

Abstract
High‐voltage‐activated calcium (CaV1/CaV2) channels translate action potentials into Ca2+ influx in excitable cells to control essential biological processes that include; muscle contraction, synaptic transmission, hormone secretion and activity‐dependent regulation of gene expression. Modulation of CaV1/CaV2 channel activity is a powerful mechanism to regulate physiology, and there are a host of intracellular signalling molecules that tune different aspects of CaV channel trafficking and gating for this purpose. Beyond normal physiological regulation, the diverse CaV channel modulatory mechanisms may potentially be co‐opted or interfered with for therapeutic benefits. CaV1/CaV2 channels are potently inhibited by a four‐member sub‐family of Ras‐like GTPases known as RGK (Rad, Rem, Rem2, Gem/Kir) proteins. Understanding the mechanisms by which RGK proteins inhibit CaV1/CaV2 channels has led to the development of novel genetically encoded CaV channel blockers with unique properties; including, chemo‐ and optogenetic control of channel activity, and blocking channels either on the basis of their subcellular localization or by targeting an auxiliary subunit. These genetically encoded CaV channel inhibitors have outstanding utility as enabling research tools and potential therapeutics.
Keywords: calcium channel, ion channel regulation, nanobody, RGK, ubiquitin
RGK proteins are small Ras‐like GTPases that potently inhibit voltage‐gated calcium (CaV) channels by binding their auxiliary b subunits. Mechanistic insights into how RGK proteins inhibit CaV channels has been exploited to develop novel genetically‐encoded CaV channel inhibitors that can be acutely activated by small molecules or light, or produce constitutive inhibition via targeted ubiquitination using CaVb‐binding nanobodies. Advantages of such genetically‐encoded CaV channel inhibitors include their ability to be selectively targeted to specific tissue, cell types, sub‐cellular localization, and distinct CaV channel macromolecular complexes.
Voltage‐gated calcium channels: basic structure, function and regulation
Ca2+ is a universal second messenger that regulates numerous biological functions in virtually all cells (Berridge et al. 2000). Cytoplasmic Ca2+ in cells is kept low (100 nM) but rises in response to diverse stimuli (to ∼1 μM) to initiate functional responses through the action of a variety of Ca2+‐dependent proteins. The source of signalling Ca2+ is from either intracellular stores or the extracellular milieu. There are a variety of integral membrane proteins on the plasma membranes of diverse cell types that permit the entry of Ca2+ in response to specific stimuli. Amongst these are the family of voltage‐dependent Ca2+ channels (VDCCs) which gate Ca2+ entry into cells in response to changes in membrane potential. VDCCs are sub‐divided into two categories based on the threshold voltage for activation – low‐voltage‐activated (LVA) and high‐voltage‐activated (HVA) Ca2+ channels, respectively. There are three distinct LVA (CaV3.1 – CaV3.3) and seven HVA (CaV1.1 – CaV1.4; CaV2.1 – CaV2.3) (Catterall, 2011; Zamponi et al. 2015). VDCCs play many essential roles in the biology of excitable cells. As examples, Ca2+ influx through VDCCs: contributes to pacemaking in many cell types including the sino‐atrial node of the heart and substantia nigra (CaV3; CaV1.3) (Guzman et al. 2010; Mesirca et al. 2015); regulates neuronal excitability by coupling to Ca2+‐activated K+ channels (CaV1.2; CaV2.1; CaV2.2) (Marrion & Tavalin, 1998; Womack et al. 2004); controls the heartbeat by coupling electrical excitation to muscle contraction in cardiomyocytes (CaV1.2) (Bers, 2002); enables communication among neurons by triggering presynaptic neurotransmitter release (CaV2.1‐CaV2.3) (Sudhof, 2012); promotes the release of hormones, e.g. insulin, adrenaline (epinephrine), essential for metabolic and physiological homeostasis (CaV1.2, CaV1.3, CaV2) (Braun et al. 2008); and engenders long‐term changes in cellular function by regulating gene expression (CaV1.2, CaV1.3) (Wheeler et al. 2012).
Functional HVA Ca2+ channels in vivo are multi‐subunit complexes comprising distinct pore‐forming α1 subunits (α1A – CaV2.1; α1B – CaV2.2; α1C – CaV1.2; α1D – CaV1.3; α1E – CaV2.3; α1F – CaV1.4; and α1S – CaV1.1) assembled with calmodulin and auxiliary β (CaVβ1 – CaVβ4), α2δ (α2δ‐1 – α2δ‐3), and γ subunits (Zamponi et al. 2015). In heterologous expression studies, co‐expression with CaVβ is necessary for efficient α1‐subunit trafficking to the plasma membrane (Buraei & Yang, 2010). Consistent with an essential in vivo role, β1‐null mice die at birth due to asphyxiation (Gregg et al. 1996) and β2 knock‐out is embryonic lethal due to cardiac defects (Weissgerber et al. 2006). Nevertheless, recent in vivo data in adult cardiomyocytes indicate an exception to the absolute necessity for CaVβ to enable trafficking of α1C to the surface membrane of adult heart cells. Cardiac‐specific excision of CaVβ2, the dominant CaVβ isoform in heart, reduced CaVβ2 protein by 96% while decreasing CaV1.2 current amplitude by only 26% (Meissner et al. 2011). Further, a transgenic mouse expressing a dihydropyridine‐resistant α1C mutant that does not bind CaVβ displayed ample DHP‐resistant CaV1.2 current, indicating a robust CaVβ‐independent trafficking to the sarcolemma (Yang et al. 2019). It remains to be determined whether and to what extent CaVβ‐independent trafficking happens in other cell types and other CaV1/CaV2 isoforms at different developmental stages. Beyond their impact on CaV1/CaV2 trafficking, CaVβ isoforms alter multiple channel gating properties – shift the voltage dependence of channel activation to the left, increase single channel open probability, impart distinctive rates of inactivation, and endow different steady‐state inactivation profiles (Buraei & Yang, 2010). α2δ subunits promote surface trafficking and can alter biophysical properties of particular CaV1/CaV2 channels (Dolphin, 2012). γ subunits are associated with CaV1.1 channels (Kang & Campbell, 2003; Wu et al. 2016); their association with other CaV1/CaV2 channels in vivo is unclear. Multiple CaM binding sites have been described at different locations in distinct CaV1/CaV2 channels (Van Petegem et al. 2005; Dick et al. 2008; Mori et al. 2008; Ben‐Johny & Yue, 2014). CaM binds to the C‐terminus of most CaV1/CaV2 channels in a fairly conserved region containing an IQ motif (Erickson et al. 2001; Kim et al. 2008, 2004, 2010; Mori et al. 2008). Binding of apoCaM to this region has been shown to enhance the open probability, P o, of CaV1.3 channels (Adams et al. 2014). Cryo electron microscopy structures of CaV1.1 and CaV3.1 channels have yielded invaluable insights into CaV channel structure, three‐dimensional assembly and modulation by ligands (Wu et al. 2016; Zhao et al. 2019 a, b ).
An important feature of HVA CaV channels is that their activity is not static but is dynamically regulated both by stably associated proteins as well as transiently interacting signalling molecules. Typically, these regulatory mechanisms have profound physiological importance; their dysregulation can cause pathology, and they can be co‐opted or interfered with for therapy. Examples of these regulatory mechanisms include: Ca2+‐dependent inactivation of CaV1.2 channels mediated by preassociated CaM, a negative feedback mechanism which when disrupted leads to prolonged cardiac action potentials and life‐threatening cardiac arrhythmias (Peterson et al. 1999; Zuhlke et al. 1999; Alseikhan et al. 2002); protein kinase A mediated up‐regulation of cardiac CaV1.2, essential for the physiologically critical fight‐or‐flight response (Kamp & Hell, 2000); voltage‐dependent inhibition of CaV2 channels by Gβγ subunits (Dolphin, 2003), a mechanism for tuning synaptic strength that is important for the analgesic effects of opiates.
RGK GTPase inhibition of CaV channels: discovery and mechanisms
The seminal report of the functional interaction between RGK proteins and CaV1/CaV2 channels was in 2001 – a yeast two‐hybrid screen of MIN6 cells using CaVβ3 as bait fished out Gem/Kir as an interacting protein (Beguin et al. 2001). Co‐expressing Gem with recombinant CaV1.3 or CaV1.2 in Xenopus oocytes resulted in a marked inhibition of calcium channel current. Gem was initially discovered as a mitogen‐induced gene in human T cells (Maguire et al. 1994) and belongs to a sub‐family of Ras‐like monomeric G‐proteins with three other members: Rad (Ras associated with diabetes), originally discovered as a protein over‐expressed in skeletal muscle of diabetic patients (Reynet & Kahn, 1993); Rem, first identified using a degenerate cloning strategy based on homology to Gem and Rad (Finlin & Andres, 1997); and Rem 2, cloned from a rat brain cDNA library (Finlin et al. 2000). Subsequent to the original report of Gem inhibition of CaV1.2 and CaV1.3, it was shown that this phenomenon also extended to Rad and Rem, which both potently inhibited CaV1.2 channels (Finlin et al. 2003), and Rem 2 (Chen et al. 2005; Finlin et al. 2005). Over‐expressing any RGK protein markedly suppresses endogenous CaV1/CaV2 channels in native cells including cardiac myocytes, neurons and skeletal muscle (Murata et al. 2004; Chen et al. 2005; Bannister et al. 2008; Wang et al. 2010; Xu et al. 2010; Puckerin et al. 2018). A recent elegant study revealed that endogenous Rad in cardiomyocytes constitutively exerts a gating brake on a fraction of CaV1.2 channels. This inhibition is relieved by protein kinase A phosphorylation of Rad, and is the long sought‐after mechanism by which β‐adrenergic agonists increase cardiac CaV1.2 to enhance inotropy during the fight‐or‐flight response (Liu et al. 2020).
How do RGK proteins inhibit CaV1/CaV2 channels? The answer to this seemingly simple question turned out to be surprisingly complex. The whole‐cell current (I) is related to microscopic channel properties by the relation I = F A × N × i × P o; where F A is the fraction of activatable channels, N is the total number of channels, i is the unitary current amplitude, and P o is the open probability. In principle, RGK proteins could inhibit I by reducing any of the four parameters or a combination of them. We found that Rem inhibits CaV1.2 channels reconstituted in HEK293 cells in at least three distinct ways (Fig. 1) (Yang et al. 2010). First, in this system, Rem reduced CaV1.2 surface density (N) by 65%, an effect that was reversed by co‐expressing dominant negative dynamin. The second mechanism involved a reduction in channel P o, which occurred without an impact on CaV1.2 voltage sensor movement, suggesting an impairment in coupling between voltage sensors and opening of the channel pore. This mechanism specifically required simultaneous association of Rem with the plasma membrane (mediated by a polybasic distal C‐terminus) and CaVβ in the channel complex (via the guanine nucleotide binding domain). Finally, a third mechanism entailed a reduction in CaV1.2 maximal gating charge (Q max) that was not accounted for by a change in channel surface density, suggesting an immobilization of one or more voltage sensors. This third mechanism required GTP bound to Rem and would have the practical effect of diminishing both F A and P o. While these three mechanisms of Rem inhibition of CaV1.2 can be observed in HEK293 cells, their relative prevalence may differ in other cell types. For example, over‐expression of Rem in cardiac myocytes markedly depresses CaV1.2 whole‐cell current without an apparent change in channel surface density as indicated by immunofluorescence, and the acute rescue of near‐maximal current with BAYK 8644 (Xu et al. 2010).
Figure 1. Rem inhibition of reconstituted CaV1.2 channels.
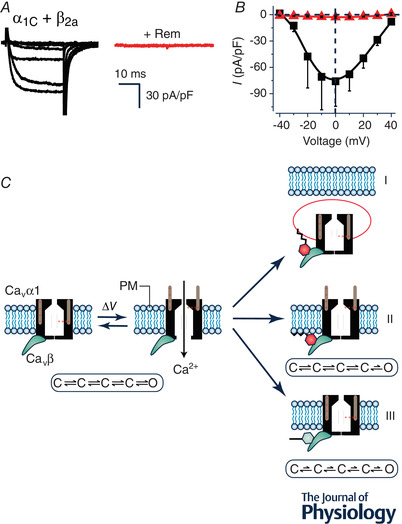
A, exemplar family of whole‐cell Ba2+ currents from recombinant CaV1.2 channels (α1C + β2a) reconstituted in HEK293 cells either without (left) or with (right) co‐expression of Rem. B, population I‐V curves from CaV1.2 channels in the absence (■) or presence ( ) of co‐expressed Rem. C, schematic diagram showing three distinct mechanisms (I‐III) utilized by Rem to inhibit recombinant CaV1.2 channels. In mechanism I, co‐expressed Rem results in a decrease in the number of channels at the cell surface (N) due to enhanced CaV1.2 endocytosis. Mechanism II involves a reduction in the open probability (P
o) of channels residing on the plasma membrane without impacting on voltage sensor movement as measured by total gating charge (Q
max). This mechanism requires Rem simultaneously binding to the CaVβ subunit (using the guanine nucleotide binding domain) and the plasma membrane (via the polybasic distal C‐terminus). Mechanism III involves an impaired movement of the voltage sensor movement of surface channels as measured by a decreased Q
max (observed even when N is completely rescued by co‐expressing dominant negative dynamin). Mechanism III is blocked by a mutation (T94N) that favours GDP over GTP binding to Rem, suggesting it requires GTP‐bound Rem.
) of co‐expressed Rem. C, schematic diagram showing three distinct mechanisms (I‐III) utilized by Rem to inhibit recombinant CaV1.2 channels. In mechanism I, co‐expressed Rem results in a decrease in the number of channels at the cell surface (N) due to enhanced CaV1.2 endocytosis. Mechanism II involves a reduction in the open probability (P
o) of channels residing on the plasma membrane without impacting on voltage sensor movement as measured by total gating charge (Q
max). This mechanism requires Rem simultaneously binding to the CaVβ subunit (using the guanine nucleotide binding domain) and the plasma membrane (via the polybasic distal C‐terminus). Mechanism III involves an impaired movement of the voltage sensor movement of surface channels as measured by a decreased Q
max (observed even when N is completely rescued by co‐expressing dominant negative dynamin). Mechanism III is blocked by a mutation (T94N) that favours GDP over GTP binding to Rem, suggesting it requires GTP‐bound Rem.
From a macroscopic perspective all four RGKs profoundly inhibit all CaV1/CaV2 channels when over‐expressed. Nevertheless, underneath this apparent uniformity, there are important distinctions in the mechanisms of inhibition that extend to both the different RGKs as well as to individual channel types (Yang & Colecraft, 2013). Rem2 was found to inhibit CaV1.2 channels in mouse insulinoma MIN6 cells (Finlin et al. 2005) and also CaV2.2 channels in tsA201 cells without reducing the number of channels at the cell surface (Chen et al. 2005). By reconstituting channels with either wild type CaVβ or a mutant CaVβ that loses binding to RGK proteins, we found that Rem and Rad could inhibit CaV1.2 and CaV2.2 (but not the other CaV1/CaV2 channel types) using either β‐binding‐dependent or β‐binding‐independent mechanisms (Yang et al. 2012; Puckerin et al. 2018, 2016). In the particular case of Rem inhibition of CaV1.2, the β‐binding‐independent mechanism of inhibition is mediated by an interaction of the Rem distal C‐terminus with the α1C N‐terminus region just upstream of the first transmembrane spanning segment of the channel (Yang et al. 2012). By contrast, Gem and Rem2 utilize solely a β‐binding‐dependent mechanism to inhibit CaV1/CaV2 channels. Overall, insights into the mechanisms and physical determinants of RGK inhibition of CaV1/CaV2 channels has proven invaluable to the broad objective of drawing inspiration from these proteins as prototype molecules to design next‐generation genetically encoded CaV channel inhibitors as research tools and potential therapeutics.
RGK‐inspired genetically encoded CaV channel inhibitors
Blocking CaV1/CaV2 channels with small molecules or toxins is a prevailing or prospective therapeutic strategy for many serious diseases including hypertension, chronic pain, cardiac arrhythmias, Parkinson's disease and stroke (Zamponi et al. 2015; Zamponi, 2016). While convenient, small molecule CaV channel blockers have limitations, some of which may be circumvented by genetically encoded inhibitors (Xu & Colecraft, 2009). First, they lack tissue specificity since small molecules are typically widely distributed in the body after administration, and distinct VDCCs are present across many different tissues, organs and cell types. Second, VDCCs show an immense molecular and functional diversity stemming from their organization into distinct macromolecular complexes, and sub‐cellular localizations that are poorly discriminated by small molecules. These two gap areas could potentially be filled by novel genetically encoded CaV channel inhibitors designed to target molecularly distinct VDCC macromolecular complexes in a tissue‐ or cell‐specific manner. While RGK proteins themselves are potent VDCC inhibitors, their usefulness as research tools or therapeutics is limited by several factors: (1) they are non‐selective, as they indiscriminately inhibit all CaV1/CaV2 channel types; (2) they are constitutive inhibitors, thus providing poor temporal and spatial control of channel block; and (3) they are non‐specific as they interact with and regulate other proteins such as enzymes and the cytoskeleton in cells (Yang & Colecraft, 2013). Over the last few years, using RGK proteins themselves as inspiration, we and others have explored different ways to engineer new genetically encoded CaV channel inhibitors that improve on various aspects of functional CaV channel block that are lacking in wild‐type RGK proteins.
Our finding that Rem specifically inhibits CaV1.2 using both a β‐binding‐dependent and α1C‐binding‐dependent mechanism but used only a β‐binding‐dependent mechanism to block other CaV1/CaV2 channel types suggested a simple method to create a CaV1.2‐selective genetically encoded CaV channel inhibitor – introduce mutations in Rem that weaken its interaction with CaVβ without altering the tertiary structure of the protein. Indeed, such mutations (Rem[R200A/L227A]) were identified by an extensive mutagenesis study (Beguin et al. 2007). Consistent with the hypothesis, Rem[R200A/L227A] selectively inhibited CaV1.2, but not other CaV1/CaV2 channels, reconstituted in HEK293 cells (Puckerin et al. 2018). The ability of Rem[R200A/L227A] to discriminate between CaV1.2 and CaV1.3 was especially notable given the difficulty of identifying small molecules that can effectively distinguish between these two L‐type channel subtypes (Zamponi et al. 2015). Using a similar logic, we found that Rad[R208A/L235A] selectively blocked CaV1.2 and CaV2.2, consistent with the finding that Rad inhibits these two channels using both β‐binding‐dependent and β‐binding‐independent mechanisms (Puckerin et al. 2018). Importantly, both Rem[R200A/L227A] and Rad[R208A/L235A] strongly inhibited CaV1.2 channels in cardiomyocytes, indicating that the β‐binding‐independent mechanism of inhibition is operational in this native environment. Similarly, the two proteins inhibited HVA CaV channel currents in dorsal root ganglion (DRG) neurons to different extents, reflecting their varying selectivity for CaV1.2 and CaV1.2/CaV2.2 channels, respectively (Puckerin et al. 2018).
Rem associates with the plasma membrane via the 32‐residue distal C‐terminus (DCT) using hydrophobic and electrostatic interactions. Deletion of the DCT abolishes both Rem membrane targeting and inhibition of CaV1/CaV2 channels (Finlin et al. 2003; Yang et al. 2007). The requirement for Rem binding to the plasma membrane for CaV channel inhibition has been exploited to engineer Rem derivatives that enable chemo‐ and optogenetic control of channel inhibition, and also subcellular specificity (Fig. 2). We replaced Rem DCT with the C1 domain from protein kinase γ, creating Rem1‐265‐C1PKCγ which when expressed in cells was primarily distributed in the cytosol but could be rapidly recruited to the plasma membrane with a small molecule, phorbol‐12,13‐dibuytrate (PdBu). The PdBu‐induced recruitment of Rem1‐265‐C1PKCγ caused a concomitant rapid inhibition of CaV1/CaV2 channel currents (Fig. 2B ) (Yang et al. 2013, 2007). The generality of this chemogenetic regulation was demonstrated by development of a FK506‐binding protein (FKBP)‐tagged Rem265 version that could be recruited to the membrane to inhibit CaV1/CaV2 channels using rapamycin‐mediated heterodimerization in cells that also expressed constitutively membrane‐targeted FRB (a fragment of mTOR) (Crabtree & Schreiber, 1996; Inoue et al. 2005; Yang et al. 2007). Similarly, a 490 nm blue light‐mediated heterodimerization strategy was utilized to develop optogenetic control of Rem inhibition (Fig. 2C ). The approach is based on a light‐induced protein‐protein interaction created by inserting a bacterial peptide, ssrA, into a naturally occurring photoswitch, light‐oxygen‐voltage 2 (LOV2) domain from Avena sativa (Guntas et al. 2015). In the dark, SsrA is sterically obstructed from interacting with a binding partner, sspB. With blue light, this steric inhibition is relieved, allowing SsrA to bind SspB. Extensive bioengineering of LOV2‐SsrA yielded an improved light inducible dimer (iLID) in which the affinity of the photoswitch for SspB changes > 50‐fold with light illumination (Guntas et al. 2015). Ma et al. (2018) replaced Rem DCT with SspB (creating optoRGK) and anchored iLID constitutively to the plasma membrane using Lyn11, a plasma membrane‐tethering peptide from the tyrosine protein kinase, Lyn. Exposure of cells to blue light led to rapid recruitment of optoRGK to the plasma membrane and resulted in CaV channel inhibition that was quickly reversed in the dark (Ma et al. 2018) (Fig. 2C ). Finally, as a demonstration of inhibiting CaV channels with subcellular specificity, replacing the Rem C‐terminus with a caveolin‐targeting peptide enabled selective inhibition of caveolae‐localized CaV1.2 in cardiac myocytes, without significantly affecting non‐caveolae CaV1.2 channels responsible for excitation‐contraction coupling (Fig. 2D ) (Makarewich et al. 2012).
Figure 2. Replacing Rem distal C‐terminus for novel spatio‐temporal control of CaV channel inhibition.
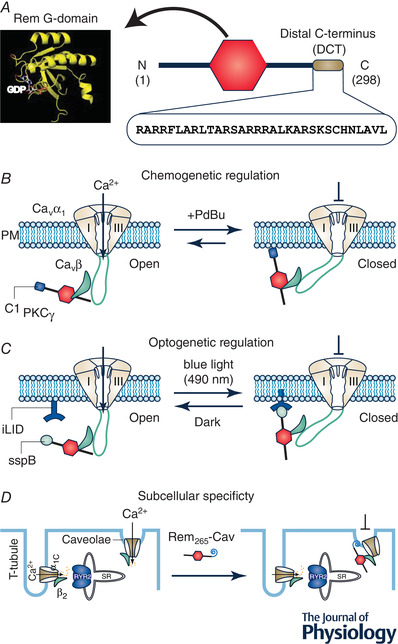
A, Rem structure consists of a guanine nucleotide binding domain (G‐domain) flanked by N‐ and C‐termini. The Rem distal C‐terminus (DCT), comprising the last 32 residues of the protein, is a polybasic peptide that mediates binding to the plasma membrane and is necessary for CaV channel inhibition. B, replacing Rem DCT with C1 domain from protein kinase C γ (C1PKCγ) enables acute recruitment of the engineered Rem to the plasma membrane with a small molecule phorbol ester, PdBu. Co‐expressed CaV1/CaV2 channels are inhibited concomitantly with Rem265‐C1PKCγ association with the plasma membrane. This chemogenetic configuration provides acute temporal control over CaV channel inhibition that is slowly reversible. C, optogenetic control of Rem inhibition of CaV channels was achieved using the photodimerizer pair, iLID (LOV2‐ssrA) and sspB. The Rem DCT was replaced with sspB via varying linkers (creating optoRGK) while iLID was constitutively anchored to the plasma membrane. Exposure of cells to blue light (470 nm) enabled acute recruitment of optoRGK to the plasma membrane and inhibition of CaV1.2 channels. Both plasma membrane association of optoRGK and CaV1.2 channel inhibition were reversed in the dark. D, replacing Rem DCT with a caveolae‐targeting peptide enabled selective inhibition of caveolae‐targeted CaV1.2 channels in cardiomyocytes while sparing dyadic CaV1.2 channels that mediate cardiac excitation‐contraction coupling.
The next conceptual advance came from further consideration of why Rem inhibition of CaV1.2 P o had the dual requirement for CaVβ binding and plasma membrane association? We hypothesized that Rem binding to the plasma membrane ‘pulled’ on the I‐II loop via the associated CaVβ subunit and induced a conformation of the channel with a low P o. This hypothesis led to a testable prediction that we could potentially evoke a similar low‐P o channel conformational state by directly attaching a membrane‐targeting module to auxiliary CaVβ subunits, thereby bypassing the need for an RGK altogether (Yang et al. 2013). To accomplish this, we fused the C1PKCγ onto the C‐terminus of CaVβ3 (generating β3‐C1PKCγ) which enabled a PdBu‐induced association of β3 with the plasma membrane (Yang et al. 2013). Channels reconstituted with β3‐C1PKCγ yielded robust baseline whole‐cell currents that were inhibited by exposure to PdBu. The kinetics and extent of inhibition could be tuned by serial truncations of the disordered β3 C‐terminus (shortening the β3 C‐terminus sped up the onset and deepened the extent of inhibition) (Yang et al. 2013). While this result was in accord with the stated hypothesis, it was, nevertheless surprising, because β2a and β2e subunits are naturally membrane‐associated via their N‐termini (Chien et al. 1998; Takahashi et al. 2003). β2a is palmitoylated, while the N‐terminus of β2e forms a helix that associates with the plasma membrane using electrostatic and hydrophobic interactions (Miranda‐Laferte et al. 2014). However, neither β2a nor β2e constitutively inhibit channels (rather, they both slow down voltage‐dependent inactivation of CaV1/CaV2 channels) (Takahashi et al. 2003). An apparent explanation for this discrepancy arose from the finding that placing the C1PKCγ module on the β3 N‐terminus yielded a construct that did not effectively inhibit CaV channels in response to PdBu, indicating that the phenomenon is sensitive to the polarity of the membrane‐targeting module on CaVβ (Yang et al. 2013). This suggests a geometric constraint to this mode of inhibition. Based on these results, we probed whether other cytosolic proteins that bound other intracellular loops of CaV channels could be transformed into CaV1/CaV2 inhibitors simply by introducing a membrane binding module to them. Indeed, we found that 14‐3‐3, a protein previously reported to bind to CaV2.2 C‐terminus (Li et al. 2006), could be turned into either a PdBu‐inducible or constitutive inhibitor by attaching C1PKCγ or a palmitoylated peptide, respectively (Yang et al. 2013). Unexpectedly, we found that 14‐3‐3‐C1PKCγ also effectively inhibited CaV1.2 and CaV2.1 channels in a phorbol ester‐dependent manner, revealing that these other channels also interacted with 14‐3‐3. We termed this general mechanism ChIMP, an acronym for ‘channel inactivation by membrane‐tethering an associated protein’ (Yang et al. 2013). Beyond CaV1/CaV2 channels, ChIMP may also be used either as an investigational tool or method to develop genetically encoded modulators for other ion channels. In this regard, we exploited ChIMP to reveal that calmodulin is preassociated with TMEM16A and TMEM16B Ca2+‐activated chloride channels and mediates Ca2+‐dependent sensitization of activation as well as Ca2+‐dependent inactivation of particular splice variants (Yang et al. 2014).
Deployment of genetically encoded CaV channel inhibitors derived from endogenous proteins (such as Rem1‐265‐C1PKCγ, β3‐C1PKCγ, and 14‐3‐3‐C1PKCγ) in vivo could potentially have unwanted effects owing to over‐expression of these modified natural proteins. As such, we sought to develop genetically encoded CaV channel inhibitors that would have limited off‐target effects relative to their inhibition of HVA CaV channels. Given the importance of CaVβ‐binding in RGK‐mediated CaV1/CaV2 inhibition, we first isolated nanobodies targeted to auxiliary CaVβ subunits. We immunized a llama with purified β1 and β3 subunits, isolated lymphocytes, amplified nanobodies by PCR, and cloned into a phagemid vector to generate a VHHS phage library. Several nanobody binders to β1 were isolated using phage display and an ELISA assay. One of these nanobodies, termed nb.F3, bound all four CaVβ isoforms when expressed in cells (Morgenstern et al. 2019), which was not surprising given the high homology among these auxiliary subunits in their conserved src homology 3 (SH3) and guanylate kinase (GK) domains (Chen et al. 2004; Opatowsky et al. 2004; Van Petegem et al. 2004). Purified nb.F3 bound CaVβ with high affinity (∼12 nM) and 1:1 stoichiometry as assessed by isothermal calorimetry. When expressed with reconstituted CaV2.2 and CaV1.2 channels in HEK293 cells, nb.F3 appeared functionally inert, as it had no impact on channel trafficking to the plasma membrane or on whole‐cell currents. Therefore, nb.F3 provided an ideal CaVβ‐targeting module that could potentially be modified to generate a genetically encoded CaV channel inhibitor exploiting the mechanisms we had identified for RGK proteins. We first sought to mimic the impact of RGKs on decreasing the channel surface density by fusing the catalytic HECT domain of the ubiquitin ligase, Nedd4L, onto nb.F3. The rationale for this approach is that in many ion channels and membrane proteins, ubiquitination typically reduces surface density and, often, enhances protein degradation as well (Abriel & Staub, 2005; Jespersen et al. 2007; MacGurn et al. 2012; Kanner et al. 2017). In heterologous cells, nb.F3‐Nedd4L decreased the surface density of reconstituted CaV2.2 and CaV1.2 channels without enhancing the degradation of the pore‐forming α1B and α1C subunits, respectively (Fig. 3A and B ) (Morgenstern et al. 2019). Whole‐cell patch clamp experiments demonstrated that nb.F3‐Nedd4L essentially eliminated reconstituted CaV1.2, CaV1.3 and CaV2.1‐CaV2.3 channel currents (Fig. 3C ). Therefore, we named nb.F3‐Nedd4L as CaV‐aβlator, reflecting it's exceptional efficacy to inhibit HVA CaV channels by targeting auxiliary CaVβ subunits. CaV‐ablator also proved effective in eliminating endogenous CaV1/CaV2 channels in pancreatic β‐cells, dorsal root ganglion (DRG) neurons and cardiac myocytes (Morgenstern et al. 2019). Examination of how CaV‐ablator eliminated CaV1.2 currents in ventricular cardiomyocytes indicated that pore‐forming α1C subunits were re‐directed from dyadic junctions to intracellular compartments, specifically Rab 7‐positive late endosomes (Morgenstern et al. 2019).
Figure 3. Mimicking RGK‐mediated CaV inhibition mechanisms with an engineered CaVβ‐targeted nanobody.
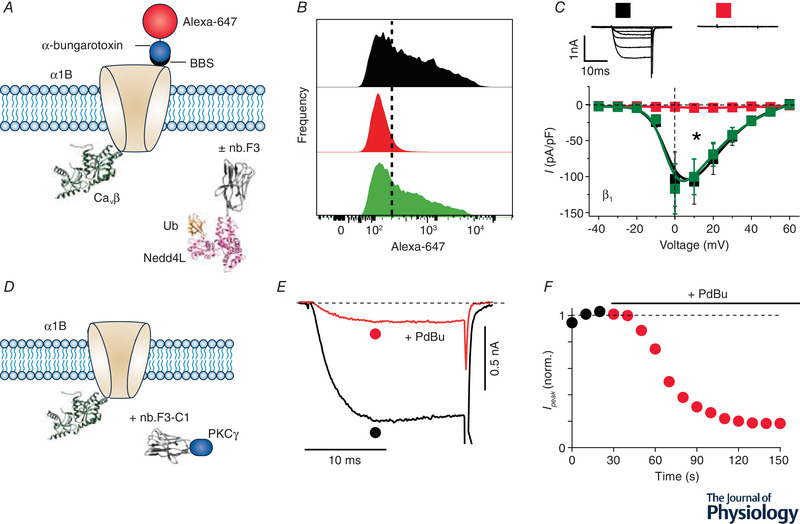
A, schematic diagram of experimental paradigm. Recombinant CaV2.2 (α1B) with an extracellular bungarotoxin‐binding site (BBS) epitope is co‐expressed CaVβ without (control) or with a CaVβ‐targeting nanobody (nb.F3) fused to catalytic HECT domain of the E3 ubiquitin ligase, NEDD4L. Surface channels are measured by exposing non‐permeabilized transfected cells to Alexa 647‐conjugated bungarotoxin. B, histograms of surface BBS‐α1B assessed by flow cytometry in cells expressing no nanobody (top), nb.F3‐NEDD4L (middle), or nb.F3‐NEDD4L*, a catalytically dead variant (bottom). Results show a substantial decline in surface density when the channel is co‐expressed with nb.F3‐NEDD4L. C, exemplar (top) and population I‐V curves (bottom) in cells expressing α1B + β1b alone (■) or with either nb.F3‐NEDD4L ( ) or nb.F3‐NEDD4L* (
) or nb.F3‐NEDD4L* ( ) co‐expression. D, schematic diagram of experimental paradigm. HEK293 cells are co‐transfected with recombinant CaV2.2 (α1B + β3) and nb.F3‐C1PKCγ. E and F, exemplar currents and diary plot showing rapid and deep PdBu‐induced inhibition of CaV2.2 currents in cells expressing α1B + β3 + nb.F3‐C1PKCγ.
) co‐expression. D, schematic diagram of experimental paradigm. HEK293 cells are co‐transfected with recombinant CaV2.2 (α1B + β3) and nb.F3‐C1PKCγ. E and F, exemplar currents and diary plot showing rapid and deep PdBu‐induced inhibition of CaV2.2 currents in cells expressing α1B + β3 + nb.F3‐C1PKCγ.
We have also explored whether we could also use nb.F3 to create a small‐molecule‐inducible genetically encoded CaV channel inhibitor that exploited the ChIMP mechanism. We generated nb.F3‐ C1PKCγ and co‐expressed it with recombinant CaV1.2 channels. Exposure of cells to phorbol ester resulted in a rapid decline in current that was not observed in control cells lacking nb.F3‐C1PKCγ, indicating that nb.F3 permits inducible inhibition of CaV1/CaV2 channels via the ChIMP method (Fig. 3D–F ).
Conclusion
In summary, this review highlights work focused on understanding the mechanisms by which RGK proteins potently inhibit CaV1‐ and CaV2‐family channels and exploiting mechanistic insights to create novel genetically encoded CaV channel inhibitors. This work has led to the development of intracellular acting genetically encoded CaV channel inhibitors that can be controlled by either small molecules or light, and that have the capacity to block CaV1.2 channels in cardiac myocytes with subcellular specificity. Genetically encoded CaV channel inhibitors have potential utility as therapeutics for indications such as chronic pain, with the advantage that their expression can be restricted to target tissues or cell types of interest, thereby circumventing off‐target effects. The viability of such gene therapy approaches has been advanced by continually improved development of viral and non‐viral gene delivery methods in vivo. For such potential therapeutic applications, it would be important to develop variants whose potency can be controlled either through dosage or with a small molecule. The nanobody‐based approach offers opportunities to design novel genetically encoded CaV channel inhibitors that can eliminate or modulate CaV channel complexes on the basis of identity of the associated β subunit isoform. This would be a key enabling tool to probe the potential role of auxiliary β subunits in organizing distinct CaV channels into distinct signalling complexes that permit functional diversification of Ca2+ influx via CaV channels in individual cells. Finally, some of the approaches described here may be generalizable to develop genetically encoded inhibitors or modulators for other ion channels and membrane proteins. Indeed, we have previously shown that the nanobody‐based targeted ubiquitination approach can be used to inhibit KCNQ1 channels by eliminating them from the cell surface (Kanner et al. 2017).
Additional information
Competing interests
None declared.
Funding
This work was supported by grants from the National Institutes of Health (RO1‐GM107585, RO1‐HL121253, and 1RO1‐HL122421) to H.M.C.
Acknowledgements
Thanks to all the talented trainees (Tingting Yang, Linling He, Akil Puckerin, Donald Chang, Zunaira Shuja, Scott Kanner and Travis Morgenstern) who have contributed to this research direction over the past few years.
Biography
Henry M. Colecraft, PhD, is the John C. Dalton Professor of Physiology and Cellular Biophysics at Columbia University Vagelos College of Physicians and Surgeons. Dr Colecraft's laboratory focuses on modulation of voltage‐gated ion channels (by intracellular signalling proteins, auxiliary subunits and posttranslational modifications) and understanding molecular/biophysical mechanisms underlying diseases caused by ion channel mutations (ion channelopathies). His lab has used protein engineering approaches to develop genetically encoded molecules to inhibit or modulate the activity of ion channels for customized applications.

Edited by: Ian Forsythe & Reinhold Penner
This review was presented at the symposium ‘FASEB Calcium and Cell Function meeting’, which took place at Granlibakken Resort, Tahoe City, CA USA, 10–15 June 2018.
This is an Editor's Choice article from the 1 May 2020 issue.
References
- Abriel H & Staub O (2005). Ubiquitylation of ion channels. Physiology (Bethesda) 20, 398–407. [DOI] [PubMed] [Google Scholar]
- Adams PJ, Ben‐Johny M, Dick IE, Inoue T & Yue DT (2014). Apocalmodulin itself promotes ion channel opening and Ca2+ regulation. Cell 159, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alseikhan BA, DeMaria CD, Colecraft HM & Yue DT (2002). Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A 99, 17185–17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister RA, Colecraft HM & Beam KG (2008). Rem inhibits skeletal muscle EC coupling by reducing the number of functional L‐type Ca2+ channels. Biophys J 94, 2631–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T & Seino S (2001). Regulation of Ca2+ channel expression at the cell surface by the small G‐protein kir/Gem. Nature 411, 701–706. [DOI] [PubMed] [Google Scholar]
- Beguin P, Ng YJ, Krause C, Mahalakshmi RN, Ng MY & Hunziker W (2007). RGK small GTP‐binding proteins interact with the nucleotide kinase domain of Ca2+‐channel beta‐subunits via an uncommon effector binding domain. J Biol Chem 282, 11509–11520. [DOI] [PubMed] [Google Scholar]
- Ben‐Johny M & Yue DT (2014). Calmodulin regulation (calmodulation) of voltage‐gated calcium channels. J Gen Physiol 143, 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P & Bootman MD (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation‐contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR & Rorsman P (2008). Voltage‐gated ion channels in human pancreatic beta‐cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628. [DOI] [PubMed] [Google Scholar]
- Buraei Z & Yang J (2010). The β subunit of voltage‐gated Ca2+ channels. Physiol Rev 90, 1461–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2011). Voltage‐gated calcium channels. Cold Spring Harb Perspect Biol 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, 3rd Puhl HL, , Niu SL, Mitchell DC & Ikeda SR (2005). Expression of Rem2, an RGK family small GTPase, reduces N‐type calcium current without affecting channel surface density. J Neurosci 25, 9762–9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L & Yang J (2004). Structural basis of the alpha1‐beta subunit interaction of voltage‐gated Ca2+ channels. Nature 429, 675–680. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Gao T, Perez‐Reyes E & Hosey MM (1998). Membrane targeting of L‐type calcium channels. Role of palmitoylation in the subcellular localization of the beta2a subunit. J Biol Chem 273, 23590–23597. [DOI] [PubMed] [Google Scholar]
- Crabtree GR & Schreiber SL (1996). Three‐part inventions: intracellular signaling and induced proximity. Trends Biochem Sci 21, 418–422. [DOI] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W & Yue DT (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2003). G protein modulation of voltage‐gated calcium channels. Pharmacol Rev 55, 607–627. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2012). Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci 13, 542–555. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ & Yue DT (2001). Preassociation of calmodulin with voltage‐gated Ca2+ channels revealed by FRET in single living cells. Neuron 31, 973–985. [DOI] [PubMed] [Google Scholar]
- Finlin BS & Andres DA (1997). Rem is a new member of the Rad‐ and Gem/Kir Ras‐related GTP‐binding protein family repressed by lipopolysaccharide stimulation. J Biol Chem 272, 21982–21988. [DOI] [PubMed] [Google Scholar]
- Finlin BS, Crump SM, Satin J & Andres DA (2003). Regulation of voltage‐gated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci U S A 100, 14469–14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlin BS, Mosley AL, Crump SM, Correll RN, Ozcan S, Satin J & Andres DA (2005). Regulation of L‐type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J Biol Chem 280, 41864–41871. [DOI] [PubMed] [Google Scholar]
- Finlin BS, Shao H, Kadono‐Okuda K, Guo N & Andres DA (2000). Rem2, a new member of the Rem/Rad/Gem/Kir family of Ras‐related GTPases. Biochem J 347, 223–231. [PMC free article] [PubMed] [Google Scholar]
- Gregg RG, Messing A, Strube C, Beurg M, Moss R, Behan M, Sukhareva M, Haynes S, Powell JA, Coronado R & Powers PA (1996). Absence of the beta subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the alpha 1 subunit and eliminates excitation‐contraction coupling. Proc Natl Acad Sci U S A 93, 13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G, Hallett RA, Zimmerman SP, Williams T, Yumerefendi H, Bear JE & Kuhlman B (2015). Engineering an improved light‐induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci U S A 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez‐Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT & Surmeier DJ (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ‐1. Nature 468, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Heo WD, Grimley JS, Wandless TJ & Meyer T (2005). An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods 2, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baro I & Abriel H (2007). The KCNQ1 potassium channel is down‐regulated by ubiquitylating enzymes of the Nedd4/Nedd4‐like family. Cardiovasc Res 74, 64–74. [DOI] [PubMed] [Google Scholar]
- Kamp TJ & Hell JW (2000). Regulation of cardiac L‐type calcium channels by protein kinase A and protein kinase C. Circ Res 87, 1095–1102. [DOI] [PubMed] [Google Scholar]
- Kang MG & Campbell KP (2003). Gamma subunit of voltage‐activated calcium channels. J Biol Chem 278, 21315–21318. [DOI] [PubMed] [Google Scholar]
- Kanner SA, Morgenstern T & Colecraft HM (2017). Sculpting ion channel functional expression with engineered ubiquitin ligases. eLife 6, e29744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F & Minor DL, Jr. (2008). Structures of CaV2 Ca2+/CaM‐IQ domain complexes reveal binding modes that underlie calcium‐dependent inactivation and facilitation. Structure 16, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Van Petegem F, Arant RJ, Findeisen F, Cooley ES, Isacoff EY & Minor DL, Jr (2010). Multiple C‐terminal tail Ca2+/CaMs regulate Ca(V)1.2 function but do not mediate channel dimerization. EMBO J 29, 3924–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Nunziato DA & Pitt GS (2004). Identification of the components controlling inactivation of voltage‐gated Ca2+ channels. Neuron 41, 745–754. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu Y & Zhou Y (2006). Modulation of inactivation properties of CaV2.2 channels by 14‐3‐3 proteins. Neuron 51, 755–771. [DOI] [PubMed] [Google Scholar]
- Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, Dangas K, Gygi SP, Pitt GS, Colecraft HM, Ben‐Johny M, Kalocsay M & Marx SO (2020). Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577, 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G, Liu J, Ke Y, Liu X, Li M, Wang F, Han G, Huang Y, Wang Y & Zhou Y (2018). Optogenetic control of voltage‐gated calcium channels. Angew Chem Int Ed Engl 57, 7019–7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC & Emr SD (2012). Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem 81, 231–259. [DOI] [PubMed] [Google Scholar]
- Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J & Kelly K (1994). Gem: an induced, immediate early protein belonging to the Ras family. Science 265, 241–244. [DOI] [PubMed] [Google Scholar]
- Makarewich CA, Correll RN, Gao H, Zhang H, Yang B, Berretta RM, Rizzo V, Molkentin JD & Houser SR (2012). A caveolae‐targeted L‐type Ca(2)+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res 110, 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV & Tavalin SJ (1998). Selective activation of Ca2+‐activated K+ channels by co‐localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905. [DOI] [PubMed] [Google Scholar]
- Meissner M, Weissgerber P, Londono JE, Prenen J, Link S, Ruppenthal S, Molkentin JD, Lipp P, Nilius B, Freichel M & Flockerzi V (2011). Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte‐specific excision of the cacnb2 gene. J Biol Chem 286, 15875–15882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P, Torrente AG & Mangoni ME (2015). Functional role of voltage gated Ca2+ channels in heart automaticity. Front Physiol 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda‐Laferte E, Ewers D, Guzman RE, Jordan N, Schmidt S & Hidalgo P (2014). The N‐terminal domain tethers the voltage‐gated calcium channel beta2e‐subunit to the plasma membrane via electrostatic and hydrophobic interactions. J Biol Chem 289, 10387–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern TJ, Park J, Fan QR & Colecraft HM (2019). A potent voltage‐gated calcium channel inhibitor engineered from a nanobody targeted to auxiliary CaVbeta subunits. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori MX, Vander Kooi CW, Leahy DJ & Yue DT (2008). Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high‐resolution mechanistic implications for channel regulation by Ca2+ . Structure 16, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Cingolani E, McDonald AD, Donahue JK & Marban E (2004). Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res 95, 398–405. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP & Hirsch JA (2004). Structural analysis of the voltage‐dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42, 387–399. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP & Yue DT (1999). Calmodulin is the Ca2+ sensor for Ca2+ ‐dependent inactivation of L‐type calcium channels. Neuron 22, 549–558. [DOI] [PubMed] [Google Scholar]
- Puckerin AA, Chang DD, Shuja Z, Choudhury P, Scholz J & Colecraft HM (2018). Engineering selectivity into RGK GTPase inhibition of voltage‐dependent calcium channels. Proc Natl Acad Sci U S A 115, 12051–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckerin AA, Chang DD, Subramanyam P & Colecraft HM (2016). Similar molecular determinants on Rem mediate two distinct modes of inhibition of CaV1.2 channels. Channels (Austin) 10, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynet C & Kahn CR (1993). Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science 262, 1441–1444. [DOI] [PubMed] [Google Scholar]
- Sudhof TC (2012). Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol 4, a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi SX, Mittman S & Colecraft HM (2003). Distinctive modulatory effects of five human auxiliary beta 2 subunit splice variants on L‐type calcium channel gating. Biophysical Journal 84, 3007–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC & Minor DL, Jr. (2005). Insights into voltage‐gated calcium channel regulation from the structure of the CaV1.2 IQ domain‐Ca2+/calmodulin complex. Nat Struct Mol Biol 12, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC & Minor DL, Jr (2004). Structure of a complex between a voltage‐gated calcium channel beta‐subunit and an alpha‐subunit domain. Nature 429, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, Wang Y, Chen C, Song R, Cao C, Zhang J, Wu C, Liu J & Cheng H (2010). Rad as a novel regulator of excitation‐contraction coupling and beta‐adrenergic signaling in heart. Circ Res 106, 317–327. [DOI] [PubMed] [Google Scholar]
- Weissgerber P, Held B, Bloch W, Kaestner L, Chien KR, Fleischmann BK, Lipp P, Flockerzi V & Freichel M (2006). Reduced cardiac L‐type Ca2+ current in Ca(V)beta2‐/‐ embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ Res 99, 749–757. [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P & Tsien RW (2012). Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca2+ signaling to control CREB‐dependent gene expression. Cell 149, 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C & Khodakhah K (2004). Calcium‐activated potassium channels are selectively coupled to P/Q‐type calcium channels in cerebellar Purkinje neurons. J Neurosci 24, 8818–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q & Yan N (2016). Structure of the voltage‐gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 537, 191–196. [DOI] [PubMed] [Google Scholar]
- Xu X & Colecraft HM (2009). Engineering proteins for custom inhibition of Ca(V) channels. Physiology (Bethesda) 24, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Marx SO & Colecraft HM (2010). Molecular mechanisms, and selective pharmacological rescue, of Rem‐inhibited CaV1.2 channels in heart. Circ Res 107, 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Katchman A, Kushner J, Kushnir A, Zakharov SI, Chen BX, Shuja Z, Subramanyam P, Liu G, Papa A, Roybal D, Pitt GS, Colecraft HM & Marx SO (2019). Cardiac CaV1.2 channels require beta subunits for beta‐adrenergic‐mediated modulation but not trafficking. J Clin Invest 129, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T & Colecraft HM (2013). Regulation of voltage‐dependent calcium channels by RGK proteins. Biochim Biophys Acta 1828, 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, He LL, Chen M, Fang K & Colecraft HM (2013). Bio‐inspired voltage‐dependent calcium channel blockers. Nat Commun 4, 2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Hendrickson WA & Colecraft HM (2014). Preassociated apocalmodulin mediates Ca2+‐dependent sensitization of activation and inactivation of TMEM16A/16B Ca2+‐gated Cl‐ channels. Proc Natl Acad Sci U S A 111, 18213–18218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Puckerin A & Colecraft HM (2012). Distinct RGK GTPases differentially use alpha(1)‐ and auxiliary beta‐binding‐dependent mechanisms to inhibit Ca(V)1.2/Ca(V)2.2 channels. PLoS One 7, e37079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Suhail Y, Dalton S, Kernan T & Colecraft HM (2007). Genetically encoded molecules for inducibly inactivating CaV channels. Nat Chem Biol 3, 795–804. [DOI] [PubMed] [Google Scholar]
- Yang T, Xu X, Kernan T, Wu V & Colecraft HM (2010). Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol 588, 1665–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW (2016). Targeting voltage‐gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov 15, 19–34. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Huang G, Wu J, Wu Q, Gao S, Yan Z, Lei J & Yan N (2019a). Molecular Basis for Ligand Modulation of a Mammalian Voltage‐Gated Ca2+ Channel. Cell 177, 1495–1506 e1412. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Huang G, Wu Q, Wu K, Li R, Lei J, Pan X & Yan N (2019b). Cryo‐EM structures of apo and antagonist‐bound human Cav3.1. Nature 576, 492–497. [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW & Reuter H (1999). Calmodulin supports both inactivation and facilitation of L‐type calcium channels. Nature 399, 159–162. [DOI] [PubMed] [Google Scholar]