

Abstract
Hepatocellular carcinoma (HCC) is a deadly form of liver cancer with limited treatment options. The c-Myc transcription factor is a pivotal player in hepatocarcinogenesis, but the mechanisms underlying c-Myc oncogenic activity in the liver remain poorly delineated. Mammalian target of rapamycin complex 2 (mTORC2) has been implicated in cancer by regulating multiple AGC kinases, especially AKT proteins. In the liver, AKT1 and AKT2 are widely expressed. While AKT2 is the major isoform downstream of activated phosphoinositide 3-kinase and loss of phosphatase and tensin homolog-induced HCC, the precise function of AKT1 in hepatocarcinogenesis is largely unknown. In the present study, we demonstrate that mTORC2 is activated in c-Myc-driven mouse HCC, leading to phosphorylation/activation of Akt1 but not Akt2. Ablation of Rictor inhibited c-Myc-induced HCC formation in vivo. Mechanistically, we discovered that loss of Akt1, but not Akt2, completely prevented c-Myc HCC formation in mice. Silencing of Rictor or Akt1 in c-Myc HCC cell lines inhibited phosphorylated forkhead box o1 expression and strongly suppressed cell growth in vitro. In human HCC samples, c-MYC activation is strongly correlated with phosphorylated AKT1 expression. Higher expression of RICTOR and AKT1, but not AKT2, is associated with poor survival of patients with HCC. In c-Myc mice, while rapamycin, an mTORC1 inhibitor, had limited efficacy at preventing c-Myc-driven HCC progression, the dual mTORC1 and mTORC2 inhibitor MLN0128 effectively promoted tumor regression by inducing apoptosis and necrosis. Conclusion: Our study indicates the functional contribution of mTORC2/Akt1 along c-Myc-induced hepatocarcinogenesis, with AKT1 and AKT2 having distinct roles in HCC development and progression; targeting both mTORC1 and mTORC2 may be required for effective treatment of human HCC displaying c-Myc amplification or overexpression.
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death worldwide.(1) In the United States, the incidence of HCC has been on the rise over the past decade.(2) According to the American Cancer Society, it is estimated that in 2019 42,030 new HCC cases will be diagnosed and that 31,780 patients will die from HCC. When unresectable, treatment options for HCC are very limited. Multikinase inhibitors, such as sorafenib, are of limited efficacy.(3) Immunotherapy with checkpoint inhibitors is a promising therapeutic strategy and may be effective in a subset of patients with HCC.(4) Clearly, more efficacious therapies are needed for HCC treatment.
Mammalian target of rapamycin (mTOR) is one of the essential complexes regulating tumor cell proliferation, metabolism, and survival.(5) mTOR is assembled into two distinct complexes: mTORC1 and mTORC2.(6) Raptor is the unique subunit of mTORC1, whereas Rictor and mSin1 are the specific subunits for mTORC2. mTORC1 modulates cell growth through S6 kinase/ribosomal protein S6 (RPS6) and 4E binding protein 1 (4EBP1)/eukaryotic translation initiation factor 4E cascades, and it has been well characterized to play a pivotal role in tumorigenesis,(7) whereas mTORC2 has been shown to regulate AGC proteins, including AKT, serum/glucocorticoid-regulated kinase (SGK), and protein kinase C.(8) Among them, AKT is considered the prominent kinase modulating cell growth and survival. In mammalian cells, three AKT isoforms have been identified: AKT1, AKT2, and AKT3. In addition to AKT, recent studies have implicated SGK3, one of the SGK family members, as a mediator of helical mutant forms of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA).(9) Unlike mTORC1, the functional role of mTORC2 in hepatocarcinogenesis remains poorly characterized. In the liver, AKT1 and AKT2, but not AKT3, are expressed.(10) Studies have shown that Akt2 is the pivotal Akt protein transducing insulin signals in the liver as well as the main Akt oncogenic isoform responsible for HCC formation in mice in the presence of activated PIK3CA mutant or loss of the phosphatase and tensin homolog (Pten) tumor suppressor.(11–13) The precise role of Akt1 in liver homeostasis is instead less defined, although it is clear that Akt1 and Akt2 may compensate each other during liver development and regeneration.(14,15) Intriguingly, in a recent study, it was found that hepatic deletion of Akt1 and Akt2 induces early onset of spontaneous HCC and that mice are highly sensitive to chemically induced HCC formation.(10) Obviously, further investigation is necessary to better delineate the functions of AKT1 and AKT2 during hepatocarcinogenesis.
Because the mTOR cascade is found to be activated in a substantial percentage of human tumor samples, significant efforts in developing mTOR inhibitors for cancer treatment have been made.(16) The first generation of mTOR inhibitors, such as everolimus and temsirolimus, only partially inhibited mTORC1 and displayed very limited antitumor activity in clinical trials.(17) In HCC, everolimus failed to improve overall patient survival.(18) The second generation of mTOR inhibitors are adenosine triphosphate-competitive pan-mTOR inhibitors, able to concomitantly suppress both mTORC1 and mTORC2,(19) and show improved efficacy in vitro as well as in preclinical models.(20)
c-Myc is a well-characterized oncoprotein and has been implicated in multiple tumor types.(21) In human HCC, c-Myc is found to be frequently overexpressed, and high levels of c-Myc are associated with poor prognosis.(22) In mice, overexpression of c-Myc alone leads to formation of poorly differentiated HCC, sup porting c-Myc as a major driver oncogene in hepatocarcinogenesis.(23,24) c-Myc is currently considered to be “undruggable.” Thus, significant efforts are required to identify downstream effectors of the c-Myc oncogene, whose inhibition is detrimental for the growth of c-Myc-driven tumors. In a recent study, we demonstrated that c-Myc activates mTORC1 by directly inducing the expression of selected amino acid transporters.(22) In the present study, we characterize the functional role of mTORC2 in c-Myc-driven HCC.
Materials and Methods
PLASMIDS
The plasmids used in the project, including pT3-elongation factor 1 alpha (EF1α)-myeloid cell leukemia 1 (MCL1), pT3-EF1α-c-Myc, phosphorylated cytomegalovirus (pCMV)-cyclization recombination (Cre), and pCMV/sleeping beauty transposase, have been described in our previous publications.(22,25) Plasmids for in vivo studies were purified using the Endotoxin Free Maxi Prep Kit (Sigma-Aldrich, St. Louis, MO).
HYDRODYNAMIC TAIL VEIN INJECTION AND MOUSE TREATMENT
Wild-type FVB/N mice, Akt1fl/fl mice,(26) Akt2fl/+ mice,(27) and Rictorfl/fl mice(28) (all in C57BL/6 background) were purchased from Jackson Laboratory (Bar Harbor, ME). Sgk3+/− mice(29) (in C57BL/6 background) were kindly provided by Dr. David Pearce from the University of California, San Francisco. AKT2fl/+ mice were crossed together to generate Akt2fl/fl mice; Sgk3+/− mice were crossed together to generate Sgk3−/− mice and Sgk3+/+ control littermates. Hydrodynamic injection was performed as described,(30) and detailed information is available in Supporting Table S1. Mice were monitored for liver tumor development as palpable abdominal masses and euthanized as described in Results. MLN0128 (1 mg/kg/day) and rapamycin (6 mg/kg/day) were prepared as described(31) and administered by either oral gavage or intraperitoneal injection, respectively, 6 days a week. Animal experiments were performed according to protocols approved by the Committee for Animal Research at the University of California, San Francisco.
HUMAN LIVER TISSUE SPECIMENS
A collection of formalin-fixed, paraffin-embedded HCC samples (n = 108) was used in the present study. Tumors were divided into groups of HCC with shorter survival/poorer prognosis (n = 56) and longer survival/better prognosis (n = 52), characterized by <3 and >3 years’ survival following partial liver resection, respectively. The clinicopathological features of patients with liver cancer are summarized in Supporting Table S2. HCC specimens were collected at the Medical Universities of Greifswald (Greifswald, Germany) and Sassari (Sassari, Italy). Institutional review board approval was obtained from the local ethical committee of the Medical Universities of Greifswald and Sassari. Informed consent was obtained from all individuals.
STATISTICAL ANALYSIS
All data were analyzed using Prism 6 (GraphPad, San Diego, CA). Comparisons between two groups were performed with a two-tailed unpaired t test. Comparisons among three or more groups were performed with analysis of variance. P < 0.05 was considered significant. Data are expressed as mean ± standard error of the mean for each group.
Additional materials and methods can be found in the Supporting Information.
Results
mTORC2 IS ACTIVATED IN c-Myc-DRIVEN HCC
First, we investigated whether mTORC2 is activated in mouse c-Myc HCC. Our analysis showed that Rictor, the unique subunit of mTORC2, was expressed in normal and tumor tissues (Fig. 1). Levels of phosphorylated/activated p-Akt(S473/S474) were higher in c-Myc HCC tumor tissues than normal liver tissues from wild-type mice, leading to more pronounced levels of phosphorylated/inactivated forkhead box o1 (p-Foxo1), a well-known target of Akt as well as of the downstream mTORC1 cascade, as indicated by p-4EBP1 and phosphorylated/activated S6 expression (Fig. 1). Intriguingly, a distinct expression pattern was detected for phosphorylated/activated Akt1 and Akt2 proteins, with p-Akt1(S473) exhibiting up-regulation and p-Akt2(S474) down-regulation, in c-Myc HCCs when compared with wild-type livers (Fig. 1). No differences were detected in total levels of Akt1 and Akt2 proteins between normal livers from wild-type mice and c-Myc hepatocellular lesions (Fig. 1). SGK3 is another known target of mTORC2 and may be the major kinase downstream of PIK3CA mutants.(9) Unfortunately, we were unable to determine the phosphorylated/activated form of Sgk3 due to the lack of a commercially available anti-p-SGK3-specific antibody able to detect endogenous p-Sgk3 levels. However, we found that the total amount of Sgk3 protein was higher in c-Myc HCC (Fig. 1).
FIG. 1.
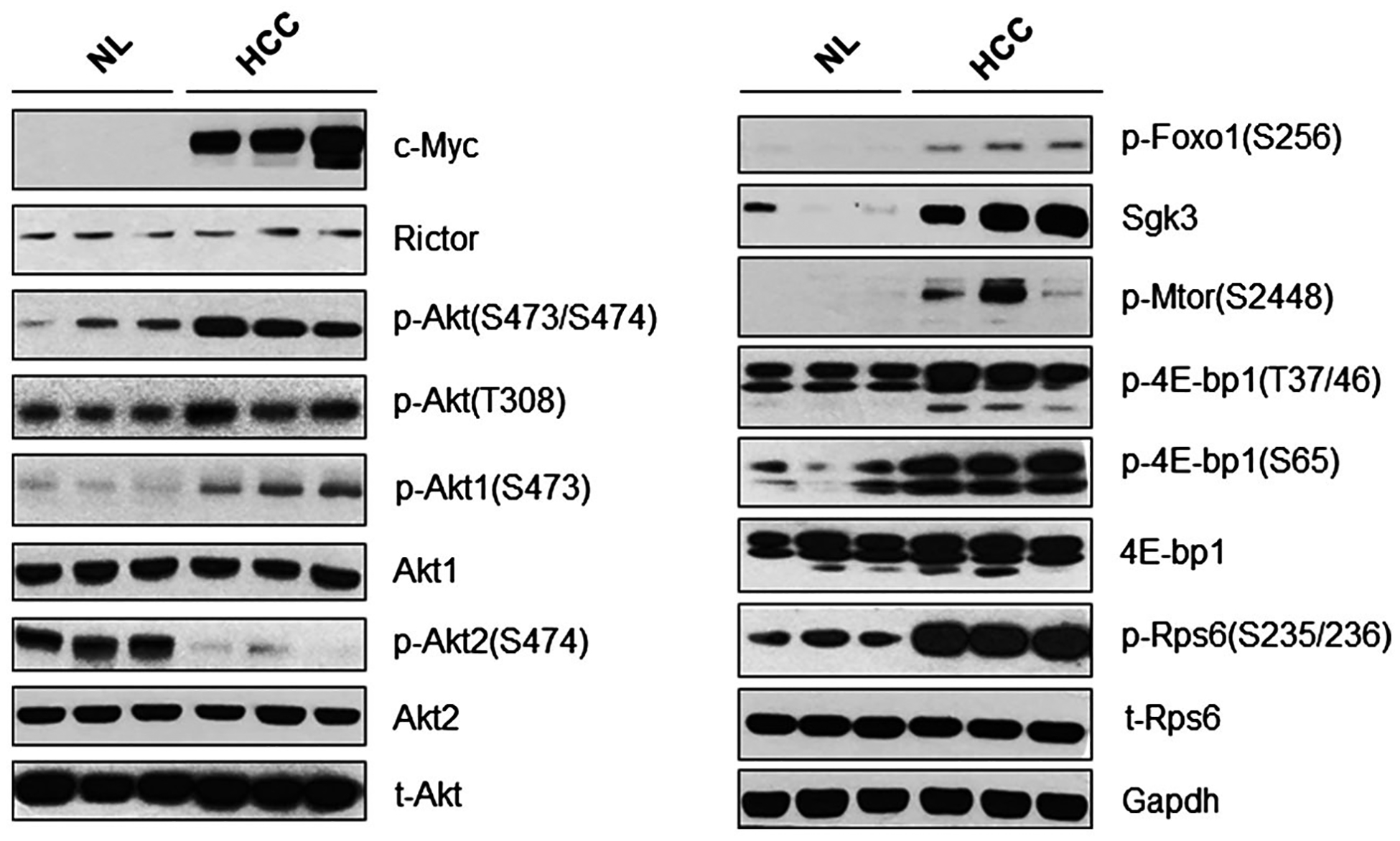
The mTORC2 pathway is activated in c-Myc-induced HCC. Representative western blotting from normal liver tissues and c-Myc-injected mouse HCC tissues. Abbreviations: Gapdh, glyceraldehyde 3-phosphate dehydrogenase; NL, normal liver; t-, total.
Subsequently, the mTORC2 status was determined in two c-Myc-derived HCC cell lines: HCC3–4 and HCC4–4.(32) We found high levels of p-Akt and p-Foxo1 in both cell lines. The level of p-Akt1 was high in both cell lines; however, unlike c-Myc mouse tissues, p-Akt2 expression was also robust (Supporting Fig. S1A). The difference between tumor tissues and in vitro cell lines may be attributable to the cell lines being cultured in the presence of fetal bovine serum, which contains high levels of growth factors and may lead to the higher levels of p-Akt2 in HCC3–4 and HCC4–4 cells. Next, we investigated whether p-Akt1 expression was dependent on c-Myc expression. Thus, we silenced c-Myc in HCC3–4 and HCC4–4 cell lines. While total levels of the two Akt isoforms were not affected by c-Myc suppression, p-Akt1, but not p-Akt2, levels were down-regulated in both cell lines (Supporting Fig. S1B,C). Of note, silencing of c-Myc did not influence the levels of Akt1 and Akt2 mRNA in the same cells, indicating that c-Myc regulates Akt1 at the posttranscriptional level (Supporting Fig. S1D,E).
In summary, our study demonstrates that c-Myc promotes mTORC2/Akt1 activation in hepatocarcinogenesis.
HCC DEVELOPMENT IN c-Myc MICE IS mTORC2-DEPENDENT
To elucidate the role of mTORC2 in c-Myc-induced HCC, we silenced Rictor in two mouse HCC cell lines derived from c-Myc-induced mouse HCC.(32) A significant decrease of c-Myc HCC cell growth accompanied Rictor suppression (Supporting Fig. S2A,B). At the biochemical level, as expected, short hairpin Rictor decreased the expression of p-Akt, leading to down-regulation of p-Foxo1. Intriguingly, short hairpin Rictor had little effect on the mTORC1 pathway effectors p-4EBP1 and p-S6. Concerning the Akt isoforms, knockdown of Rictor consistently inhibited p-Akt1, but not p-Akt2, in c-Myc HCC cell lines (Supporting Fig. S2C).
Next, we investigated whether ablation of Rictor affected c-Myc-induced HCC formation in mice using conditional Rictor knockout (KO) mice. Rictorfl/fl mice are in the C57BL/6 genetic background; our previous studies showed that hydrodynamic transfection of c-Myc alone is unable to induce HCC formation in C57BL/6 mice.(25) Coexpression of c-Myc with MCL1 consistently leads to HCC formation in C57BL/6 mice, and c-Myc/MCL1 liver tumor lesions share highly similar histological, gene expression, and metabolic features with c-Myc HCC induced in FVB/N mice.(25) Thus, we hydrodynamically injected c-Myc/MCL1 with pCMV (empty vector control; c-Myc/MCL1/pCMV) or with pCMV-Cre (c-Myc/MCL1/Cre) into Rictorfl/fl mice (Fig. 2A; Supporting Table S3). All c-Myc/MCL1/pCMV-injected Rictorfl/fl control mice developed a lethal burden of liver tumors and had to be euthanized between 5 and 12 weeks postinjection. In contrast, none of the c-Myc/MCL1/Cre-injected mice showed any sign of liver tumor development, even at 19 weeks postinjection (Fig. 2B,C; Supporting Table S3). Histologically, poorly differentiated and highly proliferative HCC nodules were detected throughout the liver of c-Myc/MCL1/pCMV mice, whereas c-Myc/MCL1/Cre mouse livers appeared to be completely normal with few proliferating cells (Fig. 2D). In summary, our data indicate that mTORC2 is required for c-Myc-induced HCC formation.
FIG. 2.
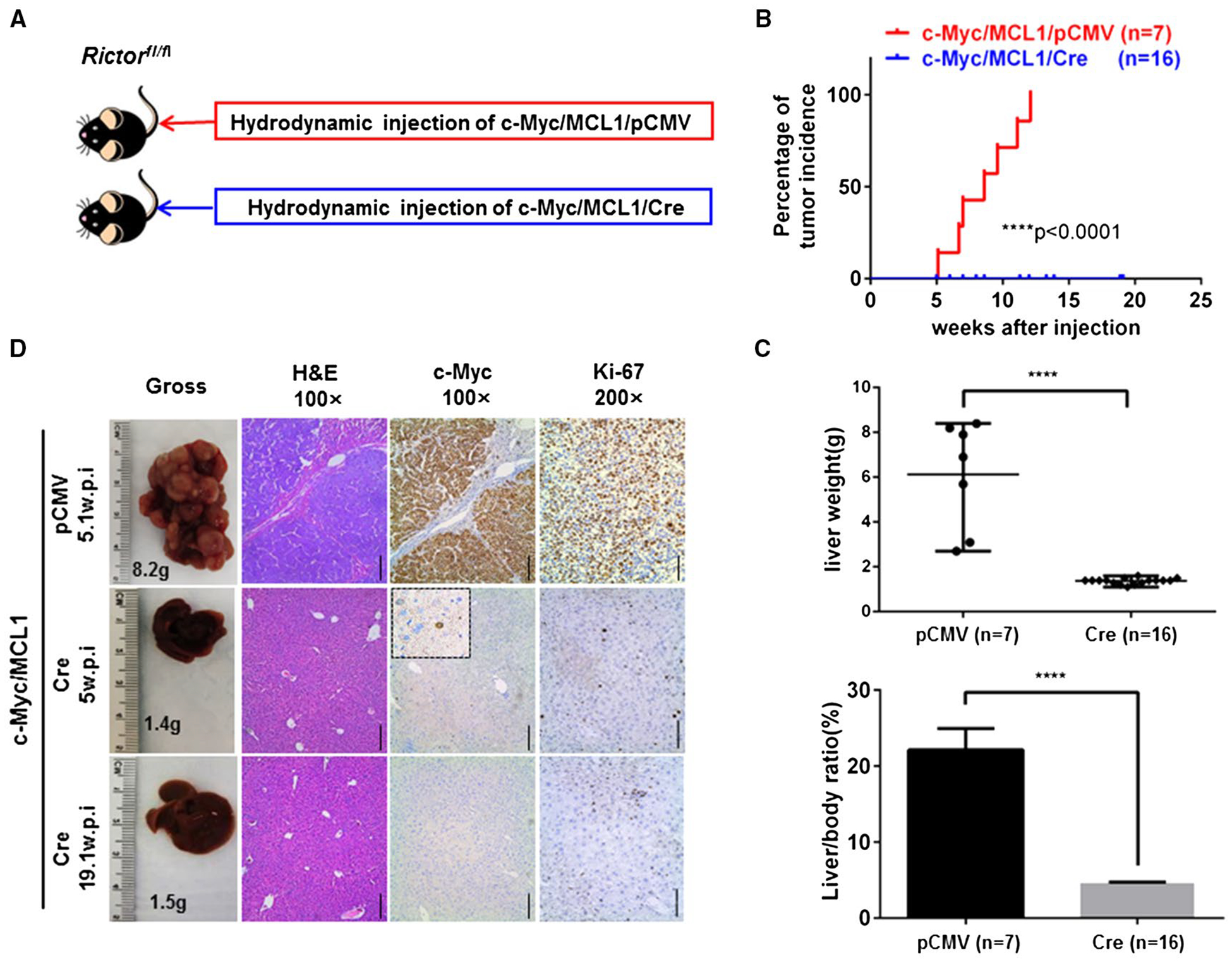
mTORC2 is required for c-Myc-driven HCC formation. (A) Study design. (B) Tumor incidence curve. (C) Liver weight and liver/body ratio of Rictorfl/fl mice injected with c-Myc/MCL1/pCMV or c-Myc/MCL1/Cre constructs, respectively. **** P < 0.0001. (D) Gross images of livers, hematoxylin and eosin, c-Myc, and Ki-67 staining in Rictorfl/fl mice injected with c-Myc/MCL1/pCMV or c-Myc/MCL1/Cre constructs at specific weeks postinjection. Magnifications ×100 (hematoxylin and eosin, c-Myc), scale bar, 200 μm; ×200 (Ki-67), scale bar, 100 μm. Abbreviations: H&E, hematoxylin and eosin; w.p.i., weeks postinjection.
Akt1 IS THE MAJOR KINASE DOWNSTREAM OF mTORC2 IN c-Myc-DRIVEN HCC DEVELOPMENT
Our data indicate that Akt1, but not Akt2, is strongly activated by phosphorylation in c-Myc HCC (Fig. 1). We hypothesized that Akt1 is the major Akt isoform downstream of mTORC2 in c-Myc-driven hepatocarcinogenesis. We applied conditional Akt1 KO mice to investigate whether Akt1 is required for c-Myc-induced HCC in vivo. For this purpose, we hydrodynamically injected c-Myc/MCL1 with pCMV (empty vector control; c-Myc/MCL1/pCMV) or pCMV-Cre (c-Myc/MCL1/Cre) into Akt1fl/fl mice (Fig. 3A). Consistent with our hypothesis, all c-Myc/MCL1/pCMV-injected Akt1fl/fl control mice developed a lethal burden of liver tumors and had to be euthanized between 5 and 12 weeks postinjection. In contrast, none of the c-Myc/MCL1/Cre-injected mice showed any sign of liver tumors, even at 19 weeks postinjection (Fig. 3B,C; Supporting Table S4). Histologically, poorly differentiated and highly proliferative HCC nodules were detected in c-Myc/MCL1/pCMV Akt1fl/fl mice, whereas c-Myc/MCL1/Cre mouse livers were completely normal (Fig. 3D), recapitulating the phenotype observed when c-Myc/MCL1/Cre constructs were injected into Rictorfl/fl mice.
FIG. 3.
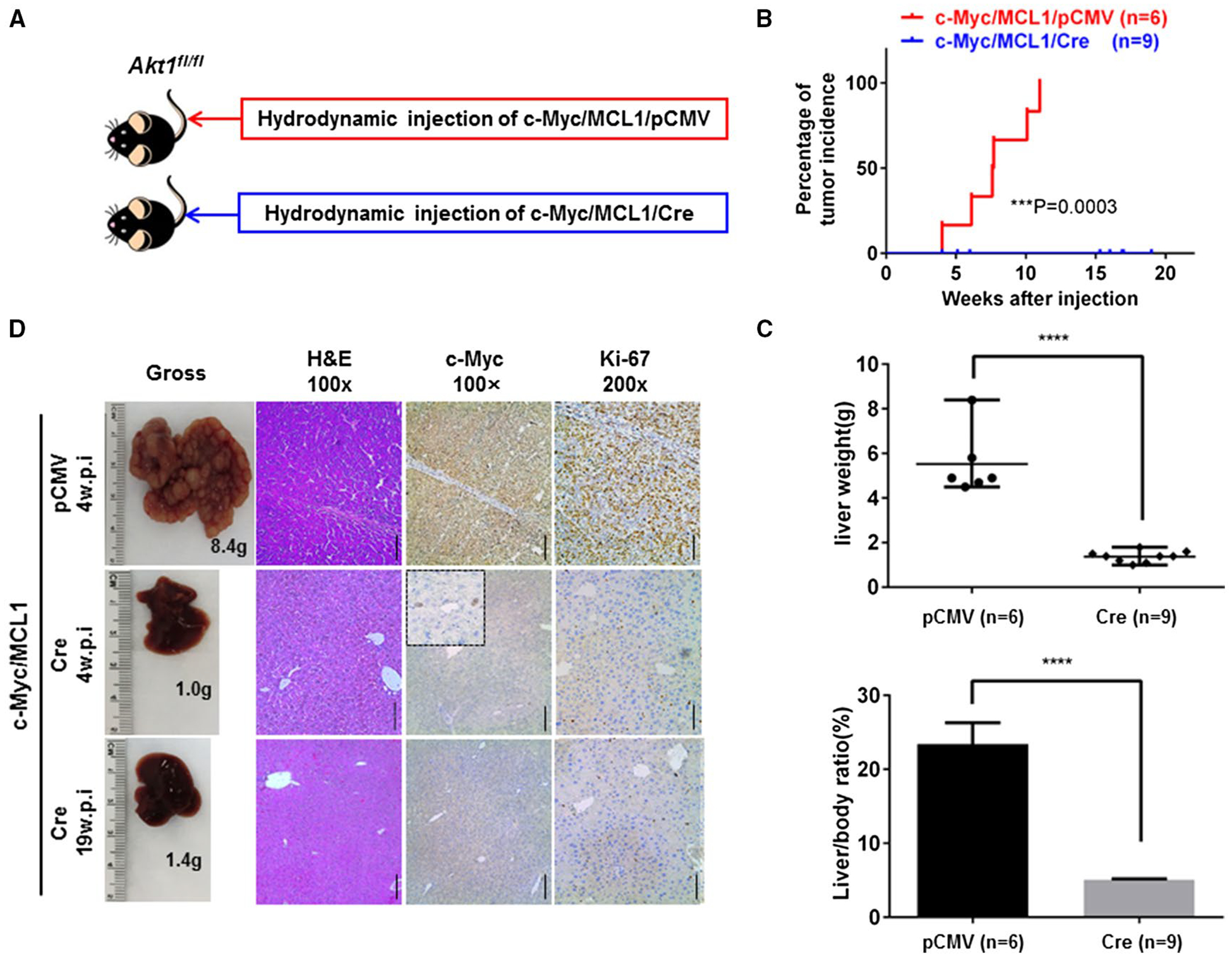
Akt1 is required for c-Myc-driven HCC formation. (A) Study design. (B) Tumor incidence curve. (C) Liver weight and liver/body ratio of Akt1fl/fl mice injected with c-Myc/MCL1/pCMV or c-Myc/MCL1/Cre constructs, respectively. ****P< 0.0001. (D) Gross images of livers, hematoxylin and eosin, c-Myc, and Ki-67 staining in Akt1fl/fl mice injected with c-Myc/MCL1/pCMV or c-Myc/MCL1/Cre constructs at specific weeks postinjection. Magnifications ×100 (hematoxylin and eosin, c-Myc), scale bar, 200 μm; ×200 (Ki-67), scale bar, 100 μm. Abbreviations: H&E, hematoxylin and eosin; w.p.i., weeks postinjection.
Previously, we found that c-Myc can induce HCC formation in Akt2 KO mice.(22) The experiments were performed using whole-body KO mice. Because Akt1 may be up-regulated in response to the loss of Akt2 during development, based on the experiments previously conducted, we cannot exclude that Akt2 may still have a role in regulating c-Myc oncogenic potential in the liver. To address this important issue, we used conditional Akt2 KO mice. Thus, c-Myc/MCL1 with pCMV (empty vector control; c-Myc/MCL1/pCMV) and pCMV-Cre (c-Myc/MCL1/Cre) were hydrodynamically injected into Akt2fl/fl mice (Fig. 4A). We found that ablation of Akt2 moderately delayed c-Myc/MCL1 HCC development. At 15 weeks postinjection, all c-Myc/MCL1/Cre-injected Akt2fl/fl mice developed liver tumors, although the tumor burden was lower in c-Myc/MCL1/Cre mice than in c-Myc/MCL1/pCMV mice (Fig. 4B–D; Supporting Table S5). At the biochemical level, we confirmed the deletion of Akt2 in c-Myc/MCL1/Cre HCC lesions, but p-Akt(S473/S474) and p-Akt1(S473) as well as p-Foxo1 levels remained high in the tumor tissues (Fig. 4E).
FIG. 4.
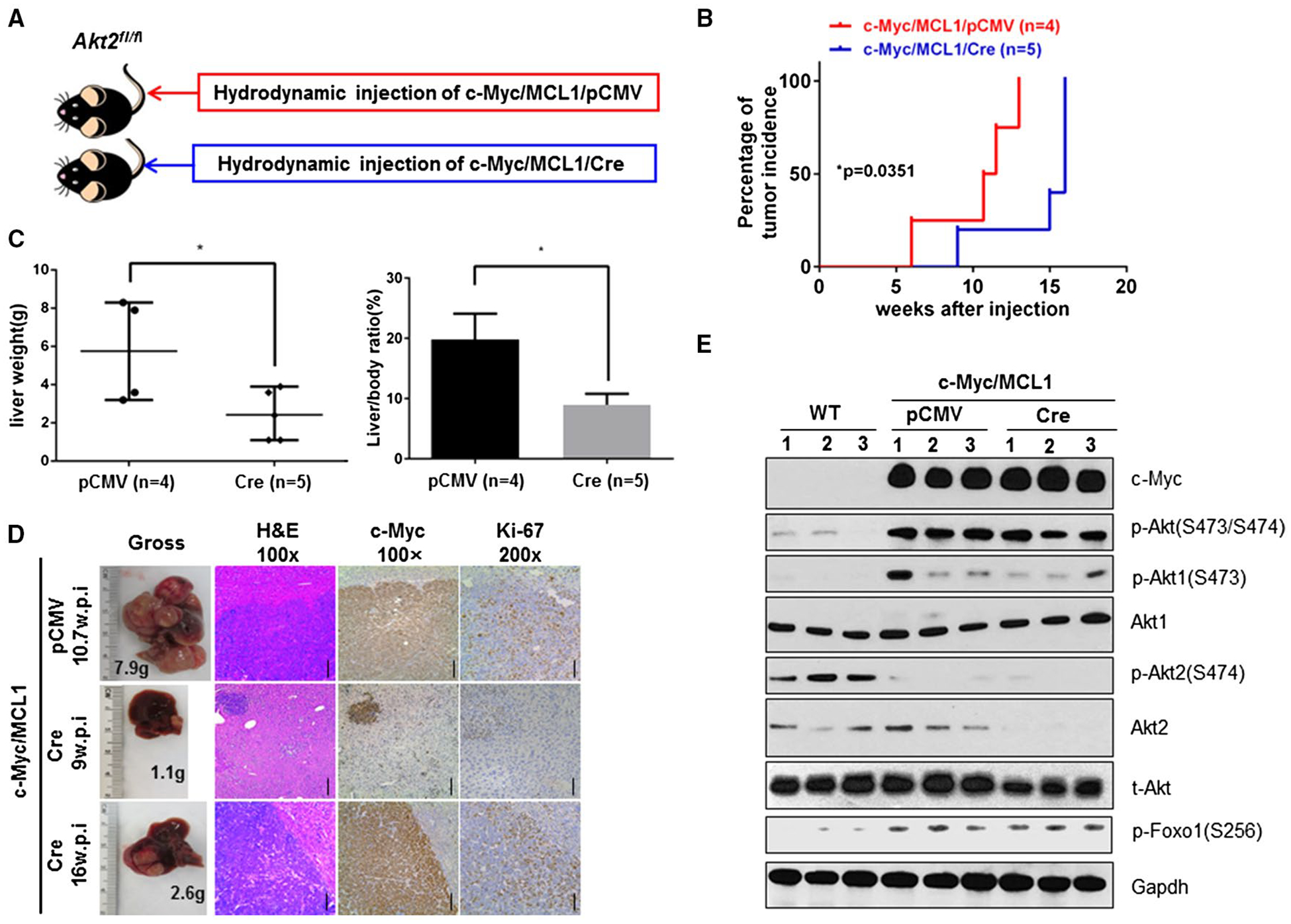
Delayed c-Myc-driven HCC formation in Akt2 conditional KO mice. (A) Study design. (B) Tumor incidence curve. (C) Liver weight and liver/body ratio of Akt2fl/fl mice injected with c-Myc/MCL1/pCMV and c-Myc/MCL1/Cre constructs, respectively. *P < 0.05. (D) Gross images of livers, hematoxylin and eosin, c-Myc, and Ki-67 staining in Akt2fl/fl mice injected with c-Myc/MCL1/pCMV or c-Myc/MCL1/Cre constructs at specific weeks postinjection. Magnifications ×100 (hematoxylin and eosin, c-Myc), scale bar, 200 μm; ×200 (Ki-67), scale bar, 100 μm. (E) Representative western blotting from Akt2fl/fl mouse liver tissue or Akt2fl/fl mouse liver tissue injected with c-Myc/MCL1/pCMV or c-Myc/MCL1/Cre. Abbreviations: Gapdh, glyceraldehyde 3-phosphate dehydrogenase; H&E, hematoxylin and eosin; t-, total; w.p.i., weeks post injection.
Finally, as SGK3 has been shown to be an mTORC2 effector alternative to AKT proteins and it is required for PIK3CA helical domain mutant PIK3CA tumorigenesis,(9) we assessed the role of Sgk3 in c-Myc-driven carcinogenesis. As reported above, Sgk3 protein expression was found to be up-regulated in c-Myc HCC samples (Fig. 1). Thus, we bred Sgk3+/− mice to generate Sgk3−/− mice, and their Sgk3+/+ littermates were used as control. Subsequently, c-Myc/MCL1 plasmids were hydrodynamically injected into Sgk3−/− and Sgk3+/+ mice. We found that all mice developed a lethal burden of liver cancer irrespective of the Sgk3 genotype (Supporting Fig. S3 and Table S6). Thus, the results suggest that Sgk3 is not a major downstream effector of mTORC2 in c-Myc-driven hepatocarcinogenesis.
To investigate the cellular and signaling pathways downstream of mTORC2/Akt1 in c-Myc-driven HCC development, we silenced Akt1 and/or Akt2 in two c-Myc mouse HCC cell lines using specific small interfering RNAs. Consistent with the in vivo results, we found that small interfering Akt1 (siAkt1) strongly inhibited HCC3–4 and HCC4–4 cell growth, whereas siAkt2 moderately decreased cell growth (Supporting Fig. S4A,B). At the cellular level, this was due to the stronger effects of siAkt1 in inhibiting cell proliferation and inducing apoptosis (Supporting Fig. S5). Equivalent results in terms of growth restraint were obtained when HCC3–4 and HCC4–4 cells were subjected to treatment with the AKT1 selective inhibitor A-674563 and the AKT2-specific inhibitor CCT128930 (Supporting Fig. S6). Noticeably, the same pattern of changes in proliferation and apoptosis was also detected in Huh7, Hep3B (with low c-Myc expression), SNU182, and SNU449 (with high c-Myc expression) human HCC cell lines, with the AKT1 inhibitor A-674563 inducing a much more pronounced antigrowth effect only in the cell lines with elevated basal levels of c-Myc (Supporting Fig. S7). Mechanistically, we found that, consistent with Rictor-silenced cells, siAkt1 inhibited p-Foxo1, but not the mTORC1 effector p-Rps6, in c-Myc HCC cells (Supporting Fig. S4C,D). It is worth noting that silencing of Akt2 resulted in a strong reduction of total Akt levels in mouse HCC cell lines, whereas knockdown of Akt1 had limited impact on total Akt levels (Supporting Fig. S4C,D). This is consistent with previous data indicating that Akt2 consists of ~80% of total Akt proteins in the liver and Akt1 consists of ~15%. Nevertheless, in c-Myc HCC cell lines, Akt1 is highly activated and responsible for most of the Akt activity.
Altogether, the data demonstrate that c-Myc-induced liver carcinogenesis requires an active mTORC2/Akt1 cascade. Foxo1, but not mTORC1, may be the major signaling event downstream of Akt1 in c-Myc HCC.
COORDINATED ACTIVATION OF c-MYC AND AKT1 IN HUMAN HCC
Next, we investigated c-MYC, RICTOR, AKT1, and AKT2 expression in human HCC samples. Using The Cancer Genome Atlas data set,(33) we found that high c-MYC mRNA expression had a trend to be associated with poor survival, although it did not reach statistical significance (P = 0.058). Overexpression of RICTOR and AKT1 correlated with worse outcome, whereas AKT2 expression levels did not associate with HCC patient survival (Supporting Fig. S8).
As c-MYC is known to be regulated at the protein level and p-AKT1/2 levels are both indicators for their activation status, we investigated c-MYC, p-AKT1, and p-AKT2 protein expression in a large collection of human HCC specimens (n = 108) using immunohistochemistry (Fig. 5A). Nuclear immunoreactivity for c-MYC was detected in 44 (40.7%) HCC specimens. Of note, while 39 of 44 HCCs (88.6%) displaying c-Myc nuclear accumulation concomitantly showed strong immunoreactivity for p-AKT1, only 7 of the 44 (15.9%) c-Myc-positive HCCs exhibited intense p-AKT2 immunostaining (Fig. 5B). Also, of the seven HCC specimens displaying robust immunolabeling for c-Myc and p-AKT2, only two were p-AKT1 negative. Furthermore, although the overall number of HCC samples with up-regulation of p-AKT1 and p-AKT2 staining was similar (50/108, 46.3%, and 55/108, 50.9%, respectively) (Fig. 5C), most of the p-AKT1-positive samples also showed c-MYC up-regulation (39/48, 81.2%), while the vast majority of p-AKT2-positive samples (48/55, 87.3%) were negative for c-MYC nuclear accumulation (Fig. 5D). Thus, the immunohistochemical pattern observed in human HCC lesions recapitulates the crosstalk between c-Myc and AKT1 (but not AKT2) in mice. When evaluating the relationship with patients’ prognosis, strong immunoreactivity for c-MYC, p-AKT1, and p-AKT2 was found in 32 of 44 (72.7%), 34 of 50 (68%), and 14 of 55 (25.4%) HCCs with the poorest outcome, respectively, suggesting that c-MYC overexpression and AKT1 activation may have prognostic value (Fig. 5E). No association between the levels of c-MYC, p-AKT1, or p-AKT2 and other clinicopathologic features of the patients, including age, gender, etiology, presence of cirrhosis, tumor size, and tumor differentiation, was detected (Supporting Table S2).
FIG. 5.
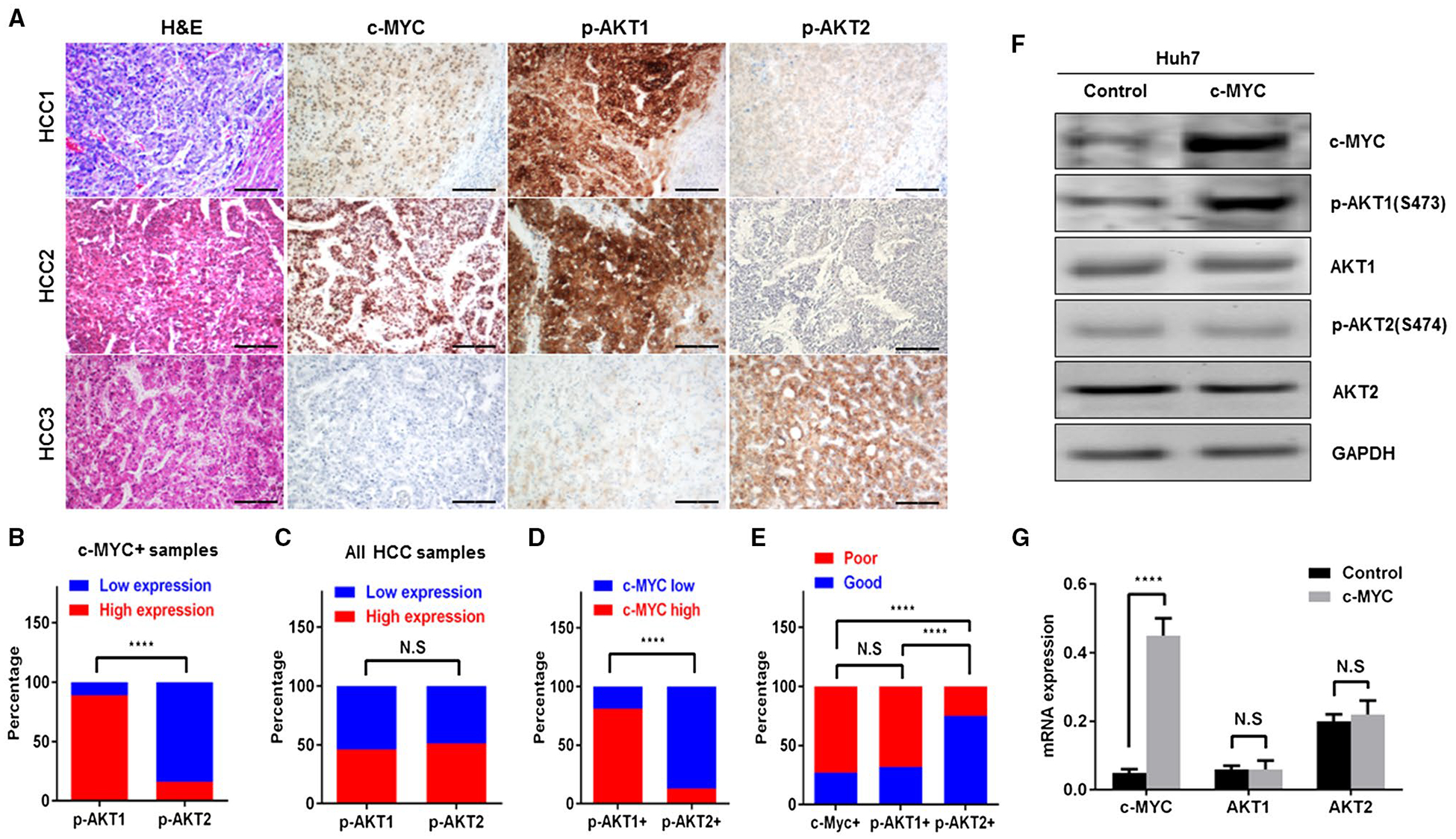
Coordinated activation of c-MYC and AKT1 in human HCC. (A) Representative expression patterns of c-MYC, activated p-AKT1, and p-AKT2 in human HCC as detected by immunohistochemistry. Upper panels: HCC case showing up-regulation of c-Myc, p-AKT1, and p-AKT2 in the tumor part compared with nontumorous surrounding liver (lower right corner). Middle panels: HCC case displaying concomitant induction of c-MYC and p-AKT1, whereas p-AKT2 immunoreactivity is absent. Lower panels: HCC exhibiting low levels of c-MYC and p-AKT1 in the presence of strong immunoreactivity for p-AKT2. Magnifications ×100 (hematoxylin and eosin, c-MYC, p-AKT1, and p-AKT2), scale bar, 200 μm. (B) Percentage of high and low p-AKT1 or p-AKT2 expressing HCC samples in c-MYC-positive HCC specimens. ****P < 0.0001. (C) Percentage of high and low p-AKT1 or p-AKT2 expressing HCC samples in all HCC specimens. (D) Percentage of high and low c-MYC expressing HCC samples in p-AKT1-positive or p-AKT2-positive HCC specimens, respectively. ****P < 0.0001. (E) Percentage of patients with good and poor prognosis in c-MYC-positive, p-AKT1-positive, or p-AKT2-positive HCC samples. ****P < 0.0001. (F,G) Forced overexpression of c-MYC gene in the Huh7 human HCC cell line triggers activation of AKT1, but not of AKT2, phosphorylation, without affecting total protein (F) and mRNA (G) levels of AKT1. ****P < 0.0001. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; H&E, hematoxylin and eosin staining; N.S, no significance.
Next, we transiently transfected c-MYC into Huh7 human HCC cells. We found that forced expression of c-MYC led to up-regulation of p-AKT1, but not p-AKT2, in Huh7 cells, in accordance with mouse data (Fig. 5F). Furthermore, no differences were detected in AKT1 and AKT2 expression at both protein (Fig. 5F) and mRNA (Fig. 5G) levels.
In summary, our study indicates coordinated activation of c-MYC and AKT1 in human HCC.
THE DUAL mTORC1/2 INHIBITOR MLN0128 INDUCES HCC REGRESSION IN c-Myc MICE
Recently, we demonstrated that intact mTORC1 is required for c-Myc-dependent hepatocarcinogenesis.(22) Here, we provide evidence that mTORC2 is indispensable for c-Myc liver tumor formation. We compared the therapeutic potential of rapamycin, a partial mTORC1 inhibitor, with MLN0128, a pan-mTOR1/2 inhibitor, for the treatment of late-stage c-Myc HCC. Specifically, we hydrodynamically injected c-Myc into FVB/N mice (n = 39) and monitored mouse liver tumor development through abdominal palpation. We harvested the cohort of mice when the mice showed mild abdominal enlargement (n = 9). The average liver weight of the mice was around 4.5 g, and this cohort was used as a “pretreatment” cohort. The remaining mice were randomly divided into three groups and treated with vehicle (n = 9), rapamycin (10), or MLN0128 (n = 11) for up to 3 weeks (Fig. 6A). Following the guidelines from the Institutional Animal Care and Use Committee protocol, mice were harvested if they became moribund or demonstrated significant abdominal masses. Of note, HCC lesions continued to grow in all mice treated with vehicle or rapamycin. Most of the mice needed to be euthanized ~1 to 2 weeks post-drug treatment (Fig. 6B,C). In striking contrast, all MLN0128-treated mice appeared to be healthy. During the 3-week treatment course, among the 11 mice treated with MLN0128, only one showed significant tumor growth (Fig. 6B,C). Using total liver weight as the measurement of tumor burden, we found that the overall liver weight was significantly higher in vehicle-treated and rapamycin-treated cohorts compared with the pretreatment cohort (Fig. 6D), suggesting that mice in the two cohorts underwent tumor progression and growth. Rapamycin-treated mice showed a lower tumor burden than vehicle-treated mice, suggesting its limited efficacy in preventing HCC progression. In contrast, MLN0128 treatment significantly decreased the tumor burden when compared to the vehicle and rapamycin cohorts and, importantly, lower tumor burden than the pretreatment group (Fig. 6D; Supporting Table S7). These findings indicate that MLN0128 treatment causes liver tumor regression in c-Myc mice.
FIG. 6.
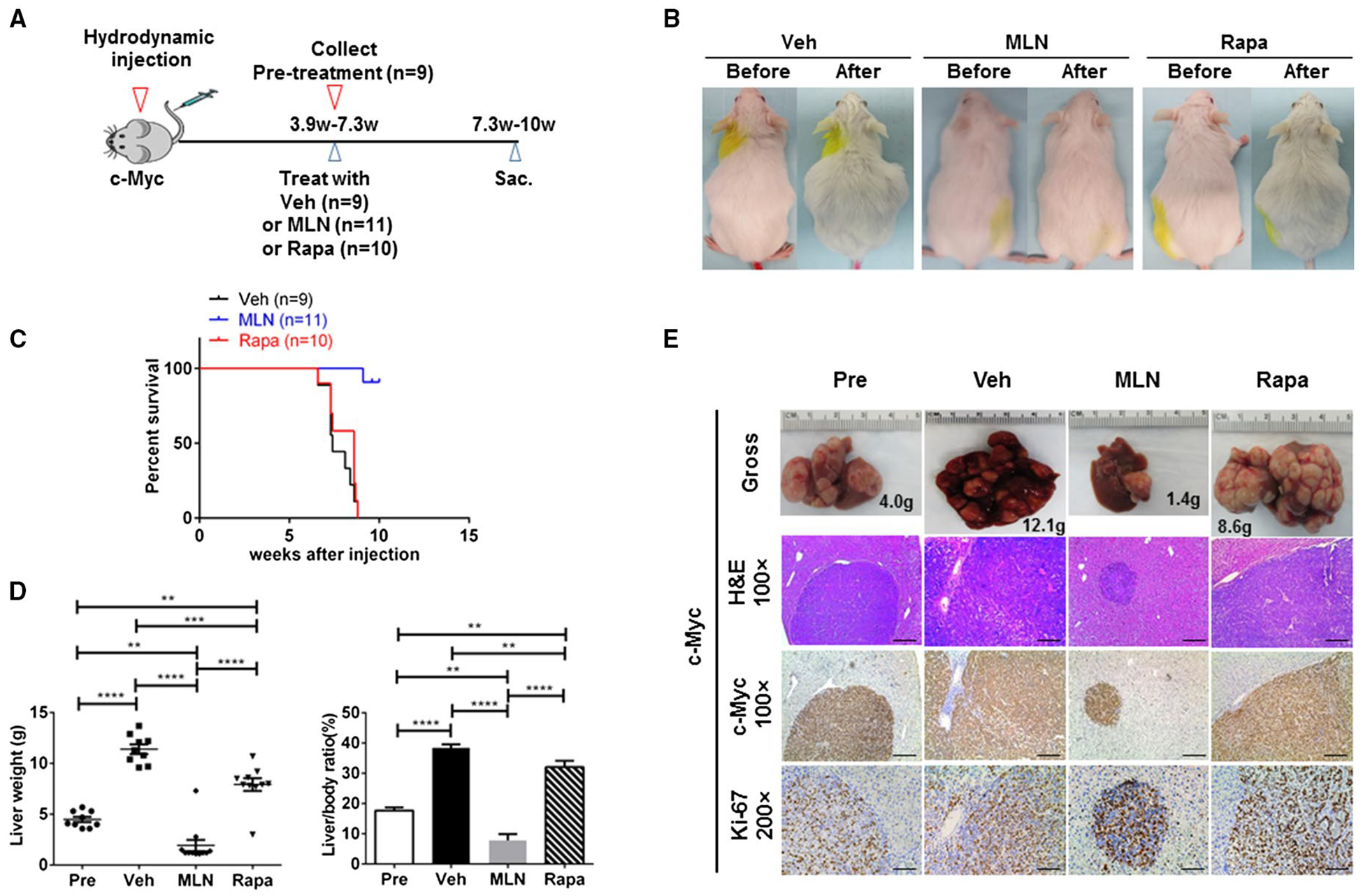
MLN0128, but not rapamycin, effectively induces regression of late-stage, progressed c-Myc mouse liver tumors. (A) Study design. (B) Gross images in vehicle-treated, rapamycin-treated, and MLN0128-treated FVB mice injected with the c-Myc construct. (C) Survival curve. (D) Liver weight and liver/body ratio of c-Myc-injected mice treated with vehicle, rapamycin, or MLN0128 for 3 weeks. Data are presented as mean ± standard error of the mean. **P < 0.01, ***P < 0.001, ****P < 0.0001. (E) Gross, hematoxylin and eosin, and c-Myc and Ki-67 staining images in vehicle-treated, rapamycin-treated, and MLN0128-treated mouse livers injected with the c-Myc construct. Magnifications ×100 (hematoxylin and eosin, c-Myc), scale bar, 200 μm; ×200 (Ki-67), scale bar, 100 μm. Abbreviations: H&E, hematoxylin and eosin; MLN, MLN0128; Rapa, rapamycin; Sac, Sacrifice; Veh, vehicle.
Grossly, numerous tumor nodules were detected in the liver of mice from pretreatment, vehicle treatment, and rapamycin treatment cohorts (Fig. 6E). In striking contrast, except for the one mouse with a high tumor burden, only a few small tumor nodules were observed in livers from MLN0128-treated mice (Fig. 6E). Histologically, poorly differentiated HCC lesions characterized the livers of mice from the pretreatment, vehicle-treated, and rapamycin-treated groups (Fig. 6E). In striking contrast, only small tumor lesions occurred in MLN0128-treated mice (Fig. 6E). All tumor nodules in all cohorts were confirmed to express the ectopically transfected c-Myc oncogene and were highly proliferative, as shown by Ki-67 immunostaining (Fig. 6E).
At the biochemical level, we investigated the AKT/mTOR pathway in nonnecrotic lesions from vehicle-treated, rapamycin-treated, and MLN0128-treated mice (Fig. 7C). Rapamycin treatment was associated with inhibition of p-Rps6 expression, without affecting p-Akt(S473/S474) and p-4Ebp1 levels. In contrast, MLN0128 effectively inhibited p-Rps6, p-4Ebp1, and p-Akt(S473/S474) (Fig. 7C), leading to decreased expression of proliferating cell nuclear antigen and survivin (Fig. 7C). In our previous studies, blocking the 4Ebp1 cascade could not completely inhibit c-Myc HCC development in vivo.(22) Thus, the therapeutic efficacy of MLN0128 is likely mediated by its ability to inhibit mTORC2/Akt1 as well as 4Ebp1 in this murine HCC model.
FIG. 7.
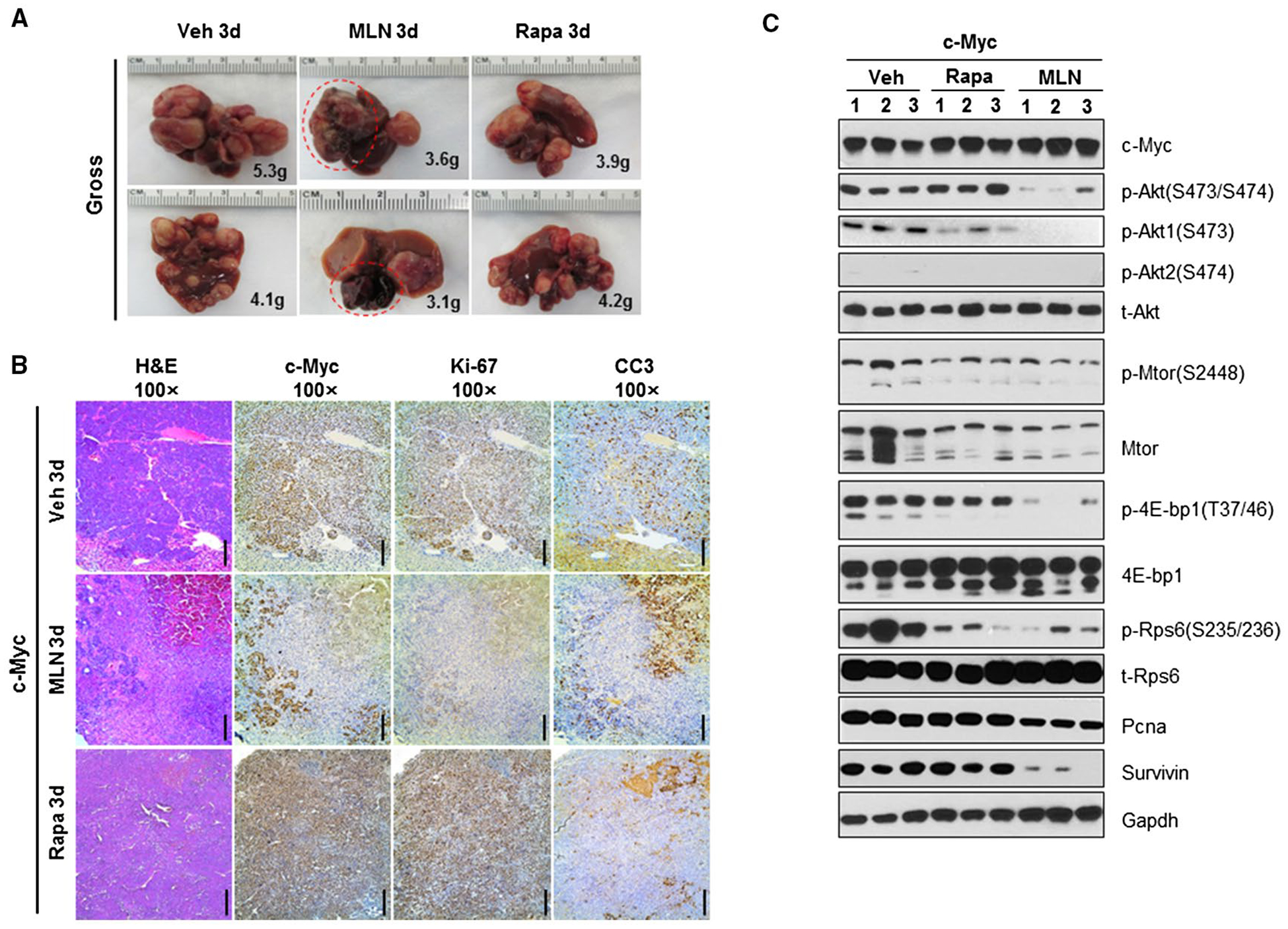
MLN0128 induces necrosis in progressed c-Myc HCC. (A) Gross images in vehicle-treated, rapamycin-treated, or MLN0128-treated FVB mice injected with the c-Myc constructs for 3 days. (B) Hematoxylin and eosin, c-Myc, Ki-67, and CC3 staining images in 3-day vehicle-treated or MLN0128-treated mouse livers injected with c-Myc. (C) Representative western blotting from FVB mice liver tissues and 3-day vehicle-treated, rapamycin-treated, or MLN0128-treated mouse livers injected with c-Myc. Magnifications ×100 (hematoxylin and eosin, c-Myc, cleaved caspase-3, and Ki-67), scale bar, 200 μm. Abbreviations: CC3, cleaved caspase-3; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; H&E, hematoxylin and eosin; MLN, MLN0128; Pcna, proliferating cell nuclear antigen; Rapa, rapamycin; t-, total; Veh, vehicle.
Importantly, 10 of 11 MLN0128-treated mice displayed few tumor liver lesions, whereas all pretreatment cohort mice exhibited numerous tumor lesions (Fig. 6E; Supporting Table S7), suggesting that MLN0128 induced the regression of most lesions. To further investigate this hypothesis, we treated tumor-bearing mice with vehicle, rapamycin, or MLN0128 for 3 days and harvested the mice (Fig. 7A; Supporting Table S8). We found that macroscopically, in MLN0128-treated mice, some lesions turned a dark color (Fig. 7A). Histologically, the lesions were shown to be replaced by large necrotic areas (Fig. 7B). Surrounding the necrotic regions, most tumor cells stained positive for cleaved caspase-3, while losing Ki-67 staining. The results are consistent with the fact that MLN0128 treatment led to tumor cell death, necrosis, and, eventually, tumor regression.
To further investigate the observed phenotype, we injected the mice with c-Myc/MCL1 constructs. MCL1 is a well-characterized antiapoptosis protein. Notably, when c-Myc/MCL1 mice were treated with vehicle, rapamycin, or MLN0128 and apoptosis was inhibited (by MCL1), MLN0128 was still able to reduce the tumor burden in mice, although at much lower efficiency (Supporting Fig. S9 and Table S9).
In summary, the present data indicate that pan-mTOR inhibition, but not partial mTORC1 suppression, is effective at inducing tumor regression in c-Myc HCC.
Discussion
c-Myc is a well-characterized oncogene for multiple tumor types, including HCC. In our recent investigation, we showed that mTORC1 induction in HCC developed in c-Myc mice.(22) In the present study, we found that mTORC2 is activated in c-Myc liver tumors, as indicated by elevated p-Akt(S473) expression. Using genetic approaches, we demonstrated that mTORC2 is essential for the initiation of c-Myc-driven hepatocarcinogenesis. The results add important data supporting the major role of mTORC2 in inducing liver tumor development and progression. Intriguingly, we discovered that Akt1, but not Akt2, is activated in c-Myc HCC. The precise mechanisms by which Akt1, but not Akt2, is induced by c-Myc are not clear. As activated phosphoinositide 3-kinase (PI3K) is a well-characterized upstream regulator of AKT, we treated two mouse c-Myc HCC cell lines with the PI3K inhibitor wortmannin. We found that wortmannin did not affect c-Myc HCC cell growth or p-Akt1 expression (Supporting Fig. S10A,B). Wortmannin treatment indeed decreased p-Akt2 expression in these cells (Supporting Fig. S10B). We also cultured the cells in serum-free medium. We found that it resulted in a decrease of HCC cell growth and p-Akt1 expression (Supporting Fig. S10C,D). These findings suggest that c-Myc-induced p-Akt1 activation might be triggered by extracellular signals, independent of PI3K. However, it remains to be determined whether the same mechanisms apply in vivo. Nonetheless, we found that AKT1 total proteins and mRNA levels were not affected by c-Myc modulation in mouse and human HCC cell lines, thus indicating a posttranscriptional regulation. Specifically, our data support a crucial role played by c-Myc in either favoring phosphorylation of AKT1 or suppressing the cellular inhibitors of AKT1 activity. Some of these inhibitors have been identified, including pleckstrin homology-like domain family A members 1–3 (PHLDA1–3), protein phosphatase 2A (PP2A), and pleckstrin homology domain leucine-rich repeat protein phosphatases (PHLPPs).(34) Whether c-Myc impairs the activity of PHLDA1–3, PP2A, and/or PHLPPs, thus inducing the unrestrained activation of AKT1, remains to be determined.
Using conditional Akt1 and Akt2 KO mice, we demonstrated that Akt1, but not Akt2, is required for c-Myc-driven hepatocarcinogenesis. In contrast, previous studies have shown that Akt2 is the major AKT isoform required for activated PI3K(12) or loss-of-Pten(13)-induced HCC formation in vivo. Together, our data provide solid evidence that different AKT isoforms may have distinct roles downstream of oncogenic signals and that they may play distinct roles in hepatocarcinogenesis. Importantly, a recent study revealed that combined deletion of Akt1 and Akt2 in the mouse liver led to spontaneous HCC formation due to liver damage and inflammation.(10) Thus, the use of pan-AKT inhibitors for cancer treatment might not be ideal and even dangerous. In contrast, isoform-specific AKT inhibitors could be developed and tested for cancer therapy. Mechanistically, our study suggests that Foxo1, rather than mTORC1, might be the major signaling cascade downstream of mTORC2/Akt1 in c-Myc-dependent hepatocarcinogenesis (Supporting Fig. S11). Indeed, Foxo1 has been implicated as the major signaling molecule in loss of Akt1/Akt2-driven HCC formation,(10) as well as Akt1/Akt2-dependent liver regeneration.(14) Clearly, additional experiments, which are beyond the scope of the present study, are required to further investigate the precise signaling cascades downstream of mTORC2/AKT during hepatocarcinogenesis and the precise role of FOXO1 in this process.
While multiple studies have investigated the therapeutic potential of dual mTORC1/2 inhibitors in HCC, all of these studies were limited to using HCC cell lines in vitro or in xenograft models.(35,36) It is well known that those cell line-based studies rarely translate into clinically relevant efficacies. Therefore, the use of genetic murine models is important to provide critical preclinical data to support the investigation of the drugs during cancer treatment. In the present study, we evaluated the therapeutic potential of rapamycin and the dual mTORC1/2 inhibitor MLN0128 in the c-Myc preclinical HCC model. We found that rapamycin had limited efficacy, similar to what has been reported in human HCC clinical studies.(18) In contrast, MLN0128 treatment led to profound tumor regression in c-Myc HCC. Mechanistically, MLN0128 effectively inhibited p-Rps6 and p-4Ebp1 downstream of mTORC1, as well as p-Akt(S473/S474) downstream of mTORC2 (Fig. 7C), leading to decreased tumor cell proliferation and increased apoptosis. Thus, our investigation suggests that dual mTORC1/2 inhibitors might be useful for the treatment of human HCCs with c-Myc amplification and/or overexpression. Currently, dual mTORC1/2 inhibitors are in clinical trials for advanced-stage solid tumors,(37) including HCC. For instance, CC-223, a dual mTORC1/2 inhibitor similar to MLN0128, is now being tested in a phase 2 clinical trial in HBV-positive advanced HCC subjects (NCT03591965). It remains to be determined whether these dual mTORC1/2 inhibitors are efficacious in clinics. However, it is important to note that most of these trials are conducted in an unselected patient population. In our study, we suggest that dual mTORC1/2 inhibitors may be effective in HCC patients with c-MYC amplification and/or overexpression. However, our investigation also suggests that the antitumor properties of MLN0128 were greatly limited if an antiapoptosis gene was coexpressed. In another study, it was shown that HCCs with mutations in tuberous sclerosis complexes 1 and 2 may be sensitive to mTOR inhibitors.(38) Altogether these studies support the need of establishing reliable biomarkers to select patients who may benefit from dual mTORC1/2 inhibition.
Supplementary Material
Acknowledgment:
We thank Dr. Dean W. Felsher of Stanford University for HCC3-4 and HCC4-4 mouse c-Myc mouse liver tumor cell lines and Dr. David Pearce from University of California, San Francisco, for Sgk3+/− mice.
Supported by grants from the National Institutes of Health (R01CA136606, R21CA198490, and P30DK026743), the National Natural Science Foundation of China (81860439), and scholarships from the China Scholarship Council (201408525080, 201606280273, 201706240075, and 201703170154).
Abbreviations:
- Cre
cyclization recombination
- CMV
cytomegalovirus
- 4EBP1
4E binding protein 1
- Foxo1
forkhead box o1
- HCC
hepatocellular carcinoma
- KO
knockout
- MCL1
myeloid cell leukemia 1
- mTOR
mammalian target of rapamycin
- mTORC2
mTOR complex 2
- p-
phosphorylated
- PI3K
phosphoinositide 3-kinase
- PIK3CA
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- RPS6
ribosomal protein S6
- SGK
serum/glucocorticoid-regulated kinase
- siAkt1
small interfering Akt1
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.30697/suppinfo.
REFERENCES
- 1).Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 2).El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? HEPATOLOGY 2014;60:1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–390. [DOI] [PubMed] [Google Scholar]
- 4).Greten TF, Sangro B. Targets for immunotherapy of liver cancer. J Hepatol 2018;68:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017;169:361–371. [DOI] [PubMed] [Google Scholar]
- 6).Xie J, Proud CG. Signaling crosstalk between the mTOR complexes. Translation (Austin) 2014;2:e28174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol 2017;45:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Gaubitz C, Prouteau M, Kusmider B, Loewith R. TORC2 structure and function. Trends Biochem Sci 2016;41:532–545. [DOI] [PubMed] [Google Scholar]
- 9).Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009;16:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, et al. Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell 2016;29:523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab 2009;10:405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Wang C, Che L, Hu J, Zhang S, Jiang L, Latte G, et al. Activated mutant forms of PIK3CA cooperate with RasV12 or c-Met to induce liver tumour formation in mice via AKT2/mTORC1 cascade. Liver Int 2016;36:1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA, et al. Expansion of hepatic tumor progenitor cells in Ptennull mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 2010;139:2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Pauta M, Rotllan N, Fernandez-Hernando A, Langhi C, Ribera J, Lu M, et al. Akt-mediated foxo1 inhibition is required for liver regeneration. HEPATOLOGY 2016;63:1660–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 2012;18:388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci 2015;36:124–135. [DOI] [PubMed] [Google Scholar]
- 17).Everolimus Hasskarl J.. Recent Results Cancer Res 2014;201:373–392. [DOI] [PubMed] [Google Scholar]
- 18).Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA 2014;312:57–67. [DOI] [PubMed] [Google Scholar]
- 19).Feldman ME, Shokat KM. New inhibitors of the PI3K-Akt-mTOR pathway: insights into mTOR signaling from a new generation of Tor kinase domain inhibitors (TORKinibs). Curr Top Microbiol Immunol 2010;347:241–262. [DOI] [PubMed] [Google Scholar]
- 20).Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 2010;17:249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Dang CV. MYC on the path to cancer. Cell 2012;149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Liu P, Ge M, Hu J, Li X, Che L, Sun K, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. HEPATOLOGY 2017;66:167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Chow EK, Fan LL, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. HEPATOLOGY 2012;56:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004;431:1112–1117. [DOI] [PubMed] [Google Scholar]
- 25).Mendez-Lucas A, Li X, Hu J, Che L, Song X, Jia J, et al. Glucose catabolism in liver tumors induced by c-MYC can be sustained by various PKM1/PKM2 ratios and pyruvate kinase activities. Cancer Res 2017;77:4355–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Wan M, Easton RM, Gleason CE, Monks BR, Ueki K, Kahn CR, et al. Loss of Akt1 in mice increases energy expenditure and protects against diet-induced obesity. Mol Cell Biol 2012;32:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Bell GI, Horita S, Karam JH. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 1984;33:176–183. [DOI] [PubMed] [Google Scholar]
- 28).Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012;11:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).McCormick JA, Feng Y, Dawson K, Behne MJ, Yu B, Wang J, et al. Targeted disruption of the protein kinase SGK3/CISK impairs postnatal hair follicle development. Mol Biol Cell 2004;15:4278–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Zhang S, Song X, Cao D, Xu Z, Fan B, Che L, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol 2017;67:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011;71:2286–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 35).Shao H, Gao C, Tang H, Zhang H, Roberts LR, Hylander BL, et al. Dual targeting of mTORC1/C2 complexes enhances histone deacetylase inhibitor-mediated anti-tumor efficacy in primary HCC cancer in vitro and in vivo. J Hepatol 2012;56:176–183. [DOI] [PubMed] [Google Scholar]
- 36).Chen BW, Chen W, Liang H, Liu H, Liang C, Zhi X, et al. Inhibition of mTORC2 induces cell-cycle arrest and enhances the cytotoxicity of doxorubicin by suppressing MDR1 expression in HCC cells. Mol Cancer Ther 2015;14:1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Tian T, Li X, Zhang J. mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int J Mol Sci 2019;20:E755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Ho DWH, Chan LK, Chiu YT, Xu IMJ, Poon RTP, Cheung TT, et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017;66:1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.