

Graphical abstract

Keywords: Cyclophilin D, Inhibitor, Fragment-screening, X-ray structure, Surface plasmon resonance, Structure-based design, Fragment-based lead discovery
Highlights
-
•
6 Cyclophilin D crystal structures with SPR binding confirmed fragment hits having millimolar potencies were discovered.
-
•
One fragment was merged with a potent reference Cyclophilin D inhibitor resulting in single-digit nanomolar potency.
-
•
A second fragment provided the basis for replacing the urea linker in the reference inhibitor scaffold by an oxalyl group.
-
•
Two fragment hits bound in the S1′ and S2 pockets were linked via an amide group resulting in submicromolar potencies.
Abstract
Fragment-based screening by SPR enabled the discovery of chemical diverse fragment hits with millimolar binding affinities to the peptidyl-prolyl isomerase Cyclophilin D (CypD). The CypD protein crystal structures of 6 fragment hits provided the basis for subsequent medicinal chemistry optimization by fragment merging and linking yielding three different chemical series with either urea, oxalyl or amide linkers connecting millimolar fragments in the S1′ and S2 pockets. We successfully improved the in vitro CypD potencies in the biochemical FP and PPIase assays and in the biophysical SPR binding assay from millimolar towards the low micromolar and submicromolar range by >1000-fold for some fragment derivatives. The initial SAR together with the protein crystal structures of our novel CypD inhibitors provide a suitable basis for further hit-to-lead optimization.
Cyclophilins are peptidyl-prolyl isomerases (PPIase) catalyzing the cis–trans interconversion of proline peptide bonds during protein folding.1, 2 The human cyclophilin protein family consists of 17 highly conserved isoforms, which are abundantly and ubiquitously present in a wide range of tissue types and organisms with functions beyond protein maturation.2, 3 Among them, Cyclophilin A (CypA) is the major cellular target of the immunosuppressive drug cyclosporin A (CsA, Fig. 1 ) forming a ternary complex with calcineurin and thus preventing regulation of cytokine gene transcription.4, 5, 6 Cyclophilins also play a key role in pathophysiological processes such as inflammation and vascular dysfunction, wound healing, innate HIV immunity, hepatitis C infection, host-parasite interactions and tumor biology.7 Cyclophilin D (CypD) is the mitochondrial isoform of the enzyme, and a key regulator of the mitochondrial permeability transition pore. Mitochondrial dysfunction has been implicated in a cascade of cellular processes linked to multiple sclerosis and cardiovascular disease, making CypD a therapeutic drug target.7, 8, 9, 10 The crystal structures of several cyclophilins have been determined and show a common fold consisting of two α-helices packing against an eight-stranded anti-parallel P-barrel structure.11 The cyclophilins contain a large active binding groove composed by several highly conserved hydrophobic, aromatic and polar residues including the catalytic Arg55 located at the entrance of the S1′ proline pocket.2, 12 A second S2 pocket has been identified nearby: it is deep and relatively non-specific, with access controlled by a set of gatekeeper residues.2 The cyclic peptide CsA binds via specific interactions involving both S1′ and S2 pockets with nanomolar potency to cyclophilins, e.g. to CypD with a PPIase IC50 of 20 nM.13 However, CsA and its semisynthetic analogues such as Debio 025 and NIM811 have unfavorable drug-like properties due to high molecular weight, limited solubility and poor bioavailability.14, 15 Only few small and non-peptidic CypD inhibitors have been published including urea derivatives such as 2, which were discovered by fragment-based lead discovery (Fig. 1).10, 16, 17 These urea derivatives demonstrated in vitro PPIase inhibitory activity and antiviral activity against hepatitis C virus, human immunodeficiency virus and coronaviruses.16 Protein crystallography of 2 in CypD revealed specific binding of the pyrrolidine ring in the S1′ pocket, while the aniline substituent is bound in the S2 pocket (Supporting information).13 Our aim was to identify novel chemical hit matter from HTS and fragment screening approaches to develop CypD inhibitors with drug-like properties for prevention of mitochondrial dysfunction in multiple sclerosis.
Fig. 1.

Published CypD inhibitors (1–2).
We started our hit identification efforts by high-throughput screening on our corporate compound library with ~650,000 compounds using an FP biochemical assay, which resulted in only a small hit rate of 178 hits with IC50s < 10 µM. Disappointingly, none of these hits could be confirmed in orthogonal biophysical CypD binding assays using surface plasmon resonance (SPR) and protein-based NMR studies. Due to this outcome, we conducted an additional fragment-based screening campaign using our internal fragment library with 2688 structurally diverse fragments (Supporting information). The fragments were screened by SPR at fixed concentrations of 2 mM using immobilized CypD protein and yielded 168 primary hits. For subsequent hit confirmation, we used CsA at 200 nM for SPR-based competition experiments in compound titration series of 10 concentrations up to 10 mM. The affinity determination by SPR confirmed 58 hits with steady state dissociation constants (KD,ss) in the range of 1 mM to >10 mM. The identified fragments represented a large chemical diversity consisting of different aromatic as well as saturated rings as potential proline-mimicking motifs. However, the fragments had only millimolar potencies and overall low ligand efficiencies (LEs 0.1–0.3 kcal/heavy atom) beyond the high LE range of >0.3 kcal/heavy atom considered as optimal starting point for fragment optimization.18, 19 We therefore aimed to determine the binding mode in the CypD binding groove for as many fragments as possible by protein crystallography for structure-guided optimization. We evaluated 52 fragments by co-crystallization and by soaking into apo crystals of the CypD K175I mutant and obtained 6 crystal structures with clearly defined fragment electron densities in the active site at resolutions of 1.15–2.0 Å (Table 1 and Supporting information).20 The 6 fragments displayed a certain variety of binding modes within the CypD binding groove: 3 and 4 are bound in the gatekeeper S2 pocket, 5–7 are located in the proline S1′ pocket and 8 is targeting both S1′ and S2 pockets (Supporting information). All fragment X-ray structures were superimposed with published CypD structures in complex with CsA and urea derivatives such as 2 to define promising fragment linking and merging strategies for hit optimization. These considerations provided the basis of three hit series followed up by medicinal chemistry to improve potency in the biochemical FP and SPR binding assays.
Table 1.
Overview of SPR-confirmed hits from fragment screening against human CypD confirmed by X-ray crystallography.
| Compound | Structure | CypD |
Binding modec | Crystal structure PDB ID | |
|---|---|---|---|---|---|
| KD (mM)a | LEb | ||||
| 3 | 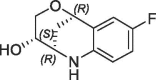 |
7.1 | 0.2 | S2 pocket | 6R9S |
| 4 | 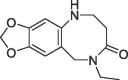 |
7.5 | 0.16 | S2 pocket | 6R9U |
| 5 | 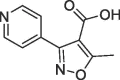 |
>10 | S1′ pocket | ||
| 6 | 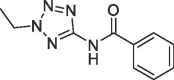 |
3.9 | 0.21 | S1′ pocket | |
| 7 | 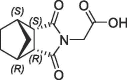 |
>10 | S1′ pocket | 6RA1 | |
| 8 | 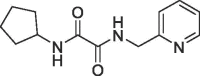 |
1.1 | 0.22 | S1′ & S2 pocket | 6R9X |
CypD SPR binding assay.
Ligand binding efficiencies (LE) based on the SPR KDs.
Fragment binding location in the CypD pocket from protein crystallization.
The superimposition of CypD crystal structures in complex with the three-dimensional fragment 3 (KD = 7.1 mM, LE = 0.2) and the published urea derivative 2 (PDB-ID: 4J5B) indicated almost perfect matching of the aniline rings in the S2 pocket. The annulated tetrahydropyran ring of 3 binds in a lipophilic sub pocket, which becomes accessible by side chain movement of Arg124 (Fig. 2 ). Furthermore, the fluorine atom of 3 provided a suitable exit vector towards the urea for fragment merging. We prepared a series of urea derivatives by incorporating the bicyclic fragment 3 in combination with the pocket S1′ substituent of 2 as different stereoisomers according to Scheme 1 .21
Fig. 2.
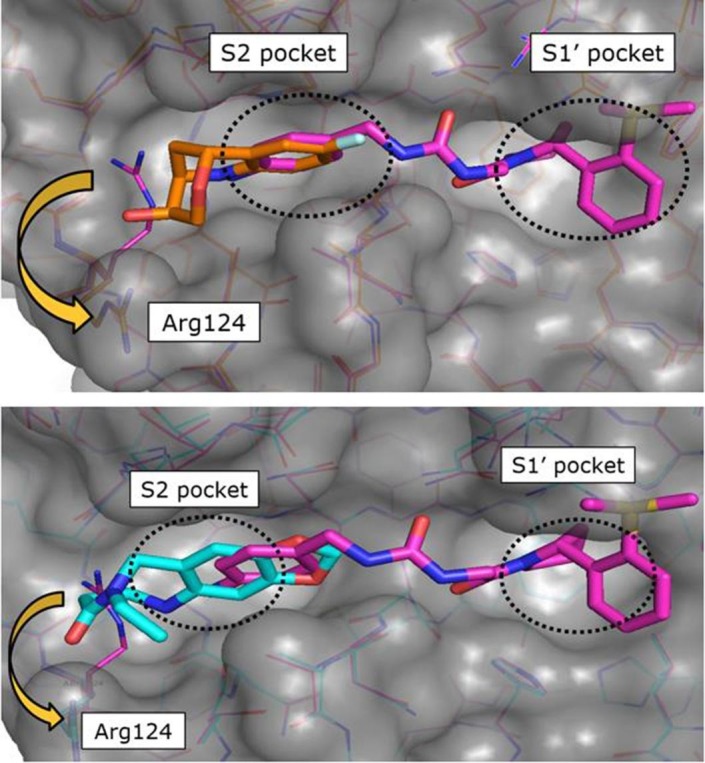
Superimpositions of CypD crystal structures in complex with published CypD inhibitor 2 (magenta, PDB-ID: 4J5B) and fragment hits 3 (orange, PDB-ID: 6R9S) and 4 (cyan, PDB-ID: 6R9U) indicated identical binding positions of the aniline rings in the S2 pocket. The annulated rings of 3 and 4 bind in a lipophilic sub pocket, which becomes accessible by side chain movement of Arg124.
Scheme 1.

Reagents and conditions: (a) Montmorillonite in MeCN at RT for 5 d; (b) step 1: lutidine, TBDMSOTf in DCM at 0 °C → RT for 4 h, step 2: TFA in DCM at 0 °C → RT for 1 h; (c) CDI in DCM at RT for 1 h, 12 in THF at 60 °C for 16 h; (d) HF.Pyr in THF at RT for 5 h.
The resulting urea derivatives 14–19 showed a clear SAR in the biochemical FP assay as well as in the CypD SPR assay regarding stereochemistry (Table 2 ). The most potent derivative 14 (FP IC50 = 60 nM, PPIase IC50 = 4 nM, SPR KD = 6 nM) had the same stereochemistry as the bicyclic fragment 3 (2R,3S,6R-enantiomer) and the S-methylphenyl substituent of 2 (R-enantiomer), which corresponds to their binding modes in the crystal structure. Fragment merging towards urea 14 resulted in significant potency improvements compared to the starting urea 2 (FP IC50 = 750 nM, SPR KD = 230 nM) of 12- or 40-fold referring to the FP and SPR assays respectively. The corresponding derivative 15 with the S-methylphenyl substituent as S-enantiomer was significantly less potent (FP IC50 = 3.6 µM, SPR KD = 630 nM). Also, the combination of the bicyclic fragment 3 as different stereo isomer (2S,3S,6S-enantiomer) and the S-methylphenyl substituent of 2 as R-enantiomer resulted in a clear potency drop (16: FP IC50 = 1.7 µM, SPR KD = 670 nM). For further analysis, we solved the CypD X-ray structures in complex with 14 (PDB-ID: 6R8O) and 16 (PDB-ID: 6R8L) at 1.4 Å and 1.6 Å resolution respectively (Fig. 3 ). Superimposition of both structures showed complete overlap of the urea groups including direct and water-mediated H-bonds to Arg97 and His96 as well as of the pocket S1′ substituents. The bicyclic rings of 14 and 16 also overlap regarding the NH-groups, which form H-bonds to the backbone carbonyl O-atom of Thr149 on one side and to ‘water molecule 1’ on the other side. In the crystal structure with 14, the ‘water molecule 1’ forms five H-bonds to the backbone atoms of Ser152, Gln153, Gln116 as well as to the OH group of the bicyclic ring, which has also H-bonds to Ser123 and Ser152. In contrast, the position of the 16 OH-group is completely shifted and not in H-bond contact to ‘water molecule 1’, which is involved in a more ideal tetrahedral H-bond network. In the structure with 16, the OH-group forms two H-bonds to Ser123 and to ‘water molecule 2’.
Table 2.
CypD SAR of optimized ureas derived from merging of fragments 3 or 4 with the reference inhibitor 2.
| # | Structure | Biochemical CypD FP assay |
Biochemical PPIase assay IC50 (µM)c | SPR binding CypD |
||
|---|---|---|---|---|---|---|
| IC50 (µM)a | LEb | KD (µM)d | LEe | |||
| 14 | 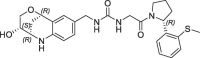 |
0.060 | 0.28 | 0.004 | 0.006 | 0.31 |
| 15 | 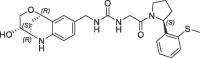 |
3.6 | 0.21 | nt | 0.630 | 0.23 |
| 16 | 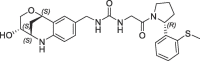 |
1.7 | 0.23 | nt | 0.670 | 0.23 |
| 17 | 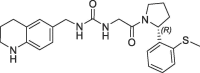 |
3.5 | 0.24 | 7.4 | 1.10 | 0.25 |
| 18 | 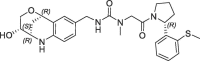 |
10 | 0.19 | nt | 7.0 | 0.19 |
| 19 | 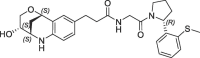 |
54 | 0.17 | nt | 39 | 0.17 |
| 20 | 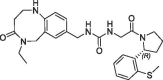 |
0.190 | 0.26 | nt | 0.180 | 0.25 |
| 21 | 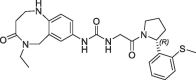 |
>100 | nt | 260 | 0.13 | |
| 22 |  |
4.1 | 0.21 | nt | 2.3 | 0.21 |
| 23 | 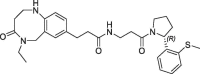 |
>100 | nt | 420 | 0.12 | |
a,bCypD biochemical FP and PPIase assays. c,eLigand binding efficiencies (LE) based on biochemical FP IC50s or SPR KDs. dCypD SPR binding assay. nt = not tested.
Fig. 3.

Overlay of CypD X-ray structures in complex with 14 (orange, PDB-ID: 6R8O) and 16 (magenta, PDB-ID: 6R8L) at 1.4 Å and 1.6 Å resolution respectively. H-bonds are displayed as dashed lines (hydrogen atoms are omitted and only water molecules in H-bond contact to the inhibitors are shown for clarity). The different stereochemistry of the bicyclic rings results in altered H-bond networks involving the OH-groups and water molecules 1 and 2.
This altered H-bond network and water co-ordination might contribute to the potency differences observed for 14 (SPR KD = 6 nM) and 16 (SPR KD = 670 nM). The potency contribution of the 3-OH-tetrahydropyrane part of the bicyclic ring is demonstrated by the tetrahydroquinoline derivate 17, which is significantly less active (FP IC50 = 3.5 µM, PPIase IC50 = 7.4 µM). Modifications of the urea linker either by N-methyl substitution (18: FP IC50 = 1.7 µM) or by amide replacement (19: FP IC50 = 54 µM) also resulted in large potency drops.
We also evaluated the second three-dimensional fragment 4 (KD = 7.5 mM, LE = 0.16) for similar merging with the urea derivative 2 by analyzing the superimposed crystal structures (Fig. 2). In this case, the 8-membered lactam ring of 4 is again located in a lipophilic sub cavity formed by induced fit of Arg124 within the S2 pocket. However, the aniline rings have significantly shifted positions (N-atom distance ~1.8 Å) resulting in suboptimal exit vectors for fragment merging with the urea group. Consequently, we prepared derivatives 20–23 with urea and amide linkers of different length merging the lactam ring of 4 with 2 as R-enantiomer (Table 2). The most potent analogue was the methylene urea derivative 20 (FP IC50 = 190 nM, SPR KD = 180 nM), while the urea directly attached to the aromatic ring of 4 resulted in complete loss of binding affinity (21: FP IC50 > 10 µM). The amide derivatives 22 and 23 with different chain lengths were also less potent (22: FP IC50 = 4.1 µM, 23: FP IC50 > 10 µM).
Fragment hit 8 (KD = 1.1 mM, LE = 0.22) binds with its cyclopentyl and 2-pyridine substituents in the S1′ and S2 pockets respectively, which are linked by an oxalyl group in H-bond contact to the Asn144 backbone atoms and the His168 side chain on one side (Supporting information). The side chain of Gln105 is not oriented in optimal angle towards the oxalyl carbonyl and NH-groups on the other side, but forms H-donor π-interactions as calculated with the software tool ViewContacts.22 The oxalyl group of fragment hit 8 overlaps with the urea of 2 and provides therefore an alternative strategy for linking the three-dimensional fragments 3 and 4 with S2 pocket substituents (Supporting information). The crystal structure superimposition of fragment hits 3 and 8 indicated a certain positional shift of the aniline and pyridine rings. We therefore prepared a series of oxalyl derivatives 27–32 from merging the bicyclic ring of fragment 3 with the cyclopentyloxalyl core of 8 with methylene and ethylene linkers according to Scheme 2 .23
Scheme 2.

Reagents and conditions: (a) Montmorillonite in MeCN at RT for 3 d; (b) TFA in DCM at 0 °C → RT for 1 h; (c) EDCI, HOPO and DIPEA in DMF at RT for 24 h.
The most potent analogue 27 achieved a >1000-fold potency enhancement in the biochemical assays (FP IC50 = 2 µM, PPIase IC50 = 1.5 µM) as well as in the SPR assay (KD = 2.8 µM) compared to the millimolar potencies of the individual fragments 3 and 8 (Table 3 ).
Table 3.
CypD SAR of optimized oxalamides derived from merging the fragment hits 3 or 4 with fragment hit 8.
| # | Structure | Biochemical CypD FP assay |
Biochemical PPIase assay IC50 (µM)c | SPR binding CypD |
||
|---|---|---|---|---|---|---|
| IC50 (µM)a | LEb | KD (µM)d | LEe | |||
| 27 | 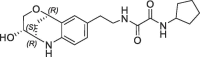 |
2 | 0.29 | 1.5 | 2.8 | 0.28 |
| 28 | 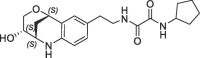 |
44 | 0.22 | nt | 23 | 0.21 |
| 29 | 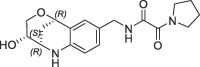 |
>100 | nt | >1500 | ||
| 30 | 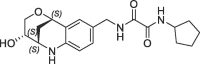 |
870 | 0.16 | nt | 217 | 0.19 |
| 31 | 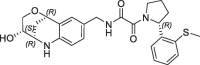 |
>100 | nt | 184 | 0.16 | |
| 32 | 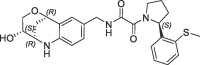 |
>30 | nt | >240 | ||
a,bCypD biochemical FP and PPIase assays. c,eLigand binding efficiencies (LE) based on biochemical FP IC50s or SPR KDs. dCypD SPR binding assay. nt = not tested.
Derivative 27 contains the bicyclic fragment 3 in the preferred stereochemistry (2R,3S,6R-enantiomer) as observed for 14 and an ethylene linker attached to the oxalyl group. The alternative analogues 28–32 with different bicyclic stereochemistry and/or methylene linkers are less potent or inactive (28: FP IC50 = 44 µM, 29–30: FP IC50 > 100 µM). We also evaluated a similar merging strategy of the lactam fragment 4 in combination with 8, but the analogues were again less potent (data not shown). Finally, we replaced the cyclopentyl ring of 27 by the S-methylphenyl substituent of 2 as R-enantiomer to gain additional potency by more optimized interactions in the S1′ pocket. But the resulting analogues did not show improved potencies (31: FP IC50 > 100 µM, 32: FP IC50 > 30 µM).
The protein crystal structure determination of the bicyclic fragment hit 7 (KD > 10 mM) failed by soaking into apo-crystals and by co-crystallization, but we succeeded to solve the structure by soaking of 7 into CypD co-crystals with 4. In the crystal structure, the fragment 7 binds as (1S,2R,6S,7R)-enantiomer in the S1′ proline pocket and forms H-bonds via one of the maleimide carbonyl O-atoms to Ala143 and His168 (Supporting information). The carboxylic acid of 7 is in ionic and H-bond contact to Arg97 and Gln105 respectively and points towards the S2 pocket. The shortest distance between one of the carboxylic O-atoms of 7 and the 1,3-benzodioxole C-atom of the co-crystallized fragment 4 is 3.3 Å, which promptly suggested fragment linking by 1–2 atom linkers (Fig. 4 ). We considered a more chemically stable amide instead of an ester for linking the bicyclic fragment 7 and the lactam fragment 4 and prepared a series of amides according to Scheme 3 .24
Fig. 4.
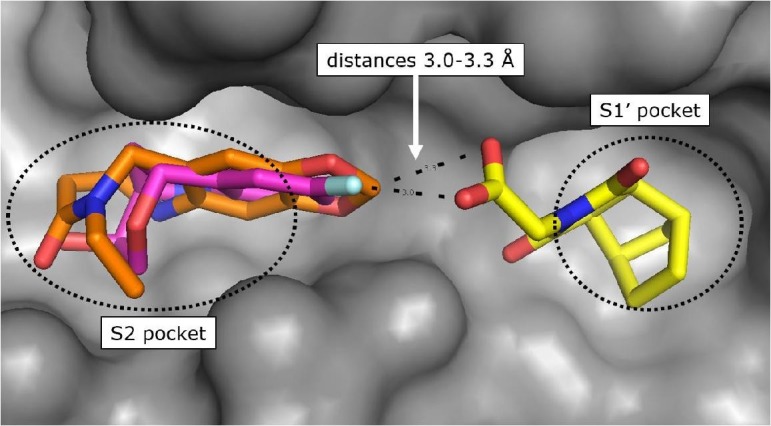
CypD X-ray structure in ternary complex with fragment hits 7 (yellow) and 4 (orange, PDB-ID: 6R9U) superimposed with X-ray structure in complex with fragment hit 3 (magenta, PDB-ID: 6R9S).
Scheme 3.

Reagents and conditions: (a) EDCI, HOPO and DIPEA in DMF at 50 °C for 24 h; (b) TBAF in THF at RT for 20 h.
The benzolactam analogue 34 linked by a N-methylamide group to the bicyclic ring of fragment 7 showed a modest potency improvement (Table 4 , 34: FP IC50 = 80 µM, SPR KD = 51 µM), while the shorter aniline derivative 35 was inactive (FP IC50 > 1000 µM). For further fine tuning of the optimal linker length, we evaluated the simple amides 36 and 37 with a methylene or ethylene spacer and an aniline substituent in the S2 pocket. In this case, the shorter methylene derivative 36 showed already a >1000-fold improvement (FP IC50 = 2.8 µM, PPIase IC50 = 28 µM, SPR KD = 4.9 µM) compared to the millimolar potencies of 7 and 4. In contrast, the longer ethylene analogue 37 was only weakly potent (FP IC50 = 490 µM). This result corresponds well with the distance of 3.0 Å between the 7 carboxylic O-atom and the 3-fluorine atom in the overlay of the CypD·7·4 ternary complex with CypD·3 (Fig. 4).
Table 4.
CypD SAR of optimized amides derived from fragment linking of fragment hits 3 or 4 with fragment hit 7.
| # | Structure | Biochemical CypD FP assay |
Biochemical PPIase assay IC50 (µM)c | SPR binding CypD |
||
|---|---|---|---|---|---|---|
| IC50 (µM)a | LEb | KD (µM)d | LEe | |||
| 34 | 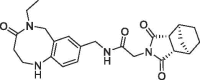 |
80 | 0.18 | nt | ND | |
| 35 | 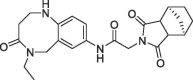 |
ND | nt | >6000 | ||
| 36 | 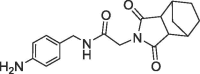 |
2.8 | 0.32 | 28 | 4.9 | 0.29 |
| 37 | 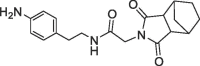 |
490 | 0.18 | nt | >200 | |
| 38 | 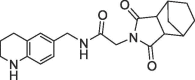 |
1.35 | 0.30 | 0.6 | 1.6 | 0.28 |
| 39 | 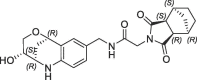 |
1.7 | 0.26 | nt | 0.660 | 0.26 |
| 40 | 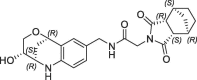 |
0.735 | 0.27 | nt | 1.2 | 0.25 |
a,bCypD biochemical FP and PPIase assays. c,eLigand binding efficiencies (LE) based on biochemical FP IC50s or SPR KDs. dCypD SPR binding assay. nt = not tested, ND = not detected.
Finally, we replaced the aniline of 36 with a tetrahydroquinoline substituent and the bicyclic fragment core 3 in the preferred stereochemistry (2R,3R,6S) yielding compounds 38–40 (Table 4).
The tetrahydroquinoline derivative 38 showed a 2-fold potency improvement (FP IC50 = 1.35 µM, PPIase IC50 = 0.6 µM, SPR KD = 1.6 µM) compared with 36, but 38 has been tested as racemic mixture of the different bicyclic norbornane maleimides (exo and endo forms). The separated stereoisomers 39 (endo) and 40 (exo) indeed showed slight potency improvements regarding either the biochemical or SPR binding assays (39: FP IC50 = 1.7 µM, SPR KD = 660 nM, 40: FP IC50 = 735 nM, SPR KD = 1.2 µM). We co-crystallized 40 in CypD (PDB-ID: 6R8W) and confirmed the fragment linking approach by clear identification of the bicyclic rings of 3 and 7 in the S2 and S1′ pockets respectively (Fig. 5 ). The binding orientation of the annulated tetrahydropyran ring of 40 in the S2 pocket corresponds with the fragment hit 3 or with the optimized urea 14. In contrast, the electron density indicated two alternative conformations of the norbornane maleimide ring of 40 in the S1′ pocket. Both orientations are distinguished by 180° rotation of the maleimides ring (exo form) in the S1′ pocket resulting in nearly identical positions of the carbonyl O-atom in H-bond contact to Asn144 and His168. Consequently, the bicyclic norbornane ring adopts two alternative orientations in the S1′ proline pocket with vdW-interactions to Phe102, Phe155 and Met103. The amide linker of 40 forms H-bonds directly to Arg97 and Gln105 and water mediated H-bonds to Gly114 and His96.
Fig. 5.

CypD X-ray structure in complex with the amide derivative 40 (PDB-ID: 6R8W) at 1.4 Å resolution, which was derived from fragment linking of 3 and 7. The bicyclic ring of 3 is bound in the S2 pocket with specific H-bonds involving also water molecules 1 and 2. The bicyclic norbornane maleimide ring of fragment 7 binds in two alternative conformations (green and yellow) distinguished by 180° rotation in the S1′ pocket. In both orientations, the maleimide carbonyl O-atom is in H-bond contact to Asn144 and His168. The amide group connecting both fragments forms H-bonds to Arg97 (direct) and Gln116 and His (water mediated).
In summary, our fragment-based approaches yielded three different series with either urea, oxalyl or amide linkers connecting millimolar fragments in the S1′ and S2 pockets. We successfully improved the in vitro CypD potencies in the biochemical FP assay and the biophysical SPR binding assays from millimolar towards the low and submicromolar range by >1000-fold for some fragments. The initial SAR together with the protein crystal structures of our novel CypD inhibitors provide a suitable basis for further hit-to-lead optimization.
Accession codes
Coordinates and structure factors have been deposited to the protein data bank with codes 6R9S, 6R9U, 6RA1, 6R9X, 6R8L, 6R8O and 6R8W.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2019.126717.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Viaud J., Subra G., Lin Y.-L. Structure-based design, synthesis, and biological evaluation of novel inhibitors of human cyclophilin A. J Med Chem. 2006;49(3):900–910. doi: 10.1021/jm050716a. [DOI] [PubMed] [Google Scholar]
- 2.Davis T.L., Walker J.R., Campagna-Slater V. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010;8(7) doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang P., Heitman J. The cyclophilins. Genome Biol. 2005;6:7. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y., Jiang J., Richardson P.L., Reddy R.D., Johnson D.D., Kati W.M. A fluorescence polarization-based assay for peptidyl prolyl cis/trans isomerase cyclophilin A. Anal Biochem. 2006;356(1):100–107. doi: 10.1016/j.ab.2006.04.040. [DOI] [PubMed] [Google Scholar]
- 5.Handschumacher R.E., Harding M.W., Rice J., Drugge R.J., Speicher D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science (80-) 1984;226(4674):544. doi: 10.1126/science.6238408. LP – 547. [DOI] [PubMed] [Google Scholar]
- 6.Huai Q., Kim H.-Y., Liu Y. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc Natl Acad Sci USA. 2002;99(19):12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giorgio V., Soriano M.E., Basso E. Cyclophilin D in mitochondrial pathophysiology. Biochim Biophys Acta. 2010;1797(6–7):1113–1118. doi: 10.1016/j.bbabio.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forte M., Bernardi P. Genetic dissection of the permeability transition pore. J Bioenergy Biomembr. 2005;37(3):121–128. doi: 10.1007/s10863-005-6565-9. [DOI] [PubMed] [Google Scholar]
- 9.Elrod J.W., Molkentin J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77:1111–1122. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panel M., Ruiz I., Brillet R. Small-molecule inhibitors of cyclophilins block opening of the mitochondrial permeability transition pore and protect mice from hepatic ischemia–reperfusion injury. Gastroenterology. 2019 doi: 10.1053/J.GASTRO.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 11.Dornan J., Taylor P., Walkinshaw M.D. Structures of immunophilins and their ligand complexes. Curr Top Med Chem. 2003;3(12):1392–1409. doi: 10.2174/1568026033451899. [DOI] [PubMed] [Google Scholar]
- 12.Ke H., Huai Q. Crystal structures of cyclophilin and its partners. Front Biosci. 2004;9:2285–2296. doi: 10.2741/1396. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed-Belkacem A., Colliandre L., Ahnou N. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat Commun. 2016;7 doi: 10.1038/ncomms12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopkins S., Gallay P. Cyclophilin inhibitors: an emerging class of therapeutics for the treatment of chronic hepatitis C infection. Viruses. 2012;4(11):2558–2577. doi: 10.3390/v4112558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ni S., Yuan Y., Huang J. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach. J Med Chem. 2009;52(17):5295–5298. doi: 10.1021/jm9008295. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed-Belkacem A., Colliandre L., Ahnou N. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat Commun. 2016;7:12777. doi: 10.1038/ncomms12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shore E.R., Awais M., Kershaw N.M. Small molecule inhibitors of cyclophilin D to protect mitochondrial function as a potential treatment for acute pancreatitis. J Med Chem. 2016;59(6):2596–2611. doi: 10.1021/acs.jmedchem.5b01801. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins A.L., Keseru G.M., Leeson P.D., Rees D.C., Reynolds C.H. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–121. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- 19.Siegal G., Ab E., Schultz J. Integration of fragment screening and library design. Drug Discov Today. 2007;12(23–24):1032–1039. doi: 10.1016/j.drudis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Schlatter D., Thoma R., Küng E. Crystal engineering yields crystals of cyclophilin D diffracting to 1.7 Å resolution. Acta Crystallogr Sect D Biol Crystallogr. 2005;61(5):513–519. doi: 10.1107/S0907444905003070. [DOI] [PubMed] [Google Scholar]
- 21.Jorand-Lebrun C, Johnson TL, Graedler U, et al. Preparation of urea and amide compounds for the inhibition of cyclophilins. PCT Int Appl 2017;(WO2017173048A1):95pp.
- 22.Kuhn B., Fuchs J.E., Reutlinger M., Stahl M., Taylor N.R. Rationalizing tight ligand binding through cooperative interaction networks. J Chem Inf Model. 2011;51(12):3180–3198. doi: 10.1021/ci200319e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorand-Lebrun C, Johnson TL, Graedler U, et al. Preparation of oxalyl compounds for the inhibition of cyclophilins. PCT Int Appl 2017;(WO2017173049A1):61pp.
- 24.Jorand-Lebrun C, Johnson TL, Graedler U, Jiang X, Kulkarni S. Preparation of dioxo-hexahydroisoindolyl compounds for the inhibition of cyclophilins. PCT Int Appl 2017;(WO2017173052A1):56pp.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.