

Abstract
Background/Objectives:
Interplay between the Hedgehog (HH) and epidermal growth factor receptor (EGFR) pathways modulating the outcome of their signaling activity have been reported in various cancers including pancreatic ductal adenocarcinoma (PDAC). Therefore, simultaneous targeting of these pathways may be clinically beneficial. This Phase I study combined HH and EGFR inhibition in metastatic PDAC patients.
Methods:
Combined effects of HH and EGFR inhibition using Vismodegib and Erlotinib with or without gemcitabine in metastatic solid tumors were assessed by CT. Another cohort of patients with metastatic PDAC was evaluated by FDG-PET and tumor biopsies-derived biomarkers.
Results:
Treatment was well tolerated with the maximum tolerated dose cohort experiencing no grade 4 toxicities though 25% experienced grade 3 adverse effects. Recommended phase II dose of Vismodegib and Erlotinib were each 150 mg daily. No tumor responses were observed although 16 patients achieved stable disease for 2-7 cycles. Paired biopsy analysis before and after first cycle of therapy in PDAC patients showed reduced GLI1 mRNA, phospho-GLI1 and associated HH target genes in all cases. However, only half of the cases showed reduced levels of desmoplasia or changes in fibroblast markers. Most patients had decreased phospho-EGFR levels.
Conclusions:
Vismodegib and Erlotinib combination was well-tolerated although overall outcome in patients with metastatic PDAC was not significantly impacted by combination treatment. Biomarker analysis suggests direct targets inhibition without significantly affecting the stromal compartment. These findings conflict with pre-clinical mouse models, and thus warrant further investigation into how upstream inhibition of these pathways is circumvented in PDAC.
Keywords: Pancreatic cancer, Hedgehog, EGFR, GLI1, Erlotinib, Vismodegib
Introduction
Pancreatic ductal adenocarcinoma (PDAC), the most frequent and aggressive form of pancreatic cancer, is a devastating disease with an overall 5-year survival rate less than 8% (1). It is the third leading cause of death by cancer in the United States and predicted to be 2nd by 2030 (2). Surgical resection represents the best option for curative treatment, but is only available in 10% of cases as PDAC is typically locally advanced (50%) or metastatic (40%) at diagnosis (3). Furthermore, PDAC has significant resistance to traditional radiation and chemotherapeutic modalities. Gemcitabine, a nucleoside analog, was approved for use in 1996 based on a randomized trial demonstrating improvement in survival over 5-fluorouracil (4). Several gemcitabine-based combinations failed to confer survival benefit maintaining monotherapy as the cornerstone for the next decade despite median survival of only about six months (5–9). In 2011, FOLFIRINOX supplanted gemcitabine as standard of care given improved median overall survival (11.1 vs 6.8 months) (10). More recently, addition of nab-paclitaxel to gemcitabine also improved survival over single agent therapy (11). However, due to toxicity profiles of these regimens, their utilization remains restricted to patients with good performance status. Thus, there is an unmet need for novel therapeutics for patients with advanced PDAC.
Elucidation of PDAC molecular pathogenesis prompted development of targeted therapies including the blockade of the Hedgehog (HH) signaling. The canonical HH pathway regulated by the transmembrane proteins Smoothened (SMO) and Patched (PTCH1) has been implicated in PDAC development through activation of oncogenic transcription factors (the glioma-associated oncogenes (GLI)). One or more genes in the HH pathway are typically dysregulated in PDAC. HH ligands are frequently overexpressed, and growth and survival rely on sustained HH signaling with GLI1 expression associated with PDAC development and worse outcomes (12–18). Therefore, the HH signaling axis represents an attractive therapeutic target although clinical translation has proven complicated since clinical trials with SMO inhibitors failed to confer survival benefit (19,20).
Such shortcomings are explained in part by signaling crosstalk conferring HH ligand-independent regulation of GLI transcription factors. For example, extensive evidence supports GLI1 activation downstream of PI3K/AKT/mTOR (21–25) and MEK/ERK signaling (26–28) with the common upstream driver being epidermal growth factor receptor (EGFR). Interestingly, EGFR signaling is known to promote PDAC proliferation through activation of GLI1 (23,29–34). Furthermore, an increase in EGFR expression in PDAC is associated with a more aggressive phenotype and poorer prognosis (35,36). This was the rationale for a phase III study. Gemcitabine in combination with EGFR tyrosine kinase inhibitor, Erlotinib, was superior to gemcitabine alone, although benefit was minimal (37). Not only can EGFR signaling confer resistance to pharmacologic SMO abrogation, but HH signaling has been identified as a resistance mechanism to anti-EGFR therapy in head and neck cancer and lung cancer (38,39). Furthermore, concurrent HH and EGFR inhibition demonstrates synergistic effects while individual inhibition in preclinical and clinical studies frequently resulted in resistance (27,40–41). Further, there is also evidence for a role of HH and EGFR signaling crosstalk in modulating the stromal compartment of PDAC. Secreted by PDAC cells, HH ligand acts on fibroblasts to activate GLI1-dependent transcription (42–44). GLI1 activity in fibroblasts promotes IL-6 secretion, which feeds back to promote tumor growth (16). While HH inhibitors decrease activated stromal cells and desmoplasia with associated enhanced gemcitabine exposure in preclinical models (45,46), genetic deletion of HH ligand causes not only stromal depletion, but also an aggressive PDAC phenotype (47). However, there is evidence that the type and level of GLI1 activity in fibroblasts may also be at least partially regulated on MAPK/ERK signaling (48), which could occur as a result of EGFR activation. Taken together, combinatorial inhibition of HH and EGFR signaling represents a logical therapeutic approach for PDAC but has never been tested in a clinical setting.
Targeting both the HH and EGFR pathways is clinically feasible as small molecule inhibitors directed against these pathways are currently available. Vismodegib is an orally bioavailable first-in-class small-molecule antagonist of the HH pathway which binds to and inhibits the HH receptor SMO. Erlotinib is an orally active quinazoline small molecule that acts as a selective, reversible EGFR tyrosine kinase inhibitor. Thus, we suspect gemcitabine and erlotinib will likely act on the tumor cells while Vismodegib cooperates by impacting the stromal fibroblasts. Therefore, a phase I trial was initiated to evaluate the safety and adverse-effect profile of the combination of Vismodegib and Erlotinib with and without gemcitabine in patients with advanced solid tumors. The primary goals of this trial were to determine the maximally tolerated dose (MTD) of the combination of Vismodegib and Erlotinib (cohort I), as well as the MTD of Vismodegib, Erlotinib and gemcitabine (cohort II) in patients with unresectable solid tumors. The effect of Erlotinib and Vismodegib on select biomarkers and FDG PET was assessed in patients with metastatic pancreatic cancer in cohort III with a primary endpoint of dose limiting toxicity analysis. Secondary goals were to describe the adverse event profile and patient response to each combination. To further explore the molecular consequences of this therapy, we assessed alterations in biomarkers associated with these pathways in tumor tissue samples obtained before and after one month of therapy.
Materials and Methods
Patient Selection
Patients were eligible for inclusion in cohorts I and II if they had histologic proof of unresectable solid tumors. Patients were eligible for inclusion in cohort III if they had unresectable PDAC not amenable to any other standard therapies or if the patient refused standard therapy. Common inclusion criteria were age ≥18 years; life expectancy ≥12 weeks; ECOG performance status ≤2; adequate hematologic (hemoglobin ≥9.0 g/dL, ANC ≥ 1.5 x 109 cells/L, and platelets ≥ 100 x 109 cells/L), hepatic (total bilirubin ≤ upper limit of normal [ULN], AST ≤ 3 x ULN) and renal function (creatinine ≤1.5 x ULN); no chemotherapy, immunotherapy, biologic or radiation therapy within 4 weeks prior to study entry; and no mitomycin C or nitrosourea chemotherapy within 6 weeks prior to study entry. Patients were deemed ineligible in the event of other, potentially curative or life extending therapeutic options. Also excluded were patients who had received radiation therapy to > 25% of their bone marrow; uncontrolled concurrent illness; seizure disorders; or central nervous system metastases if not stable for at least 2-3 months. The study was approved by the Institutional Review Board, and all patients provided written informed consent under federal and institutional guidelines.
Compounds and materials
Vismodegib (GDC-0449) is a first-in-class small-molecular antagonist of the HH receptor developed by Genentech. Erlotinib (OSI-774; Tarceva) is an orally active quinazoline small molecule that acts as a selective, reversible EGFR tyrosine kinase inhibitor also developed by Genentech.
Pretreatment and Follow-up Evaluation:
Complete patient histories, physical examination, performance status, toxicity assessment, complete blood counts (CBCs), serum electrolytes, chemistries and CA-19-9 were obtained within 7 days of study registration and prior to each treatment cycle. Electrocardiogram, PT/INR and serum pregnancy test in women of childbearing potential were also obtained at baseline. CBCs were obtained weekly. Evaluation of indicator lesion (computed axial tomographic scans or magnetic resonance imaging, etc) for patients with measurable disease was performed at baseline. Each participant in cohort III underwent baseline biopsy of metastasis within 7 days of therapy. Baseline FDG-PET was performed on day of biopsy.
MTD Study Design and Treatments:
Patients in cohort I received a fixed Vismodegib dose combined with increasing doses of Erlotinib (Table 1A). Patients in cohort II received the same fixed dose of Vismodegib but escalating doses of Erlotinib and gemcitabine (Table 1B). A third cohort, started after the recommended phase 2 doses of Vismodegib and Erlotinib were defined (150 mg for each drug daily), consisted of patients with metastatic pancreatic cancer and is described in further detail below.
Table 1A—
Cohort I dose escalation (all cycles)
| Dose levels | Erlotinib (mg/day)* | Vismodegib (mg/day) |
|---|---|---|
| −1 | 25 | 150 |
| 1 | 50 | 150 |
| 2 | 75 | 150 |
| 3 | 100 | 150 |
| 4** | 150 | 150 |
As assigned by the Registration Office upon enrollment
Dose level used in cohort III
Cycle length = 28 days; for all cycles, Erlotinib and Vismodegib taken orally on days 1-28.
Erlotinib and Vismodegib taken on an empty stomach (1 hour prior to or 2 hours after eating)
Table 1B—
Cohort II dose escalation (all cycles)
| Dose level | Erlotinib (mg/day)* | Vismodegib (mg/day) | Gemcitabine (mg/m2/day)* |
|---|---|---|---|
| −2 | 75 | 150 | 600 |
| −1 | 75 | 150 | 750 |
| 1 | 75 | 150 | 1000 |
| 2 | 100 | 150 | 750 |
| 3 | 100 | 150 | 1000 |
As assigned by the Registration office upon enrollment
Cycle 1 length = 28 days
Erlotinib and Vismodegib taken orally daily throughout each cycle; gemcitabine administered on days 1, 8 and 15, followed by 1 week of rest
The cohorts-of-three phase I design was used for dose escalation to assess the MTD of each combination. The dose escalation schemes are outlined in tables 1A and 1B. Erlotinib and Vismodegib were given orally once daily for the 28 days of each cycle in each cohort. In the three-drug cohort, the initial protocol design included intravenous administration of gemcitabine on days 1, 8, 15, 22, 29, 36 and 43, followed by a one week rest for cycle 1, then on days 1, 8 and 15 in all subsequent cycles. Due to significant hematologic toxicities with this gemcitabine schedule, the protocol was later amended and the length of cycle 1 in cohort II was reduced to 28 days. Gemcitabine was subsequently administered on days 1, 8 and 15, followed by one week of rest for all cycles.
All toxicities were graded according to the National Cancer Institute Common Toxicity Criteria (version 3.0). Dose-limiting toxicity (DLT) was defined as an adverse event attributed (definitely, probably or possibly) to the study treatment during cycle 1 and meeting established hematologic, renal and other non-hematologic criteria. Dose-limiting hematologic criteria were grade 4 neutropenia lasting 5 or more days, febrile neutropenia, grade 4 anemia, or platelet count ≤ 25 x 109/L. Serum creatinine ≥ 2 times baseline or ≥ 2 x upper limit of normal if the patient’s baseline was less than the upper limit of baseline were the dose-limiting renal toxicities. Other non-hematologic toxicities were considered dose-limiting if ≥ grade 3, including nausea, vomiting or diarrhea with maximum supportive treatments. However, alopecia and weight loss were not considered DLTs. The following two events were also considered DLTs: two straight dose reductions occurring during cycle 1, and persistent toxicities that resulted in dose delays of > 14 days after cycle 1 (even if they are < grade 3 in severity). The MTD was defined as the dose below the lowest dose that induced DLT in at least one-third of patients (at least 2 of a maximum of 6 new patients). Three patients were initially treated at a given dose level combination and assessed for at least 4 weeks in cohort I and 8 weeks in cohort II (4 weeks following protocol amendment, as noted previously) from start of treatment to assess for dose-limiting toxicity (DLT). The next three patients were accrued and placed on either: i) one dose level higher (if DLT was not observed in any of the 3 patients); ii) one dose level lower (if DLT was seen in 2 or 3 of 3 patients); or iii) the same dose level (if DLT was seen in 1 of 3 patients). For the latter group, if DLT was not seen in any of the three additional patients, 3 new patients were accrued and treated at the next higher dose level. If DLT was seen in at least one of these three additional patients (≥2 of 6), the MTD would have been exceeded and further accrual to this dose level would have ceased. The MTD would then be defined as the previous dose, unless only 3 patients were treated at that lower dose level. In that case, 3 additional patients would have been treated to result in a total of 6 patients being treated at the MTD to more fully assess the toxicities associated with the MTD.
Supportive care was administered as necessary, Routine or prophylactic use of colony-stimulation factors (G-CSF or GM-CSF) was not allowed, though therapeutic use in patients with serious neutropenic complications was allowed at the physician’s discretion.
Follow-up Evaluation:
Follow-up evaluation in participants in cohorts I and II was performed by CT imaging prior to every other cycle (every 8 weeks) to assess tumor response. Each patient in cohort III underwent repeat biopsy of the same metastatic lesion on day 28 +/− 3 days of initiation of therapy. FDG-PET was performed on patients in cohort III to assess metabolic effect on days biopsy was obtained. All toxicities were graded according to the National Cancer Institute Common Toxicity Criteria (version 3.0). Response to therapy was determined using RECIST (1.1).
FDG-PET Analysis
Standard uptake values (SUV) were calculated for each lesion noted for each patient. For each paired data point a change in the SUV from baseline was calculated. To account for patients with multiple lesions, the paired data were analyzed 2 ways. For each patient, the pair that led to the biggest change from baseline was used. In addition, for each patient the mean change was used. For each analysis a Student’s t-test was performed to test whether the mean of these changes was equal to 0. The alternative hypothesis was that the mean change was not equal to 0.
Biomarker Studies:
Up to four core needle biopsies were cut into thirds, with the 1/3 from each end placed in 10% neutral buffered formalin (Sigma, St. Louis, MO) and the center 1/3 of the sample flash frozen in liquid nitrogen or placed in RNAlater (QIAGEN, Gaithersburg, MD) solution. The formalin-fixed samples were paraffin embedded, and sections of 4-6 microns in thickness were cut to charged slides. The first and the last sections cut were stained with H&E and were evaluated by a pathologist to assess tissue quality, tumor characteristics, and extent of the tumor. All sections in between were reserved for immunohistochemical (IHC) analysis of select cell signaling pathway markers. IHC evaluation of AKT, Phospho-AKT, ERK, Phospho-ERK (Cell Signaling, Danvers, MA); alpha-SMA, vimentin, EGFR (Dako); and Phospho-EGFR (Epitomics 1727-1, Burlingame, CA) was performed on a Bond (Leica, Buffalo Grove, IL) automated staining unit by the Cancer Center Research Pathology Core at Mayo Clinic, Rochester, Minnesota. Briefly, tissue sections were deparaffinized in xylene, dipped into decreasing concentrations of ethyl alcohol, and rehydrated in distilled water. P-GLI1-S84 IHC was performed as previously described (28). Antibody against phospho-S84-GLI1 was produced using the synthetic phosphorylated peptide KLTKKRALpSISPLSDA (Peptide 2.0 Inc, Chantilly, VA) as antigen and purified on a phosphopeptide column (EZBiolab Inc, Carmel, IN). The EGFR IHC was performed using PharmDx kit (#K1494, Dako, Carpinteria, CA) according to kit instructions on a Bond autostainer. Positive and negative control cell pellets provided with this kit were also stained. All IHC specimens were reviewed by a pathologist and scored on the basis of cells stained, nuclear and/or cytoplasmic staining, membranous staining, extent of cells stained, and the intensity of staining (weak, 1+, 2+, or 3+). For paired biopsy samples, H-scores ranging from 0-300 for each sample were calculated using the following algorithm: 1x(%cells staining 1+) + 2x(%cells staining 2+) + 3x(%cells staining 3+).
Tissue that was preserved for RNA evaluation was stored at −80°C under monitored conditions and kept frozen until extraction of the RNA. RNA was isolated from the tissues that were flash frozen via Trizol (Invitrogen, Grand Island, NY) extraction and purification via QIAcube (QIAGEN) by the Biospecimens Accessioning and Processing Core. Tissues that were preserved in RNAlater were treated with 500 μl Trizol and subjected to homogenization in 30 seconds on and 30 seconds off cycles using a rotor-stator homogenizer (PRO Scientific, Oxford, CT) for 10 minutes. Tissue was maintained on ice throughout homogenization. The homogenized samples were then subjected to RNA isolation and purification using the RNeasy Mini Protocol (QIAGEN) exactly as described following the homogenization step. RNA was eluted with 30 ul of RNase-free water and the concentration was measured using a Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA).
RNA levels of pathway biomarkers were evaluated using quantitative reverse-transcription polymerase chain reaction (qRT-PCR). 500 ng of RNA isolated from the patient tissue samples was used as input for the cDNA reaction using the High-Capacity cDNA kit (Applied Biosystems, Grand Island, NY) and a DNA Engine Peltier thermocycler (Bio-Rad, Hercules, CA). Quantitative PCR was performed from the cDNA using Taqman pre-developed assays and master mix (Applied Biosystems) for GLI1 (Hs_01110766), PTCH1 (Hs_00181117), SHH (Hs_01123832), COL1A1 (Hs_00164004), and BCL2 (Hs_00608023). PCR was performed on the ABI 7900HT or ViiA7 systems. Results were evaluated against two separate housekeeping genes (18S and UBC) to ensure consistency of results. Changes were calculated using the ΔΔCT method with normalization to the housekeeping gene.
Results
Patient characteristics
A total of 69 patients were enrolled in this study, 15 to cohort I, 29 to cohort II and 25 to cohort III. Basic patient demographics are outlined in Table 2. It should be noted that most major tumor groups were represented in cohorts I and II. The median age was 62 years and there were 25 males and 44 females. The majority (56.5%) had a performance score of 1.
Table 2—
Patient Characteristics
| Variable | Overall | Cohort I | Cohort II | Cohort III | |
|---|---|---|---|---|---|
| Age, median (range) | 62 (37,83) | 57 (44,78) | 61 (37,83) | 68 (40,81) | |
| Gender | Female | 44 (63.8%) | 10 (66.7%) | 20 (69%) | 14 (56%) |
| Male | 25 (36.2%) | 5 (33.3%) | 9 (31%) | 11 (44%) | |
| Race | White | 68 (98.6%) | 15 (100%) | 28 (96.6%) | 25 (100%) |
| American Indian or Alaska Native | 1 (1.4%) | 1 (3.4%) | |||
| Performance Score | 0 | 26 (37.7%) | 5 (33.3%) | 13 (44.8%) | 8 (32%) |
| 1 | 39 (56.5%) | 9 (60%) | 13 (44.8%) | 17 (68%) | |
| 2 | 4 (5.8%) | 1 (6.7%) | 3 (10.3%) | ||
| Prior Treatments | Chemotherapy | 58 (84.1%) | 15 (100%) | 25 (86.2%) | 18 (72%) |
| Radiation Therapy | 30 (43.5%) | 7 (46.7%) | 13 (44.8%) | 10 (40%) | |
| Surgery | 67 (97.1%) | 14 (93.3%) | 29 (100%) | 24 (96%) | |
Adverse events
The combination of Vismodegib and Erlotinib was generally well tolerated with no grade 3 or 4 hematologic-related toxicity. Of the patients in cohort III, 25% experienced a non-hematologic-related toxicity. The most common nonhematologic toxicities were anorexia, nausea, vomiting, abdominal pain, and rash.
MTD determination
In cohort I, there were no DLTs observed in any of the three patients that were treated at each of the first three dose levels. There was one grade 4 rash observed among six patients at dose level 4 during cycle 1. The rash was considered definitely related to treatment and was a DLT by definition. Therefore, the MTD was determined at dose level 4; that is, Vismodegib 150 mg orally daily in combination with Erlotinib 150 mg orally daily. Grade 3 events that were related to the study treatment were nausea, anorexia, hiccups, abdominal pain, and hyponatremia. These events all occurred during cycle 1 by the same patient who had the dose limiting grade 4 rash. There were no observed treatment-related grade 5 events. At the starting dose level in cohort II, one patient experienced a grade 3 right eye thrombus that was possibly related to treatment and was dose limiting. A second patient experienced grade 3 nausea and bladder infection also thought to possibly be related to treatment and, therefore, considered dose limiting. Given the presence of DLTs in two of three patients at this starting dose level, patients were then accrued at dose level −1. Of the first five patients enrolled, two experienced grade 4 thrombocytopenia. Therefore, the MTD had still already been exceeded and the decision was then made to halt accrual and the protocol was amended. It was found that the patients with dose limiting thrombocytopenia were heavily pretreated and so eligibility criteria were revised to include patients with only one prior line of chemotherapy. The length of cycle 1 was reduced from 56 to 28 days and dose level −2 was introduced. Thirteen additional patients were enrolled on the amended protocol. DLTs were observed in two patients (grade 3 rash and grade 3 hyponatremia) at dose level −1. However, only 1 of 6 evaluable patients at dose level −1 experienced a DLT (grade 4 thrombocytopenia). Therefore, the MTD was determined at a dose level −2; that is, Vismodegib 150 mg orally daily in combination with Erlotinib 75 mg orally daily and gemcitabine 600 mg/m2 intravenously on days 1, 8 and 15 (Table 1B).
Best response & antitumor activity
There were no tumor responses. A total of thirteen patients had stable disease (SD). One patient in cohort I dose level 1 had SD. One patient from cohort II at dose level −1 had SD for 3 cycles and two patients had SD for two cycles. At dose level −2 one patient had SD for seven cycles and one had SD for 3 cycles. Amongst patients in cohort III, seven patients had SD for two cycles.
Thirty-four patients progressed after the first or second cycle of treatment and had no prior higher response. F18-FDG PET imaging was performed on patients with metastatic pancreatic cancer. No significant changes in SUV (p ≥ 0.05) were seen after 28 days of treatment when compared to the baseline measurements from the individual patients.
Histopathologic evaluation of paired tumor biopsies
Evaluation of markers important for HH and EGFR signaling was performed on tumor biopsies. A total of 10-paired samples were obtained from the patients enrolled into cohort III who had PDAC and were treated with Erlotinib and Vismodegib. Biopsies were obtained at time of enrollment and at the end of cycle I of therapy (day 28). Of these, ten had tissue available to perform immunohistochemical analysis for evaluation of protein expression, nine of the paired samples were evaluated by H&E staining to confirm the presence of tumor and note its histologic features, and seven had tissue available for RNA extraction to evaluate gene expression. In most cases, the tumors had characteristics associated with undifferentiated PDACs and had abundant desmoplastic reactions with collagen deposition. Two of the nine paired samples with notable changes in the tumor microenvironment are depicted in Figure 1A. In one case (top panels), there was a decrease in the apparent collagen deposition noted and in the second there was a decrease in desmoplasia (bottom panels). Only the second sample set (patient 4) had tissue available for evaluation of RNA. The expression of COL1A1, encoding collagen in this sample set showed a decrease from baseline, consistent with decreased desmoplasia noted on H&E. Three of the seven samples available for RNA analysis demonstrated significantly reduced COL1A1 expression from pre-treatment to post-treatment (Figure 1B). Furthermore, five of nine paired samples reviewed by a Pathologist demonstrated reduced relative extent of desmoplasia after treatment (Figure 1C). In addition, given Vismodegib may preferentially act on cells of the stromal compartment of PDAC tumors, relative levels of fibroblast markers, α-smooth muscle actin (α-SMA) and vimentin, were compared pre- and post-treatment. No significant changes in these markers were observed (Supplementary Figures 1 and 2).
Figure 1. Alterations in tumor desmoplasia following combined Vismodegib and Erlotinib treatment.
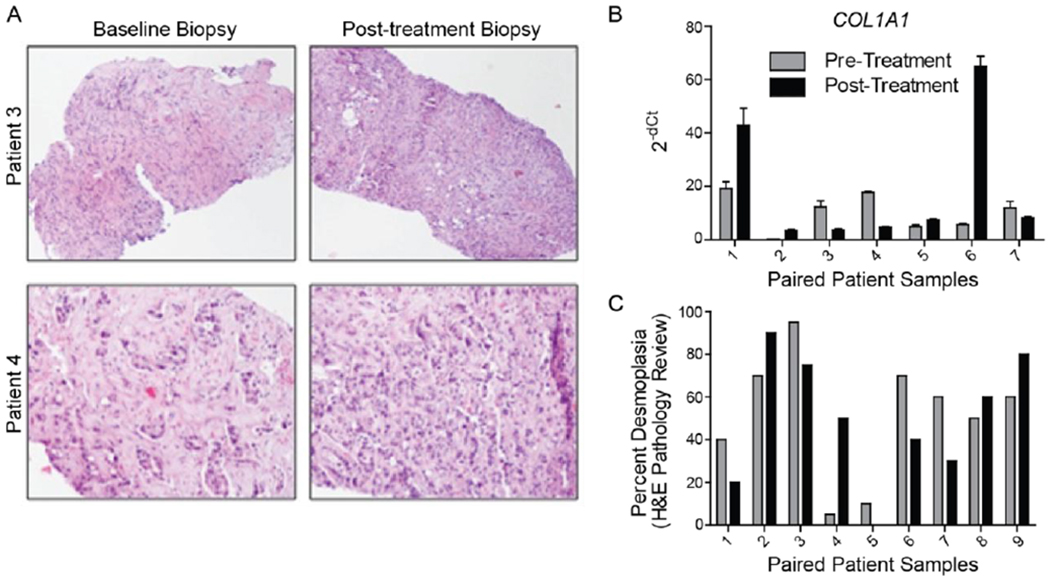
A, H&E staining of tumors from two patients with undifferentiated pancreatic adenocarcinomas demonstrates changes in the stromal reaction following treatment. There was a decrease in the amount of collagen deposition in the tumor from patient 3 and a decrease in the overall percentage of desmoplasia in the tumor from patient 4. B, Quantitative RT-PCR of COL1A1 gene expression from paired patient samples. The H&E stained sample from patient 4 in A correlates with patient 4 in B. C, Pathology review of H&E of biopsies from paired patient samples characterizing extent of desmoplasia.
Evaluation of downstream pathway targets
GLI1 is the downstream effector of the HH pathway, and as such, we evaluated the expression of phosphorylated serine 84 of GLI1 (P-GLI1-S84) by immunohistochemistry (28). A high level of P-GLI1-S84 was observed in 6 of 9 baseline samples, but this expression was decreased in the post-treatment biopsy in 5 of these 6 originally high expressing baseline samples (Figure 2A). Additionally, 2 of 3 samples that had low expression levels of P-GLI1-S84 still showed a further reduction upon treatment. A representative example of a tumor with high P-GLI1-S84 expression before treatment and reduction after treatment is shown in Figure 2B. Total GLI protein expression levels was not assessed. Thus, the identified decrease in P-GLI1-S84 levels could be due to overall decreased levels of total GLI1 protein or due to a decrease in its phosphorylation.
Figure 2. Expression of HH pathway components in paired patient biopsy samples.
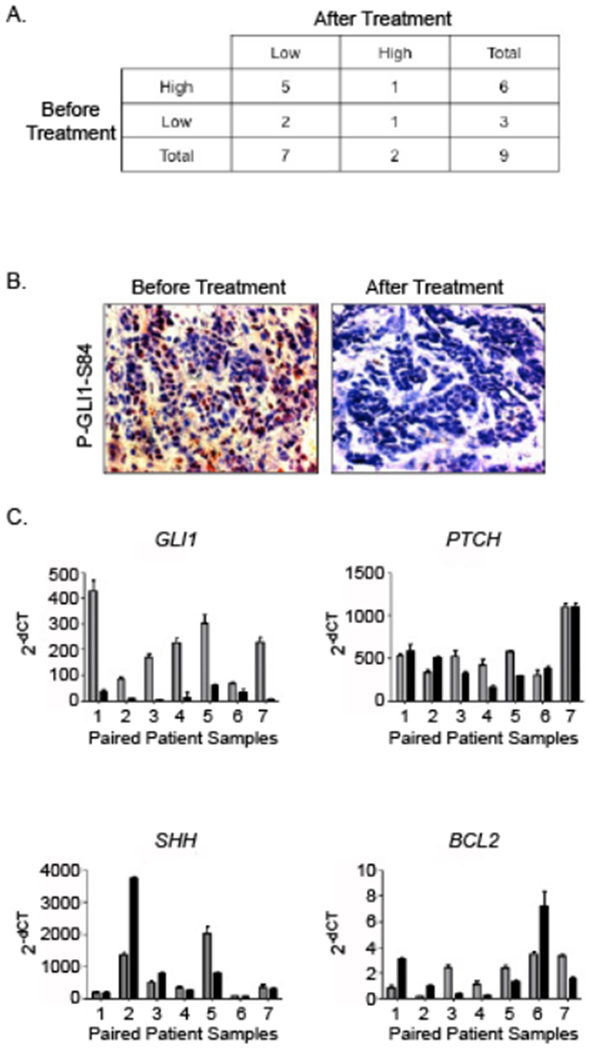
A. The table represents expression patterns of P-GLI1-S84 in pre- and post-treatment paired tumor biopsy samples. The level of P-GLI1-S84 expression was significantly decreased following combined Vismodegib and Erlotinib treatment in the majority of paired patient samples. B. A representative IHC image of change in P-GLI1-S84 expression from pre- to post-treatment. P-GLI1-S84 is mainly seen in the stroma. C. Expression levels of various HH pathway components measured by qRT-PCR before (gray bars) and after (black bars) treatment. Expression is normalized to the 18S housekeeping gene. RNA expression levels of GLI1 in all paired patient biopsies evaluated were decreased following treatment when compared to baseline. Most samples showed no change in levels from baseline for PTCH1 or SHH. BCL2, a downstream target of GLI1, was decreased in four of the seven paired samples.
We followed these findings with qRT-PCR from RNA extracted from these samples and looked at the changes in the total levels of GLI1 mRNA expression. In all seven paired samples available for evaluation of the RNA, GLI1 levels decreased following treatment (Figure 2C). We further evaluated the effect of GLI1 reduction by looking at HH pathway components that are expressed upon pathway activation or are targets of GLI1, including PTCH1 and BCL2. Only 3 of 7 paired samples showed a decrease in PTCH1 mRNA expression while 1 of 7 showed a decrease in SHH mRNA expression (Figure 2C). In most cases there was no change and rarely an increase in expression was actually seen. In the case of the GLI1 target gene BCL2, 4 of 7 showed a decrease in expression while the remaining 3 actually showed an increase in BCL2 (Figure 2C). In summary, treatment with Vismodegib and Erlotinib decreased both GLI1 expression and phosphorylation but did not affect the levels of downstream HH signaling molecules.
We further looked at the effects of the combination treatment on EGFR and the activated, phosphorylated form of EGFR by performing IHC for total EGFR and phosphorylated EGFR on the paired patient samples. While in most cases the total expression of EGFR did not change from the baseline to the post-treatment samples, the expression of the phosphorylated form decreased from the baseline levels (Figure 3, far left panel). We determined the H-score for the total and phosphorylated EGFR (Tables 3A and 3B). Six of ten paired samples showed a 25% or greater decrease in phosphorylated EGFR in the post-treatment biopsy sample when compared to the baseline (Figure 3). We additionally evaluated changes in the expression of proteins downstream of the EGFR pathway, including ERK, phospho-ERK, AKT, and phospho-AKT. Interestingly, there was little to no change in the expression of these proteins when the post-treatment biopsy samples were compared to their respective baseline samples. Representative examples of these IHCs are shown in Figure 3 (middle and far right panels). H-scores were calculated for these proteins (Figure 3, Table 3). There was no change in total AKT expression and only one of the 10 paired samples had a greater than 25% decrease in phospho-AKT expression. Four of 10 paired samples showed a 25% or greater decrease in phospho-ERK but only two of 10 showed a similar decrease in total ERK expression. While the decrease in phospho-EGFR in tumor samples post-treatment was modest, no or minimal decrease in the phosphorylation of downstream proteins was appreciated. These results mirror what was observed in evaluation of the HH pathway in that while there was inhibition of activation of the primary target, the downstream components were not affected.
Figure 3. Immunohistochemical evaluation of EGFR and downstream signaling proteins in paired patient biopsies.
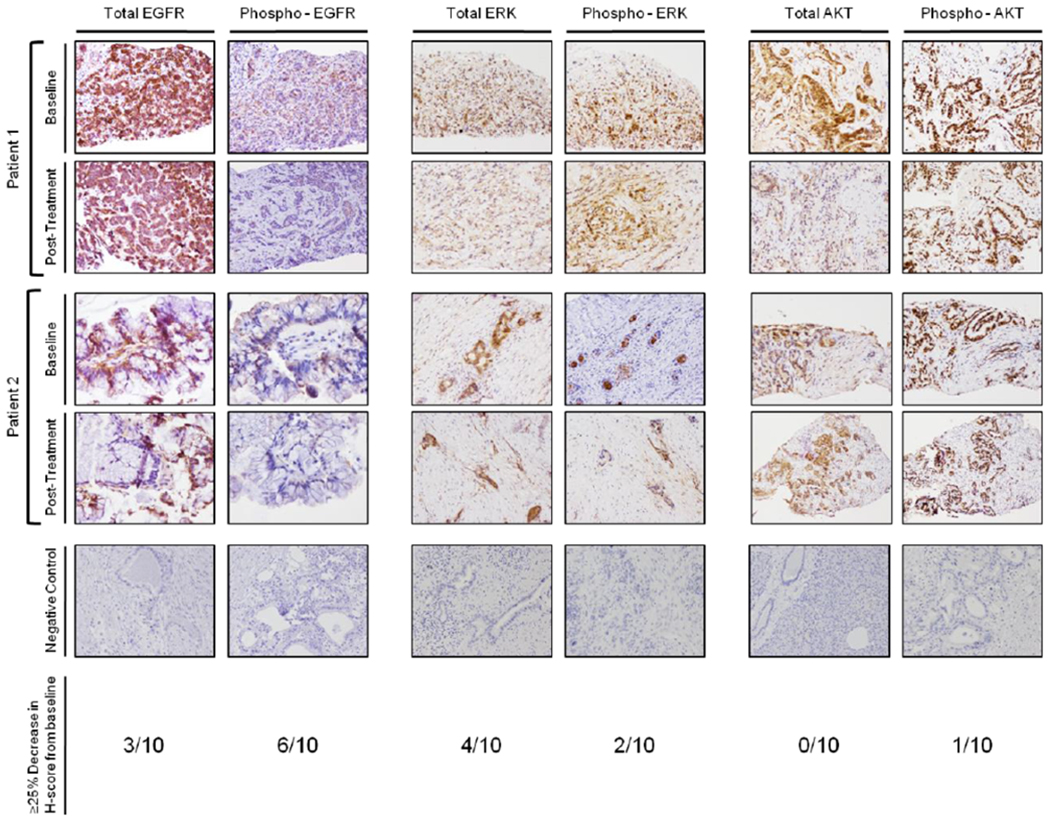
Total and phospo-EGFR IHC in two representative paired biopsies (far left panel). Both total and phospho-EGFR have a decrease in extent and intensity of staining in the post-treatment samples compared to the baseline. Total and phosphorylated ERK IHC in two representative paired biopsies. In most biopsies, the levels and extent of staining was relatively unchanged (middle panel). Total and phosphorylated AKT IHC in two representative paired biopsies (far right panel). In the patient 1 pair, there is a decrease in staining intensity of total ERK. However, the phosphorylated levels do not change. No changes were observed in the second example.
Table 3A.
Total EGFR IHC H-scores from paired patient biopsies
| Patient Pair | Baseline H-score | Post-Treatment H-score |
|---|---|---|
| 1 | 300 | 400 |
| 2 | 270 | 140 |
| 3 | 190 | 285 |
| 4 | 150 | 150 |
| 5 | 300 | 400 |
| 6 | 60 | 285 |
| 7 | 270 | 140 |
| 8 | 210 | 240 |
| 9 | 300 | 300 |
| 10 | 300 | 270 |
Table 3B.
Phospho-EGFR IHC H-scores from paired patient biopsies
| Patient Pair | Baseline H-score | Post-Treatment H-score |
|---|---|---|
| 1 | 70 | 5 |
| 2 | 0 | 0 |
| 3 | 5 | 5 |
| 4 | 30 | 1 |
| 5 | 270 | 10 |
| 6 | 0 | 50 |
| 7 | 60 | 60 |
| 8 | 100 | 10 |
| 9 | 80 | 50 |
| 10 | 60 | 10 |
Discussion
Previously reported literature has demonstrated synergistic activity between the HH and EGFR pathways. For example, one study demonstrated transactivation of EGFR and subsequent activation of ERK1/2 occurs with active HH signaling in neural stem cells during development (49). Given both pathways play important roles in pathogenesis of PDAC, concomitant inhibition was hypothesized to improve responses.
These observations combined with established crosstalk between the two pathways led to our interest in the use of a combinatorial approach for treatment of PDAC. This was investigated in a phase I clinical trial. Overall, the combination of Vismodegib and Erlotinib was well-tolerated by patients with minimal toxicity, and an MTD was established. A recent study investigated the combination of Vismodegib with gemcitabine in patients with metastatic PDAC. In this study, most patients demonstrated upregulated HH signaling with increased expression levels of HH ligand prior to treatment. Similar to our results, treatment with Vismodegib resulted in decreased levels of GLI1, but no significant improvement in survival relative to gemcitabine alone (19).
The ability to evaluate patient samples both before and after initiation of therapy has provided us with insight in the molecular biology of these tumors. We focused our evaluation on components of the HH and EGFR pathways to assess effectiveness of functional inhibition by Vismodegib and Erlotinib respectively. Comparing baseline and post-treatment paired samples, GLI1 mRNA levels were decreased more than 90% consistent with successful HH abrogation. In line with these findings, phosphorylation of GLI1 on serine 84 was also decreased in paired samples. These results indicate that Vismodegib effectively suppressed the HH-GLI1 pathway during this study.
Evaluation of EGFR and its activated, phosphorylated form generally demonstrated successful inhibition though the effect appeared modest with erlotinib treatment resulted in approximately 25% inhibition in 6 of 10 patients. Surprisingly, three of the remaining paired samples showed no change, low initial phospho-EGFR, or increased expression. We also assessed downstream components ERK, AKT and their phosphorylated forms, but minimal to no effect was observed between samples. Taken together, this suggests a few possibilities. First, the degree of change in phosphorylated EGFR is variable among samples, and so levels in some samples suggest that EGFR is not activated in all PDAC samples. Thus, incomplete inhibition of EGFR could result from an absense of initial activation. In addition, other mechanisms of crosstalk may bypass upstream EGFR inactivation. EGFR is able to induce downstream signaling through the Ras-Raf-MAPK, PI3K/AKT and other pathways not investigated in this study (50,51). Increases in EGFR and its ligand have been identified in PDAC and are associated with poor prognosis presumably through any number of these downstream effectors (35,36). However, there is evidence pathways downstream from EGFR may be independently activated. For example, activating KRAS mutations, the most common driver mutations found in PDAC, activate ERK while AKT has been found to be overexpressed in 60% of PDAC, occasionally secondary to gene amplification (35). Finally, the degree of desmoplasia, commonly found in PDAC, may limit tumor exposure to Erlotinib. While these possibilities may explain our findings, it is unclear what level of EGFR inhibition results in a biologically relevant decrease in pathway activation and if this level of inhibition was reached in the present study.
Interesting findings in the tumor microenvironment were observed in this study. The desmoplastic reaction associated with PDAC is profound and well-described. There is evidence that desmoplasia confers protection from therapy (45). Interestingly, HH signaling is not only important in cancer cells, but also has significant involvement in regulating the stromal compartment. Hence, a therapeutic approach targeting both the stromal cells and the tumor cells made HH pathway abrogation an attractive option. In all pre-treatment biopsies evaluated, a significant desmoplastic reaction was identified. In several paired samples we observed significant decrease in not only the desmoplastic reaction but also RNA levels encoding collagen with treatment relative to baseline. These findings support our hypothesis that EGFR and HH abrogation would have beneficial effects on both tumor and stromal compartments. Nonetheless, one technical limitation to this study is gene expression being assessed from whole biopsy specimen-derived RNA as opposed to a microdissection-based method to separate these compartments. While this limitation and a small sample size do not allow us to make any definitive conclusions regarding the effect of Erlotinib and Vismodegib, these findings appear promising.
In summary, based upon biomarker analysis Vismodegib effectively inhibits phosphorylated, activated forms of GLI1. This appears to significantly alter the stromal compartment of PDAC. The desmoplastic reaction appears to be more supportive of fully-formed PDAC tumors and thus, targeting the stromal compartment may be a useful therapeutic strategy and effectively achieved with Vismodegib. Erlotinib, on the other hand, appeared effective if abrogating EGFR activity based on biomarker assessments. However, given ongoing activation of downstream effectors, findings suggest the combination of EGFR and HH crosstalk alone is insufficient to abrogate signaling in the tumor compartment completely. Thus, a better understanding of PDAC signaling networks and stromal dynamics for more tactical targeting is necessary.
Supplementary Material
Table 3C.
Total AKT IHC H-scores from paired patient biopsies
| Patient Pair | Baseline H-score | Post-Treatment H-score |
|---|---|---|
| 1 | 100 | 80 |
| 2 | 80 | 80 |
| 3 | 0 | 0 |
| 4 | 60 | 50 |
| 5 | 80 | 160 |
| 6 | 180 | 160 |
| 7 | 180 | 180 |
| 8 | 140 | 70 |
| 9 | 210 | 300 |
| 10 | 270 | 160 |
Table 3D.
Phospho-AKT IHC H-scores from paired patient biopsies
| Patient Pair | Baseline H-score | Post-Treatment H-score |
|---|---|---|
| 1 | 285 | 285 |
| 2 | 285 | 270 |
| 3 | 285 | 210 |
| 4 | 285 | 270 |
| 5 | 285 | 285 |
| 6 | 270 | 285 |
| 7 | 285 | 285 |
| 8 | 285 | 285 |
| 9 | 300 | 300 |
| 10 | 300 | 270 |
Acknowledgements
The authors thank all of the patients that participated in this study as well as their providers and caregivers for their participation. We also thank the clinical trial coordinators and personnel, including Janet Lensing and Daniel Satele for assisting with this trial. No potential conflicts of interest were disclosed by any of the authors. Grant support for this study was provided through CA136526, AACR-PANCAN, CA15083, CA186686, Mayo Clinic Pancreatic Cancer SPORE CA102701, Kellen Foundation, Georgeson Professorship fund, Mayo Clinic Cancer Center.
Footnotes
Conflict of Interest: The authors declared no conflicts with this submission.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: a cancer journal for clinicians. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver and pancreas cancers in the United States. Cancer Research. 2014;74:4006. [DOI] [PubMed] [Google Scholar]
- 3.Chames P, Kerfelec B, Baty D. Therapeutic antibodies for the treatment of pancreatic cancer. TheScientificWorldJournal. 2010;10:1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1997;15:2403–13. [DOI] [PubMed] [Google Scholar]
- 5.Colucci G, Giuliani F, Gebbia V, Biglietto M, Rabitti P, Uomo G, et al. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer. 2002;94:902–10. [PubMed] [Google Scholar]
- 6.Rocha Lima CM, Green MR, Rotche R, Miller WH, Jr., Jeffrey GM, Cisar LA, et al. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22:3776–83. [DOI] [PubMed] [Google Scholar]
- 7.Oettle H, Richards D, Ramanathan RK, van Laethem JL, Peeters M, Fuchs M, et al. A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2005;16:1639–45. [DOI] [PubMed] [Google Scholar]
- 8.Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:3509–16. [DOI] [PubMed] [Google Scholar]
- 9.Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:5513–8. [DOI] [PubMed] [Google Scholar]
- 10.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine. 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 11.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine. 2013;369:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy SJ, Hart SN, Halling G, Johnson SH, Smadbeck JB, Drucker T, et al. Integrated genomic analysis of pancreatic ductal adenocarcinomas reveals genomic rearrangement events as significant drivers of disease. Cancer research. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajurkar M, De Jesus-Monge WE, Driscoll DR, Appleman VA, Huang H, Cotton JL, et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci U.S.A. 2012;109:E1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills LD, Zhang Y, Marler RJ, Herreros-Villaneueva M, Zhang L, Almada LL, et al. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J Biol Chem. 2013;288:11786–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheng W, Dong M, Zhou J, Li X, Liu Q, Dong Q, et al. The clinicopathological significance and relationship of Gli1, MDM2 and p53 expression in resectable pancreatic cancer. Histopathology. 2014;64(4):523–535. [DOI] [PubMed] [Google Scholar]
- 18.Marechal R, Bachet JB, Calomme A, Demetter P, Delpero JR, Svrcek M, et al. Sonic hedgehog and Gli1 expression predict outcome in resected pancreatic adenocarcinoma. Clin Cancer Res. 2015;21(5):1215–1224. [DOI] [PubMed] [Google Scholar]
- 19.Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, et al. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riobo NA, Haines GM, Emerson CP Jr., Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer research. 2006;66:839–45. [DOI] [PubMed] [Google Scholar]
- 22.Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr., Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer research. 2009;69:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rovida E, Stecca B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin Cancer Biol. 2015;35:154–167. [DOI] [PubMed] [Google Scholar]
- 25.Syu LJ, Zhao X, Zhang Y, Grachtchouk M, Demitrack E, Ermilov A, et al. Invasive mouse gastric adenocarcinomas arising from Lgr5+ stem cells are dependent on crosstalk between the Hedgehog/GLI2 and mTOR pathways. Oncotarget. 2016;7:10255–10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Molecular and cellular biology. 2006;26:6283–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO molecular medicine. 2012;4:218–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21:374–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131:337–45. [DOI] [PubMed] [Google Scholar]
- 30.Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD, et al. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132:335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bigelow RL, Jen EY, Delehedde M, Chari NS, McDonnell TJ. Sonic hedgehog induces epidermal growth factor dependent matrix infiltration in HaCaT keratinocytes. The Journal of investigative dermatology. 2005;124:457–65. [DOI] [PubMed] [Google Scholar]
- 32.Algul H, Adler G, Schmid RM. NF-kappaB/Rel transcriptional pathway: implications in pancreatic cancer. Int J Gastrointest Cancer. 2002;31:71–78. [DOI] [PubMed] [Google Scholar]
- 33.Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeu DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–2081. [DOI] [PubMed] [Google Scholar]
- 34.Pan X, Arumugam T, Yamamoto T, Levin PA, Ramachandran V, Ji B, et al. Nuclear factor-kappaB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin Cancer Res. 2008;14:8143–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemoine NR, Hughes CM, Barton CM, Poulsom R, Jeffery RE, Kloppel G, et al. The epidermal growth factor receptor in human pancreatic cancer. The Journal of pathology. 1992;166:7–12. [DOI] [PubMed] [Google Scholar]
- 36.Yamanaka Y, Friess H, Kobrin MS, Buchler M, Kunz J, Beger HG, et al. Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Human pathology. 1993;24:1127–34. [DOI] [PubMed] [Google Scholar]
- 37.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:1960–6. [DOI] [PubMed] [Google Scholar]
- 38.Keysar SB, Le PN, Anderson RT, Jason Morton J, Bowles DW, Paylor JJ, et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013;73:3381–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maria Della Corte C, Bellevicine C, Vicidomini G, Vitagliano D, Malapelle U, Accardo M, et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin Cancer Res. 2015;21:4686–4697. [DOI] [PubMed] [Google Scholar]
- 40.Hu WG, Liu T, Xiong JX, Wang CY. Blockade of sonic hedgehog signal pathway enhances antiproliferative effect of EGFR inhibitor in pancreatic cancer cells. Acta pharmacologica Sinica. 2007;28:1224–30. [DOI] [PubMed] [Google Scholar]
- 41.Rucki AA, Xiao Q, Muth S, Chen J, Che X, Kleponis J, et al. Dual inhibition of hedgehog and c-Met pathways for pancreatic cancer treatment. Mol Cancer Ther. 2017;16:2399–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10. [DOI] [PubMed] [Google Scholar]
- 43.Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walter K, Omura N, Hong SM, Griffith M, Vincent A, Borges M, et al. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:1781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, et al. An orally bioavailable small-molecule inhibitor of hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu F, Feng XX, Zhu SL, Huang HY, Chen YD, Pan YF, et al. Sonic hedgehog signaling pathway mediates proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis via MAPK/ERK signaling pathway. Front Immunol. 2018;9:2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reinchisi G, Parada M, Lois P, Oyanadel C, Shaughnessy R, Gonzalez A, et al. Sonic Hedgehog modulates EGFR dependent proliferation of neural stem cells during late mouse embryogenesis through EGFR transactivation. Frontiers in cellular neuroscience. 2013;7:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21:2787–99. [DOI] [PubMed] [Google Scholar]
- 51.Blasco MT, Navas C, Martin-Serrano G, Grana-Castro O, Lechuga CG, Martin-Diaz L, et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and c-RAF. Cancer Cell. 2019;35:573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.