

Abstract
Background
Several agents are used to clear secretions from the airways of people with cystic fibrosis. Mannitol increases mucociliary clearance, but its exact mechanism of action is unknown. The dry powder formulation of mannitol may be more convenient and easier to use compared with established agents which require delivery via a nebuliser. Phase III trials of inhaled dry powder mannitol for the treatment of cystic fibrosis have been completed and it is now available in Australia and some countries in Europe. This is an update of a previous review.
Objectives
To assess whether inhaled dry powder mannitol is well tolerated, whether it improves the quality of life and respiratory function in people with cystic fibrosis and which adverse events are associated with the treatment.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group Trials Register which comprises references identified from comprehensive electronic databases, handsearching relevant journals and abstracts from conferences.
Date of last search: 12 December 2019.
Selection criteria
All randomised controlled studies comparing mannitol with placebo, active inhaled comparators (for example, hypertonic saline or dornase alfa) or with no treatment.
Data collection and analysis
Authors independently assessed studies for inclusion, carried out data extraction and assessed the risk of bias in included studies. The quality of the evidence was assessed using GRADE.
Main results
Six studies (reported in 36 unique publications) were included with a total of 784 participants.
Duration of treatment in the included studies ranged from 12 days to six months, with open‐label treatment for an additional six months in two of the studies. Five studies compared mannitol with control (a very low dose of mannitol or non‐respirable mannitol) and the final study compared mannitol to dornase alfa alone and to mannitol plus dornase alfa. Two large studies had a similar parallel design and provided data for 600 participants, which could be pooled where data for a particular outcome and time point were available. The remaining studies had much smaller sample sizes (ranging from 22 to 95) and data could not be pooled due to differences in design, interventions and population.
Pooled evidence from the two large parallel studies was judged to be of low to moderate quality and from the smaller studies was judged to be of low to very low quality. In all studies, there was an initial test to see if participants tolerated mannitol, with only those who could tolerate the drug being randomised; therefore, the study results are not applicable to the cystic fibrosis population as a whole.
While the published papers did not provide all the data required for our analysis, additional unpublished data were provided by the drug's manufacturer and the author of one of the studies.
Pooling the large parallel studies comparing mannitol to control, up to and including six months, lung function (forced expiratory volume at one second) measured in both mL and % predicted was significantly improved in the mannitol group compared to the control group (moderate‐quality evidence). Beneficial results were observed in these studies in adults and in both concomitant dornase alfa users and non‐users in these studies. In the smaller studies, statistically significant improvements in lung function were also observed in the mannitol groups compared to the non‐respirable mannitol groups; however, we judged this evidence to be of low to very low quality.
For the comparisons of mannitol and control, we found no consistent differences in health‐related quality of life in any of the domains except for burden of treatment, which was less for mannitol up to four months in the two pooled studies of a similar design; this difference was not maintained at six months. It should be noted that the tool used to measure health‐related quality of life was not designed to assess mucolytics and pooling of the age‐appropriate tools (as done in some of the included studies) may not be valid so results were judged to be low to very low quality and should be interpreted with caution. Cough, haemoptysis, bronchospasm, pharyngolaryngeal pain and post‐tussive vomiting were the most commonly reported side effects in both treatment groups. Where rates of adverse events could be compared, statistically no significant differences were found between mannitol and control groups; although some of these events may have clinical relevance for people with CF.
For the comparisons of mannitol to dornase alfa alone and to mannitol plus dornase alfa, very low‐quality evidence from a 12‐week cross‐over study of 28 participants showed no statistically significant differences in the recorded domains of health‐related quality of life or measures of lung function. Cough was the most common side effect in the mannitol alone arm but there was no occurrence of cough in the dornase alfa alone arm and the most commonly reported reason of withdrawal from the mannitol plus dornase alfa arm was pulmonary exacerbations.
In terms of secondary outcomes of the review (pulmonary exacerbations, hospitalisations, symptoms, sputum microbiology), evidence provided by the included studies was more limited. For all comparisons, no consistent statistically significant and clinically meaningful differences were observed between mannitol and control treatments (including dornase alfa).
Authors' conclusions
There is moderate‐quality evidence to show that treatment with mannitol over a six‐month period is associated with an improvement in some measures of lung function in people with cystic fibrosis compared to control. There is low to very low‐quality evidence suggesting no difference in quality of life for participants taking mannitol compared to control. This review provides very low‐quality evidence suggesting no difference in lung function or quality of life comparing mannitol to dornase alfa alone and to mannitol plus dornase alfa.
The clinical implications from this review suggest that mannitol could be considered as a treatment in cystic fibrosis; but further research is required in order to establish who may benefit most and whether this benefit is sustained in the longer term. Furthermore, studies comparing its efficacy against other (established) mucolytic therapies need to be undertaken before it can be considered for mainstream practice.
Plain language summary
Mannitol, an inhaled drug, for treating lung disease in cystic fibrosis
Review question
We reviewed the evidence about the effect of inhaling mannitol to treat lung disease in people with cystic fibrosis.
Background
Cystic fibrosis is a genetic disorder that affects the exocrine glands (sweat glands and others). Lung infections produce thick mucus (phlegm) which can block air passages and cause more infection and repeated inflammation. In turn, this progressively damages the lungs and can eventually cause respiratory failure. There are several drugs that are used to clear mucus from the airways of people with cystic fibrosis and inhaled dry powder mannitol is a new one that may improve their lung function. The dry powder formulation of mannitol may be more convenient and easier to use compared with established agents delivered by a nebuliser (e.g. hypertonic saline). Mannitol is available in Australia and some European countries. This is an updated version of the review.
Search date
The evidence is current to: 12 December 2019.
Study characteristics
We included six studies (with a total of 784 adults and children) in this review. Five studies compared a standard dose of mannitol with control (a very low dose of mannitol or a version of mannitol which did not allow the active drug to reach the lungs) and the sixth study compared mannitol with nebulised recombinant human deoxyribonuclease (dornase alfa), both alone and taken together. Participants could continue using dornase alfa and other standard therapies, but were excluded from the five of the six studies if they were using hypertonic saline. Treatment in these studies lasted from 12 days to six months. Five studies provided the treatments to people as outpatients and in one study, the children treated were in hospital due to pulmonary exacerbations (flare ups of disease).
Key results
It was difficult to combine evidence from the studies in this review due to differences in the designs of the studies, treatments examined and the settings (hospital or outpatients). Some additional information was obtained from the drug manufacturer and one study author to aid the review.
The review found low‐ to very low‐quality evidence that there is no difference between mannitol and control treatments or mannitol given either with or without additional dornase alfa in terms of quality of life. There was moderate‐quality evidence of improvements in some measures of lung function across the larger studies comparing mannitol to control. Beneficial effects were also seen in the subgroup of adults and in both those who were using dornase alfa and those who were not. Cough (including coughing up blood), contraction of the airways, pain in the pharynx or larynx and post‐treatment vomiting were the most commonly reported side effects on both treatments, but there was no evidence to suggest that these side effects occurred more on mannitol than on control treatments or on dornase alfa.
None of the studies compared mannitol to nebulised hypertonic saline and so we can not comment on which agent is better for airway clearance. More research is needed to answer this question.
Quality of the evidence
We judged the quality of the evidence from this review to be of very low to moderate quality, depending on the outcome measured. We do not think that the way the studies were designed affected the results. We judged that everyone taking part had equal chances of being in either of the treatment groups and would not have known in advance or during the study which treatment they were receiving. However, the numbers of people who dropped out of the studies might affect how the results are interpreted, as well as how many people were recruited into the studies and how they were selected from all people with cystic fibrosis who could have been included.
Although some of these issues were resolved when the drug's manufacturer (who also sponsored the studies) provided some additional information. It is important to realise that before people started the study, they took a test to see if they could tolerate mannitol and only those who did could carry on. This means that the results of the studies only apply to those people with cystic fibrosis who can tolerate mannitol.
Summary of findings
Background
Description of the condition
Cystic fibrosis (CF) is the most prevalent inherited, life‐limiting disorder in populations of Northern European origin. The disorder is caused by a gene defect on the long arm of chromosome 7 which encodes for a conductance‐regulating protein called the cystic fibrosis transmembrane conductance regulator (CFTR) (Dinwiddie 2000). It is a relatively common autosomal recessive disease occurring in approximately 1 in 2500 live births (Ratjen 2003) and in the USA and the UK, approximately 1 in 25 people of Northern European origin are carriers of a CFTR abnormality (CFF 2000; Hodson 2000). It is estimated that the present number of CF cases is 35,000 in Europe, 30,000 in North America and 3000 in Canada (CCFF 2002; CFF 2005).
Although CFTR is primarily a chloride ion channel, it is also involved in the regulation of other membrane channels and transport of molecules which may be important in mucociliary clearance and innate defence mechanisms (McAuley 2000). The interaction of CFTR with the epithelial sodium channel is also important and it is up‐regulated in CF. Thus, defective CFTR in CF causes reduced chloride secretion and excess absorption of sodium across the epithelia, leading to thickened secretions in organs such as the pancreas and lung (McAuley 2000). These viscous secretions lead to airway obstruction, infection and chronic inflammation in a vicious cycle resulting in progressive lung damage with bronchiectasis and eventual respiratory failure (Dinwiddie 2000). Although with advances in treatment, life expectancy has increased dramatically over the past six decades from a median of two years to a projected median survival of over 50 years of age (Dodge 2007), lung disease remains the most common cause of death in people with CF (Accurso 2007).
Several agents are used to clear secretions from the airways of people with CF. These agents include inhaled n‐acetylcysteine, hypertonic saline and recombinant human deoxyribonuclease (dornase alfa) (Bye 2007; Tam 2013). A Cochrane Review demonstrated that dornase alfa reduces pulmonary exacerbations and improves lung function (Yang 2018). A second Cochrane Review showed that although regular treatment with hypertonic saline led to only modest improvements in lung function in adults and children, there were substantial reductions in pulmonary exacerbations (Wark 2018). Although n‐acetylcysteine was used as a mucolytic drug in CF for many years, a Cochrane Review found little evidence for any beneficial effect (Tam 2013). These three treatments have to be given using a nebuliser, which can be time‐consuming, inconvenient and unpopular. Sawicki noted that the level of daily treatment activity is high for adults with CF regardless of age or disease severity and that increasing numbers of nebulised therapies are associated with higher perceived treatment burden (Sawicki 2009). The authors emphasised the importance of evaluating treatment burden as an outcome in the development of new CF treatments as many of these are designed to be added to existing treatment regimens. In addition, dornase alfa is expensive; in a study in children, treatment with daily dornase alfa significantly increased lung function compared with hypertonic saline but significantly increased healthcare costs (Suri 2002). In the Cochrane Review it was noted in a cost analysis of a second study in adults and children, that healthcare costs relating to respiratory tract infections were less in the treated participants, but this cost saving did not offset the cost of therapy itself (Yang 2018). Inhaled dry powder mannitol is now being investigated as an alternative treatment for airway clearance.
Description of the intervention
Mannitol is a dry powder contained in 40 mg capsules for inhalation. It is inhaled orally via a breath‐activated hand‐held inhalation device. Clinical studies have used varying doses of mannitol (315 mg to 635 mg), but generally around a dose of 400 mg (10 capsules) twice daily has been administered. Since mannitol is inhaled as a dry powder, it does not generally require a nebuliser; although a non‐randomised study conducted in Iran has compared nebulised mannitol to nebulised hypertonic saline (Sadr 2019). However, it is not as quick to use as many other dry powder medications because the dose is divided into multiple capsules which must be loaded and inhaled individually.
How the intervention might work
Mannitol is a naturally occurring six‐carbon monosaccharide (sugar alcohol) which is not actively transported and is poorly absorbed after inhalation (Anderson 1997). It appears to act by inducing an influx of water into the airway lumen and evidence from in vitro and animal studies suggests a number of mechanisms by which it may increase mucociliary clearance, including improving hydration of airway secretions and reducing sputum viscosity (Bye 2007). However, Daviskas examined ex vivo sputum and was only able to demonstrate an improvement in hydration with no substantial change in viscoelasticity (Daviskas 2010). Another suggested mechanism of action is stimulating release of mediators that increase ciliary beat frequency (Brannan 2003; Wanner 1983). Regardless of the exact mechanism by which it is achieved, mannitol appears to increase mucociliary clearance in people with CF (Robinson 1999).
Why it is important to do this review
Inhaled dry powder mannitol is available for the treatment of CF and is licensed for use in adults in the European Union and for adults and children over the age of six years in Australia. A Cochrane Review of inhaled hyperosmolar agents in people with bronchiectasis identified five randomised controlled trials (RCTs) and demonstrated that inhaled mannitol improved airway clearance and reduced exacerbations, but had no effect on quality of life outcome (Hart 2014); these findings were supported by additional observational studies (Daviskas 1997; Daviskas 2001; Daviskas 2002; Daviskas 2005). A number of studies looking at mannitol in CF have now been completed. In addition, the dry powder formulation of mannitol may be more convenient and easier to use compared with established agents which require delivery via a nebuliser; anecdotal reports suggest that once individuals become familiar with the technique, administration time for inhaled mannitol compares well with the time taken to administer nebulised dornase alfa. Thus, the evidence in CF should be combined and presented in a systematic review. The UK National Institute for Health and Care Excellence (NICE) has made a recommendation for the the use of mannitol in certain individuals with CF; particularly those with rapid decline in lung function and those for which dornase alfa and other osmotic agents have failed or are not considered appropriate (NICE 2012).
This is an update of a previously published review (Nolan 2015; Nevitt 2018).
Objectives
To assess whether inhaled dry powder mannitol is well tolerated, whether it improves the quality of life and respiratory function in people with CF and which adverse events are associated with the treatment.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) of parallel or cross‐over design, blinded or open‐label.
Types of participants
Adults (18 years old and over) and children (under 18 years old) with CF (diagnosed clinically and by sweat or genetic testing and including all degrees of disease severity).
Types of interventions
We compared orally inhaled dry powder mannitol (either alone or with another agent) with either control, active inhaled comparators (for example, hypertonic saline or dornase alfa) or with no treatment. We included all doses and regimens of mannitol administration. We also included studies of all durations of treatment and follow‐up.
Types of outcome measures
Primary outcomes
Health‐related quality of life (HRQoL) as measured by a validated tool such as the Cystic Fibrosis Questionnaire‐Revised version (CFQ‐R (Quittner 2009)) or Cystic Fibrosis Quality of Life Questionnaire (CFQoL (Gee 2000))
-
Lung function
forced expiratory volume in one second (FEV1) (mL or % predicted)
forced vital capacity (FVC) (mL or % predicted)
forced expiratory flow between 25% and 75% expired volume (FEF25-75) (mL/s or % predicted)
-
Adverse events relating to treatment
-
type and number of adverse events defined as follows according to perceived effect on the participant
mild (not requiring extra treatment, noted but tolerable)
moderate (requiring extra treatment and cessation of the intervention)
severe (life‐threatening or results in death, requiring hospitalisation, results in persistent or significant disability)
number of participants who ceased inhalations because of poor tolerability e.g. cough or bronchoconstriction
-
Secondary outcomes
-
Pulmonary exacerbations
number of participants free of pulmonary exacerbations
time to first pulmonary exacerbation
Time off school or work
-
Need for additional non‐routine antibiotics
intravenous
oral
nebulised
-
Hospitalisations
number of hospitalisations
duration
Assessment of symptoms (including cough, sputum volume, ease of expectoration and dyspnoea)
Sputum microbiology (change in numbers of pathogens, emergence of new pathogens)
Burden of treatment (using a validated measure)
Search methods for identification of studies
There were no restrictions regarding language or publication status.
Electronic searches
We identified relevant studies from the Group's Cystic Fibrosis Trials Register using the terms: mannitol [bronchitol].
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cochrane Cystic Fibrosis and Genetic Disorders Group website.
Date of last search: 12 December 2019.
We searched the trials database clinicaltrials.gov (https://clinicaltrials.gov/) using the terms: "mannitol" AND "cystic fibrosis".
We also searched the WHO ICTRP (http://apps.who.int/trialsearch/) using the terms: ["mannitol" OR "bronchitol"] AND "cystic fibrosis".
Date of most recent search of trial databases: 19 December 2019. No starting date limit was set for the searches of trial databases to allow for identification of retrospectively registered studies.
Searching other resources
We checked the reference lists of any studies identified for further relevant studies. We also contacted primary authors of identified studies and research institutions or biotech companies for unpublished studies.
Data collection and analysis
Selection of studies
All authors independently reviewed all potential studies for inclusion and examined the title and abstract of potential publications to remove those that did not meet inclusion criteria (e.g. single case reports, reviews, etc.). We examined the full‐text publications of the remaining studies to determine if they met the eligibility criteria. If we were unable to reach agreement regarding the determination of eligibility by discussion, we resolved this by approaching a colleague to act as arbiter. We examined publications which were potentially eligible for inclusion to identify instances of duplication by comparing author, institution, study detail (intervention, dosing, timing, etc.) and participant demographics.
Data extraction and management
Three authors (JT, CM and TD) independently extracted study characteristics and outcome data from the studies onto a standard data extraction form. If there was disagreement, we resolved this by discussion. The fourth author (SJN) checked data and extracted additional data from the files which were provided by the manufacturers of mannitol.
We originally planned to report data at one month, three months and one year; however, in a post hoc change which grouped data into more clinically appropriate time points, we reported data at the time points of 'up to and including one month', 'up to and including two months', 'up to and including three months', 'up to and including four months', 'up to and including five months', 'up to and including six months', and then at six‐monthly intervals thereafter. If any studies had followed participants beyond this time, we also planned to report these data.
We planned the following comparisons (in all cases mannitol either alone or in combination with another agent): mannitol versus no treatment; mannitol versus control; mannitol versus n‐acetylcysteine; mannitol versus hypertonic saline; and mannitol versus dornase alfa. We were only able to present results for the randomised comparisons; the comparisons made within this review are listed in Effects of interventions.
Assessment of risk of bias in included studies
We assessed the risk of bias using the 'Risk of bias' assessment tool as documented in section 8.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We defined low and high risk of bias as in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Sequence generation
We determined if the allocation sequence was adequately generated. An adequate generation of the allocation sequence constitutes a low risk of bias, inadequate generation constitutes a high risk of bias and if it is unclear how the sequence was generated, this constitutes an unclear risk of bias.
Allocation concealment
We determined if the allocation sequence was adequately concealed. An adequate concealment of the allocation sequence constitutes a low risk of bias, inadequate concealment constitutes a high risk of bias and if it is unclear how allocation was concealed, this constitutes an unclear risk of bias.
Blinding (or masking) of participants, personnel and outcome assessors
We assessed whether knowledge of the allocated intervention was adequately prevented during the study. If knowledge of allocation was adequately prevented, this constitutes a low risk of bias; if knowledge of allocation was not adequately prevented, this constitutes a high risk of bias; and if it is unclear whether knowledge of allocation was adequately concealed, this constitutes an unclear risk of bias.
Incomplete outcome data
We assessed whether incomplete outcome data were adequately addressed. For example, if missing data from a large proportion of the total number of participants resulted from participants dropping out of only one arm of the study (e.g. the mannitol arm), this would constitute a high risk of bias. However, if there are small numbers of participants with missing data spread evenly across the study arms, then this would constitute a low risk of bias. If it is unclear how investigators addressed the issue of incomplete outcome data, this constitutes an unclear risk of bias.
Selective outcome reporting
We assessed whether the reports of the studies are free of suggestions of selective outcome reporting. If a study appears to be free of selective outcome reporting, this constitutes a low risk of bias; evidence of selective outcome reporting constitutes a high risk of bias; and if it is unclear whether there is selective outcome reporting, this constitutes an unclear risk of bias.
Other potential sources of bias
We assessed whether the studies were apparently free of other problems that could put them at a high risk of bias. For example, we planned to report on whether the investigators performed a sample size calculation, for which outcome(s) and the method used.
Measures of treatment effect
For binary (dichotomous) outcome measures (numbers of: participants who ceased inhalations because of poor tolerability; participants free of pulmonary exacerbations; participants requiring additional antibiotics; and hospitalisations), we aimed to calculate a pooled estimate of treatment effect for each outcome across studies using the risk ratio (RR) where appropriate and the corresponding 95% confidence intervals (CIs). In a post hoc change to account for type I statistical errors when analysing the number of participants with any adverse events of interest, the authors used RRs and 99% CIs.
For continuous outcome measures (HRQoL, lung function, time off school or work, duration of hospitalisations, assessment of symptoms, sputum microbiology, burden of treatment), we calculated a pooled estimate of treatment effect by calculating the mean difference (MD) and the corresponding 95% CIs. Where studies reported standard errors (SE), we converted these to standard deviations (SD).
The manufacturer of mannitol, Pharmaxis, provided the us with additional data for all included studies. In two of the included studies, investigators analysed outcomes of lung function data via a mixed model repeated measures analysis (MMRM) based on the average effect across the measured time points (Aitken 2012; Bilton 2011). Such an analysis is longitudinal and uses all available data at every visit. The data presented within this review are the estimates from the MMRM model for the effect at each time point derived from the treatment by visit term in the model (so data presented as 'up to six months' are the estimates from the model at the six‐month time point); it also allows for covariate adjustment (age, gender, disease severity, previous dornase alfa use, country or region, etc.). The models in the Aitken and Bilton studies were nearly identical in terms of covariates; the only difference was that the Bilton study also included treatment by visit and treatment by dornase alfa use interaction terms (Aitken 2012; Bilton 2011). We felt it was most informative and appropriate to pool the MDs and SEs from the adjusted mixed models from the two studies via generic inverse variance (Aitken 2012; Bilton 2011).
Currently, we have not needed to analyse any count data, but we may undertake such an analysis in future updates. When analysing count data, we plan to decide whether to treat this as dichotomous, continuous, time‐to‐an‐event or as a rate when we access the data. Although we accept that it is preferable to decide how count data will be analysed in advance, we believe that the choice will be determined by the format of the available data and thus we are not be able to decide which method to use until the relevant data have been reviewed. We plan to analyse such data as described in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011).
We analysed time‐to‐event data (time to first pulmonary exacerbation) using the hazard ratio (HR) and 95% CIs.
Where studies measured data longitudinally (other than for the Pharmaxis MMRM analysis described above), we based the analysis on the final time‐point results, which is a common method used in Cochrane Reviews (Jones 2005). We accept that this analysis treats the data as independent, although in reality they are not. However, methods are not yet available to carry out a meta‐analysis of aggregate longitudinal data, unless we are able to obtain individual patient data.
Unit of analysis issues
When conducting a meta‐analysis combining results from cross‐over studies, we planned to use the methods recommended by Elbourne, which take account of the correlation between measurements taken from the same group of participants via paired analyses (Elbourne 2002). However, if only limited data were available, we would only be able to either use the first‐period data only or to treat the cross‐over studies as if they are parallel studies. Elbourne states that this approach will produce conservative results as it does not take into account within‐patient correlation (Elbourne 2002). Also each participant will appear in both the treatment and control group, so the two groups will not be independent.
One cross‐over study adjusted for the cross‐over design via mixed effects analysis of variance, but the paper did not present the results in a way that allowed paired analyses in this review (Jaques 2008). Therefore, we have presented adjusted results from the paper narratively or in additional tables. For the remaining two cross‐over studies, continuous data (e.g. lung function and sputum weight) were presented from adjusted paired analyses, therefore we have used a generic inverse variance method to analyse continuous outcomes where possible, but present the remaining data from this study in narrative form (de Boeck 2017; Minasian 2010).
We did not plan to combine data from parallel studies with data from cross‐over studies and have analysed these separately.
Dealing with missing data
In the event of missing, incomplete, or unclear data, we contacted the original investigators and the manufacturer of mannitol (Pharmaxis). If we could not obtain the necessary data for analysis, we reported the outcomes in a narrative way.
Assessment of heterogeneity
For the pooled analyses, we assessed the degree of statistical heterogeneity between studies using the I² statistic (Higgins 2003). This measure describes the percentage of total variation across studies that are caused by heterogeneity rather than by chance (Higgins 2003). The values of I² lie between 0% and 100%, and a simplified categorisation of heterogeneity that the we used is of low (I² value of less than 25%), moderate (I² value of between 25 and 50%), and high (I² value of over 50%) (Higgins 2003).
We assessed clinical heterogeneity between studies by comparing study characteristics and participant demographics and planned to assess any differences found between studies via subgroup analyses.
Assessment of reporting biases
The manufacturer of mannitol, Pharmaxis, sponsored all the studies included in the review. We did not assess publication bias among the studies as there were insufficient studies (i.e. fewer than 10). In future updates, we may do this using the funnel plot method discussed in the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). If asymmetry is present, we will explore possible causes including publication bias, methodological quality, and true heterogeneity.
Data synthesis
We entered the data extracted from included studies into the Review Manager software (Review Manager 2014).
We computed pooled estimates of the treatment effect for each outcome using a fixed‐effect model. We planned to use a random‐effects model to analyse outcomes with high heterogeneity (I² value of over 50%) in the data. We used the inverse variance method to pool continuous data and the Mantel‐Haenszel method to pool dichotomous data, unless stated above that we would use the generic inverse variance method (seeMeasures of treatment effect and Unit of analysis issues).
Subgroup analysis and investigation of heterogeneity
For the comparison of mannitol versus control (where possible), we performed the following subgroup analyses for all primary outcomes (and for the secondary outcome of pulmonary exacerbations) in order to investigate potential differences in treatment effect across the subgroups:
age: children (under 18 years) and adult (18 years and over);
dornase alfa: participants using or not using dornase alfa;
In future updates of this review where there are sufficient studies included, we plan to investigate the possible causes of heterogeneity further by performing the additional subgroup analyses:
sex;
disease severity: FEV1 % predicted over 90%, 70% and over but less than 90%, 40% and over but less than 70%, up to 40%;
infection with Pseudomonas aeruginosa.
Sensitivity analysis
There were insufficient data to test the robustness of the review results through a sensitivity analysis on the basis of the risk of bias of the included studies, e.g. unpublished studies, allocation concealment, assessor blinding, and loss to follow‐up on the results. In future updates of the review, we may do this if we are able to include sufficient studies.
Summary of findings and the quality of the evidence (GRADE)
In a post hoc change, we have presented five summary of findings tables, one for each comparison (Table 1; Table 2; Table 3; Table 4; Table 5).
Summary of findings 1. Summary of findings ‐ 400 mg inhaled mannitol compared with 50 mg inhaled mannitol for cystic fibrosis.
| 400 mg inhaled mannitol compared with 50 mg inhaled mannitol for CF | ||||||
|
Patient or population: adults, children and young people with CF Settings: outpatients Intervention: 400 mg inhaled mannitol Comparison: 50 mg (sub‐therapeutic) inhaled mannitol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| 50 mg inhaled mannitol | 400 mg inhaled mannitol | |||||
|
HRQoL ‐ all domains (change from baseline) Scale: age‐appropriate versions of the CFQ‐R questionnaire Follow‐up: up to 6 months |
There were no consistent statistically significant differences between treatment groups in changes from baseline for any domains of the CFQ‐R at any of the time points for which data were available. | NA | 324 ‐ 507 participants (variable by domains) 2 studies |
⊕⊕⊝⊝ low1,2 | ||
|
Lung function: FEV1 mL (change from baseline) Follow‐up: up to 6 months, repeated measures |
The mean change from baseline in FEV1 mL ranged across the 50 mg mannitol groups from 26.0 to 32.5. | The mean change from baseline in FEV1 mL in the 400 mg mannitol groups was on average 86.5 higher (95% CI 45.2 to 127.9 higher). | NA | 600 participants 2 studies |
⊕⊕⊕⊝ moderate1 | Data provided by mannitol manufacturer Pharmaxis were analysed via a MMRM analysis. |
|
Lung function: FEV1 % predicted (change from baseline) Follow‐up: up to 6 months, repeated measures |
The mean change from baseline in FEV1 % predicted ranged across the 50 mg mannitol groups from 0.62 to 1.63. | The mean change from baseline in FEV1 % predicted in the 400 mg mannitol groups was on average 3.89 higher (95% CI 1.69 to 6.08 higher). | NA | 600 participants 2 studies |
⊕⊕⊕⊝ moderate1 | Data provided by mannitol manufacturer Pharmaxis were analysed via a MMRM analysis. |
|
Lung function: FVC mL (change from baseline) Follow‐up: up to 6 months, repeated measures |
The mean change from baseline in FVC mL ranged across the 50 mg mannitol groups from 15.9 to 47.5. | The mean change from baseline in FVC mL in the 400 mg mannitol groups was on average 102.2 higher (95% CI 48.4 to 155.9 higher). | NA | 600 participants 2 studies |
⊕⊕⊕⊝ moderate1 | Data provided by mannitol manufacturer Pharmaxis were analysed via a MMRM analysis. |
|
Lung function: FEF25-75 mL/s (change from baseline) Follow‐up: up to 6 months, repeated measures |
The mean change from baseline in FEF25-75 mL/s ranged across the 50 mg mannitol groups from 10.87 to 46.7. | The mean change from baseline in FEF25-75 mL/s in the 400 mg mannitol groups was on average 42.67 higher (95% CI ‐28.07 lower to 113.42 higher). | NA | 600 participants 2 studies |
⊕⊕⊕⊝ moderate1 | Data provided by mannitol manufacturer Pharmaxis were analysed via a MMRM analysis. |
|
Adverse events relating to treatment Scale: mild, moderate, severe and total Follow‐up: up to 6 months |
The most commonly adverse events reported were cough and haemoptysis (in 5% and 2% of participants respectively). | The most commonly adverse events reported were cough and haemoptysis (in 10% and 5% of participants respectively). | See comment | 600 participants 2 studies |
⊕⊕⊕⊝ moderate1 | We found no statistically significant differences in rates of adverse events related to treatment (of all severities) between treatment groups. |
| *For lung function outcomes, the basis for the assumed risk is the range of mean values in the control group and the corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). For Health related Quality of Life and Adverse events, the basis of the assumed risk and the corresponding risk is described in the comments CF: cystic fibrosis;CFQ‐R: Cystic Fibrosis Questionnaire‐Revised version, CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; HRQoL: health‐related quality of life; MMRM: mixed model repeated measures; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Evidence downgraded due to indirectness: the participant population included only those with CF who passed the tolerance test and not all potential participants with CF.
2. Evidence downgraded due to indirectness: the CFQ‐R tool used in the studies was not designed to assess mucolytics. Also, pooling of the age‐appropriate tools may not be valid so results should be interpreted with caution.
Summary of findings 2. Summary of findings ‐ Inhaled mannitol compared with control (non‐respirable mannitol) for cystic fibrosis.
| Inhaled mannitol compared with control (non‐respirable mannitol) for CF | ||||||
|
Patient or population: adults, children and young people with CF Settings: outpatients Intervention: inhaled mannitol Comparison: non‐respirable mannitol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Non‐respirable mannitol | Inhaled mannitol | |||||
|
HRQoL ‐ all domains (change from baseline) Scale: age‐appropriate versions of the CFQ‐R questionnaire Follow‐up: 2 weeks |
At the end of the study there were no significant differences between mannitol and control for the respiratory, health, physical and vitality domains. | NA | 391 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2,3 | ||
|
Lung function: FEV1 mL (absolute change from baseline) Follow‐up: 2 weeks |
A statistically significant improvement on mannitol compared to control was observed. | NA | 391 1 study |
⊕⊕⊝⊝ low1,2 | ||
|
Lung function: FEV1 % predicted (change from baseline) Follow‐up: 2 weeks to 8 weeks |
One study showed a statistically significant improvement in absolute change from baseline on mannitol compared to control at 2 weeks. The second study showed statistically significant improvement in both absolute and relative change from baseline on mannitol compared to control at 8 weeks. |
NA. | 1261 2 cross‐over studies |
⊕⊕⊝⊝ low1,2 | ||
|
Lung function: FVC mL or % predicted (change from baseline) Follow‐up: 2 weeks to 8 weeks |
No statistically significant differences in absolute or relative change from baseline in FVC (mL or % predicted) were found in either study. | NA | 1261 2 cross‐over studies |
⊕⊕⊝⊝ low1,2 | ||
|
Lung function: FEF25-75 mL/s or % predicted (change from baseline) Follow‐up: 2 weeks to 8 weeks |
One study showed a statistically significant improvement in absolute change from baseline in FEF25-75 (mL/S) on mannitol compared to control at 2 weeks. The other study showed statistically significant improvement in both absolute and relative change from baseline in in FEF25-75 (% predicted) on mannitol compared to control at 8 weeks. |
NA | 1261 2 cross‐over studies |
⊕⊕⊝⊝ low1,2 | ||
|
Adverse events relating to treatment Scale: mild, moderate, severe and total Follow‐up: 2 weeks to 8 weeks |
The most commonly reported adverse events in both groups in the two studies were cough, haemoptysis, headache, nasopharyngitis and lung infections. | NA | 123‐1254 2 cross‐over studies |
⊕⊕⊝⊝ low1,2 | Frequencies of adverse events according to severity and association to treatment only were reported, a statistical comparison was not made in either study. | |
| *The basis of the assumed risk and the corresponding risk is described in the comments. The study authors adjusted for the cross‐over design of the study via a mixed model of analysis of variance when analysing and presenting results, however the format of the presented data does not allow us to perform analyses in this review. Published results from the study paper are presented CF: cystic fibrosis;CFQ‐R: Cystic Fibrosis Questionnaire‐Revised version; CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; HRQoL: health‐related quality of life; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1. In one of the studies it was stated that 39 participants were randomised, unclear how many were evaluated for each outcome. In the other study, the study may have been underpowered and imputation of missing data may have introduced bias (evidence downgraded due to risk of bias of incomplete outcome data).
2. Evidence downgraded due to indirectness: the participant population included only those with CF who passed the tolerance test and not all potential participants with CF.
3. Evidence downgraded due to indirectness: the CFQ‐R tool used in the studies was not designed to assess mucolytics. Also, pooling of the age‐appropriate tools may not be valid so results should be interpreted with caution.
4. One of the studies, adverse event data available for 38 and 36 participants in the mannitol and control groups respectively.
Summary of findings 3. Summary of findings ‐ Inhaled mannitol compared with control (non‐respirable mannitol) for people with cystic fibrosis, hospitalised due to pulmonary exacerbations.
| Inhaled mannitol compared with control (non‐respirable mannitol) for people with cystic fibrosis, hospitalised due to pulmonary exacerbations | ||||||
|
Patient or population: children and young people with CF Settings: children and young people hospitalised due to pulmonary exacerbations (14 days) and up to 1 month of outpatient follow‐up Intervention: inhaled mannitol Comparison: non‐respirable mannitol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Non‐respirable mannitol | Inhaled mannitol | |||||
|
HRQoL ‐ change from baseline in CFCS total score and CFQ‐R respiratory domain Follow‐up: up to 1 month |
There was no statistically significant difference in the change from baseline in HRQoL (CFCS total score or CFQ‐R respiratory domain) at hospital discharge or at 1 month follow‐up. | NA | 22 1 study |
⊕⊝⊝⊝ very low1,2,3 | ||
|
Lung function: FEV1 mL% predicted (change from baseline) Follow‐up: NA |
Outcome not reported. | NA | ||||
|
Lung function: FEV1 % predicted (change from baseline) Follow‐up: up to 1 month |
The mean difference in the change from baseline in FEV1 (% predicted in the mannitol group compared to the control group was 4.60% higher (3.80% lower to 13.00% higher) at hospital discharge and 5.40% higher (2.70% lower to 13.50% higher) after 1‐month follow‐up. | NA | 22 1 study |
⊕⊕⊝⊝ low1,2 | ||
|
Lung function: FVC % predicted (change from baseline) Follow‐up: up to 1 month |
The mean difference in the change from baseline in FVC (% predicted in the mannitol group compared to the control group was 2.80% higher (3.60% lower to 9.20% higher) at hospital discharge and 1.70% higher (4.10% lower to 7.50% higher) after 1‐month follow‐up. | NA | 22 1 study |
⊕⊕⊝⊝ low1,2 | ||
|
Lung function: FEF25-75 % predicted (change from baseline) Follow‐up: up to 1 month |
The mean difference in the change from baseline in FEV1 (% predicted in the mannitol group compared to the control group was 12.80% higher (3.30% lower to 28.90% higher) at hospital discharge and 3.90% higher (‐10.70% lower to 18.50% higher) after 1‐month follow‐up. | NA | 22 1 study |
⊕⊕⊝⊝ low1,2 | ||
|
Adverse events relating to treatment Follow‐up: up to 1 month |
Vomiting (mucous) haemoptysis (flecks only) and blurred vision each occurred in one participant in the control group. | Vomiting (mucous), dizziness and headaches each occurred in one participant in the 400mg mannitol group. | NA | 22 1 study |
⊕⊕⊝⊝ low1,2 | Also stated that no serious adverse events occurred. Severity and relationship to treatment not stated. |
| *The basis for the assumed risk is the range of mean values in the control group and the corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CF: cystic fibrosis; CFCS: Cystic Fibrosis Clinical Score;CFQ‐R: Cystic Fibrosis Questionnaire‐Revised version, CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; HRQoL: health‐related quality of life; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1. Evidence downgraded due to indirectness: the participant population included only those with CF who passed the tolerance test and not all potential participants with CF. The population recruited into the study were hospitalised, so results may not be applicable to all individuals with CF.
2. Evidence downgraded due to imprecision: small sample size and wide confidence intervals around the effect size.
3. Evidence downgraded due to indirectness: the CFQ‐R tool used in the studies was not designed to assess mucolytics. Also, pooling of the age‐appropriate tools may not be valid so results should be interpreted with caution.
Summary of findings 4. Summary of findings ‐ Inhaled mannitol compared with dornase alfa for cystic fibrosis.
| Inhaled mannitol compared with dornase alfa for CF | ||||||
|
Patient or population: children and young people with CF Settings: outpatients Intervention: inhaled mannitol Comparison: dornase alfa | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Dornase alfa | Inhaled mannitol | |||||
|
HRQoL ‐ all domains (change from baseline) Scale: age‐appropriate versions of the CFQ‐R questionnaire Follow‐up: up to 3 months |
No significant differences were found between treatment groups for any domains of the CFQ‐R. | NA | up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2,3 | ||
|
Lung function: FEV1 mL (percentage change from baseline) Follow‐up: up to 3 months |
The mean (SD) absolute change from baseline in the dornase alfa group was 84 (273) mL. | The mean (SD) absolute change from baseline in the mannitol group was ‐1 (279) mL. |
MD 2.80% (95% CI: ‐4.80% to 10.40%). |
up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Only the relative effect of percentage change from baseline could be analysed*. |
|
Lung function: FEV1 % predicted Follow‐up: NA |
Outcome not reported. | NA | ||||
|
Lung function: FVC mL (percentage change from baseline) Follow‐up: up to 3 months |
The mean (SD) absolute change from baseline in the dornase alfa group was 7 (415) mL. | The mean (SD) absolute change from baseline in the mannitol group was ‐58 (361) mL. |
MD 0.14% (95% CI: ‐0.02% to 0.30%). |
up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Only the relative effect of percentage change from baseline could be analysed*. |
|
Lung function: FEF25-75 mL/s (percentage change from baseline) Follow‐up: up to 3 months |
The mean (SD) absolute change from baseline in the dornase alfa group was 173 (310) mL/s. | The mean (SD) absolute change from baseline in the mannitol group was 55 (282) mL/s. |
MD ‐0.01% (95% CI: ‐0.23 to 0.21%). |
up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Only the relative effect of percentage change from baseline could be analysed*. |
|
Adverse events relating to treatment Scale: mild, moderate, severe and total Follow‐up: up to 3 months |
CF exacerbation was the most commonly reported adverse event (5% of participants). | Cough and CF exacerbation were the most commonly reported adverse events (22% and 17% of participants respectively). | See comment. | up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Frequencies of adverse events according to severity only were reported, a statistical comparison was not made. |
| *The basis of the assumed risk and the corresponding risk is described in the comments. For lung function outcomes, absolute data was not presented in a format which could be analysed due to the cross‐over design of the study, therefore only analyses of percentage change from baseline were included in this review CF: cystic fibrosis;CFQ‐R: Cystic Fibrosis Questionnaire‐Revised version, CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; HRQoL: health‐related quality of life; MD: mean difference; NA: not applicable; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Stated that 28 participants were randomised, unclear how many participants dropped out and how many were evaluated for each outcome (evidence downgraded due to incomplete outcome data). Evidence also downgraded due to imprecision, study is known to be underpowered.
2. Evidence downgraded due to indirectness: the participant population included only those with CF who passed the tolerance test and not all potential participants with CF.
3. Evidence downgraded due to indirectness: the CFQ‐R tool used in the studies was not designed to assess mucolytics. Also, pooling of the age‐appropriate tools may not be valid so results should be interpreted with caution.
Summary of findings 5. Summary of findings ‐ Inhaled mannitol plus dornase alfa compared with dornase alfa for cystic fibrosis.
| Inhaled mannitol plus dornase alfa compared with dornase alfa for CF | ||||||
|
Patient or population: children and young people with cystic fibrosis Settings: outpatients Intervention: inhaled mannitol plus dornase alfa Comparison: dornase alfa | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Dornase alfa | Inhaled mannitol plus dornase alfa | |||||
|
HRQoL ‐ all domains (change from baseline) Scale: age‐appropriate versions of the CFQ‐R questionnaire Follow‐up: up to 3 months |
No significant differences were found between treatment groups for any domains of the CFQ‐R. | NA | up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2,3 | ||
|
Lung function: FEV1mL (percentage change from baseline) Follow‐up: up to 3 months |
The mean (SD) absolute change from baseline in the dornase alfa group was 84 (273) mL. | The mean (SD) absolute change from baseline in the mannitol group was ‐31 (306) mL. |
MD ‐4.30% (95% CI: ‐14.10% to 5.50%). |
up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Only the relative effect of percentage change from baseline could be analysed*. |
|
Lung function: FEV1 % predicted Follow‐up: NA |
Outcome not reported. | NA | ||||
|
Lung function: FVC mL (percentage change from baseline) Follow‐up: up to 3 months |
The mean (SD) absolute change from baseline in the dornase alfa group was 7 (415) mL. | The mean (SD) absolute change from baseline in the mannitol group was ‐103 (394) mL. |
MD ‐0.07% (95% CI: ‐0.30% to 0.16%). |
up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Only the relative effect of percentage change from baseline could be analysed*. |
|
Lung function: FEF25-75 mL/s (percentage change from baseline) Follow‐up: up to 3 months |
The mean (SD) absolute change from baseline in the dornase alfa group was 173 (310) mL/s. | The mean absolute change from baseline in the mannitol group was 68 (489) mL/s. |
MD ‐0.03% (95% CI: ‐0.18 to 0.24%). |
up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Only the relative effect of percentage change from baseline could be analysed*. |
|
Adverse events relating to treatment Scale: mild, moderate, severe and total Follow‐up: up to 3 months |
CF exacerbation was the most commonly reported adverse event (5% of participants). | Cough and CF exacerbation were the most commonly reported adverse events (9% and 30% of participants respectively). | See comment. | up to 231 1 cross‐over study |
⊕⊝⊝⊝ verylow1,2 | Frequencies of adverse events according to severity only were reported, a statistical comparison was not made. |
| *The basis of the assumed risk and the corresponding risk is described in the comments. For lung function outcomes, absolute data was not presented in a format which could be analysed due to the cross‐over design of the study, therefore only analyses of percentage change from baseline were included in this review CF: cystic fibrosis;CFQ‐R: Cystic Fibrosis Questionnaire‐Revised version, CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; HRQoL: health‐related quality of life; MD: mean difference; NA: not applicable; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1 Stated that 28 participants were randomised, unclear how many participants dropped out and how many were evaluated for each outcome (evidence downgraded due to incomplete outcome data). Evidence also downgraded due to imprecision, study is known to be underpowered.
2. Evidence downgraded due to indirectness: the participant population included only those with CF who passed the tolerance test and not all potential participants with CF.
3. Evidence downgraded due to indirectness: the CFQ‐R tool used in the studies was not designed to assess mucolytics. Also, pooling of the age‐appropriate tools may not be valid so results should be interpreted with caution.
Primary outcomes at the latest‐reported time point are presented in the summary of findings tables (HRQoL, lung function (FEV1 (mL and % predicted separately), FVC (mL or % predicted), FEF25-75 (mL/s or % predicted) and adverse events from treatment).
For clarity in the tables, HRQoL and adverse events are not presented according to the sub‐domains in Effects of interventions; instead we have inserted a general statement about the summary of findings for these outcomes and the evidence is graded based on all of the sub‐domains combined.
We determined the quality of the evidence using the GRADE approach; and downgraded evidence in the presence of a high risk of bias in at least one study, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results, high probability of publication bias. They downgraded evidence by one level if they considered the limitation to be serious and by two levels if very serious.
Results
Description of studies
Results of the search
For the original version of the review, searches identified 51 unique references to 10 studies (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Middleton 2015; Minasian 2010; NCT02134353; Robinson 1999; Sadr 2019; Teper 2011). 10 of these papers referred to more than one of the included studies and are listed under all three study identifiers (Aitken 2012; Bilton 2011; NCT02134353). Pharmaxis also highlighted three publications relating to mannitol which were not eligible for inclusion (and are not even listed as excluded) as they were not randomised (Bilton 2012; Button 2013a; Button 2013b).
In the previous version of the review, four studies (36 references) were included (Aitken 2012; Bilton 2011; Jaques 2008; Minasian 2010), two studies (four references) were excluded (Robinson 1999; Teper 2011), two studies were listed as ongoing (now referred to as de Boeck 2017; NCT02134353) and and one study was listed as awaiting classification (Middleton 2015).
The search for the 2017 update identified five new records; one additional reference to an excluded study in the previous version of the review (Teper 2011), one additional reference to an included study in the previous version of the review (Aitken 2012) and three references to a study which was ongoing at the time of the previous review (de Boeck 2017). A total of 14 relevant trial registry entries were identified for the 2017 update from a search of Clinicaltrials.gov and WHO ICTRP (see Electronic searches).
The search of the Cystic Fibrosis Trials Register for the 2020 update identified four new records; three conference abstracts of an ongoing study (NCT02134353) and one reference from a trials registry (further investigation linked this entry to two published full papers) which were excluded (Sadr 2019). Searches of trial registries identified 318 records; all of which were clearly irrelevant or had been identified in previous versions of the review.
Hence, the current version of the review includes six studies (62 records, 50 published references) (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Middleton 2015; Minasian 2010), excludes three studies (eight records) (Robinson 1999; Sadr 2019; Teper 2011) and one study remains ongoing (NCT02134353). This is shown in a study flow chart in the figures (Figure 1).
1.
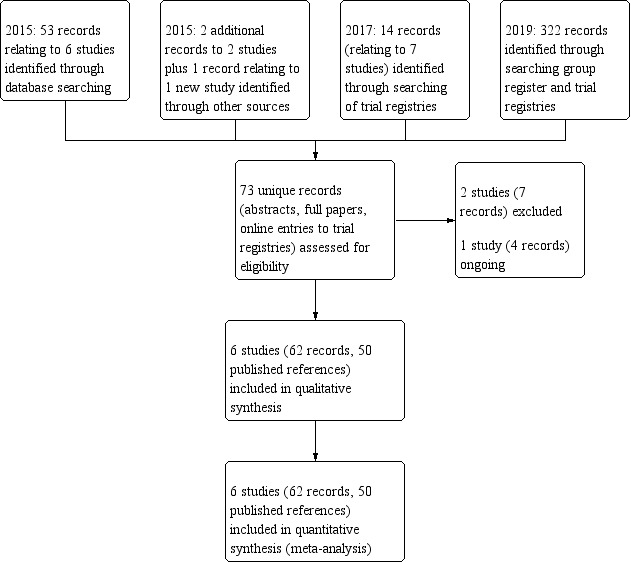
Study flow diagram.
Included studies
Six studies met the review's inclusion criteria; all were available as full papers and related abstracts, posters and unpublished presentation slides were consulted for additional information (Aitken 2012; Bilton 2011; de Boeck 2017Jaques 2008; Middleton 2015; Minasian 2010). One of these included studies had 14 publications (Aitken 2012), one had 18 publications (Bilton 2011) (10 publications discussed both the Aitken and Bilton studies and are listed under both studies in this review), one had four publications (de Boeck 2017), one had five publications (Jaques 2008), one had three publications (Middleton 2015) and one had had two publications (Minasian 2010).
Trial design
All six studies were randomised, three were of parallel design (Aitken 2012; Bilton 2011; Middleton 2015) and three had a cross‐over design (de Boeck 2017; Jaques 2008; Minasian 2010).
The duration of the studies ranged from two weeks (Jaques 2008) to one year (consisting of a 26‐week randomised treatment period with a further 26‐week open‐label follow‐up period; data from the follow‐up period are not eligible for consideration in this review) (Aitken 2012; Bilton 2011). Five studies were described as double‐blind (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Middleton 2015) and one study was open‐label (Minasian 2010) (see Risk of bias in included studies). Five studies were described as were multicentre and conducted in Australia, New Zealand, Europe, and North and South America (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Minasian 2010). One study was conducted in Australia, but it was not stated whether this study was single or multicentre (Middleton 2015).
Participants
A total of 784 randomised participants, children, young people and adults aged six to 56 years from CF clinics, were included in the six studies. In three studies the mean age was late teens or early 20s (Aitken 2012; Bilton 2011; Jaques 2008) and in three studies the mean age was between 12 and 14 years (de Boeck 2017; Middleton 2015; Minasian 2010). Two studies had roughly equal numbers of male and female participants (Aitken 2012; Bilton 2011) and the remaining four studies recruited more females than males; 59% females in two studies (Jaques 2008; Middleton 2015), 60% females in one study (de Boeck 2017) and 64% females in the final study (Minasian 2010).
In all studies, the diagnosis of CF was made by clinical assessment and by sweat or genetic testing, or both (information available in study publication or provided by study sponsor Pharmaxis). In five studies the participants were described as having CF and being clinically stable at the start of treatment (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Minasian 2010); the mean baseline FEV1 ranged from 62% (Bilton 2011) to 72.23% predicted (de Boeck 2017). In the final study, participants within the study were hospitalised with a pulmonary exacerbation (as defined by Fuchs criteria (Fuchs 1994)) (Middleton 2015). Within this study, compared to the mannitol group, the control group had lower mean (SD) admission FEV1 (58.0 (13.0)% predicted versus 73.1 (15.6)% predicted, P < 0.05) and mean (range) FEF25-75 (25.7 (20.1 – 71.8)% predicted versus 44.1 (22.8 – 92.43)% predicted, P < 0.05).
The following table summarises participant baseline characteristics in the included studies.
|
Design / Characteristic |
Aitken 2012 (n = 305) |
Bilton 2011 (n = 295) |
de Boeck 2017 (n = 95) |
Jaques 2008 (n = 39) |
Middleton 2015 (n = 22) |
Minasian 2010 (n = 28) |
| Design | Parallel study | Parallel study | Cross‐over study | Cross‐over study | Parallel study | Cross‐over study |
| Interventions | 400 mg mannitol (n = 184) vs 50 mg mannitol (n = 121) |
400 mg mannitol (n = 177) vs 50 mg mannitol (n = 118) |
400mg mannitol vs control Baseline characteristics reported for 92 participants Results reported for 87 participants |
Mannitol vs non‐respirable mannitol Results reported for 39 participants |
400 mg mannitol (n = 11) vs control (n = 11) | Mannitol or mannitol plus dornase alfa vs dornase alfa alone Results reported for 20 participants who completed the study |
| Age | Mean: 20 years Range: 6 ‐ 53 years Aged < 18 years: n = 154 |
Mean (SD): 23 (11.3) years Aged < 18 years: n = 105 |
Mean age (SD): 12.0 (3.0) years Range 6 to 17 years |
Mean: 19.1 years Range: 8 ‐ 48 years |
Mean age (SD): Mannitol group = 13.2 (3.7) years; Control group = 14.4 (3.0) years Range 8 to 18 years . |
Mean (SD) age: 13.3 (2.24) years For 20 who completed Mean (SD) age: 13.2 (2.4) years |
| Gender | 51.5% male (n = 157) 48.5% female (n = 148) |
55.3% male (n = 163) 44.7% female (n = 132) |
40% male (n = 37) 60% female (n = 55) |
41% male (n = 16) 59% female (n = 23) |
Mannitol group = 45% male (n=5), 55% female (n=6); Control group = 36% male (n=4); 64% female (n=7) |
36% male (n = 10) 64% female (n = 18) For 20 who completed 30% male (n = 6) 70% female (n = 14) |
| FEV1 | Mean (SD) 63.8 (15.9)% predicted | Mean (SD): 62.0 (16.3)% predicted | Mean (SD): 72.23 (11.6) % predicted |
Mannitol group
Mean (SD) 64.9 (13.0)% predicted Control group Mean (SD) 64.4 (11.8)% predicted |
Mannitol group Mean (SD): 73.1 (15.6)% predicted Control group Mean (SD): 58.0 (13.6)% predicted |
For 20 who completed Mean (SD): 1670 (500) mL Mean (SD): 64 (10)% predicted |
| Dornase alfa use | 75.1% (n = 229) of total participants received concomitant dornase alfa 74.5% (n = 137) of the 400 mg mannitol group 76% (n = 92) of the 50 mg mannitol group |
55.3% (n = 163) of total participants received concomitant dornase alfa 54.2% (n = 96) of the 400 mg mannitol group 56.8% (n = 67) of the 50 mg mannitol group |
68.5% (n = 63) had previously used or were using dornase alfa at screening | 46.2% (n = 18) received concomitant dornase alfa | Stated that dornase alfa was used regularly in 59% of the total cohort | NA (dornase alfa is an intervention in the study) |
FEV1: forced expiratory volume in one second; NA: not applicable; SD: standard deviation; vs: versus.
Interventions
Five studies compared mannitol to a control treatment; two studies used low‐dose mannitol 50 mg as the control (Aitken 2012; Bilton 2011), one study used 'very low dose' (50 mg) non‐respirable mannitol (Middleton 2015), one study used non‐respirable mannitol with a fine particle fraction over 2% (Jaques 2008) and one study used 10 mg of non‐spray dried, non‐respirable raw material mannitol (de Boeck 2017). One study compared mannitol to mannitol plus dornase alfa and to dornase alfa alone (Minasian 2010).
In the Aitken study, 74.5% of participants randomised to mannitol and 76% of participants randomised to control were concomitant dornase alfa users (Aitken 2012). In the Bilton study, 54.2% of participants randomised to mannitol and 56.8% of participants randomised to control were concomitant dornase alfa users (Bilton 2011). The the remaining studies 68.5% (de Boeck 2017); 64% (Jaques 2008) and 59% (Middleton 2015) had previously used or were concomitant dornase alfa users. In three of the studies, randomisation was stratified according to existing dornase alfa use and participants were instructed under protocol not to change their dornase alfa use from baseline (Aitken 2012; Bilton 2011; de Boeck 2017).
Five studies evaluated inhaled mannitol 400 mg twice daily (Aitken 2012; Bilton 2011; de Boeck 2017; Middleton 2015; Minasian 2010) and one study evaluated mannitol 420 mg twice daily (Jaques 2008). Treatment periods in the randomised phase were 12 days (Middleton 2015), two weeks (Jaques 2008), eight weeks (de Boeck 2017), 12 weeks (Minasian 2010) and 26 weeks (Aitken 2012; Bilton 2011). After completion of the randomised phase of the study, two studies had an open‐label follow‐up phase lasting up to 52 weeks (Aitken 2012; Bilton 2011).
Outcomes
In terms of the Primary outcomes of this review; all six included studies reported at least one measure of lung function and reported details of adverse events; five studies reported HRQoL, but one did not (de Boeck 2017).
For some of the included studies, additional information was provided on these outcomes by Pharmaxis (see Characteristics of included studies for details of additional data provided).
The majority of the Secondary outcomes defined in the review were also reported in the published papers or information was provided by Pharmaxis, only 'Time off school or work' was not recorded in any included study.
Excluded studies
Three studies (nine references), each published as full papers, were excluded (Robinson 1999; Sadr 2019; Teper 2011). One was a single‐dose study which examined the effect of mannitol on bronchial mucus clearance (Robinson 1999). The second study did not randomise selection to interventions, but describes using random sampling to identify participants (Sadr 2019). The final study was a dose‐response type study, and we could not incorporate any data on a clinically‐used dose (Teper 2011).
Ongoing studies
One randomised, double‐blind and parallel study run by Pharmaxis, the manufacturer of mannitol, is ongoing (NCT02134353). Participants over 18 years of age have been recruited, and the design is multicentre across North and Central America, Europe and South Africa. Inhaled mannitol 400 mg twice daily is being compared to inhaled control twice daily and the study duration is reported as 26 weeks. The primary outcome measure is mean change in FEV1 (mL). The study commenced in October 2014 and investigators are due to collect final data for the primary outcome measure in March 2016 (NCT02134353).
Risk of bias in included studies
Several details relating to data and study design were not available from the published papers so we requested this missing information from the pharmaceutical company (Pharmaxis) or authors, or both. Pharmaxis provided analysis results from the studies and answered some specific follow‐up queries, so we were able to complete risk of bias assessments. Additional data were also received from Dr Minasian. A visual summary of the risk of bias judgements is presented as a figure (Figure 2).
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Generation of sequence
We judge the risk of bias in five studies to be low (Aitken 2012; Bilton 2011; Jaques 2008; Middleton 2015; Minasian 2010). It was stated in one study that the randomisation code used was externally generated in a small block design, stratified by site and dornase alfa use (Jaques 2008). In a second study, it was stated that randomisation was performed (1:1) ratio via random number generation without stratification (Middleton 2015). For the remaining three studies, there was not sufficient information in the original publications to allow a clear judgement. For the Aitken and Bilton studies Pharmaxis provided additional details; in both studies, the master randomisation lists were prepared by an external company; randomisation numbers were generated, stratified by country and dornase alfa use, and randomisation blocking by country was done in paired blocks (Aitken 2012; Bilton 2011). In the final study, Dr Minasian confirmed that participants were each allocated a unique randomisation number and treatment schedule with equal probability for assignment to treatment sequences. Randomisation was carried out in balanced blocks with separate schedules created for each of the two recruiting centres (Minasian 2010).
One study was judged to be at unclear risk of bias (de Boeck 2017). It was stated that randomisation was conducted in a 1:1 ratio and stratified according to age (6 to 11 years or 12 to 17 years) and dornase alfa use (user or non‐user), but no information was provided with regards to how the random sequence was generated.
Concealment of allocation
Pharmaxis provided additional information for two studies describing how an interactive voice response system was used to manage randomisation and the risk of bias for these studies was judged to be low (Aitken 2012; Bilton 2011). No information regarding allocation concealment was available for the remaining four studies, so these are judged to be at an unclear risk of bias (de Boeck 2017; Jaques 2008; Middleton 2015; Minasian 2010).
Blinding
The risk of bias from blinding is judged to be low for participants, clinicians and outcome assessors in three studies (Aitken 2012; Bilton 2011; Jaques 2008). The Aitken study was described as double‐blind and the mannitol and control capsules were identical in appearance, taste and method of administration, so the risk of bias was judged to be low for participants and clinicians. It was unclear from the publication whether outcome assessors were blinded; however, Pharmaxis have confirmed that investigators and all study staff were blinded (Aitken 2012). In the Bilton study, participants, clinicians and outcome assessors were also blinded (the control group received sub‐therapeutic doses of mannitol); Pharmaxis confirmed this study likewise used mannitol and control capsules which were identical in appearance, taste and method of administration (Bilton 2011). In the Jaques study, participants were blinded as mannitol and control were also identical in taste and appearance.The study publication states that "study staff and investigators" were blinded; Pharmaxis confirmed that the statistician was part of the "study staff" and hence also blinded, we therefore consider the risk of bias to be low (Jaques 2008).
In one study, the risk of bias for participants and clinicians was judged to be low risk of bias as the double‐blind was achieved with a placebo also containing a low dose of mannitol (de Boeck 2017). However, it was not stated if outcome assessors were blinded in this study so the risk of bias for outcome assessors was unclear.
One study was described as 'double‐blind' but no information was given about how the blinding was achieved or who was blinded, therefore the risk of bias for participants, clinicians and outcome assessors was judged to be unclear (Middleton 2015).
The final study was an open study, so we judge the risk of bias from lack of blinding to be high for participants, clinicians and outcome assessors (Minasian 2010).
Incomplete outcome data
Three studies were judged to have a low risk of bias (Aitken 2012; Bilton 2011; Middleton 2015). In one study, there was higher drop‐out rate with mannitol (17% in the 400 mg mannitol group versus 12% for 50 mg mannitol) due to adverse events and other reasons, e.g. withdrawal of consent (Aitken 2012); and in a second study 37% during 400 mg mannitol treatment and 28% in the 50 mg mannitol group, for which the authors of the study considered one possible reason to be the high burden of treatment (Bilton 2011). Sensitivity analyses conducted by Pharmaxis (methods of imputation of missing data for withdrawals) showed a consistent treatment effect in favour of mannitol and no change to conclusions. In the third study judged to have low risk of bias, results were reported for all 22 randomised participants (Middleton 2015).
One study was judged to have unclear risk of bias; even though reasons for withdrawal were clearly documented in a flow diagram and primary and secondary efficacy endpoints were reported to have been analysed on an intention‐to‐treat (ITT) basis, it is also stated that missing data were imputed using 'last observation carried forward' (de Boeck 2017). Such a method does not capture change over time, so may have introduced bias into results. However, it is not stated how much data have been imputed and for how many participants, therefore the risk of bias is unclear.
The remaining two studies were judged to have a high risk of bias for this domain (Jaques 2008; Minasian 2010). In the Jaques study, three participants withdrew because of adverse effects and there was also an unexplained withdrawal of participants by the physician (Jaques 2008). It is unclear how many participants were evaluated for outcomes and, in addition it is not stated why the 56‐day follow‐up data were not reported. In the Minasian study, there were many dropouts during the study, but little information was provided about why this occurred; eight participants withdrew because of adverse events or treatment burden. Additionally, Minasian only reported results for participants who completed all three arms of the cross‐over study (per protocol analysis) (Minasian 2010).
Selective reporting
Limited information regarding the outcomes considered in this review was available in the publications for four included studies, particularly for HRQoL, for measures of lung function other than FEV1 and for the review's secondary outcomes (Aitken 2012; Bilton 2011; Jaques 2008; Minasian 2010). However, for three of the studies detailed additional information was provided on request for all of the primary outcomes and the majority of the secondary outcomes included in this review; for two studies information was provided by Pharmaxis (Aitken 2012; Bilton 2011) and for one study by the principle investigator (Minasian 2010). Therefore these three studies were judged to be at low risk of bias.
Some additional information was also supplied by Pharmaxis for the fourth study (Jaques 2008); however, as there was insufficient detail to include data for many of the secondary outcomes of this review, this study is judged to be at unclear risk of bias.
For the remaining two studies which were phase 2 and pilot studies respectively, examining efficacy and safety, all important outcomes relating to efficacy and safety appear to have been reported, therefore the two studies were judged to be at low risk of selective reporting bias (de Boeck 2017; Middleton 2015).
Other potential sources of bias
We identified a number of other potential sources of bias in the included studies and all studies are judged to be at high risk of 'other bias'.
As there is a risk of bronchoconstriction with mannitol, participants in all studies underwent a mannitol tolerance test at screening; those who failed the test or in whom the test was incomplete did not enter the study. Thus, the participant population included only those people with CF who passed the tolerance test and not all potential participants with CF.
All studies were funded and sponsored by Pharmaxis, and the manufacturer participated in the study design, data collection, data analysis, data interpretation and writing of the reports for five of the studies, therefore the risk of bias for all studies is high (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Minasian 2010).
Three studies had a cross‐over design but two of these studies reported little information on potential carry‐over effects between arms of the study (Jaques 2008; Minasian 2010).
One pilot study notes a relatively small sample size which prevents "strong conclusions about clinical utility in this setting" (Middleton 2015). The remaining five studies reported sample size calculations and were reported to be powered to detect changes in FEV1; however, one study did not manage to recruit a sufficient number of participants and a priori and post‐hoc power calculations are based on different sizes of treatment difference (de Boeck 2017); while a further study may have been underpowered (Minasian 2010).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Five studies compared mannitol with control (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Middleton 2015). Two studies comparing mannitol to low‐dose (sub‐therapeutic) mannitol had similar designs and homogeneous populations, so we were able to pool the results analyse these for the time points up to two months, up to four months and up to six months (Aitken 2012; Bilton 2011). Two studies were cross‐over in design and of shorter duration with different control treatments; therefore we report these studies under a separate comparison (de Boeck 2017; Jaques 2008). The fifth study comparing mannitol to low‐dose (sub‐therapeutic) mannitol in individuals with CF who had been hospitalised due to a pulmonary exacerbation was a pilot study (Middleton 2015). Due to the difference in the population recruited within this study (hospitalised patients) compared to the other studies (outpatients); we report this study under a third separate comparison (Middleton 2015).
The final study included in the review compared mannitol to mannitol plus dornase alfa and to dornase alfa alone; and the two comparisons are reported separately below (Minasian 2010).
In summary, the following comparisons are presented in this review.
Mannitol compared with control ‐ parallel studies of people with CF
Mannitol compared with control ‐ cross‐over studies of people with CF
Mannitol compared with control ‐ parallel study of people with CF hospitalised due to pulmonary exacerbations
Mannitol compared with dornase alfa ‐ cross‐over study of people with CF
Mannitol plus dornase alfa compared with dornase alfa ‐ cross‐over study of people with CF
For all comparisons, we have taken the results we present from the final published papers, previously published abstracts and additional data provided by the manufacturer (Pharmaxis) and the authors (Dr Minasian). We note that where data have been converted to another measure for entry into analysis (for example, MDs and 95% CIs converted into MDs and SEs to enter via generic inverse variance), some conversions have lead to very slight differences (to one or two decimal places) between results in published papers and results in this review. We assume this is due to rounding of numbers within the published papers and we note no differences in statistical significance or conclusions between our analyses and the published papers.
Mannitol compared with control ‐ parallel studies of individuals with cystic fibrosis
Two parallel studies with 600 participants in the ITT population reported data for this comparison and additional data were provided by Pharmaxis (Aitken 2012; Bilton 2011). We refer to the intervention as 400 mg mannitol and the control treatment (sub‐therapeutic mannitol) as 50 mg mannitol throughout this section.
Primary outcomes
1. HRQoL
Both studies measured HRQoL using the age‐appropriate versions of the CFQ‐R questionnaire and reported on differing numbers of participants for different domains at different time points (for exact numbers please see the graphs in Data and analyses) (Aitken 2012; Bilton 2011). Although 'Burden of treatment' is part of the QoL assessment, these results are presented under a separate outcome below (secondary outcome 7). We note that results presented for the domains of respiratory, physical, emotion, eating, social, body, weight and digestion are the pooled results for all individuals (results from age‐appropriate version of the questionnaire combined and such a combination of results across questionnaires may not be valid (Quittner 2015 [pers comm]), therefore we encourage caution when interpreting the results from any of these domains. The domains of vitality, health, role and weight were assessed only in individuals over the age of 14 years. Furthermore, we are aware that the HRQoL tool used in the studies was not designed to be used to assess mucolytics and therefore the results for some domains, e.g. mucus production, should be interpreted in light of the mechanism of action of the drug.
In the publications, Bilton reported some limited data from the respiratory domain and Aitken reported that there were no differences between treatments for any of the QoL domains (Aitken 2012; Bilton 2011). Pharmaxis supplied additional data for the respiratory domain for both studies. When data were combined at the time points 'up to four months' and 'up to six months', no statistically significant differences were observed (Analysis 1.1; low‐quality evidence). It should be noted that as part of its mechanism of action mannitol increases sputum production; however, this has a negative result in the CFQ‐R questionnaires.
1.1. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 1: HRQoL ‐ respiratory (change from baseline)
Pharmaxis also supplied additional data for the change from baseline for other domains of the CFQ‐R for the Aitken and Bilton studies at up to four months and up to six months. For domains of physical, emotion, eating, health, social, body, role, weight and digestion, none of the results were statistically significant (Analysis 1.3; Analysis 1.4; Analysis 1.5; Analysis 1.6; Analysis 1.7; Analysis 1.8; Analysis 1.9; Analysis 1.10; Analysis 1.11), although the results favoured 400 mg mannitol for the vitality domain at six months, MD 4.84 (95% CI 0.86 to 8.82; P = 0.02) (Analysis 1.2).
1.3. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 3: HRQoL ‐ physical (change from baseline)
1.4. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 4: HRQoL ‐ emotion (change from baseline)
1.5. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 5: HRQoL ‐ eating (change from baseline)
1.6. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 6: HRQoL ‐ health (change from baseline)
1.7. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 7: HRQoL ‐ social (change from baseline)
1.8. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 8: HRQoL ‐ body (change from baseline)
1.9. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 9: HRQoL ‐ role (change from baseline)
1.10. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 10: HRQoL ‐ weight (change from baseline)
1.11. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 11: HRQoL ‐ digestion (change from baseline)
1.2. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 2: HRQoL ‐ vitality (change from baseline)
Some heterogeneity was observed between the two studies at six months in the domains of respiratory, physical, social and role; for all four domains the Aitken study showed an advantage to the 50 mg mannitol group and the Bilton study showed an advantage to the 400 mg mannitol group (of varying magnitudes), but not statistically significant. Given the subjective nature of this outcome and the limited number of studies included, we have not conducted any subgroup analyses to explore this heterogeneity and encourage caution when interpreting the numerical results from these domains.
Subgroup analyses ‐ dornase alfa use and age
Subgroup‐specific data for HRQoL were not available for either study (Aitken 2012; Bilton 2011).
2. Lung function
Pharmaxis provided additional lung function data from a MMRM for the two studies (seeMeasures of treatment effect for further information). The primary comparison in the original publications was on the average effect across the three time points up to and including six months. However, the results at each time point (which correspond to our analyses) were also presented.
No consistent heterogeneity was found for any of the outcomes at any of the time points.
a. FEV1 (mL or % predicted)
Data were available for the absolute change from baseline in FEV1 (mL) at up to two, four and six months for 600 participants in the ITT populations of both studies. We found a significant benefit for 400 mg mannitol compared with 50 mg mannitol at: up to two months, pooled MD 71.27 mL (95% CI 36.00 to 106.54, P < 0.0001); up to four months, pooled MD 64.54 mL (95% CI 26.65 to 102.42, P = 0.0008); and up to six months, pooled MD 86.50 (95% CI 45.15 to 127.86, P < 0.0001) (Analysis 1.12, moderate‐quality evidence).
1.12. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 12: FEV1 ml (repeated measures, change from baseline)
Data were also available for the relative change from baseline in FEV1 % predicted at up to two, four and six months for 600 participants in the ITT populations of both studies (Aitken 2012; Bilton 2011). Pooling these data, we found a significant benefit for 400 mg mannitol compared with 50 mg mannitol at: up to two months, pooled MD 2.98% (95% CI 1.04 to 4.92, P = 0.003); up to four months, pooled MD 3.26% (95% CI 1.16 to 5.35, P = 0.002); and up to six months, MD 3.89% (95% CI 1.69 to 6.08, P = 0.0005) (Analysis 1.13, moderate‐quality evidence).
1.13. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 13: FEV1 % predicted (repeated measures, change from baseline)
b. FVC (mL or % predicted)
Data were available for the absolute change from baseline in FVC (mL) at up to two, four and six months for 600 participants in the ITT populations of both studies (Aitken 2012; Bilton 2011). There was a statistically significant benefit for 400 mg mannitol compared with 50 mg mannitol at all three time points; MD 87.81 mL (95% CI 41.96 to 133.66, P = 0.0002), MD 89.24 mL (95% CI 40.55 to 137.92, P = 0.0003) and MD 102.17 mL (95% CI 48.40 to 155.94, P = 0.0002), respectively (Analysis 1.14, moderate‐quality evidence).
1.14. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 14: FVC mL (repeated measures, change from baseline)
FVC % predicted was not recorded in either of the studies.
c. FEF25-75 (mL/s or % predicted)
Data were available for the absolute change from baseline in FEF25-75 (mL/s) at up to two, four and six months for 600 participants in the ITT populations of both studies; there was no statistically significant difference between 400 mg mannitol and 50 mg mannitol at any time‐point (Analysis 1.15, moderate‐quality evidence).
1.15. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 15: FEF25-75 mL/s (repeated measures, change from baseline)
FEF25-75 % predicted was not recorded in either of the studies (Aitken 2012; Bilton 2011).
Subgroup analysis ‐ dornase alfa use
Subgroup data were available for the two studies for all measures of lung function at all time points of interest in the review. For brevity, the results of the subgroup analysis are summarised in the additional tables (Table 6). As in the primary analysis above, generally the change from baseline of the measures of lung function was significantly improved in the 400 mg mannitol group compared to the 50 mg mannitol group in both subgroups (dornase alfa users and non‐users). The MD in the change from baseline was larger in the non‐user subgroup than the user subgroup for the majority of measures and time points; however, there was no significant difference between the subgroups for any of the measures (Table 6).
1. Mannitol versus control ‐ subgroup analysis by dornase alfa use (lung function).
| Outcome | Time point | Dornase alfa users (n = 373)* | Dornase alfa non users (n = 196)* |
P value for test of subgroup differences |
||||
| MD | 95% CI | P value | MD | 95% CI | P value | |||
| FEV1 (mL) | up to 2 months | 54.13 | 6.67 to 101.95 | 0.03 | 104.90 | 42.36 to 167.44 | 0.001 | 0.21 |
| up to 4 months | 49.25 | ‐5.87 to 104.37 | 0.08 | 82.77 | 8.38 to 157.16 | 0.03 | 0.48 | |
| up to 6 months | 90.76 | 28.32 to 153.19 | 0.004 | 103.22 | 19.82 to 186.63 | 0.02 | 0.82 | |
| FEV1 (% predicted) |
up to 2 months | 2.29 | ‐0.49 to 5.07 | 0.11 | 4.69 | 1.11 to 8.27 | 0.01 | 0.30 |
| up to 4 months | 2.05 | ‐10.6 to 5.17 | 0.20 | 4.49 | 0.44 to 8.54 | 0.03 | 0.35 | |
| up to 6 months | 4.66 | 1.46 to 7.86 | 0.004 | 4.05 | ‐0.11 to 8.21 | 0.06 | 0.82 | |
| FVC (mL) | up to 2 months | 70.99 | 9.98 to 132.00 | 0.02 | 117.82 | 33.59 to 202.05 | 0.01 | 0.38 |
| up to 4 months | 59.45 | ‐8.08 to 126.99 | 0.08 | 127.34 | 31.20 to 223.48 | 0.01 | 0.26 | |
| up to 6 months | 114.56 | 38.30 to 190.82 | 0.003 | 121.93 | 15.76 to 228.11 | 0.02 | 0.91 | |
| FEF25-75 (mL/s) | up to 2 months | 22.59 | ‐68.62 to 113.80 | 0.63 | 129.14 | 12.95 to 245.33 | 0.03 | 0.16 |
| up to 4 months | 6.52 | ‐87.14 to 100.18 | 0.89 | 99.17 | ‐22.97 to 221.30 | 0.11 | 0.24 | |
| up to 6 months | 47.83 | ‐58.72 to 154.38 | 0.38 | 44.70 | ‐97.52 to 186.92 | 0.54 | 0.97 | |
| Outcome | Time point | RR | 95% CI | p value | RR | 95% CI | P value |
P value for test of subgroup differences |
| Exacerbations* | up to 6 months | 0.85 | 0.58 to 1.25 | 0.41 | 0.46 | 0.25 to 0.87 | 0.02 | 0.1 |
CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; MD: mean difference; RR: risk ratio.
MD and 95% CI for measures of lung function is change from baseline pooled across two studies (Aitken 2012; Bilton 2011).
RR and 95% CI represents the proportion of participants experiencing pulmonary exacerbations pooled across two studies (Aitken 2012; Bilton 2011).
* Intention‐to‐treat population analysed for lung function outcomes, all randomised participants were included for the outcome of pulmonary exacerbations: dornase alfa users (n = 392) and non‐users (n = 208).
Subgroup analysis ‐ age
Subgroup data were available for the two studies for all measures of lung function at all time points of interest in the review. For brevity, the results of the subgroup analysis are summarised in the additional tables (Table 7). The statistically significant benefits for 400 mg mannitol compared to 50 mg mannitol shown in the primary analyses above were also shown in the adult subgroup; however, this was not shown in the subgroup of children. There were no significant differences between the subgroups for the majority of the measures at most time points (Table 7).
2. Mannitol versus control ‐ subgroup analysis by age (lung function).
| Outcome | Time point | Adults (n = 317) | Children (n = 258) |
P value for test of subgroup differences |
||||
| MD | 95% CI | P value | MD | 95% CI | P value | |||
| FEV1 (mL) | up to 2 months | 89.3 | 38.96 to 139.64 | 0.001 | 50.24 | ‐7.82 to 108.30 | 0.09 | 0.32 |
| up to 4 months | 87.83 | 29.08 to 146.58 | 0.003 | 25.89 | ‐40.72 to 92.50 | 0.45 | 0.17 | |
| up to 6 months | 123.12 | 56.43 to 189.81 | 0.0001 | 62.52 | ‐11.65 to 136.69 | 0.09 | 0.23 | |
| FEV1 (% predicted) |
up to 2 months | 3.72 | 0.82 to 6.64 | 0.012 | 2.64 | ‐0.73 to 6.02 | 0.13 | 0.63 |
| up to 4 months | 4.23 | 0.98 to 7.48 | 0.01 | 1.34 | ‐2.42 to 5.10 | 0.49 | 0.25 | |
| up to 6 months | 5.74 | 2.36 to 9.13 | 0.001 | 3.03 | ‐0.78 to 6.84 | 0.12 | 0.30 | |
| FVC (mL) | up to 2 months | 101.66 | 35.15 to 168.16 | 0.003 | 69 | ‐5.0 to 142.99 | 0.07 | 0.52 |
| up to 4 months | 111.82 | 36.75 to 186.89 | 0.004 | 46.16 | ‐35.53 to 127.84 | 0.27 | 0.25 | |
| up to 6 months | 158.44 | 74.87 to 242.01 | 0.001 | 71.21 | ‐19.1 to 161.51 | 0.12 | 0.17 | |
| FEF25-75 (mL/s) | up to 2 months | 95.09 | 0.56 to 189.63 | 0.05 | 15.44 | ‐95.62 to 126.51 | 0.79 | 0.28 |
| up to 4 months | 108.42 | 10.23 to 206.63 | 0.03 | ‐53.07 | ‐165.97 to 59.84 | 0.36 | 0.03 | |
| up to 6 months | 59.27 | ‐55.59 to 174.12 | 0.31 | 31.29 | ‐96.72 to 159.30 | 0.63 | 0.75 | |
| Outcome | Time point | RR | 95% CI | P value | RR | 95% CI | P value |
P value for test of subgroup differences |
| Exacerbations | up to 6 months | 0.76 | 0.52 to 1.13 | 0.18 | 0.62 | 0.35 to 1.09 | 0.10 | 0.55 |
CI: confidence interval; FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; MD: mean difference; RR: risk ratio.
MD and 95% CI for measures of lung function is change from baseline pooled across two studies (Aitken 2012; Bilton 2011).
RR and 95% CI represents the proportion of participants experiencing pulmonary exacerbations pooled across two studies (Aitken 2012; Bilton 2011).
* Intention‐to‐treat population analysed for lung function outcomes, all randomised participants were included for the outcome of pulmonary exacerbations: adults (n = 341) and children (n = 259).
3. Adverse effects relating to treatment
a. type and number of adverse events (defined as mild, moderate or severe in this review)
Both studies used the following researcher‐defined classification when reporting adverse events: mild (awareness of sign or symptom but easily tolerated); moderate (discomfort causes interference with usual activity); and severe (incapacitating, or unable to do usual activities) (Aitken 2012; Bilton 2011). Both studies further defined serious adverse events as a reaction that results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, or is a congenital anomaly or birth defect. We have reported the classification applied in the Pharmaxis studies within the framework of our outcomes (seeTypes of outcome measures for review definitions of adverse event classifications). Aitken and Bilton both considered that the reported adverse events presented here were possibly or probably related to treatment. All data and analyses relate to the 'up to six months' time point; some data are taken from the published papers and other data were supplied additionally by Pharmaxis. A wide range of different adverse events were reported by both Aitken and Bilton relating to respiratory, gastrointestinal and musculoskeletal events, amongst others. We report the number of participants experiencing one or more of these events and the quality of the evidence for this outcome was judged to be moderate.
i. mild
The most common 'mild' adverse effects experienced by participants in both treatment arms were cough (3% of participants in the 400 mg mannitol group and 1.7% of participants in the 50 mg mannitol group), haemoptysis (1.7% of participants in the 400 mg mannitol group and 0.8% of participants in the 50 mg mannitol group) and pharyngolaryngeal pain (1.4% of participants in the 400 mg mannitol group and 1.3% of participants in the 50 mg mannitol group), with no clinically or statistically significant difference between the two study arms (Analysis 1.16). Three participants in the 400 mg mannitol group (0.8%) and two participants in the 50 mg mannitol group (0.8%) experienced an aggravation of their condition. Most other side effects were only reported in one or two participants per treatment arm. None of the results for mild adverse effects were statistically significant, although they may have clinical relevance for people with CF.
1.16. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 16: Adverse events at up to 6 months(mild)
ii. moderate
Considering 'moderate' adverse effects, 5.3% of participants in the 400 mg mannitol treatment group and 1.4% of participants in the 50 mg mannitol group experienced cough. Also, more participants experienced haemoptysis in the 400 mg mannitol group compared with those in the 50 mg mannitol group (2.8% compared to 0.4%). Pharyngolaryngeal pain was experienced in the 400 mg mannitol and 50 mg mannitol groups by 1.4% and 0.4% of participants, respectively. A total of 11 participants (3.0%) in the 400 mg mannitol group and four participants (1.7%) in the 50 mg mannitol group experienced aggravation of their condition. None of the results for moderate adverse effects were statistically significant (Analysis 1.17), although they may have clinical relevance for people with CF.
1.17. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 17: Adverse events at up to 6 months (moderate)
iii. severe
Combined data from the two studies showed that at least one treatment‐related adverse event (defined in the trials as serious and presented in line with our definition of severe) occurred in 10 out of 361 participants (2.8%) in the 400 mg mannitol group and four out of 239 participants (1.7%) in the 50 mg mannitol group (Aitken 2012; Bilton 2011).
The most common 'severe' adverse effect was cough with 1.9% of participants from the 400 mg mannitol group and 1.7% of participants in the 50 mg mannitol group experiencing this; this was not statistically significant (Analysis 1.18). One or more report of haemoptysis occurred in 0.8% and 0.4% participants in the 400 mg mannitol and 50 mg mannitol arms, respectively. There were reports of pharyngolaryngeal pain in 1.1% of in the 400 mg mannitol group but in none of the participants in the 50 mg mannitol group. An aggravation of CF was reported in 0.2% of participants taking 400 mg mannitol and 0.4% of participants taking 50 mg mannitol. None of these results were statistically significant, although they may have clinical relevance for people with CF.
1.18. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 18: Adverse events at up to 6 months (severe)
iv. total
Considering all treatment‐related adverse events from the two studies (whether defined as mild, moderate or severe), the most common adverse effects were cough (occurring 10% and 5% respectively in the 400 mg and 50 mg mannitol groups) and haemoptysis (occurring 5% and 2% respectively in the 400 mg and 50 mg mannitol groups). Other common adverse events were pharyngolaryngeal pain, occurring in 4% of the 400 mg mannitol group compared to 2% of the 50 mg mannitol group, and aggravation of condition, occurring in 4% of the 400 mg mannitol group compared to 3% of the 50 mg mannitol group. Differences between the rates of all adverse events, were not statistically significant; however, such events may have clinical relevance for people with CF and further long‐term studies with post hoc monitoring are required to determine the clinical importance of adverse effects associated with inhaled mannitol (Analysis 1.19).
1.19. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 19: Adverse events at up to 6 months (total)
Subgroup analysis ‐ dornase alfa use
Subgroup data were available for the two studies for all treatment‐related adverse events of interest in the review. For brevity, the results of the subgroup analysis are summarised in the additional tables (Table 8). As in the primary analysis, 99% CIs are used to allow for multiple testing of different adverse events of interest. The most commonly occurring adverse events in the dornase alfa users were cough, haemoptysis, aggravation of condition and infections and infestations. The most commonly occurring adverse events in the non dornase alfa users were cough, haemoptysis, pharyngolaryngeal pain and aggravation of condition. As in the primary analysis, in the subgroups there were no significant differences between treatments in the rates of adverse events. There were also no significant differences across the subgroups (Table 8).
3. Mannitol versus control ‐ subgroup analysis by dornase alfa use (adverse events).
| Adverse event |
Dornase alfa users (n = 392) |
Dornase alfa non users (n = 208) |
P value for test of subgroup differences |
||
| RR | 99% CI | RR | 99% CI | ||
| Cough | 1.63 | 0.68 to 3.92 | 4.82 | 0.53 to 43.70 | 0.24 |
| Haemoptysis | 3.06 | 0.68 to 13.67 | 2.52 | 0.15 to 43.23 | 0.88 |
| Pharyngolaryngeal pain | 1.66 | 0.33 to 8.33 | 2.71 | 0.27 to 27.12 | 0.65 |
| Throat irritation | 1.14 | 0.08 to 16.23 | 3.13 | 0.06 to 161.52 | 0.59 |
| Productive cough | 1.59 | 0.13 to 19.36 | 1.90 | 0.03 to 124.54 | 0.93 |
| Wheezing | 0.52 | 0.07 to 3.62 | 0.63 | 0.05 to 8.13 | 0.88 |
| Asthma | 1.40 | 0.06 to 31.86 | 0.21 | 0.00 to 13.84 | 0.35 |
| Bronchospasm | 2.10 | 0.03 to 138.37 | 1.90 | 0.03 to 124.54 | 0.97 |
| Condition aggravated | 1.63 | 0.43 to 6.28 | 0.94 | 0.09 to 9.55 | 0.59 |
| Chest discomfort | 1.38 | 0.23 to 8.29 | 0.62 | 0.02 to 22.39 | 0.60 |
| Chest pain | 0.14 | 0.00 to 7.43 | NA | NA | NA |
| Vomiting | 0.69 | 0.07 to 6.84 | 4.44 | 0.09 to 212.27 | 0.29 |
| Post‐tussive vomiting | 4.72 | 0.10 to 228.19 | 2.52 | 0.15 to 43.83 | 0.74 |
| Headache | 3.87 | 0.40 to 37.22 | 1.26 | 0.06 to 28.54 | 0.45 |
| Decreased appetite | 2.10 | 0.03 to 138.37 | 1.90 | 0.03 to 124.54 | 0.97 |
| Infections and infestations | 0.39 | 0.10 to 1.51 | 1.46 | 0.12 to 17.63 | 0.23 |
| Musculosketal and connective tissue disorders | 2.06 | 0.18 to 22.87 | 1.90 | 0.03 to 124.54 | 0.97 |
| Skin and subcutaneous disorders | 1.61 | 0.13 to 19.39 | 3.17 | 0.06 to 167.04 | 0.71 |
CI: confidence interval; NA: not estimable (no events reported in the subgroup); RR: risk ratio.
RR and 99% CI represents the proportion of participants experiencing pulmonary exacerbations pooled across two studies (Aitken 2012; Bilton 2011) (except asthma, bronchospasm, chest pain and decreased appetite reported in Bilton 2011 only).
Subgroup analysis ‐ age
Subgroup data were available for the two studies for all treatment‐related adverse events of interest in the review. For brevity, the results of the subgroup analysis are summarised in the additional tables (Table 9). As in the primary analysis, 99% CIs are used to allow for multiple testing of different adverse events of interest. The most commonly occurring adverse events in adults were cough, haemoptysis, aggravation of condition and pharyngolaryngeal pain. The most commonly occurring adverse events in children were cough, pharyngolaryngeal pain, headache and infections and infestations. As in the primary analysis, in the subgroups there were no significant differences between treatments in the rates of adverse events. There were also no significant differences across the subgroups (Table 9).
4. Mannitol versus control ‐ subgroup analysis by age (adverse events).
| Adverse event | Adults (n = 341) | Children (n = 259) |
P value for test of subgroup differences |
||
| RR | 99% CI | RR | 99% CI | ||
| Cough | 2.05 | 0.75 to 5.57 | 2.03 | 0.48 to 8.67 | 0.99 |
| Haemoptysis | 1.83 | 0.46 to 7.28 | 5.48 | 0.36 to 82.41 | 0.35 |
| Pharyngolaryngeal pain | 2.18 | 0.35 to 13.47 | 1.77 | 0.26 to 11.92 | 0.84 |
| Throat irritation | 0.97 | 0.09 to 10.24 | 2.05 | 0.11 to 39.42 | 0.61 |
| Productive cough | 0.65 | 0.08 to 5.16 | 3.48 | 0.07 to 183.99 | 0.33 |
| Wheezing | 0.32 | 0.04 to 3.00 | 1.13 | 0.08 to 15.79 | 0.35 |
| Asthma | 0.13 | 0.00 to 7.11 | 3.36 | 0.06 to 175.87 | 0.14 |
| Bronchospasm | 3.35 | 0.06 to 177.81 | NA | NA | |
| Condition aggravated | 1.30 | 0.33 to 5.17 | 1.73 | 0.21 to 14.32 | 0.77 |
| Chest discomfort | 1.08 | 0.17 to 6.89 | 1.13 | 0.08 to 15.79 | 0.97 |
| Chest pain | NA | 0.13 | 0.00 to 7.03 | NA | |
| Vomiting | 0.67 | 0.05 to 8.52 | 2.73 | 0.16 to 47.76 | 0.34 |
| Post‐tussive vomiting | 4.39 | 0.09 to 210.79 | 2.73 | 0.16 to 47.76 | 0.80 |
| Headache | 3.90 | 0.25 to 61.93 | 1.70 | 0.20 to 14.30 | 0.54 |
| Decreased appetite | 3.35 | 0.06 to 177.81 | NA | NA | |
| Infections and infestations | 0.43 | 0.08 to 2.21 | 0.69 | 0.12 to 4.07 | 0.61 |
| Musculosketal and connective tissue disorders | 3.24 | 0.20 to 53.67 | 0.68 | 0.05 to 8.93 | 0.29 |
| Skin and subcutaneous disorders | 1.31 | 0.15 to 11.79 | 2.05 | 0.11 to 39.42 | 0.75 |
CI: confidence interval; NA: not estimable (no events reported in the subgroup); RR: risk ratio.
RR and 99% CI represents the proportion of participants experiencing pulmonary exacerbations pooled across two studies (Aitken 2012; Bilton 2011) (except asthma, bronchospasm, chest pain and decreased appetite reported in Bilton 2011 only).
b. number of participants who ceased inhalations because of poor tolerability, e.g. cough or bronchoconstriction
Pharmaxis provided additional data on tolerability and in both the Aitken and Bilton studies, respiratory and thoracic side effects were the most common treatment‐related reasons for stopping treatment. These side effects were asthma, bronchospasm, cough, haemoptysis, hyperventilation, obstructive airways disorder, pharyngolaryngeal pain, productive cough, throat irritation and wheezing. Our analysis showed no statistically significant differences between the 400 mg mannitol and 50 mg mannitol groups (Analysis 1.20). However, we were not able to simply pool the data to give a total as some participants ceased inhalations for multiple reasons. Pharmaxis confirmed there were a total of 31 out of 361 (8.6%) participants who dropped out of the 400 mg mannitol group and 7 out of 239 (2.9%) participants who dropped out of the 50 mg mannitol group due to respiratory and thoracic side effects. Pharmaxis also confirmed that a total of 37 out of 361 (10.2%) participants in the 400 mg mannitol group and eight out of 239 (3.3%) participants in the 50 mg mannitol group discontinued treatment due to any treatment‐related adverse event.
1.20. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 20: Participants with treatment‐related respiratory and thoracic adverse events leading to study discontinuation
Secondary outcomes
1. Pulmonary exacerbations
a. number of participants free of pulmonary exacerbations
Both studies included a pre‐specified definition of a pulmonary exacerbation (Aitken 2012; Bilton 2011). Investigators using the criteria specified by Fuchs which define exacerbations as when participants are treated with intravenous antibiotics for four or more of the following signs or symptoms: change in sputum production; dyspnoea; new or increased haemoptysis; malaise; fatigue or lethargy; fever (38°C or above); anorexia or weight loss; sinus pain or tenderness; change in sinus discharge; pulmonary function decreased by at least 10% from previous recorded value; radiographic signs indicative of pulmonary infection; increased cough; or changes in physical auscultation of the chest (Fuchs 1994).
The number of participants who had at least one protocol‐defined pulmonary exacerbation (PDPE) at up to six months was reported for both studies in the data from Pharmaxis. Combining data from the two studies showed a statistically significant benefit with 400 mg mannitol compared to 50 mg mannitol, pooled RR 0.71 (95% CI 0.51 to 0.98, P = 0.04), but the CIs are wide due to the low numbers of events, which shows that the average effect of 400 mg mannitol may reduce the exacerbation risk by as much as 49% or by as little as only 2% (Analysis 1.21). Hence, we are unsure of the clinical relevance of this result.
1.21. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 21: Number of patients with protocol defined pulmonary exacerbations
Subgroup analysis ‐ dornase alfa use
Subgroup data were available for the two studies for the number of participants experiencing pulmonary exacerbations. For brevity, the results of the subgroup analysis are summarised in the additional tables (Table 6). The statistically significant benefits for 400 mg mannitol compared to 50 mg mannitol shown in the primary analyses above was also shown in participants who were not dornase alfa users, but this was not the case for participants using dornase alfa. There was, however, no significant difference between the subgroups (Table 6).
Subgroup analysis ‐ age
Subgroup data were available for the two studies for the number of participants experiencing pulmonary exacerbations. For brevity, the results of the subgroup analysis are summarised in the additional tables (Table 7). The statistically significant benefits for 400 mg mannitol compared to 50 mg mannitol shown in the primary analyses above was not shown in either subgroup and there was no significant difference between the subgroups (Table 7).
b. time to first exacerbation
Both studies showed a slight increase in the time to first PDPE in the 400 mg mannitol group compared with the 50 mg mannitol group. Combining data from the two studies showed a slight increase in the time to first PDPE, but this increase was not statistically significant (Analysis 1.22)
1.22. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 22: Time to first protocol defined pulmonary exacerbation (PDPE)
2. Time off school or work
Neither study recorded time off school or work (Aitken 2012; Bilton 2011).
3. Need for additional non‐routine antibiotics
a. intravenous antibiotics
In both studies, PDPEs included the requirement for intravenous antibiotics (Analysis 1.21). Pharmaxis provided full data for intravenous antibiotic use for all pulmonary exacerbations (protocol‐defined or otherwise) at the 'up to six months' time point for two studies; significantly fewer participants in the 400 mg mannitol groups compared to the 50 mg mannitol groups required additional intravenous antibiotics, pooled RR 0.81 (95% CI 0.70 to 0.95, P = 0.01) (Analysis 1.23). Additional information was also provided for the Aitken study where a total of 377 courses of intravenous antibiotics were administered in 102 participants in the 400 mg mannitol group and 280 courses in 74 participants in the 50 mg mannitol group (Aitken 2012).
1.23. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 23: Number of patients needing additional antibiotics
b. oral antibiotics
Neither study reported the use of additional oral antibiotics (Aitken 2012; Bilton 2011).
c. inhaled antibiotics
Neither study reported the use of additional inhaled antibiotics (Aitken 2012; Bilton 2011).
4. Hospitalisations
a. number of hospitalisations
Both Aitken and Bilton provided data for the need for hospitalisation due to both PDPE and all pulmonary exacerbations at the 'up to six months' time point. Although in both studies fewer participants receiving 400 mg mannitol were hospitalised for pulmonary exacerbations and PDPEs compared with those in the 50 mg mannitol group, there was no statistically significant difference in the number of participants requiring hospitalisation due to PDPE or all pulmonary exacerbations (Analysis 1.24).
1.24. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 24: Number of participants requiring hospitalisation
b. duration of hospitalisation
Pharmaxis also provided data on the duration of hospitalisation for both pulmonary exacerbations and PDPEs at the 'up to six months' time point for the two studies. For all pulmonary exacerbations and PDPE, the mean duration of hospitalisation seemed to be slightly reduced in the 400 mg mannitol compared to 50 mg mannitol group, but no significant difference was seen between the groups (Analysis 1.25).
1.25. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 25: Duration of hospitalisation
5. Assessment of symptoms (including cough, sputum volume, ease of expectoration and dyspnoea)
Results for respiratory domain of the CFQ‐R are reported in the HRQoL section above (Analysis 1.1).
a. sputum weight (g): post‐treatment and change from baseline
Sputum was collected for 30 minutes after treatment administration at baseline and then again at 14 weeks for the assessment of sputum weight.
At 14 weeks (post‐treatment), one study showed no differences between the treatment groups in terms of sputum weight (Aitken 2012) while the second study showed a significant difference between the groups with the 400 mg mannitol group producing over twice as much sputum as the 50 mg mannitol group (Bilton 2011). When pooling the data from the two studies, the difference between the 400 mg mannitol and 50 mg mannitol groups was not significant and heterogeneity between the studies was high, MD 1.02 (95% CI ‐0.10 to 2.14), I² = 65% (Analysis 1.26).
1.26. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 26: Sputum weight (g)
This heterogeneity may have originated from the differences in sputum weight at 'baseline;' we note that the 'baseline' measurement was taken after the first administration of the study drug and therefore may have been influenced by treatment. The mean 'baseline' sputum weights in the 400 mg mannitol and 50 mg mannitol groups in the one study were 4.9 g and 3.5 g respectively (Aitken 2012) and 6.28 g and 2.26 g respectively in the second study (Bilton 2012). Therefore, we carried out an additional analysis examining the change from baseline in sputum weight in the two studies. Neither study showed a significant difference in the change in sputum weight from baseline between the 400 mg mannitol and 50 mg mannitol groups. The pooled analysis also showed no significant difference between groups and no heterogeneity was present between studies in this analysis, MD ‐0.95 (95% CI ‐2.05 to 0.15), I² = 0% (Analysis 1.27).
1.27. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 27: Sputum weight (g) (change from baseline)
6. Sputum microbiology (change in numbers of pathogens, emergence of new pathogens)
Pharmaxis provided data for both studies on the pathogens present at the 'up to six months' time‐point (Aitken 2012; Bilton 2011); the pathogens assessed were Pseudomonas aeruginosa (mucoid, non‐mucoid and not specified), Pseudomonas spp (other), Staphylococcus aureus and methicillin‐resistant Staphylococcus aureus, Burkholderia cepacia, Aspergillus spp and Candida spp. There were no statistically significant differences in the prevalence of any pathogens isolated (Analysis 1.28).
1.28. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 28: Microbiology: pathogens present at end of study
7. Burden of treatment
Additional CFQ‐R data (change from baseline and final scores) on the burden of treatment domain were provided by Pharmaxis. When the data were pooled at the 'up to four months' time‐point, there was a larger decrease in the burden of treatment in the 400 mg mannitol group compared with the 50 mg mannitol group, giving a statistically significant result, MD ‐4.60 (95% CI ‐7.90 to ‐1.30, P = 0.006); however, this effect was not sustained to six months (Analysis 1.29). We note that due to the blinding procedures of the two studies, participants in both 400 mg mannitol group and 50 mg mannitol group would have had the same number of additional capsules, therefore results should be interpreted accordingly. We further note that not all participants contributed data to the burden of treatment domain at each time point, therefore we recommend caution when interpreting results of these analyses.
1.29. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 29: Burden of treatment (change from baseline)
At the 'up to six month' time‐point, one study assessed treatment adherence by counting returned unused medication and blister packaging and defined compliance as the use of 60% or more of drug dispensed; 152 out of 184 participants (83%) were compliant in the 400 mg mannitol group and 107 out of 121 participants (88%) in the 50 mg mannitol group (Aitken 2012). The paper reported compliance was good in both arms; in the 400 mg mannitol arm, mean (SD) use of drug dispensed was 85.2% (23.81%) and in the 50 mg mannitol arm, mean (SD) 88.7% (17.66%).
At the same time‐point, the second paper recorded adherence to protocol of at least 60% for 111 out of 177 participants in the 400 mg mannitol group (62.7%) and 89 out of 118 participants in the 50 mg mannitol group (75.4%) (Bilton 2011). The paper used the same definition of compliance and stated that the "median compliance was 89% and 91% for 400 mg mannitol and 50 mg mannitol groups, respectively."
Pooling this data shows that significantly fewer participants were compliant with 400 mg mannitol compared to 50 mg mannitol treatment, pooled RR 0.89, (95% CI 0.82 to 0.97, P = 0.006) (Analysis 1.30).
1.30. Analysis.

Comparison 1: Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis, Outcome 30: Adherence ≥ 60%
Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis
Two cross‐over studies (n = 134) compared mannitol with control treatments (de Boeck 2017; Jaques 2008).
In one of the studies (n = 95), each treatment arm of the study lasted eight weeks separated by an eight‐week washout period (de Boeck 2017). The control arm of this study used 10 mg of non‐spray dried, non‐respirable raw material mannitol which is different to the low‐dose of mannitol used in the two parallel studies described above. This study adjusted for the cross‐over design of the study using an analysis of covariance model including terms for participant, treatment period, treatment and adjusting for baseline measurements (de Boeck 2017). Investigators presented MDs and 95% CIs from this model for lung function outcomes and these data are entered into the analysis in this review.
In the second study (n = 39), each treatment arm of the study lasted two weeks separated by a two‐week washout period (Jaques 2008). The control arm in this study used non‐respirable mannitol, which is also different to the low‐dose of mannitol used in the two parallel studies described above. Investigators stated that they adjusted for the cross‐over design of the study via a mixed model of analysis of variance when analysing and presenting results, but the format of the data presented in the paper, only allows us to present these data as if this was a parallel study. Therefore we have not performed an analysis of the difference between treatment groups and we have instead presented results narratively or in additional tables (Table 10; Table 11; Table 12).
5. Jaques 2008 ‐ health‐related quality of life (change from baseline at 2 weeks).
| Domain | Mannitol (n = 35) | Control (n = 34) | P value* | ||
| Mean | SD | Mean | SD | ||
| Respiratory | 4.7 | 19.62 | ‐0.7 | 19.34 | 0.09 |
| Health | 5.2 | 17.20 | 0.6 | 16.96 | 0.25 |
| Physical | 3.2 | 27.76 | ‐3.0 | 27.37 | 0.16 |
| Vitality | 2.7 | 18.71 | 0.5 | 18.74 | 0.54 |
SD: standard deviation.
Based on Table 3 of the primary publication (Jaques 2008). P value for difference in means taken from F test of Least Squares Generalised mixed model (analysis of variance), see Jaques 2008 for further details.
6. Jaques 2008 ‐ lung function data (change from baseline at 2 weeks).
| Measure | Mannitol (n = 36) | Control (n = 36) | P Value | ||
| Mean | SD | Mean | SD | ||
| FEV1 (mL) | 121 | 198 | 2 | 198 | 0.01 |
| FEV1 (% predicted) | 3.86 | 6.48 | ‐0.09 | 6.48 | <0.01 |
| FVC (mL) | 110 | 240 | 40 | 240 | 0.16 |
| FEF25-75 (mL/s) | 150 | 300 | ‐10 | 300 | 0.03 |
FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; mL: millilitres; mL/s: millilitres per second; SD: standard deviation.
Based on additional data provided by Pharmaxis and the primary publication (Jaques 2008). P value for difference in means taken from F test of Least Squares Generalised mixed model (analysis of variance), see Jaques 2008 for further details.
7. Jaques 2008 ‐ adverse events at 2 weeks.
|
Adverse event (possibly or probably related to treatment) |
Mannitol group (n = 38) |
Control group (n = 36) |
||
| Not severe | Severe | Not severe | Severe | |
| Upper respiratory tract infection | 1 | 0 | 1 | 0 |
| Diarrhoea | 0 | 0 | 0 | 1 |
| Gastro‐oesophageal reflux | 0 | 0 | 1 | 0 |
| Vomiting | 2 | 0 | 0 | 0 |
| Chest discomfort | 2 | 0 | 1 | 0 |
| Aggravation of CF symptoms | 1 | 0 | 0 | 0 |
| Fatigue | 1 | 0 | 0 | 0 |
| Anorexia | 1 | 0 | 0 | 0 |
| Chest wall pain | 2 | 0 | 0 | 0 |
| Dysgeusia | 0 | 0 | 0 | 1 |
| Cough | 2 | 2 | 0 | 0 |
| Haemoptysis | 2 | 0 | 2 | 0 |
| Nasal congestion | 0 | 0 | 1 | 0 |
| Pharynolaryngeal pain | 3 | 0 | 0 | 0 |
| Respiratory tract congestion | 0 | 0 | 1 | 0 |
| Wheeze | 0 | 0 | 1 | 0 |
| Total events | 17 | 2 | 8 | 2 |
CF: cystic fibrosis Study definition of "severe": incapacitating, or unable to do usual activities.
Primary outcomes
1. HRQoL
Only one study reported on HRQoL (Jaques 2008).
As stated above, it should be noted that while 'Burden of treatment' is part of the QoL assessment, these results are presented under a separate outcome below (secondary outcome 7). Jacques measured HRQoL using the age‐appropriate CFQ‐R; for participants aged six to 13 years, either the parent or child questionnaire was completed as appropriate, and those participants aged 14 years and over used the teen or adult questionnaire with a two‐week recall period (Jaques 2008). The results (adjusted for the cross‐over design) as reported in the paper are presented in the additional tables (Table 10).
At the end of the study there were no significant differences between mannitol and control for the respiratory, health, physical and vitality domains (very low‐quality evidence). As stated above, mannitol may have a negative effect on QoL scores due to its mechanism of action being to increase sputum production (scored as a negative outcome in the respiratory domain). Furthermore, we are aware that the HRQoL tool used in the studies was not designed to be used to assess mucolytics.
2. Lung function
One study presented results (adjusted from cross‐over design) for absolute and relative change in FEV1, FVC and FEF25-75 (% predicted) (de Boeck 2017). This study also presented results for pre‐planned subgroup analyses based on age and dornase alfa use. The results are presented below for each lung function measure.
The results for lung function measures (adjusted for cross‐over design) as reported in the second study (Jaques 2008) or provided by Pharmaxis are presented in the additional tables and summarised below (Table 11).
a. FEV1 (mL or % predicted)
Absolute change from baseline
One study reported a statistically significant advantage for 400 mg mannitol over control in terms of the absolute change from baseline in FEV1 (% predicted), MD 3.42 (95% CI 1.13 to 5.71, P = 0.003) (Analysis 2.1) (low‐quality evidence) (de Boeck 2017). The second study recorded FEV1 as the absolute change from baseline in mL and % predicted (Jaques 2008). A statistically significant improvement in participants on mannitol compared to control was observed for both measures; MD FEV1 119 mL, P value for difference = 0.01 and MD FEV1 3.95% predicted, P value for difference < 0.01 (Table 11, low‐quality evidence).
2.1. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 1: FEV1 % predicted (absolute change from baseline)
Subgroup analysis ‐ age (absolute change from baseline)
The de Boeck study also reported a statistically significant advantage in the absolute change from baseline in FEV1 (% predicted) for 400 mg mannitol over control in both age subgroups (de Boeck 2017); aged 6 to 11 years, MD 3.78 (95% CI 0.13 to 7.43, P = 0.04) and aged 12 to 17 years, MD 3.35 (95% CI 0.14 to 6.56, P = 0.04) (Analysis 2.2).
2.2. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 2: FEV1 % predicted (absolute change from baseline ‐ subgroup analysis by age)
Subgroup analysis ‐ dornase alfa use (absolute change from baseline)
The de Boeck study also reported a statistically significant advantage in the absolute change from baseline in FEV1 (% predicted) for 400 mg mannitol over control in both dornase alfa subgroups (de Boeck 2017); dornase alfa users, MD 3.29 (95% CI 0.28 to 6.30, P = 0.03) and dornase alfa non‐users, MD 3.92 (95% CI 0.42 to 7.42, P = 0.03) (Analysis 2.3).
2.3. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 3: FEV1 % predicted (absolute change from baseline ‐ subgroup analysis by dornase alfa use
Relative change from baseline
One study reported a statistically significant advantage for 400 mg mannitol over control in terms of the relative change from baseline in FEV1 (% predicted) (de Boeck 2017), MD 4.97% (95% CI 1.52 to 8.42, P = 0.005) (Analysis 2.4) (low‐quality evidence). The study also reported that subgroup results (including age and dornase alfa use) were consistent with the overall treatment difference for this endpoint, but did not present numerical results (de Boeck 2017).
2.4. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 4: FEV1 % predicted (relative change from baseline)
b. FVC (mL or % predicted)
Absolute change from baseline
One study reported no statistically significant difference between the treatment groups in terms of the absolute change from baseline in FVC (% predicted) (de Boeck 2017), MD 1.80% (95% CI ‐0.72 to 4.32, P = 0.16) (Analysis 2.5) (low‐quality evidence). Furthermore, de Boeck also reported that subgroup results (including age and dornase alfa use) were consistent with the overall treatment difference for this endpoint, but did not present numerical results (de Boeck 2017).
2.5. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 5: FVC % predicted (absolute change from baseline)
The second study found no statistically significant difference in absolute change in FVC (mL) between mannitol and control (Jaques 2008), MD 70 mL, P value for difference = 0.16 (Table 11) (low‐quality evidence).
Relative change from baseline
One study reported no statistically significant difference between the treatment groups in terms of the relative change from baseline in FVC (% predicted) (de Boeck 2017), MD 2.54% (95% CI ‐0.72 to 5.80, P = 0.13) (Analysis 2.6) (low‐quality evidence). Investigators also reported that subgroup results (including age and dornase alfa use) were consistent with the overall treatment difference for this endpoint, but did not present numerical results (de Boeck 2017).
2.6. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 6: FVC % predicted (relative change from baseline)
c. FEF25-75 (mL/s or % predicted)
Absolute change from baseline
One study reported a statistically significant advantage in the absolute change from baseline in FEF25-75 (% predicted) for 400 mg mannitol over control (de Boeck 2017), MD 5.75% (95% CI 1.81 to 9.69, P = 0.004) (Analysis 2.7) (low‐quality evidence). Investigators also reported that subgroup results (including age and dornase alfa use) were consistent with the overall treatment difference for this endpoint, but did not present numerical results (de Boeck 2017).
2.7. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 7: FEF25-75 % predicted (absolute change from baseline)
The second study reported a statistically significant improvement in absolute FEF25-75 (mL/s) during mannitol treatment compared with control (Jaques 2008 ), MD 160 mL/s, P value for difference = 0.03 (Table 11) (low‐quality evidence).
Relative change from baseline
One study reported a statistically significant advantage for 400 mg mannitol over control in terms of the relative change from baseline in FEF25-75 (% predicted) (de Boeck 2017), MD 10.52% (95% CI 2.31 to 18.73, P = 0.01) (Analysis 2.8) (low‐quality evidence). Investigators also reported that subgroup results (including age and dornase alfa use) were consistent with the overall treatment difference for this endpoint, but did not present numerical results (de Boeck 2017).
2.8. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 8: FEF25-75 % predicted (relative change from baseline)
3. Adverse events relating to treatment
Due to the cross‐over designs of both of the studies, no data regarding adverse events is entered into analysis for either study (de Boeck 2017; Jaques 2008). Adverse events are summarised narratively in additional tables (Table 12; Table 13; Table 14) (low‐quality evidence).
8. de Boeck 2017 ‐ participants with adverse events and serious adverse events.
| Event | 400 mg mannitol (n = 87) | Control (n = 87) |
| At least one treatment‐emergent AE | 54 (62.1%) | 52 (59.8%) |
| At least one treatment‐related AE | 16 (18.4%) | 11 (12.6%) |
| At least one SAE | 10 (11.5%) | 13 (14.9%) |
| At least one treatment‐related SAE | 0 (0%) | 1 (1.1%) |
| AEs leading to withdrawal from the study | 2 (2.3%) | 1 (1.1%) |
| Treatment‐related AEs leading to withdrawal from the study | 2 (2.3%) | 0 (0%) |
AE: adverse event; SAE :serious adverse event
9. de Boeck 2017 ‐ treatment‐emergent and treatment‐related adverse events.
| Event | 400 mg mannitol (n = 87) | Control (n = 87) |
| Treatment‐emergent adverse events defined by MeDRa preferred term | ||
| Cough | 14 (16.1%) | 14 (16.1%) |
| Infective pulmonary exacerbation of cystic fibrosis | 10 (11.5%) | 14 (16.1%) |
| Headache | 6 (6.9%) | 7 (8.0%) |
| Nasopharyngitis | 6 (6.9%) | 6 (6.9%) |
| Lung infection | 2 (2.3%) | 5 (5.7%) |
| Treatment related AEs leading to withdrawal from the study | ||
| Dizziness | 1 (1.1%) | 0 (0%) |
| Cough | 2 (2.3%) | 0 (0%) |
| Oropharnygeal pain | 1 (1.1%) | 0 (0%) |
| Total number of AEs | 114 | 117 |
Treatment emergent adverse events reported in de Boeck 2017 occurring in at least 5% of participants on either treatment
a. type and number of adverse events defined as mild, moderate or severe
The de Boeck study reported treatment‐emergent adverse events (TEAEs) and serious adverse events (SAEs) during the study period (de Boeck 2017); there were similar rates in the 400 mg mannitol and control groups, 62.1% versus 59.8% respectively for TEAEs and 11.5% versus 14.9% respectively for SAEs (Table 13). Rates of treatment‐related adverse events during the study period were also similar in the 400 mg mannitol and control groups, 18.4% versus 12.6% respectively; and treatment‐related SAEs were rare,only one occurred in the control group (Table 13).
In terms of specific TEAEs, investigators reported on the 'most common' events which occurred in at least 5% of participants in either treatment group (de Boeck 2017). Cough, headache, nasopharyngitis and lung infections occurred in either equal or greater frequency in the control arm compared to the 400 mg mannitol arm (Table 14). Rates of pulmonary exacerbations are discussed below in 'Secondary outcomes'.
Study investigators reported that the majority of adverse events were mild or moderate with only four (4.6%) participants in the mannitol arm and three (3.4%) participants in the placebo arm experiencing severe adverse events (de Boeck 2017). The severe events reported included abdominal pain, anal fistula, pyrexia, tonsillitis, gingivitis, headache, migraine, cough and respiratory distress. No instances of severe bronchospasm were reported and haemoptysis was uncommon, occurring in three participants taking mannitol and two taking placebo. All haemoptysis events were either scant or mild.
Jacques reported adverse events considered possibly or probably related to treatment and classified in the published paper as severe or not severe, but the classifications are not defined (Jaques 2008). It was later confirmed by Pharmaxis that the definitions used in this study were the same as in the two later parallel studies and as detailed above (Aitken 2012; Bilton 2011). In the published paper, a total of 19 adverse events were reported in 38 participants during mannitol treatment, 17 of these were classed as not severe and two as severe. A further 10 adverse events were reported in 36 participants receiving the control treatment, eight of these were classed as not severe and two as severe. The most common adverse events measured across both groups were cough, haemoptysis, pharyngolaryngeal pain and chest discomfort (Table 12). During mannitol treatment, cough was the most common adverse event and during the control period haemoptysis was the most common. The paper reported that seven serious adverse events occurred during the study, but none were considered by the investigator to be related to treatment and none resulted in death.
Pharmaxis further stated that a total of five (13.2%) participants reported six severe adverse events (all adverse events ‐ not just those considered to be possibly or probably treatment‐related and defined as incapacitating or leading to participants being unable to do usual activities) and two (5.6%) participants reported one severe adverse event each in the control group. There were three participants who withdrew due to adverse events; one each for lower respiratory tract infection and liver transplant in the mannitol group, and one in the control group due to their condition being aggravated. Pharmaxis confirmed that there were no treatment‐related serious adverse events (defined as a reaction that results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, or is a congenital anomaly or birth defect).
b. number of participants who ceased inhalations because of poor tolerability, e.g. cough or bronchoconstriction
It was reported by de Boeck that three participants (two in the 400 mg mannitol group and one in the control group) withdrew due to adverse events (de Boeck 2017); and that these adverse events were treatment‐related in the two participants in the 400 mg mannitol group (see Table 13). Specifically, reasons listed for withdrawal from the study were dizziness, cough and oropharyngeal pain (Table 14).
The published paper by Jacques states that none of the reasons given for participants stopping treatment (a liver transplant and a lower respiratory tract infection in the mannitol arm; and aggravation of CF symptoms in the placebo arm) were considered to be related to tolerability (Jaques 2008).
Secondary outcomes
1. Pulmonary exacerbations
One study reported on infective pulmonary exacerbations of CF (de Boeck 2017); these occurred less frequently in the 400 mg mannitol group (11.5%) compared to the control arm (16.1%) (Table 14).
The second study did not report the occurrence of pulmonary exacerbations, but the published paper did note the aggravation of CF symptoms in three out of 38 (7.9%) participants being treated with mannitol (none were considered related to treatment) and no participants in the control treatment phase (Jaques 2008). No additional data were available; however, in such a short study, it would be unlikely that this outcome would be assessed.
2. Time off school or work
Neither study reported either time off work or school (de Boeck 2017; Jaques 2008).
3. Need for additional non‐routine antibiotics
Neither study reported the need for additional intravenous, oral or inhaled antibiotics (de Boeck 2017; Jaques 2008).
4. Hospitalisations
Neither study reported details of hospitalisations (de Boeck 2017; Jaques 2008).
5. Assessment of symptoms
a. sputum weight
One study reported a statistically significant advantage for 400 mg mannitol over control in terms of sputum weight post‐initial treatment (de Boeck 2017), MD 1.33 g (95% CI 0.29 to 2.37, P = 0.01) (Analysis 2.9).
2.9. Analysis.

Comparison 2: Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis, Outcome 9: Sputum weight (grams) (post‐initial treatment)
b. other measures
Results for respiratory domain of the CFQ‐R from the Jacques study are reported in the HRQoL section above (Jaques 2008).
Investigators assessed symptoms using a standard set of questions (produced specifically for the study and not validated) (Jaques 2008). The questionnaire concentrated on participant‐perceived severity of respiratory symptoms (e.g. breathlessness, cough, mucus production, congestion, fatigue and discomfort) using a two‐week recall period and also sinus and nasal symptoms (e.g. post‐nasal drip, sore throat, sinus headache and pressure, nasal itch, nasal blockage, loss of smell, and mucus colour and production) using a 24‐hour recall period. Responses were obtained before and after each treatment arm. The published paper stated that after adjusting for the cross‐over design, the change in respiratory symptom scores for mannitol was MD ‐0.91 (95% CI ‐2.1 to 0.2) and for control it was MD 0.39 (95% CI ‐0.8 to 1.5) and that there were no significant differences between treatment arms.
6. Sputum microbiology
Only one study reported on this outcome (Jaques 2008). Investigators noted that there was no difference from baseline in either treatment arm after two weeks of treatment with regards to the acquisition or loss of the following micro‐organisms: Pseudomonas aeruginosa, Staphyloccocus aureus, Haemophilis influenzae, Aspergillus spp, Candida albicans and Mycobacterium. There was no increase or decrease in the qualitative bacterial growth of respiratory isolates from baseline. Specific data per pathogen were not recorded.
7. Burden of treatment
Neither study reported on burden of treatment according to a validated measure (de Boeck 2017; Jaques 2008).
Only one study reported on adherence which was measured by counting returned empty capsules and defined as more than 70% of the maximum dose used during the two‐week treatment period (Jaques 2008). According to this definition, 37 out of 39 participants taking mannitol and 33 out of 39 taking the control treatment were adherent to treatment. The median time to complete treatment with either mannitol or control was five minutes (interquartile range three to 10 minutes) (Jaques 2008).
Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations
One parallel pilot study of 22 participants compared 400 mg mannitol (twice daily) to a control treatment (very low‐dose (50 mg) non‐respirable mannitol) (Middleton 2015). Treatment was administered for 12 consecutive days and outcome measures were reported at 14 days (hospital discharge) and at one month of follow‐up.
Primary outcomes
1. HRQoL
Investigators measured HRQoL using the Cystic Fibrosis Clinical Score (CFCS) and the CFQ‐R respiratory domain (Middleton 2015). There were no statistically significant differences between the treatment groups in terms of CFCS total score at hospital discharge, MD 1.10 (95% CI ‐0.50 to 2.70, P = 0.18) or at one month follow‐up, MD ‐1.00 (95% CI ‐4.10 to 2.10, P = 0.53) (Analysis 3.1) (very low‐quality evidence). There were also no statistically significant differences in the CFQ‐R respiratory domain at hospital discharge, MD 1.30 (95% CI ‐10.50 to 13.10, P = 0.83) or at one month follow‐up, MD 3.50 (95% CI ‐10.70 to 17.70, P = 0.63) (Analysis 3.2) (very low‐quality evidence).
3.1. Analysis.

Comparison 3: Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations, Outcome 1: HRQoL ‐ change from baseline in CFCS total score
3.2. Analysis.

Comparison 3: Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations, Outcome 2: HRQoL ‐ change from baseline in CFQ‐R respiratory domain
2. Lung function
The only study in this comparison reported three measures of lung function (% predicted) (Middleton 2015).
a. FEV1 (% predicted)
There were no statistically significant differences between the treatment groups in terms of FEV1 % predicted at hospital discharge, MD 4.60% predicted (95% CI ‐3.80 to 13.00, P = 0.28) or at one month follow‐up, MD 5.40% predicted (95% CI ‐2.70 to 13.50, P = 0.19) (Analysis 3.3) (low‐quality evidence).
3.3. Analysis.

Comparison 3: Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations, Outcome 3: FEV1 % predicted (change from baseline)
b. FVC (mL or % predicted)
There were no statistically significant differences between the treatment groups in FVC % predicted at hospital discharge, MD 2.80% predicted (95% CI ‐3.60 to 9.20, P = 0.39) or at one month follow‐up, MD 1.70% predicted (95% CI ‐4.10 to 7.50, P = 0.57) (Analysis 3.4) (low‐quality evidence).
3.4. Analysis.

Comparison 3: Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations, Outcome 4: FVC % predicted (change from baseline)
c. FEF25-75 (mL/s or % predicted)
There were no statistically significant differences between the treatment groups in terms of FEF25-75 % predicted at hospital discharge, MD 12.80% predicted (95% CI ‐3.30 to 28.90, P = 0.12) or at one month follow‐up, MD 3.90% predicted (95% CI ‐10.70 to 18.50, P = 0.60) (Analysis 3.5) (low‐quality evidence).
3.5. Analysis.

Comparison 3: Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations, Outcome 5: FEF25-75 % predicted (change from baseline)
3. Adverse events relating to treatment
The included study reported that inhaled dry‐powder mannitol was well tolerated and that no serious adverse events occurred (Middleton 2015).
The authors provided unpublished presentation slides of the study which outline specific adverse events. Vomiting (mucous) occurred in one participant in each treatment group, haemoptysis (flecks only) and blurred vision each occurred in one participant in the control group and dizziness and headaches occurred in one participant in the 400 mg mannitol group (low‐quality evidence).
Due to the small number of adverse events and as we had no information regarding the severity of these reported events and whether the events could be related to treatment, we did not enter these data into the analysis.
Secondary outcomes
1. Pulmonary exacerbations
The participants recruited into the study were hospitalised during the first 14 days of the study due to pulmonary exacerbation (Middleton 2015). It was not reported whether any further pulmonary exacerbations occurred in any participants while in hospital or during the one month follow‐up.
2. Time off school or work
The included study did not report time off school or work (Middleton 2015).
3. Need for additional non‐routine antibiotics
The included study did not report the use of additional non‐routine antibiotics (Middleton 2015).
4. Hospitalisations
The participants recruited into the study were hospitalised during the first 14 days of the trial due to pulmonary exacerbation and investigators did not report if any participants were re‐admitted to hospital during the one month follow‐up (Middleton 2015).
5. Assessment of symptoms (including cough, sputum volume, ease of expectoration and dyspnoea)
The study did not report any measures of assessment of symptoms (Middleton 2015).
6. Sputum microbiology (change in numbers of pathogens, emergence of new pathogens)
The study did not report on sputum microbiology (Middleton 2015).
7. Burden of treatment
The study did not report on burden of treatment (Middleton 2015).
Mannitol versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis
One cross‐over study compared 400 mg mannitol (twice daily) to 2.5 mg dornase alfa (once daily) (Minasian 2010). This study also had a third arm, described below, which compared mannitol plus dornase alfa to dornase alfa alone. Each arm lasted 12 weeks.
Note: this was a per protocol analysis, 28 participants were randomised to treatment and 20 completed all three treatment periods in the three‐arm cross‐over study.
Primary outcomes
1. HRQoL
Again, it should be noted that while 'Burden of treatment' is part of the QoL assessment, these results are presented under a separate outcome below (Secondary outcome 7). As stated above, mannitol has a negative effect on some QoL scores due to its mode of action with regards to sputum production.
The included study measured this outcome using the CFQ‐R, but did not report results in the published paper (Minasian 2010). Pharmaxis provided additional data on the respiratory domain for the age‐specific questionnaires: adolescent and adult; children 12 to 13 years; children six to 11 years; parents and caregivers; and overall. However, these additional data did not allow any further comparison between treatment arms (Table 15, very low‐quality evidence).
10. Minasian 2010 ‐ quality of life questionnaire (respiratory domain): change from start of treatment at 2 weeks (additional data from Pharmaxis).
| Treatment arm | Adolescent and adult | Children 12 ‐ 13 years | Children 6 ‐ 11 years | Parents and caregivers | Overall |
| Mannitol | n = 7 | n = 6 | n = 4 | n = 10 | n = 27 |
| Mean change | 0.0 | ‐5.6 | 0.0 | ‐3.3 | ‐2.5 |
| Dornase alfa | n = 4 | n = 9 | n = 3 | n = 13 | n = 29 |
| Mean change | 8.3 | ‐3.7 | 13.9 | 0.4 | 1.6 |
| Dornase alfa & mannitol | n = 6 | n = 5 | n = 4 | n = 9 | n = 24 |
| Mean change | 1.9 | ‐15.0 | ‐10.4 | ‐12.2 | ‐9.0 |
2. Lung function
We were only able to use the data on % change from baseline in lung function from this study in our analysis (Minasian 2010). We note there may be minor discrepancies between our results (no difference in statistical significance) and the publication, these are likely due to the analysis of FEV1 using a log scale. Please see the additional tables for further absolute data provided by Pharmaxis which could not be analysed in this review (Table 16).
11. Minasian 2010 ‐ lung function data: change from baseline at 12 weeks (additional data from Pharmaxis).
| Measure | Mannitol (n = 23) | Dornase alfa (n = 21) | Mannitol + dornase alfa (n = 23) | |||
| Mean | SD | Mean | SD | Mean | SD | |
| FEV1 (mL) | ‐1 | 279.2 | 84 | 237.3 | ‐31 | 305.7 |
| FVC (mL) | ‐58 | 360.7 | 7 | 414.7 | ‐103 | 393.8 |
| FEF25-75 (mL/s) | 55 | 282.1 | 173 | 309.6 | 68 | 488.8 |
FEF25-75: mid‐expiratory flow; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; mL: millilitres; mL/s: millilitres per second; SD: standard deviation.
a. FEV1 (mL or % predicted)
Using data reported in the published paper, our analysis found no significant difference in the mean % change in FEV1 (mL) between the mannitol and dornase alfa groups at up to three months, MD 2.80 (95% CI ‐4.80 to 10.40, P = 0.47) (Analysis 4.1) (very low‐quality evidence); data for FEV1 % predicted were not available. The published paper reported that mannitol was as effective as dornase alfa (Minasian 2010).
4.1. Analysis.

Comparison 4: Mannitol versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis, Outcome 1: FEV1 (% change from baseline)
b. FVC (mL or % predicted)
Using data reported in the published paper, we found no significant difference in the mean % change in FVC (mL) between the mannitol and dornase alfa groups at up to three months, MD 0.14 (95% CI ‐0.02 to 0.30, P = 0.08) (Analysis 4.2) (very low‐quality evidence). Again, the published paper reported that mannitol was as effective as dornase alfa (Minasian 2010).
4.2. Analysis.

Comparison 4: Mannitol versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis, Outcome 2: FVC (% change from baseline)
c. FEF25-75 (mL/s or % predicted)
Using data reported in the published paper, we found no significant difference in the mean % change in FEF25-75 (mL/s) between the mannitol and dornase alfa groups at up to three months (MD ‐0.01, 95% CI ‐0.23 to 0.21, P = 0.93) (Analysis 4.3) (very low‐quality evidence). The published paper reported that mannitol was as effective as dornase alfa (Minasian 2010).
4.3. Analysis.

Comparison 4: Mannitol versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis, Outcome 3: FEF25-75 (% change from baseline)
3. Adverse events relating to treatment
a. type and number of adverse events defined as mild, moderate or severe
The included study used the MedDRA classification of mild, moderate and severe adverse events and considered that the reported adverse events were possibly or probably related to treatment (Minasian 2010). In the original publication, investigators reported that six out of the eight participants who withdrew reported a troublesome cough (three of these also had evidence of a chest exacerbation) and one of the eight reported nausea (Minasian 2010).
Dr Minasian also provided some additional data, but in the form of the total number of adverse events rather than the number of participants experiencing an adverse event. Hence, we are unable to present these data in the meta‐analysis (Minasian 2010). However, we also received additional data on the number of events from Pharmaxis which we have presented in the additional tables (Table 17; Table 18, very low‐quality evidence); we note that in Table 18 for brevity some adverse effects are grouped into system organ class definitions and therefore do not match exactly the definitions of individual adverse effects as described in Table 17. A total of 12 out of 23 participants in the mannitol arm and one out of 21 participants in the dornase alfa arm experienced a TEAE. The most common treatment‐related adverse events in the mannitol arm were cough and CF exacerbation (Table 17). Additionally, Pharmaxis confirmed that most events were either mild or moderate.
12. Minasian 2010 ‐ treatment‐emergent adverse events at 12 weeks.
| Adverse event | Mannitol (n = 23) n (%) | Dornase alfa (n = 21) n (%) | Mannitol + dornase alfa (n = 23) n (%) | ||||||
| Mild | Moderate | Severe | Mild | Moderate | Severe | Mild | Moderate | Severe | |
|
Number of participants with any adverse event (some may have more than one adverse event) |
4 (17.4) | 6 (26.1) | 2 (8.7) | 0 | 0 | 1 (4.8) | 5 (21.7) | 5 (21.7) | 4 (17.4) |
| CF exacerbation (CF lung) | 1 (4.3) | 2 (8.7) | 1 (4.3) | 0 | 0 | 1 (4.8) | 0 | 3 (13.0) | 4 (17.4) |
| Nausea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.3) | 0 |
| Ear infection | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pharyngitis | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Viral infection | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Viral upper respiratory tract infection | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.3) | 0 | 0 |
| Musculoskeletal pain | 0 | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.3) | 0 | 0 |
| Cough | 3 (13.0) | 2 (8.7) | 0 | 0 | 0 | 0 | 2 (8.7) | 0 | 0 |
| Productive cough | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rash | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.3) | 0 | 0 |
CF: cystic fibrosis
13. Minasian 2010 ‐ all adverse events at 12 weeks.
| Adverse event | Mannitol (n = 23) n (%) | Dornase alfa (n = 21) n (%) | Mannitol + dornase alfa (n = 23) n (%) | ||||||
| Mild | Moderate | Severe | Mild | Moderate | Severe | Mild | Moderate | Severe | |
|
Number of participants with any adverse event (some may have more than one adverse event) |
8 (34.9) | 8 (34.9) | 4 (17.4) | 10 (47.6) | 9 (42.9) | 7 (33.3) | 10 (43.4) | 11 (47.8) | 6 (26.1) |
| CF exacerbation (CF lung) | 1 (4.3) | 4 (17.4) | 3 (13.0) | 1 (4.8) | 2 (9.5) | 5 (23.8) | 0 | 7 (30.4) | 5 (21.7) |
| Gastrointestinal events (nausea, diarrhoea, abdominal pain, gastritis) |
0 | 0 | 0 | 0 | 3 (14.3) | 0 | 1 (4.3) | 2 (8.7) | 0 |
| Ear infection | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pharyngitis | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Infections and infestations (ABPA, S aureus, viral) |
4 (17.4) | 0 (4.3) | 0 | 7 (33.3) | 3 (14.3) | 0 | 3 (13.0) | 0 | 0 |
| Injury, poisoning, procedure (contusion, wrist fracture, implant site infection) |
0 | 0 | 0 | 1 (4.8) | 0 | 0 | 1 (4.3) | 1 (4.3) | 0 |
| Musculoskeletal pain | 0 | 0 | 1 (4.3) | 0 | 1 (4.8) | 0 | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 | 2 (9.5) | 0 | 0 | 2 (8.7) | 0 | 0 |
| Cough | 3 (13.0) | 2 (8.7) | 0 | 4 (19.0) | 0 | 0 | 2 (8.7) | 1 (4.3) | 0 |
| Productive cough | 0 | 1 (4.3) | 0 | 1 (4.8) | 0 | 0 | 0 | 0 | 0 |
| Pharyngolaryngeal pain | 0 | 0 | 0 | 1 (4.8) | 0 | 0 | 0 | 0 | 0 |
| Rash | 1 (4.3) | 0 | 0 | 0 | 0 | 0 | 1 (4.3) | 0 | 0 |
| Immune (anaphylactic reaction or hypersensitivity) | 0 | 0 | 0 | 0 | 1 (4.8) | 2 (9.5) | 0 | 0 | 0 |
| CFRD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.3) |
| Surgical and medical procedures (central venous catheterization) |
0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.3) |
ABPA: Allergic bronchopulmonary aspergillosis; CF: cystic fibrosis; CFRD: cystic fibrosis‐related diabetes; S aureus : Staphylococcus aureus.
b. number of participants who ceased inhalations because of poor tolerability, e.g. cough or bronchoconstriction
The flow diagram in the published papers states that eight participants withdrew from the study, mainly because of troublesome cough associated with mannitol treatment (Minasian 2010). However, the text of the paper states that eight participants withdrew during the study; the primary reason cited for six participants was troublesome cough (in three of these participants there was evidence of a chest exacerbation at the time of withdrawal), in one child the primary reason was nausea and in another child the primary reason was treatment burden. Furthermore, treatment burden was reported as a contributory factor in two of the eight participants in whom cough was the main problem. In contrast, the additional information provided by Pharmaxis states that eight participants withdrew from the study, six of whom cited troublesome cough. Two participants withdrew before receiving randomised treatment and of the remaining six participants, not all of them recorded an adverse event which led to withdrawal. For these participants withdrawal was described as "subject decision" and all participants who stopped treatment withdrew from the study.
Secondary outcomes
1. Pulmonary exacerbations
The additional Pharmaxis data stated that by 12 weeks, three out of 23 (13%) participants in the mannitol group and three out of 21 (14.3%) participants in the dornase alfa arm experienced an exacerbation and that there was no significant difference between mannitol and dornase alfa. We did not have sufficient information to adjust for the cross‐over design in our analysis.
2. Time off school or work
The study did not report time off school or work (Minasian 2010).
3. Need for additional non‐routine antibiotics
a. intravenous antibiotics
The additional Pharmaxis data for pulmonary exacerbations (defined as requiring treatment with intravenous antibiotics) suggests that during the 12‐week study period, three out of 23 (13.0%) participants in the mannitol arm of the study and three out of 21 (14.3%) participants in the dornase alfa arm required additional intravenous antibiotics. We did not have sufficient information to adjust for the cross‐over design in our analysis.
b. oral antibiotics
The study did not report the use of additional oral antibiotics (Minasian 2010).
c. inhaled antibiotics
The study did not report the use of additional inhaled antibiotics (Minasian 2010).
4. Hospitalisations
a. number of hospitalisations
No data relating to the hospitalisation of participants were published (Minasian 2010). From the additional Pharmaxis data, nine out of 26 participants who received the study drug were hospitalised for CF exacerbations and treatment with intravenous antibiotics, but it is not stated how these were distributed between the groups.
b. duration of hospitalisation
Pharmaxis reported that the duration of hospitalisation for the nine participants ranged from four to 21 days.
5. Assessment of symptoms (including cough, sputum volume, ease of expectoration and dyspnoea)
Pharmaxis provided additional data from the study on the respiratory domain for the following age‐specific questionnaires: adolescent and adult; children 12 to 13 years; children six to 11 years; parents and caregivers; and overall (Minasian 2010). However, these additional data did not allow any further analyses.
Symptoms were also assessed in a 'Respiratory Symptom and Treatment Effects' questionnaire devised by the investigators (a copy has been provided to the authors of this review), but the results were not published. Pharmaxis provided symptom scores for the Minasian study at 12 weeks and the net change from baseline (number improved minus number worsened) was determined. The net change was in favour of mannitol for chest symptoms, tiredness, amount of sputum and coughing. The net change was in favour of the dornase alfa arm for breathlessness.
6. Sputum microbiology (change in numbers of pathogens, emergence of new pathogens)
The Pharmaxis data reported "no notable changes"; further details are presented in the additional tables (Table 19). We did not have sufficient information to adjust for the cross‐over design in our analysis.
14. Minasian 2010 ‐ sputum microbiology at 12 weeks (additional data from Pharmaxis).
| Pathogen |
Mannitol n (%) |
Dornase alfa n (%) |
Mannitol + dornase alfa n (%) |
| P aeruginosa non‐mucoid | 5 (33.3) | 2 (16.7) | 3 (25.0) |
| P aeruginosa mucoid | 1 (6.7) | 5 (41.7) | 6 (50.0) |
| Stenotrophomonas maltophilia | 1 (6.7) | 5 (41.7) | 1 (8.3) |
| Aspergillus spp | 4 (26.7) | 5 (41.7) | 6 (50.0) |
| Candida spp | 3 (20.0) | 5 (41.7) | 4 (33.3) |
Total number in each treatment group was not reported
P aeruginosa : Pseudomonas aeruginosa
7. Burden of treatment
Burden of treatment would have been determined as part of the CFQ‐R in the Minasian study, but the results from this questionnaire were not reported (Minasian 2010). One participant withdrew from the study because of the burden of treatment, but it is not reported in the paper at which stage of study this occurred.
Adherence over the 12 weeks of treatment was estimated using returned empty packs and vials.The published paper states that adherence was greater than 70% in 14 out of 19 participants receiving mannitol and in 13 out of 19 participants receiving dornase alfa; there was no significant difference between treatment arms. One participant did not return the pack (Minasian 2010). We did not have sufficient information to adjust for the cross‐over design in our analysis.
Mannitol plus dornase alfa compared with dornase alfa ‐ cross‐over study of individuals with cystic fibrosis
The Minasian study also compared 400 mg of mannitol (twice daily) plus 2.5 mg dornase alfa (once daily) with 2.5 mg dornase alfa alone (once daily) as part of the same cross‐over study with 12 weeks in each arm (Minasian 2010).
Note: this was a per protocol analysis, 28 participants were randomised to treatment and 20 completed all three treatment periods in the three‐arm cross‐over study. We present data below for the mannitol plus dornase alfa arm versus the dornase alfa arm.
Primary outcomes
1. HRQoL
Again, please note that while 'Burden of treatment' is part of the QoL assessment, these results are presented under a separate outcome below (secondary outcome 7) and that as already stated mannitol has a negative effect on QoL scores due to its mode of action with regards to sputum production.
Although investigators measured HRQoL using the CFQ‐R, they did not publish the results (Minasian 2010). Pharmaxis provided additional data on the respiratory domain for age‐specific questionnaires, but these did not allow any further comparison between treatment arms (Table 15; very low‐quality evidence).
2. Lung function
We were only able to use the data reporting % change from baseline in lung function from this study in our analysis (Minasian 2010). We note there may be minor discrepancies between our results (no difference in statistical significance) and the publication, these are likely due to the analysis of FEV1 using a log scale. We present the additional data provided by Pharmaxis which could not be analysed in the additional tables (Table 16).
a. FEV1 (mL or % predicted)
Using data reported in the published paper, we found no significant difference in the mean % change in FEV1 (mL) between the mannitol plus dornase alfa group and the group receiving dornase alfa alone, MD ‐4.30 (95% CI ‐14.10 to 5.50, P = 0.39) (Analysis 5.1) (very low‐quality evidence).
5.1. Analysis.

Comparison 5: Mannitol plus dornase alfa versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis, Outcome 1: FEV1 (% change from baseline)
b. FVC (mL or % predicted)
Using data reported in the published paper, we found no significant difference in the mean % change in FVC (mL) between the mannitol plus dornase alfa group and the group receiving dornase alfa alone, MD ‐0.07 (95% CI ‐0.30 to 0.16, P = 0.55) (Analysis 5.2) (very low‐quality evidence).
5.2. Analysis.

Comparison 5: Mannitol plus dornase alfa versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis, Outcome 2: FVC (% change from baseline)
c. FEF25-75 (mL/s or % predicted)
Using data reported in the published paper, we found no significant difference in the mean % change in FEF25-75 (mL/s) between the mannitol plus dornase alfa group and the group receiving dornase alfa alone (MD 0.03, 95% CI ‐0.18 to 0.24, P = 0.78) (Analysis 5.3) (very low‐quality evidence).
5.3. Analysis.

Comparison 5: Mannitol plus dornase alfa versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis, Outcome 3: FEF25-75 (% change from baseline)
3. Adverse events relating to treatment
a. type and number of adverse events defined as mild, moderate or severe
The study used the MedDRA classification of mild, moderate and severe adverse events and considered that the reported adverse events were possibly or probably related to treatment (Minasian 2010). Pharmaxis provided additional data for the total number of participants experiencing an adverse event for this study, which we have presented in the additional tables (Table 17; Table 18) (very low‐quality evidence); we note that in Table 18 for brevity some adverse effects are grouped into system organ class definitions and therefore do not match exactly the definitions of individual adverse effects as described in Table 17. A total of 14 out of 23 participants in the mannitol plus dornase alfa arm experienced treatment‐emergent adverse events and one out of 21 participants in the dornase alfa alone arm (Table 17).
b. number of participants who ceased inhalations because of poor tolerability, e.g. cough or bronchoconstriction
Eight participants withdrew from the study because of troublesome cough associated with mannitol treatment. However, the additional data from Pharmaxis stated that while two participants from the mannitol arm plus dornase alfa arm had adverse events (one each of cough and gastrointestinal disorder) leading to withdrawal, this was not the case for any participant in the dornase alfa arm.
Secondary outcomes
1. Pulmonary exacerbations
Minasian recorded pulmonary exacerbations, but did not publish these results in the paper (Minasian 2010). Additional data provided by Pharmaxis, stated that six out of 23 (26.1%) participants in the mannitol plus dornase alfa arm experienced an exacerbation compared with three out of 21 (14.3%) in the dornase alfa alone arm and that there was no significant difference between mannitol and dornase alfa. We did not have sufficient information to adjust for the cross‐over design in our analysis.
2. Time off school or work
The study did not report time off school or work (Minasian 2010).
3. Need for additional non‐routine antibiotics
a. intravenous antibiotics
Investigators measured the need for additional antibiotics but did not report any data (Minasian 2010). The additional Pharmaxis data for pulmonary exacerbations (defined as requiring treatment with intravenous antibiotics) suggests that three out of 23 participants (13.0%) in the mannitol plus dornase alfa arm and three out of 21 participants (14.3%) receiving dornase alfa alone required additional antibiotics during the 12‐week study. We did not have sufficient information to adjust for the cross‐over design in our analysis.
b. oral antibiotics
The study did not report the use of additional oral antibiotics (Minasian 2010).
c. inhaled antibiotics
The study did not report the use of additional inhaled antibiotics (Minasian 2010).
4. Hospitalisations
From the additional Pharmaxis data, nine of 26 participants who received the study drug were hospitalised for a CF exacerbation and treated with intravenous antibiotics, but it is not stated how these cases were distributed between the groups. We did not have sufficient information to adjust for the cross‐over design in our analysis.
5. Assessment of symptoms (including cough, sputum volume, ease of expectoration and dyspnoea)
Symptoms were assessed in a 'Respiratory Symptom and Treatment Effects' questionnaire devised by the study investigators (a copy has been provided to the authors of this review), but the results were not reported (Minasian 2010). Pharmaxis provided symptom scores at 12 weeks and the net change from baseline (number improved minus number worsened) was determined. This favoured mannitol plus dornase alfa for a reduction in coughing, but favoured dornase alfa alone for a reduction in chest symptoms and tiredness.
6. Sputum microbiology (change in numbers of pathogens, emergence of new pathogens)
Sputum microbiology was measured, but not published (Minasian 2010); the additional data from Pharmaxis reported "no notable changes". Further details are presented in the additional tables (Table 19), We did not have sufficient information to adjust for the cross‐over design in our analysis.
7. Burden of treatment
Burden of treatment would have been determined as part of the CFQ‐R, but the results from this questionnaire were not available (Minasian 2010). One participant withdrew from the study because of the burden of treatment, but it is not reported at which stage of study this occurred or which treatment arm this participant was in or had completed. In addition, two participants who withdrew from the study reported that although cough had been the main problem, treatment burden was a contributory factor (treatment arms ongoing or completed were not stated).
Adherence over 12 weeks of treatment was estimated using returned empty packs and vials. The published paper reported that adherence was greater than 70% in 17 out of 19 participants receiving mannitol plus dornase alfa and in 13 out of 19 participants receiving dornase alfa alone (Minasian 2010). One participant did not return the pack. We did not have sufficient information to adjust for the cross‐over design in our analysis.
Discussion
Lung disease remains the most common cause of mortality and morbidity in people with CF (Accurso 2007; Robinson 2001). Currently, nebulised dornase alfa and hypertonic saline are the main pharmacological treatments used to manage secretions. However, both agents require nebulization and some participants are unable to tolerate one or both, or find them unpleasant to use. Potential alternatives or supplementary treatments should be considered and investigated. Mannitol is one possible alternative treatment.
Summary of main results
The key studies included in the review were two large randomised controlled studies of similar parallel design comparing mannitol with control in adults, children and young people and reporting results at our specified time point "up to six months" (Aitken 2012; Bilton 2011). Additional data were provided by the manufacturer and study sponsor (Pharmaxis) and we were able to pool data in our analyses. Below, we summarise the primary outcomes 'HRQoL', 'lung function' and 'adverse events' and important secondary outcomes 'number of pulmonary exacerbations' and 'sputum microbiology' for each of the comparisons made in this review.
Mannitol versus control for individuals with cystic fibrosis (parallel studies)
Pharmaxis provided additional information for HRQoL, one of the primary outcomes in our review. In our pooled analyses of the parallel studies, we found no consistent significant differences for any domains of the CFQ‐R at any of the time points for which data were available (Analysis 1.1 to Analysis 1.11) (low‐quality evidence). We do note that results presented for many of the HRQoL domains are taken from the pooled results for all individuals (results from age‐appropriate version of the questionnaire combined) and that such a combination of results across questionnaires may not be valid, so encourage caution in the interpretation of these results. Also, for the respiratory domain it should be noted that as stated above, mannitol has a negative effect on HRQoL scores due to its mode of action causing increased sputum production.
From the additional HRQoL information provided by Pharmaxis which was pooled for the two parallel studies, we found a statistically significant reduction in the burden of treatment compared to baseline at four months in the mannitol group compared to the control group, MD ‐4.60 (95% CI ‐7.90 to ‐1.30); however, the difference between the groups was no longer significant by six months, MD ‐2.12 (95% CI ‐5.70 to 1.46). Combining data from the two parallel studies did show significantly fewer participants to be adherent to treatment with mannitol (defined as more than 60% compliance) than to control, RR 0.89 (95% CI 0.82 to 0.97), but the data from the cross‐over study (see below) showed that more participants were adherent (defined as above 70% compliance) to mannitol than to control. We do note that such measures are subjective to participants and study personnel and therefore encourage caution when interpreting numerical results.
Lung function was a further primary outcome of this review. Pharmaxis provided data from their original analysis via a MMRM model, appropriate for the longitudinal design of the studies and adjusted for participant factors (see Measures of treatment effect for further details). Our pooled analyses of the two parallel studies showed moderate quality evidence of significant improvements in lung function as measured by FEV1 (mL), FEV1 (% predicted) and FVC (mL) for participants receiving mannitol compared to those receiving control at up to two months, up to four months and up to six months (Analysis 1.12; Analysis 1.13; Analysis 1.14).
Cough, haemoptysis, bronchospasm, pharyngolaryngeal pain and post‐tussive vomiting were the most commonly reported side effects (moderate‐quality evidence). Haemoptysis, bronchospasm, pharyngolaryngeal pain and cough were the most common reasons for stopping treatment. There was a small but significant reduction in the incidence of PDPE, the need for additional antibiotics and the number of hospitalisations in favour of mannitol compared to control from pooling the two parallel studies (Analysis 1.21; Analysis 1.23).
Considering subgroups of dornase alfa users and non‐users and adults and children, we found that benefits in terms of lung function and exacerbations of 400 mg mannitol compared to 50 mg mannitol tend to be greater in adults and non dornase alfa users, but that there was no significant differences between the subgroups for any measures of lung function at any time point (Table 6; Table 7). We also found a similar distribution of adverse events across the subgroups and no significant differences in rates of adverse events between the subgroups (Table 8; Table 9).
No significant differences were found between mannitol and control in terms of assessment of symptoms of sputum microbiology.
Mannitol versus control for individuals with cystic fibrosis (cross‐over studies)
Two cross‐over studies compared mannitol to control (non‐respirable mannitol) over two weeks treatment duration (Jaques 2008) and over eight weeks duration (de Boeck 2017).
There were no statistically significant differences in any HRQoL domains in the two‐week cross‐over study of mannitol compared to control (non‐respirable mannitol) included in the review (Jaques 2008; Table 10). As for the previously mentioned parallel trials, we are aware of issues (related to age of participants and the mechanism of action of mannitol) which should be considered when interpreting the results presented in the review and we consider this evidence to be of very low quality.
Analysis of absolute and relative change from baseline in lung function in the two cross‐over studies showed low‐quality evidence of a significant improvement in FEV1 (mL) and FEV1 (% predicted) and FEF25-75 mL/s in favour of mannitol, but no statistically significant differences between the groups in terms of FVC (mL or % predicted) (Table 11; Analysis 2.1 to Analysis 2.8).
The most commonly reported adverse events in both groups in the two studies were cough, haemoptysis, headache, nasopharyngitis and lung infections (Table 12; Table 13; Table 14) (low‐quality evidence) and in the eight‐week cross‐over study, pulmonary exacerbations were more commonly observed in the control arm compared to the mannitol arm (Table 14).
Finally, as for the two afore‐mentioned parallel studies, no significant differences were found between mannitol and control in terms of assessment of symptoms of sputum microbiology.
Mannitol versus control for individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations (parallel study)
A parallel study recruiting children and young people hospitalised due to pulmonary exacerbations compared mannitol to control (low‐dose non‐respirable mannitol) for 12 consecutive days of treatment and followed up for one month following hospital discharge (Middleton 2015). It was not reported in the study whether any further pulmonary exacerbations occurred in any participants while in hospital or during the one month follow‐up.
There were no statistically significant differences in any measure of HRQoL at hospital discharge or after one month of follow‐up (Analysis 3.1; Analysis 3.2). As for the previously mentioned parallel trials, we are aware of issues (related to age of participants and the mechanism of action of mannitol) which should be considered when interpreting the results presented in the review and we consider this evidence to be of very low quality.
In terms of lung function, the study provided low‐quality evidence demonstrating no statistically significant differences in FEV1, FVC or FEF25-75 (% predicted) at hospital discharge or after one month of follow‐up (Analysis 3.3; Analysis 3.4; Analysis 3.5). No information regarding sputum microbiology was reported.
Adverse events were uncommon and no serious adverse occurred, however due to the small number of participants and limited duration of follow‐up, we consider this evidence to be of low quality.
Mannitol alone versus mannitol plus dornase alfa versus dornase alfa alone (cross‐over study)
A cross‐over study compared 12 weeks of treatment with mannitol, dornase alfa or mannitol plus dornase alfa in children and young people (Minasian 2010). No significant differences were found in the recorded domains of HRQoL or the measures of lung function (FEV1, FVC or FEF25-75 ) between the three treatment arms. There seemed to be a higher rate of pulmonary exacerbations in the mannitol plus dornase alfa arm compared with dornase alfa alone (although this was not statistically significant) and this was the most common reason for stopping treatment in this arm. Cough was the most common side effect in the mannitol alone arm, but there was no occurrence of cough in the dornase alfa alone arm. No significant differences were found between mannitol and control in terms of assessment of symptoms of sputum microbiology.
Overall completeness and applicability of evidence
Applicability of interventions and controls
Two studies used sub‐therapeutic doses of mannitol as the control treatment (Aitken 2012; Bilton 2011), while the other studies comparing mannitol to a control treatment used non‐respirable mannitol as the control treatment (de Boeck 2017; Jaques 2008; Middleton 2015). Use of a sub‐therapeutic dose of mannitol was justified by citing a dose‐escalation study which showed that 50 mg mannitol had no effect (Teper 2011). However, it was also acknowledged that this dose may have had some degree of effect as an improvement in FEV1 was noted in the control arm of one study (Aitken 2012).
In three studies, between 44% and 75% of participants were also using dornase alfa before randomisation and continued their treatment regimen throughout the study (Aitken 2012; Bilton 2011; de Boeck 2017; Jaques 2008; Middleton 2015). Comparing the subgroups of dornase alfa users to non‐users in the two large parallel studies, generally a beneficial effect in terms of lung function was observed in both subgroups; however, the benefit seems to be larger in non‐dornase alfa users than users (Aitken 2012; Bilton 2011). Again, we note that these studies were not designed to test subgroup differences and results do not show any significant evidence of a difference between the subgroups. This suggests that mannitol may be an effective treatment as single therapy or in combination with dornase alfa.
One cross‐over study compared mannitol with and without dornase alfa with dornase alfa alone, but the study was underpowered which prevents any conclusions being drawn (Minasian 2010). The mechanism of action of dornase alfa is to cleave extracellular DNA from inflammatory cells in the CF mucus, thus it works in a quite different way to the rehydrating properties of mannitol. It, therefore, seems reasonable to presume that people with CF may benefit from the addition of mannitol to dornase alfa therapy, and to include participants on dornase alfa in the study design.
While it is known that the addition of mannitol to an already intense treatment regimen will be time‐consuming and could cause problems with adherence, the studies collected little information on the burden of treatment and adherence. Although the CFQ‐R was used in all studies, only the two longer‐term studies reported the results of the treatment burden domain (as would be expected) (Aitken 2012; Bilton 2011). Four studies reported information on adherence to treatment which was measured through pack counts; the two larger studies used a threshold of at least 60% and the smaller studies used a threshold of 70% (Jaques 2008; Minasian 2010).
Adherence to other CF medications was not measured during any of the studies and therefore the impact of an additional treatment (i.e. mannitol or control) on the use of other CF medication is unknown. In addition, it is accepted that participants in clinical trials are usually more adherent to treatment regimens than those receiving the same treatment but as part of usual care. In the open study by Minasian, treatment burden was reported as a contributory factor to withdrawal (Minasian 2010). Although adding mannitol capsules for inhalation to current treatment regimens appeared to have little long‐term effect on the burden of treatment or adherence throughout the studies, it is hard to compare this fully as in the three studies comparing the active mannitol treatment to a control treatment both were administered using the same inhalers. An arm with no treatment is needed to determine the additional size of the treatment burden.
Hypertonic saline acts as an airway rehydrating agent and therefore people currently using hypertonic saline were excluded from five out of six of these studies; in one included study, 14% of participants randomised in the study were reported to have been using hypertonic saline in addition to their randomised treatment, but the split by treatment group was not reported (Middleton 2015). No studies compared the use of mannitol (alone) to the use of hypertonic saline (alone) therefore this review does not inform a comparison between these two treatments and future research should address this.
Applicability of participant populations and study design
As there is a risk of bronchoconstriction with mannitol, participants in all six included studies underwent a mannitol tolerance test at screening; those who failed the test or in whom the test was incomplete did not enter into the study. Approximately 12% of individuals failed the test dose demonstrating that mannitol, even if effective, will not be suitable for everyone with CF.
Three studies recruited a mixture of adults, children and young people (Aitken 2012; Bilton 2011; Jaques 2008) and three recruited only children and young people under the age of 18 (de Boeck 2017; Middleton 2015; Minasian 2010). The mean age of the two large studies was approximately 20 years of age, around 43% of participants were under 18 years of age and around 20% of participants were under 12 years of age (Aitken 2012; Bilton 2011). The evidence from these studies in this review suggests a significantly beneficial effect in terms of lung function for adults treated with mannitol compared to control but this effect has not been shown in children (Aitken 2012; Bilton 2011). We do note that the studies included in the review were not designed to test subgroup differences and the results in the subgroup of children have wide confidence intervals which does not exclude the presence of a treatment effect.
The two large studies recruited participants with a range of lung function (% predicted FEV1 between 30% and 90%) and mean FEV1 was around 60% of predicted (Aitken 2012; Bilton 2011). In the remaining three studies recruiting individuals with CF as outpatients (two cross‐over studies) participants had a baseline mean FEV1 % predicted ranging between 64% and 72% and it was stated that participants were clinically stable at the start of the study (de Boeck 2017; Jaques 2008; Minasian 2010). The final study recruited participants with CF who were hospitalised with a pulmonary exacerbation (Middleton 2015) and within this study, the control group had a significantly lower FEV1 and FEF25-75 than the mannitol group. For these reasons, results reported within this study may not be applicable to all individuals with CF (Middleton 2015).
The randomised phases of the two large studies lasted for 26 weeks; and although they continued for a further 26 weeks, this phase had an open design (Aitken 2012; Bilton 2011). The treatment duration of the cross‐over studies lasted two weeks (Jaques 2008), eight weeks (de Boeck 2017) and 12 weeks (Minasian 2010); and in the third parallel study participants were treated for only 12 days (Middleton 2015). In studies of people with chronic conditions, it is important that the duration of the study is sufficient to demonstrate differences in long‐term outcome measures. Clearly the smaller studies included in this review are likely of too short a duration to adequately measure the effectiveness of mannitol of too short a duration (de Boeck 2017; Jaques 2008; Middleton 2015; Minasian 2010).
The two longer studies incorporated an open‐label phase as the second six months of the total 12‐month study (open‐label results not presented in this review), which makes the longer‐term reporting of more subjective outcomes such as HRQoL and burden of treatment less meaningful (Aitken 2012; Bilton 2011). However, it is of note that in the two longer studies, any improvements shown in lung function were apparent at the six‐week follow‐up visit and appeared to be maintained through to the 12‐month follow‐up visit (Aitken 2012; Bilton 2011).
As stated above, one of the cross‐over studies was underpowered which prevents conclusions from being drawn (Minasian 2010). In an additional study, the recruitment of participants was halted early after potential candidate pools were exhausted and 95 out of a target 160 participants had been recruited (de Boeck 2017). Post‐hoc power calculations were reported, but using a different sizes of treatment difference (3% or 3.5% change in FEV1 % predicted), therefore it is unclear whether this study was adequately powered and whether conclusions can be drawn from these results.
Finally, although two of the studies had a similar design and results could be pooled, one was run in Europe (UK and Ireland), Australia and New Zealand (Bilton 2011) while the other was run in Europe (Germany, France, Belgium and the Netherlands) and North and South America (Aitken 2012). It is possible that standard clinical practice may vary between these areas.
Quality of the evidence
The quality of the evidence provided in this review ranges from moderate quality to very low quality, varying by comparison and by outcome (Table 1; Table 2; Table 3; Table 4; Table 5). The main issues influencing the quality of the evidence within this review were that all six studies included in the review were sponsored by the manufacturer of mannitol (Pharmaxis); some study authors declared financial interests. We also note that the study populations of all included studies were somewhat selective; excluding participants who failed a mannitol tolerance test, therefore the individuals included in this review may not represent the wider CF population.
Furthermore, we note that for the assessment of HRQoL, the CFQ‐R tool was frequently used and such a tool was not designed to assess mucolytics. Also, where a range of age groups had been included in studies and age appropriate tools had been used, pooling the results across age‐groups may be not be valid. Therefore results for HRQoL are judged to be low to very low quality for all comparisons of the review and should be interpreted with caution.
The two largest studies were of high methodological quality, generally low risk of bias and well‐powered, each including approximately 300 participants (Aitken 2012; Bilton 2011). Evidence from these studies was consistent and mostly moderate quality (except for HRQoL), with very little heterogeneity observed between the study results (Table 1).
The remaining studies were smaller, randomising 95 participants (de Boeck 2017), 39 participants (Jaques 2008), 28 participants (Minasian 2010) and 22 participants (Middleton 2015); furthermore, in three of these studies, results were not reported for all randomised individuals and the studies may be at risk of bias due to incomplete outcome data (de Boeck 2017; Jaques 2008; Minasian 2010). In addition, one study was open label with no attempt at allocation concealment or blinding (Minasian 2010) and other studies did not report clear information regarding either randomisation or allocation concealment methods (de Boeck 2017; Jaques 2008; Middleton 2015). Evidence provided from these studies was more limited and judged to be of low to very low quality (Table 2; Table 3; Table 4; Table 5).
Potential biases in the review process
We are grateful for the additional information provided to us by Pharmaxis to supplement the information in the published papers. We acknowledge that the studies were designed by Pharmaxis with a primary outcome of change in FEV1 therefore may not be powered to detect a difference in our first primary outcome HRQoL, an outcome we deemed to be the most important to people with CF. Therefore an absence of evidence of a difference in HRQoL found in this review does not rule out that mannitol may improve HRQoL compared to control treatments including dornase alfa. We also note that the combination of results across all ages for HRQoL may not be valid; however, we were not able to separate age groups in the analyses. Furthermore, we are aware that the HRQoL tool used in the studies was not designed to be used to assess mucolytics and therefore the results for some domains, e.g. mucus production, should be interpreted in light of the mechanism of action of the drug.
Given the complex structures of the studies included in this review (two longitudinal studies, three cross‐over studies and a pilot study), in many instances we felt it would be more appropriate to summarise results narratively rather than perform our own analyses. We felt that the statistical methodology used originally was the most appropriate for the design of the included studies and attempting to adjust original results in order to perform meta‐analysis for this review would not have been appropriate.
Agreements and disagreements with other studies or reviews
The Aitken and Bilton studies have been combined and their results published as a pooled analysis (Bilton 2013; Flume 2015). It was possible to compare these results to the results of our pooled analyses of these studies; we found that the results are numerically similar (we assumed that any minor differences are due to this review pooling summary results from the statistical models, while the pooled analyses combine data and then fit the statistical models) and all conclusions are the same.
There are no other published systematic reviews of mannitol to our knowledge. A technology appraisal of mannitol has been published by the UK National Institute for Health and Care Excellence (NICE) and recommends mannitol dry powder for inhalation as an option for treating CF in adults fulfilling the following criteria (NICE 2012):
unable to use dornase alfa because of ineligibility, intolerance or inadequate response to this agent; and
rapid decline in lung function (FEV1 decline greater than 2% annually); and
other osmotic agents not considered appropriate.
There are a few differences in approach between the NICE review and our review. The NICE review considered only adults, whereas we considered adults, children and young people. The NICE review also considered the sub‐populations of dornase alfa users and non‐users separately, whereas we have considered all participants as a single group regardless of concomitant treatment in our primary analysis. Finally, the NICE review included an analysis of cost‐effectiveness.
We also identified an online publication by the European Medicines Agency (EMA) which considered information submitted by Pharmaxis on five phase I studies, three phase II studies and two phase III studies. Two of the phase II studies (Jaques 2008; Minasian 2010) and the phase III studies (Aitken 2012; Bilton 2011) are also included in this review; the third phase II dose‐response study is listed in this review as excluded (Teper 2011). The EMA report granted marketing authorisation for 400 mg mannitol inhalation powder for the treatment of CF in adults aged 18 years and above (EMA 2011). Similar to our review, this report found small improvements in FEV1 % predicted and recognised the difficulty in determining the clinical benefit while simultaneously acknowledging that even a small effect may be of relevance given the deterioration of FEV1 seen in the progression of CF. The report also agreed that the most commonly observed side effect was cough, with other important adverse effects of mannitol being bronchospasm and haemoptysis.
A document published by the Scottish Medicines Consortium (SMC) agreed that using mannitol at a dose of 400 mg per day led to superior lung function compared to control (sub‐therapeutic mannitol), but also found that there was an insufficiently robust economic analysis to allow acceptance by the SMC (SMC 2013). This Cochrane Review did not include an analysis of cost‐effectiveness.
A Dutch report undertook an indirect comparison with hypertonic saline, which this review did not do. The report agreed that there was an improvement in FEV1 with mannitol; however, it judged mannitol to have limited applicability and to be more often associated with adverse effects in comparison to nebulised hypertonic saline (Zorginstituut 2014).
Authors' conclusions
Implications for practice.
In this review, we were able to combine results from two large, well‐powered studies comparing 400 mg twice‐daily inhaled mannitol to low‐dose, sub‐therapeutic (50 mg inhaled mannitol) in people with cystic fibrosis (CF) (Aitken 2012; Bilton 2011). Pooled evidence from these studies demonstrates moderate quality evidence of efficacy for 400 mg mannitol in terms of improved lung function (forced expiratory volume at one second (FEV1)), both mL and % predicted) at two, four and six months. This efficacy is shown in adults and both dornase alfa users and non‐users.
We found no clear evidence in this review of an association between health‐related quality of life (HRQoL) and the use of inhaled mannitol. We also found no consistent evidence of the association between inhaled mannitol and adverse effects.
When compared to non‐respirable mannitol as a control treatment in four small studies of short duration, this review provides only low‐ to very low‐quality evidence regarding differences in HRQoL, lung function and adverse events associated with treatment.
However, results of this review do not provide a definitive argument for the universal use of mannitol in all people with cystic fibrosis (CF). This review provides limited information regarding the effectiveness of inhaled mannitol in different severities of CF. Stakeholders need to be aware of this evidence base when assessing the use of inhaled mannitol for CF.
Implications for research.
It is important that future studies collect, analyse and publish outcomes to determine the long‐term use of inhaled mannitol in terms of both efficacy and tolerability. It is essential that future studies are powered to detect a difference in both efficacy outcomes and clearly defined HRQoL outcomes, outcomes which are of great importance to people with CF. Existing HRQoL tools used in CF are not designed to assess mucolytics; it is important that tools developed in future take into account the mechanism of action of mucolytic drugs such as mannitol. Information on burden of treatment is particularly important as the impact of adding a further inhaled treatment to people's current regimens is unclear. In the two large studies, participants in the control arms received a matched product administered using the same inhalers and taking the same amount of time to administer; such study designs make it difficult to determine the true size of any additional burden. In addition, adherence to other medications for CF was not measured during any of the published studies; future research should specifically examine this issue.
As it is accepted that participants in clinical trials are usually more adherent to treatment regimens than people receiving usual care, pragmatic studies may be more appropriate for examining burden of treatment and adherence in real‐life situations compared with randomised controlled studies. The two large included studies in this review had a six‐month open‐label period after the randomised period and data from these extension periods suggest that the gains in lung function remain. However, some self‐selection often occurs with open‐label extensions to studies, i.e. only participants who perceive a benefit will continue to take the study treatment.
Future studies should be large enough to enable detailed analysis of the effectiveness of mannitol in population subgroups such as adults and children, varying severities of CF or concomitant use of dornase alfa. Currently, data are pooled from study participants treated across a range of countries and we have already commented that routine clinical practice for treating CF may vary between the different settings, so sufficient numbers of participants are needed in each setting. Currently no studies compare mannitol to treatment with hypertonic saline; future research should address this.
Determination of resource use and the cost of mannitol treatment are important and cost‐effectiveness evaluations to relate these to benefits in local settings are needed.
What's new
| Date | Event | Description |
|---|---|---|
| 29 April 2020 | New citation required but conclusions have not changed | Since no new studies or data have been included at this update, our conclusions remain the same. |
| 29 April 2020 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Review Group's Cystic Fibrosis Trials Register identified four references. Three were conference abstracts related to an ongoing study (NCT02134353) (one of these additionally related to two already included studies (Aitken 2012; Bilton 2011)) and the fourth reference was a trial registry entry to which we linked a full paper, this study was excluded as it was not randomised (Sadr 2019). Searches of trial registries identified 318 records. No new studies were included in the review. |
History
Protocol first published: Issue 8, 2010 Review first published: Issue 10, 2015
| Date | Event | Description |
|---|---|---|
| 4 January 2018 | New search has been performed | A search of the Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Trials Register identified five new references which were potentially eligible for inclusion in this review. One reference was an additional reference to an already excluded study (Teper 2011) and one was an additional reference to an already included study (Aitken 2012). Three references were to a previously identified ongoing study (de Boeck 2017) now included. A study previously classified as awaiting classification is now also included (Middleton 2015). |
| 4 January 2018 | New citation required but conclusions have not changed | Despite the inclusion of results from two studies (one previously listed as ongoing and one as awaiting classification), our conclusions remain the same. |
Notes
Sarah J Nolan (lead author of the 2015 version of the review) is now Sarah J Nevitt
Acknowledgements
Contacts at Pharmaxis who kindly supplied the additional data for the analyses: Frazer Chidwick, Kristen Morgan, Joanna Leadbetter and Brett Charlton.
Dr Mark Elkins, who commented on and contributed to the published protocol and who also commented on drafts of the full review.
Nikki Jahnke, Managing Editor at the Cochrane Cystic Fibrosis and Genetic Disorders Review Group who advised and helped throughout the review and particularly with the analysis of the data.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Glossary
| Term | Explanation |
| bronchiectasis | a congenital or acquired disorder of the large bronchi of the lungs, characterized by permanent, abnormal dilation and destruction of bronchial walls. In this condition some of the bronchi and bronchioles have lost their elasticity and have expanded and filled with fluid. It may be caused by recurrent inflammation or infection of the airways |
| ciliary beat frequency | the rate at which the cilia beat (cilia are fine hair‐like projections from certain kinds of cells; they line the respiratory tract and move in rhythmic unison to "sweep" away fluids and particles within the lungs) |
| epithelial | to do with the outside layer of cells that covers all the free, open surfaces of the body including the skin and mucous membranes |
| hyperosmolar agents | agents causing the abnormal increase in the concentration of a solution, especially a body fluid, as occurs in dehydration |
| mucociliary clearance | the movement of the mucous covering of the surface of the respiratory tract by the beating of cilia: rapid, forward (effective) stroke and slow, return (recovery) stroke |
| mucolytic | capable of dissolving, digesting, or liquefying mucus |
Data and analyses
Comparison 1. Mannitol versus control ‐ parallel studies of individuals with cystic fibrosis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 HRQoL ‐ respiratory (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 Up to 4 months | 2 | 507 | Mean Difference (IV, Fixed, 95% CI) | ‐1.54 [‐4.69, 1.61] |
| 1.1.2 Up to 6 months | 2 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐0.99 [‐4.50, 2.52] |
| 1.2 HRQoL ‐ vitality (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 Up to 4 months | 2 | 361 | Mean Difference (IV, Fixed, 95% CI) | 3.42 [‐0.21, 7.04] |
| 1.2.2 Up to 6 months | 2 | 325 | Mean Difference (IV, Fixed, 95% CI) | 4.84 [0.86, 8.82] |
| 1.3 HRQoL ‐ physical (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 Up to 4 months | 2 | 505 | Mean Difference (IV, Fixed, 95% CI) | ‐1.80 [‐4.72, 1.11] |
| 1.3.2 Up to 6 months | 2 | 465 | Mean Difference (IV, Fixed, 95% CI) | 0.52 [‐2.75, 3.79] |
| 1.4 HRQoL ‐ emotion (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.4.1 Up to 4 months | 2 | 506 | Mean Difference (IV, Fixed, 95% CI) | ‐2.11 [‐4.56, 0.34] |
| 1.4.2 Up to 6 months | 2 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐1.27 [‐3.74, 1.20] |
| 1.5 HRQoL ‐ eating (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.5.1 Up to 4 months | 2 | 505 | Mean Difference (IV, Fixed, 95% CI) | 0.81 [‐1.96, 3.58] |
| 1.5.2 Up to 6 months | 2 | 466 | Mean Difference (IV, Fixed, 95% CI) | 0.68 [‐2.29, 3.65] |
| 1.6 HRQoL ‐ health (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.6.1 Up to 4 months | 2 | 360 | Mean Difference (IV, Fixed, 95% CI) | ‐0.43 [‐4.18, 3.32] |
| 1.6.2 Up to 6 months | 2 | 325 | Mean Difference (IV, Fixed, 95% CI) | ‐0.21 [‐4.14, 3.72] |
| 1.7 HRQoL ‐ social (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.7.1 Up to 4 months | 2 | 504 | Mean Difference (IV, Fixed, 95% CI) | ‐1.20 [‐3.70, 1.30] |
| 1.7.2 Up to 6 months | 2 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐1.47 [‐4.25, 1.32] |
| 1.8 HRQoL ‐ body (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.8.1 Up to 4 months | 2 | 500 | Mean Difference (IV, Fixed, 95% CI) | ‐3.10 [‐6.49, 0.29] |
| 1.8.2 Up to 6 months | 2 | 461 | Mean Difference (IV, Fixed, 95% CI) | ‐1.19 [‐4.51, 2.13] |
| 1.9 HRQoL ‐ role (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.9.1 Up to 4 months | 2 | 358 | Mean Difference (IV, Fixed, 95% CI) | 1.22 [‐2.21, 4.66] |
| 1.9.2 Up to 6 months | 2 | 324 | Mean Difference (IV, Fixed, 95% CI) | ‐1.43 [‐4.87, 2.00] |
| 1.10 HRQoL ‐ weight (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.10.1 Up to 4 months | 2 | 360 | Mean Difference (IV, Fixed, 95% CI) | ‐4.23 [‐10.28, 1.83] |
| 1.10.2 Up to 6 months | 2 | 325 | Mean Difference (IV, Fixed, 95% CI) | ‐3.27 [‐9.84, 3.31] |
| 1.11 HRQoL ‐ digestion (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.11.1 Up to 4 months | 2 | 505 | Mean Difference (IV, Fixed, 95% CI) | ‐1.49 [‐4.77, 1.78] |
| 1.11.2 Up to 6 months | 2 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐1.07 [‐5.04, 2.90] |
| 1.12 FEV1 ml (repeated measures, change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.12.1 up to 2 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 71.27 [36.00, 106.54] |
| 1.12.2 Up to 4 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 64.54 [26.65, 102.42] |
| 1.12.3 Up to 6 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 86.50 [45.15, 127.86] |
| 1.13 FEV1 % predicted (repeated measures, change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.13.1 up to 2 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 2.98 [1.04, 4.92] |
| 1.13.2 up to 4 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 3.26 [1.16, 5.35] |
| 1.13.3 up to 6 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 3.89 [1.69, 6.08] |
| 1.14 FVC mL (repeated measures, change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.14.1 Up to 2 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 87.81 [41.96, 133.66] |
| 1.14.2 Up to 4 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 89.24 [40.55, 137.92] |
| 1.14.3 up to 6 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 102.17 [48.40, 155.94] |
| 1.15 FEF25-75 mL/s (repeated measures, change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.15.1 Up to 2 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 47.53 [‐16.42, 111.48] |
| 1.15.2 Up to 4 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 31.30 [‐34.78, 97.37] |
| 1.15.3 Up to 6 months | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | 42.67 [‐28.07, 113.42] |
| 1.16 Adverse events at up to 6 months(mild) | 2 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.16.1 Cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.82 [0.41, 8.07] |
| 1.16.2 Haemoptysis | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.73 [0.26, 11.62] |
| 1.16.3 Pharyngolaryngeal pain | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.04 [0.18, 5.97] |
| 1.16.4 Throat irritation | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.30 [0.06, 176.31] |
| 1.16.5 Productive cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.66 [0.15, 46.93] |
| 1.16.6 Wheezing | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.07, 6.59] |
| 1.16.7 Asthma | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.00, 14.79] |
| 1.16.8 Bronchospasm | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.16.9 Condition aggravated | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.99 [0.10, 10.33] |
| 1.16.10 Chest discomfort | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.40 [0.03, 5.66] |
| 1.16.11 Chest pain | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.00, 14.79] |
| 1.16.12 Vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.99 [0.25, 63.83] |
| 1.16.13 Post‐tussive vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.99 [0.10, 38.69] |
| 1.16.14 Headache | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.43 [0.23, 25.79] |
| 1.16.15 Decreased appetite | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.34 [0.06, 178.67] |
| 1.16.16 Infections and infestations | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.37 [0.05, 2.78] |
| 1.16.17 Musculoskeletal and connective tissue disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.66 [0.15, 46.93] |
| 1.16.18 Skin and subcutaneous disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.33 [0.20, 55.34] |
| 1.17 Adverse events at up to 6 months (moderate) | 2 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.17.1 Cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.15 [0.78, 12.77] |
| 1.17.2 Haemoptysis | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.66 [0.50, 43.49] |
| 1.17.3 Pharyngolaryngeal pain | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.43 [0.23, 25.79] |
| 1.17.4 Throat irritation | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.01 [0.03, 133.11] |
| 1.17.5 Productive cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.02, 24.81] |
| 1.17.6 Wheezing | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.44 [0.04, 4.57] |
| 1.17.7 Asthma | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.33 [0.06, 30.80] |
| 1.17.8 Bronchospasm | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.01 [0.03, 133.11] |
| 1.17.9 Condition aggravated | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.82 [0.41, 8.03] |
| 1.17.10 Chest discomfort | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.99 [0.25, 16.10] |
| 1.17.11 Chest pain | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.00, 14.79] |
| 1.17.12 Vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.13 [0.00, 7.15] |
| 1.17.13 Post‐tussive vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.62 [0.10, 224.14] |
| 1.17.14 Headache | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.99 [0.18, 22.39] |
| 1.17.15 Decreased appetite | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.17.16 Infections and infestations | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.04 [0.18, 5.95] |
| 1.17.17 Musculoskeletal and connective tissue disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.10 [0.08, 15.63] |
| 1.17.18 Skin and subcutaneous disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.22 [0.00, 14.59] |
| 1.18 Adverse events at up to 6 months (severe) | 2 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.18.1 Cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.17 [0.24, 5.69] |
| 1.18.2 Haemoptysis | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.55 [0.13, 18.99] |
| 1.18.3 Pharyngolaryngeal pain | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.33 [0.20, 55.34] |
| 1.18.4 Throat irritation | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.66 [0.05, 8.70] |
| 1.18.5 Productive cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.6 Wheezing | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.7 Asthma | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.8 Bronchospasm | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.01 [0.03, 133.11] |
| 1.18.9 Condition aggravated | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.67 [0.02, 25.14] |
| 1.18.10 Chest discomfort | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.11 Chest pain | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.12 Vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.13 Post‐tussive vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.01 [0.03, 133.11] |
| 1.18.14 Headache | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.98 [0.03, 131.35] |
| 1.18.15 Decreased appetite | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.16 Infections and infestations | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.28 [0.02, 3.47] |
| 1.18.17 Musculoskeletal and connective tissue disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 1.18.18 Skin and subcutaneous tissue disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.01 [0.03, 133.11] |
| 1.19 Adverse events at up to 6 months (total) | 2 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.19.1 Cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.05 [0.90, 4.67] |
| 1.19.2 Haemoptysis | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.88 [0.77, 10.85] |
| 1.19.3 Pharyngolaryngeal pain | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.13 [0.54, 8.44] |
| 1.19.4 Throat irritation | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.99 [0.18, 22.42] |
| 1.19.5 Productive cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.99 [0.18, 22.30] |
| 1.19.6 Wheezing | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.10, 2.95] |
| 1.19.7 Asthma | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.67 [0.05, 8.60] |
| 1.19.8 Bronchospasm | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.34 [0.06, 178.67] |
| 1.19.9 Condition aggravated | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.44, 4.52] |
| 1.19.10 Chest discomfort | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.16 [0.23, 5.74] |
| 1.19.11 Chest pain | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.13 [0.00, 7.15] |
| 1.19.12 Vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.46 [0.21, 10.28] |
| 1.19.13 Post‐tussive vomiting | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.64 [0.30, 72.37] |
| 1.19.14 Headache | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.31 [0.46, 24.06] |
| 1.19.15 Decreased appetite | 1 | 295 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.34 [0.06, 178.67] |
| 1.19.16 Infections and infestations | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.53 [0.16, 1.75] |
| 1.19.17 Musculoskeletal and connective tissue disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.43 [0.23, 25.79] |
| 1.19.18 Skin and subcutaneous tissues disorders | 2 | 600 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.44 [0.23, 25.82] |
| 1.20 Participants with treatment‐related respiratory and thoracic adverse events leading to study discontinuation | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.20.1 Asthma | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [0.08, 48.82] |
| 1.20.2 Bronchospasm | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [0.08, 48.82] |
| 1.20.3 Cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.26 [0.85, 6.02] |
| 1.20.4 Haemoptysis | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.33 [0.39, 28.25] |
| 1.20.5 Hyperventilation | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [0.08, 48.17] |
| 1.20.6 Obstructive airways disorder | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [0.08, 48.17] |
| 1.20.7 Pharynolaryngeal pain | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.66 [0.30, 23.62] |
| 1.20.8 Productive cough | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [0.08, 48.17] |
| 1.20.9 Throat irritation | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.09, 4.70] |
| 1.20.10 Wheezing | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.01, 2.76] |
| 1.21 Number of patients with protocol defined pulmonary exacerbations | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.21.1 Up to 6 months | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.51, 0.98] |
| 1.22 Time to first protocol defined pulmonary exacerbation (PDPE) | 2 | 600 | Hazard Ratio (IV, Fixed, 95% CI) | 0.70 [0.48, 1.02] |
| 1.23 Number of patients needing additional antibiotics | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.23.1 Intravenous | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.70, 0.95] |
| 1.24 Number of participants requiring hospitalisation | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.24.1 With pulmonary exacerbation, up to 6 months | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.60, 1.15] |
| 1.24.2 With PDPE, up to 6 months | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.53, 1.15] |
| 1.25 Duration of hospitalisation | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.25.1 Duration of hospitalisation with pulmonary exacerbation, up to 6 months (days) | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | ‐1.04 [‐2.58, 0.50] |
| 1.25.2 Duration of hospitalisation with PDPE, up to 6 months (days) | 2 | 600 | Mean Difference (IV, Fixed, 95% CI) | ‐0.64 [‐1.87, 0.60] |
| 1.26 Sputum weight (g) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.26.1 at 14 weeks | 2 | 460 | Mean Difference (IV, Fixed, 95% CI) | 1.02 [‐0.10, 2.14] |
| 1.27 Sputum weight (g) (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.27.1 Up to 4 months | 2 | 439 | Mean Difference (IV, Fixed, 95% CI) | ‐0.95 [‐2.05, 0.15] |
| 1.28 Microbiology: pathogens present at end of study | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.28.1 Pseudomonas aeruginosa (mucoid) | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.69, 1.17] |
| 1.28.2 Pseudomonas aeruginosa (non‐mucoid) | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.53, 1.03] |
| 1.28.3 Pseudomonas aeruginosa (not specified) | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.80 [0.45, 31.97] |
| 1.28.4 Pseudomonas spp (other) | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.42, 3.17] |
| 1.28.5 Staphylococcus aureus | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.81, 1.28] |
| 1.28.6 Methicillin‐resistant Staphylococcus aureus | 1 | 263 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.58, 2.39] |
| 1.28.7 Burkholderia cepacia (cenocepacia) | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.32, 1.48] |
| 1.28.8 Aspergillus spp | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.51, 1.30] |
| 1.28.9 Candida spp | 2 | 461 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.49, 1.33] |
| 1.29 Burden of treatment (change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.29.1 Up to 4 months | 2 | 505 | Mean Difference (IV, Fixed, 95% CI) | ‐4.60 [‐7.90, ‐1.30] |
| 1.29.2 Up to 6 months | 2 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐2.12 [‐5.70, 1.46] |
| 1.30 Adherence ≥ 60% | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.30.1 Up to 6 months | 2 | 600 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.82, 0.97] |
Comparison 2. Mannitol versus control ‐ cross‐over studies of individuals with cystic fibrosis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 FEV1 % predicted (absolute change from baseline) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 3.42 [1.13, 5.71] |
| 2.2 FEV1 % predicted (absolute change from baseline ‐ subgroup analysis by age) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.2.1 age 6 to 11 years | 1 | 78 | Mean Difference (IV, Fixed, 95% CI) | 3.78 [0.13, 7.43] |
| 2.2.2 age 12 to 17 years | 1 | 106 | Mean Difference (IV, Fixed, 95% CI) | 3.35 [0.14, 6.56] |
| 2.3 FEV1 % predicted (absolute change from baseline ‐ subgroup analysis by dornase alfa use | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.3.1 Dornase alfa user | 1 | 126 | Mean Difference (IV, Fixed, 95% CI) | 3.29 [0.28, 6.30] |
| 2.3.2 Dornase alfa non‐user | 1 | 58 | Mean Difference (IV, Fixed, 95% CI) | 3.92 [0.42, 7.42] |
| 2.4 FEV1 % predicted (relative change from baseline) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 4.97 [1.52, 8.42] |
| 2.5 FVC % predicted (absolute change from baseline) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 1.80 [‐0.72, 4.32] |
| 2.6 FVC % predicted (relative change from baseline) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 2.54 [‐0.72, 5.80] |
| 2.7 FEF25-75 % predicted (absolute change from baseline) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 5.75 [1.81, 9.69] |
| 2.8 FEF25-75 % predicted (relative change from baseline) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 10.52 [2.31, 18.73] |
| 2.9 Sputum weight (grams) (post‐initial treatment) | 1 | 174 | Mean Difference (IV, Fixed, 95% CI) | 1.33 [0.29, 2.37] |
Comparison 3. Mannitol versus control ‐ parallel study of individuals with cystic fibrosis, hospitalised due to pulmonary exacerbations.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 HRQoL ‐ change from baseline in CFCS total score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1.1 at hospital discharge (14 days) | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [‐0.50, 2.70] |
| 3.1.2 at 1 month follow‐up | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐1.00 [‐4.10, 2.10] |
| 3.2 HRQoL ‐ change from baseline in CFQ‐R respiratory domain | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.2.1 at hospital discharge (14 days) | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [‐10.50, 13.10] |
| 3.2.2 at 1 month follow‐up | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 3.50 [‐10.70, 17.70] |
| 3.3 FEV1 % predicted (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.3.1 at hospital discharge (14 days) | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 4.60 [‐3.80, 13.00] |
| 3.3.2 at 1 month follow‐up | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 5.40 [‐2.70, 13.50] |
| 3.4 FVC % predicted (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.4.1 at hospital discharge (14 days) | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 2.80 [‐3.60, 9.20] |
| 3.4.2 at 1 month follow‐up | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 1.70 [‐4.10, 7.50] |
| 3.5 FEF25-75 % predicted (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.5.1 at hospital discharge (14 days) | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 12.80 [‐3.30, 28.90] |
| 3.5.2 at 1 month follow‐up | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 3.90 [‐10.70, 18.50] |
Comparison 4. Mannitol versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 FEV1 (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1.1 Up to 3 months | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | 2.80 [‐4.80, 10.40] |
| 4.2 FVC (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.2.1 Up to 3 months | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐0.02, 0.30] |
| 4.3 FEF25-75 (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.3.1 Up to 3 months | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.23, 0.21] |
Comparison 5. Mannitol plus dornase alfa versus dornase alfa ‐ cross‐over study of individuals with cystic fibrosis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 FEV1 (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1.1 Up to 3 months | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐4.30 [‐14.10, 5.50] |
| 5.2 FVC (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.2.1 Up to 3 months | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐0.07 [‐0.30, 0.16] |
| 5.3 FEF25-75 (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.3.1 Up to 3 months | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.18, 0.24] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aitken 2012.
| Study characteristics | ||
| Methods | Double‐blind, randomised parallel trial. Duration: intervention for 26 weeks, followed by 26‐week open‐label treatment with mannitol. Multicentre: 53 sites. Location: USA, Canada, Argentina, Europe. |
|
| Participants | 305 participants with CF: mannitol group (n = 184) and control group (n = 121). Mean age: 20 years, range: 6 ‐ 53 years, 154 aged < 18 years. Mannitol group: 19% aged 6 ‐ 11 years; 30.4% aged 12 ‐ 17 years. Control group: 19.8% aged 6 ‐ 11 years; 32.2% aged 12 ‐ 17 years. Gender split: 51.5% male, 48.5% female. All participants clinically stable at start of study, mean (SD) baseline FEV1 % predicted 63.8 (15.9). 75.1% received concomitant dornase alfa. Participants receiving hypertonic saline excluded. |
|
| Interventions |
Treatment: inhaled dry powder mannitol, 400 mg 2x daily, 10 x 40 mg capsules. Administered using RS01 inhaler model 7, Plastiape, Italy. Control: subtherapeutic mannitol 50 mg 2x daily, 10 x 50 mg capsules. Administered using RS01 inhaler model 7, Plastiape, Italy (same inhaler as for active intervention). |
|
| Outcomes |
Primary outcome: FEV1 (absolute value over the 26‐week study). Secondary outcomes: FEV1 % predicted at 26 weeks; FVC; FEF25-75; PDPE; sputum weight; HRQoL (as assessed by CFQ‐R, including burden of treatment and assessment of symptoms provided by Pharmaxis). Other outcome measures: adverse events and sputum microbiology (also additional non‐routine antibiotic use and hospitalisations provided by Pharmaxis) |
|
| Funding Source | Supported by Pharmaxis Limited. | |
| Conflicts of Interest | No specific conflicts of interest of the authors of the manuscript reported. Stated that the study sponsor participated in the study design, data collection, data analysis, data interpretation, and writing of the report. After completion of the trial, the data were held and analysed by the sponsor. |
|
| Notes | Additional data supplied by manufacturer of mannitol (Pharmaxis) for the majority of outcomes reported in this review. Pharmaxis stated that the lung function results in the paper were presented as per the primary analysis method also described in the publication. The FEV1 and FVC data were presented in full, however they acknowledged that in this study the outcome FEF25-75 was mentioned in the methodology and not presented in the results section. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Further details were provided by Pharmaxis: the master randomisation list, stratified by country and dornase alfa user (yes/no) for a parallel design was prepared using SAS Version 8.1 by an external company. 300 randomisation numbers (180 active and 120 control) were generated for each country and each dornase alfa user/non‐user group. Randomization blocking by country was done in paired blocks of 5, 1 block for dornase alfa users and 1 block for non‐users. |
| Allocation concealment (selection bias) | Low risk | Pharmaxis confirmed randomisation managed via an IVRS, therefore the investigator was unaware prior to randomising the participant which specific blinded pack of treatment they would be allocated to. This provided an extra level of security (over and above the blinded nature of the study) against selection bias. |
| Blinding (performance bias and detection bias) Participants | Low risk | Described as double blind ‐ mannitol and low‐dose mannitol control administered as capsules identical in taste and appearance with identical methods of administration. |
| Blinding (performance bias and detection bias) Clinicians | Low risk | Described as double blind ‐ mannitol and low‐dose mannitol control administered as capsules identical in taste and appearance and with identical methods of administration. Pharmaxis confirmed investigators and study staff, including statisticians and all outcome assessors at investigator sites e.g. spirometry technicians were blinded. Both Bilton 2011 and Aitken 2012 used the same low‐dose mannitol control that was identical in taste and appearance to the 400 mg mannitol active intervention. |
| Blinding (performance bias and detection bias) Outcome assessors | Low risk | Described as double blind ‐ mannitol and low‐dose mannitol control administered as capsules identical in taste and appearance and with identical methods of administration. Pharmaxis confirmed blinding of investigators and study staff including statisticians and all outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Higher dropout rate with mannitol, 17% versus 12% for control, for adverse events and other reasons e.g. withdrawal of consent. However, paper provides flow diagram with timing and reasons for drop out and which group the participants were in. Sensitivity analyses conducted by Pharmaxis (methods of imputation of missing data for withdrawals) showed a consistent treatment effect in favour of mannitol and no change to conclusions. |
| Selective reporting (reporting bias) | Low risk | Limited information was reported in the study publication; particularly HRQoL and lung function. Additional data were provided by Pharmaxis on request for all primary outcomes of this review and many secondary outcomes. |
| Other bias | High risk | Participants underwent a mannitol tolerance test at screening; those who failed the test or in whom the test was incomplete were not entered into the study and thus, the participant population included only those with CF who passed the tolerance test and not all potential participants with CF. Sponsored by manufacturer of mannitol (Pharmaxis); authors or study staff worked for Pharmaxis or had financial interest. |
Bilton 2011.
| Study characteristics | ||
| Methods | Double‐blind, randomised parallel trial, stratified according to concurrent dornase alfa use. Duration: intervention for 26 weeks compared with control followed by optional 26‐week open‐label phase. Multicentre. Location: Australia, New Zealand, UK, Ireland. |
|
| Participants | 295 participants with CF; mannitol group (n = 177), control group (n = 118). Mean (SD) age: 23 (11.3) years. Gender split: 55.3% male, 44.7% female. All participants clinically stable at start of study, mean (SD) baseline FEV1 % predicted 62.0 (16.3). 55.3% received concomitant dornase alfa. Participants receiving hypertonic saline excluded. |
|
| Interventions |
Treatment: inhaled dry powder mannitol, 400 mg 2x daily administered using RS01 Monodose Inhaler model 7, Plastiape, Italy. Control: subtherapeutic mannitol (mannitol 50 mg), 2x daily administered using RS01 Monodose Inhaler model 7, Plastiape, Italy (same inhaler as for active intervention). |
|
| Outcomes |
Primary outcome: FEV1 (change over 26 weeks). Secondary outcomes: % of responders to treatment at 26 weeks; FVC; FEF25-75; PDPEs; PEs; rescue antibiotics; HRQoL scores (using age‐appropriate CFQ‐R, including burden of treatment and assessment of symptoms provided by Pharmaxis). Other outcome measures: adverse events; haematology; liver and renal function; sputum microbiology (also additional non‐routine antibiotic use and hospitalisations provided by Pharmaxis) Safety and efficacy endpoints were examined in cohorts according to dornase alfa use as well as within total cohort. |
|
| Funding Source | This work was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust, and Imperial College London (London, UK). The study was supported by Pharmaxis Limited. |
|
| Conflicts of Interest | Conflicts of interest of the all authors of the manuscript are reported on the website of the European Respiratory Journal. Stated that the study sponsor participated in the study design, data collection, data analysis, data interpretation, and writing of the report. After completion of the trial, the data were held and analysed by the sponsor. |
|
| Notes | Additional data supplied by manufacturer of mannitol (Pharmaxis) for the majority of outcomes reported in this review. Pharmaxis stated that results for investigator‐defined PEs were not reported in the paper; however, the more appropriate PDPEs were included. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised using 3:2 ratio (mannitol versus control) and stratified according to current dornase alfa use. Pharmaxis confirmed qualifying participants were randomised to a treatment arm via an IVRS using the site‐subject identification number, date of birth, initials and dornase alfa use as requisites. A master randomisation list, stratified by region (Australia and Europe) and dornase alfa use (yes/no), was prepared by an external company. Randomisation numbers (for both mannitol and control) were generated for each stratum, in blocks of 5. The randomisation number was assigned sequentially within each stratum. |
| Allocation concealment (selection bias) | Low risk | Pharmaxis confirmed randomisation was managed via an IVRS, therefore the investigator was unaware prior to randomising the participant which specific blinded pack of treatment they would be allocated to. |
| Blinding (performance bias and detection bias) Participants | Low risk | Participants blinded to treatment allocation through using a subtherapeutic does of mannitol and the same inhaler devices were used for both treatment arms (not specifically stated in the published paper but Pharmaxis confirmed use of same inhaler device with 10 capsules for both 400 mg mannitol and control (50 mg mannitol)). |
| Blinding (performance bias and detection bias) Clinicians | Low risk | Study personnel blinded to treatment allocation. Pharmaxis confirmed that investigators and study staff including statisticians were blinded. Low‐dose mannitol used as control which was identical in taste and appearance to the 400 mg mannitol. |
| Blinding (performance bias and detection bias) Outcome assessors | Low risk | Pharmaxis confirmed investigators and study staff, including statisticians and all outcome assessors at investigator sites e.g. spirometry technicians were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | High dropout rates in blinded phase of study in both arms: 37% in mannitol arm and 28% in control arm. However, sensitivity analyses conducted by Pharmaxis (methods of imputation of missing data for withdrawals) showed a consistent treatment effect in favour of mannitol and no change to conclusions. |
| Selective reporting (reporting bias) | Low risk | Limited information was reported in the study publication; particularly HRQoL and lung function. Additional data were provided by Pharmaxis on request for all primary outcomes of this review and many secondary outcomes. |
| Other bias | High risk | Participants underwent a mannitol tolerance test at screening; those who failed the test or in whom the test was incomplete were not entered into the study and thus, study population included only those with CF who passed the tolerance test and not all potential participants with CF. Sponsored by Pharmaxis; authors or study staff worked for Pharmaxis or had financial interest. |
de Boeck 2017.
| Study characteristics | ||
| Methods | Double‐blind, randomised, phase 2, cross‐over study. Duration (total): 24 weeks (2x 8‐week cross‐over periods, separated by an 8‐week washout period). Location: multicentre (39 centres) in UK, France, Belgium, Canada, Germany, Switzerland, Italy, Netherlands. |
|
| Participants | 95 children or adolescents aged 6 ‐ 17 years with CF were randomised and 84 completed the study. Inclusion criteria: aged between 6 and 18 years with a confirmed diagnosis of CF and FEV1 (% predicted) between 30% and 90%. Mean age (SD): 12.0 (3.0) years (reported for 92 children). Gender split: 55 females (60%); 37 males (40%) (reported for 92 children). Mean (SD) FEV1 (% predicted) at screening: 72.23 (11.6), range (38.3 to 89.9). Use of dornase alfa and maintenance antibiotics (if established 3 months before screening) were permitted during the study. 63 participants (68.5%) had previously used or were using dornase alfa at screening. Other CF therapies except nebulised hypertonic saline were also permitted during the study. Exclusion criteria: failing the mannitol tolerance test, design to identify participants with bronchial hyper‐reactivity. |
|
| Interventions |
Treatment: inhaled mannitol 400 mg 2x daily. Control: 10 mg of non‐spray dried, non‐respirable raw material mannitol. Interventions administered 2x daily in a cross‐over fashion. Treatment duration: 8 weeks for each treatment arm, separated by an 8‐week wash‐out period. |
|
| Outcomes |
Primary outcome: FEV1 % predicted (absolute change from each treatment period baseline). Relative change from each treatment period baseline of FEV1 % predicted was also measured. Secondary outcomes: FVC % predicted (absolute and relative change from each treatment period baseline); FEF25-75% predicted (absolute and relative change from each treatment period baseline); safety (physical examination and adverse event data); sputum weight. |
|
| Funding Source | The study was funded by Pharmaxis Limited and an employee of Pharmaxis was among the authors. Pharmaxis participated in the study design, data collection, data interpretation and and writing of the study report. |
|
| Conflicts of Interest | The authors declare that Pharmaxis provided travel support for investigator meetings, but that no investigator received any personal funding to participate in the study. | |
| Notes | The original planned sample size was 160 participants, calculated to have 90% power of detecting a difference of 3% absolute change in FEV1 % predicted. Recruitment was halted early after 95 participants were recruited (centres exhausted potential candidate pools). Post hoc power calculations showed that the recruited sample size had 80% power of detecting a difference of 3.5% absolute change in FEV1 % predicted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was conducted in a 1:1 ratio and stratified according to age (6 ‐ 11 years or 12 ‐ 17 years) and dornase alfa use (user or non‐user). No information given of how random sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding (performance bias and detection bias) Participants | Low risk | Double‐blind achieved with a placebo also containing a low dose of mannitol. |
| Blinding (performance bias and detection bias) Clinicians | Low risk | Double‐blind achieved with a placebo also containing a low dose of mannitol. |
| Blinding (performance bias and detection bias) Outcome assessors | Unclear risk | No information provided regarding blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 95 participants were randomised and 84 completed the study. Reasons for withdrawal were clearly documented in a flow diagram and primary and secondary efficacy endpoints were analysed on an intention‐to‐treat basis. Missing data were imputed using 'last observation carried forward;' such a method does not capture change over time so may have introduced bias into results. However, it is not stated how much data has been imputed and for how many participants, therefore the risk of bias is unclear. |
| Selective reporting (reporting bias) | Low risk | All important outcomes of the phase 2 efficacy and safety trial seem to be reported. |
| Other bias | High risk | Participants underwent a mannitol tolerance test at screening; those who failed the test or in whom the test was incomplete were not entered into the study and thus, study population included only those with CF who passed the tolerance test and not all potential participants with CF. Sponsored by Pharmaxis; authors or study staff worked for Pharmaxis or had financial interest. The study may be underpowered; a priori and post‐hoc power calculations are based on different sizes of treatment difference (3% or 3.5% change in FEV1 % predicted). |
Jaques 2008.
| Study characteristics | ||
| Methods | Double‐blind, randomised, cross‐over study. Duration (total): 6 weeks; each arm of cross‐over study lasted 2 weeks with 2 week wash‐out period in between. Location: Australia, New Zealand. (Multicentre) |
|
| Participants | 49 participants with CF, 39 randomised. Mean age: 19.1 years, range 8 ‐ 48 years. Gender split: 41% male, 59% female. All participants clinically stable at start of study, mean (SD) baseline FEV1 % predicted 64.9 (13.6) for the mannitol group and 64.4 (11.8) for the control group. 46.2% received concomitant dornase alfa. No hypertonic saline within 2 weeks of start of study. Group A (received mannitol first, then control) and group B (received control first, then mannitol). |
|
| Interventions |
Treatment: inhaled dry powder mannitol 420 mg 2x daily,14 x 30 mg capsules. Fine particle fraction > 40%. Children < 12 years: administered via low resistance dry powder inhaler RS01 Plastiape, Osnago, Italy. Children ≧ 12 years and adults: administered via higher resistance dry powder inhaler, Inhalator, Boeringer‐Ingelheim, Ingelheim, Germany. Control: non‐respirable mannitol with a fine particle fraction < 2%. Identical in appearance and taste to mannitol capsules. 14 capsules 2x daily using same inhaler devices as for mannitol. |
|
| Outcomes |
Primary outcome: FEV1 (change over 2 weeks). Secondary outcomes: FEF25-75; FVC; FEV1/FVC ratio and PEF; HRQoL scores (using age appropriate CFQ‐R, including burden of treatment); respiratory symptoms; sputum microbiology and safety. |
|
| Funding Source | Supported by Pharmaxis Limited and and an employee of Pharmaxis was among the authors. | |
| Conflicts of Interest | Conflicts of interest of all authors outlined in the manuscript | |
| Notes | Some additional data supplied by Pharmaxis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation code externally generated in small block design stratified to site and dornase alfa. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding (performance bias and detection bias) Participants | Low risk | Described as double‐blind; mannitol and control capsules identical in taste and appearance. Same inhaler devices for mannitol and control. |
| Blinding (performance bias and detection bias) Clinicians | Low risk | Described as double‐blind; mannitol and control capsules identical in taste and appearance. Same inhaler devices for mannitol and control. |
| Blinding (performance bias and detection bias) Outcome assessors | Low risk | Described as double‐blind, it was stated that study staff and investigators were blinded. Same inhaler devices for mannitol and control. Pharmaxis confirmed the statistician is part of the study staff and therefore was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 56 days follow‐up data not reported. 4 participants withdrew and one of these was due to "Unexplained withdrawal" by physician. Unclear how many participants were evaluated for each outcome. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes specified were reported, but little detail. |
| Other bias | High risk | Participants underwent a mannitol tolerance test at screening; those who failed the test or in whom the test was incomplete were not entered into the study and thus, the study population included only those with CF who passed the tolerance test and not all potential participants with CF. Sponsored by Pharmaxis; authors or study staff worked for Pharmaxis or had financial interest. |
Middleton 2015.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, pilot study. Parallel design. Duration: 1 month. Location: Australia. |
|
| Participants | Inclusion criteria: young people hospitalised with a pulmonary exacerbation (defined by Fuchs criteria). Exclusion criteria: FEV1 on admission < 40%, oxygen requirement, oral corticosteroids, recent commencement of mucolytic agents and failure to tolerate a test dose of the randomised study drug. 23 participants (8 ‐ 18 years) recruited. Both groups had 11 participants. Mean (SD) age: mannitol group = 13.2 (3.7) years; control group = 14.4 (3.0) years. Gender split: mannitol group = 45% male, 55% female; control group = 36% male; 64% female. Control group had lower mean (SD) admission FEV1 (58.0 (13.0)% versus 73.1 (15.6)% predicted, P < 0.05) and mean (range) FEF25-75 (25.7 (20.1 – 71.8)% versus 44.1 (22.8 – 92.43)% predicted, P < 0.05) than the IDPM group. All other parameters were equivalent on admission. |
|
| Interventions |
Treatment: inhaled dry‐powder mannitol (10x 40 mg capsules) 2x daily. Control: very low‐dose inhaled dry‐powder mannitol (10x 5 mg non‐respirable mannitol) 2x daily. Administered for 12 consecutive days prior to airway clearance treatments supervised by a physiotherapist. Used as an adjunct to intravenous antibiotics. |
|
| Outcomes | As this was a pilot study, primary and secondary outcomes were not specified, outcomes measured were: clinical status (CFCS and CFQ‐R); lung function and cardiopulmonary exercise parameters (peak oxygen consumption, ventilation efficacy, carbon dioxide production). Outcome parameters were assessed at day 7, day 14 (discharge from hospital) and 1 month (follow‐up). |
|
| Funding Source | Funded by Pharmaxis Australia but all aspects of the research study, design and analysis were conducted without input from Pharmaxis. | |
| Conflicts of Interest | The study authors declare no conflicts of interest. | |
| Notes | Additional unpublished information and results provided from conference presentation slides from the authors. A study‐drug tolerance test was conducted and deemed acceptable if baseline FEV1 decreased by < 50% immediately or < 20% 15 min after the test dose, or if oxygen saturation remained > 89%. Inhaled dry‐powder mannitol was tolerated well with 22 participants completing the study; 1 participant was excluded due to vomiting after the test dose. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Blinded randomisation was performed (1:1) ratio via random number generation without stratification. |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was described as 'blinded' but not discussed how this was achieved. |
| Blinding (performance bias and detection bias) Participants | Unclear risk | The study was described as 'double‐blind' but not discussed exactly who was blinded and how the blind was achieved. |
| Blinding (performance bias and detection bias) Clinicians | Unclear risk | The study was described as 'double‐blind' but not discussed exactly who was blinded and how the blind was achieved. |
| Blinding (performance bias and detection bias) Outcome assessors | Unclear risk | The study was described as 'double‐blind' but not discussed exactly who was blinded and how the blind was achieved. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 22 out of 23 participants completed the study and contributed data to analysis. 1 participant failed the tolerance test and was excluded. |
| Selective reporting (reporting bias) | Low risk | All important outcomes of the pilot study seem to be reported. |
| Other bias | High risk | The authors note a relatively small sample size which prevents 'strong conclusions about clinical utility in this setting. The study population included only those with CF who passed the tolerance test and not all potential participants with CF. |
Minasian 2010.
| Study characteristics | ||
| Methods | Open‐label, randomised, cross‐over study. Duration (total): 40 weeks; each arm of cross‐over study lasted 12 weeks with a 2‐week wash‐out period in between. Location: UK. (multicentre, two centres) |
|
| Participants | 45 were recruited but only 28 were randomised, 8 participants withdrew. Considering randomised participants (results different from those who completed treatment in all arms and were analysed): 28 participants with CF: Mean (SD) age: 13.3 (2.24) years. Gender split: 36% male, 64% female. All participants clinically stable at start of study. Excluded participants using hypertonic saline Characteristics of the 20 participants who completed treatment: Mean (SD) age: 13.2 (2.4) years. Gender split: 30% male, 70% female. Mean (SD) baseline FEV1: 1.67 (0.50) litres, 64 (10)% of predicted FEV1. |
|
| Interventions | Pre‐treated with participant's usual bronchodilator 15 minutes beforehand. Arm 1 Mannitol 400 mg 2x daily, 10 x 40 mg capsules. Manufacturer of mannitol not reported but assumed to be Pharmaxis as study was sponsored by this company. Administered via breath‐actuated device Osmohaler, Plastiape, Osnago, Italy. Arm 2 Dornase alfa alone ‐ 2.5 mg Pulmozyme® 2x daily via participant's usual device. Arm 3 Mannitol (as above) plus dornase alfa (dose unclear). |
|
| Outcomes |
Primary outcome: FEV1. Secondary outcomes: FVC; FEF25-75; frequency of pulmonary exacerbations; sputum microbiology; exercise tolerance; HRQoL scores; respiratory symptom questionnaires; and adverse events. |
|
| Funding Source | The study was funded by Pharmaxis Limited. | |
| Conflicts of Interest | No specific conflicts of interest are declared, other than the funding source of the study. | |
| Notes | Pulmonary exacerbations, respiratory symptom and treatment effects questionnaire, sputum microbiology and burden of treatment not reported in the publication, Pharmaxis and primary investigator (Dr Minasian) supplied some additional information for most outcomes included in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised, but details of randomisation process not discussed in paper. Dr Minasian provided additional information ‐ participants were allocated a unique randomisation number and treatment schedule with equal probability for assignment to treatment sequences. Randomisation was carried out in balanced blocks with separate schedules created for each of the 2 recruiting centres. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding (performance bias and detection bias) Participants | High risk | Open study. |
| Blinding (performance bias and detection bias) Clinicians | High risk | Open study. |
| Blinding (performance bias and detection bias) Outcome assessors | High risk | Open study. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Results in the paper only reported for participants who completed all 3 arms of the study. The desired sample size was 48 participants: 45 were recruited but only 28 were randomised and 8 participants withdrew. |
| Selective reporting (reporting bias) | Low risk | Limited information was reported in the study publication regarding outcomes of HRQoL, pulmonary exacerbations, sputum microbiology and no information reported on: time off work or school, non‐routine antibiotics, hospitalisations, tolerability or burden of treatment. Additional data were provided by Pharmaxis and primary investigator Dr Minasian on request for all primary outcomes of this review and many secondary outcomes |
| Other bias | High risk | Participants underwent a mannitol tolerance test at screening; participants who failed the test or in whom the test was incomplete were not entered into the study and thus, the participant population included only those participants with CF who passed the tolerance test and not all potential participants with CF. Underpowered study, many dropouts (48% of required sample size were analysed in the published analysis). Actual dropout rate was 8/28 = 29% (as stated above). Cross‐over design ‐ state that no carryover effect observed, but more details needed. Sponsored by Pharmaxis; authors or study staff worked for Pharmaxis or had financial interest. |
CF: cystic fibrosis CFCS: Cystic Fibrosis Clinical Score CFQ‐R: cystic fibrosis questionnaire (revised) HRQoL: health‐related quality of life FEF25-75: mid‐expiratory flow FEV1: forced expiratory volume at one second FVC: forced vital capacity IVRS: interactive voice response system PDPE: protocol‐defined pulmonary exacerbation PE: pulmonary exacerbation PEF: peak expiratory flow SD: standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Robinson 1999 | Single treatment study and examined effect on bronchial mucus clearance, not clinical. |
| Sadr 2019 | Not randomised selection to interventions, used random sampling to identify participants. |
| Teper 2011 | Dose‐response study and data on clinically‐used dose cannot be incorporated. |
Characteristics of ongoing studies [ordered by study ID]
NCT02134353.
| Study name | Long‐Term Administration of Inhaled Mannitol in Cystic Fibrosis ‐ A Safety and Efficacy Trial in Adult Cystic Fibrosis Subjects |
| Methods | Randomised, double‐blind, parallel study. Treatment duration: 26 weeks. Multicentre in USA, Argentina, Australia, Belgium, Canada, Czech Republic, Hungary, Israel, Italy, Mexico, New Zealand, Poland, Romania, Russian Federation, Slovakia, South Africa, Spain, Ukraine. |
| Participants | Estimated enrolment: 440 adults with CF (18 years or older). |
| Interventions | Inhaled mannitol 400 mg twice daily versus inhaled control twice daily for 26 weeks. |
| Outcomes | Primary outcome: FEV1 (mean absolute change in mL) over weeks 6, 14 and 26 with a REML‐based repeated measures approach. Secondary outcomes: FVC (mean change from baseline in mL); time to first pulmonary exacerbation; rate of pulmonary exacerbations; number of days in hospital due to pulmonary exacerbation; incidence of pulmonary exacerbations; days on antibiotics (oral, inhaled or IV) due to pulmonary exacerbation; ease of expectoration measured using a visual analogue scale; CFQ‐R respiratory domain score. |
| Starting date | October 2014. |
| Contact information | April 2016 (March 2016 is final data collection date for primary outcome measure). |
| Notes | Contact: Brett Charlton (brett.charlton@pharmaxis.com.au). |
CF: cystic fibrosis CFQ‐R: cystic fibrosis questionnaire (revised) FEV1: forced expiratory volume at one second FVC: forced vital capacity IV: intravenous REML: restricted maximum likelihood
Differences between protocol and review
The initial inclusion criteria were for mannitol used as a single agent. We later changed the inclusion criteria for studies to include mannitol used in conjunction with another agent (i.e. mannitol plus dornase alfa compared with dornase alfa alone) in the review as this is most likely how it will be used in clinical practice.
We originally planned to report data at one month, three months and one year; however, in a post hoc change which grouped data into more clinically appropriate time points, we reported data at time points of up to one month, up to two months, up to three months, up to six months and up to one year. If any studies followed participants beyond this time, we also planned to report these data.
A post hoc change was made to 'Measures of Treatment effect' to account for type I statistical errors when analysing number of participants with any adverse events of interest, risk ratios and 99% confidence intervals were used.
The authors have moved the outcome 'Adverse events' from the first secondary outcome to the third primary outcome and moved 'Pulmonary exacerbations' from the third primary outcome to the first secondary outcome, in line with Cochrane guidance that adverse events should be listed as one of the primary outcomes (maximum of three primary outcomes) in any review. This has also led to the 'Objectives' being revised. The authors note that pulmonary exacerbations are still considered an important and relevant outcome to individuals with CF and therefore have placed more emphasis on this secondary outcome than other secondary outcomes in the results, discussion and conclusion.
In a post hoc change, in line with Cochrane guidance, the authors have presented five summary of findings tables; one for each comparison including the primary outcomes of the review at the latest reported time point.
Contributions of authors
| Roles and responsibilities | |
| Task | Who undertook the task? |
| Protocol stage: draft the protocol | JT CM ME TD |
| Review stage: select which trials to include | JT CM TD |
| Review stage: if necessary, arbitrate on which trials to include | ME |
| Review stage: extract data from trials | JT CM TD SN |
| Review stage: enter data into RevMan | JT SN |
| Review stage: carry out the analysis | JT SN |
| Review stage: interpret the analysis | JT CM SN |
| Review stage: draft the final review | JT CM SN |
| Review stage: comment on the final review | ME TD |
| Update stage: update the review | JT CM TD SN |
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Sarah Nevitt has no potential conflict of interest to declare.
Dr Murray was local principal investigator (and thus recruiting participants) for an inhaled mannitol study run by Pharmaxis. Her institution was paid on a per‐participant basis for participants recruited to the study, but she herself did not benefit financially for running the study.
Dr Thornton was pharmacist at one site for a clinical study of inhaled mannitol in children and young people with CF in the UK. Dr Thornton’s main employment is with NICE, Centre for Guidelines. NICE Centre for Health Technology Evaluation, have published an appraisal of mannitol in CF. The Centre for Guidelines have published a guideline for the management of CF which includes mannitol. Dr Thornton is also a member of the GRADE working group.
Tiffany Dwyer declares she has worked on a trial of hypertonic saline in people with cystic fibrosis and another trial in non‐cystic fibrosis bronchiectasis. Hypertonic saline is a mucolytic agent that works similarly to inhaled mannitol.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Aitken 2012 {published and unpublished data}CF302
- Aitken M, Bilton D, Fox H, Charlton B, CF-301 &CF-302SI. Bronchitol (inhaled dry powder mannitol) in adult patients with cystic fibrosis. Journal of Cystic Fibrosis 2012;11 Suppl 1:S68. [ABSTRACT NO.: 47] [CENTRAL: 962111] [CFGD REGISTER: BD198f/BD152h] [Google Scholar]
- Aitken M, Flume PA, Geller DE, Lapey A, Zuckerman J, De Boeck K, et al. Phase III study [CF-302] of inhaled dry powder mannitol (Bronchitol™) in cystic fibrosis - results from the 6 month open label phase. Journal of Cystic Fibrosis 2011;10 Suppl 1:S20. [ABSTRACT NO.: 77] [CENTRAL: 962109] [CFGD REGISTER: BD198b] [Google Scholar]
- Aitken MI, Flume PA, Geller DE, Lapey A, Zuckerman J, De Boeck K, et al. Efficacy and safety by age group from the phase III studies of bronchitol (inhaled mannitol) in patients with CF. Pediatric Pulmonology 2011;46 Suppl 34:297. [ABSTRACT NO.: 236] [CENTRAL: 991565] [CFGD REGISTER: BD198j /BD152o] [Google Scholar]
- Aitken ML, Bellon G, De Boeck K, Flume PA, Fox HG, Geller DE, et al. Long-term inhaled dry powder mannitol in cystic fibrosis: an international randomized study. American Journal of Respiratory and Critical Care Medicine 2012;185:645-52. [CENTRAL: 881656] [CFGD REGISTER: BD198c] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Aitken ML, Flume PA, Geller DE, Lapey A, Zuckerman J, Fox H, et al. Six-month administration of inhaled mannitol in cystic fibrosis - a safety and efficacy study. Pediatric Pulmonology 2010;45 Suppl 33:299. [ABSTRACT NO.: 225] [CENTRAL: 962107] [CFGD REGISTER: BD198a] [Google Scholar]
- Bilton D, Aitken M, De Boeck K, Fox H, Charlton B, CF-301 &CF-302SI. Inhaled dry powder mannitol in cystic fibrosis (CF): impact on pulmonary exacerbations (PEs) in the Phase III studies (CF-301 & CF-302). Journal of Cystic Fibrosis 2012;11 Suppl 1:S11. [ABSTRACT NO.: WS5.2] [CENTRAL: 962110] [CFGD REGISTER: BD198g/BD152i] [Google Scholar]
- Bilton D, Aitken M, Flume PA, Geller DE, Lapey A, Zuckerman J, et al. Combined data from the two phase III studies of bronchitol (inhaled dry powder mannitol) in adult cystic fibrosis (CF) patients. Journal of Cystic Fibrosis 2011;10 Suppl 1:S20. [ABSTRACT NO.: 78] [CENTRAL: 962114] [CFGD REGISTER: BD198e/BD152g] [Google Scholar]
- Bilton D, Aitken M, Fox H, Charlton B, for the CF301 and CF 302 SI. Inhaled dry powder mannitol in cystic fibrosis (CF): the microbiology demographics and results from the phase III studies (CF301 and CF302). Journal of Cystic Fibrosis 2011;10 Suppl 1:S20. [ABSTRACT NO.: 79] [CFGD REGISTER: BD198i/BD152n] [Google Scholar]
- Bilton D, Aitken ML, Fox H, Charlton B. Improvements in lung function in Pseudomonas colonised patients treated with inhaled dry powder mannitol (Bronchitol™). Pediatric Pulmonology 2012;47 Suppl 35:314. [ABSTRACT NO.: 258] [CENTRAL: 962112] [CFGD REGISTER: BD198h/BD152j] [Google Scholar]
- Bilton D, Bellon G, Charlton B, Cooper P, De Boeck K, Flume PA, et al. Pooled analysis of two large randomised phase III inhaled mannitol studies in cystic fibrosis. Journal of Cystic Fibrosis 2013;12(4):367-76. [CENTRAL: 867634] [CFGD REGISTER: BD198d/BD152d] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Flume PA, Aitken ML, Bilton D, Agent P, Charlton B, Forster E, et al. Optimising inhaled mannitol for cystic fibrosis in an adult population: a review. Breathe 2015;11(1):39-48. [CENTRAL: 1043587] [CFGD REGISTER: BD152q // BD198l ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00630812. Long Term Administration of Inhaled Mannitol in Cystic Fibrosis. ClinicalTrials.gov/show/NCT00630812 .
- Pharmaxis. Media release: Pharmaxis announces results for second phase III trial of bronchitol in cystic fibrosis. www.pharmaxis.com.au/assets/documents/pdf/2010/2010_06_22_cf302_top_line_results.pdf 2010. [CFGD REGISTER: BD198m]
- Toward TJ, Van Engen A, Bilton D. An analysis of medical resource utilisation (MRU) associated with hospitalised pulmonary exacerbations (PulmEx) in the DPM-CF-301 and DPM-CF-302 registration trials of inhaled mannitol (Bronchitol®) in patients with cystic fibrosis (CF). Journal of Cystic Fibrosis 2013;12 Suppl 1:S125. [ABSTRACT NO.: 301] [CENTRAL: 991566] [CFGD REGISTER: BD198k/BD152p] [Google Scholar]
Bilton 2011 {published and unpublished data}CF301
- Aitken M, Bilton D, Fox H, Charlton B, CF-301 & CF-302SI. Bronchitol (inhaled dry powder mannitol) in adult patients with cystic fibrosis. Journal of Cystic Fibrosis 2012;11 Suppl 1:S68. [ABSTRACT NO.: 47] [CENTRAL: 962111] [CFGD REGISTER: BD152h/BD198f] [Google Scholar]
- Aitken MI, Flume PA, Geller DE, Lapey A, Zuckerman J, De Boeck K, et al. Efficacy and safety by age group from the phase III studies of bronchitol (inhaled mannitol) in patients with CF. Pediatric Pulmonology 2011;46 Suppl 34:297. [ABSTRACT NO.: 236] [CENTRAL: 991565] [CFGD REGISTER: BD152o/BD198j ] [Google Scholar]
- Bilton D, Aitken M, De Boeck K, Fox H, Charlton B, CF-301 & CF-302 SI. Inhaled dry powder mannitol in cystic fibrosis (CF): impact on pulmonary exacerbations (PEs) in the Phase III studies (CF-301 & CF-302). Journal of Cystic Fibrosis 2012;11 Suppl 1:S11. [ABSTRACT NO.: WS5.2] [CENTRAL: 962110] [CFGD REGISTER: BD152i/BD198g] [Google Scholar]
- Bilton D, Aitken M, Flume PA, Geller DE, Lapey A, Zuckerman J, et al. Combined data from the two phase III studies of bronchitol (inhaled dry powder mannitol) in adult cystic fibrosis (CF) patients. Journal of Cystic Fibrosis 2011;10 Suppl 1:S20. [ABSTRACT NO.: 78] [CENTRAL: 962114] [CFGD REGISTER: BD152g/BD198e] [Google Scholar]
- Bilton D, Aitken M, Fox H, Charlton B, for the CF301 and CF302 SI. Inhaled dry powder mannitol in cystic fibrosis (CF): the microbiology demographics and results from the phase III studies (CF301 and CF302). Journal of Cystic Fibrosis 2011;10 Suppl 1:S20. [ABSTRACT NO.: 79] [CFGD REGISTER: BD152n/BD198i ] [Google Scholar]
- Bilton D, Aitken ML, Fox H, Charlton B. Improvements in lung function in Pseudomonas colonised patients treated with inhaled dry powder mannitol (Bronchitol™). Pediatric Pulmonology 2012;47 Suppl 35:314. [ABSTRACT NO.: 258] [CENTRAL: 962112] [CFGD REGISTER: BD152j/BD198h] [Google Scholar]
- Bilton D, Bellon G, Charlton B, Cooper P, De Boeck K, Flume PA, et al. Pooled analysis of two large randomised phase III inhaled mannitol studies in cystic fibrosis. Journal of Cystic Fibrosis 2013;12(4):367-76. [CENTRAL: 867634] [CFGD REGISTER: BD152d/BD198d] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Charlton B. Randomised, double blind, placebo-controlled phase III study of bronchitol (inhaled powder mannitol) in Cystic Fibrosis CF. Journal of Cystic Fibrosis 2009;8 Suppl 2:S25. [ABSTRACT NO.: 95] [CFGD REGISTER: BD152a] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Charlton B. Randomised, double-blind, placebo-controlled phase III study of inhaled dry powder mannitol in cystic fibrosis. In: European Respiratory Society Annual Congress; 2009 Sep 12-16; Vienna, Austria. 2009. [ABSTRACT NO.: 1619] [CENTRAL: 758813] [CFGD REGISTER: BD152k]
- Bilton D, Robinson P, Cooper P, Charlton B. Randomized, double blind, placebo-controlled phase III study of inhaled dry powder mannitol (bronchitol) in CF. Pediatric Pulmonology 2009;44 Suppl 32:286. [ABSTRACT NO.: 216] [CFGD REGISTER: BD152b] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Gallagher C, Kolbe J, Charlton B. Phase III study of inhaled dry powder mannitol (Bronchitol") in cystic fibrosis results from the 6 month open label phase. American Journal of Respiratory and Critical Care Medicine 2010;181(Meeting Abstracts):A2338. [CENTRAL: 758927] [CFGD REGISTER: BD152l] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Gallagher CG, Klobe J, Fox H, et al. Inhaled dry powder mannitol in cystic fibrosis: an efficacy and safety study. European Respiratory Journal 2011;38(5):1071-80. [CFGD REGISTER: BD152c] [DOI] [PubMed] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Kolbe J, Gallagher CG, Fox H, et al. Inhaled dry powder mannitol in cystic fibrosis (CF): microbiology results from the 6 month double-blind CF301 study. European Respiratory Journal 2010;36 Suppl:631s. [ABSTRACT NO.: E3500] [CENTRAL: 991567] [CFGD REGISTER: BD152m ] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Kolbe J, Gallagher CG, Fox H, et al. Long term administration of inhaled dry powder mannitol in CF: results from the open label phase III DPM-CF-301 study. Pediatric Pulmonology 2010;45 Suppl 33:321. [ABSTRACT NO.: 286] [CENTRAL: 962108] [CFGD REGISTER: BD152f] [Google Scholar]
- Bilton D, Robinson P, Cooper P, Kolbe J, Gallagher CG, Fox H, et al. Phase III study of inhaled dry powder mannitol (Bronchitol™) in cystic fibrosis - results from the 6 and 12 month open label phase. Journal of Cystic Fibrosis 2010;9 Suppl 1:S21. [ABSTRACT NO.: 79] [CENTRAL: 977629] [CFGD REGISTER: BD152e] [Google Scholar]
- EUCTR2006-004078-28-GB. Long Term Administration of Inhaled Dry Powder Mannitol In Cystic Fibrosis: A Safety and Efficacy Study. https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2006-004078-28 .
- EUCTR2007-001412-23-IE. Long Term Administration of Inhaled Dry Powder Mannitol In Cystic Fibrosis: A Safety and Efficacy Study. https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2007-001412-23 .
- EUCTR2008-002740-42-DE. Long Term Administration of Inahled Mannitol in Cystic Fibrosis - A Safety and Efficacy Study. https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2008-002740-42 .
- EUCTR2013-005357-79-CZ. Long Term Administration of Inhaled Mannitol in Cystic Fibrosis: A Safety and Efficacy Trial in Adult Cystic Fibrosis Subjects. https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2013-005357-79 .
- Flume PA, Aitken ML, Bilton D, Agent P, Charlton B, Forster E, et al. Optimising inhaled mannitol for cystic fibrosis in an adult population: a review. Breathe 2015;11(1):39-48. [CENTRAL: 1043587] [CFGD REGISTER: BD152q // BD198l ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00446680. Long Term Administration of Inhaled Dry Powder Mannitol In Cystic Fibrosis - A Safety and Efficacy Study. https://ClinicalTrials.gov/show/NCT00446680 .
- Toward TJ, Van Engen A, Bilton D. An analysis of medical resource utilisation (MRU) associated with hospitalised pulmonary exacerbations (PulmEx) in the DPM-CF-301 and DPM-CF-302 registration trials of inhaled mannitol (Bronchitol®) in patients with cystic fibrosis (CF). Journal of Cystic Fibrosis 2013;12 Suppl 1:S125. [ABSTRACT NO.: 301] [CENTRAL: 991566] [CFGD REGISTER: BD152p/BD198k ] [Google Scholar]
de Boeck 2017 {published data only}NCT01883531
- Crossover trial determining the efficacy of dry powder mannitol to improve lung function in subjects aged 6-17 years. clinicaltrials.gov/ct2/show/NCT01883531 (accessed 21 April 2015). [PHARMAXIS ID: CF204]
- De Boeck C, Haarman E, Hull J, Lands LC, Moeller A, Munck A, et al. A phase II, randomised, double-blind, placebo-controlled study of dry powder mannitol in children with CF (overall and subgroup analyses). Pediatric Pulmonology 2016;51 Suppl 45:273-4. [ABSTRACT NO.: 215] [CFGD REGISTER: BD234b] [Google Scholar]
- De Boeck K, Haarman E, Hull J, Lands LC, Moeller A, Munck A, et al. A phase II, randomised, double-blind, placebo-controlled, crossover study of dry powder mannitol in children with cystic fibrosis (CF). Journal of Cystic Fibrosis 2016;15 Suppl 1:S5. [ABSTRACT NO.: WS03.3] [DOI] [PubMed] [Google Scholar]
- De Boeck K, Haarman E, Hull J, Lands LC, Moeller A, Munck A, et al. Inhaled dry powder mannitol in children with cystic fibrosis: a randomised efficacy and safety trial. Journal of Cystic Fibrosis 2017;16(3):380-7. [CFGD REGISTER: BD234c] [DOI] [PubMed] [Google Scholar]
- EUCTR2012-002699-14-GB. Trial of inhaled mannitol in children with cystic fibrosis. https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2012-002699-14 .
- NCT01883531. Crossover Trial Determining the Efficacy of Dry Powder Mannitol to Improve Lung Function in Subjects Aged 6-17 Years. https://ClinicalTrials.gov/show/NCT01883531 .
- NTR4453. A trial to determine the efficacy of dry powder mannitol in improving lung function in subjects with Cystic Fibrosis aged six to seventeen years. http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=4453 .
Jaques 2008 {published and unpublished data}
- Charlton B, Lassig A, CF-201 Study Investigators. Inhaled dry powder mannitol (Bronchitol) improves FEV1 in cystic fibrosis. Journal of Cystic Fibrosis 2006;5 Suppl 1:S11. [CFGD REGISTER: BD120b] [Google Scholar]
- Charlton B, Lassig A, CF-201 Study Investigators. Inhaled dry powder mannitol (bronchitol) improves FEV1 in cystic fibrosis. In: Proceedings of TSANZ 2006 Annual Scientific Meeting. 2006:A11. [CFGD REGISTER: BD120c]
- Charlton B, Lassig A. A randomized, blinded, controlled, crossover study of inhaled, dry powder mannitol (bronchitol) in CF. In: Proceedings of American Thoracic Society Conference; 2006 May 19-24; California, USA. 2006:A727. [CFGD REGISTER: BD120a]
- Daviskas E, Anderson SD, Jaques A, Charlton B. Inhaled mannitol improves the hydration and surface properties of sputum in patients with cystic fibrosis. Chest 2010;137:861-8. [CFGD REGISTER: BD120e] [DOI] [PubMed] [Google Scholar]
- Jaques A, Daviskas E, Turton JA, McKay K, Cooper P, Stirling RG, et al. Inhaled mannitol improves lung function in cystic fibrosis. Chest 2008;133(6):1388-96. [CFGD REGISTER: BD120d] [DOI] [PubMed] [Google Scholar]
- NCT00455130. A Phase 2 Study to Determine the Safety and Efficacy of Inhaled Dry Powder Mannitol in Cystic Fibrosis. https://ClinicalTrials.gov/show/NCT00455130 .
Middleton 2015 {published data only}
- ACTRN12612001167853. A study examining the utility of inhaled mannitol as an additional therapy during an admission to hospital for an acute pulmonary exacerbation in children with cystic fibrosis. http://www.anzctr.org.au/ACTRN12612001167853.aspx .
- Middleton A, Robinson P, McKay K, Selvadurai H. Pilot study of inhaled dry powder mannitol in young people with CF hospitalised with pulmonary exacerbation. Pediatric Pulmonology 2012;47 Suppl 35:355. [ABSTRACT NO.: 368] [CENTRAL: 962113] [CFGD REGISTER: BD199a] [Google Scholar]
- Middleton A, Robinson P, McKay K, Selvadurai H. Pilot study of inhaled dry powder mannitol in young people with cystic fibrosis hospitalised with pulmonary exacerbation. Department of Respiratory Medicine, Children's Hospital at Westmead (www.thoracic.org.au) (accessed 21 Oct 2014) 2014. [CENTRAL: 1012538] [CFGD REGISTER: BD199b]
- Middleton A, Robinson PD, McKay K, Jaffe A, Selvadurai H. A pilot study of inhaled dry-powder mannitol during cystic fibrosis-related pulmonary exacerbation [letter]. European Respiratory Journal 2015;45(2):541-4. [CENTRAL: 1048444] [CFGD REGISTER: BD199c] [EMBASE: 2015713766] [DOI] [PubMed] [Google Scholar]
Minasian 2010 {published and unpublished data}
- EUCTR2004-001888-21-GB. A cross-over comparative study of inhaled mannitol, alone and in combination with daily rhDNase, in children with cystic fibrosis - Inhaled Mannitol in Cystic Fibrosis. https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2004-001888-21 .
- Minasian C, Wallis C, Metcalfe C, Bush A. Comparison of inhaled mannitol, daily rhDNase and a combination of both in children with cystic fibrosis: a randomised trial. Thorax 2010;65(1):51-6. [CFGD REGISTER: BD133b] [DOI] [PubMed] [Google Scholar]
- Minasian CC, Wallis C, Metcalfe C, Bush A. A crossover comparative study of inhaled mannitol, alone and in combination with daily rhDNase, in children with cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:301. [CFGD REGISTER: BD133a] [Google Scholar]
- NCT00117208. Comparison of Inhaled Mannitol and rhDNase in Children With Cystic Fibrosis. https://ClinicalTrials.gov/show/NCT00117208 .
References to studies excluded from this review
Robinson 1999 {published data only}
- Robinson M, Daviskas E, Eberl S, Baker J, Anderson SD, Bye PT. Effect of inhaling a dry powder of mannitol on mucociliary clearance in adults with cystic fibrosis. Pediatric Pulmonology 1998;26 Suppl 17:280. [CFGD REGISTER: BD84a] [Google Scholar]
- Robinson M, Daviskas E, Eberl S, Baker J, Chan HK, Anderson SD, et al. The effect of inhaled mannitol on bronchial mucus clearance in cystic fibrosis patients: a pilot study. European Respiratory Journal 1999;14(3):678-85. [CFGD REGISTER: BD84b] [DOI] [PubMed] [Google Scholar]
- Robinson M, Daviskas E, Eberl S, Baker J, Chan HK, Anderson SD, et al. The effect of inhaled mannitol on bronchial mucus clearance in cystic fibrosis patients: a pilot study. Pediatric Pulmonology 2000;29(6):484. [CFGD REGISTER: BD84c] [DOI] [PubMed] [Google Scholar]
Sadr 2019 {published data only}
- IRCT20180307038994N1. Effect of mucolytic in treatment of cystic fibrosis [Comparison of the nebulized mannitol with nebulized hypertonic saline on pulmonary function in patients with cystic fibrosis]. apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT20180307038994N1 (first registered 15 April 2018).
- Sadr S, Kiani M, Rezaei M, Khanbabaee G, Ahmad S. The efficacy of nebulized soluble mannitol and comparison with 5% hypertonic saline on pulmonary function of children with cystic fibrosis. Age 2019;4(3):11-4. [Google Scholar]
- Sadr S, Kiani M, Rezaei M, Khanbabaee G, Tabatabaee SA, Hosseini A. The efficacy of nebulized soluble mannitol and comparison with 5% hypertonic saline on pulmonary function of children with cystic fibrosis. Journal of Comprehensive Pediatrics 2019;10(3):e85616. [DOI: 10.5812/compreped.85616] [DOI] [Google Scholar]
Teper 2011 {published data only}
- NCT00251056. Mannitol Dose Response Study in Cystic Fibrosis. https://ClinicalTrials.gov/show/NCT00251056 .
- Teper A, Charlton B. A dose response trial of inhaled mannitol in patients with cystic fibrosis. Canadian Respiratory Journal 2010;17:13B. [CFGD REGISTER: BD200b] [DOI] [PubMed] [Google Scholar]
- Teper A, Jaques A, Charlton B. Inhaled mannitol in patients with cystic fibrosis: a randomised open-label dose response trial. Journal of Cystic Fibrosis 2011;10:1-8. [CFGD REGISTER: BD200a] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT02134353 {published data only}NCT02134353
- A safety and efficacy trial of inhaled mannitol in adult cystic fibrosis subjects. clinicaltrials.gov/ct2/show/NCT02134353 (accessed 21 April 2015). [PHARMAXIS ID: CF303]
- Amelina E, Flume P, Krasko V, Carryer B, Charlton B, Leadbetter J, et al. Phase 3 randomised controlled study of the efficacy and safety of inhaled mannitol in adults with cystic fibrosis. Journal of Cystic Fibrosis 2018;17(Suppl 3):S25. [ABSTRACT NO.: WS14.2] [CENTRAL: CN-01778473] [CFGD REGISTER: BD251a] [EMBASE: 622931002] [Google Scholar]
- Flume P, Amelina E, Krasko V, Carryer B, Charlton B, Leadbetter J, et al. The efficacy and safety of inhaled mannitol in adults with cystic fibrosis. Pediatric Pulmonology 2017;52(Suppl 47):311. [ABSTRACT NO.: 257] [CENTRAL: CN-01430873] [CFGD REGSISTER: BD251b] [EMBASE: 619069770] [Google Scholar]
- Flume P, Bilton D, Charlton B, Dell'Anna C, Guasconi A, Miller JE, et al. Inhaled dry powder mannitol improves lung function in adults with cystic fibrosis-an integrated analysis. Pediatric Pulmonology 2019;54(S2):329-30. [ABSTRACT NO.: 464] [CENTRAL: CN-02011319] [CFGD REGISTER: BD152r // BD198n // BD266] [EMBASE: 629387702] [Google Scholar]
Additional references
Accurso 2007
- Accurso FJ. Update in cystic fibrosis 2006. American Journal of Respiratory and Critical Care Medicine 2007;175(8):754-7. [DOI: 10.1164/rccm.200701-160UP] [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderson 1997
- Anderson SD, Brannan J, Spring J, Spalding N, Rodwell LT, Chan K, et al. A new method for bronchial-provocation testing in asthmatic subjects using a dry powder of mannitol. American Journal of Respiratory and Critical Care Medicine 1997;156(3 Pt 1):758-65. [DOI] [PubMed] [Google Scholar]
Bilton 2012
- Bilton D, Aitken ML, Fox H, Charlton B. Predicting sustained response to Bronchitol™ treatment in patients with cystic fibrosis. Pediatric Pulmonology 2012;47 Suppl 35:289-90. [ABSTRACT NO.: A192] [Google Scholar]
Bilton 2013
- Bilton D, Bellon G, Charlton B, Cooper P, De Boeck K, Flume PA, et al. Pooled analysis of two large randomised phase III inhaled mannitol studies in cystic fibrosis. Journal of Cystic Fibrosis 2013;12(4):367-76. [CENTRAL: 867634] [CFGD REGISTER: BD152d/BD198d] [PMID: ] [DOI] [PubMed] [Google Scholar]
Brannan 2003
- Brannan JD, Gulliksson M, Anderson SD, Chew N, Kumlin M. Evidence of mast cell activation and leukotriene release after mannitol inhalation. European Respiratory Journal 2003;22(3):491-6. [DOI] [PubMed] [Google Scholar]
Button 2013a
- Button BM, Finlayson F, Borg B, Ivulich S, Wilson JW. Inhaled mannitol as an adjunct to airway clearance therapy in an adult cystic fibrosis population and clinical lessons learned. Pediatric Pulmonology 2013;48 Suppl 36:356-7. [Google Scholar]
Button 2013b
- Button BM, Finlayson F, Borg B, Ivulich S, Wilson JW. Clinical impact of inhaled mannitol in an adult cystic fibrosis population. Journal of Cystic Fibrosis 2013;12 Suppl 1:S98. [ABSTRACT NO.:: 194] [Google Scholar]
Bye 2007
- Bye PTP, Elkins MR. Other mucoactive agents for cystic fibrosis. Paediatric Respiratory Reviews 2007;8(1):30-9. [DOI] [PubMed] [Google Scholar]
CCFF 2002
- Canadian Cystic Fibrosis Foundation. Report of the Canadian Patient Data Registry. Canadian Patient Data Registry 2002.
CFF 2000
- Cystic Fibrosis Foundation. National Patient Registry Annual Data Report 2000. Cystic Fibrosis Foundation, Bethesda, Maryland 2000.
CFF 2005
- Cystic Fibrosis Foundation. National Patient Registry Annual Data Report 2005. Cystic Fibrosis Foundation, Bethesda, Maryland 2005.
Daviskas 1997
- Daviskas E, Anderson SD, Brannan JD, Chan H-K, Eberl S, Bautovich G. Inhalation of dry powder mannitol increases mucociliary clearance. European Respiratory Journal 1997;10(11):2449-54. [DOI] [PubMed] [Google Scholar]
Daviskas 2001
- Daviskas E, Anderson SD, Eberl S, Chan H-K, Young IH. The 24-h effect of mannitol on the clearance of mucus in patients with bronchiectasis. Chest 2001;119(2):414-21. [DOI] [PubMed] [Google Scholar]
Daviskas 2002
- Daviskas E, Anderson SD, Eberl S, Chan H-K, Young IH, Seale JP. Effects of terbutaline in combination with mannitol on mucociliary clearance. European Respiratory Journal 2002;20(6):1423-9. [DOI] [PubMed] [Google Scholar]
Daviskas 2005
- Daviskas E, Anderson SD, Gomes K, Briffa P, Cochrane B, Chan H-K, et al. Inhaled mannitol for the treatment of mucociliary dysfunction in patients with bronchiectasis: effect on lung function, health status and sputum. Respirology 2005;10(1):46-56. [DOI] [PubMed] [Google Scholar]
Daviskas 2010
- Daviskas E, Anderson SD, Jaques A, Charlton B. Inhaled mannitol improves the hydration and surface properties of sputum in patients with cystic fibrosis. Chest 2010;137(4):861-8. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG. Chapter 9 Analysing data and undertaking meta-analysis. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Dinwiddie 2000
- Dinwiddie R. Pathogenesis of lung disease in cystic fibrosis. Respiration 2000;67(1):3-8. [DOI] [PubMed] [Google Scholar]
Dodge 2007
- Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK 1947-2003. European Respiratory Journal 2007;29(3):522-26. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
EMA 2011
- European Medicines Agency. Bronchitol. www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001252/human_med_001475.jsp&mid=WC0b01ac058001d124 (accessed 18 June 2015).
Flume 2015
- Flume PA, Aitken ML, Bilton D, Agent P, Charlton B, Forster E, et al. Optimising inhaled mannitol for cystic fibrosis in an adult population. Breathe 2015;11(1):39-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fuchs 1994
- Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. New England Journal of Medicine 1994;331(10):637-42. [DOI] [PubMed] [Google Scholar]
Gee 2000
- Gee L, Abbott J, Conway S, Etherington C, Webb A. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000;55(11):946-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hart 2014
- Hart A, Sugumar K, Milan SJ, Fowler SJ, Crossingham I. Inhaled hyperosmolar agents for bronchiectasis. Cochrane Database of Systematic Reviews 2014, Issue 5. [DOI: 10.1002/14651858.CD002996.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011).The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Hodson 2000
- Hodson ME, Geddes DM. Cystic Fibrosis. Second edition. Arnold Press, 2000. [Google Scholar]
Jones 2005
- Jones AP, Riley R, Williamson PR, Whitehead A. Meta-analysis of longitudinal data. In: Royal Statistical Society Annual Conference. 2005.
McAuley 2000
- McAuley DF, Elborn JS. Cystic fibrosis: basic science. Paediatric Respiratory Reviews 2000;1(2):93-100. [DOI] [PubMed] [Google Scholar]
NICE 2012
- National Institute for Health and Clinical Excellence. Mannitol dry powder for inhalation for treating cystic fibrosis (TA266). National Institute for Health and Clinical Excellence 2012.
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Quittner 2015 [pers comm]
- Quittner A (Department of Psychology, Miami College of Arts and Sciences, USA). [personal communication]. Conversation with: T Dwyer (Sydney Medical School, Australia) February 2015.
Ratjen 2003
- Ratjen J, Doring G. Cystic Fibrosis. Lancet 2003;361:681-689. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Robinson 2001
- Robinson P. Cystic Fibrosis. Thorax 2001;56(3):237-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sawicki 2009
- Sawicki GS, Sellers DE, Robinson WM. High treatment burden in adults with cystic fibrosis: challenges to disease self-management. Journal of Cystic Fibrosis 2009;8:91-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
SMC 2013
- Scottish Medicines Consortium. Mannitol, 400mg, inhalation powder, hard capsule (Bronchitol®) SMC No. (837/13). www.scottishmedicines.org.uk/files/advice/inhaled_mannitol_Bronchitol_FINAL_January_2013_for_website.pdf (accessed 18 June 2015).
Sterne 2011
- Sterne JA, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S (editors). Cochrane Handbook forSystematic Reviews of Interventions Version 5.1 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Suri 2002
- Suri R, Grieve R, Normand C, Metcalfe C, Thompson S, Wallis C, et al. Effects of hypertonic saline, alternate day and daily rhDNase on healthcare use, costs and outcomes in children with cystic fibrosis. Thorax 2002;57(10):841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tam 2013
- Tam J, Nash EF, Ratjen F, Tullis E, Stephenson A. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD007168.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wanner 1983
- Wanner A, Maurer D, Abraham WM, Szepfalusi Z, Sielczak M. Effects of chemical mediators of anaphylaxis on ciliary function. Journal of Clinical Immunology 1983;72(6):663-7. [DOI] [PubMed] [Google Scholar]
Wark 2018
- Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 9. [DOI: 10.1002/14651858.CD001506.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2018
- Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 9. [DOI: 10.1002/14651858.CD001127.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zorginstituut 2014
- Zorginstituut Nederland. Mannitol (Bronchitol®) for the indication 'adult patients with cystic fibrosis'. Pharmacotherapeutic report, summary. www.zorginstituutnederland.nl/binaries/content/documents/zinl-www/documenten/publicaties/publications-in-english/2014/1407-mannitol-bronchitol-for-the-indication-adult-patients-with-cystic-fibrosis-pharmacotherapeutic-report-summary/Mannitol+(Bronchitol)+for+the+indication+'adult+patients+with+cystic+fibrosis'+(Pharmacotherapeutic+report,+summary).pdf (accessed 18 June 2015).
References to other published versions of this review
Nevitt 2018
- Nevitt SJ, Thornton J, Murray CS, Dwyer T. Inhaled mannitol for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 2. [DOI: 10.1002/14651858.CD008649.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nolan 2015
- Nolan SJ, Thornton J, Murray CS, Dwyer T. Inhaled mannitol for cystic fibrosis. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD008649.pub2] [DOI] [PubMed] [Google Scholar]