

Summary
Despite advances in single-cell and molecular techniques, it is still unclear how to best quantify phenotypic heterogeneity in cancer cells that evolved beyond normal, known classifications. We present an approach to phenotypically characterize cells based on their activities rather than static classifications. We validated the detectability of specific activities (epithelial-mesenchymal transition, glycolysis) in single cells, using targeted RT-qPCR analyses and in vitro inductions. We analyzed 50 established activity signatures as a basis for phenotypic description in public data and computed cell-cell distances in 28,513 cells from 85 patients and 8 public datasets. Despite not relying on any classification, our measure correlated with standard diversity indices in populations of known structure. We identified bottlenecks as phenotypic diversity reduced upon colorectal cancer initiation. This suggests that focusing on what cancer cells do rather than what they are can quantify phenotypic diversity in universal fashion, to better understand and predict intra-tumor heterogeneity dynamics.
Subject Areas: Biological Sciences, Mathematical Biosciences, Cancer Systems Biology, Cancer
Graphical Abstract
Highlights
-
•
Cells categorized as having the same identity can perform different activities
-
•
Single-cell expression data can be used to infer the activities cells take part in
-
•
Activity profiles provide a basis to measure phenotypic cell-cell divergence
-
•
Cell activity can quantify intra-tumor heterogeneity more fully than identity
Biological Sciences; Mathematical Biosciences; Cancer Systems Biology; Cancer
Introduction
Somatic evolution naturally occurs in all multicellular organisms, as cells accumulate genetic alterations upon replication and exposure to mutagenic environments (Gatenby and Brown, 2017). This can eventually select for highly adapted cells breaking free of the constraints imposed by homeostatic regulation on proliferation and motility, leading to cancer (Greaves and Maley, 2012, Trigos et al., 2018). This evolutionary nature implies that cancer cells originating from a common ancestor can display extensive diversity at both the genetic and phenotypic levels (Gerlinger et al., 2012). This diversity, known as intra-tumor heterogeneity (ITH) (McGranahan and Swanton, 2015), can foster resistance and facilitate adaptation upon the environmental changes induced by therapeutic regimens (Nowell, 1976). To limit the risk of resistant populations emerging upon treatment and predict cancer evolution, it is thus necessary to better understand the dynamics of ITH (Lässig et al., 2017, Maley et al., 2006).
Being able to follow the evolution of ITH first implies that one should be able to reliably quantify it. Although there exist multiple methods for genetic ITH thanks to alteration frequencies in the population (Nik-Zainal et al., 2012, Andor et al., 2014, Fischer et al., 2014, Martinez et al., 2017, Williams et al., 2018), phenotypic ITH is more challenging. Many studies have relied on the identification of static classifications (Frazer et al., 2007, Patel et al., 2014, Zhang et al., 2019), often based on lineage markers (Almendro et al., 2014, Nguyen et al., 2018), allowing the calculation of standard diversity metrics such as the Shannon (Bertucci et al., 2019), Simpson (Martinez et al., 2016), or GINI (Ferrall-Fairbanks et al., 2019) indices. Although these classifications make perfect sense in the context of normal tissue homeostasis, they may not be relevant in cancer cells bypassing the host's regulatory mechanisms through abnormal transcriptional programs. Cancer cells drift away from well-characterized normal phenotypes according to evolutionary trajectories specific to each tumor. They, however, display strong convergence at the phenotypic level, with key pathways and cellular activities recurrently dysregulated across both patients and tumor types (Hanahan and Weinberg, 2000, Hanahan and Weinberg, 2011). Aside from static subtype classifications, other methods have focused on expression variation among specific gene sets (Davis-Marcisak et al., 2019) and uneven repartition of expressed transcripts per gene (Hinohara et al., 2018). Yet, there is no golden standard approach to quantify phenotypic diversity in cancer.
Here we investigated the feasibility of predicting the activities that a single cell partakes in and the relevance of considering them as traits to describe the cell's overall phenotypic profile. We performed targeted single-cell experiments on three cellular activities induced in vitro (epithelial-mesenchymal transition, DNA repair, glycolysis), which suggested that targeted panels can reliably identify the presence of a given activity from single cell RNA expression data. To expand on this limited data, we then analyzed 50 hallmark activity signatures from the Molecular Signature database (MSigDB) in eight publicly available single-cell tumor datasets. We used leave-one-out procedures to avoid overfitting, along with Principal Component and clustering analyses to account for the redundancy among the 50 activities. By using activity-based phenotypic profiles to quantify cell-cell divergence and sample-wise phenotypic diversity, we report that such an approach is relevant in pan-cancer fashion. It could furthermore recapitulate diversity indices based on known population structures, independently of tissue and cell types. Finally, such a method allowed a glimpse into the evolutionary dynamics of phenotypic diversity, hinting at the existence of evolutionary bottlenecks reducing phenotypic diversity upon colorectal cancer initiation. Although more work is necessary to provide specific and accurate quantitative tools and software, our results suggest that focusing on cell activities to measure phenotypic ITH can provide a more relevant angle than standard classification and marker-based methods.
Results
Detecting Hallmark Signatures in Single Cells
We assessed the relevance of three MSigDB hallmark gene signatures in single cells via in vitro inductions: epithelial-mesenchymal transition (EMT), DNA repair, and glycolysis. We aimed to take advantage of the higher accuracy of single-cell RT-qPCR compared with whole transcriptome scRNA-seq (Mojtahedi et al., 2016) and designed reduced panels of 9–13 marker genes to detect each activity in single cells (see Methods). To do so, we first analyzed gene expression in 1,036 cell lines samples from the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012) for marker gene discovery and 10,885 pan-cancer samples from The Cancer Genome Atlas (TCGA) (Chang et al., 2013) for cross-validation. The activity-specific markers, respectively, achieved areas under the curve (AUCs) of 0.96, 086, and 0.79 in teasing out the top and bottom scoring TCGA samples for EMT, DNA repair, and glycolysis, respectively (Table S3).This suggested that these reduced gene panels satisfactorily recapitulated the signal from whole-gene set enrichment analyses, implying that analyzing the expression of few marker genes could help quantify the presence of activity-based phenotypic traits in single cells.
We analyzed the expression of 48 selected marker genes in 48 single epithelial mammary cells (MCF10A), in which each activity had been induced or not (12 EMT-induced, 12 DNA-repair-induced, 12 glycolysis-induced, 12 control cells with no induction, Figure 1A). Significantly differentially expressed genes could be identified in all experiments (Figure 1B). We inferred Beta-Poisson expression distributions for each gene in active/inactive conditions, which we used to calculate the likelihood that expression values from marker genes corresponded to cells in which the related activity was induced (Figure 1C). Differentially expressed genes, generalized linear models, and leave-one-out procedures were used to predict cells undergoing each activity induction (see Transparent Methods). We could achieve AUCs of 0.99, 0.72, and 0.86 for, respectively, the EMT, DNA repair, and glycolysis activities (Figures 1D, S2, S3, and Table S4). The absence of expression patterns clearly separating DNA repair cells from the other three types, for most DNA repair genes, impaired prediction for this activity (Figure S3). This targeted experiment, however, suggests that the expression of adequate marker genes can be used to identify whether an activity is present in a given cell with satisfying accuracy.
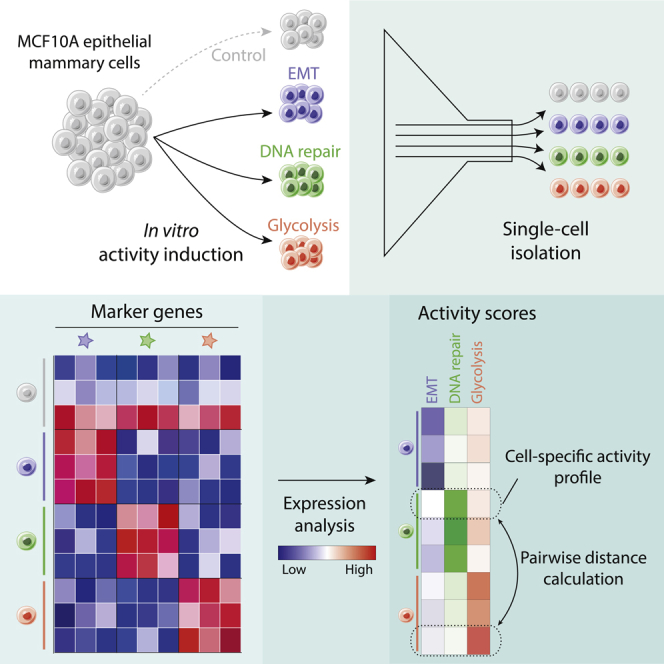
Figure 1.

Detection of Selected Activities Induced In Vitro Using Single-Cell Expression of Targeted Genes
(A) Overall scheme. EMT (blue), DNA repair (green), and glycolysis (red) activities are induced in vitro in MCF10A cells, prior to single-cell analysis and RNA quantification. Targeted marker genes expression is used to assess the likelihood that an activity, considered as a phenotypic trait, is present in a cell. All quantified traits are used to create cell-specific phenotypic profiles and serve as a basis to calculate pairwise cell-cell divergence and overall phenotypic diversity.
(B) Row-normalized single-cell expression for the marker genes of EMT (left), DNA repair (center), and glycolysis (right). Blue: lower expression; red: higher expression. Cells in which the activity was induced are on the left and indicated by colored bars below. Control cells having undergone no induction are on the right and indicated by a gray bar. Significantly differentially expressed genes in bold (p < 0.05, BPglm function).
(C) PFKM marker gene expression in glycolysis and control conditions. Blue curve: number of transcripts in cells in which glycolysis cells was induced; gray curve: control conditions. Confidence intervals around the observed values are used to calculate the probability that a value comes from glycolytic (pglyco, blue) and control (pctrl, gray) conditions. The pglyco/(pglyco + pctrl) ratio gives the likelihood that the observed value comes from a cell in which glycolysis was induced.
(D) Glycolysis prediction in single cells from all four populations: glycolysis (red), EMT (blue), and DNA repair (green) inductions and control (gray). Black and white bar underneath indicates the reported probability of each cell to be glycolytic (log10 scale). Black: missing values.
Whole Transcriptome Cell Activity Scores
Following these targeted in vitro results supporting the feasibility of predicting the activities of single cells, we investigated the relevance of an activity-centered approach to quantify phenotypic diversity in high-throughput patient datasets. In the absence of single-cell inference methods tailored to each of the 50 hallmark cell activities, we used standard tools to investigate the behavior of the related signatures in patient data. We used the AUCell (Aibar et al., 2017) software to score the enrichment of all MSigDB hallmark gene sets in all cells from eight datasets (Fan et al., 2018, Filbin et al., 2018, Li et al., 2017, Neftel et al. 2019, Patel et al., 2014, Tirosh et al., 2016, Tirosh et al., 2016, Venteicher et al., 2017). We normalized these data per set and merged them into a meta-dataset of 50 activity scores per cell in 28,513 cells from different cancer types (see Methods). No major batch effect could be observed as samples did not specifically cluster according to their sets of origin, whereas similar cell types appear to cluster together (Figure 2). However, the most common cell types (T cells, macrophages, and malignant cells) segregated into more than one cluster each. This suggests that cells with similar identity tend to behave similarly across batches and tissues but that different subset of activity profiles could also be observed among cells of identical classification.
Figure 2.

Normalized Activity Scores in the Meta-Dataset
Heatmap of activity scores in the meta-dataset, normalized per activity per set. Dendrograms highlight relationships between activities (left) and cells (top). The dataset of origin of each cell is reported by the bottom color bar. The top row below the score heatmap indicates the dataset of origin of each cell, whereas the bottom one indicates its reported type.
Our analysis, however, revealed extensive redundancy among the 50 activities scored (Figure 3A), suggesting that the signal from the hallmark signatures likely corresponded to fewer than 50 distinct activity-based phenotypic traits. We furthermore assigned cell-cycle phases (G1/S/G2M) to cells using the cyclone software (Scialdone et al., 2015). The cell-cycle phase in which a cell is influences its transcriptome, which can in turn bias cell-type assignment. However, because our approach is cancer oriented and based on cellular activities rather than identities, we considered this information as part of the phenotypic state of a cell and purposely did not correct for it. Cell-cycle phase assignment was found to correlate with the G2M Checkpoint, E2F Targets, and Mitotic Spindle signatures, highlighting that such cycle phase information was indeed taken into account in our phenotypic profiling of cells (Figure S4).
Figure 3.

Principal Component and Clustering Analyses to Circumvent Hallmark Activity Redundancy
(A) Correlation heatmap between all 50 MSigDB hallmark activities on a meta-dataset comprising 28,513 cells from 8 different datasets.
(B) Importance of the 15 Principal Components (PC) for each activity (squared cosine, indicated by increasing circle size and blueness). Below, the proportion of total variance in the dataset explained by each PC.
(C) Relative increase in measure of clustering consensus as the number of clusters is increased. CDF, cumulative distribution function.
(D) Cluster assignment of all 50 activities, for a number of 2–15 clusters.
Redundancy Reduction to Obtain Phenotypic Profiles
We designed two methods to tackle redundancy, based on Principal Component (PC) and clustering analyses (see Methods). The first three PCs of the entire meta-dataset, respectively, explained 25.9%, 12.8%, and 7.7% of the variance in the data, whereas 11 PCs explained more than 2% of the variance (Figure 3B). For the clustering analyses, we investigated the relevance of splitting the data into 2–15 clusters. Using the consensus indices from bootstrapping experiments, we defined an optimal range between 6 and 10 clusters, after which increasing the number of clusters would not improve consensus (Figures 3C and 3D).
We defined phenotypic profiles for each cell based on either the PC scores or the average activity scores per cluster. We analyzed the six sets that provided metadata describing the predicted (sub)type of each cell (see Methods), using leave-one-out procedures to prevent overfitting. In line with our observations that cells clustered according to their type rather than set of origin, defining PC weights and optimal cluster compositions on all sets but the one analyzed still allowed one to identify patterns differentiating cell types (Figures S5–S10).
Cell-Cell Divergence across Tissue and Cancer Types
Pairwise Euclidean distances between phenotypic profiles then served to measure the phenotypic divergence between cells. We used different thresholds to calculate PCA- and cluster-based divergence, respectively, based on the minimum percentage of variance for a PC to be included in phenotypic profiles (0%, 1%, 2%, 3%, and 5%) and on the numbers of clusters to summarize all 50 activities (6–10 clusters). Phenotypic heterogeneity measures were highly correlated regardless of the thresholds in both methods (all Spearman's rho ≥ 0.72, all p < 0.001, Table S5), suggesting they are nearly equivalent. However, we observed less redundancy between PC scores than between cluster scores, independently of the number of clusters (Figure S10). We therefore use PCA-based phenotypic heterogeneity measures hereafter, with a 2% minimum threshold on explained variance for PC inclusion.
We investigated the pan-cancer relevance of our activity-based phenotypic divergence measure, using the six datasets for which cell type metadata were available. We report differences in cell-cell divergence distributions, according to whether two cells are of the same type or not and what that cell type is (Figures 4 and S11). In agreement with our pan-cancer observations that cells clustered by type more than dataset, the divergence between cells of different cell types was always the highest distribution (compared with same-type distributions) in all six datasets. This suggests that our metric will assign smaller divergence scores to cells from the same cell type. Using bootstrapped clustering analyses, we also investigated if different recurrent activity profiles could be observed among cancer cells only, in each set (Figures S12–S16, see Methods). Clusters related to proliferation and immune response could be observed in most analyses, whereas the most discriminant activities, and PC scores derived from them, varied between datasets. In the Venteicher astrocytoma dataset, a discernible sub-population tied to immune activities can be distinguished on the left, with marked differences in interferon alpha and gamma signatures (Figure 5). A separate sub-population with strong proliferation signaling can be observed in the center, whereas cells on the right side do not display particularly strong proliferation or immune-related signal. This suggests that activity-based distances can separate distinct subpopulations of malignant cells presenting different phenotypic characteristics.
Figure 4.

Pan-Cancer Phenotypic Cell-Cell Divergence
Pairwise cell-cell divergence distributions per cell type in each of the six datasets with curated metadata. Inter: inter-type divergence (between cells of different subtypes). All other distributions are between cells of the reported type. Dashed horizontal line: total average; broad horizontal lines: individual distribution averages.
Figure 5.

Isolated Activity Profiles of Significant Clusters of Malignant Cells in the Venteicher et al. Astrocytoma Dataset
Top: distinct significant clusters are identified by alternating black and gray color bars. Cells are ordered left to right according to the overall cluster data including all cells, although only significant clusters of five cells or more are displayed. Middle: Heatmap of PCA-based activity scores. All principal components were used for clustering analyses, but only those explaining >3% of total variance are displayed. PCA scores are ordered top to bottom according to complete hierarchical clustering based on Euclidean distances. Bottom: Heatmap of normalized activity scores, ordered top to bottom according to complete hierarchical clustering based on Euclidean distances.
Phenotypic Diversity Quantification
We further analyzed the relevance of activity-based approaches on two subsets with extended characterization in a large number of patients: 7 non-malignant cell types (T cell, B cell, Macrophage, Endothelial, Fibroblast, NK, Undefined) in 19 patients from the Tirosh melanoma dataset; 6 malignant subtypes (AC-like, OPC-like, MES1-like, MES2-like, NPC1-like, NPC2-like) in 28 patients from the Neftel glioma dataset. The average divergence in a group of cells was used as a surrogate for the group's phenotypic heterogeneity. We observed differences across the average profiles calculated for the distinct cell types, suggesting they are each characterized by specific activity patterns. The differences between the most divergent cells in each category, however, exemplify that individual cells can strongly deviate from these overall profiles (Figures 6A, 6B, and S17). Such variability, possibly due to the stochastic nature of gene expression, would be absent from standard classifying methods.
Figure 6.

Phenotypic Diversity in Populations of Known Structure
(A and B) PCA-based phenotypic profiles of (A) seven non-malignant cell types from the Tirosh et al. melanoma dataset and (B) six glioma subtypes from the Neftel et al. H3K27M-glioma dataset. Average profiles on top were obtained by averaging all cells from a given subtype across all patients. The outlier profiles at the bottom were obtained from the same-type cell pairs displaying the highest activity-based divergence for each cell type. Only the first five principal components are shown.
(C) Barplots showing the breakdown of how non-malignant cells from melanoma samples would be re-categorized, based on the average activity profiles of each category in the Tirosh melanoma dataset.
(D) Barplots showing the breakdown of how malignant glioblastoma cells would be re-categorized, based on the average activity profiles of each category in the Neftel dataset.
(E) Relationship between mean phenotypic divergence between non-malignant cells in the melanoma dataset and the Simpson diversity index calculated on the repartition of cells into the seven non-malignant classes.
(F) Relationship between mean phenotypic divergence between malignant cells in the glioma dataset and the Simpson diversity index calculated on the classification of cells into the six glioma subtypes. Black lines: linear models.
We proceeded to reclassify all cells according to the smallest Euclidean distance between their PCA-based profiles and the average profiles of each classification in both datasets. We observed a stronger concordance (p = 0.022, Wilcoxon test) when reclassifying cells from established normal cell types in melanoma samples according to their activities (Figure 6C, 82% ± 14 correctly reclassified samples), compared with subtypes of malignant glioma cells (Figure 6D, 54% ± 23). This confirmed that cells of similar type tend to partake in similar activities. However, in the glioma samples we analyzed, the differences between marker-based malignant subtypes were not as closely reflected by activity profiles as was observed in normal cell types.
We then computed the standard Simpson diversity index on a per-patient basis, according to the repartition of all cells from a patient into the relevant categories in both subsets. We found that it correlated very significantly with our divergence-based phenotypic heterogeneity score in both non-malignant cells from melanoma samples and malignant glioma cells (Figures 6E and 6F, Spearman's rho = 0.73 and rho = 0.49; p = 0.001 and p = 0.009, respectively). This suggests that this approach, although not relying on cell classification, can accurately capture the diversity of populations whose structure is known, both for malignant and normal cells from different tissues. Similar observations were reported using cluster-based distances (Figure S18).
Using the average activity-based divergence between malignant cells, we quantified intra-tumor phenotypic heterogeneity in all samples from the six datasets with metadata and compared them (Figure 7A). The mean phenotypic divergence of colorectal cancers (Li et al.) was significantly higher than other datasets, whereas melanoma heterogeneity was significantly lower (Wilcoxon test, Benjamini-Hochberg (BH) correction, p < 0.001 and p = 0.004, respectively). We furthermore report that between-samples variation in phenotypic diversity was the highest in melanoma (i.e., most heterogeneous in heterogeneity levels) and the lowest in oligodendroglioma (Figures 7B and 7C).
Figure 7.

Differences and Dynamics of Phenotypic Diversity
(A) Distribution of phenotypic divergence between malignant cells in each sample across six datasets. Samples ordered by sample-wise phenotypic diversity (average divergence). ∗∗∗: p < 0.001; ∗: p < 0.05 (Wilcoxon test, BH correction). Boxes represent the middle quartiles; black horizontal bars represent the median of each distribution; whiskers extend up to 1.5 times the interquartile range (box height) away from the box. Outliers (beyond the whiskers) are not displayed.
(B) Per-sample phenotypic diversity in all six sets.
(C) Coefficient of variation in phenotypic diversity across samples in each set.
(D) Phenotypic divergence distributions in normal and cancerous epithelia in five patients from the Li et al. dataset. Dashed horizontal line: total average; broad horizontal lines: individual distribution averages.
Phenotypic Diversity Evolution
We finally took advantage of cancer samples paired with normal tissue in the colorectal dataset to investigate the evolution of phenotypic diversity. In the five patients with colorectal cancer from Li et al. for which we could find paired tumor-normal data, diversity stayed at similar levels in three cases (CRC04, CRC06, CRC10), whereas it decreased very significantly in the tumor material in two cases (Figure 7D, CRC05, CRC08, p < 0.001, Wilcoxon test). Such decrease in diversity was not observed in other cell types in these patients (Figure S19). This fits a scenario in which cells go through a phenotypic bottleneck at tumor initiation, followed by the expansion of few selected clones.
Discussion
Better understanding the dynamics of intra-tumor heterogeneity will help tailor better therapeutics to control and funnel cancer evolution. During malignant somatic evolution, cells drift away from their well-characterized normal ancestors by following trajectories unique to each patient (Tokutomi et al., 2019), whereas there is convergence across patients to (de)activate the necessary cellular activities (Hanahan and Weinberg, 2000, Hanahan and Weinberg, 2011). Consequently, we investigated the relevance of focusing on what cancer cells do, rather than what they are, to measure phenotypic diversity in the cancer context. We considered cellular activities as traits describing the phenotypic state of cells and used pairwise distances to quantify cell-cell divergence and overall diversity. Unlike many existing methods (Almendro et al., 2014, Ferrall-Fairbanks et al., 2019, Zhang et al., 2019), such an approach does not rely on classifying cells into putative, static identities that cancer cells drift away from in patient-specific fashion. It furthermore encompasses the temporal variability inherent to populations of cells replicating asynchronously and exhibiting stochastic differences in gene expression, which can itself foster resistance (Shaffer et al., 2017). In addition, such a method is not tissue-type specific and was relevant in all investigated datasets.
We first performed in vitro analyses, which revealed that it was possible to reliably predict in which cells a given activity had been induced, using targeted panels based on the MSigDB hallmark gene sets and the literature. This was done using single-cell RT-qPCR technology, which is more precise than RNA-seq on specific genes of interest (Mojtahedi et al., 2016). Our analysis, however, revealed that some of the best markers for activity detection were absent from the hallmark gene sets. Although this is likely to be attenuated when using entire gene sets rather than targeted panels, it exemplifies the need for more reliable gene signatures, particularly ones taking into account single-cell level specificities (Hwang et al., 2018, Larsson et al., 2019).
We then scored 50 hallmark activity signatures in 28,513 cells from eight publicly available datasets using the AUCell software. AUCell is based on a ranking procedure, which efficiently deals with normalization and is not affected by the dissimilarity in using either FPKM or TPM units across the datasets (Aibar et al., 2017). This was illustrated by cells not clustering according to their dataset of origin in the meta-dataset. “Dropouts” occurring when transcripts are not captured before sequencing can, however, affect ranking in low-expressed genes (Davis-Marcisak et al., 2019). Gene set enrichment analyses, in which multiple genes can contribute to the overall enrichment signal for an activity in each cell, are, however, less affected by dropouts than gene-specific differential expression analyses.
We reported high redundancy among the 50 activities scored, which we addressed by using PC and clustering analyses. We found that both methods were by and large equivalent. Importantly, hallmark activities do not focus on lineage-specific markers. Using their output, which summarizes multiple genes, is thus less likely to separate cells according to the expression of few highly discriminating lineage markers, such as can occur when focusing on the entire transcriptome. This is particularly relevant for cancer cells that broke free of homeostatic control and differentiation hierarchies, in which lineage markers inherited from ancestors may no longer correlate with phenotype and behavior.
We applied such an activity-based approach to investigate the divergence between and among cell types in six datasets with available metadata. We found that cells of the same type were less divergent than cells of different types. This can be explained by the fact that most reported cell types are non-malignant, with cells from the same type thus likely to partake in similar activities. We also observed that activity profiles recapitulated normal cell types better than malignant subtypes, although with very limited data (n = 1 in both cases). Furthermore, we could identify distinct clusters of malignant cells showing marked differences in their activity profiles in all datasets. Therefore, although same cell identity often implied similar activities, it was not always the case, especially in cancer cells for which our activity-based approach was aimed. This also indicated that this approach could reflect the divergence between cells similarly considered malignant using a blanket classification, but which appeared to engage in different activities.
Interestingly, the divergence between malignant cells was not recurrently higher or lower than that between normal subtypes, and patterns varied according to tumor type. In the two datasets with high numbers of both patients and cell type sub-classifications, the mean cell-cell divergence correlated significantly with standard diversity indices based on the repartition of individuals into subpopulations. These results suggest that avoiding the use of known lineage markers did not hamper the relevance of this approach across the investigated tissue types. Although we used a leave-one-out design to avoid overfitting, it is, however, worth noticing that brain cell and tumor data are likely overrepresented in this study. Finally, using this approach on five patients with paired tumor-normal data suggested the existence of evolutionary bottlenecks on phenotypic diversity at tumor initiation. This would be in agreement with the genetic diversity decrease observed at this stage in orthogonal studies (Cross et al., 2020).
In this work, we focused on the quantification of phenotypic diversity according to cancer's atavistic evolutionary nature, as cells deviate from normal healthy cell types and regress toward ancestral unicellular growth (Davies and Lineweaver, 2011). We used single-cell expression analyses to quantify activity-based traits for each cell to create individual phenotypic profiles differing from static subtype classifications. This provides an alternative to marker-based methods, which can rely on markers not relevant anymore in the cancer context and that often cannot allow quantification of the differences between cells classified similarly. Not relying on markers furthermore bypasses tissue specificity and provides a universal approach applicable to all tumor types.
Limitations of the Study
In this study we used pre-defined activity signatures based on bulk data that were not specifically designed for relevance in cancer studies. More work is therefore needed to provide standardized tools to reproducibly measure phenotypic ITH from single-cell RNA data. The development of accurate single-cell-specific expression signatures for the most recurrently dysregulated pathways in cancer would provide enhanced precision to build per-cell phenotypic profiles. This will require determination of the most relevant activities that contribute to the convergence toward the “cancer hallmarks” (Hanahan and Weinberg, 2011) dysregulation common to most cancer types. It will also be necessary to reliably assess their predictability in single cells, taking into account the specificity of single cell expression data and design methods accounting for the redundancy among them. Finally, it will also be critical to understand how intra-tumor heterogeneity at single-cell level can be extrapolated from bulk samples, how this reflects inter-patient heterogeneity, and how it ties to genetic and clinical features.
Successful implementations will improve future similar activity-based approaches to quantify phenotypic diversity in the evolutionary context of cancer. This will in turn allow one to better monitor the evolution of phenotypic diversity over time and space and facilitate the identification of therapeutic opportunities to control intra-tumor heterogeneity. This would ultimately help thwart the emergence of resistant populations and thereby enhance clinical outcomes.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors wish to thank Anne-Pierre Morel and Christelle Chassot for valuable discussions and assistance on the experimental setup.
Author Contributions
L.M. and F.F. performed in vitro experiments. F.F., A.V., A.P., and P.M. designed in vitro experiments. L.D.S., B.L., and P.M. performed bioinformatics analyses. A.V., A.P., and P.M. supervised the work. P.M. wrote the manuscript. All authors revised the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101061.
Data and Code Availability
The R scripts and data for this project are available on github: https://github.com/pierremartinez/PhDiv.
Supplemental Information
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almendro V., Kim H.J., Cheng Y.K., Gönen M., Itzkovitz S., Argani P., van Oudenaarden A., Sukumar S., Michor F., Polyak K. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014;74:1338–1348. doi: 10.1158/0008-5472.CAN-13-2357-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor N., Harness J.V., Müller S., Mewes H.W., Petritsch C. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics. 2014;30:50–60. doi: 10.1093/bioinformatics/btt622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F., Ng C.K.Y., Patsouris A., Droin N., Piscuoglio S., Carbuccia N., Soria J.C., Dien A.T., Adnani Y., Kamal M. Genomic characterization of metastatic breast cancers. Nature. 2019;569:560–564. doi: 10.1038/s41586-019-1056-z. [DOI] [PubMed] [Google Scholar]
- Chang K., Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross W. Stabilising selection causes grossly altered but stable karyotypes in metastatic colorectal cancer. bioRxiv. 2020:2020. doi: 10.1101/2020.03.26.007138. [DOI] [Google Scholar]
- Davies P.C.W., Lineweaver C.H. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys. Biol. 2011;8:015001. doi: 10.1088/1478-3975/8/1/015001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Marcisak E.F. Differential variation analysis enables detection of tumor heterogeneity using single-cell RNA-sequencing data. Cancer Res. 2019 doi: 10.1158/0008-5472.can-18-3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Lee H.O., Lee S., Ryu D.E., Lee S., Xue C., Kim S.J., Kim K., Barkas N., Park P.J., Park W.Y., Kharchenko P.V. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 2018;28:1217–1227. doi: 10.1101/gr.228080.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrall-Fairbanks M.C., Ball M., Padron E., Altrock P.M. Leveraging single-cell RNA sequencing experiments to model intratumor heterogeneity, JCO clinical cancer informatics. Am. Soc. Clin. Oncol. 2019;3:1–10. doi: 10.1200/CCI.18.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin M.G., Tirosh I., Hovestadt V., Shaw M.L., Escalante L.E., Mathewson N.D., Neftel C., Frank N., Pelton K., Hebert C.M. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360:331–335. doi: 10.1126/science.aao4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Vázquez-García I., Illingworth C.J.R., Mustonen V. High-definition reconstruction of clonal composition in cancer. Cell Rep. 2014;7:1740–1752. doi: 10.1016/j.celrep.2014.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby R.A., Brown J. Mutations, evolution and the central role of a self-defined fitness function in the initiation and progression of cancer. Biochim. Biophys. Acta. 2017 doi: 10.1016/j.bbcan.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M., Rowan A.J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M., Maley C.C. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. http://www.ncbi.nlm.nih.gov/pubmed/10647931 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hinohara K., Wu H.J., Vigneau S., McDonald T.O., Igarashi K.J., Yamamoto K.N., Madsen T., Fassl A., Egri S.B., Papanastasiou M. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. 2018;34:939–953.e9. doi: 10.1016/j.ccell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B., Lee J.H., Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018;50:96. doi: 10.1038/s12276-018-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A.J.M., Johnsson P., Hagemann-Jensen M., Hartmanis L., Faridani O.R., Reinius B., Segerstolpe Å., Rivera C.M., Ren B., Sandberg R. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565:251–254. doi: 10.1038/s41586-018-0836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lässig M., Mustonen V., Walczak A.M. Predicting evolution. Nat. Ecol. Evol. 2017;1 doi: 10.1038/s41559-017-0077. [DOI] [PubMed] [Google Scholar]
- Martinez P., Timmer M.R., Lau C.T., Calpe S., Sancho-Serra Mdel C., Straub D., Baker A.M., Meijer S.L., Kate F.J., Mallant-Hent R.C. Dynamic clonal equilibrium and predetermined cancer risk in Barretts oesophagus. Nat. Commun. 2016;7:12158. doi: 10.1038/ncomms12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Courtois E.T., Sengupta D., Tan Y., Chen K.H., Goh J.J.L., Kong S.L., Chua C., Hon L.K., Tan W.S. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017;49:708–718. doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- Maley C.C., Galipeau P.C., Finley J.C., Wongsurawat V.J., Li X., Sanchez C.A., Paulson T.G., Blount P.L., Risques R.A., Rabinovitch P.S. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- Martinez P., Kimberley C., BirkBak N.J., Marquard A., Szallasi Z., Graham T.A. Quantification of within-sample genetic heterogeneity from SNP-array data. Sci. Rep. 2017;7:3248. doi: 10.1038/s41598-017-03496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N., Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Mojtahedi M., Skupin A., Zhou J., Castaño I.G., Leong-Quong R.Y., Chang H., Trachana K., Giuliani A., Huang S. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016;14:e2000640.. doi: 10.1371/journal.pbio.2000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q.H., Pervolarakis N., Blake K., Ma D., Davis R.T., James N., Phung A.T., Willey E., Kumar R., Jabart E. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018;9:2028. doi: 10.1038/s41467-018-04334-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S., Van Loo P., Wedge D.C., Alexandrov L.B., Greenman C.D., Lau K.W., Raine K., Jones D., Marshall J., Ramakrishna M. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell P.C. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. http://www.ncbi.nlm.nih.gov/pubmed/959840 [DOI] [PubMed] [Google Scholar]
- Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialdone A., Natarajan K.N., Saraiva L.R., Proserpio V., Teichmann S.A., Stegle O., Marioni J.C., Buettner F. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods. 2015;85:54–61. doi: 10.1016/j.ymeth.2015.06.021. [DOI] [PubMed] [Google Scholar]
- Shaffer S.M., Dunagin M.C., Torborg S.R., Torre E.A., Emert B., Krepler C., Beqiri M., Sproesser K., Brafford P.A., Xiao M. Reprogramming as a mode of cancer drug resistance. Nat. Publ. Group. 2017;546:431–435. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Venteicher A.S., Hebert C., Escalante L.E., Patel A.P., Yizhak K., Fisher J.M., Rodman C., Mount C., Filbin M.G. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokutomi N., Moyret-Lalle C., Puisieux A., Sugano S., Martinez P. Quantifying local malignant adaptation in tissue-specific evolutionary trajectories by harnessing cancers repeatability at the genetic level. Evol. Appl. 2019;12:1062–1075. doi: 10.1111/eva.12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigos A.S., Pearson R.B., Papenfuss A.T., Goode D.L. How the evolution of multicellularity set the stage for cancer. Br. J. Cancer. 2018;118:145–152. doi: 10.1038/bjc.2017.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher A.S., Tirosh I., Hebert C., Yizhak K., Neftel C., Filbin M.G., Hovestadt V., Escalante L.E., Shaw M.L., Rodman C. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355:eaai8478. doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.J., Werner B., Heide T., Curtis C., Barnes C.P., Sottoriva A., Graham T.A. Quantification of subclonal selection in cancer from bulk sequencing data. Nat. Genet. 2018;50:895–903. doi: 10.1038/s41588-018-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.W., O'Flanagan C., Chavez E.A., Lim J.L.P., Ceglia N., McPherson A., Wiens M., Walters P., Chan T., Hewitson B. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods. 2019:1–9. doi: 10.1038/s41592-019-0529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R scripts and data for this project are available on github: https://github.com/pierremartinez/PhDiv.