

Abstract
Recent outbreak of COVID-19 caused by SARS-CoV-2 in December 2019 raised global health concerns. Re-purposing the available protease inhibitor drugs for immediate use in treatment in SARS-CoV-2 infections could improve the currently available clinical management. The current study, aims to predict theoretical structure for protease of COVID-19 and to explore further whether this protein can serve as a target for protease inhibitor drugs such as remdesivir, nelfinavir, lopinavir, ritonavir and α-ketoamide. While the 3D structure of protease was predicted using SWISS MODEL server, molecular interaction studies between protein and ligands were performed using AutoDock software. The predicted protease model was reasonably good based on reports generated by different validation servers. The study further revealed that all the protease inhibitor drugs got docked with negative dock energy onto the target protein. Molecular interaction studies showed that protease structure had multiple active site residues for remdesivir, while for remaining ligands the structure had only one active site residue each. From the output of multiple sequence alignment, it is evident that ligand binding sites were conserved. The current in silico study thus, provides structural insights about the protease of COVID-19 and also its molecular interactions with some of the known protease inhibitors.
Electronic supplementary material
The online version of this article (10.1007/s13337-020-00585-z) contains supplementary material, which is available to authorized users.
Keywords: Coronavirus, SARS-CoV-2, COVID-19, Protein homology modelling, Molecular docking
Introduction
Coronaviruses, members of the family Coronaviridae and subfamily Coronavirinae, are enveloped positive-stranded RNA viruses which have spikes of glycoproteins projecting from their viral envelopes, thus exhibit a corona or halo-like appearance [3]. The recent outbreak of novel coronavirus pneumonia referred to as neo-coronary pneumonia caused by severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) in December 2019 raised global health concerns. Neo-coronary pneumonia has been officially named by the World Health Organization as corona virus disease 2019 (COVID-19). SARS-CoV-2 virus has been identified as a zoonotic coronavirus, similar to severe acute respiratory syndrome (SARS) coronavirus and Middle East respiratory syndrome (MERS) coronavirus. Among all known RNA viruses, coronaviruses have the largest genomes ranging from 26 to 32 kb in length [16]. The ~ 306 aa long main protease is a key enzyme for coronavirus replication and is suitable for designing wide-spectrum inhibitors. It is responsible for processing the polypeptide into functional proteins [20]. The protease’s activity is triggered by the binding of molecules to specific points on the protease called active sites. However, the protease’s activity can also be blocked by molecules called inhibitors. When an inhibitor attaches to an active site, it prevents the binding of substrates—inhibiting the action of the protease altogether. Therefore, finding an inhibitor for COVID-19’s protease may be the first step to beating the epidemic. Thus the viral protease is a proven drug discovery target in case of severe acute respiratory syndrome coronavirus, and an attractive target for the design of anti-corona viral drugs.
Among the potential protease inhibitors, the antivirals remdesivir, nelfinavir, lopinavir, and ritonavir along with α-ketoamide are particularly attractive as therapeutics to combat the new coronavirus. Remdesivir is a broad-spectrum antiviral nucleotide pro-drug with potent in vitro antiviral activity against a diverse panel of RNA viruses such as Ebola virus, Marburg, MERS-CoV, SARS-CoV, respiratory syncytial virus, Nipah virus and Hendra virus [18]. Similarly, nelfinavir, lopinavir and ritonavir are protease inhibitors recommended for the treatment of SARS and MERS, which have similar mechanisms of action as HIV [13]. Using Vero cell lines infected with SARS-CoV, antiviral effects of nelfinavir has been studied [9, 25]. A separate investigation performed by Xu et al. [24] indicated that nelfinavir was identified as a potential inhibitor against COVID-19 main protease, based on binding free energy calculations using molecular mechanics with generalized Born and surface area solvation (MM/GBSA) model and solvated interaction energy methods.
Other protease inhibitors, lopinavir and ritonavir are currently available in both first and second-line antiretroviral therapy regimens in pediatrics and adult HIV/AIDS patients, respectively. China’s national health commission has recommended using these agents as an adhoc treatment against COVID-19. Since SARS-CoV-2 infection is an RNA virus similar to HIV, lopinavir/ritonavir is proposed for its management despite the absence of official approval of these drugs for the treatment of COVID-19. At present, lopinavir/ritonavir is used for possible treatment of SARS-CoV-2 infection in countries where the emerging infection prevails [1]. Due to the absence of crystallographic structure of SARS-CoV 3C-like protease, binding modes of these compounds have been proposed through docking studies [4, 14]. Antiviral like α-ketoamides have also been reported in the literature as inhibitors of the coronavirus main protease [11]. Because of their unique specificity and essential role in viral polyprotein processing, these proteases are suitable targets for the development of antiviral drugs [26].
Thus, with no proven antiviral agent available, current research now suggests that selecting drugs with the appropriate viral restraining mechanisms can yield promising results. Various clinical trials are being carried out on nucleotide analogue drugs such as remdesivir [17]. Re-purposing the available protease inhibitor drugs for immediate use in treatment in SARS-CoV-2 infections could improve the currently available clinical management.Thus, the twin objective of the current study is (1) to predict the 3D structure of protease of COVID-19 and (2) to carry out docking studies with protease inhibitors available in DrugBank such as remdesivir, nelfinavir, lopinavir and ritonavir along with ketoamide. This was followed by molecular interaction studies so as to identify any conserved ligand binding sites in the predicted structure.
Materials and methods
3D structure prediction and validation
Protease sequence of COVID-19 (region 1541-1858) was downloaded from GenBank database (accession no: P0C6X7.1) in FASTA format. For building the 3D model of protease of COVID-19, the target sequence information was submitted to SWISS-MODEL server [21] (http://swissmodel.expasy.org). Templates with the highest quality were selected for model building. The output of the predicted model generated as pdb file was downloaded for further analysis and visualized using SPDBV 4.10 [10]. The model was subsequently validated using Verify3D [6], ProSA [23] and PROCHECK [12] servers. The final structure was visualized and analyzed with SPDBV program.
Molecular Docking
Candidate protease inhibitors of COVID-19
Based on literature survey, the structural coordinates of 4 potential protease inhibitors namely: remdesivir (accession no: DB14761), nelfinavir (accession no: DB00220), lopinavir (accession no: DB01601) and ritonavir (accession no: DB00503) were downloaded from the DrugBank database (ref); for ketoamide, the coordinates were separated from the crystal structure of protease of SARS coronavirus in complex with α-ketoamide (PDB ID: 5N5O), available from protein data bank.
Docking studies were attempted to explore the binding mode of the suggested protease inhibitors onto the 3D model of protease of COVID-19 using AUTODOCK tools 1.5.6 [7]. Before docking, polar-H atoms were added to the COVID-19 model followed by Gasteiger charges calculation using AUTODOCK tools available from Scripps Research Institute (http://www.scripps.edu/mb/olson/doc/Autodock). The macromolecule file was then saved in pdbqt format and ready to be used for docking. Ligand centered maps were generated by AutoGrid program with a spacing of 0.375 Å and grid dimensions of 90 × 90 × 90 Å3. Gridbox center was set to coordinate − 0.074, 0.083 and − 0.013 in x y, and z respectively. Polar H charges of the Gasteiger-type were assigned and non-polar-H atoms were merged with the carbons and internal degrees of freedom and torsions were set. Default settings were used for all other parameters. PyMol package [5] was used to visualize the binding interactions between these ligands with 3D model of protease of COVID-19.
Multiple sequence alignment
Multiple sequence alignment was carried out to identify the conserved regions of protease sequence of COVID-19 with orf1ab polyprotein from Wuhan seafood market pneumonia virus (YP_009724389.1) and the best PDB template identified by the SWISS MODEL server using Clustal Omega [19].
Results
3D model of protease of COVID-19 and its validation
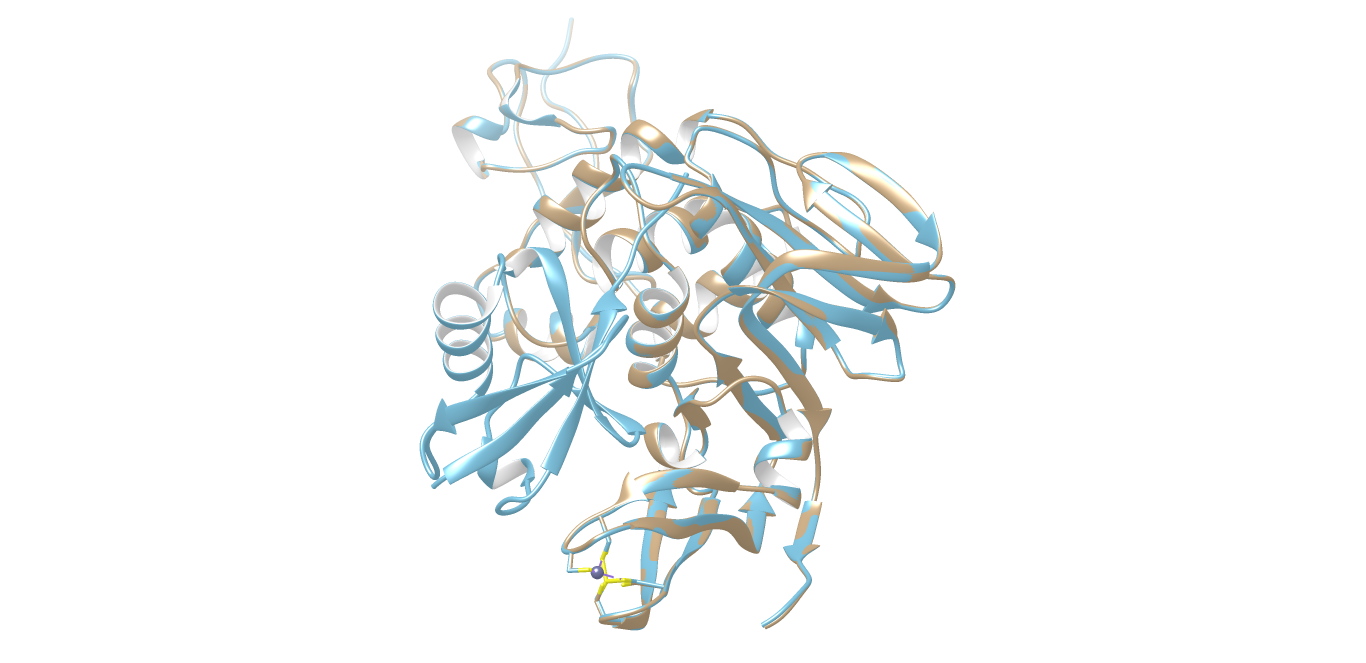
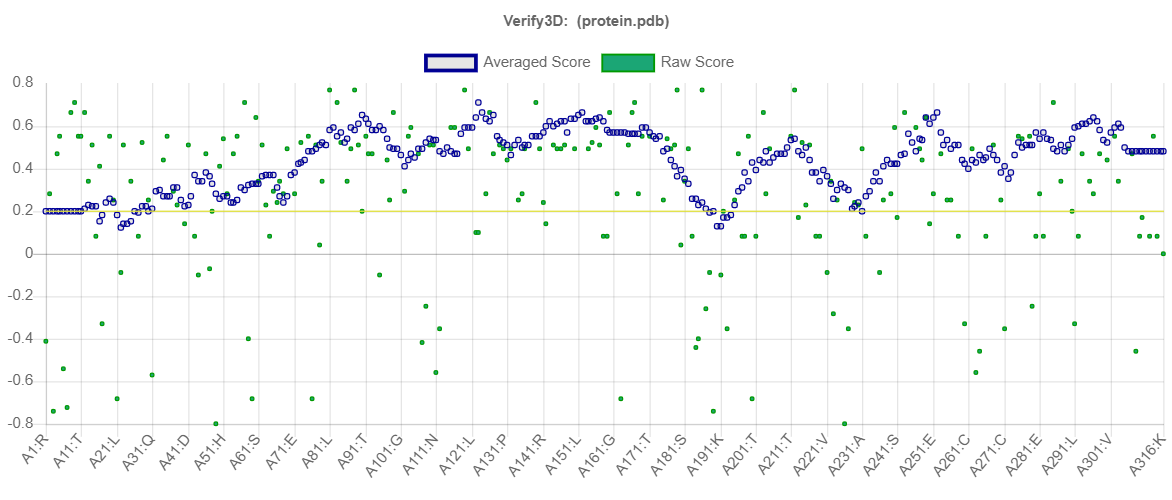
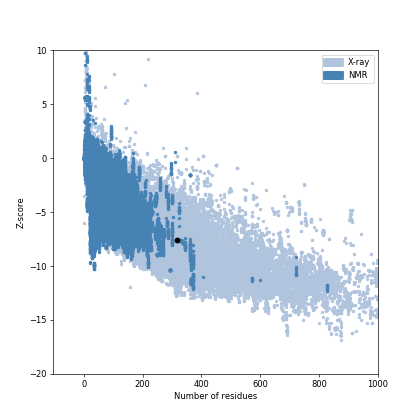
SWISS-MODEL server was successful in generating a 3D structure for protease of COVID-19 using crystal structure of SARS-CoV papain-like protease PLpro in complex with ubiquitin aldehyde (PDB ID: 4MM3_B) as the template. Chimera package was able to superimpose the 3D model of protease of COVID-19 onto the crystallographic structure of 4MM3_B. The root mean square deviation (RMSD) of Cα atoms between protease of COVID-19 and the pdb template 4MM3_B computed was 0.065Å (Supp Fig. 1).The quality factor of the residues of protease model of COVID-19 when evaluated by Verify3D server (Supp Fig. 2) showed 95.57% of the residues had an averaged 3D-1D score ≥ 0.2 which represents a good score, suggesting high compatibility of the atomic model (3D) with its amino acid sequence (1D).Validation of model using Ramachandran plot available with the PROCHECK server revealed that 86.7% residues of protease of COVID-19 model were in the most favoured regions, followed by 12.6% in additional allowed regions, 0.4% in generously allowed region and 0.4% in the disallowed regions. Overall G factor for the predicted structure was − 0.18 (Supp Fig. 3). The G-factor provides a measure of the normalcy of stereo-chemical property of a protein model. Values below − 0.5 shows unusual stereo-chemical property while values below − 1.0 show a highly unusual property. Since G value obtained for the predicted model in the present study is not less than − 0.5, it is suggestive of satisfactory quality. The main chain parameter plot statistics suggested that the overall quality of the predicted model was good. ProSA energy plot revealed negative energy distribution pattern being scored by the amino acid residues for the predicted structure (Supp Fig. 4). The Z score calculated by the ProSA tool for the model was − 7.55, which is within the range of scores typically found for NMR derived structure for the native protein of similar size. Since the structure assessment reports were reasonably good for the predicted structure of protease, it was not subjected to loop refinement.
Docking and molecular interaction studies of COVID-19 with protease inhibitors
All the 5 potential protease inhibitors viz. remdesivir, nelfinavir, lopinavir, ritonavir, and ketoamide got docked onto the predicted 3D model of protease of COVID-19 with a negative dock energy value as shown in Fig. 1. The best recorded binding energy value was obtained for nelfinavir (− 7.54 kcal mol−1) (Fig. 1). Further, molecular interaction studies showed that protease model of COVID-19 had thr75, arg141, gln175and his176” as the potential drug binding sites, with more than one drug binding site (thr75 and his176) identified with remdesivir (Fig. 1).
Fig. 1.

Docking of 5 different potential protease inhibitors of COVID-19 using AUTODOCK software. Among the 5, nelfinavir has got docked with highest biding affinity (panel A).The image has been generated using PyMOL software
Sequence conservation pattern
Multiple sequence alignment of protease of COVID-19 along with orf1ab polyprotein from Wuhan seafood market pneumonia virus (YP_009724389.1) and PDB template 4MM3_B revealed bulk of the residues were highly conserved (Fig. 2), including the ligand binding sites (thr75, arg141, gln175and his176) of protease of COVID-19.
Fig. 2.

Multiple sequence alignment of protease from COVID-19 with YP_009724389.1 and PDB template 4MM3_B chain using CLUSTALΩ. While the conserved residues have been highlighted with “*”, partially conserved residues are marked by “.” symbols. Highlighted regions show the conservation of the ligand binding sites (thr75, arg141, gln175and his176)
Discussion
The viral 3-chymotrypsin-like cysteine protease enzyme, which controls coronavirus replication and is essential for its life cycle, is a proven drug discovery target in the case of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). Recent studies revealed that the genome sequence of SARS-CoV-2 is very similar to that of SARS-CoV [15]. Given the fragile health systems, new and re-emerging disease outbreaks such as the current COVID-19 epidemic can potentially paralyse health systems. According to the Centers for Disease Control and Prevention (CDC), facilities for undergoing research for COVID-19 should strictly implement the appropriate bio-safety practices inside the laboratory, due to the extremely high virus transmissibility. These factors make it extremely difficult for researchers to work with this virus, despite an urgent need to provide a therapy quickly. Computational studies at the preliminary level can save time and resources. The current study, thus, aims to predict theoretical structure for protease of COVID-19 and to explore further whether this homology modelled protein can serve as a target for protease inhibitor drugs such as remdesivir, nelfinavir, lopinavir, ritonavir and ketoamide.
Homology modeling is a useful tool for predicting the 3D structure of proteins. Quality of the 3D structure of protease of COVID-19 generated by SWISS-MODEL server using 4MM3_B as the template was reasonably good based on the validation reports generated Verify3D, PROCHECK, and ProSA servers. Ramachandran plot analysis suggests the predicted 3D model of protease of COVID-19 as a good representation of protein structure with more than 90% of the residues located in the favourable region. The z-score calculated by ProSA tool for the model was also within the range of scores typically found for NMR derived structure for a native protein of similar size.
Docking results suggests the 5 potential protease inhibitors namely nelfinavir, remdesivir, lopinavir, ritonavir and ketoamide got docked onto the predicted 3D model of protease of COVID-19 with a negative dock energy value. However, the best recorded binding energy value was obtained for nelfinavir. Based on the lowest dock energy value scored by nelfinavir in relation to remaining ligands, this appears to be the drug of choice for treating COVID-19 infection. Since all the ligands have been docked with negative dock energy onto the target protein, it will be sensible to give equal importance to all these protease inhibitor ligands. The molecular interaction studies also showed that protease model of COVID-19 had more than 1 active site residue for remdesivir, while for remaining ligands the structure had only one active site residue each. In a recently reported study by Chang et al. [preprint; not peer reviewed], remdesivir has been identified to possess docking sites that strongly overlap with the protein pockets, and could be considered as a potential therapeutic agent. Also, one in vitro and a clinical study indicate that remdesivir, an adenosine analogue that acts as a viral protein inhibitor, has improved the condition in one patient [8, 22]. A combination of the anti-retroviral drugs lopinavir and ritonavir significantly improved the clinical condition of SARS‐CoV patients [2] and might be an option in COVID‐19 infections. From the output of multiple sequence alignment analysis, it is evident that ligand binding sites (thr75, arg141, gln175and his176) were conserved across protease sequences of COVID-19, Wuhan seafood market pneumonia virus and crystal structure of SARS-Coronovirus. Considering the global threat posed by COVID-19, and with no proven antiviral agent available for immediate relief, the current in silico study provide structural insights about the protease of COVID-19 and also its molecular interactions with some of the known protease inhibitors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We acknowledge JAIN-deemed to be University for providing the facilities necessary to carry out our work and for providing the University PhD research scholarship.
Funding
This research received no external funding.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chen ZM, Fu JF, Shu Q, Chen YH, Hua CZ, Li FB, Lin R, Tang LF, Wang TL, Wang W, Wang YS. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. World J Pediatr. 2019;2020:1–7. doi: 10.1007/s12519-020-00345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu CM, Cheng VC, Hung IF, Wong MM, Chan KH, Chan KS, Kao RY, Poon LL, Wong CL, Guan Y, Peiris JS. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59:252–256. doi: 10.1136/thorax.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dayer MR, Taleb-Gassabi S, Dayer MS. Lopinavir; A potent drug against coronavirus infection: insight from molecular docking study. Arch Clin Infect Dis. 2017;12:e13823. doi: 10.5812/archcid.13823. [DOI] [Google Scholar]
- 5.DeLano WL. Pymol: an open-source molecular graphics tool. CCP4 Newsl Prot Crystallogr. 2002;40:82–92. [Google Scholar]
- 6.Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. In: Abelson J, Simon M, Verdine G, Pyle A, editors. Methods in enzymology. New York: Academic Press; 1997. pp. 396–404. [DOI] [PubMed] [Google Scholar]
- 7.Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins Struct Funct Bioinform. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 8.Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H, Spitters C, Ericson K, Wilkerson S, Tural A, Diaz G. First case of 2019 novel coronavirus in the United States. N Engl J Med. 2020;382(10):929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsieh LE, Lin CN, Su BL, Jan TR, Chen CM, Wang CH, Lin DS, Lin CT, Chueh LL. Synergistic antiviral effect of Galanthus nivalis agglutinin and nelfinavir against feline coronavirus. Antiviral Res. 2010;88:25–30. doi: 10.1016/j.antiviral.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson MU, Zoete V, Michielin O, Guex N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012;13:173. doi: 10.1186/1471-2105-13-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Lovell S, Tiew KC, Mandadapu SR, Alliston KR, Battaile KP, Groutas WC, Chang KO. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol. 2012;86:11754–11762. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 13.Li JY, You Z, Wang Q, Zhou ZJ, Qiu Y, Luo R, Ge XY. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 2020;22:80–85. doi: 10.1016/j.micinf.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nukoolkarn V, Lee VS, Malaisree M, Aruksakulwong O, Hannongbua S. Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J Theor Biol. 2008;254:861–867. doi: 10.1016/j.jtbi.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qamar MT, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020 doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;1:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, Leist SR, Pyrc K, Feng JY, Trantcheva I, Bannister R. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9:396. doi: 10.1126/scitranslmed.aal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang F, Chen C, Tan W, Yang K, Yang H. Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Sci Rep. 2016;6:22677. doi: 10.1038/srep22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;4:1–3. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(suppl_2):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z, Peng C, Shi Y, Zhu Z, Mu K, Wang X, Zhu W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv. 2020 doi: 10.1101/2020.01.27.921627v1. [DOI] [Google Scholar]
- 25.Yamamoto N, Yang R, Yoshinaka Y, Amari S, Nakano T, Cinatl J, Rabenau H, Doerr HW, Hunsmann G, Otaka A, Tamamura H. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Lin D, Kusov Y, Nian Y, Ma Q, Wang J, von Brunn A, Leyssen P, Lanko K, Neyts J, de Wilde A. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020 doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.