

Abstract
Cystic fibrosis (CF) is the main genetic cause of death among the Caucasian population. The disease is characterized by abnormal fluid and electrolyte mobility across secretory epithelia. The first manifestations occur within hours of birth (meconium ileus), later extending to other organs, generally affecting the respiratory tract. It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR encodes a cyclic adenosine monophosphate (cAMP)-dependent, phosphorylation-regulated chloride channel required for transport of chloride and other ions through cell membranes. There are more than 2,000 mutations described in the CFTR gene, but one of them, phenylalanine residue at amino acid position 508 (p.F508del), a recessive allele, is responsible for the vast majority of CF cases worldwide. Here, we present the results of the application of genome-editing techniques to the restoration of CFTR activity in p.F508del patient-derived induced pluripotent stem cells (iPSCs). Gene-edited iPSCs were subsequently used to produce intestinal organoids on which the physiological activity of the restored gene was tested in forskolin-induced swelling tests. The seamless restoration of the p.F508del mutation resulted in normal expression of the mature CFTR glycoprotein, full recovery of CFTR activity, and a normal response of the repaired organoids to treatment with two approved CF therapies: VX-770 and VX-809.
Keywords: iPSC, Cystic Fibrosis, gene targeting, TALEN, intestinal organoids, FIS assay
Graphical Abstract
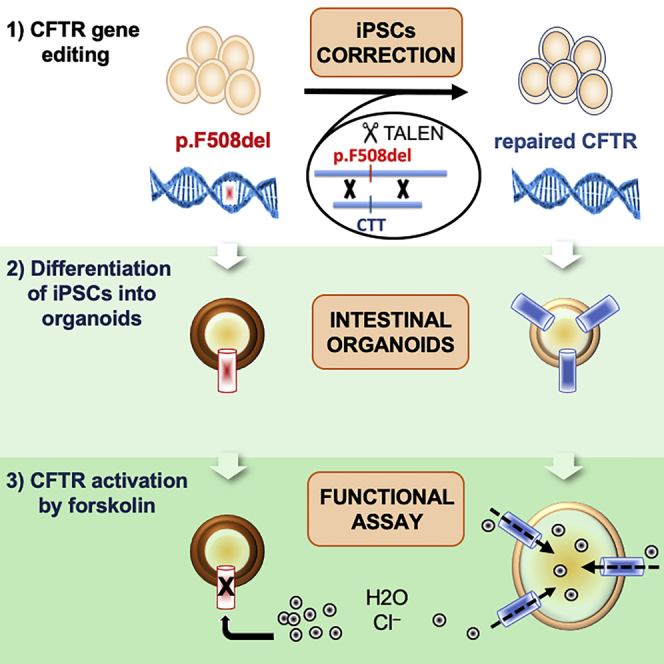
Cystic Fibrosis (CF) is the main genetic cause of death among the Caucasian population. Here, we present the results of the application of genome-editing techniques to repair the CF defect in patient-derived cells and organoids. This protocol could be the foundation for future treatments based on cell repair and transplantation.
Introduction
Cystic fibrosis (CF) is the most frequent genetic disease among the Caucasian population, affecting ca. 90,000 individuals worldwide. The first manifestations occur in early childhood and affect several organs, resulting in defects in the pancreas, liver, intestine, vas deferens, sweat ducts, and airways. The most severe and life-threatening consequences of CF occur in the lung. Three decades ago, it was shown that mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, inherited in an autosomal-recessive fashion, are the underlying cause of CF disease1. More than 2,000 different mutations have been described across the CFTR locus, but up to date, only 346 genetic variants have been identified as pathogenic, disease-causing mutations (https://www.cftr2.org/). CFTR acts as an anion channel at the apical surface of secretory epithelial tissues regulating electrolyte and fluid transport across the plasma membrane. Disruption of CFTR activity leads to acidification of luminal pH and generation of a thick, sticky mucus at the affected epithelia. Chronic pulmonary infections produced by bacterial colonization of the abnormal mucus layer still represent the primary cause of mortality. However, thanks to improved symptomatic treatments and higher success rates in lung transplantations, CF is becoming a multisystemic disease affecting lung, intestine, pancreas, and liver.2
The deletion of the CTT triplet in exon 11 of the CFTR gene is the most prevalent CF-causing mutation, representing 70% of all patients’ alleles.3 The resulting trafficking-deficient CFTR protein lacks the phenylalanine residue at amino acid (aa) position 508 (p.F508del). Despite tremendous efforts in the last years, there is still no cure for all CF patients, including p.F508del patients4. Only two types of small molecule treatments for p.F508del patients are presently available on the market. Unfortunately, the strongest possible intervention, consisting of a combination of both a potentiator to stimulate CFTR channel activity (ivacaftor) and a corrector to promote CFTR trafficking to the apical plasma membrane (tezacaftor or lumacaftor), has only modest effects on patients’ lung function5,6. Most recently, the US Food and Drug Administration (FDA) has approved a promising triple-combination therapy (ivacaftor-tezacaftor-elexacaftor) for p.F508del patients, but the real benefit has yet to be fully evaluated in the near future7,8. On the other hand, genetic-based therapies hold the promise of a complete and definitive cure for the disease, independently of the mutations carried by the patient, and are considered “the most promising strategy to cure CF” by the Cystic Fibrosis Foundation (https://www.cff.org/About-Us/Media-Center/Press-Releases/Cystic-Fibrosis-Foundation-Launches-$500-Million-Path-to-a-Cure/). It has been shown that either 6%–10% of corrected cells9 in a treated epithelium or an overall 5% level of the normal expression on the whole tissue10 is enough to restore wild-type functionality. These numbers are well within the values achievable by gene targeting and support the development of combined gene and cellular therapies, like the ones being currently tested for other diseases11,12.
Part of the slow progress in finding better treatments or even a cure for the disease has been due to the lack of suitable in vitro models that could recapitulate more closely the pathophysiology of the disease and the complexity of human organs, like lung, pancreas, liver, and intestine. Several years ago Dekkers et al.13 developed a forskolin-induced swelling (FIS) assay to monitor CFTR function in primary rectal-derived human intestinal organoids (HIOs), but the fact that variation of swelling was observed among HIOs from different CF individuals with identical CF-causing mutations indicates that the FIS assay is sensitive to the effects of modifier genes, an important issue in CF research14, 15, 16. Clearly, the results of the FIS test would be more conclusive by establishing direct comparisons between CF patient-derived organoids and, after gene correction of the mutant allele, their isogenic counterparts. However, gene editing of HIOs, although possible17, is difficult and subject to variability due to possible multiple integration sites of the recombination vector or to polyclonality of the edited organoids. Induced pluripotent stem cells (iPSCs), on the other hand, offer the possibility of unlimited cell expansion, generation of disease-affected cell lineages by directed differentiation, and easy and accurate targeted gene correction.
Here, we present an integrative approach in which CF patient-derived iPSC technology and seamless gene targeting, combined with a new and robust method for the production of intestinal organoids from iPSCs and FIS testing, provide a solid setting for the study of CFTR function. Apart from their use in disease studies and drug discovery, isogeneic CFTR-repaired organoids could also serve as the basis for future cell therapy applications, in which patients’ own cells are genetically modified and used to regenerate damage organs. The current paper demonstrates that this course of action is feasible in terms of restoring the functionality of the treated cells. It also shows that it has the potential to become the basis for an effective therapy, once proper transplantation protocols and regulatory guidelines are set in place.
Results
Seamless Correction of the CFTR Gene by TALEN-Mediated Homologous Recombination in CF iPSCs
To obtain a seamless correction of the p.F508del mutation in patient-derived CF-iPSCs, we devised a strategy based on Transcription Activator-Like Effector Nuclease (TALEN)-mediated homologous recombination (HR), followed by the total removal of the selection cassette with a piggyBac (PB) transposase system. This approach guarantees the absence of any vector fragment in the patient’s genome after the whole procedure is completed. For this purpose, we first designed a pair of TALENs that could recognize a target site nearby the p.F508del mutation (Figure 1A). The specificity of the new TALENs was initially determined in K562 and HeLa cell lines with the Surveyor nuclease assay. A high cleavage efficiency of about 50% confirmed their functionality in both cell types (Figure S1). Then, a donor vector was designed containing a functional allele of the CFTR gene. The genetic defect in p.F508del iPSCs was corrected by introducing a CTT triplet in exon 11 of the CFTR gene at the precise position where its absence causes the mutant phenotype (Figure 1A). The targeting vector contains a transposon-based, double-selection puromycin-(delta)thymidine kinase (puroΔtk) cassette driven by a phosphoglycerate kinase (PGK) promoter and flanked by PB-specific inverted terminal repeat (ITR) sequences. Once the PB transposase recognizes those sites, it efficiently catalyzes the seamless excision of the cassette. The genomic TTAA sequence located in intron 11 at 126 bp downstream of the 3′ TALEN-binding site marks the PB recognition site for integration and excision of the transposon. Two CFTR recombination arms (ca. 900 bp each) are present at both ends of the selection cassette to promote homologous recombination in the proximity of the p.F508del mutation. To prevent cutting of the targeting vector or retargeting of the edited allele by TALENs, several silent mutations were introduced into the 5′ homology arm in close proximity to the p.F508del deletion. In addition, a new BglII site was also included to facilitate the screening of recombinant clones (Figure 1A). Although silent mutations do not modify the amino acid sequence, they can introduce cryptic splice variants into the DNA with deleterious effects on CFTR activity18. This possibility was checked in silico with the Human Splicing Finder tool from Desmet et al.19, and no cryptic site was found (data not shown).
Figure 1.

Seamless Correction of the p.F508del CFTR Mutation by TALEN-Assisted HR in CF iPSCs
(A) Schematic representation of the footprint-free, TALEN-mediated HR strategy for repairing the p.F508del mutation. Target sequences of left and right TALENs, TTAA piggyBac recognition site, as well as the CTT triplet deletion are indicated on the genomic structure of the p.F508del allele (blue). The targeting vector consisted of two homology arms and a double-selection cassette (rose). The corrective CTT and eight silent mutations (red) are depicted on the 5′ homology arm. Primers (black triangles) and their respective PCR products (dashed lines) used for HR screening are shown. (B) PCR screening of puromycin-resistant representative iPSC clones from L1 and L2 iPSC lines (upper panel). Candidate clones for 5′ correction were confirmed by BglII digestion (lower panel). C−, negative control; C+, positive control. (C) PCR screening of puromycin-resistant iPSC clones from L1 and L2 iPSC lines to detect correct 3′ integration (upper panel). piggyBac-mediated excision of the selection cassette was confirmed in FIAU-resistant iPSC clones by allele-specific PCR (lower panel). C−, negative control; C+, positive control. (D) Summary of HR efficiencies in iPSC lines L1 and L2. (E) Sequence analysis of the edited allele confirms p.F508del correction and the incorporation of the eight silent mutations originally present in the recombination cassette. The newly created BglII restriction site is highlighted by a blue box. The amino acid sequence of exon 11 is shown above the DNA sequence. Underlined sequences indicate the binding sites of left (L)- and right (R)-TALEN, respectively.
Targeted repair of the p.F508del allele has been carried out on two iPSC lines, independently derived in our lab from the same CF patient (IMEDEAi001-F)20. The deletion of the CTT triplet in the targeted iPSC line was previously confirmed by PCR-directed mutagenesis and MboI digestion21. CF iPSCs were nucleofected with both TALEN plasmids and the donor vector at a ratio of 1:1:1 and seeded onto antibiotic-resistant feeders. Following 2 weeks of puromycin selection, resistant clones were isolated, expanded, and analyzed by PCR. To identify positive clones, both 5′ and 3′ ends of the recombination site were screened with combinations of primers that bound to the selection cassette and to genomic sites located outside the recombination arms (Figure 1A). Positive clones with the correct integration of the targeting construct were detected in both iPSC lines (Figures 1B and 1C). To eliminate possible recombination events 5′ to the p.F508del site that would give rise to the integration of the targeting construct into the CFTR locus, but without repairing the CTT deletion, the 5′ PCR products were digested with BglII. The newly generated BglII site is located at 28 bp 3′ of the p.F508del site, so that successful BglII digestion would very likely indicate the correct integration of the CTT triplet and therefore, the repair of the gene. 71 clones identified as having undergone homologous recombination were subjected to BglII analysis and all except one (clone 111; Figure 1B) showed two bands of 1,019 and 306 bp, corresponding to the size of the fragments expected from digesting the repaired allele (Figure 1B). The absence of BglII digestion in clone 111 indicated that this clone lacked the BglII site and very likely, the corrected CFTR sequence. This was most probably due to a recombination event that occurred 3′ of the BglII restriction site. Overall, efficient HR-mediated integration was confirmed in 60 out of 648 clones in CF iPSC line 1 (L1) and 10 out of 98 analyzed clones in CF iPSC line 2 (L2), an overall correction efficiency of approximately 10% (Figure 1D).
The next step of the procedure consisted of the excision of the recombination cassette. For that purpose, four recombined clones from L1 (L1-27, L1-43, L1-65, L1-104) and five from L2 (L2-18, L2-47, L2-61, L2-64, L2-87) were transfected with the hyperactive PB transposase. Fialuridine (2′-deoxy-2′-fluoro-β-d-arabinofuranosyl-5-iodouracil or FIAU)-resistant clones were isolated, expanded, and analyzed by PCR with primers located outside the selection cassette. To ensure specific amplification of the edited allele, the 5′ primer was designed to bind to the BglII restriction site. PCR screening demonstrated the removal of the PB-based selection cassette in approximately 53% of the analyzed clones in both previously edited CF iPSC lines (Figure 1C).
After CFTR correction and cassette excision, one subclone from L2-64 (L2-64.12) was randomly selected for further characterization. A 2.2-kb DNA fragment spanning the complete genomic region involved in HR was amplified by PCR using primers P1 and P4. PCR amplicons were cloned into the pGEM-T Easy Vector and sequenced. The detection of both wild-type and CFTR-repaired alleles confirmed CFTR repair in heterozygosity at the endogenous CFTR locus. Figure 1E shows the edited sequence around the p.F508del mutation site. The addition of the CTT triplet and the presence of the eight silent mutations originally incorporated into the targeting vector are highlighted. Alignment of the complete sequenced region with a CFTR p.F508del template is shown in Figure S2.
Characterization of CFTR-Corrected iPSCs
Once the presence of the corrected allele was verified, the repaired CF iPSC line L2-64.12 was further characterized regarding genome integrity, stemness, and pluripotency. G-banding analysis indicated a normal diploid karyotype without chromosomal aberrations (Figure 2A). To detect potential genomic imbalances at a higher resolution, array comparative genomic hybridization (aCGH) was performed by comparing on the one side the genomes of repaired and nonrepaired CF iPSCs and on the other side the genomes of repaired CF iPSCs and parental primary fibroblasts (Figure S3). All in all, only five copy number variations (CNVs) could be identified across the genome. Although two CNVs emerged during reprogramming and iPSC generation, three CNVs were generated in the course of HR-directed repair. The observed modifications could have their origin in the direct activity of the genetic material added to the cells or just be a consequence of having spent a lengthy time in culture. To test possible off-target effects of the TALENs in the L2-64.12 line, we decided to study the stability of the loci predicted by the PROGNOS software as the most likely affected by nonspecific TALEN activity. CFTR, plus the top 15 sites ranked as possible off-target regions were selected and analyzed by the Surveyor assay (Figure 2B). All of the 15 predicted off-target sites were in intergenic or intronic sequences. The analysis showed that the CFTR-specific TALENs cut exclusively at the CFTR locus without any off-target activity (Figure S4), suggesting that the three CNVs detected during HR-directed repair most probably were not originated during the TALEN-mediated correction process per se but rather, were the consequence of prolonged in vitro cell culture.
Figure 2.

Molecular and Immunohistochemical Characterization of the Corrected L2-64.12 iPSC Line
(A) G-banding analysis showing a normal diploid karyotype. (B) PROGNOS prediction of CFTR and top 15 ranked potential off targets for the CFTR-specific TALEN pair. Chromosomal localizations, type of genomic region, closest gene, and observed cleavage (+) or its absence (−) are indicated for each potential off-target site. (C) Expression of stemness markers SOX2, OCT4, NANOG, and TRA-1-60 shown by immunofluorescence. Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm. (D) Embryoid body formation by spontaneous differentiation. Markers for the three germ layers: ectoderm (NESTIN, PAX6, TUJ1), mesoderm (GATA4, ACTININ, SMA), and endoderm (SOX17, FOXA2, AFP). Nuclei were counterstained with DAPI (blue). Scale bars, 100 μm.
Stemness of the repaired iPSC line was then tested by immunofluorescent analysis with a panel of diagnostic antibodies: αSOX2, αOCT4, αNANOG, and αTRA-1-60 (Figure 2C). In all cases, the corresponding antigens were strongly expressed, indicating that the L2-64.12 line had maintained its stem cell properties during the repair process. Human pluripotent stem cells have the capacity to differentiate into three embryonic germ layers: ectoderm, mesoderm, and endoderm. To test whether our newly corrected iPSC clone L2-64.12 retained this ability, embryoid bodies (EBs) were produced, plated on gelatin-coated dishes for 20 days, and analyzed by immunostaining. The presence of ectodermal lineages was confirmed by the expression of NESTIN, Paired box protein 6 (PAX6), and Neuron-specific class III beta-tubulin (TUJ1), whereas GATA Binding Protein 4 (GATA4), α-ACTININ, and smooth muscle actin (SMA) illustrated the formation of mesoderm and Transcription factor SOX-17 (SOX17), Hepatocyte nuclear factor 3-beta (FOXA2), and α-fetoprotein (AFP) of endoderm (Figure 2D).
Directed Differentiation of the CFTR Mutant and Corrected iPSCs toward CFTR-Expressing Intestinal Organoids
Native CFTR is expressed in multiple organs, such as lungs, pancreas, liver, and gastrointestinal tract. iPSC differentiation toward lung epithelial cells is very complex and shows high variability in differentiation efficiencies22. Recently, it has been demonstrated that CFTR-expressing HIOs, derived from rectal biopsies, could be used to monitor CFTR function in a robust assay with a simple readout13. Based on this observation, we have developed a novel, stepwise, three-dimensional (3D) differentiation protocol to generate HIOs from iPSC (manuscript in preparation). Intestinal organoids, produced according to the new protocol, were used to study the restoration of CFTR function after HR-directed CFTR repair. During development, the first precursor of intestinal epithelium to appear is the multipotent definitive endoderm (DE). Thus, CF-corrected iPSCs were initially differentiated in vitro into DE by treating them with the signaling molecules ACTIVIN-A and Wingless-Type MMTV integration site family, member 3A (WNT3A)23,24. In order to ensure high differentiation frequencies, we incorporated a DMSO (dimethylsulfoxide) pretreatment step in our differentiation protocol prior to DE generation25. DE cells at day 3 of differentiation showed a typical expression pattern of endoderm transcription factors, such as FOXA2, SOX17, and GATA4 (Figure 3A). The activation of endodermal markers was accompanied by the downregulation of the pluripotent stem cell markers OCT4 and NANOG (data not shown). The efficiency of DE derivation from CF-corrected iPSCs was about 80%. The next differentiation step consisted of the production of hindgut endoderm (HGE), the direct precursor population of the more posterior digestive tract. Exposure to high concentration of WNT3A agonists for 5 days resulted in the formation of HGE lineages with the propensity to differentiate into intestinal epithelial cells26,27. A large percentage of HGE cells expressed the hindgut marker Caudal Type Homeobox 2 (CDX2), whereas the generation of the anterior foregut lineage was completely blocked, as demonstrated by the lack of FOXA2/SOX2 coexpression (Figure 3B). At that point, floating 3D HGE spheroids were embedded into Matrigel drops and cultured in the presence of intestinal growth factors. The sequential maturation of the hindgut spheroids into mature intestinal organoids could be observed during the following weeks. The resulting HIOs contained crypt-like structures and an internal lumen lined by columnar epithelium, closely resembling the morphology of native intestinal tissue. Although HIOs expressed intestinal markers, such as Kruppel Like Factor 5 (KLF5), CDX2, and VILLIN, all attempts made at this point to detect restoration of CFTR function by the FIS assay in CFTR-corrected, iPSC-derived HIOs failed (data not shown). This suggested very low to undetectable levels of CFTR expression that could be explained either by the failure in restoring the CFTR gene or by an incomplete maturation of the organoids.
Figure 3.

Directed Differentiation of CFTR-Corrected L2-64.12 iPSCs toward Human Intestinal Organoids
(A) Following 3 days of ACTIVIN-A/WNT3A treatment, CFTR-repaired iPSCs show expression of DE markers FOXA2 (green), GATA4 (red), and SOX17 (green). Scale bar, 50 μm. (B) Definitive endodermal cells were further differentiated into HGE (8 days) by additional exposure to the WNT3A agonist CHIR99021 for 5 days. At that point the cells expressed the hindgut marker CDX2 (green) and were completely negative for the anterior foregut markers FOXA2 (green) and SOX2 (red). Scale bar, 50 μm. (C) Generation of iPSC-derived intestinal organoids from the p.F508del mutant (L2) and corrected iPSCs (L2-64.12) in the absence of mesenchymal cells. Scale bars, 100 μm. (D) Immunostaining of CFTR-repaired HIO for the intestinal markers CDX2 (red), SOX9 (green), E-CAD (red), and VILLIN (green). Scale bars, 50 μm. Nuclei were counterstained with DAPI (blue) (A, B, and D).
The functionality of the FIS assay has been demonstrated in primary HIO obtained by crypt isolation during rectal biopsies28, but not in iPSC-derived organoids. We, therefore, introduced several modifications to the maturation step of the original differentiation protocol in order to generate iPSC-derived HIOs that more closely resembled biopsy HIOs (Figure 3C; manuscript in preparation). HIOs derived from CFTR-corrected and mutant iPSCs following this new protocol were able to grow without support of the mesenchymal layer that is normally found in iPSC-derived HIO cultures (Figure 3C). In addition, the growth rate was very similar to that observed in biopsy-derived HIOs29. The intestinal phenotype of iPSC-derived HIOs was confirmed by immunostaining, showing the expression of the intestinal transcription factors SOX9 and CDX2, as well as the detection of the enterocyte marker VILLIN. The epithelial origin of HIOs was confirmed by positive E-cadherin (E-CAD) staining (Figure 3D). Successful differentiation of the original CF mutant iPSC line, L2, into intestinal organoids was equally verified by immunofluorescence studies at DE, HGE, and HIO stages (Figure S5). Most importantly, CFTR protein expression, a prerequisite for the recovery of CFTR activity, was detected mainly as the complex-glycosylated form (C band) in CFTR-corrected HIOs, whereas only immature, core-glycosylated CFTR (B band) was expressed in mutant HIOs (Figure 4A). The CFTR expression pattern was similar to healthy and p.F508del primary HIOs, respectively. Moreover, corrected CFTR protein was found to be expressed on the apical membrane that lines the inner lumen of HIOs, as evidenced by colocalization of CFTR and the apical marker VILLIN (Figure 4B).
Figure 4.

Restoration of CFTR Protein Expression in Human Intestinal Organoids Derived from CFTR-Corrected iPSCs
(A) Western blot analysis. C band, mature, complex glycosylated CFTR; B band, immature CFTR. Calu-3 cells and primary rectal wild-type organoids (HUB WT) serve as positive control for the detection of the mature CFTR C band, whereas primary rectal CFTR p.F508del homozygous organoids (HUB 508/508) only express the immature B band of the CFTR protein. HIO L2 and HIO L2-64.12 intestinal organoids derived from mutant and corrected CF iPSCs, respectively. Note the inverse ratio between the immature and glycosylated forms of CFTR in the two types of organoids. α-tubulin is the protein-loading internal control. (B) Immunofluorescence analysis of CFTR expression. No CFTR protein is detected in iPSC-derived CFTR p.F508del homozygous HIOs (top row). Colocalization of CFTR (red) with Villin (green) but not with the basolateral marker E-cadherin (green/purple) at the apical surface in CFTR-corrected HIOs (middle and bottom rows). Nuclei were counterstained with DAPI (blue). Scale bars, 50 μm (top and middle rows); scale bar, 20 μm (bottom row).
Functional Restoration of CFTR Channel Activity in iPSC-Derived, CFTR-Corrected Intestinal Organoids
To determine whether heterozygous CFTR repair of the p.F508del allele could support functional recovery of CFTR channel activity, we first tested if CFTR activation by forskolin-induced elevation of cyclic AMP (cAMP) levels was able to induce fluid influx into the organoid’s lumen, thus augmenting their surface area.
To allow quantification of organoid expansion in the course of FIS assays, corrected and mutant HIOs were previously labeled with calcein green, a cell-permeant dye that exclusively marks living cells. Upon 180 min of forskolin treatment, CFTR-corrected and mutant HIOs increased their surface area to 177% (±9 SD) and 103% (±1 SD), respectively (Figures 5A–5C), where 100% represents the original surface prior to treatment. These expansion rates are in good concordance with previous findings, where primary wild-type and p.F508del mutant HIOs were used in FIS assays over a period of 60 min13. FIS response in human organoids was reported to be entirely dependent on CFTR function13, so to demonstrate CFTR dependency in our iPSC-derived HIO culture system, iPSC-derived organoids were pretreated for 16 h, with the specific CFTR inhibitor CFTRinh-17230, before starting the FIS assay. In fact, forskolin-induced expansion in CFTR-corrected HIOs was completely blocked in the presence of CFTRinh-172 (102% ± 2% SD; Figures 5A and 5C), whereas as expected, the negligible FIS response in CFTR mutant HIOs did not significantly change (96% ± 1% SD; Figures 5A and 5B). This inhibition was reversible after washing out CFTRinh-17231 (data not shown).
Figure 5.

Functional Recovery of CFTR Activity in iPSC-Derived, CFTR-Corrected Human Intestinal Organoids
(A) Representative fluorescence live-cell microscopy images of calcein green-labeled, CFTR-corrected and p.F508del mutant HIOs following 5 μM forskolin treatment, alone or in combination with pharmacological CFTR modulators for 60 min. Scale bars, 100 μm. (B and C) Normalized forskolin-induced swelling of p.F508del (B) and CFTR-corrected (C) mutant organoids upon CFTR inhibition (CFTRinh-172) and/or activation (VX-770/VX-809) for 180 min. Surface areas are calculated as percentages (mean ± SEM) relative to that at t = 0 (100%). DMSO treatment serves as negative control. (A–C) Organoids were pretreated for 16 h with 50 μM CFTRinh-172 (CFTR inhibitor) and/or 3 μM VX-809 (CFTR corrector). CFTR potentiator VX-770 (3 μM) was added simultaneously with forskolin.
To examine whether functional rescue of p.F508del mutant CFTR is feasible with a combined treatment of chemical CFTR modulators, we treated iPSC-derived HIOs simultaneously with the CFTR potentiator (VX-770) and the corrector (VX-809; preincubated for 16 h). The double treatment markedly increased the FIS response in p.F508del mutant HIOs (swelling rate: 133% ± 7% SD; Figures 5A and 5B), albeit to a lesser extent than what was observed in CFTR-corrected HIOs without VX-770/VX-809 treatment, suggesting that HR-directed CFTR repair is considerably more efficient in restoring CFTR function than the clinically approved double treatment VX-770/VX-809 (Orkambi). CFTR-corrected HIOs showed an even higher increase in surface area (206% ± 14% SD; Figures 5A and 5C) when treated with VX-770/VX-809, thus validating the impact of CFTR modulators on CFTR function in CF-corrected HIOs. Finally, organoid swelling was completely abolished in VX-770/VX-809-treated, CFTR-corrected (104% ± 6% SD) and mutant (99% ± 2% SD) HIOs when CFTR activity was blocked by CFTRinh-172 exposure (Figures 5A–5C).
Taken together, these results strongly indicate that heterozygous CFTR correction by TALEN-mediated footprint-free gene targeting resulted in full restoration of CFTR function in iPSC-derived HIOs, a result compatible with similar observations made on iPSC-derived, 2D-cultured epithelial cells32.
Discussion
Here, we describe the correction, by gene targeting in patient-derived iPSCs, of the CFTR p.F508del mutation. Gene-editing protocols are often associated with undesired and potentially harmful modifications of the genome; to minimize the possibility of this type of events, a strategy based on an antibiotic selection step, followed by a triple serial DNA screening, was devised. In addition, a new procedure to obtain intestinal organoids from iPSC-edited cells was validated, and the complete functional restoration of CFTR activity in CF-corrected, iPSC-derived intestinal organoids demonstrated using the CFTR-dependent FIS assay.
After transfection, antibiotic selection eliminated all cellular clones in which the antibiotic-resistant cassette had not been incorporated into the genome. The first PCR screening step was designed to identify cellular clones with HR-driven integration of the selection cassette into the targeted locus. Validation at both 5′ and 3′ recombination sites was necessary to avoid false positives due to the fragmentary integration of the recombination cassette. The second screening exploited a newly created, silent BglII restriction site. Due to its close proximity to the p.F508del mutation, successful digestion of the amplicon comprising the site was indicative of a very likely, correct cointegration of the restoring CTT triplet. Finally, the third screening step was carried out to confirm piggyBac-catalyzed excision of the selection cassette and therefore, a seamless repair of the CFTR gene. Due to the silent mutations introduced into the recombination cassette, allele-specific PCRs could be performed to identify recombinant CF-repaired clones in which the selection cassette had been properly cut out by PB transposase.
Our TALEN-mediated HR approach resulted in the correction of the p.F508del mutation in two iPSC lines derived from one CF patient with efficiencies between 9.3% and 10.2%. These efficiencies are superior or similar to correction rates reported in other nuclease-assisted, genome-editing studies, where the p.F508del mutation has been repaired33, 34, 35, 36. It should be noted that the efficiency of HR-driven correction depends on many factors, such as cell type, cell line, cutting efficiency of designer nuclease, nuclease retargeting, proximity of the double-strand breaks to the repair site, sequence identity between donor template and target region, as well as the type of homology-directed repair (HDR) template37. For instance, Lee et al.34 reported an overall repair efficiency of <1% in a CF lung epithelial cell line. This low level of repair was most probably due to the long distance (203 bp) between the p.F508del mutation and the Zinc Finger Nuclease (ZFN) target site, as well as to sequence variation between donor plasmid and target region. Another study used short DNA fragments and TALENs to correct the CTT deletion in CF iPSCs, with correction rates as low as 0.1%, although the efficiency increased up to 10% after six cycles of enrichment of the recombinant cells36. More recently, two other studies claimed to have obtained higher HR-driven repair efficiencies at the p.F508del locus by employing CRISPR/Cas9-mediated, gene-editing approaches in combination with a donor vector33 or single-stranded oligodeoxynucleotides (ssODNs)35 as repair templates. In the first study, editing efficiency of 16.7% was observed in a CF iPSC line, whereas the correction rate in the second study ranged from 2.5% to 22%, depending on the cell type used. Although higher HR rates can be achieved by electroporating CRISPR/Cas9 as a recombinant ribonucleoprotein35, we have opted to utilize the TALEN technology to enhance CF gene correction in iPSCs, since the CRISPR/Cas9 system, in spite of recent efforts to minimize off targeting, seems still to be associated with increased off-target effects38,39. On the contrary, TALENs generally show a higher level of specificity, which relies on their ability to recognize longer target sequences (30–36 bp)40, the limitations imposed by the required distance between target sequences41 and on the fact that they only act if both members of the pair bind to the target sequence on opposite strands.
Although small DNA fragments, including ssODNs, are nowadays widely used for HR-based gene correction of small fragments, such as single-base pair mutations, like the CTT deletion in the CFTR gene,35,36,42 this elegant and drug selection-free approach might require laborious screening analysis in order to identify targeted clones43. Additional, more serious disadvantages of ssODN-based gene editing are the generation of indel mutations at HR sites by the microhomology-mediated end-joining mechanism44 and the random insertion of ssODN throughout the genome with a frequency of up to 18%45. A recent study by Vaidyanathan et al.46 has reported recombination frequencies of around 40% at the CFTR locus in upper-airway basal stem cells from CF patients. They achieved this mark by adeno-associated virus (AAV) transduction of CRISPR/Cas9 and ssODNs. However, the results should be considered in light of new evidence suggesting the possibility of undesired rearrangements produced by ssODNs, both at the target site and at distant locations47,48. In addition, therapies based on viral vectors, including AAVs, might generate immune responses against the virus and are not applicable to individuals with preexisting neutralizing antibodies49. For all of these reasons and despite the attractiveness of the ssODN-mediated HR system, we opted instead for an ex vivo PB transposon-based gene-targeting strategy. Similar to previous reports33,50, we obtained integration-free-corrected iPSC clones with an excision efficiency of the drug-resistant cassette of 53% on already-selected edited iPSC lines. In several CFTR gene-targeting studies, seamless correction of the p.F508del allele was not employed, thereby leaving behind donor DNA remnants, such as the complete selection cassette17,34 or a residual loxP site after Causes Recombination enzyme (CRE)-mediated excision of the donor vector32, in the genome of repaired CF iPSCs. iPSCs harboring those unwanted genetic modifications have a limited application, not only in clinical settings but also for disease modeling in basic research and drug-screening programs.
Functional restoration of CFTR channel activity after gene editing has been previously demonstrated in iPSC-derived epithelial cells by using 2D differentiation monolayer systems32,33,36,42. However, due to variable differentiation efficiencies, iPSC differentiation often resulted in the derivation of heterogeneous populations in terms of CFTR expression, thereby impeding unambiguous readouts during functional assays. We, therefore, developed a novel iPSC-derived 3D organoid model that allows the precise assessment of CFTR activity in CFTR mutant and repaired HIOs by the FIS assay. Importantly, the readout of this technique has been shown to be completely dependent on CFTR in primary HIO13, thus serving as an ideal method to measure the recovery of CFTR function following CFTR gene correction. Before starting the FIS assay, CFTR expression and its localization at the plasma membrane were confirmed in CFTR-corrected HIOs by immunostaining. In addition, detection of mature, complex, glycosylated CFTR protein by western blot strongly indicated the restoration of correct CFTR processing upon CFTR gene repair and a very likely functional recovery of the gene, since only this processed form of CFTR is able to reach the plasma membrane and exert its function as an anion channel. On the contrary, p.F508del mutant HIOs only expressed the immature core-glycosylated form of CFTR that is rapidly degraded in the endoplasmic reticulum (ER) before reaching the plasma membrane. These results are in concordance with recent findings showing the localization of mature CFTR at the plasma membrane in primary wild-type but not in p.F508del homozygous HIOs13.
Finally, functional testing of CFTR protein activity in FIS assays demonstrated that p.F508del correction in one of the two mutant alleles is enough for the complete rescue of CFTR function, as indicated by similar levels of organoid swelling to those observed in wild-type13 or CFTR-corrected, primary HIOs17. Interestingly, heterozygous gene correction seems to be far more efficient in terms of restoring CFTR activity than the combinatorial treatment of homozygous p.F508del samples with the CFTR potentiator (VX-770) and corrector (VX-809) together, which is consistent with a previous report using primary HIOs17. It has been described that CF patients harboring p.F508del in homozygosity differ significantly in their response to Orkambi (VX-770/VX-809) treatment, suggesting that the CF patient in our study could belong to the low responder group6,51. Patient-specific differences were also observed in primary HIOs with identical CF-causing mutations, further indicating that individual genetic background, in addition to CFTR status, might affect the degree of FIS response in vitro and successful therapy in vivo14,15. Given the influence of genetic modifiers on CFTR functional tests and more importantly, on the course of the disease and the response of patients to treatment16, CFTR-repaired HIOs and their original mutant counterparts represent an optimal isogenic platform to use in disease modeling and drug screening. Although previous gene-targeting studies have validated the restoration of CFTR activity in iPSC-derived lung epithelial cells using a variety of techniques, such as Ussing chamber32,36. whole-cell patch-clamp33 and iodide efflux assays32, the combination of the HIO and FIS assay is presumably the easiest and most robust way to validate CFTR activity in a complex, polytypic tissue model. In addition, the possibility of expanding iPSC-derived HIOs to an unlimited number, thanks to the maintenance of an intestinal stem-cell subpopulation, makes it very attractive for high-throughput screening in 384-well plates when combined with automated live microscopy. Indeed, pharmaceutical companies have already started to integrate intestinal organoids in their drug-discovery pipeline28. It is important to keep in mind, however, that until exhaustive comparisons are performed, it cannot formally be ruled out that drug responses of certain CFTR variants can vary in a tissue-specific manner due to the recruitment of different cis-regulatory elements nearby or within the CFTR locus. In this regard, it has been demonstrated that CFTR expression is differentially regulated in lung and intestinal cells through cell type-specific enhancer elements that are located in intergenic and intronic regions18.
In summary, this report demonstrates, to our knowledge, for the first time, the integration of an efficient method for seamless CFTR correction in CF patient-derived iPSCs with a functional intestinal organoid swelling test to determine the biological activity of the resulting protein. The linkage of the two methods was made possible by a novel and efficient protocol for the production of intestinal organoids from edited iPSCs. The association of these three procedures: disease or patient-specific iPSC production, gene editing, and organoid-based functional testing, together with improved protocols for the reintroduction the targeted cells into the patients4, will have a profound impact, not only in studying the pathophysiology of CF and other human diseases but also in more accurate drug-discovery protocols and personalized medicine applications in the future, thus providing an alternative to other gene-therapy approaches being currently tested on CF4,52.
Materials and Methods
Cells and Culture Procedures
For gene-targeting experiments, we used an iPSC line (IMEDEAi001-F) recently generated in our lab from a CF patient20. Human iPSCs were cultured in KnockOut DMEM (Invitrogen, USA), supplemented with 20% KnockOut Serum Replacement (KOSR; Invitrogen, USA), 2 mM GlutaMAX, 0.1 mM β-mercaptoethanol, 1% nonessential amino acid (NEAA; Invitrogen, USA), and 10 ng/mL basic fibroblast growth factor (bFGF; PeproTech, UK). iPSC passage was carried out using recombinant trypsin (TrypLE Select; Invitrogen, USA) and the final addition of 10 μM rho-associated protein kinase (ROCK) inhibitor Y-27632 (Sigma, Spain). Mature, intestinal iPSC-derived organoids and rectal organoids were maintained in intestinal growth media and passed 1:3 to 1:6 every 5–8 days, as described previously53,54. HUB wild-type and HUB p.F508del homozygous rectal organoid lines were purchased from Hubrecht Organoid Technology (https://hub4organoids.eu/).
Generation of CFTR-Specific TALENs and Detection of Nuclease Activity
TALEN expression vectors designed to target and cleave 55–57 bp 3′ of the p.F508del mutation, were synthesized by Transposagen (USA). The length of the recognition sequence of each TALEN is 17 bp (left binding site: 5′-ACAGAAGCGTCATCAAA-3′; right binding site: 5′-AGGTAAGAAACTATGTG-3′) and is separated by a 16-bp spacer. The expression of the TALEN genes is under control of the cytomegalovirus (CMV) promoter.
The cleavage activity of the newly generated CFTR TALENs was analyzed in two highly transfectable cell lines, K562 and HeLa. 1 × 106 K562 and HeLa cells were transiently transfected with 2 μg of each TALEN vector for 48 h. Genomic DNA from the TALEN-transfected cell populations was isolated and the target site amplified by PCR using the ZFN-forward (Fw)/P6 primers (see Table S1). To assess the frequency of indels generated after cleavage by TALENs, the Surveyor nuclease assay (Integrated DNA Technologies [IDT], Belgium) was used. For this purpose, PCR amplicons were denatured and slowly reannealed. Resulting heteroduplexes were cleaved by the Surveyor nuclease, whereas homoduplexes were left intact. The heterogeneous DNA population was run on a microchip device (Bioanalyzer 2100; Agilent, USA), and the efficiency of TALEN cleavage (percentage of indels) was quantified by densitometry as a ratio between cleaved and noncleaved DNA bands.
Targeting Vector Design
Two vectors were produced. The first one (Seq_1) was designed to be used, prior to gene-targeting experiments, as a template to optimize the PCR reactions used in the screening for recombinant clones. For this purpose, specific genomic regions, immediately adjacent but external to the 5′ and 3′ recombination arms, were added to the vector to allow primer binding. The length of the 5′ and 3′ homology arms was 916 and 920 bp, respectively. In order to prevent retargeting of the recombined allele by TALENs, five silent point mutations were introduced into the 5′ recombination arm. In addition, a new silent BglII restriction site was also created to facilitate the detection of recombinant clones by PCR, followed by digestion of the amplification product with the enzyme (Figure 1A). Primers specific for the recombinant allele were also designed (Table S1). The final targeting vector, pMC3.1, was obtained by modifying Seq_1 in three cloning steps. The elimination of the external primer-binding regions attached to the homology arms was performed by EcoRI and MluI digestions. Finally, the double-selection cassette PB:PGKpuroΔtk from pMCS-AAT-PB:PGKpuroΔtk55 was cloned via NheI/NotI digestion/ligation in between the homology arms. The pMCS-AAT-PB:PGKpuroΔtk plasmid was provided by the Sanger Institute under a Material Transfer Agreement (MTA).
Electroporation of iPSCs
Nucleofection was performed with the P3 Primary Cell 4D-Nucleofector Kit (Lonza, Walkersville, MD, USA), according to the manufacturer’s instructions. To avoid feeder cell contamination, iPSCs were expanded in Matrigel-coated p100 culture plates prior to nucleofection. Adherent iPSCs were detached by using TrypLE (Gibco, USA), centrifuged at 200 × g, and resuspended in nucleofection buffer 3. For homologous recombination experiments, 2 μg of TALEN and donor plasmids were transfected per 1 × 106 cells in a 100-μL cuvette using the CB-150 program. In piggyBac excision experiments, 5 μg of piggyBac transposase (pCMV-HAhyPBase) per million cells was employed in nucleofection assays. Nucleofected iPSCs were seeded in the presence of 10 μM ROCK inhibitor onto irradiated HAF-1/W3R puromycin-resistant feeder cells, kindly provided by Dr. Chen from Johns Hopkins University School of Medicine (Baltimore, MD, USA).56
Isolation of Recombined Clones and PCR Screening
After selection with 1 μg/mL puromycin (Sigma, Spain) or 250 nM FIAU (Sigma, Spain) for 12–18 days, individual resistant colonies were manually picked and transferred to 48-well plates previously seeded with irradiated feeder cells. After 7–10 days, iPSC clones were trypsinized (TrypLE) and duplicated in either Matrigel-coated or feeder cell-containing 48-well plates. Genomic DNA was extracted from clones grown in feeder-free, Matrigel-coated plates using a standard protocol, whereas clones grown on irradiated feeders were maintained and later expanded in the case of successful recombination. Shortly, genomic DNA from 48-well plates was isolated by proteinase K digestion overnight (O.N.) at 55°C, precipitation of DNA with isopropanol, washing with 70% ethanol, and resuspension in Tris-EDTA buffer. The first round of the screening consisted of the identification of recombinant clones. For this purpose, both 5′ and 3′ ends of the recombination site were analyzed by PCR. One primer of each primer pair was located in the genomic region outside the corresponding recombination arm; the other one was situated within the selection cassette (P1/P2 and P3/P4 pairs; Table S1). To confirm positive recombinant clones, the amplicons generated by 5′ PCR were subsequently digested with BglII. The absence of random integration of the recombination cassette into the genome was verified by PCR using primers specific for the selection cassette (Reint-Fw/Reint-Rv); Table S1).
Excision of Recombination Cassette by piggyBac Transposase
Following the identification of corrected clones, the selection cassette was removed by nucleofection of 5 μg of pCMV-HAhyPBase per 1 × 106 iPSCs. Selection with FIAU (250 nM) was started 24 h after nucleofection and resistant clones picked, expanded, and duplicated, as described above. To identify clones with successful excision of the selection cassette, the correct size of the CFTR target site was confirmed by PCR using primers specific for the edited allele (P5/P6; Table S1).
Sequencing
Genomic DNA from donor cells was isolated, as described above. For sequencing, a 2,250-bp long fragment comprising the whole CFTR-targeted area, including the p.F508del mutation site, was amplified by PCR with the P1 and P4 primers (Table S1). PCR products were cleaned using the Zymoclean Gel DNA Recovery Kit (Zymo Research, CA, USA) and subcloned into the pGEM-T Easy Vector, according to the manufacturer’s recommendations (Promega, USA). Sequencing of the CFTR alleles was performed by Secugen (Madrid, Spain) using the same amplification primers.
Prediction and Analysis of Off-Target Sites
Prediction of potential off-target sites after p.F508del correction was done with PROGNOS software from Dr. Gang Bao’s laboratory.57 Parameters, such as targeted region and repeat variable domains (RVDs) for both TALENs as well as variable length of spacer (10–30 bp), were introduced to predict off-target sequences throughout the genome. The 16 most likely candidates were PCR amplified in the CFTR-corrected clone using primers designed by PROGNOS (Table S1). Subsequently, the amplicons were analyzed by the Surveyor nuclease assay (IDT, Belgium), as previously described.
Generation of Embryoid Bodies
To produce EBs, iPSC colonies were mechanically collected and transferred into a 15-mL conical tube. Once the colonies settled at the bottom of the tube, media were discarded, and iPSCs were cultured in bacteriological Petri dishes with DMEM plus 10% fetal bovine serum (FBS), 1% nonessential amino acids, and 2 mM GlutaMAX for 10 days. Cavitation was then assessed and EBs mechanically disrupted and plated onto gelatin-coated, 24-well plates. Media were changed twice weekly, and after 10–20 days of spontaneous differentiation, cells were used for immunofluorescence studies.
Differentiation toward HIOs
iPSCs were harvested by trypsinization and replated for 1 h on gelatin-coated dishes to induce the depletion of human fibroblast feeders. Then, the nonadhered iPSC suspension was centrifuged; resuspended in iPSC growth media, supplemented with 10 μM Y-27632; and plated on tissue-culture plates (100,000 cells/cm2), previously coated with growth factor-reduced Matrigel (BD Biosciences, Spain). To improve differentiation efficiency, cells were treated on the following day with 1% DMSO (Sigma, Spain) for 24 h.25 Subsequently, differentiation into definitive endoderm was performed, as previously described, with slight modifications23. Briefly, cells were washed three times with PBS (Biowest, France) and cultured in RPMI 1640, supplemented with GlutaMAX (2 mM; Life Technologies, USA), 1% NEAA (Life Technologies, USA), 1% penicillin/streptomycin (Life Technologies, USA), 0.2% FBS (Biowest, France), 100 ng/ml human activin A (R&D Systems, UK), and 25 ng/mL WNT3A (R&D Systems, UK). Following 24 h of endoderm differentiation, WNT3A was withdrawn, and cells were differentiated only in the presence of activin A. At day 2 of endoderm induction, FBS concentration was shifted to 2%.
For hindgut specification, early definitive endodermal cells from day 3 were exposed to high doses of WNT3A. Briefly, cells were washed twice with basal differentiation media (Biowest, France) and cultured in RPMI 1640, supplemented with 1xB27, GlutaMAX (2 mM; Life Technologies, USA), 1% NEAA (Life Technologies, USA), 1% penicillin/streptomycin (Life Technologies, USA), 2% FBS (Biowest, France), and 6 μM CHIR99021 (STEMCELL Technologies, France) for 5 days. Floating 3D hindgut spheroids were embedded in the Matrigel matrix (Corning, NY, USA) and cultured in the presence of R-spondin1, Noggin, epidermal growth factor (EGF), and WNT3A (PeproTech, UK) to allow further intestinal maturation.26 Finally, a novel differentiation protocol was employed to generate intestinal organoids that were able to grow without a mesenchymal niche (manuscript in preparation), thus facilitating their use in FIS assays.
FIS Assay
FIS assays were performed as recently described53 but with minor modifications. Briefly, human iPSC-derived intestinal organoids from a 5- to 7-day-old culture were harvested, mechanically broken by pipetting, resuspended in Matrigel, seeded as 4 μL drops in a 96-well culture plate (Nunc), and cultured for 24 h in intestinal growth media. Each drop contained approximately 40–80 organoids. 50 μM CFTRinh-172 (Sigma, Spain) and 3 μM VX-809 (Selleck Chemicals, USA) were added to media after seeding for CFTR inhibition and correction, respectively. After 16 h pretreatment and following preincubation for 30 min with 5 μM calcein green (Invitrogen, USA), organoids were exposed to 5 μM forskolin and analyzed with a fluorescent microscope (Leica DMI6000B, objective 5×) in vivo. Each experimental condition was measured in triplicate and during 180 min. In the case of CFTR potentiation, 3 μM VX-770 (Selleck Chemicals, USA) was added simultaneously with forskolin. Forskolin-induced organoid swelling relative to time (t) = 0 (100%) of forskolin treatment was quantified by image processing using ImageJ software (NIH), and an increase of total organoid area over time was calculated from triplicates.
Immunofluorescence
Undifferentiated iPSCs, differentiated cells, and iPSC-derived organoids were washed with PBS, fixed for 20 min with 4% paraformaldehyde, and washed again with PBS. Immunocytochemistry was performed for Nanog (1:150, rabbit immunoglobulin [Ig]G polyclonal; Abcam, Spain), Oct4 (1:100, rabbit IgG polyclonal; Santa Cruz Biotechnology, USA), Tra-1-60 (1:100, mouse IgM; Millipore, USA), AFP (1:100, rabbit IgG; Dako, Denmark), Nestin (1:500, rabbit IgG; Sigma, Spain), Tuj1 (1:500, rabbit IgG; Covance, UK), Sox17 (1:100, goat IgG; R&D Systems, UK), α-actinin (1:200, mouse IgM; Sigma, Spain), SMA (1:200, mouse; Sigma), CFTR (clone MM13-4 or M3A7, 1:100; Merck Millipore, Spain), E-CAD (1:200, rabbit; Cell Signaling Technology), SOX9 (1:100, goat; R&D Systems, UK), Villin (1:100, goat; Santa Cruz Biotechnology, USA), FOXA2 (1:100, goat IgG; Santa Cruz Biotechnology, USA), CDX2 (1:100, mouse IgG; DCS ImmunoLine, Germany), and GATA4 (1:300, mouse IgG; Santa Cruz Biotechnology, USA). After permeabilization in 0.2% Triton X-100/100 mM glycine/PBS buffer for 30 min at room temperature (RT), cells were blocked with 5% BSA in PBS for 1 h at RT. Cells were incubated with primary antibodies overnight at 4°C in 2% BSA in PBS, followed by three washing steps with PBS. Alexa Fluor 555 (1:500, donkey anti-mouse IgG or donkey anti-rabbit IgG; Invitrogen, USA), Alexa Fluor 546 (1:500, donkey anti-goat IgG; Invitrogen, USA), and Alexa Fluor 488 (1:500, donkey anti-mouse IgG, donkey anti-rabbit IgG, or donkey anti-rat IgG; Invitrogen, USA) were used as secondary antibodies, incubated 1 h at RT with 2% BSA in PBS. After washing with PBS, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; 5 min, 1 μg/mL), washed three times, and mounted with Dako fluorescent-mounting medium.
Western Blot Analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% [v/v] Nonidet P-40 [NP-40], 1% [w/v] SDS, and 50 mM Tris HCl, pH 8.0) in the presence of protease inhibitor cocktail (P8340; Sigma, Spain), incubated for 30 min at 37°C, and passed through a 22G needle to reduce viscosity. Protein samples were quantified by a modified Lowry method, and 70 μg of total protein diluted 1:1 in sample buffer (100 mM Tris HCl, 10% [v/v] β-mercaptoethanol, 4% [w/v] SDS, 0.02% [w/v] bromophenol blue, and 20% [v/v] glycerol) was separated on an 8% SDS-PAGE. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane, following a wet transfer standard protocol. Membrane was blocked with 5% (w/v) skim milk in Tris-buffered saline (TBS)-Tween (TBST) for 1 h at RT, washed three times with TBST, and incubated overnight at 4°C with a cocktail of three primary CFTR antibodies: anti-CFTR monoclonal antibody (mAb) 450 (R-domain 696–705 aa), anti-CFTR mAb 570 (R-domain 731–742 aa), and anti-CFTR mAb 596 (nucleotide-binding domain 2 [NBD2] 1204–1211 aa) (Cystic Fibrosis Foundation, https://www.cff.org), diluted 1:500 in TBST 2% (w/v) skim milk. Membrane was washed and incubated 1 h at RT in the dark with the secondary antibody (Thermo Scientific, Spain) in TBST 2% (w/v) skim milk. After repeated washes, membrane was developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Spain). As an internal loading control, α-tubulin (Sigma, Spain) was used.
Karyotyping
A standard optimized G-banding technique with minor modifications was employed to determine the karyotype of iPSC lines. Briefly, actively proliferating cells were treated with 10 μg/mL Colcemid (Sigma, Spain) for 3 h, trypsinized, washed in PBS, and incubated in hypotonic PBS (74.85 mOsm/kg H2O) for 20 min, before immersing them in Carnoy’s fixative (cold methanol/acetic acid 3:1; both from BDH) for 30 min. Nuclei were then centrifuged at 500 × g for 2 min and resuspended in fresh Carnoy’s to wash residual PBS. Fixed nuclei were spread and Gbanded by Biobanco del Sistema Sanitario Público de Andalucía, Granada (Spain).
Array CGH Analysis
Array CGH analysis was performed by Genetadi Biotech (Spain). Genomic DNA was isolated from repaired and nonrepaired CF-human iPSC (hiPSC) and from parental fibroblasts by standard techniques. Whole-genome array CGH analysis was carried out using 0.5 μg of genomic DNA and a 60-K oligonucleotide array (Agilent Technologies, Santa Clara, USA; G4450A), according to the manufacturer’s instructions. Image quantification, hybridization quality control, and CNV detection were accomplished using Agilent Feature Extraction version (v.)11.5 and Agilent Workbench v.7.0. CNVs identified in the samples were visualized using the University of California, Santa Cruz (UCSC), Genome Browser (http://genome.ucsc.edu).
Author Contributions
A.F. designed and performed experiments, analyzed the data, and wrote the manuscript. S.V.-D., J.M.M.-F, A.S.-G., S.A., and J.L.C. performed experiments and interpreted results. S.V.-D. contributed to writing the manuscript. M.C., A.d.P., A.E., E.G., J.L.P., and M.V. provided intellectual input. D.B. designed and coordinated the research, analyzed the data, and wrote the manuscript. All authors reviewed and approved the final manuscript.
Conflicts of Interest
S.V.-D., A.d.P., A.E., J.L.P., M.V., and D.B. are founders and shareholders of Karuna Good Cells Technologies S.L.
Acknowledgments
We thank M. Camarasa for her help in HR experiments and Victor Gálvez for his assistance with Figures 1 and 2. Additionally, we thank Patricia Tortosa for providing invaluable technical support along the project. We acknowledge Hubrecht Organoid Technology (https://hub4organoids.eu/) for providing the rectal organoids from healthy-donor and p.F508del/p.508del patients. We also thank the Beekman’s group for sharing their expertise in human intestinal organoids. Finally, we are indebted to Ludovic Vallier for sharing with us the pMCS-AAT-PB:PGKpuroΔtk plasmid. This work was supported by grants from the Spanish Ministry for Science and Innovation (PLE2009-0091, RTC-2014-2207-1, and IPT-2011-1402-900000); ISCIII PI14/01073, cofunded by ERDF/ESF “Investing in your future”; the Balearic Government (16023/2008); the Spanish Cystic Fibrosis Federation (Pablo Motos Grant); Federación ASEM-Telemaratón “Todos somos raros, todos somos únicos”; Fundación Salud 2000; the European Commission (H2020, PHC-667079); and an endowment from METROVACESA. J.M.M.-F. was a postdoctoral Berrikertu fellow granted by the Basque Government. A.F. was a recipient of Juan de la Cierva (JCI-2006-2675) and Torres Quevedo (PTQ-16-08496) postdoctoral fellowships from the Spanish Ministry for Science and Innovation.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.005.
Supplemental Information
References
- 1.Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Bell S.C., Mall M.A., Gutierrez H., Macek M., Madge S., Davies J.C., Burgel P.R., Tullis E., Castaños C., Castellani C. The future of cystic fibrosis care: a global perspective. Lancet Respir. Med. 2020;8:65–124. doi: 10.1016/S2213-2600(19)30337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bobadilla J.L., Macek M., Jr., Fine J.P., Farrell P.M. Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum. Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 4.Schneider-Futschik E.K. Beyond cystic fibrosis transmembrane conductance regulator therapy: a perspective on gene therapy and small molecule treatment for cystic fibrosis. Gene Ther. 2019;26:354–362. doi: 10.1038/s41434-019-0092-5. [DOI] [PubMed] [Google Scholar]
- 5.Taylor-Cousar J.L., Munck A., McKone E.F., van der Ent C.K., Moeller A., Simard C., Wang L.T., Ingenito E.P., McKee C., Lu Y. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017;377:2013–2023. doi: 10.1056/NEJMoa1709846. [DOI] [PubMed] [Google Scholar]
- 6.Wainwright C.E., Elborn J.S., Ramsey B.W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J.C., De Boeck K., Flume P.A., TRAFFIC Study Group. TRANSPORT Study Group Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heijerman H.G.M., McKone E.F., Downey D.G., Van Braeckel E., Rowe S.M., Tullis E., Mall M.A., Welter J.J., Ramsey B.W., McKee C.M., VX17-445-103 Trial Group Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Middleton P.G., Mall M.A., Dřevínek P., Lands L.C., McKone E.F., Polineni D., Ramsey B.W., Taylor-Cousar J.L., Tullis E., Vermeulen F., VX17-445-102 Study Group Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson L.G., Olsen J.C., Sarkadi B., Moore K.L., Swanstrom R., Boucher R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 10.Dorin J.R., Farley R., Webb S., Smith S.N., Farini E., Delaney S.J., Wainwright B.J., Alton E.W., Porteous D.J. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther. 1996;3:797–801. [PubMed] [Google Scholar]
- 11.Davidoff A.M., Nathwani A.C. Genetic Targeting of the Albumin Locus to Treat Hemophilia. N. Engl. J. Med. 2016;374:1288–1290. doi: 10.1056/NEJMcibr1600347. [DOI] [PubMed] [Google Scholar]
- 12.Li H., Yang Y., Hong W., Huang M., Wu M., Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020;5:1. doi: 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dekkers J.F., Wiegerinck C.L., de Jonge H.R., Bronsveld I., Janssens H.M., de Winter-de Groot K.M., Brandsma A.M., de Jong N.W., Bijvelds M.J., Scholte B.J. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013;19:939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 14.Dekkers J.F., Berkers G., Kruisselbrink E., Vonk A., de Jonge H.R., Janssens H.M., Bronsveld I., van de Graaf E.A., Nieuwenhuis E.E., Houwen R.H. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016;8:344ra84. doi: 10.1126/scitranslmed.aad8278. [DOI] [PubMed] [Google Scholar]
- 15.de Winter-de Groot K.M., Berkers G., Marck-van der Wilt R.E.P., van der Meer R., Vonk A., Dekkers J.F., Geerdink M., Michel S., Kruisselbrink E., Vries R. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2019 doi: 10.1016/j.jcf.2019.10.022. Published online November 14, 2019. [DOI] [PubMed] [Google Scholar]
- 16.Strug L.J., Stephenson A.L., Panjwani N., Harris A. Recent advances in developing therapeutics for cystic fibrosis. Hum. Mol. Genet. 2018;27(R2):R173–R186. doi: 10.1093/hmg/ddy188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwank G., Koo B.K., Sasselli V., Dekkers J.F., Heo I., Demircan T., Sasaki N., Boymans S., Cuppen E., van der Ent C.K. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Sosnay P.R., Siklosi K.R., Van Goor F., Kaniecki K., Yu H., Sharma N., Ramalho A.S., Amaral M.D., Dorfman R., Zielenski J. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013;45:1160–1167. doi: 10.1038/ng.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleischer A., Lorenzo I.M., Palomino E., Aasen T., Gómez F., Servera M., Asensio V.J., Gálvez V., Izpisúa-Belmonte J.C., Bachiller D. Generation of two induced pluripotent stem cell (iPSC) lines from p.F508del Cystic Fibrosis patients. Stem Cell Res. (Amst.) 2018;29:1–5. doi: 10.1016/j.scr.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Roqué M., Godoy C.P., Castellanos M., Pusiol E., Mayorga L.S. Population screening of F508del (DeltaF508), the most frequent mutation in the CFTR gene associated with cystic fibrosis in Argentina. Hum. Mutat. 2001;18:167. doi: 10.1002/humu.1173. [DOI] [PubMed] [Google Scholar]
- 22.Nikolić M.Z., Sun D., Rawlins E.L. Human lung development: recent progress and new challenges. Development. 2018;145:dev163485. doi: 10.1242/dev.163485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Amour K.A., Agulnick A.D., Eliazer S., Kelly O.G., Kroon E., Baetge E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005;23:1534–1541. doi: 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- 24.Kubo A., Shinozaki K., Shannon J.M., Kouskoff V., Kennedy M., Woo S., Fehling H.J., Keller G. Development of definitive endoderm from embryonic stem cells in culture. Development. 2004;131:1651–1662. doi: 10.1242/dev.01044. [DOI] [PubMed] [Google Scholar]
- 25.Chetty S., Pagliuca F.W., Honore C., Kweudjeu A., Rezania A., Melton D.A. A simple tool to improve pluripotent stem cell differentiation. Nat. Methods. 2013;10:553–556. doi: 10.1038/nmeth.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spence J.R., Mayhew C.N., Rankin S.A., Kuhar M.F., Vallance J.E., Tolle K., Hoskins E.E., Kalinichenko V.V., Wells S.I., Zorn A.M. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai Y.H., Nattiv R., Dedhia P.H., Nagy M.S., Chin A.M., Thomson M., Klein O.D., Spence J.R. In vitro patterning of pluripotent stem cell-derived intestine recapitulates in vivo human development. Development. 2017;144:1045–1055. doi: 10.1242/dev.138453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Mourik P., Beekman J.M., van der Ent C.K. Intestinal organoids to model cystic fibrosis. Eur. Respir. J. 2019;54:1802379. doi: 10.1183/13993003.02379-2018. [DOI] [PubMed] [Google Scholar]
- 29.Sato T., Stange D.E., Ferrante M., Vries R.G., Van Es J.H., Van den Brink S., Van Houdt W.J., Pronk A., Van Gorp J., Siersema P.D., Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 30.Ma T., Thiagarajah J.R., Yang H., Sonawane N.D., Folli C., Galietta L.J., Verkman A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melis N., Tauc M., Cougnon M., Bendahhou S., Giuliano S., Rubera I., Duranton C. Revisiting CFTR inhibition: a comparative study of CFTRinh -172 and GlyH-101 inhibitors. Br. J. Pharmacol. 2014;171:3716–3727. doi: 10.1111/bph.12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crane A.M., Kramer P., Bui J.H., Chung W.J., Li X.S., Gonzalez-Garay M.L., Hawkins F., Liao W., Mora D., Choi S. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4:569–577. doi: 10.1016/j.stemcr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Firth A.L., Menon T., Parker G.S., Qualls S.J., Lewis B.M., Ke E., Dargitz C.T., Wright R., Khanna A., Gage F.H., Verma I.M. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015;12:1385–1390. doi: 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee C.M., Flynn R., Hollywood J.A., Scallan M.F., Harrison P.T. Correction of the ΔF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Gene by Zinc-Finger Nuclease Homology-Directed Repair. Biores. Open Access. 2012;1:99–108. doi: 10.1089/biores.2012.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruan J., Hirai H., Yang D., Ma L., Hou X., Jiang H., Wei H., Rajagopalan C., Mou H., Wang G. Efficient Gene Editing at Major CFTR Mutation Loci. Mol. Ther. Nucleic Acids. 2019;16:73–81. doi: 10.1016/j.omtn.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki S., Sargent R.G., Illek B., Fischer H., Esmaeili-Shandiz A., Yezzi M.J., Lee A., Yang Y., Kim S., Renz P. TALENs Facilitate Single-step Seamless SDF Correction of F508del CFTR in Airway Epithelial Submucosal Gland Cell-derived CF-iPSCs. Mol. Ther. Nucleic Acids. 2016;5:e273. doi: 10.1038/mtna.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox D.B., Platt R.J., Zhang F. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merkert S., Martin U. Targeted genome engineering using designer nucleases: State of the art and practical guidance for application in human pluripotent stem cells. Stem Cell Res. (Amst.) 2016;16:377–386. doi: 10.1016/j.scr.2016.02.027. [DOI] [PubMed] [Google Scholar]
- 39.Aryal N.K., Wasylishen A.R., Lozano G. CRISPR/Cas9 can mediate high-efficiency off-target mutations in mice in vivo. Cell Death Dis. 2018;9:1099. doi: 10.1038/s41419-018-1146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boglioli E., Richard M. Rewriting the book of life: a new era in precision gene editing. Boston Consulting Group. 2015 https://www.bcg.com/publications/2015/rewriting-book-of-life-new-era-precision-gene-editing.aspx September 10, 2015. [Google Scholar]
- 41.Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merkert S., Schubert M., Olmer R., Engels L., Radetzki S., Veltman M., Scholte B.J., Zöllner J., Pedemonte N., Galietta L.J.V. High-Throughput Screening for Modulators of CFTR Activity Based on Genetically Engineered Cystic Fibrosis Disease-Specific iPSCs. Stem Cell Reports. 2019;12:1389–1403. doi: 10.1016/j.stemcr.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K.M., Gregg A., Noggle S., Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimi K., Kunihiro Y., Kaneko T., Nagahora H., Voigt B., Mashimo T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016;7:10431. doi: 10.1038/ncomms10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanza D.G., Gaspero A., Lorenzo I., Liao L., Zheng P., Wang Y., Deng Y., Cheng C., Zhang C., Seavitt J.R. Comparative analysis of single-stranded DNA donors to generate conditional null mouse alleles. BMC Biol. 2018;16:69. doi: 10.1186/s12915-018-0529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaidyanathan S., Salahudeen A.A., Sellers Z.M., Bravo D.T., Choi S.S., Batish A., Le W., Baik R., de la O.S., Kaushik M.P. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell. 2020;26:161–171.e4. doi: 10.1016/j.stem.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Codner G.F., Mianné J., Caulder A., Loeffler J., Fell R., King R., Allan A.J., Mackenzie M., Pike F.J., McCabe C.V. Application of long single-stranded DNA donors in genome editing: generation and validation of mouse mutants. BMC Biol. 2018;16:70. doi: 10.1186/s12915-018-0530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skryabin B.V., Kummerfeld D.M., Gubar L., Seeger B., Kaiser H., Stegemann A., Roth J., Meuth S.G., Pavenstädt H., Sherwood J. Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9-mediated genome editing events. Sci Adv. 2020;6 doi: 10.1126/sciadv.aax2941. eaax2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabinowitz J., Chan Y.K., Samulski R.J. Adeno-associated Virus (AAV) versus Immune Response. Viruses. 2019;11:102. doi: 10.3390/v11020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yusa K., Rad R., Takeda J., Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods. 2009;6:363–369. doi: 10.1038/nmeth.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ratjen F., Hug C., Marigowda G., Tian S., Huang X., Stanojevic S., Milla C.E., Robinson P.D., Waltz D., Davies J.C., VX14-809-109 investigator group Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017;5:557–567. doi: 10.1016/S2213-2600(17)30215-1. [DOI] [PubMed] [Google Scholar]
- 52.Alton E.W.F.W., Armstrong D.K., Ashby D., Bayfield K.J., Bilton D., Bloomfield E.V., Boyd A.C., Brand J., Buchan R., Calcedo R., UK Cystic Fibrosis Gene Therapy Consortium Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015;3:684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boj S.F., Vonk A.M., Statia M., Su J., Vries R.R., Beekman J.M., Clevers H. Forskolin-induced Swelling in Intestinal Organoids: An In Vitro Assay for Assessing Drug Response in Cystic Fibrosis Patients. J. Vis. Exp. 2017:55159. doi: 10.3791/55159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCracken K.W., Howell J.C., Wells J.M., Spence J.R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 2011;6:1920–1928. doi: 10.1038/nprot.2011.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yusa K., Rashid S.T., Strick-Marchand H., Varela I., Liu P.Q., Paschon D.E., Miranda E., Ordóñez A., Hannan N.R., Rouhani F.J. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai L., Ye Z., Zhou B.Y., Mali P., Zhou C., Cheng L. Promoting human embryonic stem cell renewal or differentiation by modulating Wnt signal and culture conditions. Cell Res. 2007;17:62–72. doi: 10.1038/sj.cr.7310138. [DOI] [PubMed] [Google Scholar]
- 57.Fine E.J., Cradick T.J., Zhao C.L., Lin Y., Bao G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 2014;42:e42. doi: 10.1093/nar/gkt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.