

Abstract
Chronic fatty liver disease is common worldwide. This disease is a spectrum of disease states, ranging from simple steatosis (fat accumulation) to inflammation, and eventually to fibrosis and cirrhosis if untreated. The fibrotic stage of chronic liver disease is primarily characterized by robust accumulation of extracellular matrix (ECM) proteins (collagens) that ultimately impairs the function of the organ. The role of the ECM in early stages of chronic liver disease is less well-understood, but recent research has demonstrated that several changes in the hepatic ECM in prefibrotic liver disease are not only present but may also contribute to disease progression. The purpose of this review is to summarize the established and proposed changes to the hepatic ECM that may contribute to inflammation during earlier stages of disease development, and to discuss potential mechanisms by which these changes may mediate the progression of the disease.
Keywords: liver disease, hepatic, acute phase
The extracellular matrix (ECM) consists of a diverse range of components that work bidirectionally with surrounding cells to create a dynamic and responsive microenvironment that regulates cell and tissue homeostasis. The basic definition of the ECM comprises fibrillar proteins (e.g., collagens, glycoproteins, and proteoglycans). The definition has more recently been extended to include ECM affiliated proteins (e.g., collagen-related proteins), ECM regulator/modifier proteins (e.g., lysyl oxidases and proteases), and secreted factors that bind to the ECM (e.g., transforming growth factor-beta and other cytokines)1; this broader definition has been coined the “matrisome.”2 The ECM not only provides structure and support for the cells in a tissue but also acts as a reservoir for growth factors and cytokines and as a signaling mechanism by which cells can communicate with their environment and vice-versa.3
Perhaps the best-characterized function of the ECM is its role as a scaffold, providing support and structure to the surrounding tissue. There are two major components of structural ECM: the interstitial matrix and the basement membrane.4 Interstitial matrix proteins, including fibronectins, elastin, and fibrillar collagens, form support networks that provide the overall superstructure that shapes and encapsulates the organ.5 The basement membrane is a thin sheet of ECM that underlies epithelial and endothelial cells. Similar to the interstitial matrix, the basement membrane comprises collagens, glycoproteins, and proteoglycans that facilitate structure and growth of the cells. In most tissues, the basement membrane is continuous and dense and is a true barrier between the epithelial/endothelial cells and the adjacent parenchymal cell layer. In contrast, the hepatic basement membrane found in the Space of Disse between endothelial cells and hepatocytes is much less dense and is fenestrated.5 Although it possesses similar ECM as more strongly define basement membranes (e.g., collagen type IV and laminin),6 this region acts more as a structural filter and facilitates bidirectional exchange of proteins and xenobiotics between the sinusoidal blood and hepatocytes. Although it is clear that liver does not have a basal lamina, whether or not the ECM found in the space of Disse should be considered a basement membrane is a subject of a histological, rather than functional, debate.4
ECM (dys)Homeostasis
As mentioned above, the ECM responds dynamically to changes. Under normal conditions, these responses assist in maintaining organ homeostasis and appropriate responses to injury/stress. The orchestrated crosstalk between the coagulation cascade and the inflammatory response during subcutaneous wound healing is an excellent example of appropriate ECM changes in response to injury/stress.7 However, failure to properly regulate these responses can lead to qualitative and/or quantitative ECM changes that are maladaptive.8 For example, “aging” of the ECM with increased crosslinking is hypothesized to contribute to dysfunction in several organ systems, including the liver.9–12 Key levels of ECM homeostasis, be it adaptive or maladaptive, include synthesis, proteolysis, and post-translational modifications.
ECM Synthesis
Under basal conditions, several hepatic cells contribute to the synthesis of ECM components, including hepatocytes, cholangiocytes, and sinusoidal endothelial cells.13 Kupffer cells do not normally synthesize fibrillar ECM, but they do produce several secreted factors (e.g., cytokines) that are associated with the ECM. The amount and content of ECM components produced by these cells change in response to injury or stress. Although it is unclear if hepatic stellate cells (HSC) generate significant ECM during normal tissue homeostasis, activated HSCs transdifferentiate into a myofibroblast-like phenotype and generate ECM.5 Furthermore, other myofibroblast-like cells have been identified, such as fibrocytes and periportal fibroblasts.14–17 The contribution of extrahepatic sources to the hepatic ECM via de novo synthesis is unclear, but these compartments clearly contribute to ECM via other mechanisms of homeostasis.
Proteolysis
Protein families that degrade ECM include matrix metalloproteinases (MMPs), a disintegrin and metalloproteinases (ADAMs), a disintegrin and metalloproteinases with thrombospondin motifs (ADAMTS), cathepsins, and plasmin.18–22 The activity of these proteases is often balanced by protease inhibitors that directly inhibit their activity. For example, MMP activities are inhibited by tissue inhibitors of metalloproteinases (TIMPs), and contribute to collagen accumulation during hepatic fibrosis.23 Similarly, macrophage-derived MMP-12 expression, activity and ratio to its inhibitor TIMP-1 regulates elastin turnover in liver injury and fibrosis.24 This mechanism is proposed to be critical for the full reversibility of liver fibrosis.25 Likewise, plasminogen activator inhibitors (e.g., PAI-1) inhibit the activity of the plasminogen activators and thereby contribute to the accumulation of fibrin ECM during hepatic injury.22 Proteases can also regulate the deposition of hepatic ECM by cleaving soluble precursors of the ECM proteins. The serine proteases of the coagulation cascade are a canonical example of an acute phase response that leads to the formation of a fibrin clot.22
Post-Translational Modifications
Post-translational modification of ECM proteins regulates the formation of polymeric, helical structures, and cross-linked complexes associated with several fibrillary ECM proteins. For example, prolyl 4-hydroxylase targets terminal proline residues on collagen monomers to facilitate the formation of collagen oligomers and triple helices.26 Recent studies indicate that lysyl oxidases and transglutaminases also contribute to ECM crosslinking.27,28 Although these events are important for stabilizing the proteins and preventing their degradation under normal conditions, their activation may contribute to excessive ECM accumulation in response to injury (e.g., fibrosis).27 Furthermore, although fibrosis is potentially reversible if the causative insult is removed,29 highly crosslinked ECM may be resistant to resolution.30,31 Crosslinking of the ECM may be altered via nonezymatic means; for example, the formation of advanced glycation end products during diabetes is hypothesized to contribute to ECM crosslinking and increased matrix “aging.”32
Inflammation as a Therapeutic Target for Chronic Liver Disease
The strategic location of the liver between the intestinal tract and the rest of the body makes it a critical physical and biochemical barrier against toxins/toxicants that enter the portal blood. However, as the main detoxifying organ in the body, the liver has a high likelihood of toxic injury.33,34 It is, therefore, not surprising that the liver has tremendous regenerative capacity.33,35 This capacity distinguishes it from other vital organs (e.g., the brain, heart, and lungs) that are far less able to replace functional tissue when damaged. Liver regeneration requires a tightly coordinated response to complement the regenerative process, so that the entire organ can be reconstituted. The complex and synchronized regenerative response in liver can be perturbed and thereby can impact normal tissue recovery from injury or damage.36 When the cycle of injury and perturbed recovery from injury is repeated, damage can accumulate and initiate the process of chronic liver disease.35
It is reported that ~30% of the US population has underlying liver disease.37 There are numerous causative factors that drive liver disease, including extrinsic (e.g., diet, alcohol abuse, and viral infection) and intrinsic (e.g., genetic disorders and auto-immune diseases) factors.38–42 No matter the etiology, chronic liver diseases share a well-documented, common natural history, which ranges initially from simple steatosis, to inflammation and necrosis (steatohepatitis), to fibrosis and cirrhosis.43–46 Fibrosis may improve with removal of insult, but reversal of severe stages of fibrosis/cirrhosis is more limited.47 Cirrhosis is often considered an end-stage liver disease and requires liver transplant. Even in the case of hepatitis C virus, where eradication of viral infection is nearly 100% with direct-acting antivirals, reportedly 30 to 60% of cirrhotic livers do not recover histologically after achieving a sustained virologic response (SVR).48,49 Moreover, vascular changes (e.g., portal hypertension) and other sequelae of severe/decompensated cirrhosis do not appear to as readily reverse even after SVR.50 Globally, over 1 million people die from complications of cirrhosis each year, and an estimated 1 million more people die from related diseases (e.g., hepatocellular carcinoma).51 Despite a clear understanding of disease progression, there is no universally accepted therapy available to halt or reverse this process in humans.52
Given the poor prognosis of treating late-stage liver disease, much of the current research focuses on identifying at-risk individuals and preventing the progression of the disease during earlier phases, especially inflammation. Inflammation plays a central role in chronic liver diseases, and comprises components of both the innate and adaptive immune responses.53,54 The net result is a chronic, low-grade inflammatory condition, in which innate immune cells are activated, and surveillance and tolerance by adaptive immune cells are dysregulated.55–59 It is this vicious cycle of cell damage/death and inflammation, when it overwhelms the repair/recover responses of the liver, which leads to the chronicity of liver diseases.
The ECM in Liver Diseases-Thinking Beyond Collagen and Before Fibrosis
Beyond Collagen
The importance of the ECM in liver disease is well established. However, research on the hepatic ECM in the context of liver disease has been largely “collagenocentric,” or primarily focused on the role of the collagen matrix. This is not necessarily surprising, given the robust collagen ECM deposition found in fibrosis and cirrhosis and the ease of visualizing this matrix with histochemical stains. However, it is well-known that there are multiple ECMs that change qualitatively and quantitatively during hepatic fibrosis.60,61 As mentioned above, the concept of the ECM as a microenvironment has been recently expanded to include nonfibrillar “matrisome” proteins that colocalize in and influence the ECM.1 This dynamic subset of over 1000 matrisome proteins varies with organ and disease state.1 It is therefore clear that changes in the hepatic ECM/matrisome during fibrosis are much more diverse than simply collagen.
Before Fibrosis
The study of the role of the ECM in liver disease has also been “fibrosocentric,” that is, focused on the dramatic ECM changes during the fibrosis stage of disease. Again, this is not necessarily a surprise, given that chronic liver disease is often asymptomatic,62 and the patient’s first presentation is often with already established late-stage disease. However, ECM changes are not solely relegated to fibrosis. As mentioned above, in some areas of research (e.g., subcutaneous wound healing), the changes to the ECM in response to acute injury have been well-understood.7
Work by this group and others has shown that the acute phase response in the liver in response to damage involves several of the ECM proteins found in subcutaneous wound healing, such as fibrin, osteopontin, and fibronectin.63–65 We recently demonstrated that the hepatic matrisome changes robustly to acute injury (e.g., acute lipopolysaccharide), even under conditions in which the ECM appears histologically unchanged.45 These subhistologic transitional changes to the matrisome appear to resolve after acute injury (66,67; ►Fig. 1); with chronic injury, the transitional matrisome is replaced by collagenous scarring in the liver, which is again in-line with subcutaneous wound healing.7 An improvement in referral practices and noninvasive tests has increased the rate of early detection of asymptomatic chronic liver diseases.68 This paradigm change opens up the opportunity for mechanism-based therapies to halt disease progression during earlier (i.e., prefibrotic) phases of the disease progression.
Fig. 1.
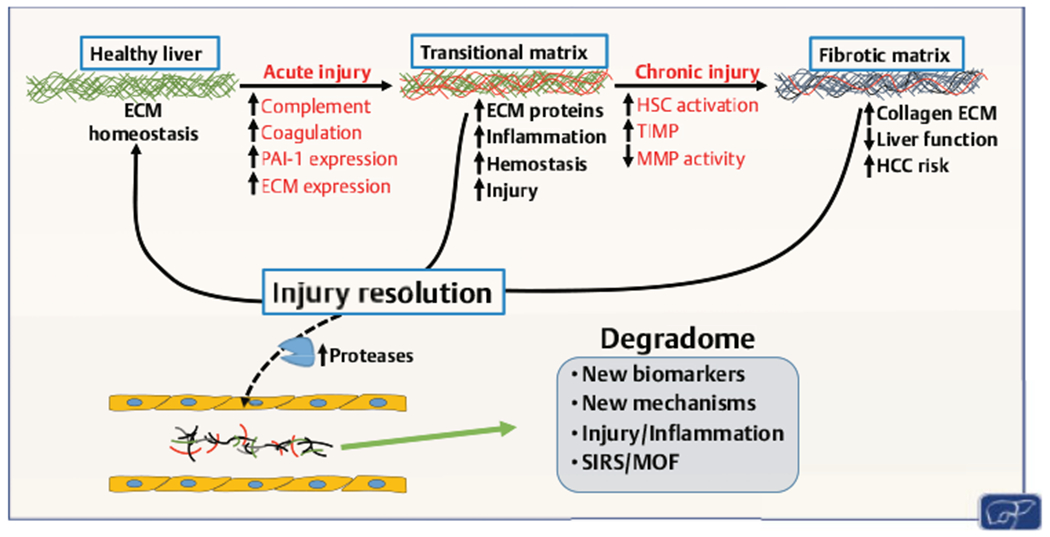
Transitional remodeling of the hepatic extracellular matrix (ECM). Acute injury causes formation of a transitional or provisional ECM. This transitional ECM may contribute to injury and inflammation. The transitional ECM often resolves after removal of the insult and may contribute to the recovery from that insult. With continued injury, the transitional matrix may progress to a fibrotic matrix, which can also resolve. Resolution of both injuries release degraded proteins into the blood that may serve as alarmins for inflammatory injury, as a well as a potential source of biomarkers for recovery from liver disease. HCC, hepatocellular carcinoma; HSC, hepatic stellate cell; MMP, matrix metalloproteinase; MOF, multiple organ failure; PAI-1, plasminogen activator inhibitor-1; SIRS, systemic inflammatory response syndrome; TIMP, tissue inhibitors of metalloproteinase.
The Hepatic Matrisome and the Control of Inflammation
The hepatic matrisome’s dynamism during inflammation represents a potential therapeutic target for liver disease. This review will explore some of these functions in the context of inflammation (►Fig. 2): adhesion, structure, presentation and storage, and sensing.
Fig. 2.
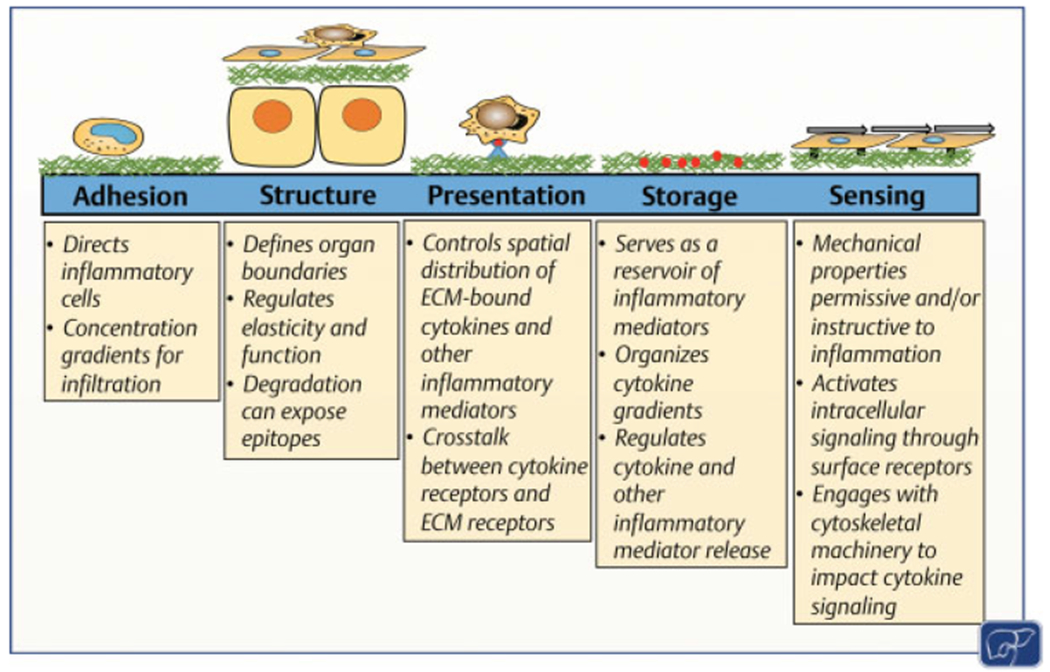
Functions of the hepatic extracellular matrix (ECM) that contribute to the roles it plays in inflammation. The ECM plays multiple roles that contribute to the regulation of inflammation and injury in the liver. These roles are related to adhesion, structure, presentation of signaling molecules and/or receptors, storage of signaling molecules, and sensing. See Section “The Hepatic Matrisome and the Control of Inflammation” for details. Figure modified with permission from Rozario and DeSimone.86
Adhesion
Hepatic inflammation after injury involves the recruitment of immune cells, including natural killer cells, natural killer T cells, dendritic cells, neutrophils, eosinophils, and monocytes.69,70 Immune cells are known to interact with many ECM proteins, including fibronectin, collagens, laminin, tenascin, and hyaluronic acid through a variety of receptors, including integrins, and surface glycoproteins(e.g., CD54, CD44, and CD26).71 These interactions have important implications in liver disease; for example, inhibition of fibronectin binding to T-cells is part of the anti-inflammatory mechanism of pentoxifylline.72
Interaction between leukocytes and the ECM is critical for the process of leukocyte adhesion and transmigration to sites of inflammation/injury.73 Leukocyte surfaces contain ECM receptors/adhesion molecules that direct their migration through interaction with the ECM.71 Regulation of these receptors is important for the rapid change between adhesive and nonadhesive states of immune cells.71 Initial leukocyte capture and rolling in the microvasculature may be mediated by selectins (CD62),73,74 although selectin-independent mechanisms have also been observed in the liver.75,76 Leukocyte activation is mediated by chemokines as described below, and arrest is mediated by the binding of leukocyte integrins to endothelial cell adhesion molecules (e.g., CD54 and CD106).73 Integrin-dependent leukocyte adhesion involves the β1- and β2-integrin.74 Alternatively, integrin-independent adhesion involves CD44 and vascular adhesion protein-1.74 Strengthening of adhesion is mediated by integrin outside-in signaling, and transmigration involves adhesion molecules (e.g., platelet and endothelial cell adhesion molecule 1 [PECAM1], Junctional adhesion molecule-A [JAM-A]) as well as barrier degradation by proteases.73 The interaction between the ECM and cell infiltration is bidirectional; as leukocytes integrate structural and biochemical cues from the ECM, they in turn release matrix-degrading proteases,77 which alter the extracellular composition and allow for easier cell migration.
The ECM not only adheres immune cells but also binds chemokines, creating a haptotactic gradient that directs immune cells to focal targets.78 After liver injury, parenchymal cells and resident leukocytes secrete chemokines.69 Chemokines bind to the glycosaminoglycan (GAG)/heparin sulfate components of basement membrane.78 An example of this is the chemokine (C-X-C motif) ligand 16 (CXCL16)-promoted recruitment and retention of CXCR6+ T cells in the liver, likely through conformational activation of β1 integrins and binding to vascular cell adhesion molecule 1 (VCAM-1).79 Chemokines also play an important role in the activation step of leukocyte adhesion.73 Indeed, it has been demonstrated that chemokines CCL2, CCL4, and CCL5 with mutations at their GAG binding sites are unable to recruit cells in vivo.80 ECM proteins themselves can also be chemotactic. For example, osteopontin has been shown to be chemotactic for natural killer T cells, neutrophils, and macrophages.81 Additionally, fibronectin secreted by T cells potently triggers macrophage agglutination and mediates monocyte and neutrophil translocation through the ECM.82 Overall, the ECM is key in the adhesion and direction of immune cells.
Structure
The ECM plays an important direct structural role, partially through definition of tissue boundaries and zonation.83 Furthermore, the ECM indirectly characterizes tissue morphology.84 For example, during branching morphogenesis, the process by which the liver and other organs develop, heparin sulfates on the cell surface and ECM regulate growth factor-epithelial cell interactions.85,86 In the mature liver, the ECM varies within the hepatic lobule and may help define zonation.87,88 The ECM defines properties permissive and/or instructive to inflammation. For example, although basement membrane may physically impede leukocyte transmigration, changes in this ECM in response to injury may also drive homing of recruited/repopulated cells within the liver.89
In addition to defining boundaries, the ECM also provides integrity and elasticity to the liver. The ECM is thereby key in maintaining or restoring normal physiology during or following insult. Remodeling of the hepatic ECM in response to acute injury can alter the super- and ultra-structure of the ECM. Acute toxic liver injury causes notable alterations in the ECM structural components (e.g., collagens I, IV, V, fibronectin, and elastin) and nonstructural proteins (e.g., olfactomedin-4 and thrombospondin-4).90 Through altering these components of the ECM, injury affects the normal elasticity provided by the ECM.90 It is known that inflammation impacts liver stiffness measurements (e.g., transient elastography), but is viewed as a “false positive” signal.49,91 However, inflammation has been shown to directly increase ECM stiffness in other organs, such as the lungs and the vasculature.92–95 It is, therefore, likely that the increase in liver stiffness measurements caused by inflammation is at least in part, a true signal. Indeed, three-dimensional magnetic resonance elastography, which measures shear stiffness, damping ratio, and magnetic resonance imaging proton density fat fraction, is an emerging technology to noninvasively differentiate inflammation from fibrosis.96 As will be discussed later (see next section), ECM stiffness can also influence inflammation.
Invasive cells can also degrade the ECM during disease and inflammation. For example, leukocytes secrete MMP-9, which mediates their transmigration during liver injury.97 This change in the ECM alters the interactions between the ECM and its receptors (e.g., integrins). Degradation can also expose self-antigens (e.g., collagen V) that can be used to promote infiltration of inflammatory cells.98 Moreover, even in cases where there is a net increase in ECM, overall ECM turnover is also increased.99 ECM turnover can also release proinflammatory ECM peptide fragments that may also serve as an alarmin/chemoattractant to recruit inflammatory cells to the site of injury (100,101; ►Fig. 1).
Storage, Presentation, and Sensing
The ECM also serves as a reservoir of signaling molecules, including growth factors during development and angiogenesis, as well as cytokines and chemokines during inflammation and disease. These interactions may serve to present or restrict access of ligands to receptors, modulate the spatial distribution of growth factors or create chemotactic gradients, or sequester a signaling molecule for later release.86 Damaged tissue can rapidly release mediators that attract components of the inflammation/wound healing response. The ECM sequesters and stores these molecules, predominantly via interactions with GAG, which protect them from normal proteolytic cleavage.102 However, these linkages can be rapidly cleaved and released by proteases (e.g., MMPs and ADAMs) induced by injury and/or inflammatory cytokines.102–105 The localized release of these mediators also creates a gradient that acts as a “homing signal” to the origin of the injury.78,105,106
The ECM is also a dynamic signaling moiety that allows the environment to interact with the cell and the cell to interact with the environment. One family of receptors that mediate these interactions is the integrins. Integrins are heterodimeric proteins composed of α and β subunits, with at least 24 different combinations having been identified in mammalian cells.107 Integrins transfer information from the ECM to the cell, allowing rapid and dynamic responses to changes in the extracellular environment. Integrins play a myriad of roles within the body, including proliferation/angiogenesis, maintenance of differentiation, as well as inflammation and apoptosis.108,109 Integrins are found on almost all cell-types in the liver, and dysregulated integrin signaling has been demonstrated to be involved in hepatic fibrogenesis in a wide variety of liver diseases, as well as inflammatory liver injury.110 There are also several nonintegrin receptors involved in signaling between the ECM and the cell. For example, CD44, a type I transmembrane glycoprotein with over 20 different isoforms, has been demonstrated to be involved in liver disease and inflammation.111,112 The canonical CD44 ligand is hyaluronic acid. Interactions between this ECM GAG and CD44 are known to facilitate migration of leukocytes to inflamed tissue, as well as the progression of inflammatory injury.113 Alternatively, CD44 has been implicated in the resolution of injury by facilitating the migration of hematopoietic stem cells to the injured liver.114
Interaction of cells with the surrounding ECM can also impact downstream signaling of both proinflammatory and restorative (e.g., growth factor) signals. This control can be at the level of receptor affinity or downstream signaling. These receptors are laterally organized on the plasma membrane in lipid/lipoprotein-rich regions (lipid rafts); this close proximity facilitates ligand binding, receptor dimerization, and cooperative downstream signaling.115 It has been recently suggested that ECM proteins contribute to this lateral organization.116 Signal integration between ECM-binding receptors (e.g., integrins) and extracellular signaling molecules is also know to vary based on ECM stratum. This has best been described for growth factor signaling and categorized as concomitant signaling, collaborative activation, direct activation, amplification, and negative regulation.117,118 Chronic inflammation has been shown to impair normal growth factor signaling, in part by altering the phenotype of the ECM.119 Moreover, ECM interactions have been shown to impact toll-like receptor and tumor necrosis factor alpha signaling to alter both effect and magnitude of the extracellular signal.120
In addition to directly and indirectly impacting downstream signaling cascades, integrin/ECM interactions foster linkage to the intracellular cytoskeleton.121,122 This interaction leads to the clustering of integrins into focal adhesions and plays key roles in influencing normal development, growth, cellular maintenance, and overall organ homeostasis.121–123 Dysregulation of adhesion signaling via ECM/integrin interactions are also key steps in cancer progression.124,125 This integration via integrins of the ECM and intracellular cytoskeleton contributes to mechanosensing, which likely influences the impact of ECM rigidity on the inflammatory response (see above123).
Inflammation and ECM remodeling can become a feed-forward cycle.104 The ECM facilitates immune cell migration and differentiation, while immune cells trigger new ECM deposition and proteolytic remodeling. Proteases subsequently cleave ECM producing proinflammatory degradation products. These processes can be adaptive, but aberrant ECM and bioactive degradation products perpetuate inflammation in a maladaptive response, such as in chronic hepatic inflammation.126 Given the key role of the ECM in mediating inflammation, it is not surprising that this compartment also plays key roles in the resolution of inflammation (i.e., catabasis127–129).
Conclusion
In conclusion, the ECM is complex and responds dynamically to internal/external stressors. Changes to the ECM in response to acute and/or inflammatory injury are well understood in some contexts. However, in the context of chronic liver diseases, most of the attention has been paid on the ECM changes associated with the end-stage of collagenic fibrosis. Although fibrosis is an important target that is relevant to clinical liver disease, it is also arguably the least sensitive to external interventions.130 In contrast, improvements in detection methods make the concept of blunting/reversing earlier stages of disease progression viable. In this context, inflammation may be a key therapeutic target. The response of the matrisome to stress is a critical component of the inflammatory response. Although changes to the ECM during hepatic inflammation are partially understood, it is an emerging area of interest. There is an opportunity to cross-fertilize our understanding from other fields in which the ECM and inflammation are more well described.131–133 These other fields may also be a source of laterally translated therapies that can be applied to chronic liver disease.134
Footnotes
Conflicts of Interest
Dr. Arteel reports grants from National Institutes of Health, during the conduct of the study.
References
- 1.Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: tools and insights for the “omics” era. Matrix Biol 2016;49:10–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 2012;11(04):014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326(5957):1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez-Hernandez A, Amenta PS. The hepatic extracellular matrix. I. Components and distribution in normal liver. Virchows Arch A Pathol Anat Histopathol 1993;423(01 ):1–11 [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL. Extracellular matrix In: Dufour JF, Clavien PA, eds. Signaling Pathways in Liver Diseases. Berlin: Springer; 2010: 93–104 [Google Scholar]
- 6.Griffiths MR, Keir S, Burt AD. Basement membrane proteins in the space of Disse: a reappraisal. J Clin Pathol 1991;44(08): 646–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun BK, Siprashvili Z, Khavari PA. Advances in skin grafting and treatment of cutaneous wounds. Science 2014;346(6212): 941–945 [DOI] [PubMed] [Google Scholar]
- 8.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 2014;15(12): 786–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol 2016;32(05):659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sessions AO, Engler AJ. Mechanical regulation of cardiac aging in model systems. Circ Res 2016;118(10):1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saccà SC, Gandolfi S, Bagnis A, et al. From DNA damage to functional changes of the trabecular meshwork in aging and glaucoma. Ageing Res Rev 2016;29:26–41 [DOI] [PubMed] [Google Scholar]
- 12.Phillip JM, Aifuwa I, Walston J, Wirtz D. The mechanobiology of aging. Annu Rev Biomed Eng 2015;17:113–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Hernandez A, Amenta PS. The extracellular matrix in hepatic regeneration. FASEB J 1995;9(14):1401–1410 [DOI] [PubMed] [Google Scholar]
- 14.Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol 2002;36(02):200–209 [DOI] [PubMed] [Google Scholar]
- 15.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 2007;282(32):23337–23347 [DOI] [PubMed] [Google Scholar]
- 16.Robertson H, Kirby JA, Yip WW, Jones DE, Burt AD. Biliary epithelial-mesenchymal transition in posttransplantation recurrence of primary biliary cirrhosis. Hepatology 2007;45(04):977–981 [DOI] [PubMed] [Google Scholar]
- 17.Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest 2008;118(10):3331–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol 201544-46:147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med 2008;29(05):258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubail J, Apte SS. Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol 201544-46:24–37 [DOI] [PubMed] [Google Scholar]
- 21.Brix K, McInnes J, Al-Hashimi A, Rehders M, Tamhane T, Haugen MH. Proteolysis mediated by cysteine cathepsins and legumain-recent advances and cell biological challenges. Protoplasma 2015;252(03):755–774 [DOI] [PubMed] [Google Scholar]
- 22.Beier JI, Arteel GE. Alcoholic liver disease and the potential role of plasminogen activator inhibitor-1 and fibrin metabolism. Exp Biol Med (Maywood) 2012;237(01):1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol 2006;21(Suppl 3):S84–S87 [DOI] [PubMed] [Google Scholar]
- 24.Pellicoro A, Aucott RL, Ramachandran P, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 2012;55(06):1965–1975 [DOI] [PubMed] [Google Scholar]
- 25.Pellicoro A, Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Fibrogenesis Tissue Repair 2012;5(Suppl 1):S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagan HM. Intra- and extracellular enzymes of collagen biosynthesis as biological and chemical targets in the control of fibrosis. Acta Trop 2000;77(01):147–152 [DOI] [PubMed] [Google Scholar]
- 27.Liu SB, Ikenaga N, Peng ZW, et al. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J 2016;30 (04):1599–1609 [DOI] [PubMed] [Google Scholar]
- 28.Tatsukawa H, Furutani Y, Hitomi K, Kojima S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death Dis 2016;7(06): e2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poynard T, McHutchison J, Manns M, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002;122(05):1303–1313 [DOI] [PubMed] [Google Scholar]
- 30.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 2004; 126(07):1795–1808 [DOI] [PubMed] [Google Scholar]
- 31.Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol 201868-69:435–451 [DOI] [PubMed] [Google Scholar]
- 32.Huijberts MS, Schaper NC, Schalkwijk CG. Advanced glycation end products and diabetic foot disease. Diabetes Metab Res Rev 2008;24(Suppl 1):S19–S24 [DOI] [PubMed] [Google Scholar]
- 33.Preziosi ME, Monga SP. Update on the mechanisms of liver regeneration. Semin Liver Dis 2017;37(02):141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147(04):765–783.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michalopoulos GK, DeFrances M. Liver regeneration. Adv Biochem Eng Biotechnol 2005;93:101–134 [DOI] [PubMed] [Google Scholar]
- 36.Forbes SJ, Newsome PN. Liver regeneration - mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol 2016;13(08):473–485 [DOI] [PubMed] [Google Scholar]
- 37.Younossi ZM, Stepanova M, Younossi Y, et al. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 2019:gutjnl-2019-318813, In press Doi: 10.1136/gutjnl-2019-318813 [DOI] [PubMed] [Google Scholar]
- 38.Kirpich IA, Marsano LS, McClain CJ. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin Biochem 2015;4813–14:923–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieber CS, Jones DP, Decarli LM. Effects of prolonged ethanol intake: production of fatty liver despite adequate diets. J Clin Invest 1965;44:1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganesan M, Poluektova LY, Kharbanda KK, Osna NA. Liver as a target of human immunodeficiency virus infection. World J Gastroenterol 2018;24(42):4728–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol 2013;10 (09):553–562 [DOI] [PubMed] [Google Scholar]
- 42.Morrison ED, Kowdley KV. Genetic liver disease in adults. Early recognition of the three most common causes. Postgrad Med 2000;107(02):147–152, 155,, 158–159 [DOI] [PubMed] [Google Scholar]
- 43.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol 2011;8(09):491–501 [DOI] [PubMed] [Google Scholar]
- 44.Schwartz JM, Reinus JF. Prevalence and natural history of alcoholic liver disease. Clin Liver Dis 2012;16(04):659–666 [DOI] [PubMed] [Google Scholar]
- 45.Poole LG, Dolin CE, Arteel GE. Organ-organ crosstalk and alcoholic liver disease. Biomolecules 2017;7(03):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seth D, Haber PS, Syn WK, Diehl AM, Day CP. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. J Gastroenterol Hepatol 2011;26(07):1089–1105 [DOI] [PubMed] [Google Scholar]
- 47.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 1998;102(03):538–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vispo E, Barreiro P, Del Valle J, et al. Overestimation of liver fibrosis staging using transient elastography in patients with chronic hepatitis C and significant liver inflammation. Antivir Ther 2009;14(02):187–193 [DOI] [PubMed] [Google Scholar]
- 49.Grgurevic I, Bozin T, Madir A. Hepatitis C is now curable, but what happens with cirrhosis and portal hypertension afterwards? Clin Exp Hepatol 2017;3(04):181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Libânio D, Marinho RT. Impact of hepatitis C oral therapy in portal hypertension. World J Gastroenterol 2017;23(26): 4669–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Byass P The global burden of liver disease: a challenge for methods and for public health. BMC Med 2014;12:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh S, Osna NA, Kharbanda KK. Treatment options for alcoholic and non-alcoholic fatty liver disease: a review. World J Gastroenterol 2017;23(36):6549–6570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hensley MK, Deng JC. Acute on chronic liver failure and immune dysfunction: a mimic of sepsis. Semin Respir CritCare Med 2018; 39(05):588–597 [DOI] [PubMed] [Google Scholar]
- 54.Li B, Selmi C, Tang R, Gershwin ME, Ma X. The microbiome and autoimmunity: a paradigm from the gut-liver axis. Cell Mol Immunol 2018;15(06):595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wree A, Marra F. The inflammasome in liver disease. J Hepatol 2016;65(05):1055–1056 [DOI] [PubMed] [Google Scholar]
- 56.Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol 2019;70(02):249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong X, Liu J, Xu Y, Cao H. Role of macrophages in experimental liver injury and repair in mice. Exp Ther Med 2019;17(05): 3835–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14(03):181–194 [DOI] [PubMed] [Google Scholar]
- 59.Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol 2016; 13(03):267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gressner OA, Weiskirchen R, Gressner AM. Evolving concepts of liver fibrogenesis provide new diagnostic and therapeutic options. Comp Hepatol 2007;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gressner AM, Bachem MG. Cellular sources of noncollagenous matrix proteins: role of fat-storing cells in fibrogenesis. Semin Liver Dis 1990;10(01):30–46 [DOI] [PubMed] [Google Scholar]
- 62.Sweet PH, Khoo T, Nguyen S. Nonalcoholic fatty liver disease. Prim Care 2017;44(04):599–607 [DOI] [PubMed] [Google Scholar]
- 63.Beier JI, Luyendyk JP, Guo L, von Montfort C, Staunton DE, Arteel GE. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology 2009;49(05):1545–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gillis SE, Nagy LE. Deposition of cellular fibronectin increases before stellate cell activation in rat liver during ethanol feeding. Alcohol Clin Exp Res 1997;21(05):857–861 [PubMed] [Google Scholar]
- 65.Thiele GM, Duryee MJ, Freeman TL, et al. Rat sinusoidal liver endothelial cells (SECs) produce pro-fibrotic factors in response to adducts formed from the metabolites of ethanol. Biochem Pharmacol 2005;70(11):1593–1600 [DOI] [PubMed] [Google Scholar]
- 66.Massey VL, Dolin CE, Poole LG, et al. The hepatic “matrisome” responds dynamically to injury: characterization of transitional changes to the extracellular matrix in mice. Hepatology 2017;65 (03):969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poole LG, Arteel GE. Transitional remodeling of the hepatic extracellular matrix in alcohol-induced liver injury. BioMed Res Int 2016;2016:3162670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Srivastava A, Jong S, Gola A, et al. Cost-comparison analysis of FIB-4, ELF and fibroscan in community pathways for non-alcoholic fatty liver disease. BMC Gastroenterol 2019;19(01):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oliveira THC, Marques PE, Proost P, Teixeira MMM. Neutrophils: a cornerstone of liver ischemia and reperfusion injury. Lab Invest 2018;98(01):51–62 [DOI] [PubMed] [Google Scholar]
- 70.Karlmark KR, Weiskirchen R, Zimmermann HW, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009;50(01): 261–274 [DOI] [PubMed] [Google Scholar]
- 71.Shimizu Y, Shaw S. Lymphocyte interactions with extracellular matrix. FASEB J 1991;5(09):2292–2299 [DOI] [PubMed] [Google Scholar]
- 72.Shirin H, Bruck R, Aeed H, et al. Pentoxifylline prevents concanavalin A-induced hepatitis by reducing tumor necrosis factor alpha levels and inhibiting adhesion of T lymphocytes to extracellular matrix. J Hepatol 1998;29(01):60–67 [DOI] [PubMed] [Google Scholar]
- 73.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007;7(09):678–689 [DOI] [PubMed] [Google Scholar]
- 74.Lee WY, Kubes P. Leukocyte adhesion in the liver: distinct adhesion paradigm from other organs. J Hepatol 2008;48(03): 504–512 [DOI] [PubMed] [Google Scholar]
- 75.Wong J, Johnston B, Lee SS, et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest 1997;99(11):2782–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fox-Robichaud A, Kubes P. Molecular mechanisms of tumor necrosis factor alpha-stimulated leukocyte recruitment into the murine hepatic circulation. Hepatology 2000;31(05):1123–1127 [DOI] [PubMed] [Google Scholar]
- 77.Woodfin A, Voisin MB, Nourshargh S. Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol 2010;17(01 ):9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Monneau Y, Arenzana-Seisdedos F, Lortat-Jacob H. The sweet spot: how GAGs help chemokines guide migrating cells. J Leukoc Biol 2016;99(06):935–953 [DOI] [PubMed] [Google Scholar]
- 79.Heydtmann M, Lalor PF, Eksteen JA, Hübscher SG, Briskin M, Adams DH. CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J Immunol 2005;174(02):1055–1062 [DOI] [PubMed] [Google Scholar]
- 80.Proudfoot AE, Handel TM, Johnson Z, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A 2003;100(04): 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ramaiah SK, Rittling S. Pathophysiological role of osteopontin in hepatic inflammation, toxicity, and cancer. Toxicol Sci 2008;103 (01):4–13 [DOI] [PubMed] [Google Scholar]
- 82.Godfrey HP. T cell fibronectin: an unexpected inflammatory lymphokine. Lymphokine Res 1990;9(03):435–447 [PubMed] [Google Scholar]
- 83.Federman S, Miller LM, Sagi I. Following matrix metalloproteinases activity near the cell boundary by infrared micro-spectroscopy. Matrix Biol 2002;21(07):567–577 [DOI] [PubMed] [Google Scholar]
- 84.Jülich D, Cobb G, Melo AM, et al. Cross-scale integrin regulation organizes ECM and tissue topology. Dev Cell 2015;34(01):33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Patel VN, Pineda DL, Hoffman MP. The function of heparan sulfate during branching morphogenesis. Matrix Biol 20175758:311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol 2010;341 (01):126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McClelland R, Wauthier E, Uronis J, Reid L. Gradients in the liver’s extracellular matrix chemistry from periportal to pericentral zones: influence on human hepatic progenitors. Tissue Eng Part A 2008;14(01):59–70 [DOI] [PubMed] [Google Scholar]
- 88.Lee-Montiel FT, George SM, Gough AH, et al. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp Biol Med (Maywood) 2017;242 (16):1617–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang S, Voisin MB, Larbi KY, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med 2006;203(06): 1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klaas M, Kangur T, Viil J, et al. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci Rep 2016;6:27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Coco B, Oliveri F, Maina AM, et al. Transient elastography: a new surrogate marker of liver fibrosis influenced by major changes of transaminases. J Viral Hepat 2007;14(05):360–369 [DOI] [PubMed] [Google Scholar]
- 92.Wu D, Birukov K. Endothelial cell mechano-metabolomic coupling to disease states in the lung microvasculature. Front Bioeng Biotechnol 2019;7:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karki P, Birukova AA. Substrate stiffness-dependent exacerbation of endothelial permeability and inflammation: mechanisms and potential implications in ALI and PH (2017 Grover Conference Series). Pulm Circ 2018;8(02):2045894018773044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mammoto A, Mammoto T, Kanapathipillai M, et al. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat Commun 2013;4:1759. [DOI] [PubMed] [Google Scholar]
- 95.Hsu JJ, Lim J, Tintut Y, Demer LL. Cell-matrix mechanics and pattern formation in inflammatory cardiovascular calcification. Heart 2016;102(21):1710–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Allen AM, Shah VH, Therneau TM, et al. The role of threedimensional magnetic resonance elastography in the diagnosis of nonalcoholic steatohepatitis in obese patients undergoing bariatric surgery. Hepatology 2018; In press . Doi: 10.1002/hep.30483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology 2008;47(01):186–198 [DOI] [PubMed] [Google Scholar]
- 98.Mak KM, Png CY, Lee DJ, Type V. Type Vcollagen in health, disease, and fibrosis. Anat Rec (Hoboken) 2016;299(05):613–629 [DOI] [PubMed] [Google Scholar]
- 99.Roderfeld M Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol 201868-69:452–462 [DOI] [PubMed] [Google Scholar]
- 100.Song KS, Kim HS, Park KE, Kwon OH. The fibrinogen degradation products (FgDP) levels in liver disease. Yonsei Med J 1993;34 (03):234–238 [DOI] [PubMed] [Google Scholar]
- 101.Santambrogio L, Rammensee HG. Contribution of the plasma and lymph degradome and peptidome to the MHC ligandome. Immunogenetics 2019;71(03):203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lipowsky HH. Role of the glycocalyx as a barrier to leukocyte-endothelium adhesion. Adv Exp Med Biol 2018;1097:51–68 [DOI] [PubMed] [Google Scholar]
- 103.Karsdal MA, Manon-Jensen T, Genovese F, et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 2015;308(10): G807–G830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sorokin L The impact of the extracellular matrix on inflammation. Nat Rev Immunol 2010;10(10):712–723 [DOI] [PubMed] [Google Scholar]
- 105.Vempati P, Popel AS, Mac Gabhann F. Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning. Cytokine Growth Factor Rev 2014;25(01):1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wasmuth HE, Tacke F, Trautwein C. Chemokines in liver inflammation and fibrosis. Semin Liver Dis 2010;30(03):215–225 [DOI] [PubMed] [Google Scholar]
- 107.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci 2006;119(Pt 19):3901–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hodivala-Dilke KM, Reynolds AR, Reynolds LE. Integrins in angiogenesis: multitalented molecules in a balancing act. Cell Tissue Res 2003;314(01):131–144 [DOI] [PubMed] [Google Scholar]
- 109.Zhou HF, Chan HW, Wickline SA, Lanza GM, Pham CT. Alphav-beta3-targeted nanotherapy suppresses inflammatory arthritis in mice. FASEB J 2009;23(09):2978–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Patsenker E, Stickel F. Role of integrins in fibrosing liver diseases. Am J Physiol Gastrointest Liver Physiol 2011;301(03):G425–G434 [DOI] [PubMed] [Google Scholar]
- 111.Seth D, Duly A, Kuo PC, McCaughan GW, Haber PS. Osteopontin is an important mediator of alcoholic liver disease via hepatic stellate cell activation. World J Gastroenterol 2014;20(36): 13088–13104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patouraux S, Rousseau D, Bonnafous S, et al. CD44 is a key player in non-alcoholic steatohepatitis. J Hepatol 2017;67(02):328–338 [DOI] [PubMed] [Google Scholar]
- 113.McDonald B, Kubes P. Interactions between CD44 and hyaluronan in leukocyte trafficking. Front Immunol 2015;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crosby HA, Lalor PF, Ross E, Newsome PN, Adams DH. Adhesion of human haematopoietic (CD34+) stem cells to human liver compartments is integrin and CD44 dependent and modulated by CXCR3 and CXCR4. J Hepatol 2009;51(04):734–749 [DOI] [PubMed] [Google Scholar]
- 115.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 2000;1(01):31–39 [DOI] [PubMed] [Google Scholar]
- 116.Sadeghi S, Vink RL. Membrane sorting via the extracellular matrix. Biochim Biophys Acta 2015;1848(02):527–531 [DOI] [PubMed] [Google Scholar]
- 117.Ivaska J, Heino J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol 2011;27:291–320 [DOI] [PubMed] [Google Scholar]
- 118.Schnittert J, Bansal R, Storm G, Prakash J. Integrins in wound healing, fibrosis and tumor stroma: highpotential targets for therapeutics and drug delivery. Adv Drug Deliv Rev 2018;129:37–53 [DOI] [PubMed] [Google Scholar]
- 119.Ozaki I, Hamajima H, Matsuhashi S, Mizuta T. Regulation of TGF-β1-induced pro-apoptotic signaling by growth factor receptors and extracellular matrix receptor integrins in the liver. Front Physiol 2011;2:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gay NJ, Gangloff M. Structure and function of toll receptors and their ligands. Annu Rev Biochem 2007;76:141–165 [DOI] [PubMed] [Google Scholar]
- 121.Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci 2009;122(Pt 2):159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Iwamoto DV, Calderwood DA. Regulation of integrin-mediated adhesions. Curr Opin Cell Biol 2015;36:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lorenz L, Axnick J, Buschmann T, et al. Mechanosensing by β1 integrin induces angiocrine signals for liver growth and survival. Nature 2018;562(7725):128–132 [DOI] [PubMed] [Google Scholar]
- 124.Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer 2018;18(09):533–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev 2005;24(03):425–439 [DOI] [PubMed] [Google Scholar]
- 126.Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15(06):349–364 [DOI] [PubMed] [Google Scholar]
- 127.Widgerow AD. Cellular resolution of inflammation-catabasis. Wound Repair Regen 2012;20(01):2–7 [DOI] [PubMed] [Google Scholar]
- 128.Franitza S, Hershkoviz R, Kam N, et al. TNF-alpha associated with extracellular matrix fibronectin provides a stop signal for chemotactically migrating T cells. J Immunol 2000;165(05): 2738–2747 [DOI] [PubMed] [Google Scholar]
- 129.Cañedo-Dorantes L, Cañedo-Ayala M. Skin acute wound healing: a comprehensive review. Int J Inflamm 2019;2019: 3706315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mehal WZ, Schuppan D. Antifibrotic therapies in the liver. Semin Liver Dis 2015;35(02):184–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hyldig K, Riis S, Pennisi CP, Zachar V, Fink T. Implications of extracellular matrix production by adipose tissue-derived stem cells for development of wound healing therapies. Int J Mol Sci 2017;18(06):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lumelsky N, O’Hayre M, Chander P, Shum L, Somerman MJ. Autotherapies: enhancing endogenous healing and regeneration. Trends Mol Med 2018;24(11):919–930 [DOI] [PubMed] [Google Scholar]
- 133.Rousselle P, Braye F, Dayan G. Re-epithelialization of adult skin wounds: cellular mechanisms and therapeutic strategies. Adv Drug Deliv Rev 2018:S0169–409X(18)30158–3, In press Doi: 10.1016/j.addr.2018.06.019 [DOI] [PubMed] [Google Scholar]
- 134.Pritchard MT, McCracken JM. Identifying novel targets for treatment of liver fibrosis: what can we learn from injured tissues which heal without a scar? Curr Drug Targets 2015;16(12): 1332–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]