhodh) is involved in pyrimidine synthesis, and DHODH inhibitors have been shown to have strong antiviral activity against influenza and other viruses (Cheung et al., 2017, Zhang et al., 2012a). Gamma-aminobutyric acid A receptor-associated protein-like 2 (Gabarapl2, also known as Gate-16) plays a role in autophagy and IFN-γ-dependent antimicrobial response (Sasai et al., 2017). Haptoglobin (Hp) is involved in MyD88-dependent inflammation, which plays an important role in primary influenza infection (Seo et al., 2010), and deficiency leads to impaired lymphocyte development and adaptive immune response (Huntoon et al., 2008, Shen et al., 2012). Despite the lack of haplotype-specific expression data, these genes may warrant further evaluation as potential candidate genes under Ari3.
Under Ari4, we identified 4 candidates of interest: Lbd2, Prom1, Cc2da2, and Tapt1. Neither Lbd2 nor Prom1 has a documented role in immunity. Cc2da2 is required for cilia biogenesis, which as previously mentioned may be relevant for influenza tropism (Smith et al., 2019, Veleri et al., 2014). Tapt1 is also important for ciliogenesis, and knockdown has been shown to decrease IAV replication (Sui et al., 2009, Symoens et al., 2015). Notably, multiple candidate genes under the Ari loci have roles in ciliary function (Hydin, Cc2da2, and Tapt1). Human patient cohorts have shown co-occurrence of primary ciliary dyskinesia and humoral immunodeficiency, both rare disorders, suggesting a possible common pathophysiological pathway between ciliary function and antibody response (Boon et al., 2014). Tapt1 has also been shown to be differentially expressed in the context of other respiratory pathogens (Denisenko et al., 2019) and was identified in a gene set associated with increased HA inhibition and neutralization titers following influenza vaccination (Ovsyannikova et al., 2016), further supporting a potential role for this gene in driving Ari4.
Following candidate identification, validation presents new challenges. This is exemplified by Ari2, under which Nlrp1b was identified as the strongest candidate gene, carrying haplotype-specific coding variants predicted to be strongly deleterious to the encoded innate immune sensor. However, analysis of this gene and interpretation of expression data is challenging because of the newly described genetic complexity of the locus; multiple Nlrp1 paralogs and haplotypes sort across mouse strains and have not been fully characterized (e.g., WSB/EiJ mice carry 5 Nlrp1 paralogs, only 2 of which are shared with the reference C57BL/6J) (Lilue et al., 2018). Upon IAV infection, Nlrp1b knockout mice did not show significant differences in antibody response (Figure S5). While this lack of phenotype might suggest that Nlrp1b is not causal for the Ari2 phenotype, it is equally likely that the specific Nlrp1b allelic variant in the CC, or one of the Nlrp1 paralogs, is responsible for the effect of Ari2. Continued work to deconvolute the mouse genome across genetically diverse strains, as well as advances in molecular and genetic tools such as CRISPR, will facilitate follow-up analyses on such complex loci.
Other potential challenges remain in the identification and validation of genes that regulate the antibody response to IAV. Notably, we studied IAV exposure in naive animals, whereas prior infection (especially with IAV) is a large factor in shaping the humoral immune response in humans (Lewnard and Cobey, 2018). Nonetheless, we identified multiple loci with genes that overlap with human mapping studies of antibody response or influenza infection. Ari1 contains kinesin family member 6 (Kif6), which was identified in a human study of antibody response to smallpox vaccination (Ovsyannikova et al., 2012). Kif6 did not meet our formal candidate criteria as its coding variant was synonymous and it did not show haplotype-specific expression in the dataset that we utilized. Additionally, Ari2 contains two genes that were identified in a human study of severe pneumonia associated with H1N1 influenza infection (Zúñiga et al., 2012), Rpain and complement component 1 Q subcomponent-binding protein (C1qbp), a high-affinity receptor for the complement protein C1q that is important for dendritic cell maturation (Gotoh et al., 2018). While Rpain met our criteria as a candidate gene, and is therefore a high-priority candidate for additional analysis, C1qbp1 had a non-coding variant, but did not show haplotype-specific expression in the lungs or blood. Lastly, as noted above, Tapt1 under Ari4 has been associated with variation in antibody responses to IAV vaccination in humans (Ovsyannikova et al., 2016), making this gene a high-priority candidate for additional study. While not all of these genes made it through our formal selection criteria, this concordance between loci mapped in our study and in human studies demonstrates the translational relevance of our work, and we are optimistic that other loci mapped here could be validated and important in humans.
Conclusions
The magnitude and kinetics of the antibody response to IAV varies greatly across a genetically diverse set of CC-F1 mice. These differences, which did not widely correlate with general susceptibility to IAV-induced disease, were leveraged to map 23 QTLs associated with variation in specific antibody isotypes across time points. Multiple significant and suggestive QTLs were identified, including Ari1–Ari4, which likely drive variation in IAV-specific antibody response through different mechanisms. Ari loci also showed effects on the antibody response to SARS-CoV and CHIKV, suggesting that these QTLs are broadly important for antibody response to multiple pathogens. Candidate genes for Ari loci have been identified and show overlap with genes identified in human studies. Overall, our findings demonstrate that multiple independent loci regulate the magnitude, kinetics, and composition of IAV-specific antibody. These results further demonstrate the utility of the CC for modeling how host genetic variation shapes pathogen-specific immunity and open the door to more robust modeling of the complex interplay between genetics and environment in promoting protective adaptive immune responses to pathogens.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HRP goat anti-mouse IgM | Southern Biotech | 1020-05; RRID: AB_2794201 |
| HRP goat anti-mouse IgG1 | Southern Biotech | 1070-05; RRID: AB_2650509 |
| HRP goat anti-mouse IgG2a | Southern Biotech | 1080-05; RRID: AB_2734756 |
| HRP goat anti-mouse IgG2b | Southern Biotech | 1090-05; RRID: AB_2794521 |
| HRP goat anti-mouse IgG2c | Southern Biotech | 1079-05; RRID: AB_2794466 |
| HRP goat anti-mouse IgG3 | Southern Biotech | 1100-05; RRID: AB_2794573 |
| HRP goat anti-mouse IgG | Southern Biotech | 1030-05; RRID: AB_2619742 |
| Biotin-conjugated goat anti-mouse IgM | Southern Biotech | RRID: AB_2794242 |
| Biotin-conjugated goat anti-mouse IgG | Southern Biotech | RRID: AB_2794296 |
| anti-CHIKV CHK-11 mAb | Diamond laboratory | Pal et al., 2013 |
| HRP-conjugated goat anti-mouse IgG | Southern Biotech | RRID: AB_2619742 |
| Bacterial and Virus Strains | ||
| Influenza A/CA/04/09 (H1N1) | Laboratory of Yoshihiro Kawaoka | N/A |
| Mouse-adapted SARS-CoV MA15 | Roberts et. al, 2007 | N/A |
| Mouse-adapted A/Hong Kong/01/68 (H3N2) | Haller et. al, 1979 | NA |
| CHIKV strain 181/25 | Dermody laboratory | Mainou et al., 2013 |
| CHIKV strain SL15649 | Morrison et. al, 2011 | icCHIKVSL15649 |
| Biological Samples | ||
| C57BL6/J immune sera | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RNAlater | Applied Biosystems/Ambion | AM7021 |
| Trizol | Invitrogen | 15596018 |
| HA antigen | BEI Resources | NR13691 |
| SARS S protein | BEI Resources | NR722 |
| TMB substrate | ThermoFisher Scientitic | 34028 |
| OPD powder | Sigma | P9029 |
| KPL TrueBlue Substrate | SeraCare | 5510-0030 |
| Streptavidin-HRP | Southern Biotech | 7100-05 |
| Critical Commercial Assays | ||
| miRNeasy mini kit | QIAGEN | 217004 |
| RNeasy Midi Kit | QIAGEN | 75144 |
| SENSE mRNA-Seq Library Prep Kit for Ion Torrent | Lexogen | 00624 |
| Ion Torrent PGM 314 chip | Life Technologies | 4482261 |
| High Sensitivity DNA chip | Life Technologies | 50674626 |
| Ion OneTouch 2 System | Life Technologies | INS1005527 |
| Ion P1 Chip | Life Technologies | A26770 |
| Whole mouse genome microarray | Agilent | 026655 |
| Deposited Data | ||
| Antibody response to IAV A/CA/04/09 in CC-F1s | This paper; Mendeley Data | https://doi.org/10.17632/kxr3t8n384.1 |
| Antibody response to SARS-CoV in CC-F1s | This paper; Mendeley Data | https://doi.org/10.17632/kxr3t8n384.1 |
| Antibody response to CHIKV in CC-RIs | This paper; Mendeley Data | https://doi.org/10.17632/kxr3t8n384.1 |
| RNaseq raw reads and normalized count matrix (lung) | GEO (Gene Expression Omnibus) | GSE136748 |
| Gene Expression array normalized expression matrix (blood) | GEO (Gene Expression Omnibus) | GSE110384 |
| Experimental Models: Cell Lines | ||
| Vero E6 cells | ATCC | CRL-1586 |
| MDCK cells | ATCC | CCL-34 |
| MDCK II cells | ATCC | CRL-2936 |
| HEK293T cells | ATCC | CRL-3216 |
| Vero cells | ATCC | CCL-81 |
| Experimental Models: Organisms/Strains | ||
| Collaborative Cross Mice | UNC SGCF | Supplemental file |
| Oligonucleotides | ||
| Forward primer for detection of IAV viral load: GACCRATCCTGTCACCTCTGAC | Ngaosuwankul et al., 2010 | WHO_CDC_Influenza A_pandemic H1N1 test |
| Reverse primer for detection of IAV viral load: AGGGCATTYTGGACAAAKCGTCTA | Ngaosuwankul et al., 2010 | WHO_CDC_Influenza A_pandemic H1N1 test |
| Probe for detection of IAV viral load: TGCAGTCCTCGCTCACTGGGCACG (FAM) | Ngaosuwankul et al., 2010 | WHO_CDC_Influenza A_pandemic H1N1 test |
| Eukaryotic 18S rRNA Endogenous Control (VIC/MGB probe, primer limited) | Applied Biosystems | 4319413E |
| Software and Algorithms | ||
| DOQTL | Gatti et al., 2014 | N/A |
| R | R Development Core Team, 2008 | N/A |
| cckit | https://github.com/kenoll/cckit | N/A |
| Variant Effect Predictor | McLaren et al., 2010 | N/A |
| C.T.L. Biospot Software | Cellular Technology Limited | V6.6.8 |
| FastQC | Andrews, 2010 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimgalore | Krueger, 2012 | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| STAR aligner | Dobin et. al, 2016 | http://github.com/alexdobin/STAR |
| RsubRead | Liao et. al, 2019 | http://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| DESeq2 | Love et. al, 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| sva | Leek, 2014 | http://bioconductor.org/packages/release/bioc/html/sva.html |
| Other | ||
| MRCA probabilities | Srivastava et al., 2017 | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark T. Heise (mark_heisem@med.unc.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The antibody datasets generated and used during this study are available at Mendeley Data (https://dx.doi.org/10.17632/kxr3t8n384.1). Lung and blood expression datasets are available through the Gene Expression Omnibus (GSE136748, GSE110384).
Experimental Model and Subject Details
Ethics statement
University of North Carolina at Chapel Hill
Mouse studies were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All mouse studies at UNC (Animal Welfare Assurance #A3410-01) were performed using protocols approved by the UNC Institutional Animal Care and Use Committee (IACUC) in a manner designed to minimize pain and suffering in infected animals. Any animals that exhibited severe disease signs was euthanized immediately in accordance with IACUC approved endpoints.
Helmholtz Centre for Infection Research
All experiments in mice were approved by an external committee according to the national guidelines of the animal welfare law in Germany (BGBl. I S. 1206, 1313 and BGBl. I S. 1934). The protocol used in these experiments has been reviewed by an ethics committee and approved by the relevant authority, the ‘Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany’ (Permit Numbers: 33.9.42502-04-051/09 and 3392 42502-04-13/1234).
Mice
IAV H1N1
Mice from 64 CC strains were purchased from the UNC Systems Genetics Core Facility (SGCF) between July 2012 and July 2016. Female mice were generated from 116 CC-F1 crosses between these 64 CC strains (see Data S1 for full list). All mice were housed in a specific pathogen free facility under standard conditions (12hr light/dark, food and water ad libitum) at UNC Chapel Hill. Female mice of 8-12 weeks of age at time of experiment were used. Nlrp1b KO and C57BL/6J mice were purchased from Jackson Laboratories and bred in our colony at UNC Chapel Hill. F1 or F2 heterozygous animals were bred to generate a single experimental cohort with littermate controls. Male and female mice were used and infected when mice were between 9-11 weeks of age.
IAV H3N2
Mice from 11 CC strains (see Data S1 for full list) were received from the UNC SGCF and then bred in the animal facility of the Helmholtz Centre for Infection Research, Braunschweig under specific pathogen free conditions. Female mice of 8-12 weeks of age at time of experiment were used.
SARS-CoV
Mice from 64 CC strains were purchased from the UNC SGCF between July 2012 and July 2016. Female F1 mice were generated from 116 CC-F1 crosses between these 64 CC strains (see Data S1 for full list). All mice were housed in a BSL-3 facility at UNC Chapel Hill. Mice were 8-12 weeks of age at time of experiment.
CHIKV
Female mice from 64 CC strains (see Data S1 for full list) were housed under BSL-3 conditions at UNC Chapel Hill. Mice were 6-7 weeks old at time of experiment.
Cell lines
HEK293T (ATCC) were used for the propagation of IAV H1N1. MDCK cells (ATCC) were used for the amplification and titration of IAV H1N1. MDCK II cells (ATCC) were used for the titration of IAV H3N2. Vero E6 cells (ATCC) were used for the titration of SARS-CoV. Vero cells (ATCC) were used for the titration of CHIKV and FRNT50 assays. All cells were maintained at 37°C in standard growth media. Serum-free media was used for propagation, amplification, and titration of IAV.
Method Details
Virus
IAV H1N1
Influenza A/CA/04/09 (H1N1) was generated by multi-plasmid transfection of HEK293T cells followed by 48 hour amplification and titration in MDCK cells as described previously (Neumann et al., 2012).
IAV H3N2
Mouse-adapted A/Hong Kong/01/68 (H3N2) virus was originally obtained from Otto Haller, University of Freiburg (Haller et al., 1979). Virus was propagated as described previously (Wilk and Schughart, 2012). Titer was determined by focus forming unit assay (FFU/ml) in Madin-Darby Canine Kidney II (MDCK) cells (ATCC).
SARS-CoV
Recombinant mouse-adapted SARS-CoV MA15 (mouse adapted SARS-CoV Urbani strain) was propagated and titered on Vero E6 cells. All experiments were performed in a certified BSL-3 laboratory.
CHIKV
The SL15649 clinical isolate of CHIKV was derived from the pMH56.2 infectious clone and titered on Vero cells, as described previously (Morrison et al., 2011).
Infections
IAV H1N1
Mice were lightly anesthetized via isoflurane inhalation and infected intranasally with either 5000 focus forming units (FFU) of influenza A/CA/04/09 in 50μL PBS or PBS only (mock-treatment). Approximately 3 mice per F1 per time point were infected with IAV. Animals were measured daily for weight loss and monitored for mortality and clinical disease scores. At 2, 4, 7, 10, 15, or 45 days post-infection, animals were euthanized via isoflurane overdose and bilateral thoracotomy. Lungs for viral load were collected at day 2 post-infection and stored in RNAlater (Applied Biosystems/Ambion, Austin, TX) at −80°C after 1 day at 4°C. Blood was collected by terminal bleed, and serum was aliquoted and stored at −40°C or −80°C. For Nlrp1b studies, all mice were infected with IAV and serum was collected by submandibular bleed at day 10 post-infection.
IAV H3N2
Mice were anesthetized by intra-peritoneal injection with Ketamine/Xylazine (85% NaCl (0.9%), 10% Ketamine, 5% Xylazine) and infected intranasally with 20μl virus solution (10 FFU) in PBS or PBS only (mock-treatment). 3-4 mice per strain per time point and treatment were infected. Body weight, survival and well-being of mice were monitored. Lungs were harvested from sacrificed mice at days 3 and 5 post-infection. Blood was collected by retro-orbital bleed at days 3, 5, and 8 post-infection.
SARS-CoV
Mice were lightly anesthetized via isoflurane inhalation and infected intranasally with 5x103 plaque forming units (PFU) of MA15 in 50μL PBS or PBS only (mock-treatment). Approximately 3 mice per F1 per time point were infected with SARS-CoV. Animals were measured daily for weight loss and monitored for mortality and clinical disease scores. At 7, 15, or 29 days post-infection, animals were euthanized via isoflurane overdose and bilateral thoracotomy. Another group of animals was re-challenged at day 28 post-infection, and euthanized at day 32 (4 days post-re-challenge). Blood was collected by terminal bleed, and serum was aliquoted and stored at −80°C.
CHIKV
Mice were infected with 100 PFU of CHIKV SL15649 in a 10μl volume via the subcutaneous route in the left rear footpad, or PBS alone (mock treatment). Three mice per group were infected and two per group received mock treatment. Mice were euthanized on day 7 post infection and serum was collected and stored at −80°C.
Viral load measurement
Lung tissue was thawed on ice and transferred to Trizol (Invitrogen, Carlsbad CA) preceding homogenization and RNA extraction using the QIAGEN miRNeasy mini kit (QIAGEN, Hilden, Germany, 217004). One-step RT-PCR was performed to quantify IAV (F: GACCRATCCTGTCACCTCTGAC. R: AGGGCATTYTGGACAAAKCGTCTA, Probe: TGCAGTCCTCGCTCACTGGGCACG, Reporter: FAM) and 18S (4319413E, Applied Biosystems, Foster City, CA) in duplex. Delta CT values were calculated as the difference between IAV and 18S CT values.
Antibody measurement
IAV H1N1 and SARS-CoV ELISA
IAV hemagglutinin (HA)-specific and SARS-CoV spike-specific antibody were quantified by ELISA. 96 well flat-bottom plates were coated with HA antigen (BEI Resources, Mannassas, VA, NR13691) or SARS S protein (BEI Resources NR722) (diluted in carbonate buffer (0.32M Na2C03, 0.68M NaHCO3) to 1 μg/mL). Half-log serum dilutions (102 to 105.5) were prepared in wash buffer (0.033% Tween-20 in PBS), added to coated plates, and incubated overnight in a humidified chamber at 4°C. Plates were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Southern Biotech, Birmingham, AL, HRP conjugated goat anti-mouse, IgM = 1020-05, IgG1 = 1070-05, IgG2a = 1080-05 pooled with IgG2c = 1079-05, IgG2b = 1090-05, IgG3 = 1100-05, Total IgG = 1030-05) for approximately 2 hours at 4°C. IAV plates were washed and developed in the dark for 30 minutes at room temperature with citrate substrate (.05M sodium citrate, 0.05M citric acid, 1mg/mL OPD, 0.216% hydrogen peroxide), then stopped with sodium fluoride (0.1M) and read immediately at 450nm. SARS-CoV plates were washed and developed in the dark for 30 minutes with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (ThermoFisher Scientific, Waltham, MA, 34028), then stopped with HCL (0.2M) and read immediately at 450nm. Background-subtracted OD measurements were set against dilution factors to calculate area under the curve (AUC) values for each sample and isotype. AUC was determined to be the most inclusive measure to robustly captured the dynamic range of antibody levels across our sample set, and performed better than other measurements such as half maximal or lowest positive titer. To monitor consistency across assays, C57BL6/J immune sera as well as serum samples from CC-F1s that were repeated across multiple cohorts were measured multiple times alongside different sets of experimental samples.
CHIKV ELISA
CHIKV-specific antibodies in mouse sera were measured using a virion-based ELISA as described (Hawman et al., 2016). CHIKV strain 181/25 grown in serum-free media (VP-SFM) was concentrated, suspended in PBS, and was adsorbed to a 96-well Immulon 4HBX plate (1.25 × 108 particles/well). Serum samples were serially diluted and added to the plate. Bound antibody was detected using biotin-conjugated goat anti-mouse IgM or IgG antibodies, followed by HRP-conjugated streptavidin, and detection with TMB substrate (Sigma-Aldrich, St. Louis, MO). Endpoint titers were defined as the reciprocal of the last dilution to have an absorbance four standard deviations greater than background. Blank wells receiving no serum were used to quantify background signal.
CHIKV Focus Reduction Neutralization Test (FRNT)
For FRNT assays, Vero cells were seeded in 96-well plates. Serum samples were heat-inactivated and serially diluted in DMEM/F12 medium with 2% FBS in 96-well plates. Approximately 100 focus-forming units (FFU) of virus stock (CHIKV strain SL15649) was added to each well and the serum plus virus mixture was incubated for 1h at 37°C. At the end of 1h, medium was removed from Vero cells and serum sample plus virus mixture was added for 2 h at 37°C. After 2 h, sample was removed and cells were overlaid with 0.5% methylcellulose in MEM/5% FBS and incubated 18h at 37°C. Cells were fixed with 1% PFA and probed with 500 ng/mL of anti-CHIKV CHK-11 mAb (Pal et al., 2013) diluted in 1X PBS/0.1% saponin/0.1% bovine serum albumin (BSA) for 2h at room temperature. After washing, cells were incubated with HRP-conjugated goat anti-mouse IgG for 1.5-2 h at room temperature. After washing, CHIKV-positive foci were visualized with TrueBlue substrate and counted using a CTL Biospot analyzer and Biospot software (Cellular Technology, Cleveland, OH). Percent infectivity was calculated compared to a virus only (no serum) control. The FRNT50 value was defined as the reciprocal of the last dilution to exhibit 50% infectivity.
Expression analysis
Lung expression
Whole lungs were stored in RNAlater solution (QIAGEN, Hilden, Germany), kept at 4°C for one day and subsequently stored at −20°C. RNA was isolated using QIAGEN Midi Kit. RNA quality was controlled on a 2100 Bioanalyzer Instrument (Agilent, Santa Clara, CA). All RNA samples had a RNA Integrity Number (RIN) of ≥ 9.5. 3 to 6 independent biological replicates were selected for each time point for subsequent RNA sequencing. 500ng of total RNA was used to prepare libraries for sequencing using the Lexogen SENSE mRNA-seq library kit for Ion Torrent. Libraries amplified for 11 cycles as the final step of library preparation. Before sequencing, 1ul aliquots of this material was pooled and sequenced on an Ion Torrent PGM 314 chip. Barcode quantification data from the PGM were used to balance the barcodes for final pooling before sequencing. Following this final pooling, the library pools were sized to a target size of 260bp on a Pippin Prep instrument using the 2% Pippin Agarose gel. The sized libraries were examined on an Agilent High Sensitivity DNA chip, quantified using real-time PCR, and used to prepare spheres using a One-Touch 2 device. These spheres were then sequenced on an Ion Torrent Proton sequencer with a P1 chip. The mean number of reads per sample was 24 million. Reads were quality checked with package FastQC (Andrews, 2010), then trimmed using Trimgalore (Krueger, 2012). Trimmed reads were mapped to mouse genome annotation mm10 (build 38.1) using STAR aligner (Dobin et al., 2016). Further analysis and visualization of expression data was performed using the R software package. Mapped reads were feature counted (per gene) using RsubRead (Liao et al., 2019). Raw counts from mouse genome and virus mapping were then combined and normalized using DESeq2 (Love et al., 2014). After normalization, single extreme outlier points were razorized, log2 transformed, normalized expression levels were then batch corrected using package sva (Leek, 2014). Extreme outlier samples were identified in principle component analyses and removed. For each group, 3 to 4 replicates were retained. Log2 transformed, normalized and batch corrected expression levels were outputted for further use.
Blood expression
Analysis of expression data from blood was previously described (Kollmus et al., 2018). Briefly, blood was collected for RNA isolation at multiple time points (days 3, 5, and 8) post-infection by retro-orbital bleed. RNA was isolated, transcribed into cDNA, amplified with incorporation of cyanine 3-labeled CTP, and hybridized to a whole mouse genome microarray (Agilent 026655).
Candidate selection
All genetic elements under each QTL were first narrowed by choosing only those containing SNPs above the association testing threshold. Association testing was performed via the assoc.map() function in DOQTL by imputing Sanger SNPs onto the CC genome builds, and thresholds were set independently for each QTL. Candidate SNPs were then run through the Ensembl Variant Effect Predictor (McLaren et al., 2010) to determine the genetic classification of the affected gene as well as the predicted consequence of the SNP itself. We selected variants in protein-coding genes only and then filtered to genes expressed in the lung, thymus, and/or spleen. Coding variants were further considered if they were predicted to have a functional consequence on the protein product (e.g., missense versus synonymous variant). Non-coding variants were further considered if haplotype-specific expression differences were observed in lung and/or blood. Genes with coding and/or non-coding variants that met these criteria were included in our final list of candidate genes.
Quantification and Statistical Analysis
Heritability
To determine heritability for single time point measurements, we fit the following linear model:
We averaged intraclass correlation [(MSCC-F1 - MSε)/(MSCC-F1 + (N-1)MSε)] and coefficient of genetic determination [ (MSCC-F1 - MSε)/(MSCC-F1 + (2N-1)MSε) ] values, using N = 3 as a representative value for group size across all CC-F1s.
Kinetics
Values for between-time point antibody measurements were determined by averaging raw AUC values for each CC-F1, then calculating ratios of between adjacent time points (day 10/day 7, day 15/day 10, day 45/day 15) for each isotype. Given only one calculated measurement per time point per CC-F1, heritability and effect size could not be calculated for between-time point phenotypes.
Normalization
QTL mapping assumes that phenotypic data is normally distributed. Data for each isotype and time point combination varied in distribution, and therefore were normalized independently based on box-cox transformation values. Normalized single-time point measurements were averaged per CC-F1 preceding QTL mapping.
Genetic mapping
We conducted genetic mapping using the DOQTL (1.18.0) (Gatti et al., 2014) package in the R statistical environment (R Development Core Team, 2008). Collaborative Cross genomes are best represented as a 36-state probability matrix (where at each marker, the values indicate the likelihood that a mouse from that strain has a given homozygous (n = 8) or heterozygous (n = 28) diplotype). We took the 36-state probability files from the CC Most Recent Common Ancestors (MRCAs) consensuses for each strain and simplified these to 8-state probabilities as previously described (Srivastava et al., 2017). For each CC-F1, we averaged the 8-state probabilities of the two parent CC strains. This approach is the most accurate for F1 animals, as they will receive only one allele from each parent CC strain. For genetic mapping in DOQTL, a multiple regression is run at each marker, running the phenotype of interest against haplotype probabilities for each strain. LOD scores are calculated based on the improvement of fit in this model compared to a null model considering only covariates and kinship. To determine significance, permutation tests were run in which phenotype and genotype data are shuffled without replacement to generate a series of null LOD scores, of which the highest is archived. Based on 1000 permutations, we determined null distributions. We calculated p value scores by empirical cumulative distribution function, comparing the maximum LOD score for a QTL to the distribution of null permutations. QTL regions were determined using a 1.5LOD drop interval.
Haplotype scoring
Once loci were identified, haplotype groups for each QTL were manually determined based on visualization of haplotype effect plots. Additive haplotype scores for CC-F1s were computed by combining the haplotype scores for the dam and sire of the F1. CC-F1s were dropped from analyses if either the dam or sire had heterozygosity or recombinations within the locus between founder haplotypes with differing effect scores. In the case of Ari2, there was an insufficient number of representative crosses for one haplotype effect group (only two F1s with an additive score of 0), and therefore this group was dropped from analyses.
Phenotypic variance
Effect sizes were computed by linear regression:
We calculated [(Sum of Squaresdam + SSsire) / (SSdam + SSsire + SSCC-F1)]. QTL effect size was multiplied by trait heritability to determine the percentage of total phenotypic variation explained by the QTL.
Phenotypic correlations
Correlations between Ari loci and other phenotypes were calculated by running the following mixed effect linear models and comparing goodness of fit between them with an ANOVA test to determine a p value. Haplotype ((0, 1) or (0, 1, 2)) was included as a numeric variable representing the dosage of the allele associated with the high response.
To determine correlations with weight loss, Mx1 haplotype was included in both models:
To determine correlations between weight loss and antibody, we ran similar models with antibody measurement values in place of haplotype scores. Since many animals did not reach maximal weight loss until after day 7, only animals from time points 10-45 were included in these models.
Differential expression analysis
Goodness of fit between the following mixed linear models was compared using an ANOVA test to determine IAV-specific (models 1&2) and haplotype-specific (models 2&3) expression.
-
1
Gene expression ∼CC-F1 + ε
-
2
Gene expression ∼day post-infection + CC-F1 + ε
-
3
Gene expression ∼day post-infection + haplotype score + CC-F1 + ε
Acknowledgments
We would like to acknowledge the Systems Genetics Core Facility at UNC for their maintenance and distribution of CC mouse strains as well as their pertinent genetic information. We thank the animal caretakers at the Central Animal Facilities of the HZI for maintaining the mice; Stefanie Edler, Karin Lammert, Christin Kurch, and Rebecka Wünsche for technical assistance; and Michael Jarek, Richard Green, Frank Fernandez, Till Lesker, and Robert Geffers (HZI) for support with the analysis of RNA sequencing (RNA-seq) data. RNA-seq has been performed by the Molecular Resource Center (MRC) at UTHSC, Memphis. This work was supported by NIH/NIAID research grants U19-AI100625, U01 AI149644, and U19 AI 109761 to R.S.B. and M.T.H.; U19 AI 109680 and R21 AI119933 to M.T.H.; R01 AI141416 to T.E.M.; intra-mural grants from the Helmholtz-Association (Program Infection and Immunity); a start-up grant from the University of Tennessee Health Science Center; and the research grant FluResearchNet (01KI1006F) from the German Ministry of Education and Research to K.S. K.E.N. was supported by NIH/NIAID grant T32 AI007419, K.S.P. was supported by NIH/NIAID grants T32 AI007151-36A1 and F32 AI126730, and M.K.M. was supported by NIH/NIAID grant F32 AI122436.
Author Contributions
These studies were designed by K.E.N., A.C.W., L.E.G., V.D.M., S.M., F.P.-M.d.V., K.S., T.E.M., R.S.B., M.T.F., and M.T.H. These experiments were conducted by K.E.N., A.C.W., A.W., M.K.M., C.R.M., K.S.P., B.K.H., H.K., C.P., S.R.L., L.E.G., V.D.M., A.S., D.M., G.S., and M.T.F. The data were analyzed by K.E.N., H.K., M.M., S.M., K.S., M.T.F., and M.T.H. The manuscript was written primarily by K.E.N., M.T.F., and M.T.H. All authors contributed to the editing of this manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: April 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107587.
Supplemental Information
List of CC Strains and CC-F1s Used across Experiments/Datasets, Related to Figures 1, 3, and 4
References
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Boon M., De Boeck K., Jorissen M., Meyts I. Primary ciliary dyskinesia and humoral immunodeficiency--is there a missing link? Respir. Med. 2014;108:931–934. doi: 10.1016/j.rmed.2014.03.009. [DOI] [PubMed] [Google Scholar]
- Brodin P., Jojic V., Gao T., Bhattacharya S., Angel C.J.L., Furman D., Shen-Orr S., Dekker C.L., Swan G.E., Butte A.J. Variation in the human immune system is largely driven by non-heritable influences. Cell. 2015;160:37–47. doi: 10.1016/j.cell.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention . Centers for Disease Control and Prevention; 2018. Estimated Influenza Illnesses, Medical Visits, Hospitalizations, and Deaths in the United States — 2017–2018 Influenza Season.https://www.cdc.gov/flu/about/burden-averted/2017-2018.htm [Google Scholar]
- Cheung N.N., Lai K.K., Dai J., Kok K.H., Chen H., Chan K., Yuen K., Yi R., Kao T. Broad-spectrum inhibition of common respiratory RNA viruses by a pyrimidine synthesis inhibitor with involvement of the host antiviral response. J. Gen. Virol. 2017;98:946–954. doi: 10.1099/jgv.0.000758. [DOI] [PubMed] [Google Scholar]
- Churchill G.A., Airey D.C., Allayee H., Angel J.M., Attie A.D., Beatty J., Beavis W.D., Belknap J.K., Bennett B., Berrettini W., Complex Trait Consortium The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Couch R.B., Atmar R.L., Franco L.M., Quarles J.M., Wells J., Arden N., Niño D., Belmont J.W. Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J. Infect. Dis. 2013;207:974–981. doi: 10.1093/infdis/jis935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisenko E., Guler R., Mhlanga M., Suzuki H., Brombacher F., Schmeier S. Transcriptionally induced enhancers in the macrophage immune response to Mycobacterium tuberculosis infection. BMC Genomics. 2019;20:71. doi: 10.1186/s12864-019-5450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicks M.D.J., Id G.B., Jimenez-guarde J.M. Multiple components of the nuclear pore complex interact with the amino-terminus of MX2 to facilitate HIV-1 restriction. PLoS Pathog. 2018;14:e1007408. doi: 10.1371/journal.ppat.1007408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diot C., Fournier G., Santos M. Dos, Magnus J. Influenza A virus polymerase recruits the RNA helicase DDX19 to promote the nuclear export of viral mRNAs. Sci. Rep. 2016;6:33763. doi: 10.1038/srep33763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Gingeras T.R., Spring C., Flores R., Sampson J., Knight R., Chia N., Technologies H.S. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics. 2016;51:586–597. doi: 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris M.T., Aylor D.L., Bottomly D., Whitmore A.C., Aicher L.D., Bell T.A., Bradel-Tretheway B., Bryan J.T., Buus R.J., Gralinski L.E. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003196. e1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forthal D.N. Functions of antibodies. Microbiol. Spectr. 2014;2:1–17. [PMC free article] [PubMed] [Google Scholar]
- Gaba A., Xu F., Lu Y., Park H.-S., Liu G., Zhou Y. The NS1 protein of influenza A virus participates in necroptosis by interacting with MLKL and increasing its oligomerization and membrane translocation. J. Virol. 2019;93:1–14. doi: 10.1128/JVI.01835-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti D.M., Svenson K.L., Shabalin A., Wu L.-Y., Valdar W., Simecek P., Goodwin N., Cheng R., Pomp D., Palmer A. Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 2014;4:1623–1633. doi: 10.1534/g3.114.013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh K., Morisaki T., Setoyama D., Sasaki K., Yagi M., Igami K., Mizuguchi S., Uchiumi T., Fukui Y., Kang D. Mitochondrial p32/C1qbp is a critical regulator of dendritic cell metabolism and maturation. Cell Rep. 2018;25:1800–1815.e4. doi: 10.1016/j.celrep.2018.10.057. [DOI] [PubMed] [Google Scholar]
- Gralinski L.E., Menachery V.D., Morgan A.P., Totura A.L., Beall A., Kocher J., Plante J., Harrison-Shostak D.C., Schäfer A., Pardo-Manuel de Villena F. Allelic variation in the Toll-like receptor adaptor protein Ticam2 contributes to SARS-coronavirus pathogenesis in mice. G3 (Bethesda) 2017;7:1653–1663. doi: 10.1534/g3.117.041434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O., Arnheiter H., Lindenmann J. Natural, genetically determined resistance toward influenza virus in hemopoietic mouse chimeras: role of mononuclear phagocytes. J. Exp. Med. 1979;150:117–126. doi: 10.1084/jem.150.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawman D.W., Fox J.M., Ashbrook A.W., May N.A., Schroeder K.M.S., Torres R.M., Crowe J.E., Jr., Dermody T.S., Diamond M.S., Morrison T.E. Pathogenic chikungunya virus evades B cell responses to establish persistence. Cell Rep. 2016;16:1326–1338. doi: 10.1016/j.celrep.2016.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz L.X., Rebsamen M., Rossi D.C., Staehli F., Schroder K., Quadroni M., Gross O., Schneider P., Tschopp J. The death domain-containing protein Unc5CL is a novel MyD88-independent activator of the pro-inflammatory IRAK signaling cascade. Cell Death Differ. 2012;19:722–731. doi: 10.1038/cdd.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntoon K.M., Wang Y., Eppolito C.A., Barbour K.W., Berger F.G., Shrikant P.A., Baumann H. The acute phase protein haptoglobin regulates host immunity. J. Leukoc. Biol. 2008;84:170–181. doi: 10.1189/jlb.0208100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B.E., Moran T.M., Bona C.A., Popple S.W., Kilbourne E.D. Immunologic response to influenza virus neuraminidase is influenced by prior experience with the associated viral hemagglutinin. II. Sequential infection of mice simulates human experience. J. Immunol. 1987;139:2010–2014. [PubMed] [Google Scholar]
- Keele G.R., Crouse W.L., Kelada S.N.P., Valdar W. Determinants of QTL mapping power in the realized Collaborative Cross. G3 (Bethesda) 2018;9:1707–1727. doi: 10.1534/g3.119.400194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimman T.G., Vandebriel R.J., Hoebee B. Genetic variation in the response to vaccination. Community Genet. 2007;10:201–217. doi: 10.1159/000106559. [DOI] [PubMed] [Google Scholar]
- Kollmus H., Pilzner C., Leist S.R., Heise M., Geffers R., Schughart K. Of mice and men: the host response to influenza virus infection. Mamm. Genome. 2018;29:446–470. doi: 10.1007/s00335-018-9750-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krištić J., Zaytseva O.O., Ram R., Nguyen Q., Novokmet M., Vučković F., Vilaj M., Trbojević-Akmačić I., Pezer M., Davern K.M. Profiling and genetic control of the murine immunoglobulin G glycome. Nat. Chem. Biol. 2018;14:516–524. doi: 10.1038/s41589-018-0034-3. [DOI] [PubMed] [Google Scholar]
- Krueger F. Babraham Institute; 2012. Trim Galore.http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ [Google Scholar]
- Kruskall M.S., Alper C.A., Awdeh Z., Yunis E.J., Marcus-Bagley D. The immune response to hepatitis B vaccine in humans: inheritance patterns in families. J. Exp. Med. 1992;175:495–502. doi: 10.1084/jem.175.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck K.F., Delmotte P., Robinson M.L., Sanderson M.J., Witman G.B. Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 2008;180:633–643. doi: 10.1083/jcb.200710162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014;42:e161. doi: 10.1093/nar/gku864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewnard J.A., Cobey S. Immune history and influenza vaccine effectiveness. Vaccines (Basel) 2018;6:28. doi: 10.3390/vaccines6020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Willingham S.B., Ting J.P.-Y., Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J. Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilue J., Doran A.G., Fiddes I.T., Abrudan M., Armstrong J., Bennett R., Chow W., Collins J., Collins S., Czechanski A. Multiple laboratory mouse reference genomes define strain specific haplotypes and novel functional loci. bioRxiv. 2018 doi: 10.1101/235838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnik J.E., Egli A. Impact of host genetic polymorphisms on vaccine induced antibody response. Hum. Vaccin. Immunother. 2016;12:907–915. doi: 10.1080/21645515.2015.1119345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainou B.A., Zamora P.F., Ashbrook A.W., Dorset D.C., Kim K.S., Dermody T.S. Reovirus cell entry requires functional microtubules. MBio. 2013;4 doi: 10.1128/mBio.00405-13. e00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizio P.L., Ferris M.T., Keele G.R., Miller D.R., Shaw G.D., Whitmore A.C., West A., Morrison C.R., Noll K.E., Plante K.S. Bayesian diallel analysis reveals Mx1-dependent and Mx1-independent effects on response to influenza A virus in mice. G3 (Bethesda) 2017;8:427–445. doi: 10.1534/g3.117.300438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri M., Crown D., Newman Z.L., Okugawa S., Eckhaus M., Cataisson C., Liu S., Sastalla I., Leppla S.H. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 2010;6:e1001222. doi: 10.1371/journal.ppat.1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison T.E., Oko L., Montgomery S.A., Whitmore A.C., Lotstein A.R., Gunn B.M., Elmore S.A., Heise M.T. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am. J. Pathol. 2011;178:32–40. doi: 10.1016/j.ajpath.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G., Ozawa M., Kawaoka Y. Reverse genetics of influenza viruses. In: Kawaoka Y., Neumann G., editors. Influenza Virus: Methods and Protocols. Humana Press; 2012. pp. 193–206. [DOI] [PubMed] [Google Scholar]
- Ngaosuwankul N., Noisumdaeng P., Komolsiri P., Pooruk P., Chokephaibulkit K., Chotpitayasunondh T., Sangsajja C., Chuchottaworn C., Farrar J., Puthavathana P. Influenza A viral loads in respiratory samples collected from patients infected with pandemic H1N1, seasonal H1N1 and H3N2 viruses. Virol. J. 2010;7:75. doi: 10.1186/1743-422X-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijnik A., Clare S., Hale C., Chen J., Raisen C., Mottram L., Lucas M., Estabel J., Ryder E., Adissu H., Sanger Mouse Genetics Project The role of sphingosine-1-phosphate transporter Spns2 in immune system function. J. Immunol. 2012;189:102–111. doi: 10.4049/jimmunol.1200282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll K.E., Ferris M.T., Heise M.T. The Collaborative Cross: a systems genetics resource for studying host-pathogen interactions. Cell Host Microbe. 2019;25:484–498. doi: 10.1016/j.chom.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovsyannikova I.G., Kennedy R.B., O’Byrne M., Jacobson R.M., Pankratz V.S., Poland G.A. Genome-wide association study of antibody response to smallpox vaccine. Vaccine. 2012;30:4182–4189. doi: 10.1016/j.vaccine.2012.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovsyannikova I.G., Salk H.M., Kennedy R.B., Haralambieva I.H., Zimmermann M.T., Grill D.E., Oberg A.L., Poland G.A. Gene signatures associated with adaptive humoral immunity following seasonal influenza A/H1N1 vaccination. Genes Immun. 2016;17:371–379. doi: 10.1038/gene.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal P., Dowd K.A., Brien J.D., Edeling M.A., Gorlatov S., Johnson S., Lee I., Akahata W., Nabel G.J., Richter M.K.S. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 2013;9:e1003312. doi: 10.1371/journal.ppat.1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team . R Foundation for Statistical Computing; 2008. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Rasmussen A.L., Okumura A., Ferris M.T., Green R., Feldmann F., Kelly S.M., Scott D.P., Safronetz D., Haddock E., LaCasse R. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346:987–991. doi: 10.1126/science.1259595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A., Deming D., Paddock C.D., Cheng A., Yount B., Vogel L., Herman B.D., Sheahan T., Heise M., Genrich G.L. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007;3:0023–0037. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogala A.R., Morgan A.P., Christensen A.M., Gooch T.J., Bell T.A., Miller D.R., Godfrey V.L., de Villena F.P.M. The Collaborative Cross as a resource for modeling human disease: CC011/Unc, a new mouse model for spontaneous colitis. Mamm. Genome. 2014;25:95–108. doi: 10.1007/s00335-013-9499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.A., Slack G.S., Marriott A.C., Kane J.A., Whittaker C.J., Silman N.J., Carroll M.W., Gooch K.E. Cellular immune response to human influenza viruses differs between H1N1 and H3N2 subtypes in the ferret lung. PLoS ONE. 2018;13:e0202675. doi: 10.1371/journal.pone.0202675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M., Sakaguchi N., Ma J.S., Nakamura S., Kawabata T., Bando H., Lee Y., Saitoh T., Akira S., Iwasaki A. Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nat. Immunol. 2017;18:899–910. doi: 10.1038/ni.3767. [DOI] [PubMed] [Google Scholar]
- Saul M.C., Philip V.M., Reinholdt L.G., Chesler E.J., Center for Systems Neurogenetics of Addiction High-diversity mouse populations for complex traits. Trends Genet. 2019;35:501–514. doi: 10.1016/j.tig.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S.-U., Kwon H.-J., Song J.-H., Byun Y.-H., Seong B.L., Kawai T., Akira S., Kweon M.-N. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J. Virol. 2010;84:12713–12722. doi: 10.1128/JVI.01675-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., Song Y., Colangelo C.M., Wu T., Bruce C., Scabia G., Galan A., Maffei M., Goldstein D.R. Haptoglobin activates innate immunity to enhance acute transplant rejection in mice. J. Clin. Invest. 2012;122:383–387. doi: 10.1172/JCI58344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.M., Do Hyang Lee D., Kulkarni H., Radhakrishnan P., Hirst R., Easton A., O’Callaghan C. Influenza virus infection of well-differentiated human airway epithelial cells by infectious aerosols: insights into the earliest stages of infection. F1000Res. 2019;8:337. [Google Scholar]
- Srivastava A., Morgan A.P., Najarian M.L., Sarsani V.K., Sigmon J.S., Shorter J.R., Kashfeen A., McMullan R.C., Williams L.H., Giusti-Rodríguez P. Genomes of the mouse Collaborative Cross. Genetics. 2017;206:537–556. doi: 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao K., McAuliffe J., Vogel L., Fahle G., Fischer S., Tatti K., Packard M., Shieh W.-J., Zaki S., Murphy B. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J. Virol. 2004;78:3572–3577. doi: 10.1128/JVI.78.7.3572-3577.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui B., Bamba D., Weng K., Ung H., Chang S., Van Dyke J., Goldblatt M., Duan R., Kinch M.S., Li W.B. The use of random homozygous gene perturbation to identify novel host-oriented targets for influenza. Virology. 2009;387:473–481. doi: 10.1016/j.virol.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symoens S., Barnes A.M., Gistelinck C., Malfait F., Guillemyn B., Steyaert W., Syx D., D’hondt S., Biervliet M., De Backer J. Genetic defects in TAPT1 disrupt ciliogenesis and cause a complex lethal osteochondrodysplasia. Am. J. Hum. Genet. 2015;97:521–534. doi: 10.1016/j.ajhg.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P.L., Jacobson R.M., Poland G.A., Jacobsen S.J., Pankratz V.S. Twin studies of immunogenicity--determining the genetic contribution to vaccine failure. Vaccine. 2001;19:2434–2439. doi: 10.1016/s0264-410x(00)00468-0. [DOI] [PubMed] [Google Scholar]
- Uv A.E., Roth P., Xylourgidis N., Wickberg A., Cantera R., Samakovlis C. members only encodes a Drosophila nucleoporin required for rel protein import and immune response activation. Genes Dev. 2000;14:1945–1957. [PMC free article] [PubMed] [Google Scholar]
- Veleri S., Manjunath S.H., Fariss R.N., May-Simera H., Brooks M., Foskett T.A., Gao C., Longo T.A., Liu P., Nagashima K. Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat. Commun. 2014;5:4207. doi: 10.1038/ncomms5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S.M., Malo D., Marquis J., Gros P. Forward genetic dissection of immunity to infection in the mouse. Annu. Rev. Immunol. 2008;26:81–132. doi: 10.1146/annurev.immunol.26.021607.090304. [DOI] [PubMed] [Google Scholar]
- Wiedermann U., Garner-Spitzer E., Wagner A. Primary vaccine failure to routine vaccines: Why and what to do? Hum. Vaccin. Immunother. 2016;12:239–243. doi: 10.1080/21645515.2015.1093263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk E., Schughart K. The mouse as model system to study host-pathogen interactions in influenza A infections. Curr. Protoc. Mouse Biol. 2012;2:177–205. doi: 10.1002/9780470942390.mo110173. [DOI] [PubMed] [Google Scholar]
- Yannay-Cohen N., Carmi-Levy I., Kay G., Yang C.M., Han J.M., Kemeny D.M., Kim S., Nechushtan H., Razin E. LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol. Cell. 2009;34:603–611. doi: 10.1016/j.molcel.2009.05.019. [DOI] [PubMed] [Google Scholar]
- Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D., Mouse ENCODE Consortium A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Xu L.-G., Han K.-J., Shu H.-B. Identification of a ZU5 and death domain-containing inhibitor of NF-kappaB. J. Biol. Chem. 2004;279:17819–17825. doi: 10.1074/jbc.M310737200. [DOI] [PubMed] [Google Scholar]
- Zhang L., Das P., Schmolke M., Manicassamy B., Wang Y., Deng X., Cai L., Tu B.P., Forst C.V., Roth M.G. Inhibition of pyrimidine synthesis reverses viral virulence factor-mediated block of mRNA nuclear export. J. Cell Biol. 2012;196:315–326. doi: 10.1083/jcb.201107058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Goldschmidt T., Salter H. Possible allelic structure of IgG2a and IgG2c in mice. Mol. Immunol. 2012;50:169–171. doi: 10.1016/j.molimm.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang K., Zhang Y., Xue J., Meng Q., Liu H., Bi C., Li C., Hu L., Yu H., Xiong T. DDX19 inhibits type I interferon production by disrupting TBK1-IKKε-IRF3 interactions and promoting TBK1 and IKKε degradation. Cell Rep. 2019;26:1258–1272.e4. doi: 10.1016/j.celrep.2019.01.029. [DOI] [PubMed] [Google Scholar]
- Zúñiga J., Buendía-Roldán I., Zhao Y., Jiménez L., Torres D., Romo J., Ramírez G., Cruz A., Vargas-Alarcon G., Sheu C.-C. Genetic variants associated with severe pneumonia in A/H1N1 influenza infection. Eur. Respir. J. 2012;39:604–610. doi: 10.1183/09031936.00020611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of CC Strains and CC-F1s Used across Experiments/Datasets, Related to Figures 1, 3, and 4
Data Availability Statement
The antibody datasets generated and used during this study are available at Mendeley Data (https://dx.doi.org/10.17632/kxr3t8n384.1). Lung and blood expression datasets are available through the Gene Expression Omnibus (GSE136748, GSE110384).