

Abstract
Mitochondria contribute to shape intraneuronal Ca2+ signals. Excessive Ca2+ taken up by mitochondria could lead to cell death. Amyloid beta (Aβ) causes cytosolic Ca2+ overload, but the effects of Aβ on mitochondrial Ca2+ levels in Alzheimer’s disease (AD) remain unclear. Using a ratiometric Ca2+ indicator targeted to neuronal mitochondria and intravital multiphoton microscopy, we find increased mitochondrial Ca2+ levels associated with plaque deposition and neuronal death in a transgenic mouse model of cerebral β-amyloidosis. Naturally secreted soluble Aβ applied onto the healthy brain increases Ca2+ concentration in mitochondria, which is prevented by blockage of the mitochondrial calcium uniporter. RNA-sequencing from post-mortem AD human brains shows downregulation in the expression of mitochondrial influx Ca2+ transporter genes, but upregulation in the genes related to mitochondrial Ca2+ efflux pathways, suggesting a counteracting effect to avoid Ca2+ overload. We propose lowering neuronal mitochondrial Ca2+ by inhibiting the mitochondrial Ca2+ uniporter as a novel potential therapeutic target against AD.
Subject terms: Mitochondria, Cell death in the nervous system, Alzheimer's disease, Neurodegenerative diseases
Calvo-Rodriguez et al. show elevated calcium levels in neuronal mitochondria in a mouse model of cerebral β-amyloidosis after plaque deposition, which precede rare neuron death events in this model. The mechanism involves toxic extracellular Aβ oligomers and the mitochondrial calcium uniporter.
Introduction
Alzheimer’s disease (AD), the most common age-related neurodegenerative disorder, is characterized by deposition of extracellular senile plaques, neurofibrillary tangles and neurodegeneration, which lead to cognitive impairment and dementia. Despite the extensive efforts to understand its pathophysiology, the exact mechanisms underlying AD remain unknown. The most influential hypothesis for AD postulates that abnormal accumulation of the amyloid β (Aβ) peptide is the precipitating event that triggers a pathogenic cascade of adverse events, and that Aβ oligomers (Aβo), rather than amyloid plaques themselves, are the neurotoxic species of Aβ1 linked to tau aggregation2. Aβ neurotoxicity has also been associated with intraneuronal Ca2+ dyshomeostasis3. The calcium hypothesis of AD4 posits that activation of the amyloidogenic pathway remodels neuronal Ca2+ signaling, altering normal Ca2+ homeostasis and the mechanisms responsible for learning and memory. Previous work from our group and others has shown that Aβ aggregates increase cytosolic Ca2+ levels in vivo both in a mouse model of AD5,6, and after application of soluble Aβo to the brain of naive wild-type (Wt) mice7,8. However, these investigations assessed Ca2+ levels in the cytosol, and whether Aβ affects Ca2+ levels in mitochondria in vivo remains elusive.
Mitochondrial malfunction has been proposed as an early event in AD and other aging-related neurodegenerative disorders9. Studies on brains from AD patients and AD mouse models have shown impaired mitochondrial function, manifested as decreased bioenergetics and ATP synthesis10, morphological abnormalities11, imbalance of mitochondrial dynamics12 and redistribution of mitochondria13. Mitochondria participate in intracellular Ca2+ signaling as modulators, buffers and sensors14. After a cytosolic Ca2+ increase, mitochondria rapidly take up Ca2+ to prevent Ca2+ overload into the cytosol. However, excessive Ca2+ taken up into mitochondria, i.e., mitochondrial Ca2+ overload, results in increased reactive oxidative species (ROS) production, ATP synthesis inhibition, mitochondrial permeability transition pore (mPTP) opening, release of cytochrome c, activation of caspases and apoptosis15. In vitro studies have reported that Aβo induce mitochondrial Ca2+ uptake16,17, and Ca2+ transfer from ER to mitochondria18 in cultured rat primary neurons, but whether these findings apply in vivo remains unknown. In the present study, we assess whether Aβ aggregates alter mitochondrial Ca2+ levels in neurons in the mouse brain in vivo. We find elevated Ca2+ levels in neuronal mitochondria in transgenic mice, but only after plaque deposition, and preceding neural death. This mitochondrial Ca2+ overload in vivo involves toxic extracellular Aβ oligomers and requires the mitochondrial Ca2+ uniporter. We propose reducing mitochondrial Ca2+ overload by blocking the mitochondrial Ca2+ uniporter as a novel potential therapeutic target for AD.
Results
Functional AAV.hSyn.2mtYC3.6 targets neuronal mitochondria
We have previously observed cytosolic Ca2+ overload5 and mitochondrial membrane potential decrease11 in the APPswe/PSEN1∆E9 (APP/PS1) Tg mouse after amyloid plaque deposition. Here we hypothesized that Aβ aggregates could also increase neuronal mitochondrial Ca2+ levels in vivo. To study free Ca2+ concentration in mitochondria ([Ca2+]mit) in neurons, we targeted the expression of the ratiometric Ca2+ indicator Yellow Cameleon 3.6 (YC3.6)19 to neuronal mitochondria (Fig. 1a). YC3.6 allows visualization of subcellular Ca2+ concentration based on Förster resonance energy transfer (FRET) imaging between CFP and YFP, quantitatively measuring absolute Ca2+ concentrations. YC3.6 is one of the brightest reporters among the YC versions, and exhibits a large dynamic range and excellent signal-to-noise ratio19. Detailed characterization of hSyn.2mtYC3.6 expression was performed in vitro using neuroblastoma Neuro2a cells (N2a) and mouse primary cortical neurons, and ex vivo after immunofluorescence of sections from a Wt adult mouse brain injected with AAV.hSyn.2mtYC3.6. The punctate and perinuclear pattern expression of 2mtYC3.6 (Fig. 1b, c) suggested its mitochondrial localization. Co-transfection of cells with hSyn.2mtYC3.6 and mRuby-Mito-7 confirmed mitochondrial colocalization of 2mtYC3.6 (Fig. 1b). Co-labeling of AAV.hSyn.2mtYC3.6 with HSP60 antibody (Heat shock protein 60, localized to the mitochondrial matrix) in tissue sections corroborated the mitochondrial and neuronal-specific expression of the 2mtYC3.6 in vivo (Fig. 1c and Supplementary Fig. 1).
Fig. 1. hSyn.2mtYC3.6 targets neuronal mitochondria and is functional in vivo.

a Diagram of construct AAV.hSyn.2mtYC3.6-WPRE. b Mitochondrial counter labeling of 2mtYC3.6 verified proper targeting to mitochondria. N2a cells (top) and primary cortical neurons (bottom) were co-transfected with hSyn.2mtYC3.6 (green) and mRuby-Mito-7 (red) and subjected to confocal microscopy imaging. Scale bar 10 µm. c Colocalization of HSP60 (red) and hSyn.2mtYC3.6 (green) in cortex shown by immunohistochemistry (top). Scale bar 20 µm. Bottom graph shows intensity profile of the ROI across the cell (inset, scale bar 10 µm). Green line represents the intensity of hSyn.2mtYC3.6 and red line represent the intensity of HSP60. d In vitro calibration of 2mtYC3.6. Primary cortical neurons and N2a cells were exposed to varying [Ca2+] solutions, and relative changes in YFP/CFP ratio vs. [Ca2+] are indicated. Left, images are pseudocolored according to the color bar. Scale bar 10 µm. Right, titration curve of 2mtYC3.6 in N2a cells, two independent experiments, number of cells: 0 µM, n = 233; 0.2 µM, n = 339; 0.575 µM, n = 436; 1 µM, n = 332; 2 µM, n = 321; 5.7 µM, n = 356; 10 µM, n = 295; 20 µM, n = 317; 53 µM, n = 372; 100 µM, n = 372; 200 µM, n = 292; 500 µM, n = 209; 1000 µM, n = 274. e Validation of AAV.hSyn.2mtYC3.6 in vivo. Left, multiphoton microscopy imaging of the expression of AAV.hSyn.2mtYC3.6 in the cortex of a C57BL/6 J mouse. Scale bar 20 µm. Middle, pseudocolored images of the mitochondria expressing AAV.hSyn.2mtYC3.6 in basal conditions and after KCl application. Right, recording of the application of KCl to the brain. Gray traces represent single-cell responses and red trace represents averaged (mean ± SEM) [Ca2+]mit responses to 300 mM KCl. Representative of three mice.
We calibrated hsyn.2mtYC3.6 in N2a cells. Cells were transfected with hSyn.2mtYC3.6, and 24 h later, cells were permeabilized and exposed to solutions containing increasing known [Ca2+] (Methods). An increase in the YFP/CFP fluorescence ratio of YC3.6 corresponds to an increase in [Ca2+]. Ratios of YFP/CFP fluorescence intensities were plotted against [Ca2+] and fitted with a sigmoidal curve (Fig. 1d). We observed nearly 4-fold increase in the ratio at high [Ca2+]mit (Rmin=0.606, Rmax = 2.6921), with an apparent Kd (Kd′) of 4.21 µM and a Hill coefficient of 1.5720. Either the mitochondrial environment or the fusion of two mitochondrial sequences shifted the affinity of YC3.6 for Ca2+ to higher values, making it more suitable for Ca2+ measurements in mitochondria. This calibration was used to obtain [Ca2+]mit in the subsequent experiments. To verify that AAV.hSyn.2mtYC3.6 was functional in vivo, we applied KCl topically onto the brain of C57BL/6J mice injected with AAV.hSyn.2mtYC3.6 to induce neuronal depolarization, and imaged the neuronal mitochondria with multiphoton microscopy. Figure 1e and Supplementary Video 1 show an increase in the YFP/CFP ratio, representing [Ca2+]mit (pseudocolored images and Ca2+ recording), immediately after KCl application. Therefore, AAV.hSyn.2mtYC3.6 specifically targets mitochondria in neurons, can be expressed in vivo and provides a quantitative readout of neuronal [Ca2+]mit.
We compared the kinetics of cytosolic Ca2+ versus mitochondrial Ca2+ in primary neurons upon KCl-induced depolarization. We loaded cortical neurons with the cytosolic Ca2+ indicator calbryte 590, and infected them with our mitochondrial reporter hSyn.2mtYC3.6, to measure Ca2+ changes in the cytosol and mitochondria simultaneously (Supplementary Fig. 2). Application of KCl increased both cytosolic Ca2+ concentration ([Ca2+]cyt) and [Ca2+]mit. However, mitochondria exhibit different kinetics, which justified the need to address the mitochondrial Ca2+ impairment independent of cytosolic Ca2+ in the disease model.
Mitochondrial Ca2+ overload in vivo after Aβ plaque deposition
To address whether there are alterations in mitochondrial Ca2+ contributing to a general Ca2+ dyshomeostasis in AD, we examined [Ca2+]mit in the APP/PS1 Tg mouse model of cerebral β-amyloidosis in vivo using multiphoton microscopy. Age-matched Wt littermates were used as controls. 8-months-old (mo-old) APP/PS1 Tg mice were intracortically injected with AAV.hSyn.2mtYC3.6 targeting layers II/III, and 5 mm diameter cranial windows were implanted over the somatosensory cortex. Imaging was conducted 3 weeks later to allow for protein expression. Methoxy X0-4 was injected intraperitoneally 24 h before to label amyloid plaques21 (Fig. 2a). [Ca2+]mit was assessed by acquiring volumes of different regions of the cortex and determining the YFP/CFP ratio in the individual mitochondria. Figure 2b shows in vivo multiphoton microscopy images of mitochondria in neurons after transduction by AAV.hSyn.2mtYC3.6 in 9-mo-old Wt (top panel) and APP/PS1 Tg mice (bottom panel). On average, 9-mo-old APP/PS1 Tg mice exhibited higher YFP/CFP ratios than 9-mo-old Wt mice (Fig. 2c).We also evaluated mitochondrial Ca2+ overload using frequency distribution histograms (Fig. 2d). A general shift towards higher YFP/CFP ratios (higher [Ca2+]mit) was observed for APP/PS1 Tg mice. Ca2+ overload was defined as YFP/CFP ratio greater than two standard deviations (SD) above the mean YFP/CFP ratio obtained for all the mitochondria in all Wt mice5,7,22 (dashed line, YFP/CFP ratio >0.99, ~1637 nM [Ca2+]mit). Twice as many mitochondria from APP/PS1 Tg mice showed Ca2+ overload compared to Wt mice (Fig. 2e). [Ca2+]mit was also higher in the APP/PS1 Tg mice in both somas and neurites (Fig. 2f).
Fig. 2. Mitochondrial Ca2+ overload after Aβ plaque deposition.

a Experimental procedure to determine [Ca2+]mit in neurons in APP/PS1 Tg mice. 8-mo-old mice were injected with AAV.hSyn.2mtYC3.6 and a cranial window was implanted. Three weeks later, [Ca2+]mit was assessed by multiphoton microscopy. b In vivo multiphoton microscopy images of mitochondria expressing AAV.hSyn.2mtYC3.6 in Wt (top) and APP/PS1 Tg mice (bottom). Field of view shows AAV.hSyn.2mtYC3.6 in mitochondria (green), and blood vessels labeled with Dextran 70,000 MW (red). FRET-based Ca2+ imaging shows pseudocolored images of the field of view (scale bar 20 µm) and the inset from the white box (scale bar 5 µm). c Nine-mo-old APP/PS1 Tg mice exhibited increased ration YFP/CFP when compared to 9-mo-old Wt mice. Scatter dot plot representing the ratio YFP/CFP of every volume acquired. Error bars represent mean ± SEM. **p = 0.0015 (0.67 ± 0.011 (~472 nM [Ca2+]mit), n = 100 z-stacks from 11 Wt mice, and 0.73 ± 0.014 (~731 nM [Ca2+]mit), n = 70 z-stacks from 7 APP/PS1 Tg mice). d Histogram of [Ca2+]mit frequency distribution (indicated by YFP/CFP ratio). The black dotted line corresponds to 2 SD above the mean [Ca2+]mit in Wt mice (YFP/CFP ratio >0.99, ~1637 nM). Ratios above this line were classified as mitochondrial Ca2+ overload. e Comparison of the percentage of Ca2+-overloaded mitochondria in 9-mo-old Wt and APP/PS1 Tg mice. APP/PS1 Tg mice exhibited significantly higher percentage of mitochondria with elevated Ca2+. Scatter dot plot represents the percentage of Ca2+-overloaded mitochondria of every volume acquired. Error bars represent mean ± SEM. ***p = 0.001 (5.4 ± 0.79% from n = 70 z-stacks from 7 APP/PS1 Tg mice vs. 2.6 ± 0.51% from n = 100 z-stacks from 11 Wt mice). f Comparison of [Ca2+]mit (YFP/CFP ratio) in the different compartments (somas and neurites) in 9-mo-old Wt and APP/PS1 Tg mice. APP/PS1 Tg mice exhibited significantly higher Ca2+ levels in mitochondria both in somas and neurites. Error bars represent mean ± SEM. ****p < 0.0001 (Somas: 0.77 ± 0.01, n = 21 z-stacks from 3 Wt mice, and 0.82 ± 0.01, n = 18 z-stacks from 3 APP/PS1 Tg mice. Neurites: 0.67 ± 0.01, n = 21 z-stacks from 3 Wt mice, and 0.76 ± 0.01, n = 18 z-stacks from 3 APP/PS1 Tg mice). a Created using modified Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Next, we assessed whether the presence of Aβ plaques was required for the increased resting [Ca2+]mit, or whether it preceded plaque formation. APP/PS1 Tg mice start developing amyloid plaques at 4–5 mo-old23. No differences in YFP/CFP ratio or Ca2+ overload were detected between Wt and APP/PS1 mice at 2.5–3-mo-old (before plaque deposition), in either somas or neurites (Supplementary Fig. 3). Histograms in Supplementary Fig. 3 show that at this age there is no shift toward higher YFP/CFP ratios in APP/PS1 Tg mice relative to Wt mice at this early age. This is consistent with previous studies reporting no changes in [Ca2+]cyt and neuronal function prior to plaque deposition5,24,25, supporting the idea that mitochondrial Ca2+ overload in neurons follows, rather than precedes, amyloid plaque deposition.
Mitochondrial Ca2+ is independent from the distance to plaques
Individual Aβ plaques were previously reported to increase the likelihood of Ca2+ overload in the cytosol of neurites and cell bodies within a range of ~20–30 µm from the plaque edge5,6 in APP/PS1 Tg mice. To test whether Aβ plaques have local effects on the [Ca2+]mit in these mice, we measured mitochondrial Ca2+ overload relative to the distance between each mitochondrion and the edge of the nearest amyloid plaque. No significant differences were found in the average mitochondrial Ca2+ overload as a function of distance to plaques in either cell bodies or neurites (Fig. 3a, b), suggesting that soluble Aβo species, rather than Aβ plaques per se, are likely responsible for the increase of neuronal [Ca2+]mit. Indeed, it has been shown that the concentration of soluble Aβo in the brain of this mouse model increases with age23,26,27, which could explain the observed age-related [Ca2+]mit increase.
Fig. 3. Mitochondrial Ca2+ overload is independent of the distance to plaques.

a In vivo multiphoton microscopy pseudocolored images of AAV.hSyn.2mtYC3.6 expressing mitochondria in 9-mo-old APP/PS1 Tg mice. Elevated [Ca2+]mit was observed in mitochondria at <30 µm distance (left), but also at >30 µm distance (right). Arrow heads show mitochondria with Ca2+ overload in neuronal soma (top, scale bar 50 µm) and neurites (bottom, scale bar 10 µm). b Within 100 µm distance from the edge of a plaque, the probability of finding mitochondrial Ca2+ overload did not change. n = 31 plaques analyzed from 5 Tg mice (mean ± SEM, n.s. non-significant). The percentage of mitochondria showing Ca2+ overload in Wt mice is shown as reference (blue dotted line).
Soluble Aβ increases mitochondrial Ca2+ in Wt mice neurons
We have previously shown that acute exposure of the brain of naive (Wt) living mice to soluble Aβo leads to disruption of cytosolic Ca2+ homeostasis in neurons7. To test the hypothesis that soluble Aβ is responsible for the [Ca2+]mit increase, we topically applied naturally secreted soluble Aβo onto the brain of Wt mice expressing AAV.hSyn.2mtYC3.6, and assessed [Ca2+]mit with multiphoton microscopy. Aβo-enriched medium was obtained from primary cortical neurons isolated from Tg2576 embryos (APP Swedish mutation) (Tg-conditioned media, TgCM), which mainly contains low molecular weight oligomers (dimers to tetramers)7,28. CM from Wt-littermate neurons (WtCM) and Aβ-immunodepleted TgCM were used as controls. The three media were subjected to an Aβ40 sandwich ELISA. Aβ40 concentration in the media was 0.3, 5 and 0.7 nM for WtCM, TgCM and Aβ-immunodepleted TgCM, respectively. Aβo concentration in TgCM represents around 10% of the total amount of Aβ407.
To assess the effects of CM on [Ca2+]mit in the healthy brain, a cranial window was implanted over the somatosensory cortex of 4- to 5-mo-old C57BL/6J mice injected with AAV.hSyn.2mtYC3.6, the mouse was imaged at baseline, the window was reopened to apply either WtCM, TgCM or Aβ-depleted TgCM onto the brain for 1 h, and finally, the mouse was reimaged in the same volumes imaged at baseline (Fig. 4a, b). Mitochondrial Ca2+ overload was defined as the YFP/CFP ratio greater than 2SD above the mean YFP/CFP ratio of all the mitochondria imaged under basal conditions (YFP/CFP ratio >0.98, (~1604 nM [Ca2+]mit)). Application of TgCM significantly increased [Ca2+]mit, causing a shift towards higher YFP/CFP ratios (higher [Ca2+]mit) in the [Ca2+]mit histogram (Fig. 4c, middle). The proportion of mitochondria with Ca2+ overload after application of TgCM was significantly different than in basal conditions (Fig. 4d, middle). By contrast, topical application of WtCM did not increase [Ca2+]mit (Fig. 4c, left). To further validate that Aβo are responsible for the mitochondrial Ca2+ overload, we immunodepleted human Aβ from the TgCM with an anti-β-Amyloid antibody (β-amyloid 1–16, 6E10). Per ELISA measurements, immunodepletion of Aβ effectively removed about 85% of Aβ present in TgCM, from 5 to 0.7 nM (final Aβ40 concentration). Application of Aβ-immunodepleted TgCM did not increase [Ca2+]mit (Fig. 4c, right) compared to basal conditions. Comparing the averaged YFP/CFP ratios in each volume (from all mitochondria in each z-stack) acquired before and after application of CM (Fig. 4e), the majority of z-stacks analyzed after the application of TgCM were increased by 5% or more (dark lines), whereas only a few z-stacks showed a ≥5% increase for WtCM and Aβ-immunodepleted TgCM. Using these ratios, we calculated the relative change in YFP/CFP ratio (∆R/R0) for each z-stack. Application of TgCM induced a relative change in the YFP/CFP ratio significantly higher than WtCM or Aβ-immunodepleted TgCM (Fig. 4f). This effect was more prominent in neurites (Fig. 4g). These relatively small, but significant differences, suggest that a small fraction of neuronal mitochondria are affected by TgCM, similar to our observation in plaque-bearing APP/PS1 Tg mice, and implicate of soluble Aβo in mitochondrial Ca2+ overload in neurons in vivo.
Fig. 4. Soluble Aβ increases mitochondrial Ca2+ concentration in neurons in vivo.

a Experimental procedure: 4-mo-old C57BL/6J mice were injected with AAV.hSyn.2mtYC3.6. A cranial window was implanted the day of the experiment (3 weeks later). [Ca2+]mit was assessed in basal conditions. Then, the window was reopened and either WtCM, TgCM or Aβ-immunodepleted TgCM was applied for 1 h. Finally, the mouse was reimaged in the exact same regions as for basal conditions. b Representative pictures of the effects of CM in the healthy living mouse brain. Arrow heads show mitochondria with high [Ca2+]. Scale bar 20 and 5 µm for the inset. c Histograms of [Ca2+]mit frequency distribution (YFP/CFP ratio) for the three conditions before (basal) and after application of CM. The black dotted line corresponds to 2SD above the mean [Ca2+]mit measured for all mice in baseline. Ratios above this line were classified as mitochondrial Ca2+ overload. d Scatter dot plot represents the percentage of mitochondria showing Ca2+ overload before and after application of CM for every volume acquired. Error bars represent mean ± SEM (WtCM: before 2.45 ± 0.43% vs. after 2.92 ± 0.49%; non-significant, n = 32 z-stacks from 5 mice. TgCM: before 2.89 ± 0.47% vs. after 7.53 ± 1.09%; ****p < 0.0001, n = 48 z-stacks from 7 mice. Aβ-depleted TgCM: before 2.43 ± 0.13% vs. after 2.66 ± 0.20%; non-significant, n = 38 from z-stacks 5 mice). The percentage of Ca2+ overload before (basal) and after CM application is also noted on the graphs. e Averaged YFP/CFP ratios before and after treatment for each z-stack acquired. The dark traces account for the z-stacks showing an increase ≥5% in YFP/CFP ratio after topical application of CM. f Scatter dot plot represents the relative change in ratio for each condition. Error bars represent mean ± SEM (WtCM: 0.20 ± 1.09%, n = 32 z-stacks; TgCM: 7.59 ± 0.92%, 48 z-stacks; depleted TgCM 1.20 ± 1.05%, 38 z-stacks from 5, 7 and 5 mice, respectively, ****p < 0.0001). g Relative increase of [Ca2+]mit (∆R/R0) in the different compartments (somas and neurites) after WtCM or TgCM application. Error bars represent mean ± SEM. **p < 0.005 (Somas: 0.79 ± 0.91, n = 8 z-stacks from 3 WtCM application mice, and 3.10 ± 0.82, n = 10 z-stacks from 3 TgCM application mice. Neurites: −0.02 ± 1.44, n = 8 z-stacks from 3 WtCM application mice, and 10.15 ± 1.05, n = 10 z-stacks from 3 TgCM application mice). a Created using modified Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
MCU is required for Aβo-driven mitochondrial Ca2+ uptake
The main Ca2+ uptake pathway in mitochondria is the mitochondrial Ca2+ uniporter (MCU) complex, which includes the MCU pore, located in the inner mitochondrial membrane29,30, and the combination of two regulatory subunits MiCU1 and MiCU2 (mitochondrial calcium uptake 1 and 2 proteins), which regulate the MCU response to extra-mitochondrial Ca2+ 31,32. Other proteins involved in the complex include the essential MCU regulator (EMRE)33, MCUb34, or MiCU335. Mitochondrial Ca2+ efflux depends mainly on the electrogenic Na+-dependent Ca2+ release exchanger (NCLX)36. Given the increased number of mitochondria with Ca2+ overload in APP/PS1 Tg mice after amyloid plaque deposition, we performed Western blots for MCU, MiCU1, MiCU2, and NCLX in mitochondrial-enriched fractions of the brain of 10-mo-old Wt and APP/PS1 Tg mice to test whether Aβ affects the expression levels of these proteins. No significant differences were detected between genotypes in any of these mitochondrial proteins (Supplementary Fig. 4).
To test whether Aβo induce mitochondrial Ca2+ increase in neurons via the MCU complex, we blocked the pore with the specific cell-permeable MCU inhibitor Ruthenium 360 (Ru360)37. As in Fig. 4a, C57BL/6J Wt mice expressing hSyn.2mtYC3.6 were subjected to a craniotomy, and [Ca2+]mit was assessed in basal conditions. The window was then reopened and the underlying brain was pre-treated with Ru360 (100 µM) for 15 min. TgCM mixed with Ru360 was next applied onto the brain for 1 h, and the mouse was reimaged in the same areas. TgCM increased neuronal [Ca2+]mit (arrow heads) as expected, but application of TgCM mixed with Ru360 did not (Fig. 5a). Pre-treatment with Ru360 did not shift the distribution of YFP/CFP ratios when compared to TgCM alone (Fig. 5b), and the proportion of mitochondria with Ca2+ overload, (YFP/CFP ratio >1.01 (~1704 nM [Ca2+]mit)), was not significantly different (Fig. 5c). Topical application of Ru360 alone did not have an effect on [Ca2+]mit. Comparing the average YFP/CFP ratio in each volume acquired before and after application of TgCM with Ru360, only a few volumes increased 5% or more (dark lines), whereas for the majority the increase was ≤5% (light lines) (Fig. 5d). Average of the relative change in YFP/CFP ratio (∆R/R0) for each volume (Fig. 5e) showed that application of TgCM without Ru360 induced a change in YFP/CFP ratio significantly different from the application of TgCM with Ru360. Importantly, Ru360 application did not inhibit the rise of Ca2+ in the cytosol induced by TgCM (as tested in C57Bl/6J mice injected with CBA.YC3.65) (Supplementary Fig. 5), suggesting that Ru360 did not render neurons insensitive to Aβo or blocked channels/influx pathways in the plasma membrane. These data suggest that MCU is required for the Aβ-driven mitochondrial Ca2+ increase in neurons in the brain in vivo.
Fig. 5. MCU is required for the Aβ-induced mitochondrial Ca2+ increase in vivo.

a Representative pictures of the effects of the MCU complex blocker Ru360 on the increase of [Ca2+]mit induced by TgCM. Arrow heads show mitochondria with high [Ca2+]. Scale bar 20 and 5 µm for the inset. b Graphs show histogram of [Ca2+]mit frequency distribution (expressed as YFP/CFP ratio) before and after topical application of TgCM in presence of Ru360 and of Ru360 alone. The percentage of Ca2+ overload before (basal) and after TgCM application in presence of Ru360 is noted. c Scatter dot plot represents the percentage of mitochondria showing Ca2+ overload before and after application of CM for every volume acquired. Error bars represent mean ± SEM (Ru360 + TgCM: before 2.37 ± 0.47% vs. after 1.89 ± 0.27%, non-significant, n = 28 z-stacks from 4 mice. Ru360: before 2.82 ± 0.86% vs. after 2.41 ± 0.78%, non-significant, n = 18 z-stacks from 3 mice). d Averaged YFP/CFP ratios before and after treatment for each z-stack acquired. The dark traces account for the z-stacks that showed an increase ≥5% in YFP/CFP ratio after topical application of the treatment. e Scatter dot plot represent the relative change in YFP/CFP ratio for each condition. Bars represent mean ± SEM (7.60 ± 0.81%, 48 z-stacks TgCM; −0.27 ± 1.05%, 28 z-stacks Ru360+TgCM; −0.92 ± 1.25%, 18 z-stacks Ru360 from 7, 4 and 3 mice, respectively, ****p < 0.0001).
Mitochondrial Ca2+ precedes neuronal death
We next asked whether the excessive Ca2+ could have a detrimental effect in Ca2+ overloaded mitochondria and, eventually, also in the neurons containing these mitochondria. We examined the time course of Aβo-driven neuronal mitochondrial Ca2+ overload in the C57BL/6 J Wt mouse brain, and specifically asked whether this was permanent or reversible and, if the latter, how long after Aβo application. At 8 h post-application of TgCM, neuronal [Ca2+]mit had already returned to basal levels (Fig. 6d–e). As expected, WtCM had no effect on [Ca2+]mit (Fig. 6a–c). Despite the relatively transient mitochondrial Ca2+ overload associated to acute topical application of Aβo, we noted altered mitochondria shape and size at 8 h only with TgCM (but not with WtCM) compared to basal conditions (Fig. 6a, d, insets; Supplementary Figs. 6 and 7), in agreement with previous reports showing mitochondrial morphological abnormalities in the human AD brain and AD Tg mice11,38.
Fig. 6. Soluble Aβ-driven mitochondrial Ca2+ increase recovers after 8 hours post-application.

a Time-lapse images showing mitochondrial [Ca2+] increase induced by WtCM, in basal conditions, 1 and 8 h after topical application. Scale bar 20 µm. b Histogram of [Ca2+]mit frequency distribution (YFP/CFP ratio) before and 1 or 8 h after application of WtCM. The black dotted line corresponds to 2SD above the mean [Ca2+]mit measured for all mitochondria before treatment. Ratios above this line were classified as mitochondrial Ca2+ overload. c Scatter dot plot represents the percentage of mitochondria with Ca2+ overload at each time point in every volume acquired. Bars errors represent mean ± SEM (Basal 2.5 ± 0.54%, 1 h 2.7 ± 0.54%; 8 h 3.3 ± 0.54%; non-significant, n = 22 z-stacks from 3 mice). d Time-lapse images showing mitochondrial [Ca2+] increase induced by TgCM, in basal conditions, 1 and 8 h after application. Scale bar 20 µm. e Histogram of [Ca2+]mit frequency distribution (YFP/CFP ratio) before and 1 or 8 h after application of TgCM. Note that [Ca2+]mit distribution is shifted to higher YFP/CFP ratios at 1 h post- TgCM application, but recovers at 8 h post-TgCM application. f Scatter dot plot represents the percentage of mitochondria with Ca2+ overload at each time point in every volume acquired. Error bars represent mean ± SEM (Basal 3.2 ± 0.60%, 1 h 6.3 ± 1.22%; 8 h 3.7 ± 0.70%; *p < 0.05 vs. basal, n = 25 z-stacks from 5 mice).
Because mitochondrial Ca2+ overload can lead to apoptosis39, we examined the viability of neurons with chronic (rather than transient) mitochondrial Ca2+ overload using longitudinal multiphoton microscopy serial imaging over a 24–48 h period in 9-mo-old APP/PS1 Tg mice. First, we evaluated whether the static mitochondrial Ca2+ levels were stable longitudinally. We observed that over 3 imaging sessions (48 h), the absolute mitochondrial Ca2+ levels were constant in the same cell, for both low and intermediate Ca2+ levels (Fig. 7a). Surprisingly, a small fraction of neurons with extremely high [Ca2+]mit in their somas at the first imaging session (on average, YFP/CFP ratio ≥1.4, (~3093 nM [Ca2+]mit)) were lost within 24 h (Fig. 7b, c). None of the neurons exhibiting YFP/CFP ratio ≤1.4 disappeared in the second imaging session. We concomitantly injected Hoechst 33342 with hSyn.2mtYC3.6 to observe the nucleus and [Ca2+]mit simultaneously. We observed that the nucleus from healthy neurons was spherical and chromatin was evenly distributed. Conversely, in neurons with high [Ca2+]mit at baseline, the nucleus became condensed (likely undergoing apoptosis), and the soma lost its of YFP/CFP fluorescence (Fig. 7c). Despite being an occasional event in this model40, this observation suggests a close correlation between high [Ca2+]mit and neuronal cell death.
Fig. 7. Mitochondrial Ca2+ overload precedes neuronal death in the APP/PS1 Tg mice.

a Time-lapse images showing that resting mitochondrial Ca2+ levels in neurons are steady over time in the 9-mo-old APP/PS1 Tg mouse. The same field of view was imaged at 3 different time points (3 consecutive days). AAV.hSyn.2mtYC3.6 (neurons, green) and Dextran Texas Red (vessels) are shown in the top row and pseudocolor images in the bottom row. Insets show somas of the same neuron during the time lapse. Scale bar 25 µm. b Time-lapse images showing mitochondrial Ca2+ overload preceding neuronal cell death in a 9-mo-old APP/PS1 Tg mouse. Arrow heads show mitochondrial Ca2+ overload in the soma of a neuron, which disappeared 24 h later. Scale bar 20 µm. Scatter dot plot represents YFP/CFP ratio of single neurons present at baseline imaging session that remained or disappeared at 24 h follow-up imaging session as a function of their baseline [Ca2+]mit. Neurons with a YFP/CFP ratio >1.4 disappeared within 24 h (mean ± SEM, n = 27 and 14 neurons from 4 APP/PS1 mice, ****p < 0.0001, Student t-test). c Images show time-lapse recordings of nuclei and mitochondrial Ca2+ levels preceding neuronal cell death in a 9-mo-old APP/PS1 Tg mouse. The same field of view was imaged at 2 different time points (2 consecutive days). AAV.hSyn.2mtYC3.6 (neurons, green) and Dextran Texas Red (vessels) are shown in the top row, and pseudocolor images are shown in the middle (scale bar 20 µm) and bottom (scale bar 5 µm) rows. 800 nm inset shows AVV (mitochondria, green) and Hoescht (nuclei, blue) acquired at 800 nm. It was observed that remaining cells (low mitochondrial Ca2+) maintained a healthy nucleus (Hoechst), whereas disappearing cells (high mitochondrial Ca2+) presented a condensed nucleus.
To further understand the effects of increased mitochondrial Ca2+ concentration and the connection to neuronal death, we performed some mechanistic studies in vitro using mouse primary cortical neurons exposed to TgCM. First, we determined that TgCM increased [Ca2+]mit in primary neurons (infected with hSyn.2mtYC3.6) (Supplementary Fig. 8A) within 1 h, corroborating the in vivo data. In addition, TgCM decreased the mitochondrial membrane potential (∆Ψm) (measured with TMRE) (Supplementary Fig. 8D), activated the mPTP (evaluated by calcein/cobalt fluorescence) (Supplementary Fig. 8C), and activated caspases 3/7 (detected with CellEvent Caspase 3/7) in primary neurons (Supplementary Fig. 8E). These effects took place in the same time window as the increase of [Ca2+]mit. WtCM did not drive any of these effects on mitochondria. Blocking of the mPTP with cyclosporine A (CsA)41, as verified by calcein/cobalt fluorescence, prevented the decrease of ∆Ψm, but did not inhibit the increase of [Ca2+]mit elicited by TgCM. These results suggest that the reduction of ∆Ψm is driven by the increase in mitochondrial Ca2+ after TgCM application. Blocking mPTP with CsA showed a trend towards decreasing the number of neurons with caspase 3/7 activation (Supplementary Fig. 8).
Mitochondrial Ca2+ dyshomeostasis in the human AD brain
Finally, we sought to test whether mitochondrial Ca2+ dyshomeostasis is relevant to the human AD brain. Expression of mitochondrial genes encoding proteins involved in Ca2+ transport was assessed using gene differential expression data publicly available at the Accelerating Medicines Partnership-Alzheimer’s Disease (AMP-AD) Knowledge Portal42. We evaluated 25 publicly available microarray and RNA-Sequencing datasets from the Mount Sinai Brain Bank (MSBB)43 and the Religious Orders Study and Memory and Aging Project (ROSMAP)44 cohorts, and compared the transcript levels of mitochondrial Ca2+-related genes between subjects with AD (Braak stages V–VI) and control individuals (Braak stages 0–I–II), specifically MCU, MCUB, MCUR1, MICU1, MICU2, MICU3 and SMDT1 for mitochondrial Ca2+ influx, and SLC8B1 (encoding NCLX) for mitochondrial Ca2+ efflux. Surprisingly, the mitochondrial Ca2+ uniporter (Mcu) and its regulatory subunits (Micu1, Micu2, Micu3 and Smdt1) are significantly downregulated (p < 0.05 and FDR < 25%) in AD compared to control subjects after adjusting for neuronal loss with the pan-neuronal marker Map2 (Supplementary Fig. 9, Supplementary Table 1). Interestingly, the Ca2+ efflux gene Slc8b1 is significantly upregulated. Mcur1 is also downregulated in the frontal pole and the superior temporal gyrus but fell just below the FDR threshold. No change was observed in Mcub. Analyses by CERAD scores (frequent versus absent neuritic amyloid plaques)45 produced similar results (Supplementary Table 2). All genes for mitochondrial Ca2+ influx were downregulated (FDR < 20%), whereas Slc8b1 was upregulated. Mcur1 was also upregulated in this case. These results suggest that the expression of most of human genes involved in mitochondrial Ca2+ transport is altered in AD patients, denoting a counteracting effect to avoid mitochondrial Ca2+ overload.
The present results suggest that Aβ aggregates (likely soluble Aβo) lead to mitochondrial Ca2+ overload in AD. Aβ-induced mitochondrial Ca2+ uptake requires MCU, as blocking MCU with the specific inhibitor Ru360 prevents the Ca2+ overload. In addition, excessive Ca2+ taken up by mitochondria leads to opening of the mitochondrial permeability transition pore (mPTP), caspase activation and, eventually, neuronal cell death (Fig. 8).
Fig. 8. Schematic representing the effects of Aβ aggregates on mitochondrial Ca2+ homeostasis.
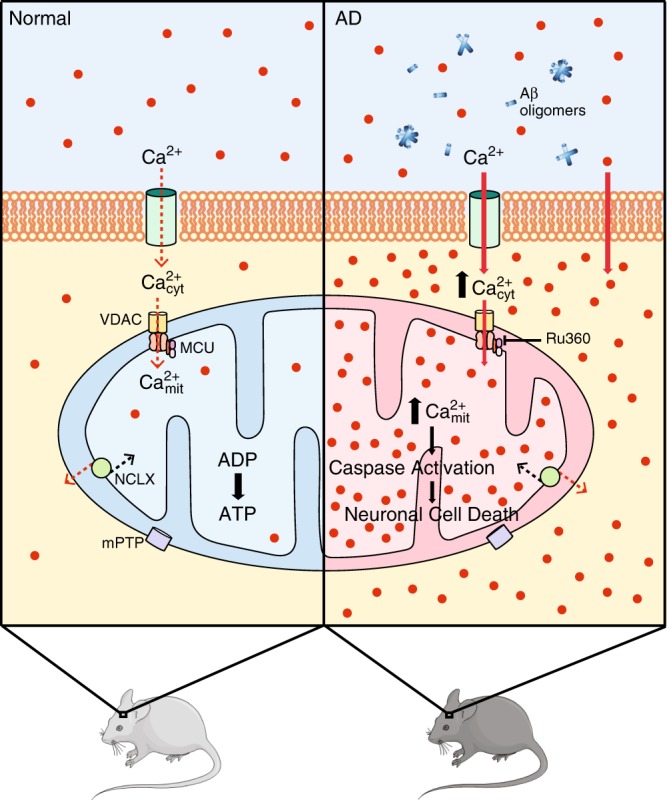
Under physiological conditions (normal), mitochondria take up Ca2+ from the cytosol, via mitochondrial Ca2+ uniporter (MCU) complex, for activation of mitochondrial metabolism, TCA cycle and OXPHOS, leading to ATP production. However, under pathological conditions (AD), Aβ aggregates (likely soluble Aβo) increase cytosolic Ca2+, thus leading to mitochondrial Ca2+ overload. Aβ-induced mitochondrial Ca2+ uptake is mediated via MCU, as blocking MCU with the specific inhibitor Ru360 prevents it. Excessive Ca2+ taken up by mitochondria could lead to opening of the mitochondrial permeability transition pore (mPTP), caspase activation and, eventually, neuronal cell death. This figure was created using modified Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Discussion
Proposed mechanisms of neurotoxicity in AD include the disruption of Ca2+ homeostasis and mitochondrial dysfunction, which contribute to synaptic and neuronal damage46. An extensive array of in vitro and in vivo studies has shown that Aβ aggregates alter cytosolic Ca2+ homeostasis. However, prior studies linking Aβ with mitochondrial Ca2+ overload16,17 have used cultured cells and high concentrations of synthetic Aβo, and whether there is mitochondrial Ca2+ overload elicited by Aβ aggregates in vivo remained an open question to date. In the present study, we provide evidence of in vivo mitochondrial Ca2+ overload in a Tg mouse model of cerebral β-amyloidosis, and after topical application of natural soluble Aβo at concentrations similar to those found in the AD brain.
We found mitochondrial Ca2+ overload in the APP/PS1 Tg mouse, a model that recapitulates an accelerated deposition of amyloid plaques. This overload was observed in the Tg mice once the Aβ plaques were present. The mitochondrial Ca2+ increase was of a higher magnitude than the cytosolic Ca2+ levels observed in the same mouse model5. Whereas Ca2+ levels in the neuronal cytosol upon exposure to Aβo were in the nM range7, mitochondrial Ca2+ levels were in the µM range. This was not surprising as mitochondria take up large amounts of Ca2+ during physiological Ca2+ elevations. Indeed, effective mitochondria Ca2+ buffering capacity was reported ~0.2–2 µM in isolated brain mitochondria47 and in neuronal mitochondria in situ48. In vivo, we found that mitochondrial Ca2+ overload only occurs at levels higher than ~1.6–1.7 µM, comparable to the reported buffering capacity in vitro.
Mitochondrial dysfunction has been suggested as an early event in AD, before plaque deposition9. However, several studies have reported no change in resting cytosolic Ca2+ levels or neuronal dysfunction before amyloid plaque deposition in AD mouse models5,24,25. In agreement with these studies, there was no appreciable mitochondrial Ca2+ overload in young mice before plaque deposition (2.5–3-mo-old), suggesting that Aβ deposition is necessary to trigger mitochondrial Ca2+ overload in neurons in AD. Further longitudinal studies will be necessary to clarify whether mitochondrial Ca2+ elevation occurs concomitantly with plaque formation. Surprisingly, no association was found between mitochondrial Ca2+ overload and distance to amyloid plaques, in contrast to cytosolic Ca2+ overload, wherein there was a higher likelihood of detecting Ca2+ overload in the immediate vicinity of individual plaques5. This suggests that diffusible soluble Aβ oligomeric species rather than plaques are likely responsible for the increase of [Ca2+]mit in the neurons of the APP/PS1 mouse, or that mitochondria are less vulnerable than cytosol to the pathophysiological insult mediated by Aβo. The intrinsic properties of mitochondria with regards to Ca2+ kinetics, and as dynamic entities that undergo axonal transport, fusion/fission, and mitophagy when damaged, could also explain this difference.
To confirm that mitochondrial Ca2+ overload is triggered by soluble Aβo, we directly applied naturally secreted Aβo (TgCM) to the naive living mouse brain. Application of TgCM to the cortical surface of the mouse brain increased [Ca2+]mit in neurons within 1 h, whereas WtCM or Aβ-immunodepleted TgCM did not. This result supports our previous findings in the APP/PS1 model, and confirms a direct effect of Aβ aggregates altering Ca2+ homeostasis in neurons. The reversibility of TgCM-induced mitochondrial Ca2+ overload supports a normal extrusion via mitochondrial Ca2+ exchangers, and/or clearance of the Aβo from the brain parenchyma.
Mitochondria have been reported as a direct site of Aβ accumulation in neurons in both human AD brains and AD Tg mouse models49. However, the mechanism(s) by which Aβ evokes mitochondrial Ca2+ uptake in vivo has not been elucidated. Proposed mechanisms include formation of Ca2+-permeable pores in the plasma membrane and also in the membranes of intracellular organelles50, or formation of a non-specific ion channel in the plasma and mitochondrial membranes51. On the other hand, Ca2+ uptake into the mitochondrial matrix is mediated by the highly conserved MCU complex29,30, which is selectively blocked by Ru360. Our results show that pre-treatment with Ru360 abolishes TgCM-induced mitochondrial Ca2+ uptake, providing evidence that the MCU complex is required for the Aβ-driven mitochondrial Ca2+ increase in neurons in the brain in vivo, and arguing against the formation of Ca2+-permeable pores or non-specific ion channel in the mitochondrial membrane. However, assessment of protein expression of the mitochondrial-enriched fraction extracted from Wt and APP/PS1 Tg mouse brains did not show any significant differences between groups. Western blot might not be sensitive enough to detect changes in protein levels that affect the small subset of Ca2+-overloaded mitochondria seen in vivo. Alternatively, Aβ may alter the levels of a component of the pore or the NCLX exchanger not examined in this model due to lack of adequate antibodies, or may impact their activity rather than expression. Along these lines, recent work by Jadiya et al.52 has observed a decrease in the expression of NCLX in mitochondria in human AD brains and in mouse models of AD, linked to impaired Ca2+ efflux from mitochondria, which could be responsible for the disease progression.
To examine the relevance of mitochondrial Ca2+ dyshomeostasis in the human AD brain, we took advantage of publicly available microarray and RNA-Seq datasets, and compared the expression levels of Ca2+-related mitochondrial genes in AD patients (Braak stages V–VI) and control subjects (Braak stages 0–I–II). Most of the genes involved in Ca2+ influx into mitochondria were significantly downregulated in AD, whereas, the only Ca2+ efflux gene was significantly upregulated. These results most certainly point towards a compensatory effect in AD to handle the mitochondrial Ca2+ overload, and confirm the existence of a mitochondrial Ca2+ dyshomeostasis in the human AD brain.
Optimal mitochondrial activity is crucial to maintain neuronal function and health. Mitochondria buffer Ca2+, and in turn, Ca2+ modulates mitochondrial activity. Excessive Ca2+ accumulation into mitochondria leads to mitochondrial function impairment53, which could lead to decreased ∆Ψm11 and increased ROS production40. Eventually, cytochrome c release and cell death via apoptosis will take place39. Remarkably, when APP/PS1 Tg mice were imaged longitudinally, the small fraction of neurons containing mitochondria with elevated [Ca2+] in their cell bodies were no longer visible 24 h later, and their nucleus was condensed, whereas neurons with physiological [Ca2+]mit and a healthy spherical nucleus remained. Surprisingly, whereas the cells with high [Ca2+]mit in the soma were likely to die, sporadic increases of mitochondrial Ca2+ within the neurites were not as lethal. This feature suggests that within the soma, high [Ca2+]mit represents an “all or none”-phenomena, and not an isolated event in individual mitochondria. The results obtained here in vivo are supported by previous studies in vitro showing neuronal cell death related to mitochondrial Ca2+ overload16,17, where a subtle or partial depolarization of mitochondria to avoid Aβ-related mitochondrial Ca2+ overload in culture cells was protective for neuronal death. Furthermore, our in vitro data show that soluble Aβ increases [Ca2+]mit, leading to activation of mPTP, ∆Ψm decrease and activation of caspases. Previous studies have already linked Aβ, mPTP and cell death in AD in neurons54–56 and astrocytes57,58, but here we show that activation of mPTP is downstream mitochondrial Ca2+ overload, as preventing mPTP activation did not forestall mitochondrial Aβo-driven Ca2+ overload, but avoided mitochondrial depolarization and caspase activation. Moreover, we have previously shown that there is a rapid cell death once Aβ-triggered oxidative stress invades neuronal cell bodies in vivo in the same mouse model40. Oxidative stress started in neurites, but in a small fraction of neurons tracked towards the cell bodies leading to rapid initiation of apoptosis. Mitochondrial Ca2+ overload has an important role in the oxidative stress-induced neuronal cell death59. Our findings of occasional rapid neuronal loss after mitochondrial Ca2+ overload support our previous findings and, more importantly, suggest a close link between high [Ca2+]mit, oxidative stress and neuronal apoptosis via mitochondria in AD.
Of note, the neurotoxic effect of Aβo on mitochondria were not restricted to Ca2+ elevation, but also involved changes in mitochondrial size and shape. Indeed, the APP/PS1 Tg mouse model exhibits structural abnormalities in mitochondria, including mitochondria swelling and fragmentation11, and impaired balance of mitochondrial fission and fusion proteins has been shown in brains from AD patients60. Aβ-induced mitochondrial fragmentation has been reported to precede neuronal cell death61. Further experiments are necessary to monitor the morphological changes and altered dynamics of mitochondria induced by Aβo in vivo.
Here we show that blocking the MCU complex in vivo prevents the Aβ-related mitochondrial Ca2+ overload, pointing to a critical role of MCU in the mitochondrial Ca2+ overload mediated by Aβo. Multiple lines of evidence support MCU as a candidate therapeutic target against AD: MCU overexpression in mitochondria increased [Ca2+]mit following excitotoxicity through activation of NMDA receptors in vitro, leading to mitochondrial membrane depolarization and cell death62. Besides, inhibition of MCU by Ru360, or silencing by siRNA or miRNA, conferred resistance to oxidative stress to the cells in vitro63. Also, reducing mitochondrial Ca2+ uptake by a null mutation in the gene encoding MCU, improved mitochondrial function and suppressed neurodegeneration in a C. elegans model of AD; and preventing mitochondrial Ca2+ uptake by pre-treatment with Ru360 decreased ROS levels in familial AD fibroblasts64. Our in vitro work shows that preventing activation of the mPTP downstream mitochondrial Ca2+ overload could also serve as a therapeutic approach against AD, to protect mitochondria from depolarization (loss of ∆Ψm) and subsequent activation of apoptosis.
In summary, our findings demonstrate that Aβ accumulation provokes mitochondrial Ca2+ overload in vivo via the MCU complex, leading to neuronal death. We have unveiled a pathophysiological link between Ca2+ dyshomeostasis and mitochondrial dysfunction hypotheses of AD pathogenesis and propose the inhibition of the MCU complex and blocking of the mPTP activation as novel therapeutic approaches against AD.
Methods
Animals
Mouse experiments were performed with the approval of the Massachusetts General Hospital Animal Care and under the guidelines of Institutional Animal Care and Use Committees (IACUC, protocol #2004N000047) for the use of experimental animals. Mice used included: (1) APPswe/PSEN1∆E9 double Tg mice (The Jackson laboratory, B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax, MMRRC Cat# 034832-JAX, RRID:MMRRC_034832-JAX) (APP/PS1, 2- to 3-mo-old and 8- to 10-mo-old) of either sex, expressing both human APP gene carrying the Swedish mutation and exon 9 deletion mutation in the PS1 gene along with age-matched Wt-littermate controls; and (2) C57BL/6J males (4- to 5-mo-old, Charles River) for the application of the conditioned media and Ru360. For preparation of the conditioned media, Tg2576 males (Taconic Farms, B6;SJL-Tg(APPSWE)2576Kha, IMSR Cat# TAC:1349, RRID:IMSR_TAC:1349), which heterozygously overexpress human APPswe under the PrP promoter, were mated with Wt females for preparation of primary cortical neurons. Mice were socially housed at 3–4 animals per cage with ad libitum access to food and water on a 12/12 h light/dark cycle with controlled conditions of temperature and humidity.
hsyn.2mtYC3.6 and CBA.YC3.6 AAV construction and production
The expression of the Ca2+ indicator Yellow Cameleon 3.6 (YC3.6)19 was targeted to mitochondria by using the mitochondrial targeting sequence cytochrome oxidase subunit 8 A (COX8A) at the N terminus, duplicated in tandem to enhance the specificity of its localization65. 2mtYC3.6 was inserted on the 5′-end and in frame with the cDNA of YC3.6, which we had previously cloned in an AAV.CBA.2mtYC3.6-WPRE (woodchuck hepatitis virus post-transcriptional regulatory element) backbone. We then cut out the 2mtYC3.6 cDNA (NheI/XhoI-fill-in) and subcloned it into an AAV-hSyn backbone obtained from the UPENN vector core, obtaining the final AAV.hSyn.2mtYC3.6-WPRE plasmid. The integrity of the inverted terminal repeats (ITRs) was verified by sequencing and SmaI digestion. Flanked by the ITRs, the expression cassette included the following components, (1) a 1.7-kb sequence containing human synapsin 1 gene promoter, (2) 2mt (mitochondrial targeting sequence), (3) YC3.6, (4) WPRE and (5) Simian virus 40 (SV40). See construct in Fig. 1a. High titers of AAV serotype 8 were produced using triple transfection in HEK293 cells by the Schepens Eye Research Institute Gene Transfer Vector Core. The virus titer was 5.7 × 1012 viral genomes copies per mL.
To measure cytosolic Ca2+ concentration we used the construct pAAV.CBA.YC3.6-WPRE5. The plasmid contained AAV terminal repeats (ITRs) and the expression cassette, which included the following components: the hybrid cytomegalovirus (CMV) immediate-early enhancer/chicken β-actin promoter/exon1/intron; yellow cameleon 3.6 cDNA and woodchuck hepatitis virus post-transcriptional regulatory element (WPRE). High titers of AAV serotype 8 were produced using triple transfection in HEK293 cells by the Schepens Eye Research Institute Gene Transfer Vector Core. The virus titer was 1.1 × 1012 viral genomes copies per mL.
Cell culture and transfection
Primary neuronal cultures were obtained from cerebral cortex of CD1 mouse embryos (Charles Rives Laboratories) at gestation day 14–16. Neurons were dissociated using Papain dissociation system (Worthington Biochemical Corporation, Lakewood, NJ, USA) and were maintained for 10–14 days in vitro (DIV) in Neurobasal medium containing 2% B27 supplement (Gibco), 1% penicillin/streptomycin (Gibco) and 1% glutamax (Gibco) in a humidified 37 °C incubator with 5% CO2 without further media exchange5.
N2a cells were maintained in OptiMEM (Gibco) supplemented with 5% Fetal Bovine Serum (FBS) (Atlanta Biologicals) and 1% penicillin and 1% streptomycin (Gibco) in a humidified 37 °C incubator with 5% CO2.
Cells were transiently transfected at 70–80% confluence with 4 µg of hSyn.2mtYC3.6 and mRuby-Mito-7 (gift from Michael Davidson Addgene, #55874) plasmids using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions.
In situ calibration of hSyn.2mtYC3.6 in N2a cells
N2a cells were plated in 35 mm glass-bottom dishes (MatTek Corporation) and transfected with lipofectamine 2000 (Life Technologies) and 4 μg of the hSyn.2mtYC3.6 construct. 24 h after transfection, cells were permeabilized for 1 min with 100 µM digitonin in intracellular medium (130 mM KCl, 5 mM NaCl, 1 mM MgCl2, 2 mM KH2PO4, 20 mM HEPES, 0.5 mM EGTA, 5 mM succinate, 1 mM ATP, pH 7), followed by three washes with intracellular medium devoid of digitonin. After permeabilization, cells were exposed to solutions containing known Ca2+ concentration (ranging from 0 µM to 1 mM) and 10 µM FCCP, to depolarize mitochondria and equilibrate the [Ca2+] in the medium and in the organelle66. Calibration solutions with free Ca2+ concentration <20 µM were prepared by mixing intracellular media with the chelators EGTA or HEDTA and 1 M CaCl2 according to MaxChelator Ca-EGTA Calculator (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/CaEGTA-NIST.htm) and to Park and Palmer67. Calibration solutions with free Ca2+ concentration >20 µM were made by adding various volumes of 1 M CaCl2 to the intracellular media solution. 2 photon excitation was used at 860 nm, and emitted light was collected in three channels in the range of 380–480, 500–540 and 560–650 nm. Images were acquired on an Olympus FluoView FV1000MPE multiphoton laser-scanning system mounted on an Olympus Bx61WI microscope, and photomultiplier settings were selected and maintained for the intravital imaging. YFP/CFP fluorescence ratios were plotted against free Ca2+ concentration and fitted with a sigmoid equation. The apparent Kd (Kd′) and Hill coefficient (n) were determined as described previously20. Custom-written ImageJ software (https://imagej.nih.gov/ij/) was used for analysis and calculation of YFP/CFP ratios. Note that ratios increased with increasing [Ca2+]. Ratios were then converted to free Ca2+ concentration with standard ratiometric equations20:
Preparation of Wt and TgCM and Aβ-immunodepleted media
Primary neuronal cultures were prepared as described above5. Neurons were plated on 35-mm culture PDL-coated dishes. Tissue from each embryo was collected for genotyping by PCR. Neurons were maintained in Neurobasal media (Gibco) containing 2% B27 supplement (Gibco), 1% penicillin/streptomycin (Gibco) and 1% glutamax (Gibco) for 14 DIV. Conditioned media from either Tg cultures (TgCM) or their Wt littermates (WtCM) was collected, and the concentration of Aβ40 was measured by mouse/human ELISA kit (Wako #294-64701 Human/Rat Aβ1–40).
Immunodepletion of human Aβ from the TgCM was performed overnight by using the mouse antibody 6E10 (Purified anti-β-Amyloid, 1–16 antibody BioLegend Cat# 803004, RRID: AB_2715854) and protein G sepharose beads (Sigma-Aldrich). Briefly, protein G beads were washed with cold Neurobasal medium in order to avoid non-specific binding. Then, 1 mL of TgCM was incubated with 40 µL of the pre-washed protein G beads and 6 µg of 6E10 antibody overnight at 4 °C under rotation. Finally, the supernatant was collected and the Aβ concentration was quantified by mouse/human ELISA kit (Wako #294-64701 Human/Rat Aβ1-40) according to the manufacturer’s instructions.
Mitochondrial and cytosolic calcium in vitro imaging
Primary cortical neurons obtained from CD1 embryos were cultured for 12–15 days in vitro (DIV) and infected with AAV.hSyn.2mtYC3.6 to target mitochondria. Experiments were performed using sister cultures obtained on the same day to control for any culture-to-culture variation. Three days after infection, cells were incubated with fresh conditioned media and subjected to multiphoton microscopy. 5–6 fields of view were imaged randomly. For some experiments cortical neurons infected with the mitochondrial reporter hSyn.2mtYC3.6 were loaded with the calcium indicator calbryte 590 (0.5 µM) for 30 min at room temperature to measure Ca2+ changes in the cytosol and mitochondria simultaneously. Images were acquired in an Olympus FluoView FV1000MPE multiphoton laser-scanning system mounted on an Olympus Bx61WI microscope. Two photon excitation was used at 800 nm (for calbryte) and 860 nm (for 2mtYC3.6), and emitted light was collected in the range of 380–480, 500–540 and 560–650 nm. Either images were acquired in basal conditions and after treatment with conditioned media, or time-lapse recordings were acquired following stimulation with KCl (50 mM) or TgCM. Background fluorescence corresponding to regions devoid of cells was subtracted. Images were analyzed using custom-written MATLAB scripts (see Image Analysis section).
Mitochondrial permeability transition pore (mPTP) activity
mPTP opening was directly assessed by the calcein/cobalt method68. Briefly, primary cortical neurons cultured for 12–15 DIV were co-loaded with calcein-AM 1 µM, CoCl2 1 mM and Hoechst 33342 1 µM for 15 min at room temperature and imaged with confocal microscopy. The effect of TgCM was evaluated longitudinally. Images were acquired in the green channel (488 nm) every 10 min. Cells were maintained at 37 °C and 5% CO2 during the recording. Fluorescence traces from mitochondria in individual cells were normalized relative to their value before addition of any solutions (t = 0 min). Background fluorescence corresponding to regions devoid of cells was subtracted. In some experiments, cells were pre-incubated with cyclosporine A 1 µM for 15 min before and co-applied during the recording.
Mitochondrial membrane potential (∆Ψm) imaging
The effects of TgCM on ∆Ψm were evaluated by confocal microscopy in cells loaded with the ∆Ψm sensitive probe TMRE. Primary cortical neurons cultured for 12–15 DIV were loaded with TMRE (10 nM) and Hoechst 33342 1 µM for 15 min at room temperature and imaged with confocal microscopy. The effect of TgCM was evaluated longitudinally. Cells were maintained at 37 °C and 5% CO2 during the recording. Excitation of the neurons was performed at 361 nm for Hoechst and 549 nm for TMRE, and images were taken every 10 min. Fluorescence traces from individual cells were normalized relative to their value before addition of any solutions (t = 0 min). Background fluorescence corresponding to regions devoid of cells was subtracted. In some experiments, cells were pre-incubated with cyclosporine A 1 µM for 15 min before and co-applied during the recording.
Caspase activation imaging
Caspase activation was detected with CellEvent Caspase 3/7 Green Detection Reagent in primary cortical neurons at 12–15 DIV. Neurons were labeled with CellEvent Caspase 3/7 5 µM and Hoechst 33342 1 µM for 15 min at room temperature. Then media was replaced by the conditioned media and green fluorescence was detected under confocal microscopy. Excitation of the neurons was performed at 361 nm for Hoechst and 488 nm for CellEvent, and images were recorded every 10 min. Cells were maintained at 37 °C and 5% CO2 during the recording. Fluorescence traces from individual cells were normalized relative to their value before addition of any solutions (t = 0 min). Background fluorescence corresponding to regions devoid of cells was subtracted. In some experiments, cells were pre-incubated with cyclosporine A 1 µM for 15 min before and co-applied during the recording.
Stereotactic intracortical injection of AAVs
Four- to five-mo-old C57BL/6 J Wt mice were injected intracortically with AAV.hSyn.2mtYC3.6 (serotype 2/8) or pAAV.CBA.YC3.6-WPRE (serotype 2/8). Stereotactic intracortical injections were performed as previously described7. Briefly, mice were anesthetized with 1.5% (vol/vol) isoflurane and positioned on a stereotactic frame (Kopf Instruments). Body temperature was maintained throughout the surgery with a heating pad. 3 µL of viral suspension were injected in the somatosensory cortex using a 33-gauge sharp needle (Hamilton Medical) attached to a 10 µL Hamilton microsyringe (Hamilton Medical), at a rate of 0.15 µL/min. Stereotactic coordinates of the injection sites were calculated from bregma (anteroposterior −1 mm, mediolateral ±1 mm and dorsoventral −0.8 mm).
Eight- to nine-mo-old APP/PS1 Tg mice were injected intracortically with AAV.hSyn.2mtYC3.6 at the moment of the cranial window implantation with 3 µL of viral suspension in the somatosensory cortex (left hemisphere) at 0.15 µL/min, followed by cranial window implantation. In some cases, 1 µl of Hoechst 33342 1uM was co-injected with AAV.hSyn.2mtYC3.6 to label nuclei in the area of injection. Mice were given buprenorphine (0.1 mg/kg) for 3 days following surgery.
Cranial window implantation
In the acute conditioned media experiments, a 6 mm cranial window was implanted in the C57BL/6J Wt mice three to four weeks after AAV.hSyn.2mtYC3.6 or pAAV.CBA.YC3.6-WPRE injection. Mice were anesthetized with 1.5% (vol/vol) isoflurane and placed in a stereotactic apparatus. A piece of skull over somatosensory cortex was removed and replaced with an 8 mm diameter glass coverslip7. Dura matter was removed before window installation and the window was sealed containing PBS. Texas Red dextran (70,000 MW; 12.5 mg/mL in PBS; Molecular Probes) was retro-orbitally injected to provide a fluorescent angiogram. An imaging session was first performed to determine the basal resting [Ca2+]mit. Then, the window was removed and sealed again with WtCM, TgCM, Aβ-immunodepleted TgCM, Ru360 (Calbiochem, Merck Millipore) or Ru360 + TgCM (final volume applied was 40 μL). In the last case, Ru360 was pre-incubated for 15 min previous application of TgCM. After 1 h, each mouse was reimaged at the same fields of view to determine the relative changes in [Ca2+] (ΔR/R0).
For the APP/PS1 Tg mice, cranial windows were implanted to the animals following the intracortical virus injection. Mice were anesthetized with 1.5% (vol/vol) isoflurane and placed in a stereotactic apparatus. A piece of skull over left somatosensory cortex was removed, replaced with a 5 mm diameter glass coverslip and fixed with Krazy Glue and dental cement7. Mice were given buprenorphine (0.1 mg/kg) for 3 days following surgery. Expression of the virus was allowed for 3–4 weeks before imaging. Methoxy-XO4 (4 mg/kg) was intraperitoneally injected 1 day before the imaging session to label amyloid plaques21.
In vivo multiphoton microscopy imaging
Mice were anesthetized with isoflurane (5% inhalation, mixed with pure O2) for induction, and a reduced concentration of isoflurane (~1.5%) was used during the imaging. The animal’s body temperature was maintained at ~37.5 °C during the imaging session with a heating pad; ophthalmic ointment was applied to protect the eyes. Mice were visually monitored throughout the duration of the imaging session (which lasted on average 2 h). Images of AAV.hSyn.2mtYC3.6 or pAAV.CBA.YC3.6-WPRE expressing neurons, amyloid pathology and Texas Dextran Red angiograms were obtained by using an Olympus FlouView FV1000MPE multiphoton laser-scanning system mounted on an Olympus Bx61WI microscope and an Olympus 25× dipping objective (NA = 1.05). A Deep-See Mai Tai Ti:Sapphire mode-locked laser (Mai Tai; Spectra-physics) generated multiphoton excitation at 860 nm, and three photomultiplier tubes (PMTs) (Hamamatsu) collected the emitted light in the range of 380–480, 500–540 and 560–650 nm69. Images were taken at 5× zoom. Five to fifteen cortical volumes (Z-series, 127 µm × 127 µm, 200–300 µm depth) were acquired per mouse, at a resolution of 512 × 512 pixels. CFP and YFP PMTs settings remained unchanged throughout the different imaging sessions. Laser power was adjusted as needed and kept below 50 mW to avoid phototoxicity70.
At the end of the last imaging session, APP/PS1 Tg mice were euthanized under CO2, perfused with phosphate buffered saline (PBS, Gibco) and brains were removed. The hemisphere injected with AAV.hSyn.2mtYC3.6 was fixed with 4% paraformaldehyde and 15% glycerol for 24 h, frozen in mounting media on ice cold isopropanol and sliced into 20 µm sections on a cryostat (Leica). The other hemisphere was flash frozen with liquid nitrogen and stored at −80 °C until use.
Image analysis
Multiphoton microscopy imaging data, acquired in vivo, were analyzed by using a custom-written MATLAB (MathWorks) script. To perform automatic segmentation of mitochondria, an adaptive thresholding procedure was applied to generate binary images and individual mitochondria were selected using a constraint on object size. Amyloid plaques were manually extracted from the z-stacks. To obtain Yellow Cameleon ratio values, the data were preprocessed by subtracting the background from the CFP and YFP channels (mean of the bottom 5% of intensity values in each slice of the z-stack) and applying a mean filter with radius 2 to each channel. The ratio for each mitochondrion was calculated by dividing the sum of the intensity values corresponding to the mitochondrion in the YFP channel by the sum of intensity values in the CFP channel. The YFP/CFP ratios were mapped to mitochondrial Ca2+ concentrations using the in situ calibration curves (see In situ calibration of 2mtYC3.6 in N2a cells section). Calcium pseudocolored images were created using MATLAB by first assigning the [Ca2+] values to a RGB (red, green, blue) colormap with the color range determined by the empirical values for Rmin and Rmax. The RGB image was then converted to an HSV (hue, saturation, value) image with the ‘value’ field set to the mean of the YFP and CFP intensity images. Distance to plaque was measured based on the centroid of the mitochondrion to the edge of the nearest plaque. To assess changes in mitochondrial morphology, a unique slice per z-stack was selected, matching the region across the three different time points (basal, 1 h, 8 h). Slices were selected within 30-60 μm depth. ROIs were drawn around individual mitochondria with the free hand selection tool in ImageJ based on the pseudocolor images. About 100–125 random individual mitochondria were taken. Matlab was used to measure the parameters of interest (area, perimeter and circularity) for each ROI. Area and perimeter were determined as the total number of pixels within the ROI and the number of pixels outlining the ROI, respectively, and then converted to physical units of μm2 and μm. Circularity was calculated using the formula, Perimeter2/4πArea. Images presented in the figures are either a single slice from the z-stack or 2D maximum intensity projections of the z-stack.
Mitochondrial-enriched fraction and Western blotting
Frozen hemibrains devoid of cerebellum from APP/PS1 mice were lysed in Tris-buffered saline (TBS) with 2% sodium dodecyl sulfate (SDS) and protease inhibitor (Roche Diagnostics) at 10% (w/vol) in a 5 mL glass dounce homogenizer. Tissue was dounce homogenized with 25 up/down strokes by hand and incubated for 30 min at 37 °C. The homogenate was then centrifuged at 100,000 × g for 30 min at 20 °C. The supernatant was collected, aliquoted and stored at −80 °C until use. Mitochondrial-enriched fraction was obtained using a mitochondrial isolation kit (mitochondrial isolation kit for mammalian tissue samples, Abcam) according to the manufacturer’s instructions. A bicinchonic acid assay (BCA, Thermo Scientific Pierce) was used to determine protein sample concentration. Supernatants were subjected to SDS-PAGE on NuPAGE Bis-tris gels (Life Technologies). Briefly, 20 µg of total protein per well were loaded on 4–12% Bis-Tris SDS-PAGE gels and run in MES buffer (Invitrogen). Proteins were then transferred to PVDF membranes (Millipore), which were incubated overnight with the primary antibodies: rabbit anti-MCU (1:1000, Sigma-Aldrich Cat# HPA016480, RRID:AB_2071893), rabbit anti-MiCU1 (1:1000, Sigma-Aldrich Cat# HPA037480, RRID:AB_10696934), rabbit anti-EFHA1 (MiCU2) (1:500, Abcam Cat# ab101465, RRID:AB_10711219), rabbit anti-SLC24A6 (NCLX) (1:250, Sigma-Aldrich Cat# SAB2102181, RRID:AB_10606696), mouse anti-VDAC1/porin (1:1000, Abcam Cat# ab14734, RRID:AB_443084), mouse anti-β-tubulin I (1: 10,000, Sigma-Aldrich Cat# T7816, RRID:AB_261770). The next day, blots were incubated with infrared secondary goat anti-mouse IgG IRDye680 or goat anti-rabbit IgG IRDye800 (1: 5,000, LICOR) for 1 h at room temperature, and imaged on an Odyssey Infrared Imaging System (LICOR). Blots were converted to gray scale and densitometry analysis was performed using Image Studio Lite Ver5.2.
Immunohistochemistry
20 µm coronal sections were subjected to antigen retrieval, permeabilized with 0.5% triton X-100 and incubated with antibodies against GFP (chicken anti-GFP IgY, 1:500, Aves Labs Cat# GFP-1020, RRID: AB_2307313), HSP60 (rabbit anti-HSP60, 1:500, Abcam Cat# ab46798, RRID:AB_881444), NeuN (mouse anti-NeuN, 1:500 Millipore Cat# MAB377, RRID:AB_2298772) and GS (rabbit anti-Glutamine Synthetase, 1:500, Abcam Cat# ab73593, RRID:AB_2247588) overnight at 4 °C. Appropriate secondary antibodies (Alexa fluor 488-conjugated goat anti-chicken IgG antibody, 1:500, Molecular Probes Cat# A-11039, RRID:AB_142924, Cy3-conjugated goat anti-rabbit IgG antibody, 1:1000, Abcam Cat# ab6939, RRID:AB_955021, Cy3-conjugated goat anti-mouse IgG antibody, 1:1000, Abcam Cat# ab97035, RRID:AB_10680176, and Cy5-conjugated goat anti-rabbit IgG antibody, 1:1000, Jackson ImmunoResearch Labs Cat# 111-175-144, RRID:AB_2338013) were applied and incubated for 1 h at room temperature. Slices were mounted with DAPI Vectashield (Vector Laboratories) and subjected to fluorescence imaging.
Analysis of publicly available human brain datasets
25 microarray and RNA-Sequencing (RNA-seq) datasets from the Mount Sinai Brain Bank (MSBB)43 and the Religious Orders Study and Memory and Aging Project (ROSMAP)44 cohorts, spanning 19 brain regions, were analyzed as described in Bihlmeyer et al.71 Four of these brain regions had both microarray and RNA-Seq data. The samples were grouped by the Braak score, which assesses the distribution of tau neurofibrillary tangles in the subject’s brain72. The expression of 8 mitochondrial genes involved in mitochondrial Ca2+ homeostasis (MCU, MCUB, MCUR1, MICU1, MICU2, MICU3, SMDT1 and SLC8B1) was compared in AD Braak stages (V–VI) versus control subjects (Braak 0–I–II) and by CERAD analysis (frequent versus absent neuritic amyloid plaques). Data were adjusted for neuronal loss with the pan-neuronal marker MAP2. Multiple comparisons corrections were performed using the Benjamini–Hochberg method73. For the purpose of selection for visual display, a FDR threshold of 0.25 was used. All results for the differential expression analysis are listed in Supplementary Tables S1 and S2.
Statistics
Statistical analyses were conducted in SAS and GraphPad 5.0. Data are reported as mean ± SEM. To assess differences between groups (Wt vs. APP/PS1; Basal vs. WtCM/TgCM/Aβ-depleted TgCM/TgCM+Ru360/Ru360), global and pair-wise, for all measurements of interest (YFP/CFP Ratio, Ca2+ overload and ∆R/R0), a linear mixed effects model was fitted with treatment group as fixed effect and mouse as random effect. The least square mean and standard error estimates and the p-values resulting from these models are presented in the figure legends. Similarly, estimates for basal, 1 h post CM application and 8 h post CM application mean and SE measurements were generated from a linear mixed effects model with a fixed effect of an intercept only and a random effect of mouse. Kruskal–Wallis test was used for the analysis of the relationship between percent of Ca2+ overload and distance to the edge of the plaque. p < 0.05 was considered significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by NIHR01AG0442603, S10 RR025645 and R56AG060974 (BJB); and by the Tosteson & Fund for Medical Discovery and the BrightFocus Foundation A2019488F (MCR). MSBB study was supported by the grants AG046170, AG054014, AG057440 and AG057907 from the NIH/National Institute on Aging (NIA). A.S.-P. was supported by the Alzheimer’s Association (AACF-17-524184) and the National Institute for Neurodegenerative Diseases and Stroke (NINDS R25NS065743). The ROSMAP project was supported by funding from the National Institute on Aging (AG034504 and AG041232). We acknowledge the Schepens Eye Research Institute Gene Transfer Vector Core for the AAV.hSyn.2mtYC3.6 and AAV.CBA.YC3.6.WPRE vector production.
Source Data
Author contributions
M.C.-R. designed experiments, collected and analyzed data and wrote the original draft. S.S.H. wrote ImageJ and Matlab macros and edited the manuscript. A.C.S. and E.K.K. helped with immunohistochemistry and mouse brain injections. S.D. analyzed microarray and RNA-seq data. Z.F. performed cloning design. A.N.R. helped preparing conditioned media and primary neurons. A.M. performed the statistical analyses. M.G.A. helped with mouse brain injections. A.S.P. provided expertise and feedback and edited the manuscript. E.H. performed cloning design, provided expertise and feedback and edited the manuscript. B.J.B. conceptualized the research, designed experiments, discussed data, edited the manuscript and secured funding. All authors read and approved the final manuscript.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon reasonable request. The source data underlying Figs. 1e, 2c–f, 3b, 4c–g, 5b–e, 6b, c, e, f, 7b, Supplementary Figs. 2A, B, 3B–E, 4B–G, 5B, 7A–F, 8A, C–E can be found in the Source Data file. The plasmids will be made available from the corresponding author upon reasonable request.
Code availability
Customized MATLAB scripts generated for this study are available from the corresponding author on request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Michael Ashby and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-16074-2.
References
- 1.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 2.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 3.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 4.Khachaturian ZS. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging. 1989;1:17–34. doi: 10.1007/BF03323872. [DOI] [PubMed] [Google Scholar]
- 5.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busche MA, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 7.Arbel-Ornath M, et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegen. 2017;12:27. doi: 10.1186/s13024-017-0169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busche MA, et al. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atamna H, Frey WH., 2nd Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion. 2007;7:297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013;33:17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santos RX, et al. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimer’s Dis. 2010;20(Suppl 2):S401–412. doi: 10.3233/JAD-2010-100666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kopeikina KJ, et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011;179:2071–2082. doi: 10.1016/j.ajpath.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 15.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 16.Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE. 2008;3:e2718. doi: 10.1371/journal.pone.0002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calvo-Rodriguez M, Garcia-Durillo M, Villalobos C, Nunez L. Aging enables Ca2+ overload and apoptosis induced by amyloid-beta oligomers in rat hippocampal neurons: neuroprotection by non-steroidal anti-inflammatory drugs and R-flurbiprofen in aging neurons. J. Alzheimer’s Dis. 2016;54:207–221. doi: 10.3233/JAD-151189. [DOI] [PubMed] [Google Scholar]
- 18.Ferreiro E, Oliveira CR, Pereira CM. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008;30:331–342. doi: 10.1016/j.nbd.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc. Natl Acad. Sci. USA. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 21.Klunk WE, et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J. Neuropathol. Exp. Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- 22.Kastanenka KV, et al. Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J. Neurosci. 2016;36:12549–12558. doi: 10.1523/JNEUROSCI.2080-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Alloza M, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Grienberger C, et al. Staged decline of neuronal function in vivo in an animal model of Alzheimer’s disease. Nat. Commun. 2012;3:774. doi: 10.1038/ncomms1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudinskiy N, et al. Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2012;15:1422–1429. doi: 10.1038/nn.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeda S, Hashimoto T, Roe AD, Hori Y, Spires-Jones TL, Hyman BT. Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease. FASEB J. 2013;27:3239–3248. doi: 10.1096/fj.13-229666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maia, L. F., et al. Changes in amyloid-beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci. Transl. Med.5, 194re192 (2013). [DOI] [PubMed]
- 28.Wu HY, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baughman JM, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perocchi F, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patron M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell. 2014;53:726–737. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sancak Y, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foskett JK, Philipson B. The mitochondrial Ca(2+) uniporter complex. J. Mol. Cell. Cardiol. 2015;78:3–8. doi: 10.1016/j.yjmcc.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plovanich M, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C. The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 1974;6:361–371. doi: 10.1016/0022-2828(74)90077-7. [DOI] [PubMed] [Google Scholar]
- 37.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 38.DuBoff B, Feany M, Gotz J. Why size matters—balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci. 2013;36:325–335. doi: 10.1016/j.tins.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Demaurex N, Distelhorst C. Cell biology. Apoptosis-the calcium connection. Science. 2003;300:65–67. doi: 10.1126/science.1083628. [DOI] [PubMed] [Google Scholar]
- 40.Xie H, Hou S, Jiang J, Sekutowicz M, Kelly J, Bacskai BJ. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl Acad. Sci. USA. 2013;110:7904–7909. doi: 10.1073/pnas.1217938110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreira PI, Santos MS, Moreno A, Oliveira C. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 2001;21:789–800. doi: 10.1023/A:1015536808304. [DOI] [PubMed] [Google Scholar]
- 42.Hodes RJ, Buckholtz N. Accelerating medicines partnership: Alzheimer’s disease (AMP-AD) knowledge portal aids Alzheimer’s drug discovery through open data sharing. Expert Opin. Therap. Targets. 2016;20:389–391. doi: 10.1517/14728222.2016.1135132. [DOI] [PubMed] [Google Scholar]
- 43.Wang M, et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci. Data. 2018;5:180185. doi: 10.1038/sdata.2018.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Jager PL, et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data. 2018;5:180142. doi: 10.1038/sdata.2018.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirra SS, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/WNL.41.4.479. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Zheng W. Ca(2+) homeostasis dysregulation in Alzheimer’s disease: a focus on plasma membrane and cell organelles. FASEB J. 2019;33:6697–6712. doi: 10.1096/fj.201801751R. [DOI] [PubMed] [Google Scholar]
- 47.Kristian T, Weatherby TM, Bates TE, Fiskum G. Heterogeneity of the calcium-induced permeability transition in isolated non-synaptic brain mitochondria. J. Neurochem. 2002;83:1297–1308. doi: 10.1046/j.1471-4159.2002.01238.x. [DOI] [PubMed] [Google Scholar]
- 48.David G, Talbot J, Barrett EF. Quantitative estimate of mitochondrial [Ca2+] in stimulated motor nerve terminals. Cell Calcium. 2003;33:197–206. doi: 10.1016/S0143-4160(02)00229-4. [DOI] [PubMed] [Google Scholar]
- 49.Lustbader JW, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 50.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl Acad. Sci. USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC. The channel hypothesis of Alzheimer’s disease: current status. Peptides. 2002;23:1311–1315. doi: 10.1016/S0196-9781(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 52.Jadiya P, et al. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019;10:3885. doi: 10.1038/s41467-019-11813-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 54.Arrazola MS, Ramos-Fernandez E, Cisternas P, Ordenes D, Inestrosa NC. Wnt signaling prevents the abeta oligomer-induced mitochondrial permeability transition pore opening preserving mitochondrial structure in hippocampal neurons. PLoS ONE. 2017;12:e0168840. doi: 10.1371/journal.pone.0168840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du H, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Briston T, Hicks AR. Mitochondrial dysfunction and neurodegenerative proteinopathies: mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018;46:829–842. doi: 10.1042/BST20180025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 2003;23:5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Angelova PR, Abramov AY. Alpha-synuclein and beta-amyloid—different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochemical Biophys. Res. Commun. 2017;483:1110–1115. doi: 10.1016/j.bbrc.2016.07.103. [DOI] [PubMed] [Google Scholar]
- 59.Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 60.Wang X, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl Acad. Sci. USA. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qiu J, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan L, et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015;16:5420–5433. doi: 10.3390/ijms16035420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sarasija S, et al. Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. eLife. 2018;7:e33052. doi: 10.7554/eLife.33052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Filippin L, Abad MC, Gastaldello S, Magalhaes PJ, Sandona D, Pozzan T. Improved strategies for the delivery of GFP-based Ca2+ sensors into the mitochondrial matrix. Cell Calcium. 2005;37:129–136. doi: 10.1016/j.ceca.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 66.Filippin L, Magalhaes PJ, Di Benedetto G, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 2003;278:39224–39234. doi: 10.1074/jbc.M302301200. [DOI] [PubMed] [Google Scholar]
- 67.Park JG, Palmer AE. Measuring the in situ Kd of a genetically encoded Ca2+ sensor. Cold Spring Harb. Protoc. 2015;2015:pdb.prot076554. doi: 10.1101/pdb.prot076554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petronilli V, Miotto G, Canton M, Colonna R, Bernardi P, Di Lisa F. Imaging the mitochondrial permeability transition pore in intact cells. BioFactors. 1998;8:263–272. doi: 10.1002/biof.5520080314. [DOI] [PubMed] [Google Scholar]
- 69.Bacskai BJ, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J. Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pagnier GJ, et al. Novel botanical drug DA-9803 prevents deficits in Alzheimer’s mouse models. Alzheimer’s Res. Ther. 2018;10:11. doi: 10.1186/s13195-018-0338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bihlmeyer NA, et al. Novel methods for integration and visualization of genomics and genetics data in Alzheimer’s disease. Alzheimer’s Dement. 2019;15:788–798. doi: 10.1016/j.jalz.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 73.Hochberg, Y. & Benjamini, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B 57, 289–300.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon reasonable request. The source data underlying Figs. 1e, 2c–f, 3b, 4c–g, 5b–e, 6b, c, e, f, 7b, Supplementary Figs. 2A, B, 3B–E, 4B–G, 5B, 7A–F, 8A, C–E can be found in the Source Data file. The plasmids will be made available from the corresponding author upon reasonable request.
Customized MATLAB scripts generated for this study are available from the corresponding author on request.