

Abstract
Apolipoprotein E (APOE) is a multifunctional protein synthesized and secreted by multiple mammalian tissues. Although hepatocytes contribute about 75% of the peripheral pool, APOE can also be expressed in adipose tissue, the kidney, and the adrenal glands, among other tissues. High levels of APOE production also occur in the brain, where it is primarily synthesized by glia, and peripheral and brain APOE pools are thought to be distinct. In humans, APOE is polymorphic, with three major alleles (ε2, ε3, and ε4). These allelic forms dramatically alter APOE structure and function. Historically, the vast majority of research on APOE has centered on the important role it plays in modulating risk for cardiovascular disease and Alzheimer’s disease. However, the established effects of this pleiotropic protein extend well beyond these two critical health challenges, with a demonstrated roles for APOE across a wide spectrum of biological conditions, including adipose tissue function and obesity, metabolic syndrome and diabetes, fertility and longevity, and immune function.
While the spectrum of biological systems in which APOE plays a role seems implausibly wide at first glance, there are some potential unifying mechanisms that could tie these seemingly disparate disorders together. In the current review, we aim to concisely summarize a wide breadth of APOE-associated pathologies and to analyze the influence of APOE in the development of several distinct disorders in order to provide insight into potential shared mechanisms implied in these various pathophysiological processes.
INTRODUCTION
Apolipoprotein E (APOE) was first identified in humans as a constituent of very low-density lipoproteins (VLDL) in 1973 by Shore and Shore (Shore and Shore, 1973). Initially termed “arginine-rich apoprotein”, Utermann named the protein apolipoprotein E in 1975 (Utermann, 1975), distinguishing it from others in the growing class of apolipoproteins. It was later found in triglyceride-rich lipoproteins, both in animal and human models, after being induced by cholesterol supplementation (Mahley et al., 1975; Shore et al., 1974).
In humans, the APOE gene is located on the long arm of chromosome 19 (locus 19q13.2). It consists of four exons and three introns, with a length of 3597 nucleotides (Das et al., 1985). The polymorphic nature of APOE was first discovered by Utermann (Utermann et al., 1977) and later clarified by Zanis and Breslow (Zannis et al., 1981). Thus, the human gene presents three common alleles: ε2, ε3 and ε4. The combination of these three alleles produces six genotypes (Table 1). The resulting proteins differ only by one or two amino acids at positions 112 and 158. APOE2 has a cysteine at both positions, APOE3 has a cysteine at 112 and an arginine at 158, and APOE4 has an arginine at both positions (Weisgraber, 1994). It is hypothesized that these residues influence the properties of the isoforms by altering the domain interaction between the N and C terminal domains (Mahley and Huang, 2012) which are presumably translated into a change in protein function. The most common isoform is APOE3, being found in the 70–80% of modern populations, whereas APOE4 is found in 10–15% and APOE2 in 5–10% of the population (CORBO and SCACCHI, 1999). Although other mammals express APOE, this allelic variation is only found in humans. The ε4 is considered as the ancestral human allele, with the ε2 (E2) and ε3 (E3) alleles arising only after the divergence of the human and primate lineages (Hanlon and Rubinsztein, 1995). Sequence analysis reveals that the primary structure of the primate APOE is identical to human APOE4 (Hanlon and Rubinsztein, 1995), although with a tertiary structure more similar to APOE3, which results in binding preferences that are functionally similar to human APOE3 (McIntosh et al., 2012).
Table 1.
APOE genotypes.
| rs429358 (codon 112) | rs7412 (codon 158) | Allele |
|---|---|---|
| T/T | T/T | ε2/ε2 |
| T/T | C/C | ε3/ε3 |
| C/C | C/C | ε4/ε4 |
| T/T | T/C | ε2/ε3 |
| T/C | T/C | ε2/ε4 |
| T/C | C/C | ε3/ε4 |
Historically, the vast majority of research on APOE has centered on the important role it plays in modulating risk for cardiovascular disease (CVD) and late onset Alzheimer’s disease (AD). However, the established effects of this pleiotropic protein extend well beyond these two critical health challenges, with demonstrated roles for APOE across a wide spectrum of biological conditions, including adipose tissue function and obesity, metabolic syndrome and diabetes, fertility and longevity, immune function, and infectious diseases.
In the current review, we aim to concisely summarize a wide breadth of APOE-associated pathologies and to analyze the influence of APOE in the development of several distinct disorders in order to provide insight into potential shared mechanisms implied in these various pathophysiological processes.
FUNCTIONS OF APOE IN THE BRAIN AND PERIPHERY
APOE is a multifunctional protein synthesized and secreted by multiple mammalian tissues. Hepatocytes contribute about 75% of the peripheral pool of APOE. In addition, APOE can also be expressed in adipose tissue, the kidney, and the adrenal glands, among other tissues (Huang and Mahley, 2014). High levels of APOE production also occur in the brain (Huang and Mahley, 2014). While brain and peripheral pools of APOE are distinct, secreted APOE shares similar systemic functions in each of these locations of synthesis, primarily regulating lipoprotein metabolism and supporting cellular differentiated function (Huang and Mahley, 2014).
Lipid transport and metabolism
APOE is involved in lipid transport through its association with chylomicron remnants, VLDL, and high-density lipoproteins (HDL) (Robert W. Mahley et al., 1979; Shore and Shore, 1973; Utermann, 1975). APOE interacts with large heparan sulfate proteoglycans (HSPGs) on the hepatocyte surface and promotes internalization of the lipoprotein particles via lipoprotein receptors of the LDL receptor family (Figure 1) (Robert W Mahley et al., 1979). APOE genotype influences plasma and brain cholesterol levels (Chernick et al., 2019; Sing and Davignon, 1985). Plasma LDL cholesterol levels are both associated with the three APOE isoforms in the order of APOE4 > APOE3 > APOE2. This association is counterintuitive as APOE4 binds to LDL receptor with a slightly higher affinity than APOE3, while APOE2 binds to the receptor with much reduced affinity (Knouff et al., 1999).
Figure 1.
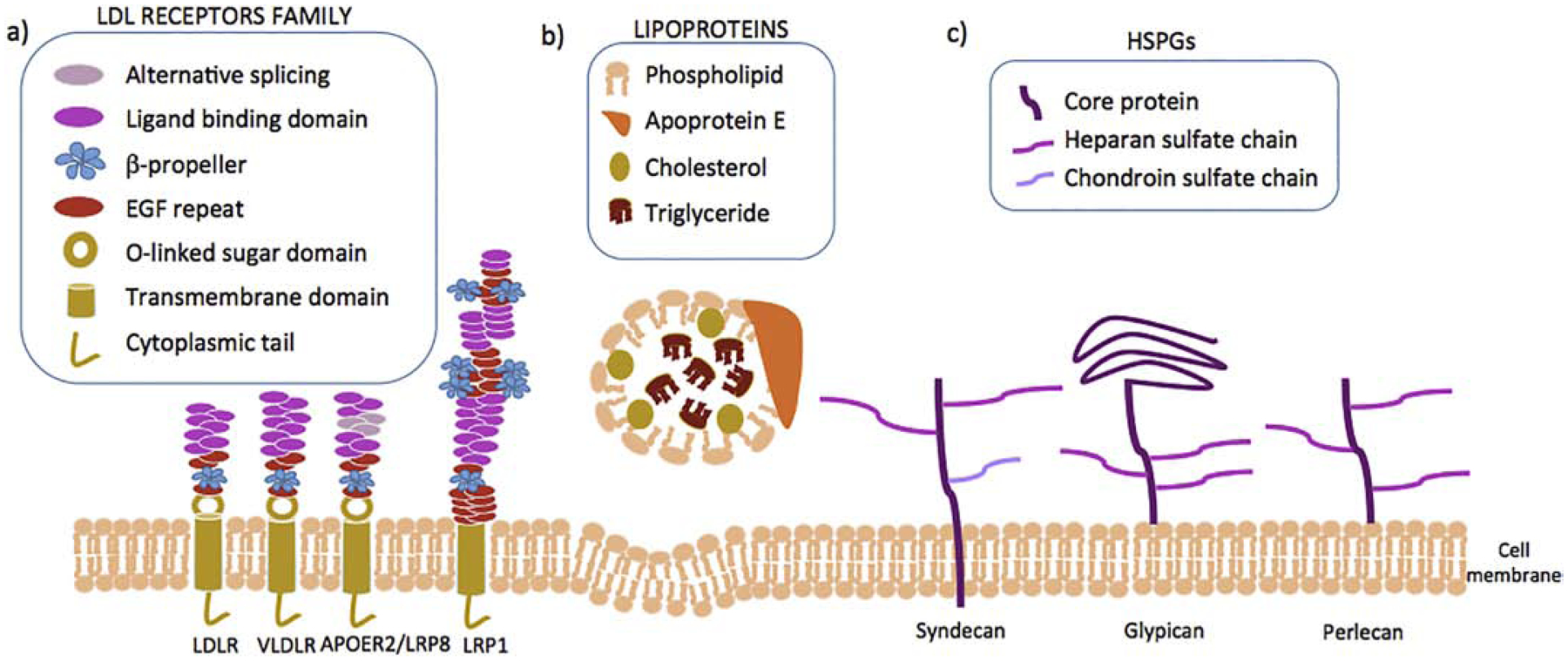
Clearance of lipoproteins is a multi-step process in which lipoproteins firstly bind to HSPGs followed by their uptaked by members of the LDL receptors family. a) APOE lipoprotein receptors. Low density lipoprotein receptor (LDLR), widely expressed in hepatocytes; Very low density lipoproteins receptor (VLDLR), widely expressed in adipose tissue, endothelium, skeletal muscle and heart; Apolipoprotein E receptor 2 or Low-density lipoprotein receptor-related protein 8 (ApoER2/LRP8), mainly expressed in brain, testis and placenta; Low density lipoprotein receptor-related protein 1 (LRP1), ubiquitously expressed and key receptor for maintaining cholesterol homeostasis. These receptors are characterized by a single transmembrane domain, a cytoplasmic tail with at least one NPxY motif, epidermal grow factor (EGF) and b-propeller domains, and one or more igand binding domains. b) Lipoproteins (CR, HDL, VLDL) are composed by a core containing triglycerides and cholesterol (esterified and free) surrounded by phospholipids and APOE. c) Heparan sulfate proteoglycans (HSPGs) are glycoproteins containing one or more covalently linked heparan sulfate chains. Some syndecans also contain a chondroitin sulfate chain.
Upon receptor-mediated endocytosis, APOE-containing lipoproteins are processed in peripheral endosomes. In human hepatoma cells and fibroblasts, lipids are targeted to the lysosomal compartment, while APOE is found in peripheral recycling endosomes to be reutilized by the cell (Heeren et al., 1999). Early studies in macrophages and hepatocytes showed that APOE degradation only occurred within lysosomes, whereas others have shown degradation in both lysosomal and proteasomal compartments [reviewed in (Maaike et al., 2008; Wenner et al., 2001)]. Secreted APOE can be re-uptaken into hepatocytes before being released into the circulation and then re-secreted, but less so in the case of APOE4 with consequent accumulation of cholesterol in the cell (Heeren et al., 2004). Of note, astrocytes preferentially degrade APOE4, leading to reduced APOE4 secretion and ultimately to reduced brain APOE levels (Riddell et al., 2008).
Liver
The liver is the major site of lipoprotein uptake and APOE is a critical ligand for the clearance of these molecules. Within the liver, hepatocytes and Kupffer cells both secrete APOE, with hepatocytes being the major source of APOE in plasma (Getz and Reardon, 2009). It has been proposed that VLDL particles in APOE4 individuals are enriched with APOE4 on the surface, leading to accelerated liver uptake. As a result, receptors are negatively regulated and LDL plasma levels increase (Gregg et al., 1986; Li et al., 2013; Weintraub et al., 1987). As a potential alternative mechanism, our group found that the high affinity of APOE4 to the LDLR enhanced VLDL sequestration on the hepatocyte surface but delayed their internalization (Altenburg et al., 2008). Conversely, the marked reduction of APOE2 in its LDL receptor binding activity translates into a low hepatic uptake of VLDL (Weisgraber, 1994): and as a consequence, E2 individuals display low LDL levels. In addition, this isoform not only has a lower capacity to promote lipolysis mediated by hepatic lipase (HL), but high E2 protein levels also inhibit lipase-mediated lipolysis (LPL) by displacing its cofactor apoC-II; concurrently, a lower processing of VLDL towards LDL has been reported (Mahley et al., 1999).
Brain
APOE is the major apolipoprotein in the central nervous system (CNS). In the brain, most cholesterol synthesis occurs in astrocytes which are also the main producers of APOE under normal physiological conditions (Fernandez et al., 2019). Upon activation microglia can dramatically upregulate APOE expression and other cell types, such as oligodendrocytes and neurons, have been also shown to produce APOE under certain conditions (Fernandez et al., 2019). It is worth noting that lipoprotein-borne APOE in the cerebrospinal fluid (CSF) is similar in size and density to peripheral HDL (LaDu et al., 1998).
The APOE isoforms also differ in promoting cholesterol efflux. APOE4 in the CSF associates with smaller APOE lipoproteins, promoting less efficient cholesterol efflux than APOE3, whereas APOE2 has the greatest cholesterol efflux efficiency. In addition to lipid homeostasis, APOE isoforms can differentially regulate multiple pathways involved in neural development, as well as plasticity and neuronal repair (Fernandez et al., 2019).
Adipose tissue
After the liver and brain, adipose tissue is a third major producer of APOE (Zechner et al., 1991), which regulates the size of adipocytes, triglyceride efflux, and the expression of genes related to fatty acid oxidation (Huang et al., 2006). This endogenous expression of APOE in adipocytes is modulated by regulators of insulin sensitivity, including the liver X receptor (LXR), peroxisome proliferator-activated receptor gamma (PPARγ), angiotensin II, inflammatory cytokines, as well as nutritional status (Espiritu and Mazzone, 2008; Huang et al., 2007; Rao et al., 2007; Yue and Mazzone, 2009). Endogenous adipocyte APOE levels decrease in response to obesity, tumor necrosis factor-α (TNF-α) and angiotensin II, whereas PPARγ agonists, fasting and weight loss increase its levels (Huang et al., 2007; Yue et al., 2004). Together, these studies may suggest that APOE protects against nutritional changes, preserving energy balance and adipose tissue functionality through its roles in lipid acquisition (Huang et al., 2007).
Triglyceride (TG) storage is one of the key functions of the adipose tissue and adipocytes lacking APOE expression display impaired acquisition of lipids from circulating VLDL through at least two separate pathways. First, APOE knockout adipocytes internalize less VLDL through the endocytic pathway due to reduced expression of LDL receptor family proteins (Huang et al., 2009). Second, lipoprotein lipase-dependent accumulation of TG is also impaired in APOE knockout adipocytes due to reduced transport of fatty acids across the adipocyte membrane, likely due to reduced expression of caveolin-1 and plasma membrane rafts (Huang et al., 2009). The APOE2 isoform also modifies adipose tissue (Huang et al., 2015). “Targeted replacement” mice ubiquitously expressing human APOE2 in place of the endogenous mouse APOE gene are hyperlipidemic, presenting with larger adipocytes and gonadal fat pads compared to APOE3 mice (Arbones-Mainar et al., 2008). Similar to APOE knockout mice, adipocytes from mice expressing APOE2 show decreased fatty acid uptake and decreased TG synthesis, likely owing to the observation that newly synthesized APOE2 is more rapidly degraded and less frequently secreted in these cells (Huang et al., 2015, 2011).
Endothelium
The vascular endothelium is an interface between circulating blood and the vessel wall. The activation of endothelial cells may result in the recruitment of circulating monocytes and accumulation of monocyte-derived macrophages (Mestas and Ley, 2008). Although endothelial cells themselves do not appear to synthesize APOE, endothelium-resident macrophages contribute to plasma APOE levels, releasing APOE at atherosclerotic lesion sites (Linton and Fazio, 1999). This locally secreted APOE inhibits the expression of the vascular cell adhesion molecule 1 (VCAM-1), which is involved in the recruitment of monocytes (Stannard et al., 2001). As a consequence, endothelial activation is suppressed and monocyte-endothelial adhesion is attenuated. This suppression likely occurs via stimulation of nitric oxide synthase (NOS), as the nitric oxide inhibitor ethyl-isothiourea blocked this effect. Alternatively, macrophage-derived APOE may also increase NO production by disrupting the inhibitory interaction of endothelial NOS with caveolin-1 (Yue et al., 2012). Therefore, APOE influences the inflammatory response by suppressing endothelial activation and the expression of adhesion molecules.
Interestingly, the effect of APOE on the endothelium is isoform-dependent. APOE3 binds to the ApoER2 receptor to stimulate endothelial NOS and endothelial cell migration, attenuating monocyte-endothelial adhesion. On the other hand, APOE4 does not stimulate endothelial NOS and rather antagonizes APOE3/ApoER2 actions, leading to a loss of the reparative and anti-inflammatory capacity of the endothelium (Ulrich et al., 2014).
Female reproductive system
APOE is expressed in different tissues of the ovary, such as the endometrium or granulosa cells. Several studies conducted with endometrial biopsies have reported increased APOE levels during the luteal phase of the menstrual cycle (Germeyer et al., 2013; Sundqvist et al., 2012). However, in vitro studies have failed to reproduce the progesterone effects on APOE levels (Germeyer et al., 2013). In the case of granulosa cells, APOE expression has been seen to be regulated by human chorionic gonadotropin (Beckmann et al., 1991). In addition, the expression of APOE by granulosa cells may account for the differences in APOE concentration between the follicular fluid and the plasma (Beckmann et al., 1991). It has also been reported that APOE levels in follicular fluid fall dramatically when the follicle approaches ovulation (Brown et al., 1989) while they increase with age, a fact that has been associated with a smaller number of mature oocytes in elderly women (Von Wald et al., 2010).
Rat studies have also found high levels of APOE expression in theca and interstitial cells of follicles in animals at all stages of the estrous cycle (Nicosia et al., 1992). Lower concentrations of APOE can stimulate theca cell androgen production (Zerbinatti and Dyer, 1999). In contrast, at higher concentrations APOE selectively inhibits androgen production without suppressing the production of progesterone (Zerbinatti and Dyer, 1999). A possible explanation for this phenomenon is that intraovarian APOE induces theca and interstitial cell apoptosis thereby controlling the production of androgen by those cells (Zerbinatti and Dyer, 1999).
PRIMARY APOE-ASSOCIATED DISEASES AND CONDITIONS
Atherosclerosis and cardiovascular disease
Atherosclerosis is a progressive disease characterized by the accumulation of lipids and fibrous elements in large arteries causing impaired endothelial function. It ranges from primary arterial atheroma (inflammation and accumulation of macrophages loaded with cholesterol in the artery wall) to the formation of plaques and inflammation of the arterial wall, with the consequent risk of suffering thrombosis (Fuster et al., 1992).
Atherosclerosis is the major cause of cardiovascular disease (CVD) and APOE is abundant in atherosclerotic lesions, where it is secreted by resident macrophages. This APOE production is atheroprotective, since it contributes to reverse cholesterol transport, inhibits the proliferation of smooth muscle cells, prevents lipid oxidation, and restricts platelet aggregation (Linton and Fazio, 1999; Reddick et al., 1994). It has been reported that APOE absence or dysfunction results in hyperlipidemia and atherosclerotic lesions, while APOE injection or hepatic overexpression protects (Plump et al., 1992; Stannard et al., 2001; Zhang et al., 1992). Similarly, APOE4 carriers have higher levels of non-HDL lipoproteins due mainly to reduced VLDL plasma clearance, which in turn contributes to an increased risk if atherosclerosis and CVD (Dallongeville et al., 1992; Knouff et al., 1999). This reduced clearance of VLDL-borne APOE4 can be explained as a downregulation of the LDLR or an enhanced VLDL sequestration on the hepatocyte, as described above in the liver section.
Interestingly, mouse models have shown dysfunctional APOE4 macrophages that have problems phagocytosing apoptotic cells and are more sensitive to cell death induced by oxidized LDL and lipopolysaccharide (LPS) (Altenburg et al., 2007). These phenomena produce hyperinsulinemia due to the inflammation of adipose tissue in mice fed a westernized diet (Cash et al., 2012), even in the absence of obesity. In addition, APOE4 increases endoplasmic reticulum stress in macrophages, leading to mitochondrial malfunction that also contributes to inflammation (Vats et al., 2006). As a consequence, and combined with an ε4-associated increase in fasting LDL levels, risk of CVD and atherosclerosis may be increased.
Dyslipidemia
Although extremely rare, humans with APOE deficiency present elevated VLDL and intermediate density lipoproteins (IDL) in plasma (Schaefer et al., 1986). On the other hand, type III hyperlipoproteinemia (HLP), characterized by the accumulation of chylomicron and VLDL remnants is consistenly associated with ε2 carriers. More than 90% of patients with HLP type III are homozygous ε2/ε2, revealing the APOE2 isoform to be the major driver of the disease (Utermann et al., 1977). The APOE2 isoform binds the APOE-receptor with lower affinity, disturbing hepatic remnant uptake, altering lipolysis of lipoproteins, and overproducing VLDL. These effects are compounded due to the increase in APOE levels in ε2 individuals (Mahley et al., 1999). However, less than 10% of homozygous APOE2 individuals develop hyperlipidemia. In fact, most ε2/ε2 subjects are normolipidemic or even hypolipidemic (Rall et al., 1982; Sing and Davignon, 1985). Therefore, it appears there are external factors that cause the change to HLP type III. Among others, the overproduction of apoB or the decrease of LDL receptors have been reported as secondary factors that lead to the development of type III HLP (Chung and Segrest, 1983). Furthermore, it has been reported that estrogen levels modify the hyperlipidemic profile of the disease. Estrogens increase both LDL receptor expression and lipolytic activity and as a result, men are more susceptible to HLP type III than women (Mahley et al., 1999). Together, these studies point to type III HLP as a multifactorial disease, requiring not only APOE2, but also genetic, hormonal or environmental factors, such as obesity, hypothyroidism, estrogen status or diabetes (Utermann, 1989).
Diabetes
Type II diabetes mellitus (T2DM) is one of the most common diseases in humans, with a high incidence and prevalence around the world (Bommer et al., 2018). CVD is the main cause of morbidity and mortality among diabetic patients (Rawshani et al., 2018). Insulin resistance and altered function of pancreatic β-cells are the two major conditions causing T2DM. Insulin resistance implies that insulin does not exert its function in insulin-sensitive tissues, such as skeletal muscle, adipose tissue, liver or endothelium. Pancreatic β-cells secrete, in turn, higher insulin levels to compensate for the reduced action on peripheral tissues and to maintain glucose levels within a normal range (Taylor, 1999).
Lipid metabolism plays an important role in this disease. Diabetic patients present an excess of circulating free fatty acids (FFAs) and TGs, as well as ectopic lipid storage in muscle and liver (McGarry, 2001). The association between APOE genotype and insulin resistance has been widely studied in subjects with or without AD although it still remains controversial (Elosua et al., 2003; Meigs et al., 2000; Profenno and Faraone, 2008; Ravona-Springer et al., 2014; Scuteri et al., 2005; Shinkuro et al., 1996; Valdez et al., 1995). Some studies found impaired glucose tolerance in APOE4 individuals (Elosua et al., 2003; Ravona-Springer et al., 2014; Scuteri et al., 2005; Valdez et al., 1995), while others did not observe this relationship (Meigs et al., 2000; Profenno and Faraone, 2008; Shinkuro et al., 1996). In addition, we reported that the expression of GLUT 4 in adipocytes correlated positively with APOE3, but not with APOE4 expression (Arbones-Mainar et al., 2008). A possible explanation is that APOE4 can reduce insulin‐ receptor substrate 1 expression and Akt phosphorylation (Ong et al., 2014), the latter required for insulin regulation of GLUT4 up-regulation and translocation (Thong et al., 2005). In contrast, the expression of a glucose transporter independent of insulin signaling (GLUT1) increased along with APOE4 expression (Arbones-Mainar et al., 2008). Decreased expression of GLUT4 with simultaneous increased expression of GLUT1 is conducive to insulin resistance in dystrophic adipose tissues (LaRosa et al., 2006). This suggests that cells expressing APOE4 have to increase the expression of GLUT1 in order to maintain glucose transport in the cell due to the lower response of GLUT4. This insensitivity to insulin action may have implications for other APOE-associated disorders where impaired glucose metabolism has been noted, such as AD (Keeney et al., 2015).
In this backgrounf of a T2DM epidemic, several antidiabetic drugs have been developed over time. A preliminary study in individuals on metformin and metformin-sulfonilurea combination therapy observed significantly improved cardiometabolic outcomes (2-hour glucose and systolic blood pressure) in APOE4 non-carriers when compared to APOE4 carriers (Sapkota et al., 2015). Likewise, evidence suggests that APOE polymorphisms impact the efficacy of rosiglitazone (a PPARγ agonist) on cognitive outcomes for AD patients (Iketani et al., 2018; Risner et al., 2006). However, pharmacogenetics studies have not found any APOE genetic variant deferentially associated with an antidiabetic response response to the most common i-diabetic drugs [reviewed in (Mannino et al., 2019)].
Obesity
According to the World Health Organization (WHO), obesity is defined as an excessive accumulation of fat that can be harmful to health. In recent years, obesity has become a serious public health problem in developed countries, increasing the risk of diabetes, dyslipidemia and hypertension. The crucial role of APOE in this disease has been demonstrated in APOE deficient (EKO) mice, which are thinner and more resistant to obesity than control mice (Chiba et al., 2003; Gao et al., 2007; Hofmann et al., 2008; Karagiannides et al., 2008). APOE’s effect on body mass may be due to its many roles in adipose tissue (described above in the “adipose tissue” section). Moreover, a leptin-sensitizing along with potential direct effects of APOE on neurons have also been proposed (Gao et al., 2007). In addition to reducing BMI, APOE deficiency also reduces some of the metabolic complications associated with obesity, including glucose intolerance and insulin resistance (Gao et al., 2007; Hofmann et al., 2008). As APOE promotes lipid accumulation, its absence might reduce ectopic fat deposition insulin sensitive tissues (Hofmann et al., 2008), which is a primary driver of impaired carbohydrate metabolism (McGarry, 2001).
However, there is some controversy regarding the relationship between APOE polymorphisms and obesity. While there are studies that show a strong association between the ε2 allele and the development of obesity (Duman et al., 2004; Tejedor et al., 2014; Volcik et al., 2006; Zeljko et al., 2011), others studies do not find any relationship (Zarkesh et al., 2012; Zhang et al., 2012). This disparity can be explained in part by the heterogeneity of the populations studied, the low number of individuals carrying the ε2 allele, and the relationship between APOE and plasma lipids, which makes it difficult to study the isoforms in relation to adiposity (Tejedor et al., 2014). In this regard, rodent models expressing human APOE2 isoform have shown a postprandial accumulation of lipids, increasing adiposity and susceptibility to obesity induced by diet (Kuhel et al., 2013; Pendse et al., 2009). In addition, an inflammatory status is promoted, accelerating tissue dysfunction and increasing infiltration of macrophages (Kuhel et al., 2013). However, unlike humans, these APOE2 mice develop hyperlipidemia even under low-fat diet conditions. Therefore, the mechanisms implied in triglyceride-rich lipoprotein metabolism seem to be different in humans versus mice (Kuhel et al., 2013). Interestingly, elderly women with APOE4 and AD lost weight while APOE4 non-carriers did not during 3.5 years of follow-up, after controlling for diabetes and exercise (Vanhanen et al., 2001).
Metabolic syndrome
Metabolic syndrome (MetS) is a combination of a number of conditions that includes central obesity, elevated blood pressure, hyperglycemia and dyslipidemia (Grundy et al., 2004). The strong heritability of these factors suggests that MetS may be regulated by genotype differences. In fact, previous studies have discovered clusters of genes related to this syndrome (including APOE) with a heritability of 30% (Bosy-Westphal et al., 2006).
The relationship between APOE4 and MetS has been evaluated in several populations and its presence has been considered either neutral (Lee et al., 2011; Miller et al., 2007; Onat et al., 2010; Teixeira et al., 2014) or a risk factor for metabolic syndrome (Das et al., 2009; Olivieri et al., 2007; Tao et al., 2011; Vučinić et al., 2014). However, a protective function for APOE4 has also been described in a study on grade III obese individuals, where APOE4 was more frequently found in carriers without MetS as compared to subjects suffering from MetS (Ferreira et al., 2011). These discrepancies might be the result of a limited number of subjects in the studies due to the low frequency of the ε2 and ε4 alleles within the population. Additionally, environmental and social factors can influence these results.
A study conducted by our group showed that the APOE4 isoform was associated with an increased risk of MetS only in overweight individuals (Torres-Perez et al., 2016). This relationship was not found in normal weight or obese subjects. This BMI-dependent effect might be explained because individuals with lower body mass index (BMI) may still overcome this adipose dysfunction while in obese subjects the mild increase of the MetS risk caused by carrying the ε4 allele would appear masked by the dramatic effect that an excessive expansion of the adipose tissue per se has on increasing the risk of developing MetS.
Alzheimer’s Disease
While this review has so far focused on the peripheral functions of APOE, there exist many parallels between the functions of APOE in the periphery and its functions in the CNS. This can be particularly appreciated in AD, where 60–80% of patients have at least one ε4 allele (Mayeux et al., 1998). AD is the most prevalent neurodegenerative disease, characterized by synapse loss and neuronal death (Hashimoto et al., 2003). Major hallmarks of the disease include deposition of amyloid β peptide into extracellular plaques, aggregation of hyperphosphorylated tau protein in neurofibrillary tangles, widespread neuroinflammation, and disruption of the blood-brain-barrier (BBB) (Wisniewski et al., 1997). APOE4 is the strongest genetic risk factor for late-onset sporadic AD (Mayeux et al., 1998) and has been implicated in each of these major hallmarks of the disease. In the context of the present review, we will briefly highlight that the role of APOE in AD is not limited solely to the CNS, but also manifests in the periphery.
The majority of APOE research in AD has focused on how the various isoforms interact with the amyloid pathway that regulates the synthesis and clearance of the amyloid β peptide (Aβ). Mechanisms implied in this neuronal dysfunction appear to be mainly two-fold: i) ε4 accelerates Aβ deposition in cholesterol-rich lipid rafts and ii) ε4 alters APOE receptor signaling to diminish protection against amyloid accumulation (Lane-Donovan and Herz, 2017). However, APOE also effects handling of Aβ in the periphery. Human ε4 targeted replacement mice injected with synthetic human Aβ42 cleared it more slowly from the plasma than ε2 or ε3 mice (Sharman et al., 2010).
A similar parallel between the periphery and CNS is seen with APOE’s effects on tau. In the CNS, APOE4 stimulates tau phosphorylation, leading to cytoskeletal disruption (Holtzman et al., 2000). However, APOE may also act upon tau in the periphery. It has been shown that young healthy APOE4 individuals had more phosphorylated tau in circulating peripheral lymphocytes than their APOE3 counterparts, which was associated with subjective cognitive impairment (Badia et al., 2013).
Inflammation is another major hallmark of AD. APOE4 is associated with increased proinflammatory cytokine production in the CNS [reviewed in (Fernandez et al., 2019)] and is intricately linked to the dysfunctional microglial phenotype prevalent in AD (Krasemann et al., 2017). Likewise, APOE4 is also associated with an increased inflammatory response in the periphery. For example, APOE4 carriers undergoing cardiopulmonary bypass surgery had increased proinflammatory IL8 and TNFa (Grocott et al., 2001), while the ε4 allele was associated with a lower antiinflammatory IL-10 serum levels in patients with coronary artery disease (Tziakas et al., 2006).
Lastly, it has been shown that APOE regulates the blood-brain barrier (BBB) integrity and that this regulation is isoform-dependent (Chernick et al., 2019). Peripheral APOE4 compromised the BBB integrity in some studies, but not all, in mice expressing human APOE isoforms [reviewed in (Yamazaki et al., 2019)]. Thus, the role of APOE in AD is clearly multifactorial, impinging upon not only the amyloid cascade hypothesis but also many of the other major hallmarks of the disease. Importantly, it is becoming clear that both peripheral and central APOE share the task of maintaining neurological health.
Fertility
Yet another site of APOE action is in the endometrium, which produces APOE and dramatically upregulates its mRNA during the implantation window (Kao et al., 2002). In addition, APOE is the major supplier of the cholesterol precursor required for estrogen and progesterone synthesis in steroids tissues (Gwynne and Strauss III, 1982). It has therefore been suggested that APOE might influence human fertility. In this regard, female E4 carriers have higher progesterone levels, implying higher fertility (Jasienska et al., 2015). Some studies have tried to measure the reproductive efficiency inferred by quantifying the offspring. This method may encounter some confounding factors, such as the use of contraceptive methods, the difficulty of covering the complete reproductive history of the subjects, and that the number of biological children in the case of men may be inaccurate. For this reason, these studies are usually carried out in subjects with advanced age or in pre-industrial populations in which the offspring is considered to correspond to reproductive efficiency. In this sense, the studies seem to point out that in the populations of Caucasian origin, the carriers of APOE3 have the greatest fertile potential (R. Corbo et al., 2004; Corbo et al., 2007; Gerdes et al., 1996), while APOE4 carriers present greater reproductive efficiency in populations with Afro-Ecuadorian origins and in the Cayapa Indians (R. M. Corbo et al., 2004). Interestingly, in all populations, the allele with the worst reproductive efficiency seems to be ε2 (R. Corbo et al., 2004; R. M. Corbo et al., 2004). However, we must be careful when drawing conclusions due to the previously mentioned confounding factors. It is worth noting that parity was found to be associated with a significantly lower AD onset age than nulliparity in E3/E3 and E2/E3 individuals, but not in women with APOE4 (Corbo et al., 2007).
Setarehbadi et al also reported a differential distribution of APOE genotype related to male fertility (Setarehbadi et al., 2012). However, in contrast to the studies mentioned above, they associated male APOE4 carriers with a higher risk for infertility. They suggest that differences between the spermatozoa lipid composition according to APOE allele affect sperm viability. The population studied and the environmental factors, among others, may explain the discrepancies observed (R. M. Corbo et al., 2004). In this regard we have observed that, when women’s race was considered, no effect of APOE isoforms on miscarriage risk was observed for black women. Interestingly we did detect increased odds for miscarrying in white pregnant women bearing APOE2 (Gamundi-Segura et al., 2016). This race-dependent effect may reconcile, at least partially, the conflicting reports of the association of APOE polymorphisms and fertility.
Longevity and aging
APOE is among the most studied genes associated with longevity. A pioneering study reported a cohort of centenarians enriched of ε2 alleles (Schächter et al., 1994). Thus, APOE2 has been associated with the lowest fertility but also the highest longevity (Figure 2). This phenomenon indicates a potential antagonistic pleiotropy and may explain the persistence of the ancestral ε4 allele, despite its indication in greater disease risk, as a sort of longevity-fertility trade-off (R. Corbo et al., 2004; Jasienska et al., 2015).
Figure 2.
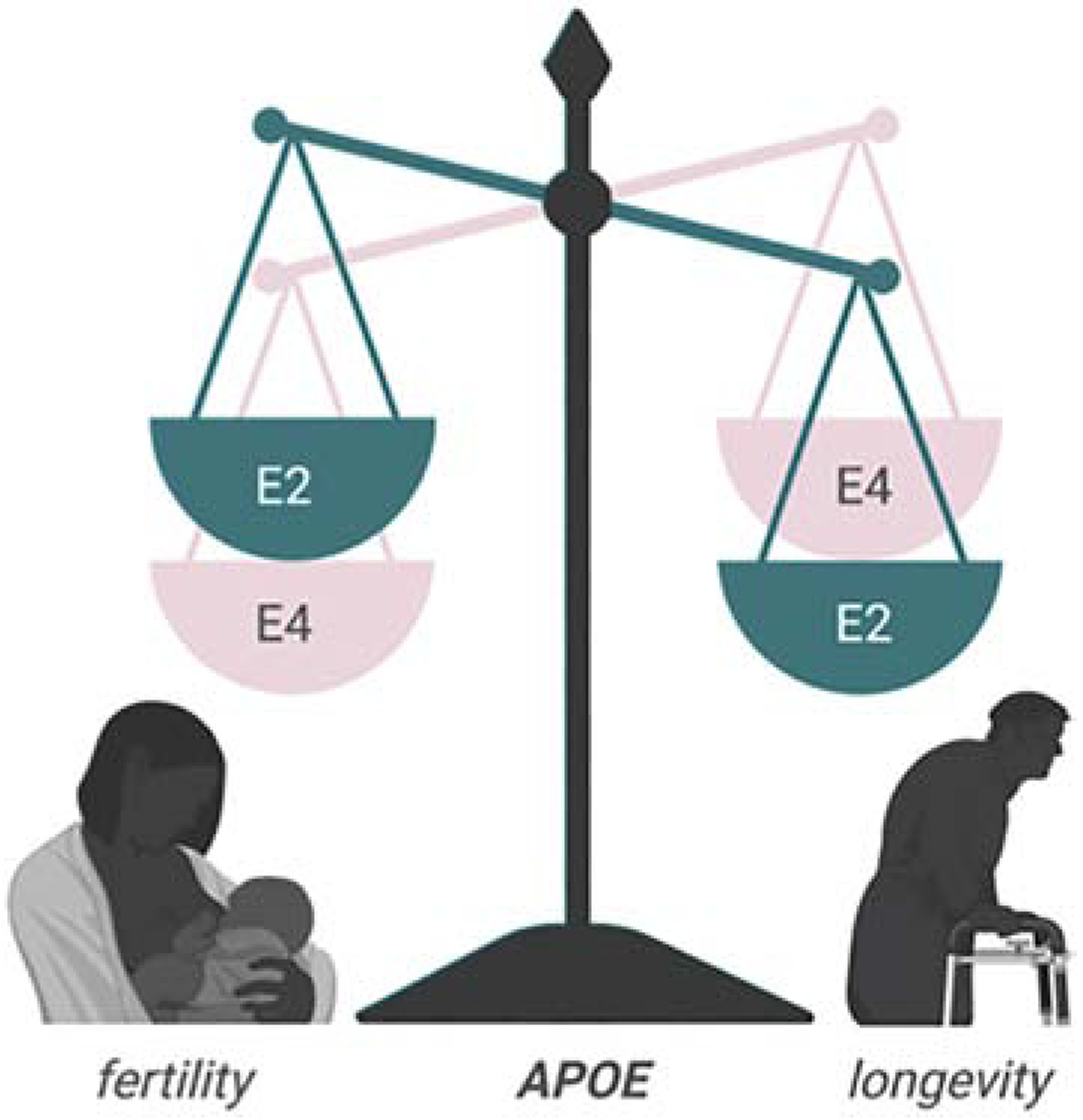
Antagonistic pleiotropy in APOE genotypes. APOE4 individuals enjoy enhanced fertility at the expense of longevity, whereas APOE2 individuals have increased longevity but reduced fertility.
Some genome-wide association studies (GWAS), but not all, have tied the ε4 allele of APOE to shortened longevity due to its role in multiple age-related diseases, whereas the ε2 allele is linked to increased longevity, mainly by its protective role against CVD and AD. Thus, APOE is an important player in aging and a major determinant of longevity (Ryu et al., 2016).
Infectious diseases
Cholesterol is essential for human immunodeficiency virus (HIV) entry and assembly (Liao et al., 2003; Mañes et al., 2000). Due to the influence of APOE in cholesterol transport, a correlation between HIV infection and APOE has been suggested. HIV infection may resemble an inflammation status akin to accelerated aging. Additionally, HIV replication in the brain is associated with several neurological disorders, including dementia. Therefore, APOE4 has been studied as a risk marker of HIV-associated neurocognitive disorders (Geffin and McCarthy, 2018). Nevertheless, this correlation has not been found in all APOE4 carriers. Becker et al suggested an age differential effect of ε4 allele in HIV patients, with elderly patients being more susceptible to neurocognitive decline (Becker et al., 2015). In concordance with this hypothesis, several studies point to an aging accelerated effect in brain HIV-infected APOE4 carriers through the interaction between HIV proteins and APOE4, increasing Aβ production and neurotoxicity to infected cells (Chang et al., 2014; Tuminello and Han, 2011; Wendelken et al., 2016).
Hepatitis C virus (HCV) associates with lipoproteins being secreted from the liver as highly infective lipoviro particles (LVP). Interestingly, APOE plays a relevant role in the assembly and production of these LVPs (Weller et al., 2017). The importance of this apolipoprotein is evidenced by the fact that APOE depletion has a significant effect in HCV particle production compared to APOB or APOA1 depletion (Benga et al., 2010). In this sense, some epidemiological studies have reported that APOE4 was associated with a reduced likelihood of HCV chronic infection (Mueller et al., 2016; Price et al., 2006). However, mixed results such as the fact that APOE4 was also associated with better histologic outcomes in recurrent HCV infection (Toniutto et al., 2004), no effect on the anti-viral response (Kim et al., 2013), or poor treatment response in HCV G1b patients (112), have likely precluded the use of APOE genotypes as biomarker in the morbidity of chronic HCV infection. Those discrepant results may reflect the limited sample size included in some studies and the failure to examine the interaction of APOE genotypes-HCV genotypes and its modulation by HCV treatments
APOE4 increases oxidative damage in the CNS and intensifies herpes simplex virus (HSV) latency (Kuhlmann et al., 2010). HSV-1 and APOE also compete to bind the same LDL receptor. HSV-1 has been detected in brain regions affected by AD, demonstrating its influence in β-amyloid production and accumulation. Burgos et al reported that the E4 allele is more permissive to the migration of HSV-1 from the adrenal gland to the brain (Burgos et al., 2006). They also showed higher latent HSV-1 DNA levels in E4 carriers. Thus, it is suggested that susceptibility for HSV-1 is increased by the presence of E4 allele. Furthermore, the combination of HSV-1 and APOE4 may increase the propensity to develop AD (Kuhlmann et al., 2010). It is hypothesized that higher LDL levels produced by E4 carriers promote accumulation of cholesterol in lipid rafts, facilitating the entry and latency of the herpes virus (Burt et al., 2008).
DISCUSSION
While the spectrum of biological systems in which APOE plays a role seems implausibly wide at first glance, there are some potential unifying mechanisms that could tie these seemingly disparate disorders together. Although liver transplantation studies have demonstrated that peripheral and brain APOE represent two distinct pools that are not exchangeable (Linton et al., 1991), APOE need not cross the blood brain barrier for its influence on the periphery to also be felt in the brain. Alterations to lipid trafficking, cellular metabolism, and immune function overlap and may act synergistically on both sides of this barrier to promote APOE’s widespread effects in the tissues, systems and disorders described in this review.
Cholesterol is the main constituent of many structural tissues in the periphery and is also critical in supporting the function of the brain, an organ which represents approximately 2% of total body weight yet contains as much as 25% of total cholesterol (Dietschy and Turley, 2001). As APOE is an important regulator of cholesterol transport and metabolism, alterations resulting from APOE isoform-specific differences have the capacity to produce greater impact, playing a role in multiple prevalent diseases. For example, the role of APOE in modulating LDL cholesterol levels in the background of CVD is well documented, and APOE isoform specific changes in glial cholesterol metabolism implicate these pathways in AD risk (Fernandez et al., 2019). Understanding the mechanisms by which APOE influences cholesterol homeostasis to confer disease risk will help to develop effective therapies for a wide range of maladies.
Although several of the molecular mechanisms through which APOE exerts its effects have been elucidated through studies in rodent models, often times contradictory results have been obtained according to the animal model studied. Human studies have reported yet even greater differences. APOE effects typically vary by age, often being more detrimental in elderly than in younger populations. For example, the magnitude of the associations between APOE genotype and plasma lipid levels differ according to sex and the population studied (Rasmussen, 2016). Results are further obfuscated by the complicated effect of different dosage levels of APOE alleles (Cacciaglia et al., 2018).
APOE-specific alterations in energy homeostasis and/or mitochondrial function are another potential area in which a single APOE isoform directed change could drive pathological changes in multiple disorders both centrally and in the periphery. The notion that APOE isoforms differentially mediate cell bioenergetics is gaining momentum as new findings challenge old assumptions (Kuehn, 2020). Along these lines, our group has shown that mice expressing human APOE4 have a global metabolic shift toward lipid oxidation and are inherently inefficient at utilizing glucose (Arbones-Mainar et al., 2016). This impaired glucose metabolism has implications for a number of disease states in which APOE plays a role. Mainly in the MetS and diabetes, as both conditions have defective glucose metabolism as their underlying cause, but also in CVD and in AD (Figure 3). The latter is characterized by a pattern of cerebral glucose hypometabolism, likely related to early mitochondrial dysfunction. This pattern is closely reflected in the brains of even young, cognitively normal individuals with APOE4 (Fukai et al., 2018). Mitochondria are essential for the regulation of the metabolic switch between lipids and glucose to fuel cellular functions. Some evidence points toward mitochondrial dysfunction as a primary event in AD onset (Weidling and Swerdlow, 2019). In this regard, APOE4 uniquely can be cleaved by a neuron-specific protease, resulting in toxic fragments of APOE4(1–272), that can bind mitochondrial complexes and disrupt mitochondrial energy balance (Nakamura et al., 2009). Alternatively, some studies propose a “systemic” mitochondrial dysfunction status as the trigger of a number of neurodegenerative and metabolic diseases such as T2DM, CVD, and AD (Bhatti et al., 2017).
Figure 3.
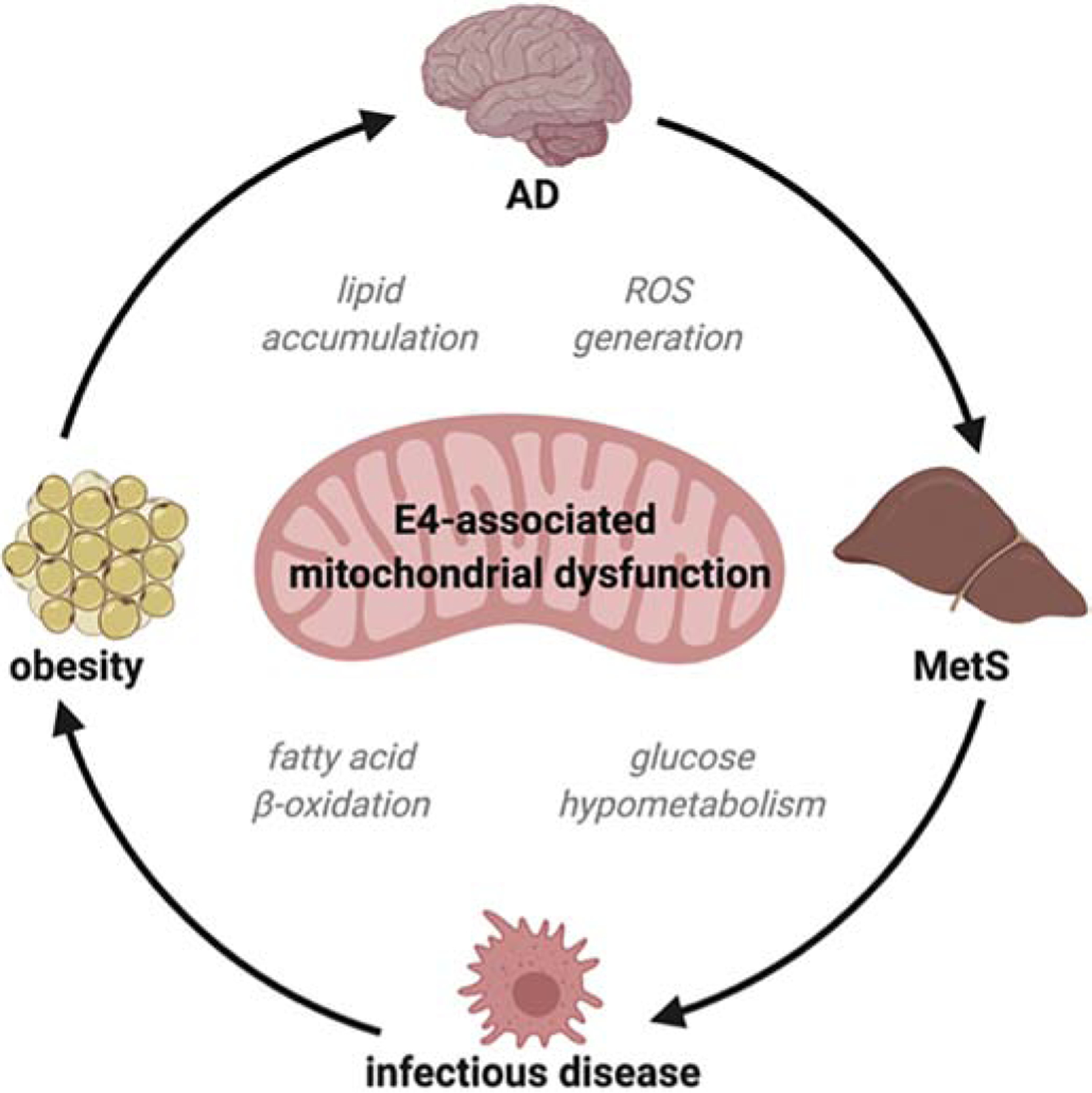
Mitochondrial dysfunction as a unifying APOE-directed mechanism.
Inflammatory responses are critical for an organism’s ability to respond to and eliminate pathogens, but widespread inflammation is also seen in the brain undergoing neurodegeneration. APOE drives inflammatory microglial phenotypes in AD, where isoform differences profoundly influence the production of inflammatory cytokines and the capacity of microglia to respond to and clear amyloid plaques and neuronal debris. Peripheral inflammatory responses are also modulated by APOE genotype, as is the case for disparate infections ranging from HIV to HCV to HSV. Furthermore, inflammation in the two compartments may interact. Peripheral inflammation damaged the integrity of the blood brain barrier in an AD mouse model with targeted replacement human APOE4, promoting cerebrovascular leakiness and reduced cerebral vessel coverage (Marottoli et al., 2017). Thus, APOE’s generalized effects on immune system function present a strong candidate for tying together the deleterious isoform-dependent differences seen in otherwise distinct pathologies and in distinct body compartments.
In summary, the effects of APOE across human biology are numerous and diverse. This critical protein has been shown to play a role in biological systems ranging from adipose tissue function and fertility to infection to neurological disease. Despite this diversity, there appear to be shared pathways that may provide insight into a unifying mechanism of action. While much remains to be discovered about this pleiotropic protein, research over the past 45 years has provided important insight into potential therapeutic targets that could have benefits across a wide range of disorders.
Acknowledgements:
We thank Prof. I. Expósito-Lopez & JM Suarez-Fernandez for their support and critical observations.
Funding: This study has been funded by project PI17/02268 (Instituto de Salud Carlos III) and by Fondo Europeo de Desarrollo Regional (FEDER): “Una manera de hacer Europa”. JMA-M is partially supported by a Miguel Servet fellowship (Instituto de Salud Carlos III) and by the DGA “Group Biology of adipose tissue and metabolic complications (B03_17R)”, co-financed with the FEDER Aragón 2014-2020: “Construyendo Europa desde Aragón”. ND is supported by funding from the National Institute of General Medical T32GM118292-03. LAJ is supported by funding from the National Institute on Aging 1R01AG060056-01, R01AG060056 02 and the National Institute of General Medical Sciences COBRE P20 GM103527
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
REFERENCES
- Altenburg M, Arbones-Mainar J, Johnson L, Wilder J, Maeda N, 2008. Human LDL receptor enhances sequestration of apoE4 and VLDL remnants on the surface of hepatocytes but not their internalization in mice. Arterioscler. Thromb. Vasc. Biol 28 10.1161/ATVBAHA.108.164863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenburg M, Johnson L, Wilder J, Maeda N, 2007. Apolipoprotein E4 in macrophages enhances atherogenesis in a low density lipoprotein receptor-dependent manner. J. Biol. Chem 282, 7817–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones-Mainar JM, Johnson LA, Altenburg MK, Maeda N, 2008. Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int. J. Obes 32 10.1038/ijo.2008.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones-Mainar JM, Johnson LA, Torres-Perez E, Garcia AE, Perez-Diaz S, Raber J, Maeda N, 2016. Metabolic shifts towards fatty acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int J Obes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badia M-C, Lloret A, Giraldo E, Dasí F, Olaso G, Alonso M-D, Viña J, 2013. Lymphocytes from young healthy persons carrying the ApoE4 allele overexpress stress-related proteins involved in the pathophysiology of Alzheimer’s disease. J. Alzheimer’s Dis 33, 77–83. [DOI] [PubMed] [Google Scholar]
- Becker JT, Martinson JJ, Penugonda S, Kingsley L, Molsberry S, Reynolds S, Aronow A, Goodkin K, Levine A, Martin E, 2015. No association between Apoε4 alleles, HIV infection, age, neuropsychological outcome, or death. J. Neurovirol 21, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann MW, Olson LM, Schreiber JR, 1991. Apolipoprotein E synthesis by cultured human ovarian granulosa cells: regulation by human chorionic gonadotropin and cholesterol. Fertil. Steril 55, 1118–1125. [DOI] [PubMed] [Google Scholar]
- Benga WJA, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C, 2010. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51, 43–53. 10.1002/hep.23278 [DOI] [PubMed] [Google Scholar]
- Bhatti JS, Bhatti GK, Reddy PH, 2017. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim. Biophys. acta. Mol. basis Dis 1863, 1066–1077. 10.1016/j.bbadis.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommer C, Sagalova V, Heesemann E, Manne-Goehler J, Atun R, Bärnighausen T, Davies J, Vollmer S, 2018. Global economic burden of diabetes in adults: projections from 2015 to 2030. Diabetes Care 41, 963–970. [DOI] [PubMed] [Google Scholar]
- Bosy-Westphal A, Onur S, Geisler C, Wolf A, Korth O, Pfeuffer M, Schrezenmeir J, Krawczak M, Muller MJ, 2006. Common familial influences on clustering of metabolic syndrome traits with central obesity and insulin resistance: the Kiel obesity prevention study. Int J Obes 31, 784–790. [DOI] [PubMed] [Google Scholar]
- Brown SA, Hay RV, Schreiber JR, 1989. Relationship between serum estrogen and level of apolipoprotein E in human ovarian follicular fluid. Fertil. Steril 51, 639–643. [DOI] [PubMed] [Google Scholar]
- Burgos JS, Ramirez C, Sastre I, Valdivieso F, 2006. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J. Virol 80, 5383–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt TD, Agan BK, Marconi VC, He W, Kulkarni H, Mold JE, Cavrois M, Huang Y, Mahley RW, Dolan MJ, McCune JM, Ahuja SK, 2008. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE ε4/ε4 genotype accelerates HIV disease progression. Proc Natl Acad Sci U S A 105 10.1073/pnas.0803526105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacciaglia R, Molinuevo JL, Falcón C, Brugulat-Serrat A, Sánchez-Benavides G, Gramunt N, Esteller M, Morán S, Minguillón C, Fauria K, Gispert JD, 2018. Effects of APOE-ε4 allele load on brain morphology in a cohort of middle-aged healthy individuals with enriched genetic risk for Alzheimer’s disease. Alzheimer’s Dement. 14, 902–912. [DOI] [PubMed] [Google Scholar]
- Cash JG, Kuhel DG, Basford JE, Jaeschke A, Chatterjee TK, Weintraub NL, Hui DY, 2012. Apolipoprotein E4 Impairs Macrophage Efferocytosis and Potentiates Apoptosis by Accelerating Endoplasmic Reticulum Stress. J. Biol. Chem 10.1074/jbc.M112.377549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Jiang C, Cunningham E, Buchthal S, Douet V, Andres M, Ernst T, 2014. Effects of APOE ε4, age, and HIV on glial metabolites and cognitive deficits. Neurology 82, 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernick D, Ortiz-Valle S, Jeong A, Qu W, Li L, 2019. Peripheral versus central nervous system APOE in Alzheimer’s disease: Interplay across the blood-brain barrier. Neurosci. Lett 708, 134306 10.1016/j.neulet.2019.134306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Nakazawa T, Yui K, Kaneko E, Shimokado K, 2003. VLDL induces adipocyte differentiation in ApoE-dependent manner. Arterioscler. Thromb. Vasc. Biol 23, 1423–9. 10.1161/01.ATV.0000085040.58340.36 [DOI] [PubMed] [Google Scholar]
- Chung BH, Segrest JP, 1983. Resistance of a very low density lipoprotein subpopulation from familial dysbetalipoproteinemia to in vitro lipolytic conversion to the low density lipoprotein density fraction. J. Lipid Res 24, 1148–1159. [PubMed] [Google Scholar]
- Corbo R, Scacchi R, Cresta M, 2004. Differential reproductive efficiency associated with common apolipoprotein e alleles in postreproductive-aged subjects. Fertil. Steril 81, 104–107. [DOI] [PubMed] [Google Scholar]
- Corbo RM, Gambina G, Ulizzi L, Monini P, Broggio E, Rosano A, Scacchi R, 2007. Combined effect of apolipoprotein e genotype and past fertility on age at onset of Alzheimer’s disease in women. Dement. Geriatr. Cogn. Disord 24, 82–85. [DOI] [PubMed] [Google Scholar]
- Corbo RM, Scacchi R, 1999. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann. Hum. Genet 63, 301–310. 10.1046/j.1469-1809.1999.6340301.x [DOI] [PubMed] [Google Scholar]
- Corbo RM, Ulizzi L, Scacchi R, Martínez-Labarga C, De Stefano GF, 2004. Apolipoprotein E polymorphism and fertility: a study in pre-industrial populations. Mol. Hum. Reprod 10, 617–620. 10.1093/molehr/gah082 [DOI] [PubMed] [Google Scholar]
- Dallongeville J, Lussier-Cacan S, Davignon J, 1992. Modulation of plasma triglyceride levels by apoE phenotype: a meta-analysis. J. Lipid Res 33, 447–454. [PubMed] [Google Scholar]
- Das HK, McPherson J, Bruns GA, Karathanasis SK, Breslow JL, 1985. Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J. Biol. Chem 260, 6240–6247. [PubMed] [Google Scholar]
- Das M, Pal S, Ghosh A, 2009. Synergistic effects of ACE (I/D) and ApoE (HhaI) gene polymorphisms among the adult Asian Indians with and without metabolic syndrome. Diabetes Res. Clin. Pract 86, e58–61. 10.1016/j.diabres.2009.09.011 [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD, 2001. Cholesterol metabolism in the brain. Curr. Opin. Lipidol 12, 105–112. [DOI] [PubMed] [Google Scholar]
- Duman BS, Oztürk M, Yilmazer S, Hatemi H, 2004. Apolipoprotein E polymorphism in Turkish subjects with Type 2 diabetes mellitus: allele frequency and relation to serum lipid concentrations. Diabetes. Nutr. Metab 17, 267–74. [PubMed] [Google Scholar]
- Elosua R, Demissie S, Cupples LA, Meigs JB, Wilson PWF, Schaefer EJ, Corella D, Ordovas JM, 2003. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes. Res 11, 1502–8. 10.1038/oby.2003.201 [DOI] [PubMed] [Google Scholar]
- Espiritu DJ, Mazzone T, 2008. Oxidative stress regulates adipocyte apolipoprotein e and suppresses its expression in obesity. Diabetes 57, 2992–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CG, Hamby ME, McReynolds ML, Ray WJ, 2019. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front. Aging Neurosci 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira DC, Costa TF, Aguiar SLF, Marques a. R.S., Ramos S. a., Gomes KB, Alvarez-Leite JI, 2011. Association of Apoliprotein E polymorphisms and metabolic syndrome in subjects with extreme obesity. Clin. Chim. Acta 412, 1559–1562. 10.1016/j.cca.2011.04.035 [DOI] [PubMed] [Google Scholar]
- Fukai M, Hirosawa T, Kikuchi M, Hino S, Kitamura T, Ouchi Y, Yokokura M, Yoshikawa E, Bunai T, Minabe Y, 2018. Different Patterns of Glucose Hypometabolism Underlie Functional Decline in Frontotemporal Dementia and Alzheimer’s Disease: FDG-PET Study. Neuropsychiatry (London). 8, 441–447. [Google Scholar]
- Fuster V, Badimon L, Badimon JJ, Chesebro JH, 1992. The pathogenesis of coronary artery disease and the acute coronary syndromes. N. Engl. J. Med 326, 310–318. [DOI] [PubMed] [Google Scholar]
- Gamundi-Segura S, Torres-Perez E, Sanz-Paris A, Arbones-Mainar JM, 2016. Interaction of apolipoprotein E gene polymorphisms on miscarriage risk in black and white American women. Fertil. Steril 105, 1554–1560.e1. 10.1016/j.fertnstert.2016.02.021 [DOI] [PubMed] [Google Scholar]
- Gao J, Katagiri H, Ishigaki Y, Yamada T, Ogihara T, Imai J, Uno K, Hasegawa Y, Kanzaki M, Yamamoto TT, Ishibashi S, Oka Y, 2007. Involvement of Apolipoprotein E in Excess Fat Accumulation and Insulin Resistance. Diabetes 56, 24–33. 10.2337/db06-0144 [DOI] [PubMed] [Google Scholar]
- Geffin R, McCarthy M, 2018. Aging and apolipoprotein E in HIV infection. J. Neurovirol 24, 529–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes LU, Gerdes C, Hansen PS, Klausen IC, Færgeman O, 1996. Are men carrying the apolipoprotein ε4- or ε2 allele less fertile than ε3ε3 genotypes? Hum. Genet 98, 239–242. 10.1007/s004390050200 [DOI] [PubMed] [Google Scholar]
- Germeyer A, Capp E, Schlicksupp F, Jauckus J, von Rango U, von Wolff M, Strowitzki T, 2013. Cell-type specific expression and regulation of apolipoprotein D and E in human endometrium. Eur. J. Obstet. Gynecol. Reprod. Biol 170, 487–491. [DOI] [PubMed] [Google Scholar]
- Getz GS, Reardon CA, 2009. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg RE, Zech LA, Schaefer EJ, Stark D, Wilson D, Brewer HB, 1986. Abnormal in vivo metabolism of apolipoprotein E4 in humans. J. Clin. Invest 78, 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grocott HP, Newman MF, El-Moalem H, Bainbridge D, Butler A, Laskowitz DT, 2001. Apolipoprotein E genotype differentially influences the proinflammatory and anti-inflammatory response to cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg 122, 622–623. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C, 2004. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 109, 433–8. 10.1161/01.CIR.0000111245.75752.C6 [DOI] [PubMed] [Google Scholar]
- Gwynne JT, Strauss JF III, 1982. The role of lipoproteins in steroidogenesis and cholesterol metabolism in steroidogenic glands. Endocr. Rev 3, 299–329. [DOI] [PubMed] [Google Scholar]
- Hanlon CS, Rubinsztein DC, 1995. Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis 112, 85–90. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Rockenstein E, Crews L, Masliah E, 2003. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med. 4, 21–35. [DOI] [PubMed] [Google Scholar]
- Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye K-A, Beisiegel U, 2004. Impaired Recycling of Apolipoprotein E4 Is Associated with Intracellular Cholesterol Accumulation. J. Biol. Chem 279, 55483–55492. 10.1074/jbc.M409324200 [DOI] [PubMed] [Google Scholar]
- Heeren J, Weber W, Beisiegel U, 1999. Intracellular processing of endocytosed triglyceride-rich lipoproteins comprises both recycling and degradation. J. Cell Sci 112, 349 LP–359. [DOI] [PubMed] [Google Scholar]
- Hofmann SM, Perez-Tilve D, Greer TM, Coburn BA, Grant E, Basford JE, Tschöp MH, Hui DY, 2008. Defective lipid delivery modulates glucose tolerance and metabolic response to diet in apolipoprotein E–deficient mice. Diabetes 57, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Hsiao Ashe K, Irizarry MC, Hyman BT, 2000. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc 47, 739–747. [PubMed] [Google Scholar]
- Huang Y, Mahley RW, 2014. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis 72, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZH, Luque RM, Kineman RD, Mazzone T, 2007. Nutritional regulation of adipose tissue apolipoprotein E expression. Am. J. Physiol. Metab 293, E203–E209. [DOI] [PubMed] [Google Scholar]
- Huang ZH, Maeda N, Mazzone T, 2011. Expression of the human apoE2 isoform in adipocytes: altered cellular processing and impaired adipocyte lipogenesis. J. Lipid Res 52, 1733–1741. 10.1194/jlr.M017160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZH, Minshall RD, Mazzone T, 2009. Mechanism for endogenously expressed ApoE modulation of adipocyte very low density lipoprotein metabolism: role in endocytic and lipase-mediated metabolic pathways. J. Biol. Chem 284, 31512–31522. 10.1074/jbc.M109.004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZH, Reardon CA, Getz GS, Maeda N, Mazzone T, 2015. Selective suppression of adipose tissue apoE expression impacts systemic metabolic phenotype and adipose tissue inflammation. J. Lipid Res 56, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZH, Reardon CA, Mazzone T, 2006. Endogenous ApoE expression modulates adipocyte triglyceride content and turnover. Diabetes 55, 3394–3402. 10.2337/db06-0354 [DOI] [PubMed] [Google Scholar]
- Iketani R, Ohno K, Kawasaki Y, Matsumoto K, Yamada H, Kishino S, 2018. Apolipoprotein E Gene Polymorphisms Affect the Efficacy of Thiazolidinediones for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Biol. Pharm. Bull 41, 1017–1023. 10.1248/bpb.b17-00929 [DOI] [PubMed] [Google Scholar]
- Jasienska G, Ellison PT, Galbarczyk A, Jasienski M, Kalemba-Drozdz M, Kapiszewska M, Nenko I, Thune I, Ziomkiewicz A, 2015. Apolipoprotein E (ApoE) polymorphism is related to differences in potential fertility in women: a case of antagonistic pleiotropy? Proc. R. Soc. London B Biol. Sci 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao LC, Tulac S, Lobo S, Imani B, Yang JP, Germeyer A, Osteen K, Taylor RN, Lessey BA, Giudice LC, 2002. Global gene profiling in human endometrium during the window of implantation. Endocrinology 143, 2119–2138. [DOI] [PubMed] [Google Scholar]
- Karagiannides I, Abdou R, Tzortzopoulou A, Voshol PJ, Kypreos KE, 2008. Apolipoprotein E predisposes to obesity and related metabolic dysfunctions in mice. FEBS J. 275, 4796–4809. [DOI] [PubMed] [Google Scholar]
- Keeney JT-R, Ibrahimi S, Zhao L, 2015. Human ApoE Isoforms Differentially Modulate Glucose and Amyloid Metabolic Pathways in Female Brain: Evidence of the Mechanism of Neuroprotection by ApoE2 and Implications for Alzheimer’s Disease Prevention and Early Intervention. J. Alzheimers. Dis 48, 411–424. 10.3233/JAD-150348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Lee K-M, Lee W-K, 2013. Association between Apolipoprotein E Genotype and Treatment Response in Chronic Hepatitis C. Ann Clin Microbiol 16, 69–74. [Google Scholar]
- Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N, 1999. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Invest 103, 1579–86. 10.1172/JCI6172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, 2017. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn BM, 2020. In Alzheimer Research, Glucose Metabolism Moves to Center Stage. JAMA. 10.1001/jama.2019.20939 [DOI] [PubMed] [Google Scholar]
- Kuhel DG, Konaniah ES, Basford JE, McVey C, Goodin CT, Chatterjee TK, Weintraub NL, Hui DY, 2013. Apolipoprotein E2 Accentuates Postprandial Inflammation and Diet-Induced Obesity to Promote Hyperinsulinemia in Mice. Diabetes 62, 382–391. 10.2337/db12-0390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann I, Minihane AM, Huebbe P, Nebel A, Rimbach G, 2010. Apolipoprotein E genotype and hepatitis C, HIV and herpes simplex disease risk: a literature review. Lipids Heal. Dis 9 10.1186/1476-511X-9-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, Holtzman DM, 1998. Nascent Astrocyte Particles Differ from Lipoproteins in CSF. J. Neurochem 70, 2070–2081. 10.1046/j.1471-4159.1998.70052070.x [DOI] [PubMed] [Google Scholar]
- Lane-Donovan C, Herz J, 2017. ApoE, ApoE receptors, and the synapse in Alzheimer’s disease. Trends Endocrinol. Metab 28, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRosa PC, Miner J, Xia Y, Zhou Y, Kachman S, Fromm ME, 2006. Trans-10, cis-12 conjugated linoleic acid causes inflammation and delipidation of white adipose tissue in mice: a microarray and histological analysis. Physiol. Genomics 27, 282–294. 10.1152/physiolgenomics.00076.2006 [DOI] [PubMed] [Google Scholar]
- Lee D-J, Kim K-M, Kim B-T, Kim K-N, Joo N-S, 2011. ApoE polymorphism may determine low-density lipoprotein cholesterol level in association with obesity and metabolic syndrome in postmenopausal Korean women. Yonsei Med. J 52, 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Dhanasekaran P, Alexander ET, Rader DJ, Phillips MC, Lund-Katz S, 2013. Molecular Mechanisms Responsible for the Differential Effects of ApoE3 and ApoE4 on Plasma Lipoprotein–Cholesterol Levels. Arterioscler. Thromb. Vasc. Biol 33, 687–693. 10.1161/ATVBAHA.112.301193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Graham DR, Hildreth JEK, 2003. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retroviruses 19, 675–687. [DOI] [PubMed] [Google Scholar]
- Linton MF, Fazio S, 1999. Macrophages, lipoprotein metabolism, and atherosclerosis: insights from murine bone marrow transplantation studies. Curr. Opin. Lipidol 10, 97–105. [DOI] [PubMed] [Google Scholar]
- Linton MF, Gish R, Hubl ST, Bütler E, Esquivel C, Bry WI, Boyles JK, Wardell MR, Young SG, 1991. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J. Clin. Invest 88, 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maaike K, Wendy J, Leonard K, 2008. Regulation of Endogenous Apolipoprotein E Secretion by Macrophages. Arterioscler. Thromb. Vasc. Biol 28, 1060–1067. 10.1161/ATVBAHA.108.164350 [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y, 2012. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Huang Y, Rall SC, 1999. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): questions, quandaries, and paradoxes. J Lipid Res. 40. [PubMed] [Google Scholar]
- Mahley Robert W., Innerarity TL, Weisgraber KH, Oh SY, 1979. Altered Metabolism (In Vivo and In Vitro) of Plasma Lipoproteins after Selective Chemical Modification of Lysine Residues of the Apoproteins. J. Clin. Invest 64, 743–750. 10.1172/JCI109518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Innerarity T, Brewer HB Jr, Assmann G, 1975. Swine lipoproteins and atherosclerosis. Changes in the plasma lipoproteins and apoproteins induced by cholesterol feeding. Biochemistry 14, 2817–2823. [DOI] [PubMed] [Google Scholar]
- Mahley Robert W, Weisgraber KH, Innerarity TL, 1979. Interaction of plasma lipoproteins containing apolipoproteins B and E with heparin and cell surface receptors. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab 575, 81–91. [DOI] [PubMed] [Google Scholar]
- Mañes S, del Real G, Lacalle RA, Lucas P, Gómez- Moutón C, Sánchez- Palomino S, Delgado R, Alcamí J, Mira E, Martínez- A C, 2000. Membrane raft microdomains mediate lateral assemblies required for HIV- 1 infection. EMBO Rep. 1, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannino GC, Andreozzi F, Sesti G, 2019. Pharmacogenetics of type 2 diabetes mellitus, the route toward tailored medicine. Diabetes. Metab. Res. Rev 35, e3109–e3109. 10.1002/dmrr.3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marottoli FM, Katsumata Y, Koster KP, Thomas R, Fardo DW, Tai LM, 2017. Peripheral inflammation, apolipoprotein E4, and amyloid-β interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro 9, 1759091417719201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux R, Saunders AM, Shea S, Mirra S, Evans D, Roses AD, Hyman BT, Crain B, Tang M-X, Phelps CH, 1998. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer’s disease. N. Engl. J. Med 338, 506–511. [DOI] [PubMed] [Google Scholar]
- McGarry JD, 2001. Banting Lecture 2001: Dysregulation of Fatty Acid Metabolism in the Etiology of Type 2 Diabetes. Diabetes 51, 7–18. 10.2337/diabetes.51.1.7 [DOI] [PubMed] [Google Scholar]
- McIntosh AM, Bennett C, Dickson D, Anestis SF, Watts DP, Webster TH, Fontenot MB, Bradley BJ, 2012. The apolipoprotein E (APOE) gene appears functionally monomorphic in chimpanzees (Pan troglodytes). PLoS One 7, e47760–e47760. 10.1371/journal.pone.0047760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meigs JB, Ordovas JM, Cupples LA, Singer DE, Nathan DM, Schaefer EJ, Wilson PW, 2000. Apolipoprotein E isoform polymorphisms are not associated with insulin resistance: the Framingham Offspring Study. Diabetes Care 23, 669 LP–674. 10.2337/diacare.23.5.669 [DOI] [PubMed] [Google Scholar]
- Mestas J, Ley K, 2008. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med 18, 228–232. 10.1016/j.tcm.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Rhyne J, Chen H, Beach V, Ericson R, Luthra K, Dwivedi M, Misra A, 2007. APOC3 Promoter Polymorphisms C-482T and T-455C Are Associated with the Metabolic Syndrome1. Arch. Med. Res 38, 444–451. 10.1016/j.arcmed.2006.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T, Fischer J, Gessner R, Rosendahl J, Böhm S, van Bömmel F, Knop V, Sarrazin C, Witt H, Marques AM, Kovacs P, Schleinitz D, Stumvoll M, Blüher M, Bugert P, Schott E, Berg T, 2016. Apolipoprotein E allele frequencies in chronic and self-limited hepatitis C suggest a protective effect of APOE4 in the course of hepatitis C virus infection. Liver Int. 36, 1267–1274. 10.1111/liv.13094 [DOI] [PubMed] [Google Scholar]
- Mueller T, Gessner R, Sarrazin C, Graf C, Halangk J, Witt H, Kottgen E, Wiedenmann B, Berg T, 2003. Apolipoprotein E4 allele is associated with poor treatment response in hepatitis C virus (HCV) genotype 1. Hepatology 38, 1592; author reply 1592–3. 10.1016/j.hep.2003.09.042 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M, 2009. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener. 4 10.1186/1750-1326-4-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicosia M, Moger WH, Dyer CA, Prack MM, Williams DL, 1992. Apolipoprotein-E messenger RNA in rat ovary is expressed in theca and interstitial cells and presumptive macrophage, but not in granulosa cells. Mol. Endocrinol 6, 978–988. [DOI] [PubMed] [Google Scholar]
- Olivieri O, Martinelli N, Bassi A, Trabetti E, Girelli D, Pizzolo F, Friso S, Pignatti PF, Corrocher R, 2007. ApoE epsilon2/epsilon3/epsilon4 polymorphism, ApoC-III/ApoE ratio and metabolic syndrome. Clin. Exp. Med 7, 164–72. 10.1007/s10238-007-0142-y [DOI] [PubMed] [Google Scholar]
- Onat A, Kömürcü-Bayrak E, Can G, Küçükdurmaz Z, Hergenç G, Erginel-Unaltuna N, 2010. Apolipoprotein A-I positively associated with diabetes in women independently of apolipoprotein E genotype and apolipoprotein B levels. Nutrition 26, 975–980. 10.1016/j.nut.2009.09.023 [DOI] [PubMed] [Google Scholar]
- Ong Q-R, Chan ES, Lim M-L, Wong B-S, 2014. Expression of human apolipoprotein E4 reduces insulin-receptor substrate 1 expression and Akt phosphorylation in the ageing liver. FEBS Open Bio 4, 260–265. 10.1016/j.fob.2014.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendse AA, Arbones-Mainar JM, Johnson LA, Altenburg MK, Maeda N, 2009. Apolipoprotein E knock-out and knock-in mice: Atherosclerosis, metabolic syndrome, and beyond. J. Lipid Res 50 10.1194/jlr.R800070-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL, 1992. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 71, 343–353. [DOI] [PubMed] [Google Scholar]
- Price DA, Bassendine MF, Norris SM, Golding C, Toms GL, Schmid ML, Morris CM, Burt AD, Donaldson PT, 2006. Apolipoprotein ε3 allele is associated with persistent hepatitis C virus infection. Gut 55, 715 LP–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profenno LA, Faraone SV, 2008. Diabetes and overweight associate with non-APOE4 genotype in an Alzheimer’s disease population. Am. J. Med. Genet. B. Neuropsychiatr. Genet 147B, 822–9. 10.1002/ajmg.b.30694 [DOI] [PubMed] [Google Scholar]
- Rall SC, Weisgraber KH, Innerarity TL, Mahley RW, 1982. Structural basis for receptor binding heterogeneity of apolipoprotein E from type III hyperlipoproteinemic subjects. Proc. Natl. Acad. Sci 79, 4696–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P, Huang ZH, Mazzone T, 2007. Angiotensin II regulates adipocyte apolipoprotein E expression. J. Clin. Endocrinol. Metab 92, 4366–4372. [DOI] [PubMed] [Google Scholar]
- Rasmussen KL, 2016. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: a review. Atherosclerosis 255, 145–155. [DOI] [PubMed] [Google Scholar]
- Ravona-Springer R, Heymann A, Schmeidler J, Sano M, Preiss R, Koifman K, Hoffman H, Silverman JM, Beeri MS, 2014. The ApoE4 genotype modifies the relationship of long-term glycemic control with cognitive functioning in elderly with type 2 diabetes. Eur. Neuropsychopharmacol 24, 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawshani Aidin, Rawshani Araz, Franzén S, Sattar N, Eliasson B, Svensson A-M, Zethelius B, Miftaraj M, McGuire DK, Rosengren A, 2018. Risk factors, mortality, and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med 379, 633–644. [DOI] [PubMed] [Google Scholar]
- Reddick RL, Zhang SH, Maeda N, 1994. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler. Thromb. a J. Vasc. Biol 14, 141–147. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y, Wagner E, Parratt A, Xu J, Li Z, Zaleska MM, Jacobsen JS, Pangalos MN, Reinhart PH, 2008. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 28 10.1523/JNEUROSCI.1972-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risner ME, Saunders AM, Altman JFB, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD, Group, for the R. in A.D.S., 2006. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254. 10.1038/sj.tpj.6500369 [DOI] [PubMed] [Google Scholar]
- Ryu S, Atzmon G, Barzilai N, Raghavachari N, Suh Y, 2016. Genetic landscape of APOE in human longevity revealed by high-throughput sequencing. Mech. Ageing Dev 155, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota B, Subramanian A, Priamvada G, Finely H, Blackett PR, Aston CE, Sanghera DK, 2015. Association of APOE polymorphisms with diabetes and cardiometabolic risk factors and the role of APOE genotypes in response to antidiabetic therapy: results from the AIDHS/SDS on a South Asian population. J. Diabetes Complications 29, 1191–1197. 10.1016/j.jdiacomp.2015.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schächter F, Faure-Delanef L, Guénot F, Rouger H, Froguel P, Lesueur-Ginot L, Cohen D, 1994. Genetic associations with human longevity at the APOE and ACE loci. Nat Genet. 6 10.1038/ng0194-29 [DOI] [PubMed] [Google Scholar]
- Schaefer EJ, Gregg RE, Ghiselli G, Forte TM, Ordovas JM, Zech LA, Brewer HB, 1986. Familial apolipoprotein E deficiency. J. Clin. Invest 78, 1206–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuteri A, Najjar SS, Muller D, Andres R, Morrell CH, Zonderman AB, Lakatta EG, 2005. ApoE4 allele and the natural history of cardiovascular risk factors. Am. J. Physiol. Endocrinol. Metab 289, E322–7. 10.1152/ajpendo.00408.2004 [DOI] [PubMed] [Google Scholar]
- Setarehbadi R, Vatannejad A, Vaisi-Raygani A, Amiri I, Esfahani M, Fattahi A, Tavilani H, 2012. Apolipoprotein E genotypes of fertile and infertile men. Syst. Biol. Reprod. Med 58, 263–267. 10.3109/19396368.2012.684134 [DOI] [PubMed] [Google Scholar]
- Sharman MJ, Morici M, Hone E, Berger T, Taddei K, Martins IJ, Lim WLF, Singh S, Wenk MR, Ghiso J, Buxbaum JD, Gandy S, Martins RN, 2010. APOE genotype results in differential effects on the peripheral clearance of amyloid-beta42 in APOE knock-in and knock-out mice. J. Alzheimers. Dis 21, 403–409. 10.3233/JAD-2010-100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkuro K, C. RD, D. CL, Oscar G, L. YJ, B. DR, R. FR, T. LE, K. WT, H.B. V, 1996. Apolipoprotein E Polymorphism in American Indians and Its Relation to Plasma Lipoproteins and Diabetes. Arterioscler. Thromb. Vasc. Biol 16, 918–925. 10.1161/01.ATV.16.8.918 [DOI] [PubMed] [Google Scholar]
- Shore VG, Shore B, 1973. Heterogeneity of human plasma very low density lipoproteins. Separation of species differing in protein components. Biochemistry 12, 502–507. [DOI] [PubMed] [Google Scholar]
- Shore VG, Shore B, Hart RG, 1974. Changes in apolipoproteins and properties of rabbit very low density lipoproteins on induction of cholesteremia. Biochemistry 13, 1579–1585. [DOI] [PubMed] [Google Scholar]
- Sing CF, Davignon J, 1985. Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am. J. Hum. Genet 37, 268. [PMC free article] [PubMed] [Google Scholar]
- Stannard AK, Riddell DR, Sacre SM, Tagalakis AD, Langer C, von Eckardstein A, Cullen P, Athanasopoulos T, Dickson G, Owen JS, 2001. Cell-derived apolipoprotein E (ApoE) particles inhibit vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J. Biol. Chem 276, 46011–46016. [DOI] [PubMed] [Google Scholar]
- Sundqvist J, Andersson KL, Scarselli G, Gemzell-Danielsson K, Lalitkumar PGL, 2012. Expression of adhesion, attachment and invasion markers in eutopic and ectopic endometrium: a link to the aetiology of endometriosis. Hum. Reprod 27, 2737–2746. [DOI] [PubMed] [Google Scholar]
- Tao MH, Liu JW, Lamonte MJ, Liu J, Wang L, He Y, Li XY, Wang LN, Ye L, 2011. Different associations of apolipoprotein E polymorphism with metabolic syndrome by sex in an elderly Chinese population. Metabolism. 60, 1488–1496. 10.1016/j.metabol.2011.03.004 [DOI] [PubMed] [Google Scholar]
- Taylor SI, 1999. Deconstructing type 2 diabetes. Cell 97, 9–12. [DOI] [PubMed] [Google Scholar]
- Teixeira AA, Marrocos MS, Quinto BMR, Dalboni MA, Rodrigues CJ de O, Carmona, S. de M, Kuniyoshi M, Batista MC, 2014. Diversity of apolipoprotein E genetic polymorphism significance on cardiovascular risk is determined by the presence of metabolic syndrome among hypertensive patients. Lipids Health Dis. 13, 174 10.1186/1476-511X-13-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor MT, Garcia-Sobreviela MP, Ledesma M, Arbones-Mainar JM, 2014. The Apolipoprotein E Polymorphism rs7412 Associates with Body Fatness Independently of Plasma Lipids in Middle Aged Men. PLoS One 9, e108605 10.1371/journal.pone.0108605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thong FSL, Dugani CB, Klip A, 2005. Turning Signals On and Off: GLUT4 Traffic in the Insulin-Signaling Highway. Physiology 20, 271–284. 10.1152/physiol.00017.2005 [DOI] [PubMed] [Google Scholar]
- Toniutto P, Fabris C, Fumo E, Apollonio L, Caldato M, Mariuzzi L, Avellini C, Minisini R, Pirisi M, 2004. Carriage of the apolipoprotein E-ε4 allele and histologic outcome of recurrent hepatitis C after antiviral treatment. Am. J. Clin. Pathol 122, 428–433. [DOI] [PubMed] [Google Scholar]
- Torres-Perez E, Ledesma M, Garcia-Sobreviela MP, Leon-Latre M, Arbones-Mainar JM, 2016. Apolipoprotein E4 association with metabolic syndrome depends on body fatness. Atherosclerosis 245, 35–42. 10.1016/j.atherosclerosis.2015.11.029 [DOI] [PubMed] [Google Scholar]
- Tuminello ER, Han SD, 2011. The apolipoprotein e antagonistic pleiotropy hypothesis: review and recommendations. Int. J. Alzheimer’s Dis 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tziakas DN, Chalikias GK, Antonoglou CO, Veletza S, Tentes IK, Kortsaris AX, Hatseras DI, Kaski JC, 2006. Apolipoprotein E genotype and circulating interleukin-10 levels in patients with stable and unstable coronary artery disease. J. Am. Coll. Cardiol 48, 2471–2481. [DOI] [PubMed] [Google Scholar]
- Ulrich V, Konaniah ES, Herz J, Gerard RD, Jung E, Yuhanna IS, Ahmed M, Hui DY, Mineo C, Shaul PW, 2014. Genetic variants of ApoE and ApoER2 differentially modulate endothelial function. Proc. Natl. Acad. Sci 111, 13493–13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utermann G, 1989. Apolipoprotein variation: Effect on plasma lipid variability, in: Human Apolipoprotein Mutants 2 Springer, pp. 9–16. [Google Scholar]
- Utermann G, 1975. Isolation and partial characterization of an arginine-rich apolipoprotein from human plasma very-low-density lipoproteins: apolipoprotein E. Hoppe-Seyleŕ s Zeitschrift für Physiol. Chemie 356, 1113–1122. [DOI] [PubMed] [Google Scholar]
- Utermann G, Hees M, Steinmetz A, 1977. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature 269, 604–607. [DOI] [PubMed] [Google Scholar]
- Valdez R, Howard BV, Stern MP, Haffner SM, 1995. Apolipoprotein E polymorphism and insulin levels in a biethnic population. Diabetes Care 18, 992–1000. [DOI] [PubMed] [Google Scholar]
- Vanhanen M, Kivipelto M, Koivisto K, Kuusisto J, Mykkänen L, Helkala E-L, Hänninen T, Kervinen K, Kesäniemi YA, Laakso MP, Soininen H, Laakso M, 2001. APOε4 is associated with weight loss in women with AD. Neurology 56, 655 LP–659. 10.1212/WNL.56.5.655 [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Greaves DR, Murray PJ, Chawla A, 2006. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volcik KA, Barkley RA, Hutchinson RG, Mosley TH, Heiss G, Sharrett AR, Ballantyne CM, Boerwinkle E, 2006. Apolipoprotein E polymorphisms predict low density lipoprotein cholesterol levels and carotid artery wall thickness but not incident coronary heart disease in 12,491 ARIC study participants. Am J Epidemiol 164, 342–348. 10.1093/aje/kwj202 [DOI] [PubMed] [Google Scholar]
- Von Wald T, Monisova Y, Hacker MR, Yoo SW, Penzias AS, Reindollar RR, Usheva A, 2010. Age-related variations in follicular apolipoproteins may influence human oocyte maturation and fertility potential. Fertil. Steril 93, 2354–2361. [DOI] [PubMed] [Google Scholar]
- Vučinić N, Djan I, Stokić E, Božin B, Obreht D, Stankov K, Djan M, 2014. Different associations of apoE gene polymorphism with metabolic syndrome in the Vojvodina Province (Serbia). Mol. Biol. Rep 41, 5221–5227. 10.1007/s11033-014-3390-4 [DOI] [PubMed] [Google Scholar]
- Weidling I, Swerdlow RH, 2019. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Biology (Basel). 8, 39 10.3390/biology8020039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub MS, Eisenberg S, Breslow JL, 1987. Dietary fat clearance in normal subjects is regulated by genetic variation in apolipoprotein E. J. Clin. Invest 80, 1571–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisgraber KH, 1994. Apolipoprotein E: structure-function relationships. Adv Protein Chem 45 10.1016/S0065-3233(08)60642-7 [DOI] [PubMed] [Google Scholar]
- Weller R, Hueging K, Brown RJP, Todt D, Joecks S, Vondran FWR, Pietschmann T, 2017. Hepatitis C virus strain-dependent usage of apolipoprotein E modulates assembly efficiency and specific infectivity of secreted virions. J. Virol. JVI 00422–17. 10.1128/JVI.00422-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendelken LA, Jahanshad N, Rosen HJ, Busovaca E, Allen I, Coppola G, Adams C, Rankin KP, Milanini B, Clifford K, 2016. ApoE ε4 is associated with cognition, brain integrity and atrophy in HIV over age 60. J. Acquir. Immune Defic. Syndr 73, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner C, Lorkowski S, Engel T, Cullen P, 2001. Apolipoprotein E in Macrophages and Hepatocytes Is Degraded via the Proteasomal Pathway. Biochem. Biophys. Res. Commun 282, 608–614. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Ghiso J, Frangione B, 1997. Biology of Aβ Amyloid in Alzheimer’s Disease. Neurobiol. Dis 4, 313–328. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G, 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol 15, 501–518. 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Bian J-T, Grizelj I, Cavka A, Phillips SA, Makino A, Mazzone T, 2012. Apolipoprotein E enhances endothelial-NO production by modulating caveolin 1 interaction with endothelial NO synthase. Hypertens. (Dallas, Tex. 1979) 60, 1040–1046. 10.1161/HYPERTENSIONAHA.112.196667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Mazzone T, 2009. Peroxisome proliferator-activated receptor {gamma} stimulation of adipocyte ApoE gene transcription mediated by the liver receptor X pathway. J. Biol. Chem 284, 10453–10461. 10.1074/jbc.M808482200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Rasouli N, Ranganathan G, Kern PA, Mazzone T, 2004. Divergent effects of peroxisome proliferator-activated receptor γ agonists and tumor necrosis factor α on adipocyte ApoE expression. J. Biol. Chem 279, 47626–47632. [DOI] [PubMed] [Google Scholar]
- Zannis VI, Just PW, Breslow JL, 1981. Human apolipoprotein E isoprotein subclasses are genetically determined. Am. J. Hum. Genet 33, 11. [PMC free article] [PubMed] [Google Scholar]
- Zarkesh M, Daneshpour MS, Faam B, Hedayati M, Azizi F, 2012. Is there any association of apolipoprotein E gene polymorphism with obesity status and lipid profiles? Tehran Lipid and Glucose Study (TLGS). Gene 7–10. 10.1016/j.gene.2012.07.048 [DOI] [PubMed] [Google Scholar]
- Zechner R, Moser R, Newman TC, Fried SK, Breslow JL, 1991. Apolipoprotein E gene expression in mouse 3T3–L1 adipocytes and human adipose tissue and its regulation by differentiation and lipid content. J. Biol. Chem 266, 10583–8. [PubMed] [Google Scholar]
- Zeljko H, Skaric-Juric T, Narancic N, Tomas Z, Baresic A, Salihovic M, Starcevic B, Janicijevic B, 2011. E2 allele of the Apolipoprotein E gene polymorphism is predictive for obesity status in Roma minority population of Croatia. Lipids Health Dis. 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Dyer CA, 1999. Apolipoprotein E peptide stimulation of rat ovarian theca cell androgen synthesis is mediated by members of the low density lipoprotein receptor superfamily. Biol. Reprod 61, 665–672. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang X, Fan P, Liu R, Huang Y, Liang S, Liu Y, Wu Y, Bai H, 2012. Distribution and Effect of apo E Genotype on Plasma Lipid and Apolipoprotein Profiles in Overweight/Obese and Nonobese C hinese Subjects. J. Clin. Lab. Anal 26, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Reddick RL, Piedrahita JA, Maeda N, 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science (80-.) 258 10.1126/science.1411543 [DOI] [PubMed] [Google Scholar]