

Abstract
Protein kinase C-θ (PKCθ) is an important component of proximal T cell receptor (TCR) signaling. We previously identified the amino-terminal C2 domain of PKCθ as a phosphotyrosine (pTyr)-binding domain. Using a mutant form of PKCθ that cannot bind pTyr (PKCθHR2A), we show that pTyr binding by PKCθ was required for TCR-induced T cell activation, proliferation, and Th2 differentiation, but not for T cell development. Using tandem mass spectrometry and co-immunoprecipitation, we identified the kinase ζ-associated protein kinase of 70 kDa (Zap70) as a ligand of the PKCθ pTyr-binding pocket. Tyr126 of Zap70 directly bound to PKCθ, and the interdomain B residues Tyr315 and Tyr319 were indirectly required for binding to PKCθ, reflecting their role in promoting the open conformation of Zap70. PKCθHR2A-expressing CD4+ T cells displayed strong defects not only in known PKCθ-dependent signaling events such as nuclear factor κB (NF-κB) activation and Th2 differentiation, but also had a defect in full activation of Zap70 itself and in the activating phosphorylation of linker of activation of T cells (LAT) and phospholipase C-γ1 (PLCγ1), signaling proteins that are traditionally considered to be activated PKC-independently. These findings demonstrate that PKCθ plays an important role in a positive feedback regulatory loop that modulates TCR-proximal signaling and, moreover, provide a mechanistic explanation for earlier reports that documented an important role for PKCθ in T cell Ca2+ signaling. This PKCθ-Zap70 interaction could potentially serve as a promising and highly selective immunosuppression drug target in autoimmunity and organ transplantation.
INTRODUCTION
T lymphocytes are a key component of the adaptive immune system. Protein kinase C-θ (PKCθ), which we have studied extensively (1), is found predominantly in T lymphocytes, where it plays non-redundant roles in cell activation, differentiation, proliferation, and survival (2, 3). PKCθ is unique among the PKC enzymes present in T cells because it undergoes peptide-MHC (pMHC)-induced translocation to the central region of the supramolecular activation cluster (cSMAC) region of the mature T cell immunological synapse (IS) (4). There it colocalizes with the T cell costimulatory molecule CD28 in a ring surrounding the central TCR (5). PKCθ is not globally required for all T cell–mediated responses but, rather, has a selective role, being required for Th2 (6, 7) and Th17 (8, 9) responses, allograft rejection (10), and graft-versus-host disease (GVHD) (11, 12), but not (or substantially less) for antiviral responses mediated by Th1 cells and cytotoxic T lymphocytes (12–15) or for graft-versus-leukemia responses (11, 12). In contrast to the positive regulatory role of PKCθ in T effector (Teff) cells, PKCθ negatively regulates the suppressive function of regulatory T (Treg) cells (16, 17). The molecular basis for the selective and context-dependent functions of PKCθ in T cells is unclear. Given the important and selective functions of PKCθ in T cells, substantial effort has been dedicated to developing selective PKCθ inhibitors for the treatment of autoimmune diseases, GVHD, and transplant patients (11, 18–20). However, it has generally been difficult to develop clinically useful, specific PKCθ catalytic inhibitors (and inhibitors of other PKC family members in general), likely due to the similarity between PKC family members, the multiple regulatory functions that PKC enzymes play in many cells and tissues, and the potential functional redundancy among PKC family members (21–23). Thus, a better understanding of the mechanisms that regulate PKCθ activation and downstream effector functions is needed.
The intracellular localization, activation, and effector functions of PKCθ are regulated by complex mechanisms, including phosphorylation (24–29), sumoylation (30), protein-protein interactions (2, 3, 31), intrinsic regulation by its regulatory domains (32–34), and catalytic activity (33). We have demonstrated the mechanism and importance of the localization of PKCθ to the cSMAC region of the IS by showing that the V3 (hinge) region between the regulatory and catalytic domains of PKCθ binds to the cytoplasmic tail of CD28, likely through the kinase Lck, and this association is critical for the downstream functions of PKCθ in T cells (31). We also demonstrated that, similar to its nearest relative within the PKC family, PKCδ (35), the N-terminal C2-like domain of PKCθ is a phosphotyrosine (pTyr)-binding domain (36). Nevertheless, the biological relevance of this function of PKCθ, as well as the identity of the pTyr-containing protein(s) that associate(s) with the PKCθ C2 domain in TCR-stimulated T cells remains to be determined. To address these unresolved questions, we functionally characterized PKCθ-deficient T cells reconstituted with a PKCθ mutant that is incapable of pTyr binding and used mass spectrometry (MS) to identify a pTyr-containing ligand of the PKCθ C2 domain.
Here, we demonstrate that pTyr binding to the PKCθ C2 domain is required for full TCR-induced, PKCθ-dependent downstream signaling and effector functions, identify the ζ chain–associated protein kinase of 70 kDa (Zap70) as a PKCθ-interacting protein, and demonstrate a bidirectional regulatory interaction between PKCθ and Zap70 that, when disrupted, causes defects in early TCR signaling events that were traditionally considered to be independent of PKCθ. This PKCθ-Zap70 interaction could provide opportunities for the development of clinically useful allosteric inhibitors of PKCθ functions and, therefore, selective immunosuppression with less adverse side effects.
RESULTS
The pTyr-binding function of the C2 domain is required for PKCθ downstream signaling
PKCθ belongs to the “novel” subfamily of PKCs (nPKCs), which do not require Ca2+ for activation, and its nearest relative within this subfamily is PKCδ, with the C2 domains of these two PKCs displaying a substantial homology not shared by other PKCs (Fig. 1A). A previous study demonstrated that the PKCδ C2 domain was a pTyr-binding domain (35) that is structurally distinct from other known pTyr-binding domains, namely, the SH2 and PTB domains (37). Sequence alignment of the C2 domains of human and mouse PKCθ, PKCδ, PKCε, and PKCη showed that critical residues in the pTyr-binding pocket of the PKCδ C2 domain (Lys48, His62 and Arg67) are conserved with PKCθ, whereas only one of these, Arg79, is found PKCε but not in PKCη (Fig. 1A). We previously demonstrated that the PKCθ C2 domain bound a pTyr-containing synthetic peptide and, furthermore, that mutation of His63 or Arg68 impaired the ability of PKCθ to stimulate a nuclear factor κB (NF-κB) reporter gene in transfected Jurkat T cells (36). To more rigorously explore the importance of pTyr binding for PKCθ’s TCR-induced downstream functions, we mutated both His63 and Arg68 in the C2 domain to alanine (HR2A mutant) and conducted a detailed functional analysis of PKCθ-deficient (Prkcq−/−) CD4+ T cells reconstituted with this mutant.
Fig. 1. Deficient activation of T cells from PKCθ knockout mice expressing the pTyr non-binding PKCθ mutant HR2A.
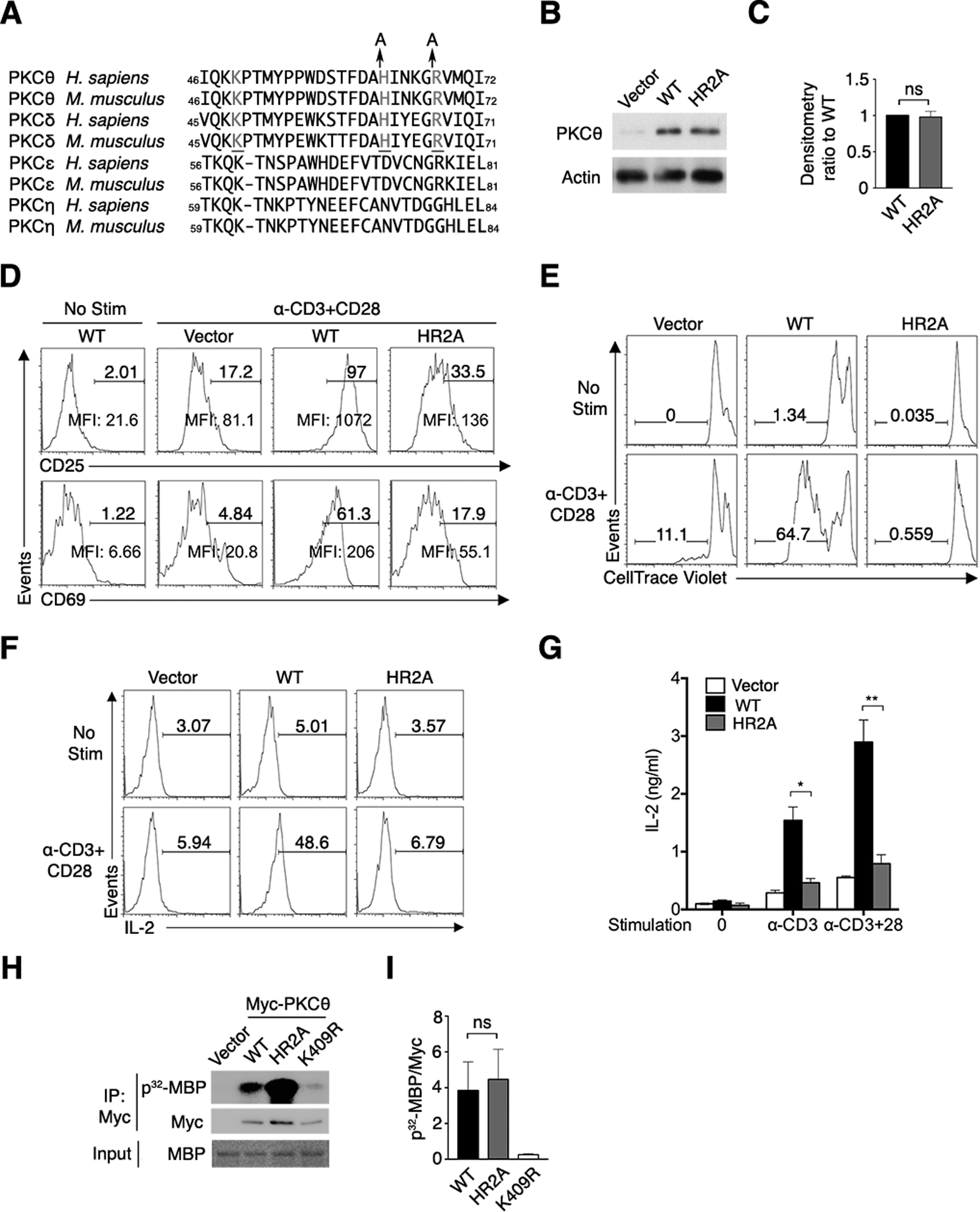
(A) Alignment of sequences surrounding the pTyr-binding site in the C2 domains of nPKC subfamily members. Residues that form the pTyr-binding pocket are in gray, and Ala mutations of His63 and Arg68 in the HR2A mutant are shown.
(B and C) Immunoblot for PKCθ in sorted (GFP+) primary CD4+ T cells that were transduced with control retrovirus (RV) encoding GFP alone (vector) or GFP and either PKCθWT or PKCθHR2A. Actin is a loading control. Results representative of 5 independent experiments are shown in (B). Quantified band intensity ratio values (C) are means ± SEM pooled from 5 independent experiments.
(D–F) Flow cytometry analysis of the surface abundance of the T cell activation markers CD25 and CD69 (D), proliferation (E), or IL-2 abundance (F) of CD4+ T cells that were either left unstimulated (No Stim) or restimulated with mAbs specific for CD3 and CD28 (α-CD3+CD28). Numbers above bracketed lines indicate % positive (D and F) or % proliferating (E) cells. Mean fluorescence intensities (MFI) of CD25 and CD69 are shown. Representative plots from 5 independent experiments are shown.
(G) Abundance of IL-2 in supernatants of sorted CD4+ T cells transduced with RV encoding PKCθWT, PKCθHR2A and left unstimulated (0), restimulated with CD3 only (α-CD3) or CD3 and CD28 mAbs (α-CD3+CD28). Data are means ± SEM of 9 biological replicates pooled from 3 independent experiments. (H and I). In vitro kinase assay with Myc-tagged PKCθWT, PKCθHR2A, or the kinase-inactive mutant PKCθK409R immunoprecipitated (IP) from transfected HEK293T cells. Immunoprecipitated proteins were mixed with radiolabeled ATP and myelin basic protein (MBP), and the reactions were separated on a gel for autoradiography and for immunoblotting for Myc and MBP. (H) is the representative blot, and (I) are band densitometry ratio (means ± SEM) pooled from 3 independent experiments. ns, not significant, *P < 0.05, **P < 0.01.
Primary CD4+ T cells from Prkcq−/− mice transduced with retrovirus (RV) expressing green fluorescent protein (GFP)-tagged wild-type PKCθ (PKCθWT) or HR2A-mutated PKCθ (PKCθHR2A) produced similar amounts of PKCθ protein (Fig. 1, B and C). Following a rest period, cells transduced with either PKCθWT or PKCθHR2A displayed very low abundance of the surface molecules CD25 and CD69, both of which are increased upon T cell activation, in the absence of restimulation. However, costimulation with monoclonal antibodies (mAbs) recognizing the TCR-associated CD3 complex and CD28 strongly increased the abundance of these two activation markers in the cells expressing PKCθWT; in contrast, CD25 and CD69 abundance increased less in CD4+ T cells expressing PKCθHR2A (Fig. 1D). The functional defects in PKCθHR2A-expressing cells were more pronounced when we assayed proliferation and IL-2 abundance in the transduced cells, as reflected by the severe reduction in both proliferation (Fig. 1E) and IL-2 abundance (Fig. 1F) in cells expressing PKCθHR2A compared to cells expressing PKCθWT. The magnitude of the decrease in IL-2 production in PKCθHR2A-expressing T cells was similar whether the cells were stimulated with the CD3 mAb alone or with a combination of both the CD3 and CD28 mAbs (Fig. 1G), implying that signals emanating from the TCR, rather than CD28 costimulation, dictate the effector function of PKCθ (see also fig. S2, B to D). These defects in T cell activation in cells that express PKCθHR2A were not the result of impaired catalytic activity, because the HR2A mutation did not reduce the catalytic activity of PKCθ in an in vitro kinase assay (Fig. 1, H and I); the kinase-inactive mutant PKCθK409R showed no activity in the same assay.
Because efficient RV transduction requires prior T cell stimulation, it was possible that PKCθ is required very early during TCR signaling, thereby accounting for the defective activation of preactivated, RV-transduced T cells (Fig. 1). To address this potential concern, we extended our analysis to CD4+ T cells derived from bone marrow (BM) mouse chimeras. These chimeras were generated by reconstituting sublethally irradiated Rag1−/− mice with BM cells isolated from Prkcq−/− mice and transduced in vitro with PKCθWT, PKCθHR2A, or empty vector (EV). Analysis of T cell development in the BM chimeras revealed similar numbers of T cells in the thymus and spleen (fig. S1A), as well as normal distribution and percentages of T cell subsets in the thymus (fig. S1B) and spleen (fig. S1C) of mice reconstituted with PKCθWT, PKCθHR2A, or EV. These results are consistent with previous reports that PKCθ is largely dispensable for T cell development (38–40) and, moreover, indicate that pTyr-binding by the PKCθ C2 domain was not required for T cell development.
Despite similar abundances of PKCθWT and PKCθHR2A in the transduced peripheral CD4+ T cells from the BM chimeras (fig. S2A), the PKCθHR2A-transduced BM chimeric CD4+ T cells displayed severe defects in the costimulation-induced increase in CD25 and CD69 abundance (Fig. 2, A and B), IL-2 production (Fig. 2, C and D), and proliferation (Fig. 2E), similar to the in vitro-transduced T cells (Fig.1). As a control, the non-transduced (GFP−) T cells from the same BM chimeras displayed similar amounts of the activation markers CD25 and CD69 (Fig. 2, A and B) and IL-2 (Fig. 2, C and D), independent of whether the mice were transduced with PKCθWT, PKCθHR2A, or EV. The modest increase in CD25 and CD69 in EV- and PKCθHR2A-expressing stimulated T cells (~30%) likely reflects the fact that increases of these activation markers are not entirely and exclusively dependent on PKCθ. We also analyzed the in vitro differentiation of sorted, transduced CD4+ T cells from the same BM chimeras into Th1 versus Th2 cells and found that Th1 differentiation, as assessed by interferon γ (IFNγ) production, of EV- or PKCθHR2A-expressing T cells was similar to that of PKCθWT-transduced T cells. In contrast, Th2 differentiation, as assayed by IL-4 production, was strongly reduced in PKCθHR2A-reconstituted T cells compared to the control PKCθWT-expressing cells (Fig. 2F and fig. S2F). These results are consistent with studies demonstrating that in vitro Th2, but not Th1, differentiation is critically dependent on PKCθ (6, 7) and, moreover, demonstrate that the pTyr-binding function of PKCθ was required for Th2, but not Th1, differentiation. Of note, CD3 stimulation alone was as effective as combined CD3 plus CD28 costimulation in increasing the abundance of CD25 (fig. S2B) and CD69 (fig. S2C), substantiating the notion that at least the increase of these activation markers by PKCθ is coupled primarily to TCR signals; however, CD28 costimulation did enhance the amount of IL-2 (fig. S2D) and the proliferation rate (Fig. S2E versus Fig. 2E) induced by CD3 stimulation alone.
Fig. 2. Deficient activation of CD4+ T cells from BM chimeric mice reconstituted with PKCθ knockout BM cells expressing PKCθHR2A.
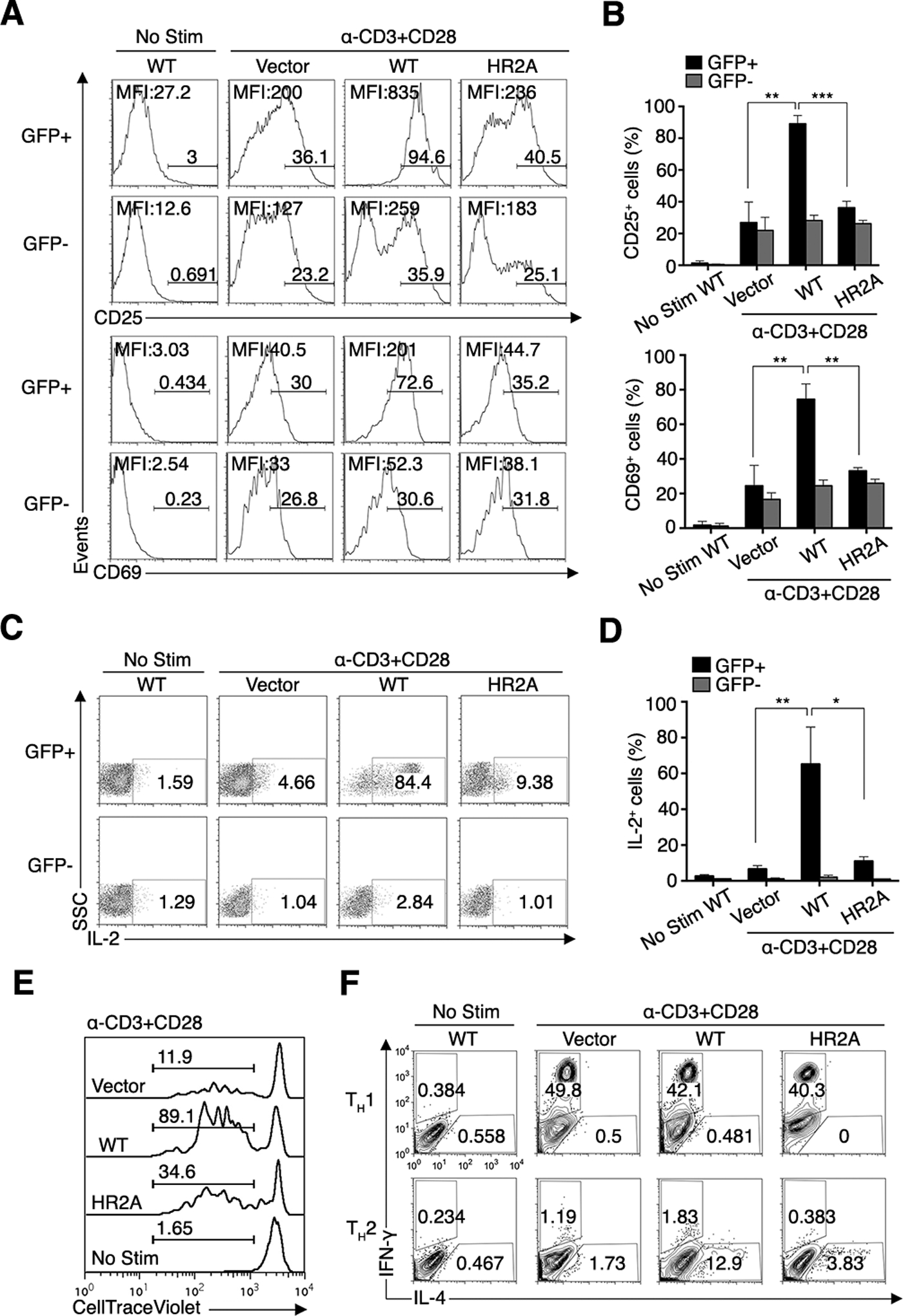
(A–D) Flow cytometry analysis of the surface abundance of CD25 and CD69 (A and B) and the production of IL-2 (C and D) by transduced (GFP+) or non-transduced (GFP−) naïve CD4+ T cells from BM chimeric mice reconstituted with PKCθ knockout BM cells expressing empty vector (vector), PKCθWT, or PKCθHR2A. Cells were left unstimulated or restimulated with CD3 and CD28 mAbs. Numbers above bracketed lines (A) and in the gated boxes (C) indicate % positive cells from a single representative mouse in each group. MFI values of CD25 and CD69 (A) or IL-2 (C) are shown, and averaged data pooled from six mice/group are shown in (B) and (D). *P < 0.05, **P < 0.01, ***P < 0.001. Results are representative of 4 independent experiments.
(E and F) Flow cytometry analysis of the proliferation (E) and the amounts of IFNγ (Th1) or IL-4 (Th2) cytokines (F) in sorted (GFP+) CD4+ cells as in (A to D). Numbers in (E) and (F) indicate the % of proliferating cells or the frequency of IFNγ- or IL-4-containing cells among gated CD4+ cells, respectively. Results are representative of 6 mice/group and 3 independent experiments.
Taken together, the above results (Figs.1, 2 and figs. S1, S2) demonstrate that pTyr binding by the PKCθ C2 domain was required for PKCθ-dependent T cell activation, proliferation, and differentiation events but not for T cell development, thereby extending our previous finding that pTyr binding by the C2 domain is also required for NF-κB activation (36), another well-established PKCθ-dependent function (39–42).
The kinase Zap70 interacts with the C2 domain of PKCθ
To identify a physiologically relevant pTyr-containing protein that associates with PKCθ in stimulated T cells, we fused glutathione S-transferase (GST) to the unmutated (C2WT) or HR2A-mutated (C2HR2A) C2 domain of PKCθ and used these recombinant fusion proteins to pull down candidate proteins from lysates of unstimulated or pervanadate (PV)-stimulated Jurkat E6.1 T cells. PV is a tyrosine phosphatase inhibitor that mimics TCR signals inducing protein tyrosine phosphorylation and, thus, greatly boosts cellular pTyr abundance in T cells. Several pTyr-containing proteins were consistently pulled down by the recombinant C2WT fusion protein, but not by the C2HR2A protein, from lysates of stimulated Jurkat cells (Fig. 3A); these protein bands were absent in lysates of unstimulated cells (Fig. 3C). Among these, protein(s) in the ~60–70-kDa molecular weight range reactive with an anti-pTyr antibody were dominant.
Fig. 3. The PKCθ C2 domain interacts with the kinase Zap70.

(A) Purified recombinant GST-tagged WT or HR2A mutant forms of the PKCθ C2 domain, GST-C2 and GST-C2-HR2A, respectively, were used for pull-down assays of lysates from unstimulated or PV-stimulated Jurkat T cells. Bound material was separated by SDS-PAGE and immunoblotted for phosphotyrosine (pTyr) and GST. The boxed area was subjected to LC-MS/MS analysis. Data are representative of 3 independent experiments.
(B) Identity of some of the peptides resolved by LC-MS/MS from PV-stimulated Jurkat T cells.
(C) Representative pull-down experiment using purified recombinant GST-C2 and GST-C2-HR2A to pull down proteins from lysates of unstimulated or PV-stimulated primary CD4+ T cells from PKCθ knockout mice. Pulled-down material (top panels) or whole-cell lysates (WCL) were analyzed by immunoblotting for the indicated proteins. Data are representative of 3 independent experiments.
(D–G) Primary B6 (D and E) or OT-II (F and G) CD4+ T cells were left unstimulated or stimulated for the indicated times with BM-derived dendritic cells (APC) pulsed with SEE or OVA peptide, respectively. PKCθ immunoprecipitates (IP) or WCL were immunoblotted for the indicated proteins. Representative immunoblots of 3 independent experiments are shown (D and F) alongside quantification of Zap70 by densitometry (E and G) pooled from 3 independent experiments. *P < 0.05, **P < 0.01.
The corresponding Coomassie-stained gel region containing the dominant Tyr-phosphorylated proteins (Fig. 3A) was excised, destained, and subjected to in-gel trypsin digestion followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). This analysis revealed eight separate Zap70 peptides that were present in the material pulled down by the PKCθ C2WT domain, but not in pull-downs with C2HR2A (Fig. 3B), potentially identifying Tyr-phosphorylated Zap70 as a ligand of the PKCθ C2 domain. A similar, single ~70-kDa protein band, which was reactive with both Zap70 and pTyr antibodies, was detected in lysates of PV-stimulated primary mouse CD4+ T cells following pull-down by GST-C2WT but not by GST-C2HR2A (Fig. 3C, upper panels). Analysis of whole cell lysates (WCL) from the same stimulated cells revealed a range of pTyr-containing proteins, including a band recognized by an antibody against phosphorylated Zap70 (Tyr319), indicative of proper T cell stimulation (Fig. 3C, lower panels).
To validate the inducible PKCθ-Zap70 interaction in T cells, we performed a co-immunoprecipitation assay on lysates of B6 (Fig. 3, D and E) and ovalbumin (OVA)-specific OT-II TCR-transgenic (Fig. 3, F and G) CD4+ T cells stimulated with superantigen E (SEE)- or OVA-pulsed antigen-presenting cells (APCs), respectively. In both cases, Zap70 co-immunoprecipitated with PKCθ as early as 1 min after stimulation, and the association was still detected after 30 min but declined at 60 min post-stimulation. A much weaker Zap70 signal was observed in unstimulated cells, indicating that the endogenous PKCθ-Zap70 association required TCR or CD28 stimulation, or both, and, hence, Zap70 tyrosine phosphorylation. Collectively, these data identify Zap70 kinase as a PKCθ C2 domain-interacting protein in stimulated T cells.
Phosphorylation of Tyr126, Tyr315, and Tyr319 in Zap70 is critical for binding to PKCθ
To identify the pTyr residue(s) in Zap70 that is (are) are required the binding of phosphorylated Zap70 to the PKCθ C2 domain, we generated five mutants in human Zap70 in a mammalian expression system with phenylalanine (F) replacements of the six Tyr (Y) residues at positions 126, 292, 315, 319, or 492 and 493 together (492/493 double mutant), all of which are known to play various important roles in Zap70 conformation, translocation, and function (43–47). We transiently expressed these mutants, as well as wild-type Zap70 (Zap70WT), in 293T cells, which do not contain endogenous Zap70 or Lck. We stimulated the cells with PV to induce cellular tyrosine phosphorylation, and then used recombinant GST-tagged PKCθ-C2WT protein (or GST protein alone as a negative control) to pull down Zap70 from these lysates. Zap70WT, Zap70Y292F, and Zap70Y492/493F bound to PKCθ-C2WT; in contrast, the Zap70Y126F, Zap70Y315F, and Zap70Y319F did not bind (Fig. 4A). These results imply that Tyr126, Tyr315, and Tyr319 are required for Zap70 to bind to PKCθ.
Fig. 4. Identification and characterization of Zap70 pTyr residues required for binding to the PKCθ C2 domain.
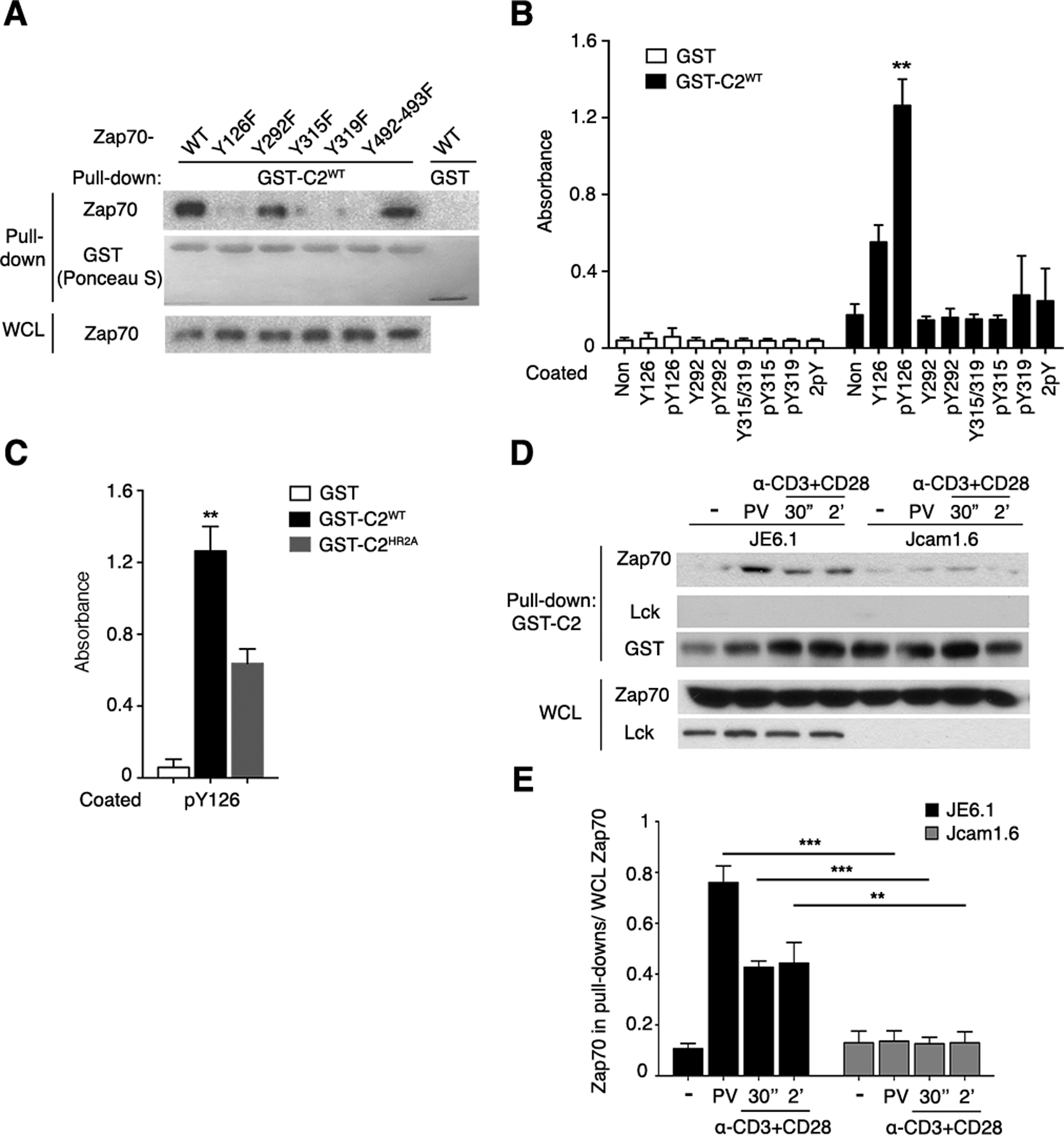
(A) Purified recombinant GST (control) or GST-C2WT was used to pull down proteins in lysates of PV-stimulated 293T cells expressing WT Zap70 or the indicated Zap70 mutants. Pulled-down material and whole-cell lysates (WCL) were separated by SDS-PAGE, stained with Ponceau S, and immunoblotted for Zap70. This experiment is representative of 3 independent experiments.
(B) Binding of recombinant GST and GST-C2WT to the indicated immobilized synthetic Zap70 peptides was assessed by an ELISA. Non, non-peptide-coated control wells.
(C) Binding of the indicated recombinant proteins to wells coated with the pTyr126 peptide determined by ELISA. Pooled data from 4 independent experiments are shown in (B) and (C), **P < 0.01.
(D and E) GST pull-down with lysates of WT (JE6.1) or Lck-deficient (Jcam1.6) Jurkat cells that were left unstimulated or stimulated with PV or CD3 and CD28 mAbs as indicated. C2WT-bound proteins or WCL were immunoblotted for the indicated proteins. Data are representative of 3 independent experiments. Pooled data from 3 independent experiments as (D) were quantitated by densitometry and shown in (E). **P < 0.01, ***P < 0.001.
To determine phosphorylation of the Tyr126, Tyr315, and/or Tyr319 residues was required for Zap70 to directly bind to the PKCθ C2 domain, we used ELISA to assess the binding of immobilized biotin-tagged 12-amino acid synthetic peptides containing each of these three residues (or Tyr292 as a negative control), in both their non-phosphorylated or phosphorylated states, to a soluble recombinant GST fusion protein of the PKCθWT C2 domain (PKCθ-C2WT), or GST alone as a negative control. Maximal binding to GST-PKCθ-C2WT (but not GST) was observed with the pTyr126 peptide, with substantially lower but detectable binding of the corresponding non-phosphorylated peptide, likely reflecting the contribution of surrounding amino acids to the binding (Fig 4B). This notion is supported by the reduced binding of the pTyr126 peptide to GST-PKCθ-C2HR2A compared to GST- PKCθ-C2WT (Fig. 4C). Binding of all other peptides was minor or barely detectable (Fig. 4B). These results strongly implicate pTyr126 in directly mediating the binding of Zap70 to the pTyr-binding pocket in the PKCθ C2 domain. As discussed below, the requirement of pTyr315 and pTyr319 for the Zap70-PKCθ association (Fig. 4A) almost certainly reflects the critical role of these phosphorylated residues in altering the conformation of resting Zap70 to a partially active “open” conformation, which would then likely expose pTyr126 for binding to the PKCθ C2 domain.
We next asked whether the kinase Lck was required for the PKCθ-Zap70 association by assessing the ability of recombinant GST-PKCθ-C2WT protein to pull down Zap70 in lysates from PV-stimulated wild-type (JE6.1) and Lck-deficient (Jcam1.6) Jurkat cells. Compared to Zap70 from JE6.1 cells, Zap70 from the Jcam1.6 cells displayed strongly reduced binding to the PKCθ C2 domain (Fig. 4, D and E). The absence of detectable Lck in the C2-bound material (Fig. 4D) rules out the possibility that Lck serves as an intermediate in forming the PKCθ-Zap70 complex, and further supports a direct interaction. These results are consistent with the well-established importance of Lck in initiating TCR signaling by phosphorylating the immunoreceptor Tyr-based activation motifs (ITAMs) of the CD3 complex, thereby enabling recruitment of Zap70 to the TCR-CD3 complex and its subsequent phosphorylation and activation.
pTyr-binding by the PKCθ C2 domain is required for full Zap70 activation and for Zap70-dependent proximal TCR signaling
To further explore the biological relevance of pTyr-binding by PKCθ, we analyzed several TCR- and CD28-proximal early signaling events in T cells from BM chimeric mice transduced with RVs encoding PKCθWT or PKCθHR2A. We first analyzed the degradation of the inhibitor of NF-κB α (IκBα), and the inducible phosphorylation of the closely related kinases IκB kinase (IKK) α and IKKβ (IKKα/β). These are surrogate markers of the activation of the conventional NF-κB pathway, which is known to require active PKCθ (39–42). As expected, stimulation of PKCθHR2A-transduced primary T cells failed to induce IKKα/β phosphorylation or IκBα degradation (Fig. 5, A and B), confirming the importance of pTyr binding by PKCθ for NF-κB activation (36).
Fig. 5. Binding of pTyr by PKCθ is required for early TCR signaling.
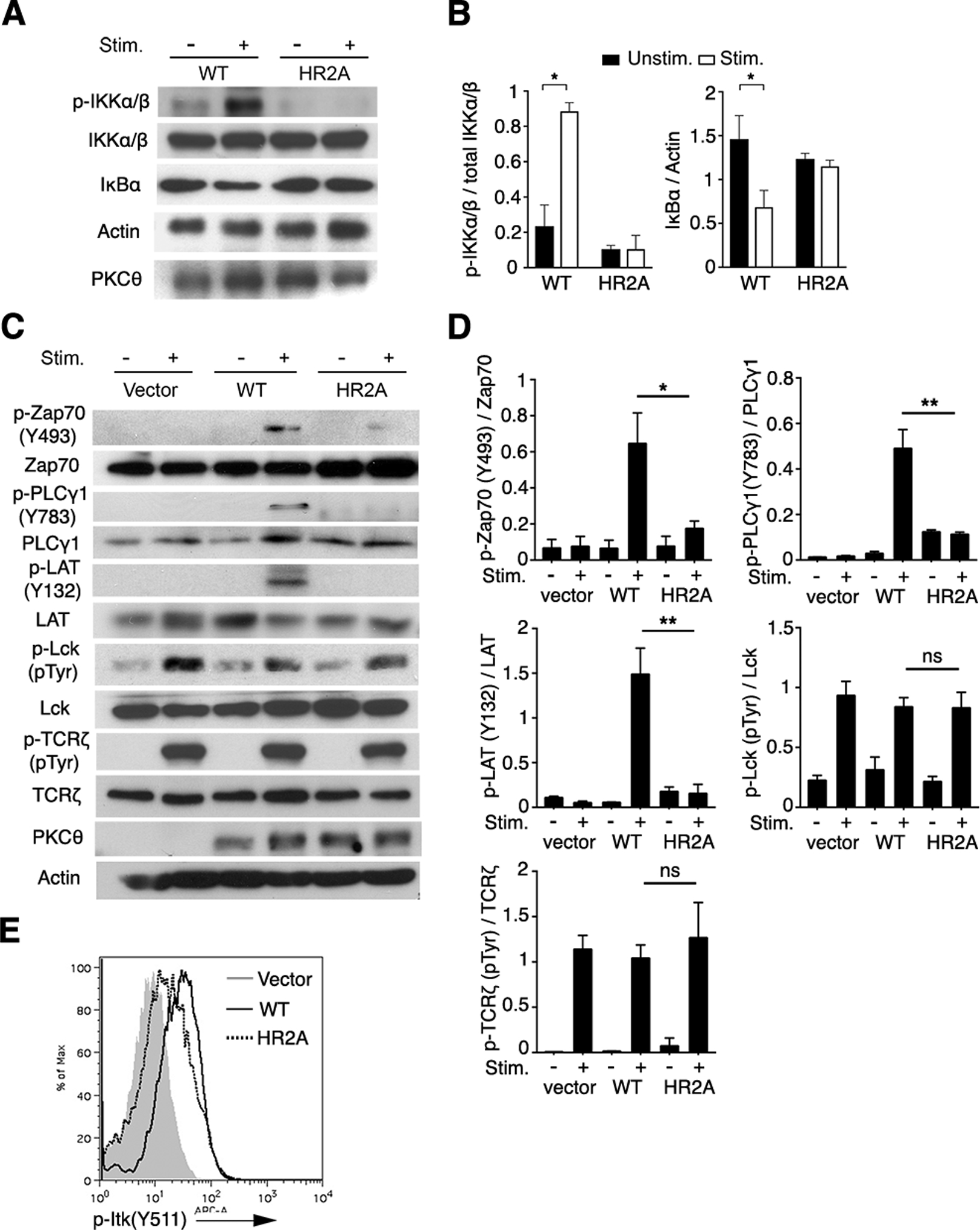
(A to D) GFP+ primary CD4+ T cells were isolated from BM chimeric mice reconstituted with PKCθ knockout BM cells transduced with RV encoding GFP and either PKCθWT or PKCθHR2A (A to C) or empty vector (C). The cells were left unstimulated or stimulated with mAbs against CD3 and CD28 for 5 min before cell lysates were subjected to SDS-PAGE and immunoblotted for the indicated proteins. p-Lck and p-TCRζ were determined by the dominant lanes of molecular weight around 55 kDa (Lck) and 23 kDa (TCRζ) from the total pTyr blot (4G10), respectively. Representative raw data are shown in (A) and (C) and pooled data from 3 independent experiments were quantitated by densitometry in (B) and (D). ns, not significant, *P < 0.05, **P < 0.01.
(E) Cells isolated and stimulated as in (A) and (C) were fixed and stained intracellularly with an APC-conjugated antibody against phosphorylated Itk (p-Itk). Plot is representative of 3 independent experiments.
To analyze the activation of more proximal TCR signaling events, we examined the phosphorylation of Zap70, phospholipase C-γ1 (PLCγ1), and the transmembrane adaptor protein linker for activation of T cells (LAT), all as of which are activated downstream of TCRs but act upstream of NF-κB. We found that stimulation of PKCθHR2A-expressing cells resulted in reduced phosphorylation of Zap70 at Tyr493, which correlates with fully active Zap70 (48), as well as the absence of detectable activating tyrosine phosphorylation of PLCγ1 and LAT (Fig. 5C and D). In contrast, the overall phosphorylation of Lck and the CD3 subunit TCRζ was largely intact as determined by a total pTyr antibody. Consistent with impaired PLCγ1 activation, the activating phosphorylation at Tyr511 on interleukin-2–inducible T-cell kinase (Itk), a tyrosine kinase that phosphorylates and activates PLCγ1 (49, 50), was also impaired in the PKCθHR2A-expressing CD4+ T cells (Fig. 5E). Similar defects were observed in CD4+ T cells from Prkcq−/− mice (fig. S3). These findings were unexpected because activating phosphorylations of Zap70, LAT, and PLCγ1 were traditionally considered to be PKCθ-independent, occurring “upstream” of the activation of PKCθ and other PKC family members (51, 52). These unexpected findings suggest that, contrary to the generally accepted dogma, active PKCθ promotes the activation of these early signal transducers.
PKCθ recruitment to the immunological synapse (IS) and the association with CD28 depend on pTyr binding by the C2 domain
The importance of pTyr-binding by the PKCθ C2 domain for PKCθ function drove us to determine if it is also required for PKCθ localization to the IS. When retrovirally expressed in Prkcq−/− OT-II CD4+ T cells, GFP-PKCθWT localized to the IS together with endogenous Zap70 following stimulation with APCs loaded with the OVA peptide (Fig. 6, A and B), consistent with the co-immunoprecipitation data (Fig. 3, D to G). In contrast, GFP-PKCθHR2A did not translocate to the IS and, instead, remained largely confined to the cytoplasm, with little or no effect on the IS recruitment of Zap70, indicating a largely intact IS formation (Fig. 6, A and B). These results suggest that binding of PKCθ C2 to pTyr was required for the selective IS localization of PKCθ, but not (or only minimally) required for general IS assembly.
Fig. 6. pTyr binding by PKCθ is required for PKCθ IS localization and association with CD28.
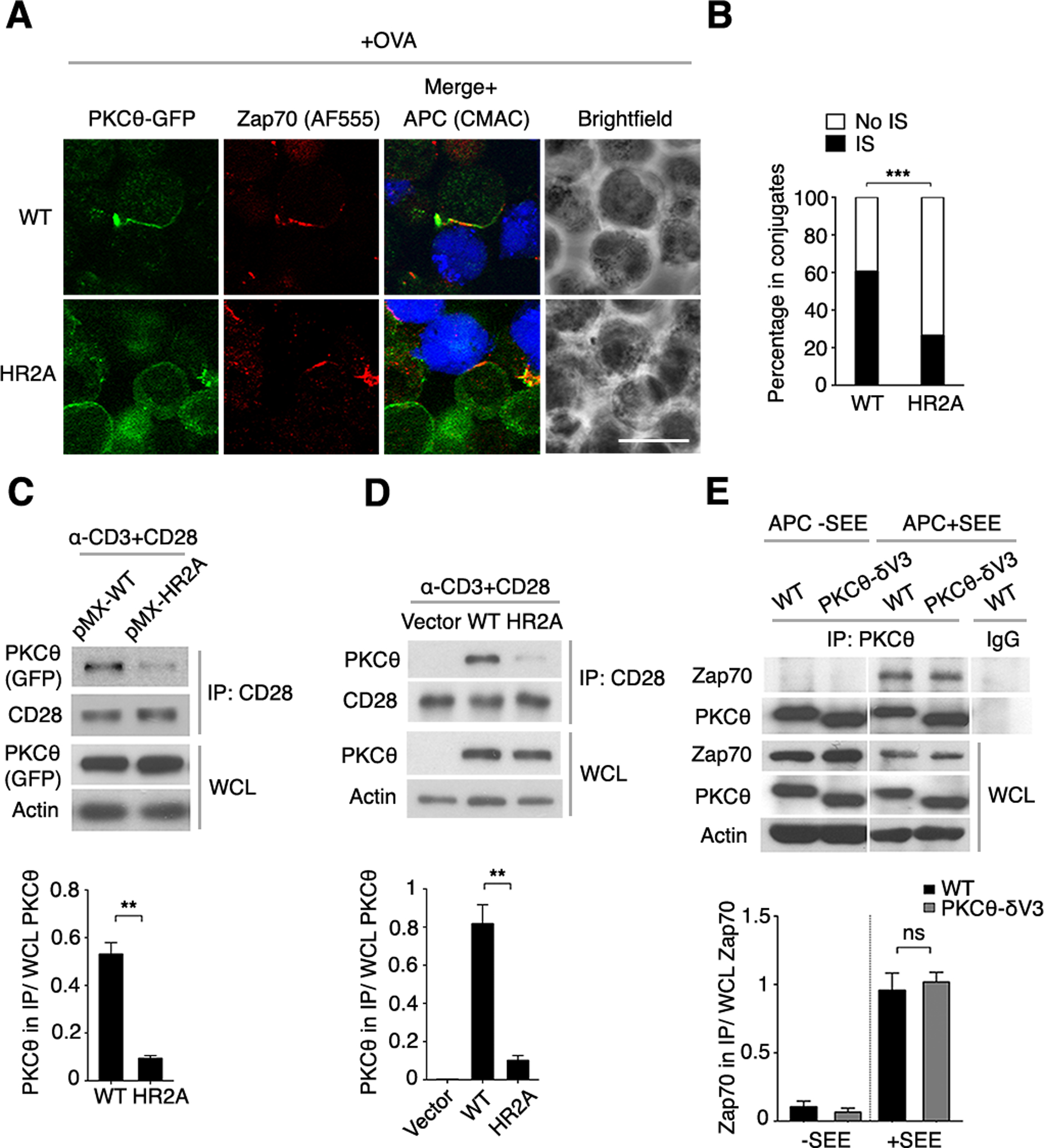
(A) Confocal imaging of Prkcq−/− OT-II CD4+ T cells that were transduced with an RV encoding GFP-tagged WT or HR2A mutant PKCθ (green), mixed with APCs labeled with the cell-tracking dye CMAC (blue), and pulsed with OVA peptide (+OVA). Fixed conjugates were stained for Zap70 plus a secondary fluorescent antibody (AF555). A representative cell is shown. Scale bar, 10 μm.
(B) Quantification of PKCθ localization within (IS) or outside of (No IS) the IS in the T cell-APC conjugates analyzed in (A). Analysis was performed only on conjugates that displayed IS-localized Zap70 and detectable PKCθ. n = ~120.
(C to E) GFP+ CD4+ T cells transduced in vitro (C and D) or derived from BM chimeras reconstituted in vivo (E) with pMIG vectors expressing PKCθWT, PKCθHR2A, or PKCθ-δV3 were stimulated with mAbs against CD3 and CD28 for 5 min. CD28 and PKCθ interaction was detected by co-immunoprecipitation (co-IP). Representative blots are shown above the corresponding band densitometry quantifications pooled from 3 independent experiments. ns, not significant, **P < 0.01, ***P < 0.001.
We demonstrated previously that inducible association of PKCθ with the cytoplasmic domain of CD28 accounts for the recruitment of PKCθ to the center of the T cell IS and, moreover, that this association is required for PKCθ-dependent downstream signaling events in TCR-stimulated T cells (31). We wished therefore to find out whether pTyr binding by PKCθ was linked to the stimulation-induced formation of the PKCθ-CD28 complex. Analysis of T cells transduced in vitro with PKCθHR2A (Fig. 6C) or obtained from PKCθHR2A-expressing BM chimeras (Fig. 6D) demonstrated that PKCθHR2A, which does not bind pTyr, was defective in its ability to associate with CD28 following T cell stimulation. This result suggests that pTyr binding by PKCθ is required for formation of the PKCθ-CD28 complex. This notion is supported by the finding that a PKCθ mutant that does not bind to CD28 (V3δ), in which the native CD28-binding V3 domain has been replaced with the corresponding domain of PKCδ (31), was still capable of efficiently associating with Zap70 after stimulation with SEE-pulsed APCs (Fig. 6E). Thus, the association of PKCθ with Zap70 appears to be largely independent of its association with CD28. We also found that, in contrast of PKCθWT expression, expression of PKCθHR2A did not alter the stimulation-induced recruitment of Zap70 to TCRζ (fig. S4A), suggesting that the binding of Zap70 to PKCθ does not exclude its recruitment to TCRζ. It is possible that two distinct pools of Zap70 associate with PKCθ and with TCRζ. However, PKCθHR2A, which did not associate with Zap70 (fig. S4B), was also impaired in its ability to co-immunoprecipitate with the adaptor protein CARMA1, a critical component of the TCR-induced NF-κB signaling pathway (53), in stimulated CD4+ T cells (fig. S4C), consistent with the other defective steps in this pathway in PKCθHR2A-expressing cells (Fig. 5, A and B).
DISCUSSION
PKCθ, a critical enzyme in T cell biology, is subject to complex regulation at several levels (2, 3). Recently we revealed that the N-terminal C2 domain of PKCθ is a pTyr-binding domain (36). As initial evidence for the importance of pTyr-binding by PKCθ, we also showed that a PKCθ mutant incapable of binding pTyr was deficient in its ability to activate an NF-κB reporter gene in Jurkat T cells (36). However, our previous study did not reveal the physiological pTyr-containing ligand that binds to PKCθ in T cells or whether this newly discovered property of the C2 domain is required for downstream effector functions of PKCθ, including proliferation, cytokine expression, and Th differentiation of primary T cells. Here we demonstrated that PKCθHR2A, a mutant that cannot bind pTyr, was unable to restore TCR-induced T cell activation in vitro or in vivo, as indicated by defective activation marker induction (CD25 and CD69), proliferation, IL-2 production, and Th2 differentiation, events all known to be PKCθ-dependent. In contrast, the in vitro Th1 differentiation of PKCθHR2A-expressing T cells remained intact, consistent with findings that Th1 differentiation is largely PKCθ-independent (6, 7). The HR2A mutation did not affect thymic T cell development, in agreement with the minor role of PKCθ in T cell development (38–40). This lack of role for PKCθ in T cell development is likely explained by redundancy with another PKC family member, PKCη (38). This finding also implies that the critical role of Zap70 kinase, the PKCθ-binding protein we identified here, in T cell development (54, 55), is independent of its interaction with PKCθ.
Using tandem MS, we identified phosphorylated Zap70 kinase as a ligand that binds to the PKCθ C2 domain in TCR-stimulated T cells and confirmed this interaction by co-immunoprecipitation of a PKCθ-Zap70 complex from stimulated T cells, including antigen (OVA)-stimulated primary OT-II CD4+ T cells. This interaction was further confirmed by the prominent IS colocalization of PKCθWT with Zap70 in stimulated T cells, which was substantially reduced in cells expressing the pTyr-non-binding PKCθHR2A mutant. PKCθ, which is known to associate with CD28 in PMA- or antigen-stimulated CD4+ T cells (5, 31), is likely to have an opportunity to physically interact with Zap70 in TCR microclusters that are formed in the periphery of the IS early after T cell stimulation because CD28 has been shown to reside in these early Zap70-containing TCR microclusters and to segregate later, together with PKCθ, from these TCR microclusters, localizing in an outer subregion of the cSMAC (5). Although we concentrated here on characterizing the PKCθ-Zap70 interaction, it is worth noting that the recombinant unmodified C2 domain (but not the HR2A-mutated C2 domain) pulled down a few unidentified pTyr-containing proteins in addition to Zap70. Thus, PKCθ may well interact with other tyrosine-phosphorylated proteins, the identity as well as importance of which remain to be established, in T cells. However, the biological relevance of the PKCθ-Zap70 interaction is supported by, and likely provides an explanation for, earlier findings that Zap70 promotes PKCθ activation (56) and that Zap70 is required for recruitment of PKCθ to the T cell IS (57).
Using a peptide-binding ELISA, we identified Zap70 Tyr126 as a highly likely site that can, when phosphorylated, mediate direct association of Zap70 with PKCθ. In contrast, peptides corresponding to pTyr315 or pTyr319 in Zap70 did not bind the PKCθ C2 domain in this assay, although mutations of these two residues to phenylalanine abolished the interaction of the mutant Zap70 proteins with the C2 domain of PKCθ in pull-down assays. This apparent discrepancy is most likely attributed to the well-established role of pTyr315 and pTyr319 in regulating the conformation and, consequently, the activity of Zap70. These two residues are important components of an autoinhibitory switch that controls the activation of Zap70. According to one model, in their unphosphorylated resting state, mimicked by mutation of these residues to phenylalanine, these residues keep the kinase in a closed, inactive state (43, 44, 48). Subsequent Lck-mediated phosphorylation or trans-autophosphorylation of these Tyr residues in TCR-bound and partially active Zap70 results in a conformational change that exposes its kinase domain, allowing trans-autophosphorylation of Tyr492/493 and other Tyr residues (such as Tyr126) and resulting in a fully active kinase (46, 48, 58). A more recent study revealed that stabilizing the open conformation of Zap70 through phosphorylation of Tyr315/319 by Lck or trans-autophosphorylation prolongs the dwell time of Zap70 at the TCR and, thereby, increases the probability of its activation through Tyr493 phosphorylation (45). On the other hand, trans-autophosphorylation of Tyr126 releases active Zap70 molecules from the TCR into the plasma membrane, thereby promoting signal amplification and propagation (47). Hence, a likely scenario is that, following TCR stimulation of resting T cells and Lck-mediated ITAM phosphorylation, Zap70 Tyr315/319 phosphorylation or TCR binding induces a conformational change that exposes Tyr126 for phosphorylation and direct binding to the C2 domain of PKCθ. This could potentially promote Zap70 release from TCR complex and allow it to efficiently phosphorylate its downstream target, LAT, and subsequently activate LAT-associated signaling proteins such as PLCγ1 and Itk (59, 60). Consistent with such a scenario, the Y126F mutation of Zap70, which would prevent its binding to the C2 domain of PKCθ, is associated with reduced LAT phosphorylation and Ca2+ signaling (47), similar to the reduced phosphorylation of LAT, PLCγ1 and Itk, which we observed here in T cells expressing the HR2A-mutated PKCθ, which cannot bind and recruit Zap70.
The association between PKCθ and Zap70 could potentially serve another purpose: facilitating integration of distinct signaling modules initiated by TCR and CD28 ligation. The TCR, which recruits Zap70, and CD28, which recruits PKCθ, have been shown to co-localize to peripheral TCR microclusters during the early (~2 min) stage of IS assembly, and to later (~20 min) segregate into distinct subdomains in the mature IS (5). Thus, the inducible PKCθ-Zap70 association might be facilitated by the colocalization of the TCR and CD28 in the same early peripheral microclusters, thereby facilitating the integration of distinct signaling modules that promote the activation of NF-κB (61) and other downstream effectors.
Unexpectedly, PKCθ-deficient T cells reconstituted with the PKCθHR2A mutant displayed substantial defects in early TCR signaling events, such as the activating phosphorylation of LAT, PLCγ1, and Itk. LAT is a direct Zap70 substrate, and it is required for the recruitment and activation of PLCγ1 and Itk. Thus, the impaired Zap70-dependent phosphorylation of LAT in the presence of PKCθHR2A provides additional, indirect support to our conclusion that full Zap70 activation or, rather, its increased ability to promote highly efficient downstream signaling due to its release from the TCR (47), is mediated by its interaction with PKCθ. More proximal TCR signaling events such as the tyrosine phosphorylation of Lck and TCRζ were unaffected in the PKCθHR2A-expressing cells. We documented similar defects in early TCR signaling in primary Prkcq−/− CD4+ T cells. These defects were surprising because the activation of these proximal signal transducers is traditionally considered to occur upstream or independently of PKC activation (51, 52). However, in retrospect, our findings are consistent with and potentially explain previous reports that PKCθ controls the activation of PLCγ1, LAT, and Itk, and that Prkcq−/− CD4+ T cells display defects in TCR-induced Ca2+ signaling and in Ca2+-dependent activation of nuclear factor of activated T cells (NFAT), a transcription factor necessary for T cell activation (39, 62, 63). Thus, rather than functioning in a linear TCR signaling scheme strictly downstream of Zap70, LAT, and PLCγ1, PKCθ likely functions in a complex feedback loop(s) triggered by TCR and CD28 stimulation, with substantial cross-regulation among components of these loops, including regulation of TCR-proximal signaling such as the activation of Zap70, LAT, and PLCγ1.
The presence of Zap70 specifically in T cells and its critical role in controlling most, if not all, TCR signaling events (43), including PKCθ function as demonstrated here, implicate this tyrosine kinase as a potentially attractive drug target in pathological conditions wherein suppression of T cell hyperactivity is desired, such as autoimmunity and transplant rejection. However, success in developing selective Zap70 catalytic inhibitors for clinical use has been limited. Our findings that pTyr binding to PKCθ is important for intact TCR signaling and effector functions and that PKCθ is required for optimal Zap70 activation raise the possibility that allosteric inhibitors that prevent binding of pTyr-containing proteins to PKCθ, which is also abundant in T cells, could represent an attractive alternative strategy for selective T cell immunosuppression. More studies are required to further explore the molecular details of the interaction and cross-regulation between PKCθ and Zap70, and to validate the potential utility of this interaction as a drug target.
MATERIALS AND METHODS
Mice, primary cells and cell lines
C57BL/6 (B6), OT-II TCR-transgenic, Rag1−/−, and Prkcq−/− mice (a gift from D. Littman) were maintained under specific pathogen-free conditions and manipulated according to a protocol approved by the La Jolla Institute for Immunology Animal Care Committee. CD4+ T cells were isolated by anti-mouse CD4 magnetic particles (L3T4; BD IMag) and cultured in RPMI-1640 medium (Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, 1 mM sodium pyruvate, 1 mM MEM nonessential amino acids, and 100 U/ml each of penicillin G and streptomycin (Life Technologies).
Jurkat E6.1 cells or the Lck-deficient Jcam1.6 T cells were maintained in complete RPMI-1640 medium supplemented with 10% FBS, penicillin, and streptomycin. HEK293T cells and Platinum-E packaging cells were cultured in complete DMEM (Gibco) containing penicillin, streptomycin, and 10% (v/v) FBS. All cell lines were obtained from the American Type Culture Collection (ATCC).
Plasmids
The cDNA for full-length human PKCθ was generated by PCR amplification and subcloned into the bicistronic IRES retroviral vector (RV) pMIG (64) for generation of bone marrow (BM) chimeras, or into the C-terminal eGFP-encoding fusion vector pMX for in vitro transduction of cells. A cDNA encoding the C2 domain of human PKCθ (aa 1–144) was subcloned into pGEX-4T2 to express PKCθ-C2 downstream of an N-terminal GST fusion tag. PKCθ-V3δ (replacement of PKCθ-V3 with amino acids 282–358 of PKCδ) was constructed by overlapping PCR (31). Full-length human Zap70 in the mammalian expression vector pEFneo was described previously (65). Point mutants were generated with the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions.
RV transduction and stimulation
Platinum-E packaging cells were plated in a 6-well plate in 2 ml RPMI-1640 medium plus 10% FBS. After 24 hours, cells were transfected with empty pMIG or pMX vector or the appropriate PKCθ-expressing vectors (3 μg) with TransIT-LT1 transfection reagent (Mirus Bio). After overnight incubation, the medium was replaced, and cultures were maintained for another 24 hours. Culture supernatants were then collected and filtered, supplemented with polybrene (8 μg/ml), and used to transduce CD4+ T cells that had been preactivated for 24 hours on plates coated with mAbs recognizing CD3 and CD28 (2 μg/ml each). After centrifuging plates for 1.5–2 hours at 2,000 rpm, supernatants were replaced by fresh FBS-containing RPMI-1640 plus recombinant IL-2 (100 U/ml; Biolegend). Cells were incubated for another 24 hours at 37°C. On day 3, cells were washed, transferred to new plates, and cultured in RPMI-1640 medium containing FBS and IL-2 without stimulation for 2 additional days before restimulation with mAbs recognizing CD3 or CD28 or both. For Th cell differentiation, CD4+ T cells transduced as described above were cultured under polarizing conditions (Th1: 100 U/ml IL-2, 20 U/ml IL-12 and 10 μg/ml IL-4 mAb; Th2: 100 U/ml IL-2, 200 U/ml IL-4, 10 μg/ml each IL-12 mAb and IFNγ mAb). After resting for 3 days, cells were restimulated for 8 hours on plates coated with CD3 and CD28 mAbs (1 μg/ml each) without additional cytokines, and intracellular cytokine staining (ICS) was performed in the presence of GolgiStop (BD Biosciences).
BM chimeras
BM cells were flushed from the femurs and tibias of Prkcq−/− mice, subjected to a lineage-negative selection with a lineage cell depletion kit (Miltenyi Biotech), and cultured for 24 hours in DMEM (Gibco) supplemented with FBS (10% v/v), IL-3 (10 ng/ml), IL-6 (20 ng/ml), and SCF (100 ng/ml; all from PeproTech) before RV transduction for 2 consecutive days as described above. RV-transduced BM cells were injected intravenously into sublethally irradiated (500 rad) Rag1−/− mice 2 days later. Recipient mice were euthanized and analyzed ~8 weeks after reconstitution.
Flow cytometry and intracellular staining
Cell proliferation was assessed by flow cytometry of T cells prelabeled with CellTrace-Violet (CTV) Cell Proliferation dye (Life Technologies), and measured by CTV dilution after stimulation for 72 hours. Abundances of CD25 and CD69 were determined by live cell surface staining on ice with mAbs recognizing these activation markers (diluted 1:200 in 1%BSA/PBS). For intracellular staining, cells were fixed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences), followed by staining with antibodies recognizing phosphorylated Itk or cytokines diluted 1:200 in Perm/Wash buffer (BD Biosciences). For PKCθ, fixed and permeabilized cells were stained with mouse primary antibodies against PKCθ (BD Biosciences) followed by an Alexa Fluor 647-conjugated anti-mouse secondary antibody. Acquisition was performed on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software (v10; FlowJo LLC).
Immunofluorescence microscopy
APCs were prepared from wild-type B6 mice by depleting splenocytes of CD4+ T cells using beads coated with a CD4 mAb (L3T4). APCs were stained with CellTracker Blue (CMAC; 20 μM) and then pulsed for 30 min at 37°C with 5 μM OVA peptide. Cells were washed twice in PBS and mixed with T cells at a ratio of 1:1, followed by incubation for 20 min at 37°C. T cell–APC conjugates were plated on poly-L-lysine–coated slides and incubated for 15 min at room temperature before fixation with 4% (w/v) paraformaldehyde and permeabilization with PBS containing BSA (1% w/v) and Triton X-100 (0.2% v/v). Nonspecific binding was blocked by incubation for 30 min with PBS/2% BSA, and cells were stained overnight with a rabbit Zap70 primary antibody at 4°C. Samples were washed three times in PBS/Tween20 and incubated for 1 hour at room temperature with Alexa Fluor 555-coupled goat anti-rabbit secondary antibody (Invitrogen). Images were captured by a Leica SP5 laser scanning confocal microscope equipped with a 63x objective lens.
Immunoprecipitation, immunoblotting, and GST pull-down assays
Cells were lysed in 1% digitonin (w/v; for CD28 co-immunoprecipitations) or 1% NP-40 (v/v; for all other immunoprecipitations and pull-downs) in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA) supplemented with protease inhibitors (10 μg/ml aprotinin, 10 μg/ml leupeptin and 1 mM PMSF) and phosphatase inhibitors (5 mM NaPPi and 1 mM Na3VO4). Cells were incubated for 2 hours with the antibody, and proteins were immunoprecipitated overnight at 4°C with protein G–Sepharose beads (GE Healthcare). Immunoprecipitated proteins were resolved by SDS–PAGE, transferred onto a PVDF membrane, and probed overnight at 4°C with primary antibodies, followed by incubation for 1 hour at room temperature with HRP-conjugated secondary antibodies. Signals were visualized by enhanced chemiluminescence (ECL; GE Healthcare) and exposed to X-ray film. Densitometry analysis was performed with ImageJ software. GST or GST fusion proteins were batch expressed in E. coli BL21 cells, purified, and dialyzed overnight against PBS/10% glycerol. GST pull-down assays were performed as previously described (66).
ELISA
Aliquots of transduced Prkcq−/− CD4+ T cells (5 × 104) were stimulated for 48 hours with CD3 or CD28 mAbs, or both, and IL-2 concentration in culture supernatants was determined by ELISA according to the manufacturer’s instructions (Biolegend). A 96-well plate (Corning Costar) was coated overnight at 4°C with an IL-2 mAb. Triplicates of IL-2 standards and supernatants from cultured cells were then added to the plate, followed by a 2-hour incubation at room temperature. A biotinylated IL-2 antibody was added to the plate, followed by incubation for 1 hour, and then streptavidin-HRP was added, followed by incubation for 30 min, all at room temperature. After washing, the amount of bound avidin was assessed with TMB peroxidase that was acidified by 2 N H2SO4. Absorbance at 450 nm was measured in a SpectraMax plate reader (Molecular Devices).
For peptide ELISA, the following N-terminal biotinylated peptides were synthesized and purified to >90% as judged by HPLC and mass spectrometry (GenScript): Y126, AMVRDYVRQTWK; pY126, AMVRDpYVRQTWK; Y292, LNSDGYTPEPAR; pY292, LNSDGpYTPEPAR; Y315/319, TSVYESPYSDPE; pY315, TSVpYESPYSDPE; pY319, TSVYESPpYSDPE; 2pY (pY315 and pY319), TSVpYESPpYSDPE. Biotinylated peptides (10 μg/ml in 100 μl PBS) were added to microtiter wells of a streptavidin-precoated plate (Pierce) and incubated overnight at 4°C. Proper and equal binding of the different peptides to the wells was confirmed by adding HRP-streptavidin to a duplicate set of wells. Non-specific binding sites were blocked by incubation with 300 μl of 5% BSA/PBS for 1 hour at room temperature. Following washing with PBST (0.5% Tween-20/PBS), GST, or GST-C2 proteins [1 μg in 1% (w/v) NP-40 lysis buffer] were added for 1 hour. After further washing, the wells were incubated for an additional hour with a mouse antibody against GST (1:2000 in PBST), followed by 1 hour incubation with HRP-conjugated anti-mouse Ig at room temperature. Samples were analyzed in triplicate in both peptide-coated and uncoated control wells, and ELISA readout was performed as described above.
PKCθ in vitro kinase assay
HEK293T cells transfected with empty vector or expression plasmids encoding Myc-tagged PKCθWT, PKCθHR2A, or kinase-dead PKCθK409R, were lysed and immunoprecipitated with an antibody against Myc. Immunoprecipitates were washed twice with lysis buffer and twice with PKCθ kinase buffer (20 mM HEPES, 10 mM MgCl2 and 0.1 mM EGTA), then resuspended in 25 μl of the corresponding kinase buffer containing 5 μCi [γ−32P] ATP (Perkin Elmer), 20 μM ATP, and 1 μg of myelin basic protein (MBP) substrate (Sigma), in the presence or absence of cofactors, phosphatidylserine (200 μg/ml; Promega) and 10 μM PMA. Samples were incubated for 30 min at 30°C with gentle shaking. The reaction was stopped by the addition of 5x loading buffer. Samples were incubated for 10 min at 95°C, and separated by SDS-PAGE. Gels were exposed to a BAS Storage Phosphor screen (GE Healthcare) and scanned with the Typhoon Trio Imager (GE Healthcare).
LC-MS/MS
Protein bands were excised from Coomassie-stained gels and destained, then subjected to overnight in-gel trypsin digestion [12.5 ng/μL in 50 mM (NH4)HCO3 at 37°C]. The resulting peptides were extracted with 50% acetonitrile/5% formic acid and dried in a vacuum centrifuge. Dried peptides were dissolved in 0.1% formic acid and analyzed by online nanoflow LC-MS/MS on an Agilent 1200 quaternary HPLC system (Agilent) connected to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) through an in-house built nanoelectrospray ion source. Peptide mixtures were pressure-loaded onto a capillary column (75-μm internal diameter) packed with 15-cm of 3-μm Aqua C18 resins (Phenomenex). Peptides were separated using a 60-min gradient from 5% to 60% acetonitrile in 0.1% formic acid at a flow rate of 300 nL/min (through split). As peptides were eluted from the analytical column, they were electrosprayed (distal 2.5 kV spray voltage) into the mass spectrometer. The MS method consisted of an FT full-scan MS analysis (400–1600 m/z, 60000 resolution) followed by data-dependent MS/MS scans of the 10 most intense precursors at a 35% normalized collision energy with dynamic exclusion for 60 seconds. Application of mass spectrometer scan functions and HPLC solvent gradients was controlled by the Xcalibur data system (Thermo Fisher Scientific). MS/MS spectra were extracted using RawXtract (version 1.9.9) (67) and searched with the ProLuCID algorithm (68) against a human UniProt database concatenated to a decoy database, in which the sequence for each entry in the original database was reversed (69). ProLuCID search results were assembled and filtered using the DTASelect (version 2.0) algorithm (70) with a 10 ppm delta mass cutoff of the peptide masses and a false positive rate below 1%. The LC-MS/MS data have been deposited in the UCSD MassIVE database, https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp, with accession number MSV000083562.
Antibodies and other reagents
The antibodies and some of the chemical reagents used in this study and their working concentrations or dilutions are listed in table S1.
Statistical analysis
Data for all experiments were analyzed with Prism software (GraphPad). Multiple t-tests were used for comparison among experimental groups. P values of less than 0.05 were considered significant.
Supplementary Material
Acknowledgments:
We thank the staff of the La Jolla Institute for Allergy and Immunology Imaging Facility for help with Confocal Microscopy.
Funding: This work was supported in part by NIH grant CA035299 (to A.A.). This is manuscript number 3265 from the La Jolla Institute for Immunology.
Footnotes
SUPPLEMENTARY MATERIALS
Fig. S1. Normal T cell development in PKCθHR2A BM chimeric mice
Fig. S2. Impaired activation of peripheral T cells expressing PKCθHR2A
Fig. S3. Defective early TCR signaling in primary CD4+ T cells from Prkcq−/− B6 mice
Fig. S4. Effect of HR2A-mutated PKCθ on Zap70 recruitment to TCRζ and on formation of the PKCθ-CARMA1 complex
Table S1. Antibodies and other reagents used in this study
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Mass spectrometry data have been deposited in the UCSD MassIVE database, https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp, with accession number MSV000083562. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Materials used in this study are available upon request.
REFERENCES AND NOTES
- 1.Baier G, Telford D, Giampa L, Coggeshall KM, Baier-Bitterlich G, Isakov N, Altman A, Molecular cloning and characterization of PKCθ, a novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J. Biol. Chem 268, 4997–5004 (1993). [PubMed] [Google Scholar]
- 2.Isakov N, Altman A, PKC-θ-mediated signal delivery from the TCR/CD28 surface receptors. Front. Immunol 3, 273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang EY, Kong K-F, Altman A, The yin and yang of protein kinase C-theta (PKCθ): a novel drug target for selective immunosuppression. Adv. Pharmacol 66, 267–312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monks CR, Kupfer H, Tamir I, Barlow A, Kupfer A, Selective modulation of protein kinase C-θ a during T-cell activation. Nature. 385, 83–86 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Yokosuka T, Kobayashi W, Sakata-Sogawa K, Takamatsu M, Hashimoto-Tane A, Dustin ML, Tokunaga M, Saito T, Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C θ translocation. Immunity. 29, 589–601 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marsland BJ, Soos TJ, Späth G, Littman DR, Kopf M, Protein kinase C-θ is critical for the development of in vivo T helper (Th)2 cell but not Th1 cell responses. J. Exp. Med 200, 181–189 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M, Differential regulation of Th2 and Th1 lung inflammatory responses by protein kinase C−θ. J. Immunol 173, 6440–6447 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Anderson K, Fitzgerald M, Dupont M, Wang T, Paz N, Dorsch M, Healy A, Xu Y, Ocain T, Schopf L, Jaffee B, Picarella D, Mice deficient in PKCθ demonstrate impaired in vivo T cell activation and protection from T cell-mediated inflammatory diseases. Autoimmunity. 39, 469–478 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Tan S-L, Zhao J, Bi C, Chen XC, Hepburn DL, Wang J, Sedgwick JD, Chintalacharuvu SR, Na S, Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase Cθ-deficient mice. J. Immunol 176, 2872–2879 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Manicassamy S, Yin D, Zhang Z, Molinero LL, M-L. Alegre, Z. Sun, A critical role for protein kinase Cθ-mediated T cell survival in cardiac allograft rejection. J. Immunol 181, 513–520 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haarberg KMK, Li J, Heinrichs J, Wang D, Liu C, Bronk CC, Kaosaard K, Owyang AM, Holland S, Masuda E, Tso K, Blazar BR, Anasetti C, Beg AA, Yu XZ, Pharmacologic inhibition of PKCθ prevents GVHD while preserving GVL activity in mice. Blood. 122, 2500–2511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valenzuela JO, Iclozan C, Hossain MS, Prlic M, Hopewell E, Bronk CC, Wang J, Celis E, Engelman RW, Blazar BR, Brevan MJ, Waller EK, Xu XZ, Beg AA, PKCθ is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice. J. Clin. Invest 119, 3774–3786 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berg-Brown NN, Gronski MA, Jones RG, Elford AR, Deenick EK, Odermatt B, Littman DR, Ohashi PS, PKCθ signals activation versus tolerance in vivo. J. Exp. Med 199, 743–752 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giannoni F, Lyon AB, Wareing MD, Dias PB, Sarawar SR, Protein kinase Cθ is not essential for T-cell-mediated clearance of murine gammaherpesvirus 68. J. Virol 79, 6808–6813 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marsland BJ, Nembrini C, Schmitz N, Abel B, Krautwald S, Bachmann MF, Kopf M, Innate signals compensate for the absence of PKC-θ during in vivo CD8+ T cell effector and memory responses. Proc. Natl. Acad. Sci. USA 102, 14374–14379 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brezar V, Tu WJ, Seddiki N, PKC-θ in regulatory and effector T-cell functions. Front. Immunol 6, 530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, Blazar BR, Abramson SB, Lafaille JJ, Dustin ML, Protein kinase C-θ mediates negative feedback on regulatory T cell function. Science. 328, 372–376 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baier G, Wagner J, PKC inhibitors: potential in T cell-dependent immune diseases. Curr. Opin. Cell Biol 21, 262–267 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Chand S, Mehta N, Bahia MS, Dixit A, Silakari O, Protein kinase C-θ inhibitors: a novel therapy for inflammatory disorders. Curr. Pharm. Des 18, 4725–4746 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Kwon M-J, Wang R, Ma J, Sun Z, PKC-θ is a drug target for prevention of T cell-mediated autoimmunity and allograft rejection. Endocr. Metab. Immune Disord. Drug Targets 10, 367–372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackay HJ, Twelves CJ, Targeting the protein kinase C family: are we there yet? Nat. Rev. Cancer 7, 554–562 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Mochly-Rosen D, Das K, Grimes KV, Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov 11, 937–957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spitaler M, Cantrell DA, Protein kinase C and beyond. Nat. Immunol 5, 785–790 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Chuang H-C, Lan JL, Chen DY, Yang CY, Chen YM, Li JP, Huang CY, Liu PE, Wang X, Tan TH, The kinase GLK controls autoimmunity and NF-κB signaling by activating the kinase PKC-θ in T cells. Nat. Immunol 12, 1113–1118 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Freeley M, Long A, Regulating the regulator: Phosphorylation of PKCθ in T Cells. Front. Immunol 3, 227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Graham C, Li A, Fisher RJ, Shaw S, Phosphorylation of the protein kinase C-θ activation loop and hydrophobic motif regulates its kinase activity, but only activation loop phosphorylation is critical to in vivo nuclear-factor-κB induction. Biochem. J 361, 255–265 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Witte S, Liu YC, Doyle M, Elly C, Altman A, Regulation of protein kinase Cθ function during T cell activation by Lck-mediated tyrosine phosphorylation. J. Biol. Chem 275, 3603–3609 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Thuille N, Heit I, Fresser F, Krumböck N, Bauer B, Leuthaeusser S, Dammeier S, Graham C, Copeland TD, Shaw S, Baier G, Critical role of novel Thr-219 autophosphorylation for the cellular function of PKCθ in T lymphocytes. EMBO J. 24, 3869–3880 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Chuang H-C, Li J-P, Tan T-H, Regulation of PKC-θ function by phosphorylation in T cell receptor signaling. Front. Immunol 3, 197 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X-D, Gong Y, Chen Z-L, Gong B-N, Xie J-J, Zhong C-Q, Wang Q-L, Diao L-H, Xu A, Han J. Altman A, Li Y, TCR-induced sumoylation of the kinase PKC-θ controls T cell synapse organization and T cell activation. Nat. Immunol 16, 1195–1203 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Kong K-F, Yokosuka T, Canonigo-Balancio AJ, Isakov N, Saito T, Altman A, A motif in the V3 domain of the kinase PKC-θ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol 12, 1105–1112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk MJ, Altman A, Antigen-induced translocation of PKC-θ to membrane rafts is required for T cell activation. Nat. Immunol 2, 556–563 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Cartwright NG, Kashyap AK, Schaefer BC, An active kinase domain is required for retention of PKCθ at the T cell immunological synapse. Mol. Biol. Cell 22, 3491–3497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Liu YC, Meller N, Giampa L, Elly C, Doyle M, Altman A, Protein kinase C activation inhibits tyrosine phosphorylation of Cbl and its recruitment of Src homology 2 domain-containing proteins. J. Immunol 162, 7095–7101 (1999). [PubMed] [Google Scholar]
- 35.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP, The C2 domain of PKCδ Is a phosphotyrosine binding domain. Cell. 121, 271–280 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Stahelin RV, Kong KF, Raha S, Tian W, Melowic HR, Ward KE, Murray D, Altman A, Cho W, Protein kinase Cθ C2 domain is a phosphotyrosine binding module that plays a key role in its activation. J. Biol. Chem 287, 30518–30528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlessinger J, Lemmon MA, SH2 and PTB domains in tyrosine kinase signaling. Sci. STKE 2003, RE12 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Fu G, Hu J, Niederberger-Magnenat N, Rybakin V, Casas J, Yachi PP, Feldstein S, Ma B, Hoerter JA, Ampudia J, Rigaud S, Lambolez F, Gavin AL, Sauer K, Cheroutre H, Gascoigne NR, Protein kinase C η is required for T cell activation and homeostatic proliferation. Sci. Signal 4, ra84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, Leitges M, Baier G, Protein kinase C-θ affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J. Exp. Med 197, 1525–1535 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, Littman DR, PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature. 404, 402–407 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Coudronniere N, Villalba M, Englund N, Altman A, NF-κB activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-θ. Proc. Natl. Acad. Sci. USA 97, 3394–3399 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin X, O’Mahony A, Mu Y, Geleziunas R, Greene WC, Protein kinase C-θ participates in NF-κB activation induced by CD3-CD28 costimulation through selective activation of IκB kinase β. Mol. Cell. Biol 20, 2933–2940 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Au-Yeung BB, Deindl S, Palacios EH, Levin SE, Kuriyan J, Weiss A, The structure, regulation, and function of ZAP-70. Immunol. Rev 228, 41–57 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Deindl S, Kadlecek TA, Brdicka T, Cao X, Weiss A, Kuriyan J, Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell. 129, 735–746 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Klammt C, Novotná L, Li DT, Wolf M, Blount A, Zhang K, Fitchett JR, Lillemeier BF, T cell receptor dwell times control the kinase activity of Zap70. Nat. Immunol 16, 961–969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freeman TS, Weiss A, ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb. Perspect. Biol 2, a002279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katz ZB, Novotná L, Blount A, Lillemeier BF, A cycle of Zap70 kinase activation and release from the TCR amplifies and disperses antigenic stimuli. Nat. Immunol 18, 86–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan Q, Barros T, Visperas PR, Deindl S, Kadlecek TA, Weiss A, Kuriyan J, Structural basis for activation of ZAP-70 by phosphorylation of the SH2-kinase linker. Mol. Cell. Biol 33, 2188–2201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao XC, Littman DR, Weiss A, Itk and Fyn make independent contributions to T cell activation. J. Exp. Med 186, 2069–2073 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu KQ, Bunnell SC, Gurniak CB, Berg LJ, T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J. Exp. Med 187, 1721–1727 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dustin ML, Chan AC, Signaling takes shape in the immune system. Cell. 103, 283–294 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Weiss A, Littman DR, Signal transduction by lymphocyte antigen receptors. Cell. 76, 263–274 (1994). [DOI] [PubMed] [Google Scholar]
- 53.Wang D, Matsumoto R, You Y, Che T, Lin XY, Gaffen SL, Lin X, CD3/CD28 costimulation-induced NF-κB activation is mediated by recruitment of protein kinase C-θ, Bcl10, and IκB kinase β to the immunological synapse through CARMA1. Mol. Cell. Biol 24, 164–171 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan A, Kadlecek TA, Elder ME, Filipovich AH, Kuo WL, Iwashima M, Parslow TG, Weiss A, ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science. 264, 1599–1601 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Elder ME, Lin D, Clever J, Chan AC, Hope TJ, Weiss A, Parslow TG, Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science. 264, 1596–1599 (1994). [DOI] [PubMed] [Google Scholar]
- 56.Herndon TM, Shan XC, Tsokos GC, Wange RL, ZAP-70 and SLP-76 regulate protein kinase C-theta and NF-κB activation in response to engagement of CD3 and CD28. J. Immunol 166, 5654–5664 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Blanchard N, Di Bartolo V, Hivroz C, In the immune synapse, ZAP-70 controls T cell polarization and recruitment of signaling proteins but not formation of the synaptic pattern. Immunity. 17, 389–399 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Au-Yeung BB, Melichar HJ, Ross JO, Cheng DA, Zikherman J, Shokat KM, Robey EA, Weiss A, Quantitative and temporal requirements revealed for Zap70 catalytic activity during T cell development. Nat. Immunol 15, 687–694 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaud G, Lesourne R, Love PE, Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol 18, 485–497 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Courtney AH, Lo W-L, Weiss A, TCR Signaling: Mechanisms of initiation and propagation. Trends Biochem. Sci 43, 108–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thaker YR, Schneider H, Rudd CE, TCR and CD28 activate the transcription factor NF-κB in T-cells via distinct adaptor signaling complexes. Immunol. Lett 163, 113–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altman A, Kaminski S, Busuttil V, Droin N, Hu J, Tadevosyan Y, Hipskind RA, Villalba M, Positive feedback regulation of PLCγ1/Ca2+ signaling by PKCθ in restimulated T cells via a Tec kinase-dependent pathway. Eur. J. Immunol 34, 2001–2011 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Manicassamy S, Sadim M, Ye RD, Sun Z, Differential roles of PKC-θ in the regulation of intracellular calcium concentration in primary T cells. J. Mol. Biol 355, 347–359 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA, Generation of T-cell receptor retrogenic mice. Nat. Protoc 1, 406–417 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Zhang Z, Elly C, Qiu L, Altman A, Liu YC, A direct interaction between the adaptor protein Cbl-b and the kinase zap-70 induces a positive signal in T cells. Curr. Biol 9, 203–206 (1999). [DOI] [PubMed] [Google Scholar]
- 66.Wysocka J, Identifying novel proteins recognizing histone modifications using peptide pull-down assay. Methods. 40, 339–343 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McDonald WH, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, Yates JR 3rd, MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom 18, 2162–2168 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Xu T, Venable JD, Park SK, Cociorva D, Lu Q-B, Liao L, Wohlschlegel J, Hewel J, Yates JR 3rd, Cociorva D, ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program (Mol. Cell. Proteomics, 2006), vol. 5. [Google Scholar]
- 69.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP, Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res 2, 43–50 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Tabb DL, McDonald WH, Yates JR 3rd, DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.