

Abstract
Type II epithelial-mesenchymal transition (EMT) plays a vital role in airway injury, repair, and remodeling. Triggered by growth factors, such as transforming growth factor beta (TGFβ), EMT induced a biological process that converts epithelial cells into secretory mesenchymal cells with a substantially increased production of extracellular matrix (ECM) proteins. Epithelial cells are not professional secretory cells and produce few ECM proteins under normal conditions. The molecular mechanism underlying the transformation of the protein factory and secretory machinery during EMT is significant because ECM secretion is central to the pathogenesis of airway remodeling. Here we report that type II EMT upregulates the protein N-glycosylation of ECMs. The mechanism study reveals that the substantial increase in synthesis of ECM proteins in EMT activates the inositol-requiring protein 1 (IRE1α)−X-box-binding protein 1 (XBP1) axis of the unfolded protein response (UPR) coupled to the hexosamine biosynthesis pathway (HBP). These two pathways coordinately up-regulate the protein N-glycosylation of ECM proteins and increase ER folding capacity and ER-associated degradation (ERAD), which improve ER protein homeostasis and protect transitioned cells from proteotoxicity. Inhibition of the alternative splicing of XBP1 or protein N-glycosylation blocks ECM protein secretion, indicating the XBP1-HBP plays a prominent role in regulating the secretion of ECM proteins in the mesenchymal transition. Our data suggest that the activation of XBP1-HBP pathways and elevation of protein N-glycosylation is an adaptive response to maintain protein quality control and facilitate the secretion of ECM proteins during the mesenchymal transition. The components of the XBP1-HBP pathways may be therapeutic targets to prevent airway remodeling.
Keywords: epithelial-mesenchymal transition, proteomics, N-glycosylation, extracellular matrix, secretome, unfolded protein response, hexosamine biosynthesis pathway
Graphical Abstract
INTRODUCTION
Airway remodeling is a structural alteration of the airways produced by epithelial injury and repair. Epithelial injury stimulates the release of growth factors, such as transforming growth factor (TGF)β to induce re-epithelialization, mucosal repair, and stem cell renewal.1,2 Triggered by TGFβ, airway epithelial cells undergo a global transcriptional reprogramming response known as epithelial-to-mesenchymal transition (EMT).3 During EMT, epithelial cells lose their adherens junctions, apical-basal polarity, reorganize their cytoskeleton, secrete extracellular matrix (ECM) proteins, and thereby transdifferentiate into motile, secretory mesenchymal cells. EMT is conceptually differentiated into type I, II, and III forms: Type I EMT occurs during normal embryonic development; Type II EMT occurs in normal (untransformed) cells; and Type III EMT is involved in the metastatic invasion of malignant epithelium.4 Although type II EMT plays a central role in normal tissue response to injury and tissue remodeling and repair,4 dysregulated (or persistent) EMT will cause disruption of the epithelial barrier, collagen over-production, and airway remodeling that plays a pathogenic role in asthma,5–7 chronic obstructive pulmonary disease (COPD),8 and lung fibrosis.9
Under normal conditions, airway epithelial cells produce few ECM proteins. Mesenchymal transition dramatically upregulates the synthesis of secretory ECM proteins, which sensitizes them to endoplasmic reticulum (ER) stress and activates the unfolded protein response (UPR).10 The UPR restores ER homeostasis by either increasing the protein folding capacity of the ER, reducing the influx of nascent proteins into the ER, and/or elevating ER-associated degradation (ERAD).11 Using an integrated genomic-proteomic approach, we recently reported that XBP1, a transcription regulator of UPR, is a potential upstream transcription regulator of EMT.12 However, the functional role, if any, of the XBP1 pathway in the initiation or maintenance of type II EMT remained elusive.
ECM proteins are frequently modified with N-glycosylation, a post-translational modification that plays a critical role in protein folding and function after secretion or cell surface presentation.13 Although it is known that N-glycosylation is dependent on the synthesis of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the sugar donor produced by the hexosamine biosynthetic pathway (HBP), the mechanisms controlling the activity of the HBP in mesenchymal cells and how HBP pathway regulates proteostasis and secretion of ECM proteins are unknown.
Here, we used a well-established cell model of type II EMT of airway epithelial cells12,14–18 and conducted an integrated omics analysis. We found that type II EMT upregulates protein N-glycosylation of ECM proteins through activation of the XBP1-HBP pathways. Activation of XBP1-HBP pathway and elevated protein N-glycosylation induced distinct proteostasis mechanisms in mesenchymal cells. Our study provides a novel mechanism on how EMT cells maintain proteostasis and regulate ECM production and also a new therapeutic approach for airway modeling diseases through targeting the XBP1-HBP pathway.
EXPERIMENTAL SECTION
Cell Culture
An immortalized primary human small airway epithelial cell (hSAEC) line was previously described.12,14–18 The immortalized hSAECs were grown in SAGM small airway epithelial cell growth medium (Lonza, Walkersville, MD) in a humidified atmosphere of 5% CO2. For induction of EMT, hSAECs were TGFβ stimulated for 14 d (10 ng/mL, PeproTech, Rocky Hill, NJ).14
Quantitative Real-Time Reverse Transcription-PCR (RT-qPCR)
Total RNA was extracted using acid guanidium phenol extraction (Tri reagent; Sigma). For gene expression analyses, 1 μg of RNA was reverse transcribed using SuperScript III in a 2-μL reaction mixture. One μL of cDNA product was amplified in a 2 μL reaction mixture containing 10 μL of SYBR green Supermix (Bio-Rad) and the final concentration of 0.4 μM (each) forward and reverse gene-specific primers (Supplemental Table S1). The reaction mixtures were aliquoted into a Bio-Rad 96-well clear PCR plate, and the plate was sealed with Bio-Rad Microseal B film. The plates were denatured for 90 s at 95 °C and then subjected to 40 cycles of 15 s at 94 °C, 60 s at 60 °C, and 1 min at 72 °C in a CFX96 Real-Time PCR Detection System (Bio-Rad). PCR products were subjected to melting curve analysis to ensure that a single amplification product was produced. Quantification of relative changes in gene expression was done using the threshold cycle (ΔΔCT) method. In brief, the ΔCT value was calculated [normalized to DNA polymerase beta (PolB)] for each sample by using the equation ΔCT = CT (target gene) − CT (PPIA). Next, the ΔΔCT was calculated by using the equation ΔΔCT = ΔCT (experimental sample) − ΔCT (control sample). Finally, the fold differences between the experimental and control samples were calculated using the formula 2−ΔΔCT.
Western Immunoblot
The cells were lysed in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Sigma P8340)]. Protein concentration was determined by bicinchoninic acid (BCA, Pierce, Thermo Scientific) and 10 μg were dissolved into SDS loading buffer (with 5% βME) and fractionated on 4−15% Mini-protean TGX gels (BioRad, Hercules CA, USA) in 1× Tris Glycine SDS (TGS) 1× running buffer at room temperature. Proteins were electro-transferred to nitrocellulose membrane (BioRad, Hercules CA, USA) in 1× TGS buffer-methanol (20%) buffer. The blots were blocked with 3% BSA in TBST (Tris-buffered saline, 0.1% Tween 20) for 1 h and incubated overnight at 4 °C in primary antibodies. Antibodies were: recombinant anti-GFPT2 antibody [EPR19095] (Abcam ab190966), recombinant anti-GFPT1 antibody [EPR4854] (Abcam ab125069), and antibeta Actin antibody [mAbcam 8226] (Abcam ab8226). Secondary antibodies were IRDye 800CW goat antirabbit IgG secondary antibody (Li-Cor 926−32211) and IRDye 680RD goat antimouse IgG secondary antibody (Li-Cor 926−68070).
Lectin Staining and Confocal Immunofluorescence Microscopy
For confocal fluorescence microscopy, 25 mm round microscope cover glasses (Fisher Scientific, Pittsburgh, PA) were first precoated with sterilized collagen solution (Roche Applied Science) in a 6-well culture plate. hSAECs or EMT-hSAECs were plated over cover glass 2 days before the experiment. The cells were then fixed with 4% paraformaldehyde in PBS for 20 min and incubated with 0.1 M ammonium chloride for 10 min. Afterward, cells were permeabilized with 0.5% Triton X-100 in PBS, followed by incubation in blocking buffer (5% goat serum, 0.1% IGEPAL CA-630, 0.05% NaN3, and 1% BSA) for 1 h, and then incubated with WGA-FITC (20 μg/mL, Sigma, St. Louis, MO) for 1 h at room temperature. After washing, cells were imaged at the wavelength of 488 nm. Nuclei were counterstained with DAPI.
Immunostaining of Protein Markers of Epithelial Cells and Mesenchymal Cells
The cells grown on the cover glasses were fixed with methanol for 5 min and permeabilized with 0.5% Triton X-100 for 5 min and then blocked with 1% BSA in TBST for 1 h. Afterward, the cells were incubated overnight at 4 °C with recombinant Anti-E-Cadherin (CDH1) antibody [EP700Y] (Alexa Fluor 647, Abcam ab194982) at 1/100 dilution. The cover glasses were washed with TBST three time and then blocked with 1% BSA in TBST for 1 h. Afterward, the cells were incubated overnight at 4 °C with anti-Vimentin antibody [V9] (Alexa Fluor 488, Abcam ab195877) at 1/250 dilution. Nuclei were counterstained with DAPI. After washing, cells were imaged at the wavelength of 405, 488, and 633 nm with Zeiss LSM 880 Confocal Laser Scanning Microscope.
Cell Viability Assay
Relative cell viabilities were determined using Cell Counting Kit-8 (CCK-8, Dojindo, Rockville, MD). In general, hSAECs and EMT-hSAECs are implanted into a 48 well plate and incubated at 37 °C overnight. Three replicates were used. On the following day, both cells are treated with Tunicamycin at a concentration of 0, 2.5, 10, 25, 50 μg/mL for 24 h. Add 25 μL of the CCK-8 solution to each well of the plate and incubate the plate for 3 h in the incubator. Measure the absorbance at 450 nm using a microplate reader. Calibration was done by implanting the cells with a relative cell density of 12.5%, 25%, 50%, 100% and 150% of the cells with experimental condition.
Proteasome Activity Measurement
Proteasome chymotryptic activities (26S and 20S) were measured according to the standard procedure (Abcam, Cambridge, MA) in cell lysis of hSAECs and EMT-hSAECs as the rate of hydrolysis of the fluorogenic peptide suc-LLVY-AMC. Briefly, cells were lysed in 0.5% of NP-40 and incubated with 50 μM suc-LLVY-AMC of in the assay buffer (26S: 50 mM Tris−HCl, pH 8, 5 mM MgCl2, 5 mM ATP, 1 mM DTT, and 10% glycerol; 20S: 50 mM Tris−HCl, pH 8, 5 mM MgCl2, 1 mM DTT, 10% glycerol and 0.02% SDS) in a total volume of 100 μL. Samples were prepared using three replicates. AMC fluorescence was read on a microplate reader using 350 nm excitation and 440 nm emission filters using free AMC as a standard every 15 min for 1.5 h at 37 °C.
siRNA-Mediated GFPT1 Silencing
Commercially obtained Silencer Select GFPT1 validated siRNA (Ambion, Thermo Scientific, Catalog number 4390824) and Negative control siRNA (Ambion, Thermo Scientific, Catalog number 4390843) were reverse transfected at a 50 nM concentration using TransIT-siQUEST transfection reagent (Mirus Bio Corp.). Forty-eight hours later, cells were treated with TGFβ for designated time.
Quantification of the Cellular UDP-GlcNac
About 1 × 106 cells were homogenized in 0.7 M perchloric acid, the protein pellets were removed by centrifuging at 15 000 rpm.19 The resulting supernatants were neutralized with 2 M sodium carbonate. The resulting nucleotide-sugars were desalted using TopTip Carbon (Hypercarb), eluted with 80% ACN and dried with speedvac. Briefly, the desalted nucleotide-sugars were resuspended in 0.01 M NH4OH-40% ACN and directly analyzed by LC-SRM-MS. The SRM parameters for UDP-GlcNAc are listed in Supplemental Table S2. LC-SRM-MS analysis was performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with a nanospray source (Thermo Scientific, San Jose, CA). The online chromatography was performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA). An aliquot of 2 μL of each of the nucleotide-sugar extract was injected on self-packed Hypercarb column (200 μm × 5 cm) at a flow rate of 5 μL/min with 50% buffer A (0.01 M NH4OH−H2O) and 50% buffer B (0.01 M NH4OH−ACN). The TSQ Vantage was operated in negative, high-resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da fwhm. All acquisition methods used the following parameters: 2100 V ion spray voltage, a 275 °C ion transferring tube temperature, a collision-activated dissociation pressure at 1.5 mTorr, and the S-lens voltage used the values in S-lens table generated during MS calibration.
Protein Extraction and Trypsin Digestion
The cells were washed three times with PBS solution before the harvest. The cells were collected and transferred to a conical tube for centrifugation. Cells were pelleted at 150g for 4 min and resuspended in 1.0 mL of Trizol reagent (Invitrogen, Carlsbad, CA). The cells were lysed thoroughly by repetitive pipetting. The proteins which are free of nucleic acids were extracted from this cell lysate using Trizol reagent according to the manufacturer’s instructions. The protein pellet was resuspended in 50 μL of 8 M Guanidine HCl. The protein concentration was measured using Bradford assay. One milligram of proteins from each sample was processed for digestion. The proteins were first reduced with 10 mM DTT at room temperature for 30 min, followed by alkylation with 30 mM iodoacetamide at room temperature for 2 h. Then, the sample was diluted with 50 μL of 100 mM triethylammonium bicarbonate (pH 8.0). An aliquot of Lys-C/Trypsin solution (Promega, Madison, WI) was added to each sample at the 100:1 protein: enzyme ratio. The samples were incubated at 37 °C for overnight, and the solutions were further diluted with 400 μL of 100 μM of triethylammonium. An aliquot of trypsin solution (Promega, Madison, WI) was added to each sample at the 50:1 protein: enzyme ratio. The samples were incubated at 37 °C for 16 h. Ten μL of 10% trifluoroacetic acid was added to each sample to stop the trypsin digestion. Tryptic peptides were desalted on reversed phase tC18 SepPak columns (Waters, Milford, MA) and evaporated to dryness in a vacuum concentrator.
Enrichment of N-Glycosylation
N-Glycosylation enrichment was done according to the N-glyco-FASP protocol of Wisniewski et al.20 Briefly, 100 μg peptides were vacuum-dried and dissolved in the binding buffer (1 mM CaCl2, 1 mM MnCl2, 0.5 M NaCl in 20 mM Tris-HCl, pH = 7.3). Samples were heated for 10 min at 95 °C and cooled down to room temperature. Lectin mixture (90 μg ConA, 90 μg WGA, 71.5 μg RCA120) was added to the sample and incubated at room temperature for 1 h. Samples were transferred to YM-30 filter units (Microcon, Millipore, Burlington, MA), and the unbound peptides were eluted by centrifugation at 10 000g for 10 min. The captured peptides were washed four times with 200 μL of binding buffer and twice with 50 μL of 40 mM NH4HCO3 in H218O. Peptides were incubated with 2 μL N-glycosidase F (Roche) in 40 μL of 40 mM NH4HCO3 in H218O at 37 °C for 3 h. The released deglycosylated peptides were eluted with 40 mM NH4HCO3. The eluted peptides were dried by speedvac and acidified with 0.01% trifluoroacetic acid. Peptides were desalted on Ziptip C18 (Waters). Three independent experiments were conducted and each sample was analyzed by LC−MS/MS twice.
Inhibition of Protein N-Glycosylation with Tunicamycin (TM) and Secretome Analysis
hSAECs and EMT-hSAECs (14 days with TGFβ treatment) were treated with 10 μM of tunicamycin for 24 h. The untreated hSAECs and EMT-hSAECs were used as controls. About 10 mL of cell culture medium (CM) were collected and processed as described previously.21 Briefly, the CM was centrifuged at 2000g for 10 min to remove dead cells and cell debris. The supernatant was added into a 3K filter unit (Millipore, Billerica, MA) and centrifuged at 14 000g for 15 min. 400 μL of 8 M urea was added into the filter unit and centrifuged at 14 000g for 15 min, and repeat this step once. The solution remained in the filter device was collected for protein digestion. Proteins were reduced with 10 mM dithiothreitol for 30 min followed by alkylation with 30 mM iodoacetamide for 60 min in the dark. The sample was diluted one time with 50 mM ammonium bicarbonate. Proteins were digested with 1.0 μg LysC-trypsin (Promega) for 12 h at 37 °C and then diluted with 50 mM ammonium bicarbonate four times. The proteins were further digested with 1.0 μg trypsin (Promega) for 16 h at 37 °C. The digestion was stopped with 0.5% trifluoroacetic acid. Peptides were desalted on reversed phase SepPak C18 cartridge (Waters). Peptides were eluted using 80% acetonitrile. The eluate was dried in a SpeedVac and the peptides were acidified with 2% acetonitrile−0.1% trifluoroacetic acid. Each experimental group has three biological replicates and each sample was analyzed by LC−MS/MS twice.
NanoLC−MS/MS Analysis
The desalted peptides were reconstituted in 20 μL 4% ACN/0.1% formic acid. All peptide samples were separated on an online nanoflow Easy nLC1000 UHPLC system (Thermo Scientific) and analyzed on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA). About 5 μg of sample was injected onto a capillary peptide trap column (Acclaim Pepmap 100, 75 μm × 2 cm, C18, 3 μm, 100 Å, Thermo Scientific). After sample injection, the peptides were separated on a 25 cm UHPLC reversed phase column (Acclaim Pepmap 100, 75 μm × 25 cm, C18, 2 μm, 100 Å, Thermo Scientific) at a flow rate of 300 nL/min. A 4-h linear gradient from 2% solvent A (0.1% formic acid in water) to 35% solvent B (0.1% formic acid in acetonitrile) was used for each LC−MS/MS run. The data-dependent acquisition was performed using the Xcalibur 2.3 software in positive ion mode at a spray voltage of 2.1 kV. Survey spectra were acquired in the Orbitrap with a resolution of 70 000, the maximum injection time of 80 ms, an automatic gain control (AGC) of 1 × 106, and a mass range from 400 to 1400 m/z. The top 15 ions in each survey scan were selected for higher-energy collisional dissociation scans with a resolution of 17 500. For all higher-energy collisional dissociation scans, collision energy was set to 30, the maximum inject time was 60 ms, and the AGC was 1 × 105. Ions selected for MS/MS were dynamically excluded for 30 s after fragmentation.
Protein Identification
Mass spectra were analyzed using MaxQuant software version 1.5.2.8 using the Andromeda search engine.22 The initial maximum allowed mass deviation was set to 10 ppm for monoisotopic precursor ions and 0.5 Da for MS/MS peaks. Enzyme specificity was set to trypsin, defined as C-terminal to arginine and lysine excluding proline, and a maximum of two missed cleavages were allowed. Carbamidomethylcysteine was set as a fixed modification and methionine oxidation as variable modifications. For identification and quantification of protein N-glycosylation, deamination with 18O label on asparagine was set as variable modification. The spectra were searched by the Andromeda search engine against the Human SWISSPORT sequence database (containing 20 193 human protein entries) combined with 248 common contaminants and concatenated with the reversed versions of all sequences. Quantifications were performed with the label-free algorithms in Maxquant.23 The “match between runs” feature of MaxQuant was used to transfer identifications to other LC−MS/MS runs based on their masses and retention time (maximum deviation 0.7 min), and this was also used in quantification experiments. The detailed parameters for label free LC−MS/MS quantification and MaxQuant data analysis were listed in Supplemental Table S3. We required at least one “razor peptide” for quantification. The required false positive rate for identification was set to 1% at the peptide level and 1% at the protein level and the minimum required peptide length was set to 6 amino acids. Contaminants, reverse identification, and proteins only identified by modified peptides were excluded from further data analysis.
Protein Quantification and Statistical Analysis
We used Perseus platform24 to analyze the Maxquant output, including statistics, Hierarchical clustering, principal component analysis (PCA), and 2D annotation analysis.25 Reversed identifications and proteins identified only by site modification were strictly excluded from further analysis. After filtering (2 valid values in at least one group), remaining missing values were imputed from a normal distribution (width: 0.3 of standard deviation; down shift: 1.8 of standard deviation). Student’s t test with Permutation-based FDR was performed to identify the significantly differentially expressed proteins. The unsupervised hierarchical clustering and heat map were based on protein expression. The rows of the heat map indicate the proteins, and the columns indicate the samples. The log2 ratios of each protein were z-score normalized for each row. Hierarchical clustering of the z-normalized log2 ratio was performed using Euclidean distances between means. The number of clusters was set as 300. Genome ontology enrichment analysis of molecular functions and biological function in differentially expressed proteins was using Panther (http://pantherdb.org/). This classification uses an evolutionary framework to infer protein functions in a species-independent manner.26 The resulting p-values were adjusted with Bonferroni correction for multiple testing. The significant hits are those with the adjusted p-value better than 0.05.
Stable Isotope Dilution-Selected Reaction Monitoring-MS
The SID-SRM-MS assays of selected proteins were developed as described previously.27 For each targeted proteins, two or three peptides were initially selected, and then the sensitivity and selectivity of these were experimentally evaluated as described previously. The peptide with best sensitivity and selectivity was selected as the surrogate for that protein. For each peptide, 3−5 SRM transitions were monitored. The signature peptides and SRM parameters are listed in Supplemental Table S2. The peptides were chemically synthesized incorporating isotopically labeled [13C615N4] arginine or [13C615N2] lysine to a 99% isotopic enrichment (Thermo Scientific, San Jose, CA). The amount of stable isotope labeled standard (SIS) peptides was determined by amino acid analysis. The proteins were trypsin digested on the beads as described above. Each experimental group has at least two biological replicates. The tryptic digests were then reconstituted in 30 μL of 5% formic acid−0.01% TFA. An aliquot of 10 μL of 50 fmol/μL diluted SIS peptides was added to each tryptic digest. These samples were desalted with a ZipTip C18 cartridge. The peptides were eluted with 80% ACN and dried. The peptides were reconstituted in 30 μL of 5% formic acid-0.01% TFA and were directly analyzed by liquid chromatography (LC)-SRM-MS. LC-SRM-MS analysis was performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with a nanospray source (Thermo Scientific, San Jose, CA). About 8−10 targeted proteins were analyzed in a single LC-SRM run. The online chromatography was performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA). An aliquot of 10 μL of each of the tryptic digests was injected on a C18 reverse-phase nano-HPLC column (PicoFrit, 75 μm × 10 cm; tip ID 15 μm) at a flow rate of 500 nL/min with a 20 min 98% A, followed by a 15 min linear gradient from 2 to 30% mobile phase B (0.1% formic acid−90% acetonitrile) in mobile phase A (0.1% formic acid). The TSQ Vantage was operated in high-resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da full-width half maximum (fwhm). All acquisition methods used the following parameters: 2100 V ion spray voltage, a 275 °C ion transferring tube temperature, a collision-activated dissociation pressure at 1.5 mTorr, and the S-lens voltage used the values in S-lens table generated during MS calibration.
All SRM data were manually inspected to ensure peak detection and accurate integration. The chromatographic retention time and the relative product ion intensities of the analyte peptides were compared to those of the stable isotope labeled standard (SIS) peptides. The variation of the retention time between the analyte peptides and their SIS counterparts should be within 0.05 min, and the difference in the relative product ion intensities of the analyte peptides and SIS peptides were below 20%. The peak areas in the extract ion chromatography of the native and SIS version of each signature peptide were integrated using Xcalibur 2.1. The default values for noise percentage and baseline subtraction window were used. The ratio between the peak area of native and SIS version of each peptide was calculated. Student’s t test was performed to determine if the changes in the protein expression was statistically significant.
RESULTS
EMT Activates Protein N-Glycosylation and Unfolded Protein Response
We have established an in vitro model of TGFβ-induced type II EMT of untransformed human airway epithelial cells.12,14–16,18 This model exhibits characteristic, time-dependent mesenchymal transition defined by microscopic imaging, genomic and proteomic analyses. Confocal immunofluorescence assays demonstrate that TGFβ stimulation inhibits the expression of the differentiation marker Epithelial E-Cadherin (CDH1), upregulates stress fiber formation, mesenchymal intermediate filament proteins (VIM) (Supplemental Figure S1). Moreover, the genomic signature distinguishing the type II EMT from a type III EMT in transformed human airway epithelial cells was demonstrated by next-generation sequencing, validating this as a well-characterized model of type II EMT.17,28
In this study, we conducted side-by-side quantitative profiles of the proteome and transcriptome of hSAECs undergoing the TGFβ-induced mesenchymal transition.12,14 The integrated bioinformatics analysis of the transcriptome and proteome profiles with a 2D annotation enrichment algorithm25 reveals 108 GO annotations and KEGG pathways that were significantly changed by mesenchymal transition (Supplemental Table S4). The upper right quadrant in Figure 1A contains annotation terms were up-regulated on the protein level as well as the mRNA levels. We found that the pathways involved in ECM disassembly, collagen structure, lamellipodia formation, and focal adhesion were up-regulated in EMT, whereas desmosomal cell adhesion components were down-regulated, a finding consistent with our previous observation of the loss of cell polarity and increased mobility in the EMT-hSAECs.12,14 The boxplots of the protein and RNA expression of ECM proteins (Figure 1B) and desmosomal proteins (Figure 1C) are shown. In this 2D analysis, we found that protein N-linked glycosylation via asparagine in EMT was up-regulated (Figure 1A–C), which was confirmed by lectin staining of glycoproteins. The confocal imaging in Figure 1D shows that the lectin staining in EMT-hSAECs display a perinuclear and cytoplasmic ER-like pattern, indicating that the abundance of intracellular glycoproteins was significantly increased in EMT cells. Interestingly, the 2D annotation analysis also reveals one KEGG pathway: activation of signaling protein involved in the UPR was remarkably up-regulated by the mesenchymal transition at both the mRNA and protein levels (Figure 1A–C). Together, these data suggest that TGFβ-induced EMT increases the production of ECM proteins, activates the UPR, and up-regulates N-glycosylation.
Figure 1.

TGFβ induced EMT activates unfolded proteins response and protein N-glycosylation. hSAECs were treated with TGFβ for 14 d to induce stable mesenchymal transition. (A) 2D annotation enrichment based on the protein and mRNA ratios (the proteome and transcriptome data used in the figure were published previously by our group1,2). (B) Box-plots of mRNA expression of the entries within representative categories identified that were up-regulated in EMT state (e.g., extracellular matrix organization, unfolded protein response, and protein N-linked glycosylation) and down-regulation in EMT (e.g., desmosome). (C) Box-plots of the protein expression of the entries within representative categories identified as significantly altered by the 2D annotation enrichment analysis. (D) Lectin staining of glycoproteins in hSAECs and EMT-hSAECs. The fixed hSAECs (left) and EMT-hSAECs (right) were stained with DAPI for nuclei (blue) and lectin for glycoproteins (green) (magnified ×63).
EMT Alters N-Glycosylation of Extracellular Matrix Proteins
We conducted a proteomics analysis to identify which protein N-glycosylation sites were regulated by TGFβ-induced type II EMT. The multiscatter plots of the MS intensity of identified N-glycosylated peptides (six replicates for each sample) are shown in Supplemental Figure S2A. The Pearson correlation of MS intensities among the replicates ranges from 0.72 to 0.86, suggesting that the quantification was robust and reproducible. We quantified 329 N-glycosylated peptides with the N-!P-[SI T]-!P (where !P is not proline) consensus motif (Supplemental Table S5). Among them, 81 N-glycosylation sites were up-regulated, and 31 were down-regulated by EMT (t test with Permutation-based FDR 0.05) (Figure 2A). GO annotation enrichment analysis of up-regulated N-glycoproteins with Panther29 found that the top GO Molecular Functions (GOMF) were related to ECM, such as fibronectin binding, collagen binding, and ECM binding (Figure 2B). For example, the N-glycosylation of FN1 Asn430 and Asn528 was up-regulated by over 900-fold in response to TGFβ. This region is critical for collagen binding.30 The magnitude of changes in the N-glycosylation of these ECM proteins was much higher than the differences in their protein expression (Figure 2C), indicating that the upregulation of protein N-glycosylation in TGFβ treated cells was primarily caused by the N-glycosylation pathway, not by the small changes in protein expression. GOMF analysis of down-regulated N-glycoproteins revealed that proteins with cation transmembrane transporter activity and O-glycosyl compounds hydrolase activity were enriched (Supplemental Figure S2B).
Figure 2.

TGFβ induced EMT alters protein N-glycosylation and activation of HBP pathway. The hSAECs were treated with TGFβ for 14 days. (A) Volcano plot for differentially abundant N-glycosylated peptides. Shown is a volcano plot of the differential protein expression. Y-axis, −log10 transformed p-value of two sample; X-axis is the log2 fold change in abundance (EMT-hSAECs relative to control hSAECs). The solid red circles are N-glycosylation sites up-regulated in EMT; solid green circles are N-glycosylation sites down-regulated in EMT; and the black circles, the difference in abundance is not statistically significant (Student’s t test, Permutation-based FDR 5%). (B) GO molecular function enrichment analysis for the proteins which N-glycosylation levels were up-regulated by EMT (p-value <0.05 with Bonferroni correction for multiple testing). Each annotation is displayed by fold enrichment (bar) and p-value (scatter plot). (C) Fold change of protein expression of ECM proteins and fold change of protein N-glycosylation of ECM proteins (the fold change of protein expression used in the figure was published previously by our group).2 (D) RNA expression of HBP enzymes measured by RNA-Seq. The raw RNA-seq data were published previously.1 (E) Protein expression of HBP enzymes measured by SID-SRM-MS. The protein expression was normalized by the expression of house-keeping protein cyclophilin A. (F). The intracellular level of UDP-GlcNAc in hSAECs and EMT-hSAECs. **, p-value of t test is below 0.05; ***, p-value of t test is below 0.001.
EMT Activates the Hexosamine Biosynthesis Pathway (HBP)
The upregulation of protein N-glycosylation in EMT led us to investigate a possible activation of HBP in mesenchymal transition. In our proteomics and RNA-seq studies, we found that the key enzymes Glutamine-fructose-6-phosphate transaminase (GFPT)-1, −2, Glucosamine-phosphate N-acetyltransferase (GNPNAT)1, and phosphoglucomutase (PGM3) were up-regulated in EMT state12,14 (Figure 2D). We confirmed this finding with SID-SRM-MS (Figure 2E) and Western blot (Supplemental Figure S2C). The final product of HBP pathway, UDP-GlcNAc, was found to be upregulated in EMT state as well (p-value = 0.007, t test) (Figure 2F). These results indicate that the HBP is activated in mesenchymal transition.
N-Glycosylation Is Essential for Secretion of Extracellular Matrix Proteins
The increased secretion and elevated level of N-glycosylation of ECM proteins led us to examine if the secretion of ECM proteins in type II EMT relied on the protein N-glycosylation. We conducted a secretome analysis of hSAECs and EMT-hSAECs treated with tunicamycin (TM),31 a protein N-glycosylation inhibitor. We quantified 1534 secreted proteins (Supplemental Table S6). The multiscatter plots of the MS intensity of proteins (six replicates for each sample) are shown in Supplemental Figure S3. The Pearson correlation of MS intensities among the replicates ranges from 0.84 to 0.97, suggesting that the quantification was robust and reproducible. We identified 495 proteins whose secretion was significantly changed by either EMT or TM, or both (t test, Permutation-based FDR 1%). Unsupervised hierarchical clustering of these 495 proteins identified four clusters (Figure 3A). Cluster 2 contains 101 proteins whose secretion was significantly upregulated by EMT and blocked by the inhibition of protein N-glycosylation. Unlike mesenchymal cells, the secretion of these proteins from hSAEC control cells was less affected by TM. The GOMF analysis of these proteins shows that the annotations related to ECM organization are enriched (Figure 3B). The abundance of some ECM proteins in the conditioned medium is displayed in Figure 3C. It showed that blocking N-glycosylation with TM inhibited ECM protein secretion in mesenchymal cells, an effect that we did not observe in hSAECs. Together, these data indicate that the secretion of ECM proteins in mesenchymal cells is dependent on protein N-glycosylation.
Figure 3.

N-Glycosylation is essential for secretion of extracellular matrix proteins. The hSAECs and EMT-hSAECs were treated with tunicamycin for 24 h. The secretome of hSAECs and EMT-hSAECs ± TM were analyzed by LC−MS/MS. (A) Unsupervised hierarchical clustering of differentially abundant secretory proteins. (B) GO molecular function enrichment analysis of the proteins which secretion was up-regulated by EMT and inhibited by TM (Cluster 2 in Figure 3A) (p-value <0.05 with Bonferroni correction for multiple testing). Each annotation is displayed by fold enrichment (bar) and p-value (scatter plot). (C) The abundance of ECM proteins in the cell culture medium of hSAECs and EMT-hSAECs ± TM. (D) The expression of GFPT1. The level of GFPT1 was knockdown with siRNA; nonspecific siRNA was used as negative control. (a) t test p-value <0.05 relative to hSAEC control + nonspecific siRNA; (b) t test p-value <0.05 in the comparison between GFPT1 knockdown and nonspecific siRNA on the same duration of TGFβ treatment. (E) The abundance of ECM proteins in the cell culture medium of hSAECs treated with GFPT1 siRNA and negative control siRNA during the time course of TGFβ treatment. (F) The cell viability analysis of hSAECs and EMT-hSAECs in response to tunicamycin. (G) The cell viability analysis of primary hSAECs and EMT-hSAECs in response to tunicamycin **, p-value of t test is below 0.05; ***, p-value of t test is below 0.001. TM, tunicamycin.
Next, we silenced GFPT1 expression, the rate-limiting enzyme of HBP, and examined how the knockdown of HBP pathway affected the secretion of ECM proteins. As shown in Figure 3D, about 37−68% of GFPT1 protein was specifically silenced by siRNA. We then measured the abundance of ECM proteins (FN1, Sparc, and Col4A1) over a time course of TGFβ stimulation. Compared to the cells treated with nonspecific siRNA, the secretion of ECM proteins was reduced by GFPT1 knockdown (Figure 3E), suggesting that the secretion of ECM proteins were dependent on the HBP pathway.
Conversely, we investigated if GlcNAc supplementation affects the secretion of ECM proteins during the mesenchymal transition. We found that GlcNAc supplement did not change the gene expression of the core EMT transcription regulators such as SNAIL1, TWIST1, and ZEB1. However, the gene and protein expression of EMT markers (FN1 and VIM) were slightly downregulated (Supplemental Figure S4A). Probably due to the reduced production of ECM proteins, the level of ER chaperone proteins PDIA6 and P4HB in the cell lysate were decreased in the cells provided with GlcNAc supplementation (Supplemental Figure S4B). Strikingly, the secretion of FN1 was increased by GlcNAc, despite the reduced mRNA expression, suggesting that exogenous activation of HBP pathway promoted ECM protein secretion (Supplemental Figure S4C).
Finally, we investigated whether the survival of mesenchymal cells was dependent on protein N-glycosylation. We measured the cell viability of mesenchymal cells vs. that of hSAECs in response to varying concentrations of TM for 24 h. As shown in Figure 3F, the cell viability of mesenchymal cells decreased with TM treatment (IC50 = 2.5 μg/mL), as compared to the hSAECs control (IC50 = 40 μg/mL). These data suggest that the survival of mesenchymal cells is more dependent on protein N-glycosylation relative to epithelial cells. We also examined if we would have the same observation in primary human SAECs isolated from normal lung. We treated primary SAECs with TGFβ and induced EMT, and then measured the cell viability in response to varying concentration of TM for 24 h. As shown in Figure 3G, the mesenchymal primary SAECs are more susceptible to TM treatment relative to SAECs, a finding consistent with that of the telomerase-immortalized hSAECs.
EMT Activates HBP Pathway via XBP1
Next, we examined the molecular mechanism underlying the upregulation of HBP pathway. In previous reports, XBP1 was identified as a transcription regulator of HBP pathway.32,33 We recently reported that XBP1 is an upstream transcription regulator of EMT with the highest activation score (z score 5.434, a p-value of overlap 8.54 × 10−26).12 The gene and protein expression of XBP1 were upregulated in EMT (Figure 4A,B). The IRE1α-XBP1s arm of UPR regulates the genes that are involved in protein folding, such as protein disulfide isomerases (PDIs). We found that PDIs were also significantly up-regulated in EMT (Figure 4C). These data confirmed the finding obtained from 2D annotation analysis described above (Figure 1A,B).
Figure 4.

XBP1-HBP in regulating protein N-glycosylation in type II EMT. hSAECs were treated with TGFβ for 14 d. (A) mRNA expression of spliced XBP1s. (B) SID-SRM-MS quantification of expression of XBP1 protein. (C) EMT up-regulated the expression of P4HB, PDIA4, and PDIA6. (D−H) The hSAECs were treated with TGFβ and/or IRE1α inhibitor, STF-083010, for 14 days. (D) The intracellular level of UDP-GlcNAc in the hSAECs and EMT-hSAECs ± STF-083010. (E) Confocal imaging of lectin staining of the glycoproteins in the hSAECs and EMT-hSAECs ± STF-083010; upper panel, lectin staining for glycoproteins; lower panel, the merge of lectin staining (green) and DAPI staining for nuclei (blue). (magnified ×63). (F) Principle component analysis of these 463 proteins (shown in Figure 4G) based on their abundance in the conditional medium. (G) Unsupervised hierarchical clustering of differentially abundant secretory proteins in the cell culture medium of the hSAECs and EMT-hSAECs ± STF-083010. (H) The abundance of ECM proteins in the cell culture medium of the hSAECs and EMT-hSAECs ± STF-083010. **, p-value of t test is below 0.05; ***, p-value of t test is below 0.001.
We tested if UPR-XBP1 and HBP were functionally linked in mesenchymal cells, and if so, whether it played a coordinating role in regulating the ECM secretion. We treated hSAECs with TGFβ for 14 days in the presence or absence of STF-083010, an imine-based small molecule that directly inhibits the IRE1α endonuclease activity and XBP1 splicing.34 We found that inhibition of XBP1 splicing significantly reduced the level of UDP-GlcNAc (p-value 8.1 × 10−6) in EMT cells, but not in hSAEC control cells (Figure 4D). Moreover, STF-083010 did not affect the lectin staining level in normal hSAECs, but significantly reduced the level of lectin staining in EMT-hSAECs (Figure 4E). These results indicate that the inhibition of XBP1 splicing blocks the activation of the HBP and the up-regulation of protein N-glycosylation in EMT. Finally, we analyzed the secretome hSAECs and EMT-hSAECs treated with STF-083010 and quantified 1629 secreted proteins (Supplemental Table S7). The multiscatter plots of the MS intensity of proteins (four replicates for each sample) are shown in Supplemental Figure S5A. The Pearson correlation of MS intensities among the replicates ranges from 0.82 to 0.97, suggesting that the quantification was robust and reproducible. We identified 463 secreted proteins whose secretion was significantly changed by either EMT or inhibition of XBP1 splicing, or both (t test, Permutation-based FDR1%). Principal component analysis (PCA) of these 463 proteins shows that EMT-hSAECs is clearly segregated from hSAECs and also from EMT-hSAEC/STF, whereas hSAECs/STF group cannot be separated from hSAECs control group (Figure 4F). One possible explanation for this phenomenon is that the activation of XBP1-HBP is an adaptive response occurred only in the mesenchymal cells upon a transient requirement to expand ER protein folding capacity. Unsupervised hierarchical clustering of these 463 proteins identified three clusters (Figure 4G). Cluster 3 has the proteins whose secretion was significantly up-regulated by mesenchymal transition and blocked by the inhibition of XBP1 splicing. GOMF analysis revealed that this cluster is enriched for the annotations that are related to ECM organization (Supplemental Figure S5B). The abundance of some ECM proteins in this cluster is displayed in Figure 4H. Taken together, these results suggest that XBP1 and HBP pathway are functionally linked in EMT, and also that the activation of XBP1-HBP is required to sustain the significantly elevated ECM secretion in mesenchymal cells.
Next, we examined the kinetics of the XBP1-HBP pathway response during the initiation of EMT. We found that the upregulation of XBP1 started from the third day of TGFβ treatment and peaked after 7 days of TGFβ treatment. The expression of the HBP enzymes (GFPT1/2, GNPNAT1, and PGM3) and intracellular UDP-GlcNAc are activated early in the process of EMT, confirming the activation of HBP during the EMT process (Supplemental Figure S6)
We also examined if the inhibition of XBP1 splicing would affect the EMT process. We treated the hSAECs with TGFβ ± STF for 0, 3, 7, and 14 days and measured expression of the core EMT transcription regulators (SNAIL1, TWIST1, and ZEB1) and EMT markers (FN1 and VIM) by Q-RT-PCR. We found that at the early stage of EMT, the inhibition of XBP1 splicing did not affect the expression of these genes (Supplemental Figure S7). However, at the later stage of EMT (14 days), inhibition of XBP1 splicing down-regulated SNAIL1, TWIST1, ZEB1, and FN1. Interpreted with the delayed onset of XBP1 splicing (Supplemental Figure S7), these data suggesting XBP1 splicing plays a role in the later stages of EMT.
EMT Activates Endoplasmic Reticulum-Associated Degradation (ERAD)
In both of our transcriptome and proteome studies, we found ERAD components such as heat shock protein 90 beta family member 1 (HSP90B1) and SYVN1 were up-regulated in EMT-hSAECs.12,14 This finding was verified by SID-SRM-MS (Figure 5A,B). Given the apparent involvement of the ERAD machinery in EMT, we tested proteasome activity in hSAECs and EMT-hSAECs. We observed a significant increase in the 26S proteasome and 20S proteasome activity in EMT cells relative to that of controls (Figure 5C). Next, we tested whether activation of ERAD in EMT cells affects protein turnover. We treated the hSAECs and EMT-hSAECs with cycloheximide, a protein synthesis inhibitor and measured changes in the abundance of FN1, ITGB1, ITGAV, and THBS1 SID-SRM-MS. These ECM proteins were selected because they were the most up-regulated proteins during the mesenchymal transition and misfolding of these proteins is the major cause of ER stress in EMT cells,10 making them likely ERAD targets. As shown in Figure 5D, the degradation of FN1, ITGB1, ITGAV, and THBS1 in EMT cells was faster than control cells, a finding consistent with elevated ERAD activity in EMT cells. Together, these data suggest that the UPR in mesenchymal transition triggers the ERAD machinery and increases the degradation of misfolded proteins to maintain proteostasis.
Figure 5.

TGFβ induced EMT activates ER associated protein degradation. (A,B) SID-SRM-MS quantification of protein expression of proteins involved in ER-associated protein degradation in hSAECs and EMT-hSAECs. (C) Proteasome activities in hSAECs and EMT-hSAECs. The left panel is the activity of the 26S proteasome; the right panel is the activity of the 20S proteasome. (D) TGFβ induced EMT affects the turnover of extracellular matrix proteins. The hSAECs and EMT-hSAECs were treated with cycloheximide. The expression of some ECM proteins was measured with SID-SRM-MS. FN1 was measured using two signature peptides. **, p-value of t test is below 0.05; ***, p-value of t test is below 0.001.
We also examined if inhibiting ECM secretion would increase the activity of ERAD. We treated EMT-hSAECs with Brefeldin A (BFA, 5 μg/mL), an inhibitor of the secretory pathway,35 and measured ECM secretion into the cell culture medium by SID-SRM-MS. As shown in the Supplemental Figure S8A, BFA significantly reduced the secretion FN1, Sparc, and ColA4. Next, we measured the turnover of FN1 in EMT-hSAECs in the presence or absence of BFA. Inhibition of FN1 secretion by BFA slightly increased the intracellular FN1 but did not affect its turnover (Supplemental Figure S8B). These results suggest that the rapid decrease in FN1 was due to enhanced intracellular degradation.
DISCUSSION
Type II EMT underlies organ injury/repair, ECM production, and tissue remodeling, playing a prominent role in the small airways in COPD (reviewed36–38), viral-induced lung inflammation16,39 and allergic airway disease.40 In this transition, the expression of ECM genes are activated via SMAD-dependent and SMAD independent signaling pathways in response to TGFβ.14,41,42 ECM proteins are folded, assembled, and N-glycosylated in ER before secretion. This process is especially important for ECM proteins because they are usually larger and more insoluble than cytosolic proteins.13 Our secretome analysis showed that the secretion of ECM proteins was dramatically increased after the mesenchymal transition. Epithelial cells are not professional secretory cells and produce little ECM proteins under normal conditions. It remains elusive how the epithelial cells transform their protein factory and secretory machinery to accommodate the demand for the production and secretion of ECM proteins in massive quantities. In this study and our previous work on the mesenchymal transition, we found EMT-hSAECs activated IRE1α-XBP1 axis of URP which up-regulated resident ER proteins and proteins involved in protein folding and translocation between ER and Golgi. The time course study indicated the activation of XBP1-HBP pathway started around the third day of TGFβ treatment when the gene expression of ECM genes was significantly up-regulated by TGFβ, supporting our hypothesis that cells activate XBP1-HBP pathway to facilitate the production and secretion of ECM proteins. That HBP pathway was functionally important was revealed by experiments silencing the rate-limiting enzyme, GFPT1, reduced the secretion of ECM proteins, and conversely, exogenous supplementation of GlcNAc promoted ECM protein secretion.
Our secretome analysis also suggests the pivotal role of XBP1 in determining the secretory phenotype of mesenchymal cells. Inhibition of XBP1 splicing significantly reduces the ECM secretion in mesenchymal cells. It has been proven that XBP1 induces a massive ER expansion when B cells differentiate into professional secretory plasma cells.43 Our findings suggest that EMT cells, like plasma cells, use an adaptive mechanism, UPR-XBP1, to increase ER folding and secretion capacity to cope with the demand for the dramatically increased ECM production.
Our study found that EMT activated HBP and up-regulated protein N-glycosylation of ECM proteins. Activation of HBP and subsequent up-regulation of protein N-glycosylation probably play two significant functional roles in EMT. First, the N-glycosylation is important in protein folding and quality control and is a pro-survival factor. The findings that inhibition of protein N-glycosylation induced a higher degree of ER-associated apoptosis in EMT cells relative to control cells suggest that EMT cells are more dependent on protein N-glycosylation to maintain ER proteostasis. Second, our secretome data indicated that protein N-glycosylation mediated the secretion of ECM proteins, a prediction confirmed by our studies inhibiting protein N-glycosylation, which blocked ECM secretion in EMT cells. Together, our data suggest that HBP is an adaptive response activated in EMT cells with the aim to improve the folding of ECM proteins and restore the ER proteostasis, and also facilitate their secretion.
HBP pathway not only plays important roles in type II EMT, growing body of evidence suggest the metabolic reprogramming process in type III EMT directs glucose flux toward HBP, which fuels aberrant protein O-GlcNAcylation that are extensively observed in cancer cells.44,45 The rate limiting enzymes of HBP, GFPTs, are overexpressed in many cancer types that present EMT features.46–49 To our knowledge, our study is to demonstrate that type II EMT activates HBP pathway and its roles in regulating the secretion of ECM proteins and proteostasis.
We also confirmed the link between UPR-XBP1 and HBP in EMT cells by inhibition of XBP1 splicing with an IRE1α inhibitor. Inhibition of XBP1 splicing with STF-083010 downregulated the production of the intracellular free UDP-GlcNAc, reduced the level of protein N-glycosylation and blocked the secretion of ECM proteins. These results suggest that XBP1-HBP plays a coordinating role in regulating the folding and secretory capacity of ER and the protein N-glycosylation of ECM proteins to restore the ER proteostasis and facilitate ECM secretion.
Accumulating evidence shows that an efficient ERAD system is required to maintain proteostasis and secretory capacity in professional secretory cells.11,13,50 We found that the EMT cells activated the ERAD pathway and enhance proteasome activities. Because ECM proteins are difficult to fold and assemble in the ER, they are likely misfolded and become the primary targets of ERAD. Our findings that the half-life of some ECM proteins is shorter in EMT cells relative to control cells are consistent with the enhanced proteasome activities in EMT. Many proteins involved in ERAD are downstream genes of IRE1α-XBP1 pathways. Therefore, the activation of UPR-XBP1 in EMT cells probably also contributed to the upregulation of ERAD. Together, our data indicated that EMT cells activate the XBP1-ERAD pathway to restore intracellular proteostasis and reduce cytotoxicity.
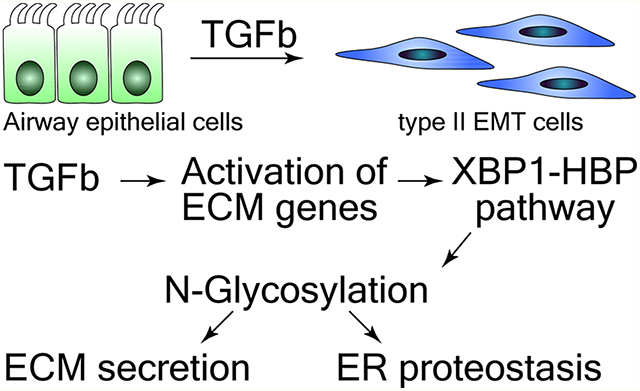
Our study here provides a novel, unifying molecular mechanism by which the XBP1-HBP axis plays a pivotal role in regulating proteostasis and ECM production and secretion in EMT. As outlined in Figure 6, TGFβ significantly activates ECM genes in epithelial cells. The elevated production of these newly synthesized ECM proteins exceed the folding capacity of ER and cause ER stress, which activates the IRE1α-XBP1 axis of UPR and increase the expression of transcription factor XBP1s. XBP1s activated the expression of resident ER proteins, expanding ER processing capacity. XBP1 also activates the HBP pathway to increase production of UDP-GlcNAc. The upregulation of protein N-glycosylation facilitates protein folding and ERAD, restoring ER hemostasis and protecting mesenchymal cells from ER-induced apoptosis. Therefore, the XBP1-HBP and protein N-glycosylation pathways are pro-survival factors for epithelial cells undergoing EMT. The secretion of ECM proteins in EMT cells is also mediated by the XBP1-HBP pathway and protein N-glycosylation.
Figure 6.

TGFβ-induced EMT activates UPR-HBP to up-regulate N-glycosylation and secretion of ECM proteins, enhance ERAD, and restore ER proteostasis.
Our study here presents a new mechanism for the pathogenesis of airway remodeling, where XBP1-HBP plays a pro-survival role in epithelial cells undergoing EMT. The injuries to lung epithelium or inflammation response induce the release of TGFβ, which induces EMT, transferring the epithelial cells into highly secretory mesenchymal cells. In response to the ER stress caused by excessive synthesis of ECM proteins, the mesenchymal cells activate XBP1-HBP pathway to expand the ER capacity and secretory machinery, upregulate protein N-glycosylation and ERAD to restore the ER hemostasis. In parallel, the upregulation of protein N-glycosylation promotes the secretion of ECM proteins and led to ECM accumulation and airway remodeling. Our study suggests that targeting XBP1-HBP pathway may provide an effective therapeutic approach for airway remodeling. Importantly, XBP1-HBP pathway is not constitutively activated without TGFβ stimulation. It is an adaptive response to a transient requirement during TGFβ-induced EMT to expand ER protein folding and secretory capacity to release ECM proteins from cells. Therefore, there may be selective toxicity of ECM-secreting mesenchymal cells to XBP1-HBP antagonists compared to normal cells. The compounds that target XBP1-HBP pathway will selectively affect mesenchymal cells that experience ER stress and require activation of the XBP1-HBP pathway for survival.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium51 via the PRIDE partner repository with the data set identifier PXD008585.
Supplementary Material
Table S1: The primers for RT-PCR (XLS)
Table S7: The proteins identified from the secretome of hSAECs and EMT-hSAECs in the presence or absence of STF-083010 (XLS)
Table S6: The proteins identified from the secretome of hSAECs and EMT-hSAECs in the presence or absence of tunicamycin (XLS)
Table S5: N-Glycosylated peptides identified and quantified in hSAECs and EMT-hSAECs (XLS)
Table S4: 2D annotation enrichment analysis of transcriptome and proteome data (XLS)
Table S3: The parameters for LC−MS/MS label-free quantification and MaxQuant analysis (XLSX)
Table S2: The parameters for LC-SRM-MS analysis (XLS)
Figure S1: Immunostaining of markers of epithelial cells and mesenchymal cells; Figure S2: TGFβ induced EMT alters protein N-glycosylation; Figure S3: Label-free quantification of the secretome of hSAECs and EMT-hSAECs in the presence or absence of tunicamycin; Figure S4: The effect of GlcNAc supplement on the EMT process and secretion of ECM; Figure S5: Label-free quantification of the secretome of hSAECs and EMT-hSAECs in the presence or absence of STF-083010; Figure S6: Activation of UPR-XBP1 and HBP during the course of EMT; Figure S7: The effect of XBP1 splicing inhibitor on the EMT process; Figure S8: The effect of Brefeldin A (BFA) on the secretion of ECMs and the turnover of fibronectin (PDF)
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grants NIAID 1R21AI133454 (to YXZ, ARB), NCATS UL1TR001439 (to ARB), DMS-1361411/DMS-1361318 (to ARB, YXZ), NIAID AI062885 (ARB), NIEHS P30 ES006676 (ARB), and pilot funding from the Sealy Center for Molecular Sciences (SCMM). Core support was from the SCMM Selected Reaction Monitoring Facility, the Optical Microscopy Core, and the Next Generation Sequencing Core. We thank Dr. Maxim Ivannikov for the assistance on confocal imaging.
ABBREVIATIONS
- AGC
automatic gain control
- ANOVA
analysis of variance
- BFA
brefeldin
- CM
culture medium
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- ERAD
ER-associated protein degradation
- FASP
filter aided sample prep
- FDR
false discovery rate
- FPKM
fragments per kilobase of transcript per million
- fwhm
full width half-maximum
- GO
gene ontology
- GOBF
gene ontology biological function
- GOCC
gene ontology cellular compartment
- GOMF
gene ontology molecular function
- HBP
hexosamine biosynthesis pathway
- HCD
high energy collisional dissociation
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LC
liquid chromatography
- MMP
metalloproteinases
- MS
mass spectrometry
- PCA
principle component analysis
- PDI
protein disulfide isomerase
- PTM
post-translational modifications
- Q
quadrupole
- Q-RT-PCR
quantitative real-time reverse transcription-PCR
- SCX
strong cation exchange
- SID
stable isotope dilution
- SIS
stable isotope labeled internal standard
- SRM
selected reaction monitoring
- TGF
transforming growth factor
- TM
tunicamycin
- UDP-GlcNAc
uradine diphosphate-N-acetyl-glucosamine
- UHPLC
ultra high performance liquid chromatography
- UPR
unfolded protein response
Footnotes
Notes
The authors declare no competing financial interest. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium51 via the PRIDE partner repository with the data set identifier PXD008585.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.9b00342.
REFERENCES
- (1).Holgate ST; Holloway J; Wilson S; Bucchieri F; Puddicombe S; Davies DE Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc. Am. Thorac. Soc 2004, 1, 93–98. [DOI] [PubMed] [Google Scholar]
- (2).Bartram U; Speer CP The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765. [DOI] [PubMed] [Google Scholar]
- (3).Xu J; Lamouille S; Derynck R TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009, 19, 156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kalluri R; Weinberg RA The basics of epithelial-mesenchymal transition. J. Clin. Invest 2009, 119, 1420−8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Holgate ST; Davies DE; Puddicombe S; Richter A; Lackie P; Lordan J; Howarth P Mechanisms of airway epithelial damage: epithelial-mesenchymal interactions in the pathogenesis of asthma. Eur. Respir. J 2003, 44, 24s–29s. [DOI] [PubMed] [Google Scholar]
- (6).Hackett TL Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr. Opin Allergy Clin Immunol 2012, 12, 53–9. [DOI] [PubMed] [Google Scholar]
- (7).Hackett TL; Warner SM; Stefanowicz D; Shaheen F; Pechkovsky DV; Murray LA; Argentieri R; Kicic A; Stick SM; Bai TR; Knight DA Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am. J. Respir. Crit. Care Med 2009, 180, 122–33. [DOI] [PubMed] [Google Scholar]
- (8).Nowrin K; Sohal SS; Peterson G; Patel R; Walters EH Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert Rev. Respir. Med 2014, 8, 547–59. [DOI] [PubMed] [Google Scholar]
- (9).Willis BC and Borok Z TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 293, L525–L534. [DOI] [PubMed] [Google Scholar]
- (10).Feng Y.-x.; Sokol ES; Del Vecchio CA; Sanduja S; Claessen JHL; Proia TA; Jin DX; Reinhardt F; Ploegh HL; Wang Q; Gupta PB Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discovery 2014, 4, 702–715. [DOI] [PubMed] [Google Scholar]
- (11).Hetz C The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol 2012, 13, 89–102. [DOI] [PubMed] [Google Scholar]
- (12).Zhao Y; Tian B; Sadygov RG; Zhang Y; Brasier AR Integrative proteomic analysis reveals reprograming tumor necrosis factor signaling in epithelial mesenchymal transition. J. Proteomics 2016, 148, 126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sitia R; Braakman I Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–4. [DOI] [PubMed] [Google Scholar]
- (14).Tian B; Li X; Kalita M; Widen SG; Yang J; Bhavnani SK; Dang B; Kudlicki A; Sinha M; Kong F; Wood TG; Luxon BA; Brasier AR Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics 2015, 16, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tian B; Hosoki K; Liu Z; Yang J; Zhao Y; Sun H; Zhou J; Rytting E; Kaphalia L; Calhoun WJ; Sur S; Brasier AR Mucosal bromodomain-containing protein 4 mediates aeroallergen-induced inflammation and remodeling. J. Allergy Clin. Immunol 2019, 143, 1380–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tian B; Liu Z; Litvinov J; Maroto R; Jamaluddin M; Rytting E; Patrikeev I; Ochoa L; Vargas G; Motamedi M; Ameredes BT; Zhou J; Brasier AR Efficacy of Novel Highly Specific Bromodomain-Containing Protein 4 Inhibitors in Innate Inflammation-Driven Airway Remodeling. Am. J. Respir. Cell Mol. Biol 2019, 60, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tian B; Widen SG; Yang J; Wood TG; Kudlicki A; Zhao Y; Brasier AR The NFkappaB subunit RELA is a master transcriptional regulator of the committed epithelial-mesenchymal transition in airway epithelial cells. J. Biol. Chem 2018, 293, 16528–16545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yang J; Tian B; Sun H; Garofalo RP; Brasier AR Epigenetic silencing of IRF1 dysregulates type III interferon responses to respiratory virus infection in epithelial to mesenchymal transition. Nat. Microbiol 2017, 2, 17086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sasai K; Ikeda Y; Fujii T; Tsuda T; Taniguchi N UDP-GlcNAc concentration is an important factor in the biosynthesis of beta1,6-branched oligosaccharides: regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology 2002, 12, 119–27. [DOI] [PubMed] [Google Scholar]
- (20).Wisniewski JR; Zielinska DF; Mann M Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal. Biochem 2011, 410, 307–9. [DOI] [PubMed] [Google Scholar]
- (21).Zhao Y; Jamaluddin M; Zhang Y; Sun H; Ivanciuc T; Garofalo RP; Brasier AR Systematic Analysis of Cell-Type Differences in the Epithelial Secretome Reveals Insights into the Pathogenesis of Respiratory Syncytial Virus-Induced Lower Respiratory Tract Infections. J. Immunol 2017, 198, 3345–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–72. [DOI] [PubMed] [Google Scholar]
- (23).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–40. [DOI] [PubMed] [Google Scholar]
- (25).Cox J; Mann M 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinf. 2012, 13, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gaudet P; Livstone MS; Lewis SE; Thomas PD Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Briefings Bioinf. 2011, 12, 449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhao Y; Brasier AR Applications of selected reaction monitoring (SRM)-mass spectrometry (MS) for quantitative measurement of signaling pathways. Methods 2013, 61, 313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tian B; Li X; Kalita M; Widen SG; Yang J; Bhavnani SK; Dang B; Kudlicki A; Sinha M; Kong F; Wood TG; Luxon BA; Brasier AR Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/ RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics 2015, 16, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mi H; Muruganujan A; Thomas PD PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2012, 41, D377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pankov R; Yamada KM Fibronectin at a glance. J. Cell Sci 2002, 115, 3861–3. [DOI] [PubMed] [Google Scholar]
- (31).Elbein AD Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains. Annu. Rev. Biochem 1987, 56, 497–534. [DOI] [PubMed] [Google Scholar]
- (32).Denzel MS; Storm NJ; Gutschmidt A; Baddi R; Hinze Y; Jarosch E; Sommer T; Hoppe T; Antebi A Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 2014, 156, 1167–78. [DOI] [PubMed] [Google Scholar]
- (33).Wang ZV; Deng Y; Gao N; Pedrozo Z; Li DL; Morales CR; Criollo A; Luo X; Tan W; Jiang N; Lehrman MA; Rothermel BA; Lee AH; Lavandero S; Mammen PP; Ferdous A; Gillette TG; Scherer PE; Hill JA Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014, 156, 1179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Papandreou I; Denko NC; Olson M; Van Melckebeke H; Lust S; Tam A; Solow-Cordero DE; Bouley DM; Offner F; Niwa M; Koong AC Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chardin P; McCormick F Brefeldin A: the advantage of being uncompetitive. Cell 1999, 97, 153–5. [DOI] [PubMed] [Google Scholar]
- (36).Jolly MK; Ward C; Eapen MS; Myers S; Hallgren O; Levine H; Sohal SS Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [DOI] [PubMed] [Google Scholar]
- (37).Eapen MS; Hansbro PM; Larsson-Callerfelt AK; Jolly MK; Myers S; Sharma P; Jones B; Rahman MA; Markos J; Chia C; Larby J; Haug G; Hardikar A; Weber HC; Mabeza G; Cavalheri V; Khor YH; McDonald CF; Sohal SS Chronic Obstructive Pulmonary Disease and Lung Cancer: Underlying Pathophysiology and New Therapeutic Modalities. Drugs 2018, 78, 1717–1740. [DOI] [PubMed] [Google Scholar]
- (38).Sohal SS Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Investig 2017, 55, 104–113. [DOI] [PubMed] [Google Scholar]
- (39).Ijaz T; Pazdrak K; Kalita M; Konig R; Choudhary S; Tian B; Boldogh I; Brasier AR Systems Biology Approaches To Understanding Epithelial Mesenchymal Transition (EMT) In Mucosal Remodeling And Signaling In Asthma. World Allergy Organ. J 2014, 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tian B; Hosoki K; Liu Z; Yang J; Zhao Y; Sun H; Zhou J; Rytting E; Kaphalia L; Calhoun WJ; Sur S; Brasier AR Mucosal Bromodomain-Containing Protein 4 (BRD4) Mediates Aeroallergen-induced Inflammation and Remodeling. J. Allergy Clin. Immunol 2019, 143 (4), 1380–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lamouille S; Xu J; Derynck R Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol 2014, 15, 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ijaz T; Pazdrak K; Kalita M; Konig R; Choudhary S; Tian B; Boldogh I; Brasier AR Systems biology approaches to understanding Epithelial Mesenchymal Transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ. J 2014, 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Reimold AM; Iwakoshi NN; Manis J; Vallabhajosyula P; Szomolanyi-Tsuda E; Gravallese EM; Friend D; Grusby MJ; Alt F; Glimcher LH Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–7. [DOI] [PubMed] [Google Scholar]
- (44).Lucena MC; Carvalho-Cruz P; Donadio JL; Oliveira IA; de Queiroz RM; Marinho-Carvalho MM; Sola-Penna M; de Paula IF; Gondim KC; McComb ME; Costello CE; Whelan SA; Todeschini AR; Dias WB Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem 2016, 291, 12917–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Freire-De-Lima L; Gelfenbeyn K; Ding Y; Mandel U; Clausen H; Handa K; Hakomori SI Involvement of O-glycosylation defining oncofetal fibronectin in epithelial-mesenchymal transition process. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 17690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Vasconcelos-Dos-Santos A; Loponte HF; Mantuano NR; Oliveira IA; de Paula IF; Teixeira LK; de-Freitas-Junior JC; Gondim KC; Heise N; Mohana-Borges R; Morgado-Diaz JA; Dias WB; Todeschini AR Hyperglycemia exacerbates colon cancer malignancy through hexosamine biosynthetic pathway. Oncogenesis 2017, 6, No. e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Dong T; Kang X; Liu Z; Zhao S; Ma W; Xuan Q; Liu H; Wang Z; Zhang Q Altered glycometabolism affects both clinical features and prognosis of triple-negative and neoadjuvant chemotherapy-treated breast cancer. Tumor Biol. 2016, 37, 8159–68. [DOI] [PubMed] [Google Scholar]
- (48).Guillaumond F; Leca J; Olivares O; Lavaut MN; Vidal N; Berthezene P; Dusetti NJ; Loncle C; Calvo E; Turrini O; Iovanna JL; Tomasini R; Vasseur S Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 3919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Shaul YD; Freinkman E; Comb WC; Cantor JR; Tam WL; Thiru P; Kim D; Kanarek N; Pacold ME; Chen WW; Bierie B; Possemato R; Reinhardt F; Weinberg RA; Yaffe MB; Sabatini DM Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Molinari M; Sitia R The secretory capacity of a cell depends on the efficiency of endoplasmic reticulum-associated degradation. Curr. Top Microbiol Immunol 2005, 300, 1–15. [DOI] [PubMed] [Google Scholar]
- (51).Vizcaino JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Rios D; Dianes JA; Sun Z; Farrah T; Bandeira N; Binz PA; Xenarios I; Eisenacher M; Mayer G; Gatto L; Campos A; Chalkley RJ; Kraus HJ; Albar JP; Martinez-Bartolome S; Apweiler R; Omenn GS; Martens L; Jones AR; Hermjakob H ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol 2014, 32, 223–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: The primers for RT-PCR (XLS)
Table S7: The proteins identified from the secretome of hSAECs and EMT-hSAECs in the presence or absence of STF-083010 (XLS)
Table S6: The proteins identified from the secretome of hSAECs and EMT-hSAECs in the presence or absence of tunicamycin (XLS)
Table S5: N-Glycosylated peptides identified and quantified in hSAECs and EMT-hSAECs (XLS)
Table S4: 2D annotation enrichment analysis of transcriptome and proteome data (XLS)
Table S3: The parameters for LC−MS/MS label-free quantification and MaxQuant analysis (XLSX)
Table S2: The parameters for LC-SRM-MS analysis (XLS)
Figure S1: Immunostaining of markers of epithelial cells and mesenchymal cells; Figure S2: TGFβ induced EMT alters protein N-glycosylation; Figure S3: Label-free quantification of the secretome of hSAECs and EMT-hSAECs in the presence or absence of tunicamycin; Figure S4: The effect of GlcNAc supplement on the EMT process and secretion of ECM; Figure S5: Label-free quantification of the secretome of hSAECs and EMT-hSAECs in the presence or absence of STF-083010; Figure S6: Activation of UPR-XBP1 and HBP during the course of EMT; Figure S7: The effect of XBP1 splicing inhibitor on the EMT process; Figure S8: The effect of Brefeldin A (BFA) on the secretion of ECMs and the turnover of fibronectin (PDF)