

Abstract
Traditional therapies have limited efficacy in hepatocellular carcinoma, pancreatic cancer, and biliary tract cancer, especially for advanced and refractory cancers. Through a deeper understanding of antitumor immunity and the tumor microenvironment, novel immunotherapies are becoming available for cancer treatment. Oncolytic virus (OV) therapy is an emerging type of immunotherapy that has demonstrated effective antitumor efficacy in many preclinical studies and clinical studies. Thus, it may represent a potential feasible treatment for hard to treat gastrointestinal (GI) tumors. Here, we summarize the research progress of OV therapy for the treatment of hepato‐bilio‐pancreatic cancers. In general, most OV therapies exhibits potent, specific oncolysis both in cell lines in vitro and the animal models in vivo. Currently, several clinical trials have suggested that OV therapy may also be effective in patients with refractory hepato‐bilio‐pancreatic cancer. Multiple strategies such as introducing immunostimulatory genes, modifying virus capsid and combining various other therapeutic modalities have been shown enhanced specific oncolysis and synergistic anti‐cancer immune stimulation. Combining OV with other antitumor therapies may become a more effective strategy than using virus alone. Nevertheless, more studies are needed to better understand the mechanisms underlying the therapeutic effects of OV, and to design appropriate dosing and combination strategies.
Keywords: biliary tract cancer, hepatocellular carcinoma, immunotherapy, oncolytic virus, pancreatic cancer
In this work, we systematically review and summarize the current status of treatment and the application of oncolytic viruses for HCC, pancreatic cancer, and biliary tract cancer. Most OV therapies exhibits potent, specific oncolysis both in cell lines in vitro and the animal models in vivo and in patients. What's more, we conclude the improvement strategy for next generation of oncolytic viruses therapies.
1. BACKGROUND
Oncolytic virus (OV) selectively replicate in and destroy tumor cells while causing virtually no damage to normal cells. The antitumor effect of oncolytic viruses is primarily achieved in two ways: (a) direct tumor lysis; and (b) induction of an antitumor immune response in animal or humans. On the one hand, because of defective anti‐viral pathways (ie interferon response pathway), tumor cells are unable to stop OV replication and undergo cell lysis and death following infection. The viral progeny are released from the infected tumor cells and spread to uninfected tumor cells, which causes a sustained antineoplastic response.1, 2 On the other hand, the infection of tumor cells express pathogen‐associated molecular pattern molecules (PAMPs) and damage‐associated molecular pattern molecules (DAMPs), which induces the innate immune response through activation of toll‐like receptors. In turn, the activation of the innate immune system facilitates presentation of viral antigens and tumor‐associated antigens (TAAs) to prime the adaptive immune response.3 Of note, several common oncolytic viruses (eg, adenovirus, herpes simplex virus [HSV], vesicular stomatitis virus [VSV], vaccinia virus, and reovirus; Figure 1) have been proven to promote antitumor immunity.4, 5, 6, 7, 8 Furthermore, oncolytic viruses armed with exogenous immune‐stimulating can exert profound antitumor effects via synergistic stimulation of anti‐cancer immunity.4
Figure 1.
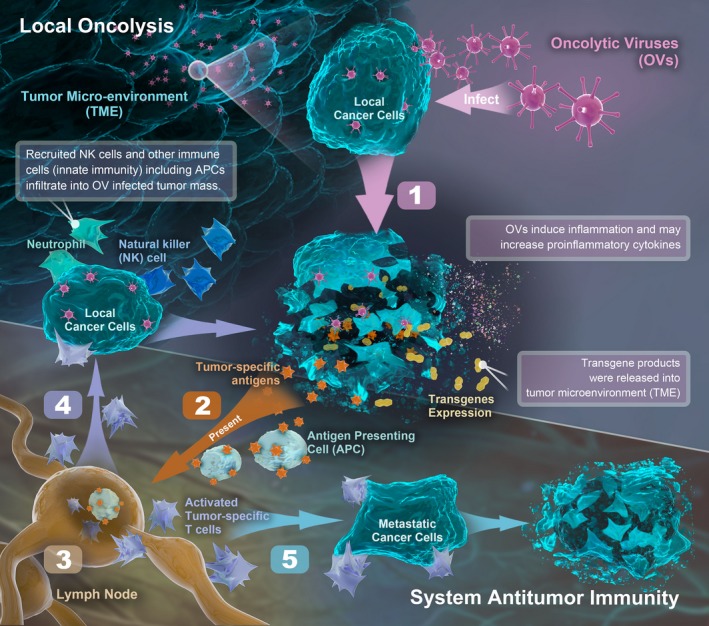
(1) OVs induce immunogenic cell death (ICD). Then oncolysis by OVs causes the release of tumor‐specific antigens (local oncolysis); (2) ~ (3) Tumor‐specific antigens uptake by APCs which migrate to lymph nodes. Antigen‐loaded APCs initiate the activation of tumor‐specific T cells; (4) ~ (5) Tumor‐specific T cells move to local tumor mass (infected) and metastatic cancer cells (uninfected) and exert antitumor effect
The traditional treatment for hepatocellular carcinoma (HCC), pancreatic cancer, and biliary tract cancer primarily include surgical resection, radiotherapy, and chemotherapy; however, the outcomes of these interventions remain unsatisfactory. Moreover although immunotherapeutic strategies including the PD‐1/CTLA4 antibody and chimeric antigen receptor T cells (CAR T cells) have been proven to have good clinical efficacy in certain tumor types,9 the complex immune microenvironment of hepato‐bilio‐pancreatic cancer appears to limit their efficacy.10 Given the broad immune‐stimulating and oncolytic activities of OV, it is tempting to speculate that they may be important components, alone and/or in combination with currently available immunotherapies, of successful treatment strategies for hard to treat gastrointestinal (GI) cancers. Here, we systematically review and summarize the current status of treatment and the application of oncolytic viruses for HCC, pancreatic cancer, and biliary tract cancer (Figure 2).
Figure 2.
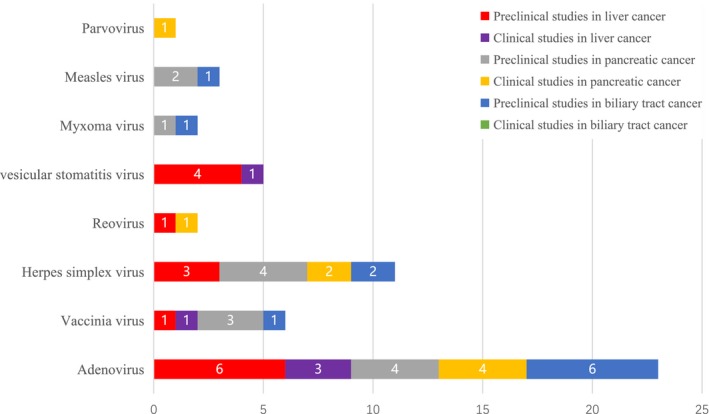
Number of published or registered preclinical and clinical studies for oncolytic virus in hepato‐bilio‐pancreatic cancer Adenovirus is the most widely used. There are few related clinical trials, and most of the existing clinical trials are only in Phase I or Phase II clinical trials
2. ONCOLYTIC VIRUSES AS A NOVEL TREATMENT FOR HCC, PANCREATIC CANCER, AND BILIARY TRACT CANCER
2.1. Adenoviruses
Adenoviruses are a non‐enveloped double‐stranded DNA viruses of approximately 36 kb genome that can be divided into 57 different serotypes (Figure 3). Among these serotypes, Ad2 and Ad5, which belong to subgroup C adenoviruses, are the most commonly used adenoviral vectors.11, 12 Subgroup C adenoviruses infect host cells by binding to the coxsackie adenovirus receptor (CAR) and their transcription requires two key viral genes (E1A and E1B).13, 14 The protein encoded by E1A orchestrates activation of the E2F transcription factor and initiates the cell cycle of the host cell during the quiescent phase.13 The protein encoded by E1B is divided into the E1B‐55 kD protein and E1B‐19 kD protein. The E1B‐55 kD protein binds to p53 and induces its degradation, whereas the E1B‐19 kD protein—similar to the anti‐apoptotic factor Bcl2, prevents the premature apoptosis of infected cells.11, 15 The advantages of using adenoviruses are their high viral titers, ease of engineering‐in of foreign genes into its moderate size (26‐46 Kbp) DNA genome, and the breadth of clinical experience around their use.16 Furthermore, adenoviruses can replicate in host cells at large quantities, and the viral genes cannot integrate into the host cell genome, which improves its safety.11
Figure 3.
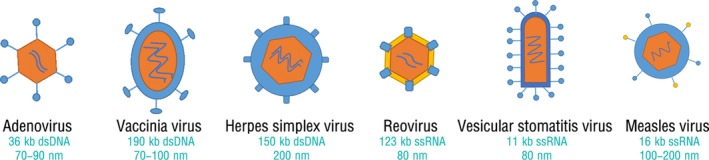
Properties of the oncolytic viruses for hepato‐bilio‐pancreatic cancer and several well‐validated oncolytic viruses are listed. The yellow region represents the capsid and the blue region represents the envelope. Both adenovirus and reovirus are non‐enveloped viruses. The values represent the range of minimum diameter of the capsid. dsDNA, double‐stranded DNA; ssRNA, single‐stranded RNA
2.1.1. Adenoviruses in liver cancer
Oncolytic adenoviruses can be designed to specifically target liver cancer cells using several different methods. The first method is to specifically target the specific signaling pathways in tumors by altering the key adenovirus replication genes.12 This design is primarily achieved by deleting either the E1A or E1B genes. ONYX‐015 is a typical engineered adenovirus (Ad2 and Ad5 hybrid), in which the E1B gene is deleted using gene editing technology to prevent the formation of the E1B‐55 kD protein without affecting the E1B‐19 kD protein, and only replicates in p53‐deficient cells.17 In both phase I and phase II clinical trials, intravenous and intratumoral injections of ONYX‐015 were shown to be safe, well tolerated, and exhibited no dose‐limiting toxicity; despite the ubiquity of anti‐adenoviral antibodies before treatment, ONYX‐015 showed some evidence of clinical benefit.18, 19, 20 Further studies have shown that the deletion of the E1B‐55 kD protein played a selective oncolytic role by mediating late viral mRNA nuclear export rather than p53 degradation.20 The second method is to control the expression of key genes required for virus replication using a specific promoter of liver cancer tissue (eg, GD55, CNHK500, and SG7011let7T).21, 22, 23, 24 ZD55 is an E1B‐deficient Ad5 similar to ONYX‐015.25 The AFP‐regulated oncolytic adenovirus (ZD55, in which the endogenous E1A promoter was replaced by the AFP promoter) and GOLPH2‐regulated oncolytic adenovirus, GD55 (ZD55, in which the endogenous E1A promoter was replaced by the GOLPH2 promoter) were designed based on the high expression of AFP and GOLPH2 in HCC, respectively. These OV demonstrated higher specificity against HCC compared to other tumors, as well as a more pronounced antitumor effect, and GD55 induced less damage to normal liver cells compared to ZD55.23, 26 The third method is to specifically target the liver cancer cell receptor by engineering adenoviral capsids.27
To enhance the antitumor effect of oncolytic adenoviruses, a variety of ZD55‐gene systems have been constructed, which preserve tumor targeting and carry different therapeutic genes (eg, ZD55‐Smac/ZD55‐TRAIL and ZD55‐IFN‐β). Tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) induces tumor cell apoptosis through the death receptor pathway and various caspases, to which HCC is highly resistant,28, 29 via in part inhibition of caspase activation by inhibitor of apoptosis proteins (IAP) and the X‐linked inhibitor of apoptosis protein (XIAP).30 Thus, ZD55‐Smac/ZD55‐TRAIL also incorporates the mitochondrial‐derived activator of caspases (Smac) which abolishes IAP inhibition of caspases and enhances tumor cell sensitivity to the TRAIL pathway.29 The combination of ZD55‐Smac and ZD55‐TRAIL proved to effectively reduce the levels of IAP and XIAP expression and promote HCC cell apoptosis in HCC cell lines in vitro and HCC xenografts in vivo, whereas the effect of either ZD55‐Smac or ZD55‐TRAIL alone was significantly attenuated.29 However, expressing pro‐apoptosis gene by an OV is never a good idea since once the gene is expressed, the cells infected will die first, which eliminates the virus replication in the cell and also inhibits virus dissemination inside of tumor mass. ZD55‐IFN‐β, a ZD55 expressing IFN‐β, showed significant antitumor effects and the high level of IFN‐β expression in the HCC cell lines, as well as HCC xenografts. In addition, no obvious cytotoxic or apoptotic effects were detected in normal cells infected with ZD55‐IFN‐β, which indicate that the toxic effect of IFN‐β did not impact the cells in normal tissues.31
2.1.2. Adenoviruses in pancreatic cancer
ONYX‐015 has been demonstrated to induce tumor‐specific cytolysis and antitumor efficacy in both pancreatic cancer cell lines in vitro and mice xenografts in vivo.32 Moreover the efficacy was significantly enhanced when combined with chemotherapy, including cisplatin and 5‐fluorouracil.32 However, a clinical trial showed limited clinical efficacy of the intratumoral endoscopic ultrasound injection of ONYX‐015 combined with an intravenous injection of gemcitabine for unresectable pancreatic cancer.33 This may be because pancreatic tumors are highly fibrotic with a high ratio of normal cells to tumor cells, which may highly limit virus dissemination in the tumor since, by definition, OVs cannot replicate in nontumor cells. Therefore, it is plausible that oncolytic virus should be designed to enhance viral spread and breakdown the extracellular matrix of pancreatic tumors.34
Several novel oncolytic adenoviruses currently being researched exhibit exciting antitumor activity against pancreatic cancer in preclinical studies. For example, Ad5ΔE1B19K, the adenoviral mutants with an anti‐apoptotic E1B‐19 kD gene deletion, could selectively kill pancreatic cancer cells in vitro and xenografts in vivo when combined with gemcitabine.35 A similar oncolytic adenovirus (AdΔΔ) with the deletions in both the E1ACR2 (another anti‐apoptotic gene) and E1B19K genes displayed enhanced antitumor efficacy combined with the chemotherapeutics, docetaxel and mitoxantrone, in pancreatic cancer xenografts in vivo and decreased levels of virus in normal cells compared to the single‐deleted AdΔCR2 mutant.36 Telomelysin (OBP‐401), in which the hTERT promoter controls viral replication with a fully functional viral E3 region, only replicates in cells with upregulated hTERT (eg, pancreatic cancer cells).37 Telomelysin effectively lysed pancreatic cancer cells and shrank xenografted tumors alone or in combination with docetaxel.38 In CRAd‐Cans, deleting the E1B‐55 kD gene and carrying canstatin (an angiogenesis inhibitor gene), showed the synergistic effects of antitumor therapy and anti‐angiogenesis therapy.39
Most pancreatic cancers demonstrate pRb hyperphosphorylation,40 and two oncolytic adenoviruses, LoAd703 and VCN‐01, have been constructed to replicate in pancreatic cancer cells with a disrupted Rb pathway and several related clinical trials are currently underway. LoAd703 is an Ad5/35 expressing human CD40L and 4‐1BBL. CD40L can induce the adaptive immune response and CD40‐mediated tumor cell apoptosis, whereas 4‐1BBL enhances immunological memory and expands T and NK cells, modulating the pancreatic cancer stroma to support the antitumor response.41 LoAd703 can efficiently lyse pancreatic cancer cell lines in vitro and reduce the established tumors in xenograft models in vivo.41 A Phase I/II clinical trial is currently underway to actively recruit patients to assess the efficacy and safety of an intratumoral injection of LOAd703 combined with gemcitabine and nab‐paclitaxel as a treatment for pancreatic cancer. Another adenovirus (VCN‐01) has been engineered to express hyaluronidase and RGD‐modified fibers, which improves the half‐life of viruses in the blood and enhances intratumoral spread, resulting in antitumor activity in mice and Syrian hamster pancreatic cancer xenografts models after intravenous or intratumoral injection.42 A Phase I dose escalation trial of VCN‐01 by intratumoral injection combined with gemcitabine and abraxane determining the safety and tolerability in patients with advanced pancreatic cancer has been completed; however, the results have yet to be reported (NCT02045589).
2.1.3. Adenoviruses in biliary tract cancer
Previous reports have revealed that an adenovirus expressing p27KIP1 induces apoptosis against cholangiocarcinoma cells by triggering extracellular Fas ligand expression.43 To further enhance the tumor specificity and viral infectivity of the adenovirus‐based therapies in cholangiocarcinoma cells, Zhu et al designed several novel recombinant adenoviruses with a survivin promoter and capsid modifications. The survivin promoter has extremely low activity in normal human tissues, especially in the liver, but exhibited higher activity in cholangiocarcinoma cells, indicating decreased toxicity to the human liver and higher tumor specificity. Moreover, three different capsid modifications (RGD, F5/3, and pk7) enhanced the levels of viral infectivity; however, the replication ratios of these adenoviruses are significantly lower than the wild type virus, suggesting that the capsid modification enhanced viral infectivity and impaired viral replication.44
AxdAdB‐3, a double‐restricted Ad with a mutant E1A and E1B‐55kD deletion, showed effective and selective replication and oncolysis of gallbladder cancer (GBC) cells in vitro and in vivo with reduced negative effects on normal cells.45 Moreover AxdAdB‐3‐F/RGD, a novel AxdAdB‐3 with RGD‐modified fibers via the incorporation of an Arg‐Gly‐Asp (RGD) motif into the HI‐loop of Ad5 fiber‐knob region, can enhance infectivity by increasing viral interaction with the integrins that are abundantly expressed in almost all biliary cancer cells.46, 47 AxdAdB3‐F/RGD exhibited efficient replication and potent oncolysis in both CAR‐positive and CAR‐negative biliary cancer cells and caused a marked inhibition of tumor growth in mouse xenografts, whereas AxdAdB3 only infected biliary cancer cells with preserved CAR expression.46 In another approach, the use of a chimeric construct with the shaft from an Ad5 serotype and the knob from Ad3 serotype enhanced transduction in several CAR‐negative cancer cells.47, 48
2.2. Vaccinia virus
Vaccinia virus is a double‐stranded DNA virus that replicates and spreads rapidly (Figure 3). Importantly, vaccinia virus harbors the thymidine kinase (TK) gene, which encodes the TK, an enzyme necessary for viral replication that is ubiquitously expressed in malignant cancer cells but rarely expressed in normal cells.49 Thus, by removing the TK gene, vaccinia virus can only target malignant cancer cells, since the defective vaccinia virus can only replicate using the TK of host cells.50 Vaccinia virus can secrete viral proteins to activate host cell EGFR‐RAS pathway to further synthesize TK, which improves the efficiency of virus infection and promotes the destruction of tumor cells.51 The disabled interferon response of malignant tumor cells is also one of the factors driving vaccinia virus to target malignant tumor cells.52 Moreover vaccinia virus exhibits effectiveness and stability when given systemically and is resistant to the effects of antibodies and complement.53 The intravenous injection of vaccinia virus has been shown to be effective in tumor xenografts, despite the presence of high antibody titers.54
2.2.1. Vaccinia virus in liver cancer
The vaccinia virus currently used in clinical liver cancer research primarily involves JX‐594. JX‐594 is a derivative of the vaccinia virus Wyeth strain (a smallpox vaccine), which was widely used in global vaccination programs to eradicate small pox due to its proven safety in humans.55 The use of a vaccinia virus with a single knockout of the TK gene (CVV) effectively removed liver cancer cells in animal models.56 JX‐594 contains two genes that were inserted into the TK gene region: (a) a gene encoding hGM‐CSF, which promotes myeloid and dendritic cell maturation to induce antitumor immunity and inhibit tumor blood vessels, but may stimulate myeloid‐derived suppressor cells (MDSCs) resulting in diminished innate and adaptive antitumor responses in numerous cancers57, 58, 59; and (b) lac‐Z, which encodes β‐galactosidase as a surrogate marker to assess viral replication.55 Current clinical trials have demonstrated that JX‐594 is safe and well‐tolerated when used to treat liver cancer patients and that intriguingly there is no correlation between serum antibody levels and JX‐594 replication, safety, and antitumor activity.60, 61, 62 Kim et al confirmed that JX‐594 was well‐tolerated in rabbits and rat liver cancer models with a significant antitumor effect.63 Park et al found that in patients with refractory primary liver cancer and metastatic liver cancer, JX‐594 could be detected in both injected and uninjected tumors, and exhibited antitumor effects.53 Grade I‐III flu‐like symptoms were common adverse reactions following an intratumoral injection of JX‐594, and several patients displayed transient grade I‐III dose‐related thrombocytopenia while Grade III hyperbilirubinemia was dose‐limiting. In addition, the maximum tolerated dose was 1 × 109 pfu.53 Subsequent studies have not found dose‐limiting toxicities for JX‐594, and suggested the maximum feasible dose is 2 × 109 pfu.60 Moreover, Heo et al demonstrated systemic and long‐term efficacy of a direct intratumoral injection of JX‐594 in patients with advanced HCC and the dose was significantly associated with overall survival; the overall survival for the high dose (1 × 109 pfu) treatment was approximately twice that of the low dose group (1 × 108 pfu) (14.1 m vs 6.7 m) with comparable safety.61 A phase II clinical trial of advanced liver cancer further confirmed that the dose of a direct intratumoral injection of JX‐594 was significantly associated with the overall survival.64 In addition, Cripe et al evaluated the safety of an intratumoral injection of JX‐594 in children with liver cancer, and found that an intratumoral injection of JX‐594 was safe and the side effects were primarily flu‐like symptoms and skin pustules.62 Since patients with liver cancer are often associated with cirrhosis and viral hepatitis, traditional treatments do not induce an antiviral response; however, JX‐594 induces high concentrations of antiviral cytokines that inhibit HBV, which may be effective for treating HBV‐related HCC.65 Compared with traditional sorafenib treatment, JX‐594 is also associated with a longer overall survival and fewer adverse reactions; however, additional clinical trials are needed to further verify the safety of JX‐594 and explore the possibilities of combining JX‐594 with other therapies. A phase III clinical trial (PHOCUS) investigating the combination of JX‐594 and sorafenib for the treatment of advanced liver cancer is currently underway (NCT02562755).
2.2.2. Vaccinia virus in pancreatic cancer
GLV‐1h68 (GL‐ONC1) is a replication‐competent virus containing the marker genes ruc‐gfp and mutations in the F14.5L, J2R, and A56R loci, which disrupts TK and hemagglutinin. GLV‐1h68 has demonstrated safety and antitumor efficacy in PANC‐1 cell lines in vitro and PANC‐1 pancreatic tumor xenografts via systemic injection in vivo.66 Moreover GLV‐1h68 combined with cisplatin or gemcitabine resulted in enhanced therapeutic efficacy.66 To enhance the role of the virus as a cancer treatment, multiple therapeutic genes have been engineered for expression in vaccinia virus. The Lister vaccinia virus vaccine strain was designed to encode the endostatin‐angiostatin fusion gene (VVhEA), and showed significant potent antitumor activity against human pancreatic tumor cells in vitro, a high selectivity for cancer cells, and inhibition of angiogenesis and tumor regression of human pancreatic tumor xenograft tumors in mice following both intravenous and intratumoral injection.67 It is important to note that VVhEA was effective against pancreatic tumors insensitive to oncolytic adenoviruses.67 Another Lister vaccine strain (VV‐IL‐10) that encoded interleukin (IL)‐10 and lacked TK has demonstrated superior antitumor efficacy in mice with subcutaneous pancreatic cancer that was also associated with long‐term antitumor immunity.68 Although, no significant difference was observed in in vitro antitumor activity when VV‐IL‐10 was compared with the control VV, the presence of IL‐10 which prevents host immune inhibition of viral replication, resulted in greater therapeutic efficacy in vivo of VV‐IL‐10.68, 69
2.2.3. Vaccinia virus in biliary tract cancer
GLV‐1h68 has also demonstrated oncolytic activity against cholangiocarcinoma cell lines in vitro, and was well tolerated and exhibited antitumor efficacy in xenograft bearing athymic nude mice.70 A phase I clinical trial is currently evaluating the safety and efficacy of GLV‐1h68, which is delivered as a bolus intravenous injection, in patients with solid organ cancers, including cholangiocarcinoma (NCT02714374).
2.3. Herpes simplex virus
Herpes simplex virus (HSV) is a double‐stranded DNA virus that can be divided into HSV‐1 and HSV‐2 according to the specific serotype (Figure 3). Currently, HSV‐1 is primarily used in clinical oncolytic therapy, whereas there are some studies which show that HSV‐2 is also effective against a variety of tumors; interestingly, a tumor cell subpopulation that only responds to treatment with HSV‐2 has been found.71, 72 Compared with other viruses, HSV can infect and quickly kill tumor cells, supporting rapid viral replication and spread of the virus within a tumor mass. Moreover the safety of HSV can be ensured through the use of anti‐HSV drugs (eg, acyclovir).71
2.3.1. HSV in liver cancer
HSV‐1 is primarily used for the treatment of liver cancer. The genome of HSV‐1 is approximately 150 kb, including many non‐essential genes that have no significant effect on viral replication and can be modified without losing the oncolytic effect of the virus.73 In the HSV‐1 OV, LCSOV, expression of the viral glycoprotein H gene is driven by the liver‐specific apolipoprotein E (apoE)‐AAT promoter and contains additional miRNA complementary sequences of miR‐122a, miR‐124a, and let‐7 inserted into the same 3′ UTR region of the modified gH gene.74 miR‐122a is a liver‐specific miRNA that is expressed at low levels only in HCC, whereas miR‐124a and let‐7 are down‐regulated or lacking in malignant cancer cells enabling translational control of gH expression in LCSOV infected normal cells.74, 75 LCSOV also displays high selectivity and effective killing in HCC xenografts and cell lines, and significantly reduces tumor volume with minimal toxicity.74 G47Δ is a third‐generation oncolytic HSV with mutations in the γ34.5, ICP6, and ICP47 genes. The γ34.5 gene production can prevent the host cell from stopping translation due to viral infection, allowing viral proteins to be continuously synthesized. Since tumor cells often lack control over translation and the antiviral response, HSV with the γ34.5 gene mutation can replicate in tumor cells.73, 76 The ICP6 gene product is required for the replication of viruses in noncycling cells, so mutations in the ICP6 gene enable HSV to selectively replicate in constantly dividing cells (eg, tumor cells).77 Moreover, an ICP47 gene mutation can amplify the targeting of tumor cells caused by a γ34.5 gene mutation and stimulate T cells to enhance the immune response and antitumor immunity to virus‐infected tumor cells.73, 78 G47Δ requires only a very low MOI to effectively kill a variety of different HCC cell lines and inhibit tumor growth in HCC xenografts with no significant toxicity.78 HSV‐1‐T‐01 is similar to G47Δ, in which the γ34.5 and ICP6 genes were deleted and the ICP6 gene was replaced with the LacZ gene. HSV‐1‐T‐01 was found to inhibit tumor growth in both HCC cell lines and xenografts, and significantly improved the antitumor effect by enhancing T cell‐mediated immunity.79
2.3.2. HSV in pancreatic cancer
Several preclinical studies have demonstrated the antitumor activity of both HSV‐1 and HSV‐2 for pancreatic cancer. G207 is a typical engineered HSV‐1 with deletions in both copies of γ34.5 and genetic inactivation of ICP6, which replicated in and lysed human pancreatic cancer cells in vitro.80 Another engineered HSV‐1, NV1020, had a deletion in only one copy of γ34.5 also displayed effective viral replication and cell lysis in human pancreatic cancer cells in vitro, as well as a higher production of viral progeny.81 Both the injection of G207 and NV1020 into athymic mice xenografts induced complete pancreatic tumor eradication in 25% and 40% of mice, respectively.81 FusOn‐H2 is an HSV‐2 construct with a deletion in the PK domain which encodes serine/threonine protein kinase activity and replicates in cells with an activated Ras signaling pathway. FusOn‐H2 could eradicate subcutaneous tumors and orthotopic tumors in human pancreatic cancer xenografts following an intratumor and systemic injection, respectively.82
A Phase I clinical trial evaluated the efficacy of HF10, a naturally mutated HSV‐1, in six patients with unresectable pancreatic cancer. No adverse effects were observed and HF10 infection stimulated the host antitumor immune responses and increased the number of NK cells, CD4+ cells, CD8+ cells, and macrophages after virus injection; four of the six patients showed clinical efficacy with longer survival.83 Additionally, talimogene laherparepvec (T‐VEC), a modified HSV‐1 with deletions in γ34.5 and ICP47, and also expressing GM‐CSF, has been approved by the FDA as the first oncolytic virus therapy for the treatment of melanoma. Unfortunately, a Phase I trial assessing the safety of T‐VEC in 17 patients with unresectable pancreatic cancer showed limited efficacy and various adverse events and only two patients completed the trial (NCT00402025).
2.3.3. HSV in biliary tract cancer
The study of oncolytic therapy for cholangiocarcinoma remains in the preclinical stage. NV1203 is an attenuated HSV with a UL56 deletion, a single copy of ICP0, ICP4, and γ34.5, as well as the insertion of the Escherichia coli lacZ marker gene into the ICP47 locus. UL56 is associated with the pathogenicity and neuroinvasiveness of HSV, and the lack of UL56 attenuates the virus.84 In addition, ICP0 is associated with both lytic and latent HSV infections, and ICP0‐null HSV‐1 is extremely sensitive to IFN and PML‐mediated disruption of the viral lifecycle; however, since IFN and PML are downregulated in most tumors, this virus can specifically target cancer cells.73, 85, 86 Combination treatment with low‐dose external radiation therapy (XRT) and NV1203 was highly tumoricidal, both in vitro and in vivo,87 though the greatest tumor volume decrease was observed in the YoMi cell line, which correlated with upregulation of growth arrest and DNA damage protein 34 (GADD34) by XRT.87 GADD34 has significant homology to γ34.5 and improves the specificity of the virus for targeting tumor cells.87
G207 is a HSV‐1 mutant with deletions in both the γ34.5 and ICP6 genes, and its safety has been demonstrated in humans.88 Nearly total cell killing was observed in five gallbladder carcinoma cell lines within 72 hours of in vitro G207 infection.89 Moreover an intratumor injection of G207 (1 × 107 pfu) in immunocompetent hamsters bearing established subcutaneous KIGB‐5 tumors displayed a significant inhibition of tumor growth and prolonged survival.89 Interestingly, a decreased antitumor effect was observed in athymic mice bearing KIGB‐5 tumors, suggesting that the systemic antitumor effect of G207 is partly mediated by T cells.89
2.4. Reovirus
Reovirus is a double‐stranded RNA virus that is ubiquitous in nature, found in untreated sewage, stagnant water, and rivers worldwide (Figure 3). In normal cells, a reovirus infection activates the PKR pathway of the host cell. PKR is a serine/threonine protein kinase involved in antiviral host defense, functioning to inhibit reovirus translation, replication, and further infection.90 In cells in which the Ras signaling pathway is activated, the PKR pathway is inhibited, resulting in viral replication and eventually destroying the host cells. Ras gene mutations have been found in various human tumors, including pancreatic, colon, and lung cancer, suggesting the potential of using reoviruses to treat tumors.91, 92 Ras transformation mediates reovirus oncolysis for cells exhibiting activated Ras signaling by enhancing viral uncoating, particle infectivity, and apoptosis‐dependent release of progeny virions.93 Reovirus also promotes direct oncolytic effects and the antitumor immune response by altering the tumor microenvironment due to the substantial secretion of inflammatory cytokines and chemokines by NK cells, DCs, and cytotoxic T cells.94 Moreover an intravenous reovirus injection is safe, displays no dose‐limiting toxicity, and has potent antitumor effects.95 Currently, although the reovirus type 3 Dearing strain is most commonly used in clinical studies, there are only a few studies using reovirus to treat liver and biliary tract cancer.
2.4.1. Reovirus in liver cancer
Most liver cancers are associated with oncogenic viral infections (eg, HBV [54%] and HCV [31%]).96 Park et al found that the oncogenic protein, HBX, produced by HBV inhibited the oncolysis of reovirus in HCC; thus, reovirus had a limited therapeutic effect on HBV‐HCC.96 In contrast, Samson et al demonstrated that reovirus could effectively inhibit HCV replication in HCV‐HCC mouse models and cell lines, demonstrating significant anti‐HCC responses through inducing innate immune activation and IFN production.97
2.4.2. Reovirus in pancreatic cancer
Approximately 90% of pancreatic cancers have K‐ras gene mutations; thus, reoviruses are oncolytic toward cancer cells displaying activated Ras signaling.92 Reovirus (serotype 3) has shown significant antitumor effects in four human pancreatic cancer cell lines with K‐ras mutations and BxPC3 with a normal Ras proto‐oncogene, and was also an effective therapy in mice with Panc1 and BxPC3 xenografts mice in vivo.98 An intraportal administration of reovirus decreased the number and size of liver metastases from pancreatic cancer without any toxicity to normal hepatic tissue in hamsters.99 Furthermore, an intraperitoneal administration of reovirus effectively controlled the peritoneal dissemination of pancreatic cancer in hamsters.100
Pelareorep (REOLYSIN®) is a proprietary isolate of the human reovirus type 3 Dearing strain and demonstrates a potential anticancer effect towards several cancers when used as a mono and/or combination therapy.101 A phase II study of pelareorep combined with gemcitabine therapy was evaluated in 34 patients with advanced pancreatic adenocarcinoma. The combined therapy was well tolerated without serious adverse events and the median progression‐free survival (PFS) was 3.4 months (median overall survival [OS]: 10.2 months) with a 1‐ and 2‐year survival rate of 45% and 24%, respectively, which were the highest observed rates in the similar studies, which demonstrated that pelareorep can complement gemcitabine treatment in pancreatic cancer.102 However, another phase II study of pelareorep combined with paclitaxel/carboplatin therapy compared with paclitaxel/carboplatin therapy alone was evaluated in 73 patients with metastatic pancreatic adenocarcinoma; no significant differences were found in the response rate, PFS, or OS between the two therapies due to insurmountable immune suppression and elevated expression of CTLA4 on T cells may be the key factor limiting the oncolytic efficacy in patients with pancreatic cancer.103
2.5. Vesicular stomatitis virus
Vesicular stomatitis virus (VSV) is a negative‐stranded RNA virus that specifically targets tumor cells due to the reduced ability of tumor cells to resist VSV infection (Figure 3). An engineered VSV that expresses β‐gal (rVSV‐β‐gal) demonstrated effective viral transduction, tumor‐selective viral replication, and extensive oncolytic effects in HCC cells, and prolonged survival of Buffalo rats bearing orthotopic HCC without significant cytotoxicity or liver damage. Further experiments showed that while the survival time of rats treated with rVSV‐β‐gal was significantly prolonged, viral spread between solid tumor cells was limited.104 To overcome this problem, recombinant virus VSV‐NDV was constructed to induce the formation of syncytia between tumor cells to promote efficient viral spread. The VSV membrane surface glycoprotein is replaced by Newcastle disease virus (NDV) hemagglutinin‐neuraminidase (HN) protein and modified fusion (F) membrane protein, which retain the ability for rapid replication while increasing the safety and the ability of virions to spread between cells and enhance the oncolytic effect.105 VSV‐NDV‐induced extensive syncytia formation and enhanced tumor cytotoxicity were observed in both in vivo and in vitro HCC models without inducing significant peripheral hepatic parenchymal damage.106 VSV (MΔ51)‐M3 is a recombinant VSV with amino acid 51 deleted from the VSV‐M protein and also expresses M3, a chemokine‐binding protein from murine gammaherpesvirus‐68. The deletion of amino acid 51 from the VSV‐M protein results in the loss of the ability of the virus to inhibit mRNA transport in host cells; therefore, IFN and cytokine expression is increased in infected cells, which increases the safety of rVSV but induces a stronger inflammatory response and reduces the oncolytic effect of rVSV.107 The addition of M3 antagonizes chemokine signaling and reduces immune infiltration allowing survival of the virus and oncolysis to continue. In an HCC‐bearing mouse model of hepatic artery perfusion, treatment with rVSV (MΔ51)‐M3 decreased infiltration of neutrophils and NK cells in the lesion, while the viral titer increased, the oncolytic effect was enhanced, and more importantly, no obvious systemic and organ toxicities were observed.107
2.6. Myxoma virus
Myxoma virus (MYXV) is a member of the poxvirus family that has a double‐stranded DNA genome and is pathogenic to rabbits, but not humans.108 In addition, a MYXV infection may be prevented through protective interferon responses induced in species other that rabbits, which results in a narrow host tropism.109 However, MYXV can replicate in cells with an activated Akt pathway, as well as p53 or Rb dysfunction.110, 111 Akt is a serine/threonine kinase that regulates cellular proliferation and death but is upregulated in several human cancer cells.112 Therefore, MYXV can be used to selectively target many cancer cells and has been shown to be effective at infecting and killing 70% of tested tumor cell lines.113 Moreover MYXV can productively infect, replicate in, and lyse human pancreatic cancer cells in vitro and prolong survival in mouse xenografts in vivo. 114 In addition, the combined use of MYXV and gemcitabine displays a robust antitumor effect.115
The effective tumor cell killing ability of MYXV has been shown in a variety of human gallbladder carcinoma cell lines.116 Both rapamycin and hyaluronan can effectively enhance the oncolytic ability of MYXV in vitro, but only hyaluronan can enhance the antitumor effects of MYXV in vivo and prolong the survival of GBC tumor‐bearing mice via the interaction between HA and CD44 which results in increased Akt signaling.117 There are no related clinical trials in human subjects are currently ongoing.
2.7. Measles virus
Measles virus (MV) is a negative‐stranded RNA paramyxovirus (Figure 3) and the vaccine strains of the virus widely used for measles prevention worldwide have demonstrated excellent safety.118 MV enters cells through the CD46 receptor, a membrane‐associated protein that protects cells against complement‐mediated lysis that is overexpressed in tumors but exhibits low levels of expression in normal cells; thus, MV preferentially infects tumor cells.119, 120 The virus kills tumor cells via cell‐to‐cell fusion and the formation of mononuclear cell aggregates.121 MV strains have demonstrated potent antitumor activity in multiple tumor models, including both solid tumors and hematologic malignancies.119
A MV expressing the sodium iodide symporter reporter gene (MV‐NIS) was found to efficiently infect human pancreatic tumor xenografts in athymic nude mice and facilitated diagnostic imaging of infection.122
MeV‐SCD is a measles vaccine virus that has been engineered to express super cytosine deaminase (SCD), a fusion protein consisting of yeast cytosine deaminase and uracil phosphoribosyl transferase.123 Since 5‐FU is commonly used for the treatment of carcinomas with low effectiveness in bile duct cancer, SCD can convert the prodrug 5‐fluorocytosine (5‐FC) to 5‐fluorouracil (5‐FU) and subsequently to 5‐fluorouridine‐monophosphate, which inhibits both DNA and protein synthesis.120, 123, 124 MeV‐SCD combined with the administration of 5‐FC displays significant oncolytic ability against cholangiocarcinoma in vitro. In vivo, the intratumoral administration of MeV‐SCD significantly reduced the tumor size and was associated with a significant survival benefit.123
2.8. Application status of OV therapy in hepato‐bilio‐pancreatic cancer
Tables 1, 2, 3 show the application situation of oncolytic virus in HCC, pancreatic cancer, and biliary tract cancer. Different OV have their unique advantages and disadvantages. For example, As the most widely used virus for research, adenovirus has broad tropism for infecting many human tissues and is conducive to clinical applications.12 However, the small genome of adenovirus only allows insertion of small portions of genetic material (not exceeding 8 kb), limiting its ability to deliver multiple antitumor or immune‐stimulating payloads. In contrast, HSV‐1 has a large genome with many genes not necessary for virus replication, thus allowing researchers to manipulate the genome to enhance the oncolytic activity without destroying the ability of virus replication.73 Importantly, HSV‐1 is highly immunogenic, directly stimulating NK cells and synergizing with IL‐15 to promote antitumor immunity.125, 126 However, HSV‐1 spreads from cell to cell, suggesting that intratumoral injection may be the best meansfor delivery while intravenous administration may be not suitable due to multiple physical (ie general “stickiness” to endothelium and blood components) and immunological barriers (inactivation by neutrophils, monocytes, neutralizing antibodies, etc).127 Vaccinia virus also has a large genome to accommodate multiple foreign genes and has high transduction efficiency.128 But different from HSV which is a neurovirulent human pathogen, the safety of vaccinia virus has been widely demonstrated through its widespread use as a vaccine to eradicate small pox. Most genetically engineered oncolytic viruses have an attenuated viral backbone improving the safety of the virus. However, the oncolysis is correspondingly weakened, so improvements are based on constructing multi‐regulated viral backbones to further increase tumor selectivity, and/or equipping the virus with co‐stimulatory factors to potentiate antitumor immunity. Different from the genetically engineered oncolytic viruses, Reolysin which has oncolytic activity and insufficient space to insert foreign genes, may necessitate combination strategies for maximal therapeutic efficacy. Almost all the combination of Reolysin were with chemotherapy, though the efficacy was not satisfactory.103, 129 Therefore, current efforts are aimed at combining Reolysin with immunotherapy.102 A common strategy is to arm virus with exogenous gene expressing cytokines such as GM‐CSF and IL‐12 to enhance antitumor immunity. It is worth noting that although GM‐CSF has been widely used as a payload in OV, it may lead to suppression of immune responses by activating MDSCs.57, 58, 59 Published preclinical studies have confirmed the effective oncolysis of oncolytic viruses in hepato‐bilio‐pancreatic cancer models both in vitro and in vivo. However, the viruses which are significantly effective in preclinical studies may not be effective in the clinical setting because of many factors within the tumor microenvironment (TME) of hard to treat GI tumors including (a) the biophysical barriers of desmoplasia, high interstitial pressures and hypoxia, and (b) tumor‐derived immunosuppressive factors such as cytokines, chemokines, and suppressor cells.128, 129
Table 1.
Application situation of oncolytic virus in HCC
| Viral type | Name | Mode of administration | Key features | Study types | Ref./Clinical trail |
|---|---|---|---|---|---|
| Adenovirus | ONYX‐015 | Intratumoral or intravenous | Disruption of the coding sequence of the E1B‐55kD protein | Phase II | [18] |
| CNHK500 | Intratumoral or intravenous | The expression of E1A gene is regulated by hTERT promoter and the expression of E1B gene is regulated by hypoxia promoter | Preclinical | [22] | |
| GD55 | Intratumoral | E1B‐55kD protein‐deficient and the endogenous E1A promoter was replaced by the GOLPH2 promoter | Preclinical | [23] | |
| AD | Intratumoral | E1B‐55kD protein‐deficient and the endogenous E1A promoter was replaced by the AFP promoter | Preclinical | [26] | |
| ZD55‐Smac/ZD55‐TRAIL | Intratumoral | E1B‐55kD protein‐deficient and arm with Smac and TRAIL genes | Preclinical | [29] | |
| ZD55‐IFN‐β | Intratumoral | E1B‐55kD protein‐deficient and arm with IFN‐β gene | Preclinical | [31] | |
| Vaccinia virus | CVV | Intratumoral | Deletion of TK gene | Preclinical | [56] |
| JX‐594 | Intratumoral or intravenous | Deletion of TK gene and insert genes encoding hGM‐CSF and β‐galactosidase | Phase II/III | [60, 63, 64] | |
| HSV‐1 | LCSOV | Intratumoral |
Additional miRNA complementary sequences of miR‐122a, miR‐124a and let‐7 inserting into the same 3′ UTR region of the modified gH gene And viral glycoprotein H gene is linked to liver‐specific apolipoprotein E (apoE)‐AAT promoter |
Preclinical | [74] |
| G47Δ | Intratumoral | The mutations of γ34.5, ICP6 and ICP47 gene | Preclinical | [78] | |
| HSV‐1‐T‐01 | Intratumoral | The ICP47 and γ34.5 loci are deleted and the LacZ gene replaces the ICP6 gene | Preclinical | [79] | |
| VSV | rVSV‐GFP | Intratumoral | Arm with the gene expressing GFP | Preclinical | [130] |
| rVSV‐β‐gal | Hepatic artery perfusion | Arm with the gene expressing β‐galactosidase | Preclinical | [104] | |
| VSV‐NDV | Hepatic artery perfusion | The membrane surface glycoprotein of VSV is replaced by Newcastle disease virus (NDV) hemagglutinin‐neuraminidase (HN) protein and modified fusion (F) membrane protein | Preclinical | [106] | |
| rVSV(MΔ51)‐M3 | Hepatic artery perfusion | Deletes amino acid 51 of the VSV‐M protein and expresses M3 | Preclinical | [107] |
Table 2.
Application situation of oncolytic virus in pancreatic cancer
| Viral type | Name | Mode of administration | Combination therapy | Key features | Study types | Ref./Clinical trail |
|---|---|---|---|---|---|---|
| Adenovirus | ONYX‐015 | Intratumoral | Gemcitabine | Disruption of the coding sequence of the E1B‐55kD protein | Phase II | [33] |
| Ad5ΔE1B19K | Intratumoral | Gemcitabine | The expression of E1A gene is regulated by hTERT promoter and the expression of E1B gene is regulated by hypoxia promoter | Preclinical | [35] | |
| AdΔΔ | Intratumoral | Docetaxel + Mitoxantrone | E1B‐55kD protein‐deficient and the endogenous E1A promoter was replaced by the GOLPH2 promoter | Preclinical | [36] | |
| OBP‐401 | Intratumoral | Docetaxel | E1B‐55kD protein‐deficient and the endogenous E1A promoter was replaced by the AFP promoter | Preclinical | [37] | |
| CRAd‐Cans | Intratumoral | — | E1B‐55kD protein‐deficient and arm with canstatin gene | Preclinical | [39] | |
| LoAd703 | Intratumoral | Gemcitabine + Nab‐paclitaxel | E1B‐55kD protein‐deficient and arm with IFN‐β gene | Phase II | NCT02705196 | |
| VCN‐01 | Intratumoral | Gemcitabine + Abraxane | Express hyaluronidase and RGD‐modified fibers | Phase I | NCT02045589/NCT02045602 | |
| Vaccinia virus | GLV‐1h68 | Intravenous | Cisplatin or Gemcitabine | The LIVP strain with mutations in F14.5L, J2R, and A56R loci | Preclinical | [66] |
| VVhEA | Intratumoral or intravenous | — | The Lister vaccine strain armed with the endostatin‐angiostatin fusion gene | Preclinical | [67] | |
| VV‐IL‐10 | Intratumoral | — | The Lister vaccine strain armed with interleukin‐10 and lacking TK | Preclinical | [68, 69] | |
| HSV‐1 | G207 | Intratumoral | — | Deletions in both copies of γ34.5 and genetic inactivation of ICP6 | Preclinical | [80] |
| NV1020 | Intratumoral | — | Deletion in only one copy of γ34.5 | Preclinical | [81] | |
| HF10 | Intratumoral | — |
Natural mutation which UL43, 49.5, 55, 56 and latency‐associated transcript are functionally inactivated |
Phase I | [83] | |
| T‐VEC | Intratumoral | — | Deletions in γ34.5 and ICP47 as well as expression of GM‐CSF | Phase I | NCT00402025 | |
| HSV‐2 | FusOn‐H2 | Intratumoral and intravenous | — | Deletion in PK domain | Preclinical | [82] |
| L1BR1 | Intratumoral | 5‐FU + Cisplatin | Anti‐apoptotic gene US3 locus‐deficient | Preclinical | [131] | |
| Reovirus | Pelareorep | Intravenous | Gemcitabine or Paclitaxel + Carboplatin | Unmodified proprietary isolate of reovirus Type 3 Dearing | Phase II | [102, 103] |
| Myxoma virus | MYXV | Intratumoral | Gemcitabine | Unmodified | Preclinical | [115] |
| Measles virus | MV‐NIS | Intratumoral | — | Arm with the sodium iodide symporter reporter gene | Preclinical | [122] |
| MV‐PNP‐anti‐PSCA | Intratumoral | Gemcitabine | Express the prostate stem cell antigen (PSCA) and the prodrug convertase purine nucleoside phosphorylase (PNP) | Preclinical | [132] | |
| Parvovirus | H‐1PV | Intravenous | — | Unmodified | Phase I/II | NCT02653313 |
Table 3.
Application situation of oncolytic virus in biliary tract cancer
| Viral type | Name | Mode of administration | Combination therapy | Key features | Cancer types | Study types | Ref./Clinical trail |
|---|---|---|---|---|---|---|---|
| Adenovirus | AxE1CAUT | Intratumoral | 5‐FU and/or Ganciclovir | Deletion of the E1A, E1B, and E3 regions and cDNAs of UPRT and HSV‐tk with the CAG promoter are inserted | Cholangiocarcinoma | Preclinical | [133] |
| AxdAdB‐3 | Intratumoral | Gene‐directed enzyme prodrug therapy | A mutant E1A and E1B‐55kD deletion | Gallbladder carcinoma | Preclinical | [45] | |
| AxdAdB‐3‐F/RGD | Intratumoral | — | A mutant E1A and E1B‐55kD deletion and the incorporation of an Arg‐Gly‐Asp motif into the HI‐loop of Ad5 fiberknob region | Gallbladder carcinoma | Preclinical | [46] | |
| AdSurp‐P53 | Intratumoral | — | Survivin promoter‐regulated as well as high expression of p53 | Gallbladder carcinoma | Preclinical | [134] | |
| SG7605‐p53‐11R | Intratumoral | — | Arm with the p53 gene and cell‐penetrating peptide 11R | Gallbladder carcinoma | Preclinical | [135] | |
| Vaccinia virus | GLV‐1h68 | Intratumoral | — | The LIVP strain with mutations in F14.5L, J2R, and A56R loci | Cholangiocarcinoma | Preclinical | [70] |
| HSV‐1 | NV1203 | Intratumoral | XRT | UL56 deletion as well as a single copy of ICP0, ICP4, γ34.5, and the Escherichia coli lacZ marker gene is inserted into the ICP47 locus | Cholangiocarcinoma | Preclinical | [87] |
| G207 | Intratumoral | — | Deletions in both copies of γ34.5 and genetic inactivation of ICP6 | Gallbladder carcinoma | Preclinical | [89] | |
| Myxoma virus | MYXV | Intratumoral | Rapamycin + Hyaluronan | Unmodified | Gallbladder carcinoma | Preclinical | [116, 117] |
| Measles virus | MeV‐SCD | Intratumoral | 5‐FC | Express super cytosine deaminase | Cholangiocarcinoma | Preclinical | [123] |
3. CONCLUSION
The current understanding of oncolytic viruses only represents the tip of the iceberg in this field. Due to the limitations of traditional treatments, oncolytic virus therapy currently represents a more promising antitumor therapy, especially for advanced refractory cancers. However, due to the above‐mentioned limitations, we consider that next generation OV therapies will need to maximize immune stimulation, antagonize immunosuppressive cells and soluble factors, and incorporate strategies to eliminate or minimize the biophysical barriers of the TME. Depending on the OV platform, these goals may be accomplished in combination with other therapeutics or by engineering the appropriate genes into the vector.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Yuwei Li and Yinan Shen contributed equally to this work. All authors contributed to the study conception and design. All authors read and approved the final manuscript.
Li Y, Shen Y, Zhao R, et al. Oncolytic virotherapy in hepato‐bilio‐pancreatic cancer: The key to breaking the log jam? Cancer Med. 2020;9:2943–2959. 10.1002/cam4.2949
Funding information
This work was financially supported by the National Key Research and Development Program of China (grant number 2019YFC1316000), the National High Technology Research and Development Program of China (grant number 2015AA020405), the National Natural Science Foundation of China (grant number 81672337), the Key Program of the National Natural Science Foundation of China (grant number 81530079), the Key Program of the National Natural Science Foundation of China (grant number 81830089), the Key Research and Development Project of Zhejiang Province (grant number 2015C03044), the Zhejiang Provincial Program for the Cultivation of High‐level Innovative Health Talents, and China Scholarship Council (grant number 201706320169), Project of Medical and Health Technology Platform of Zhejiang Province (No. 2019C03019).
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Thomson BJ. Viruses and apoptosis. Int J Exp Pathol. 2001;82(2):65‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rahal A, Musher B. Oncolytic viral therapy for pancreatic cancer. J Surg Oncol. 2017;116(1):94‐103. [DOI] [PubMed] [Google Scholar]
- 4. Prestwich RJ, Ilett EJ, Errington F, et al. Immune‐mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15(13):4374‐4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li H, Dutuor A, Tao L, Fu XP, Zhang XL. Virotherapy with a type 2 herpes simplex virus‐derived oncolytic virus induces potent antitumor immunity against neuroblastoma. Clin Cancer Res. 2007;13(1):316‐322. [DOI] [PubMed] [Google Scholar]
- 6. Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti‐tumor immunity. Hum Gene Ther. 1999;10(3):385‐393. [DOI] [PubMed] [Google Scholar]
- 7. Diaz RM, Galivo F, Kottke T, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67(6):2840‐2848. [DOI] [PubMed] [Google Scholar]
- 8. Greiner S, Humrich JY, Thuman P, Sauter B, Schuler G, Jenne L. The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti‐tumoral immunity. Clin Exp Immunol. 2006;146(2):344‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li SZ, Yang F, Ren XB. Immunotherapy for hepatocellular carcinoma. Drug Discov Ther. 2015;9(5):363‐371. [DOI] [PubMed] [Google Scholar]
- 10. Solinas C, Pusole G, Demurtas L, et al. Tumor infiltrating lymphocytes in gastrointestinal tumors: controversies and future clinical implications. Crit Rev Oncol Hematol. 2017;110:106‐116. [DOI] [PubMed] [Google Scholar]
- 11. Chu RL, Post DE, Khuri FR, Van Meir EG. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin Cancer Res. 2004;10(16):5299‐5312. [DOI] [PubMed] [Google Scholar]
- 12. Wang YG, Huang PP, Zhang R, Ma BY, Zhou XM, Sun YF. Targeting adeno‐associated virus and adenoviral gene therapy for hepatocellular carcinoma. World J Gastroentero. 2016;22(1):326‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gallimore PH, Turnell AS. Adenovirus E1A: remodelling the host cell, a life or death experience. Oncogene. 2001;20(54):7824‐7835. [DOI] [PubMed] [Google Scholar]
- 14. Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275(5304):1320‐1323. [DOI] [PubMed] [Google Scholar]
- 15. White E, Sabbatini P, Debbas M, Wold WS, Kusher DI, Gooding LR. The 19‐kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol Cell Biol. 1992;12(6):2570‐2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou W, Luo C, Zhang Z, et al. A novel oncolytic adenovirus targeting to telomerase activity in tumor cells with potent. Oncogene. 2004;23(2):457‐464. [DOI] [PubMed] [Google Scholar]
- 17. Bischoff JR, Kim DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53‐deficient human tumor cells. Science. 1996;274(5286):373‐376. [DOI] [PubMed] [Google Scholar]
- 18. Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: lessons from clinical trials. Cancer Gene Ther. 2002;9(12):979‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ganly I, Kirn D, Eckhardt G, et al. A phase I study of Onyx‐015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res. 2000;6(3):798‐806. [PubMed] [Google Scholar]
- 20. O'Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX‐015 tumor selectivity. Cancer Cell. 2004;6(6):611‐623. [DOI] [PubMed] [Google Scholar]
- 21. Jin HJ, Lv SQ, Yang JH, et al. Use of microRNA let‐7 to control the replication specificity of oncolytic adenovirus in hepatocellular carcinoma cells. PLoS ONE. 2011;6(7):e21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Q, Chen GH, Peng LH, et al. Increased safety with preserved antitumoral efficacy on hepatocellular carcinoma with dual‐regulated oncolytic adenovirus. Clin Cancer Res. 2006;12(21):6523‐6531. [DOI] [PubMed] [Google Scholar]
- 23. Wang YG, Liu T, Huang PP, et al. A novel Golgi protein (GOLPH2)‐regulated oncolytic adenovirus exhibits potent antitumor efficacy in hepatocellular carcinoma. Oncotarget. 2015;6(15):13564‐13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He GQ, Lei W, Wang SB, et al. Overexpression of tumor suppressor TSLC1 by a survivin‐regulated oncolytic adenovirus significantly inhibits hepatocellular carcinoma growth. J Cancer Res Clin. 2012;138(4):657‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang ZL, Zou WG, Luo CX, et al. An armed oncolytic adenovirus system, ZD55‐gene, demonstrating potent antitumoral efficacy. Cell Res. 2003;13(6):481‐489. [DOI] [PubMed] [Google Scholar]
- 26. Zhang KJ, Qian J, Wang SB, Yang Y. Targeting gene‐viro‐therapy with AFP driving Apoptin gene shows potent antitumor effect in hepatocarcinoma. J Biomed Sci. 2012;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mathis JM, Stoff‐Khalili MA, Curiel DT. Oncolytic adenoviruses ‐ selective retargeting to tumor cells. Oncogene. 2005;24(52):7775‐7791. [DOI] [PubMed] [Google Scholar]
- 28. Pathil A, Armeanu S, Venturelli S, et al. HDAC inhibitor treatment of hepatoma cells induces both TRAIL‐independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43(3):425‐434. [DOI] [PubMed] [Google Scholar]
- 29. Pei ZF, Chu L, Zou WG, et al. An oncolytic adenoviral vector of smac increases antitumor activity of TRAIL against HCC in human cells and in mice. Hepatology. 2004;39(5):1371‐1381. [DOI] [PubMed] [Google Scholar]
- 30. Suzuki Y, Nakabayashi Y, Nakata K, Reed JC, Takahashi R. X‐linked inhibitor of apoptosis protein (XIAP) inhibits caspase‐3 and‐7 in distinct modes. J Biol Chem. 2001;276(29):27058‐27063. [DOI] [PubMed] [Google Scholar]
- 31. He LF, Gu JF, Tang WH, et al. Significant antitumor activity of oncolytic adenovirus expressing human interferon‐beta for hepatocellular carcinoma. J Gene Med. 2008;10(9):983‐992. [DOI] [PubMed] [Google Scholar]
- 32. Heise C, SampsonJohannes A, Williams A, McCormick F, VonHoff DD, Kirn DH. ONYX‐015, an E1B gene‐attenuated adenovirus, causes tumor‐specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3(6):639‐645. [DOI] [PubMed] [Google Scholar]
- 33. Hecht JR, Bedford R, Abbruzzese JL, et al. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX‐015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin Cancer Res. 2003;9(2):555‐561. [PubMed] [Google Scholar]
- 34. Mulvihlll S, Warren R, Venook A, et al. Safety and feasibility of injection with an E1B–55 kDa gene‐deleted, replication‐selective adenovirus (ONYX‐015) into primary carcinomas of the pancreas: a phase I trial. Gene Ther. 2001;8(4):308‐315. [DOI] [PubMed] [Google Scholar]
- 35. Leitner S, Sweeney K, Oberg D, et al. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine‐induced apoptosis in pancreatic carcinoma cells and anti‐tumor efficacy in vivo. Clin Cancer Res. 2009;15(5):1730‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oberg D, Yanover E, Adam V, et al. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin Cancer Res. 2010;16(2):541‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nemunaitis J, Tong AW, Nemunaitis M, et al. A phase I study of telomerase‐specific replication competent oncolytic adenovirus (Telomelysin) for various solid tumors. Mol Ther. 2010;18(2):429‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ikeda Y, Fujiwara T, Kagawa S, et al. Enhanced antitumor efficacy of telomerase‐selective oncolytic adenoviral agent OBP‐401 with docetaxel: preclinical evaluation of chemovirotherapy. Mol Ther. 2006;13:S116‐S. [DOI] [PubMed] [Google Scholar]
- 39. He XP, Su CQ, Wang XH, et al. E1B–55kD‐deleted oncolytic adenovirus armed with canstatin gene yields an enhanced anti‐tumor efficacy on pancreatic cancer. Cancer Lett. 2009;285(1):89‐98. [DOI] [PubMed] [Google Scholar]
- 40. Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor‐suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126‐3130. [PubMed] [Google Scholar]
- 41. Eriksson E, Milenova I, Wenthe J, et al. Shaping the tumor stroma and sparking immune activation by CD40 and 4–1BB signaling induced by an armed oncolytic virus. Clin Cancer Res. 2017;23(19):5846‐5857. [DOI] [PubMed] [Google Scholar]
- 42. Rodriguez‐Garcia A, Gimenez‐Alejandre M, Rojas JJ, et al. Safety and efficacy of VCN‐01, an oncolytic adenovirus combining fiber HSG‐binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. 2015;21(6):1406‐1418. [DOI] [PubMed] [Google Scholar]
- 43. Yamamoto K, Katayose Y, Suzuki M, et al. Adenovirus expressing p27KIP1 induces apoptosis against cholangiocarcinoma cells by triggering Fas ligand on the cell surface. Hepatogastroenterology. 2003;50(54):1847‐1853. [PubMed] [Google Scholar]
- 44. Zhu ZB, Chen Y, Makhija SK, et al. Survivin promoter‐based conditionally replicative adenoviruses target cholangiocarcinoma. Int J Oncol. 2006;29(5):1319‐1329. [PubMed] [Google Scholar]
- 45. Fukuda K, Abei M, Ugai H, et al. E1A, E1B double‐restricted adenovirus for oncolytic gene therapy of gallbladder cancer. Cancer Res. 2003;63(15):4434‐4440. [PubMed] [Google Scholar]
- 46. Wakayama M, Abei M, Kawashima R, et al. E1A, E1B double‐restricted adenovirus with RGD‐fiber modification exhibits enhanced oncolysis for CAR‐deficient biliary cancers. Clin Cancer Res. 2007;13(10):3043‐3050. [DOI] [PubMed] [Google Scholar]
- 47. Tekant Y, Davydova J, Ramirez PJ, Curiel DT, Vickers SM, Yamamoto M. Oncolytic adenoviral therapy in gallbladder carcinoma. Surgery. 2005;137(5):527‐535. [DOI] [PubMed] [Google Scholar]
- 48. Kawakami Y, Li H, Lam JT, Krasnykh V, Curiel DT, Blackwell JL. Substitution of the adenovirus serotype 5 knob with a serotype 3 knob enhances multiple steps in virus replication. Cancer Res. 2003;63(6):1262‐1269. [PubMed] [Google Scholar]
- 49. Hengstschlager M, Knofler M, Mullner EW, Ogris E, Wintersberger E, Wawra E. Different regulation of thymidine kinase during the cell cycle of normal versus DNA tumor virus‐transformed cells. J Biol Chem. 1994;269(19):13836‐13842. [PubMed] [Google Scholar]
- 50. Yang HL, Kim SK, Kim M, et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest. 2005;115(2):379‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirus JX‐594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20(4):749‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kirn DH, Wang YH, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon‐beta to produce a specific, multi‐mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4(12):2001‐2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Park BH, Hwang T, Liu TC, et al. Use of a targeted oncolytic poxvirus, JX‐594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533‐542. [DOI] [PubMed] [Google Scholar]
- 54. Thorne SH, Liang W, Sampath P, et al. Targeting localized immune suppression within the tumor through repeat cycles of immune cell‐oncolytic virus combination therapy. Mol Ther. 2010;18(9):1698‐1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vilarinho S, Taddei TH. New frontier in liver cancer treatment: oncolytic viral therapy. Hepatology. 2014;59(1):343‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoo SY, Jeong SN, Kang DH, Heo J. Evolutionary cancer‐favoring engineered vaccinia virus for metastatic hepatocellular carcinoma. Oncotarget. 2017;8(42):71489‐71499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor‐cells engineered to secrete murine granulocyte‐macrophage colony‐stimulating factor stimulates potent, specific, and long‐lasting antitumor immunity. P Natl Acad Sci USA. 1993;90(8):3539‐3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Breitbach CJ, Paterson JM, Lemay CG, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15(9):1686‐1693. [DOI] [PubMed] [Google Scholar]
- 59. Tamadaho RSE, Hoerauf A, Layland LE. Immunomodulatory effects of myeloid‐derived suppressor cells in diseases: role in cancer and infections. Immunobiology. 2018;223(4–5):432‐442. [DOI] [PubMed] [Google Scholar]
- 60. Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi‐mechanistic cancer‐targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99‐U102. [DOI] [PubMed] [Google Scholar]
- 61. Heo J, Reid T, Ruo L, et al. Randomized dose‐finding clinical trial of oncolytic immunotherapeutic vaccinia JX‐594 in liver cancer. Nat Med. 2013;19(3):329‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cripe TP, Ngo MC, Geller JI, et al. Phase 1 study of intratumoral Pexa‐Vec (JX‐594), an oncolytic and immunotherapeutic vaccinia virus in pediatric cancer patients. Mol Ther. 2015;23(3):602‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX‐594, a targeted poxvirus expressing GM‐CSF. Mol Ther. 2006;14(3):361‐370. [DOI] [PubMed] [Google Scholar]
- 64. Breitbach CJ, Moon A, Burke J, Hwang TH, Kirn DH. A phase 2, open‐label, randomized study of Pexa‐Vec (JX‐594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol Biol. 2015;1317:343‐357. [DOI] [PubMed] [Google Scholar]
- 65. Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX‐594 demonstrates antitumoral, antivascular, and anti‐HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16(9):1637‐1642. [DOI] [PubMed] [Google Scholar]
- 66. Yu YA, Galanis C, Woo Y, et al. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV‐1h68. Mol Cancer Ther. 2009;8(1):141‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tysome JR, Briat A, Alusi G, et al. Lister strain of vaccinia virus armed with endostatin‐angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 2009;16(10):1223‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chard LS, Maniati E, Wang PJ, et al. A vaccinia virus armed with interleukin‐10 Is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res. 2015;21(2):405‐416. [DOI] [PubMed] [Google Scholar]
- 69. Couper KN, Blount DG, Riley EM. IL‐10: the master regulator of immunity to infection. J Immunol. 2008;180(9):5771‐5777. [DOI] [PubMed] [Google Scholar]
- 70. Pugalenthi A, Mojica K, Ady JW, et al. Recombinant vaccinia virus GLV‐1h68 is a promising oncolytic vector in the treatment of cholangiocarcinoma. Cancer Gene Ther. 2015;22(12):591‐596. [DOI] [PubMed] [Google Scholar]
- 71. Fu X, Tao L, Wang PY, Cripe TP, Zhang X. Comparison of infectivity and spread between HSV‐1 and HSV‐2 based oncolytic viruses on tumor cells with different receptor expression profiles. Oncotarget. 2018;9(30):21348‐21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fu XP, Tao LH, Cai R, Prigge J, Zhang XL. A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus. Mol Ther. 2006;13(5):882‐890. [DOI] [PubMed] [Google Scholar]
- 73. Peters C, Rabkin SD. Designing herpes viruses as oncolytics. Mol Ther‐Oncolytics. 2015;2:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fu XP, Rivera A, Tao LH, De Geest B, Zhang XL. Construction of an oncolytic herpes simplex virus that precisely targets hepatocellular carcinoma cells. Mol Ther. 2012;20(2):339‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let‐7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17(1):F19‐36. [DOI] [PubMed] [Google Scholar]
- 76. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double‐stranded RNA‐activated protein kinase. Proc Natl Acad Sci USA. 1997;94(3):843‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Goldstein DJ, Weller SK. Factor(S) present in herpes‐simplex virus type‐1‐infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase ‐ characterization of an Icp6 deletion mutant. Virology. 1988;166(1):41‐51. [DOI] [PubMed] [Google Scholar]
- 78. Wang JN, Xu LH, Zeng WG, et al. Treatment of human hepatocellular carcinoma by the oncolytic herpes simplex virus G47delta. Cancer Cell Int. 2014;14:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nakatake R, Kaibori M, Nakamura Y, et al. Third‐generation oncolytic herpes simplex virus inhibits the growth of liver tumors in mice. Cancer Sci. 2018;109(3):600‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee JH, Federoff HJ, Schoeniger LO. G207, modified herpes simplex virus type 1, kills human pancreatic cancer cells in vitro. J Gastrointest Surg. 1999;3(2):127‐133. [DOI] [PubMed] [Google Scholar]
- 81. McAuliffe PF, Jarnagin WR, Johnson P, Delman KA, Federoff H, Fong YM. Effective treatment of pancreatic tumors with two multimutated herpes simplex oncolytic viruses. J Gastrointest Surg. 2000;4(6):580‐587. [DOI] [PubMed] [Google Scholar]
- 82. Fu XP, Tao LH, Li M, Fisher WE, Zhang XL. Effective treatment of pancreatic cancer xenografts with a conditionally replicating virus derived from type 2 herpes simplex virus. Clin Cancer Res. 2006;12(10):3152‐3157. [DOI] [PubMed] [Google Scholar]
- 83. Nakao A, Kasuya H, Sahin TT, et al. A phase I dose‐escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non‐resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011;18(3):167‐175. [DOI] [PubMed] [Google Scholar]
- 84. Kehm R, Rosen‐Wolff A, Darai G. Restitution of the UL56 gene expression of HSV‐1 HFEM led to restoration of virulent phenotype; deletion of the amino acids 217 to 234 of the UL56 protein abrogates the virulent phenotype. Virus Res. 1996;40(1):17‐31. [DOI] [PubMed] [Google Scholar]
- 85. Sobol PT, Hummel JL, Rodrigues RM, Mossman KL. PML has a predictive role in tumor cell permissiveness to interferon‐sensitive oncolytic viruses. Gene Ther. 2009;16(9):1077‐1087. [DOI] [PubMed] [Google Scholar]
- 86. Boutell C, Everett RD. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol. 2013;94:465‐481. [DOI] [PubMed] [Google Scholar]
- 87. Jarnagin WR, Zager JS, Hezel M, et al. Treatment of cholangiocarcinoma with oncolytic herpes simplex virus combined with external beam radiation therapy. Cancer Gene Ther. 2006;13(3):326‐334. [DOI] [PubMed] [Google Scholar]
- 88. Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867‐874. [DOI] [PubMed] [Google Scholar]
- 89. Nakano K, Todo T, Chijiiwa K, Tanaka M. Therapeutic efficacy of G207, a conditionally replicating herpes simplex virus type 1 mutant, for gallbladder carcinoma in immunocompetent hamsters. Mol Ther. 2001;3(4):431‐437. [DOI] [PubMed] [Google Scholar]
- 90. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PWK. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. Embo J. 1998;17(12):3351‐3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Coffey MC, Strong JE, Forsyth PA, Lee PWK. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282(5392):1332‐1334. [DOI] [PubMed] [Google Scholar]
- 92. Bos JL. Ras oncogenes in human cancer ‐ a Review. Cancer Res. 1989;49(17):4682‐4689. [PubMed] [Google Scholar]
- 93. Marcato P, Shmulevitz M, Pan D, Stoltz D, Lee PWK. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis‐dependent release. Mol Ther. 2007;15(8):1522‐1530. [DOI] [PubMed] [Google Scholar]
- 94. Steele L, Errington F, Prestwich R, et al. Pro‐inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF‐kappa B mediated and supports innate and adaptive anti‐tumour immune priming. Mol Cancer. 2011;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lolkema MP, Arkenau HT, Harrington K, et al. A phase I study of the combination of intravenous reovirus type 3 dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011;17(3):581‐588. [DOI] [PubMed] [Google Scholar]
- 96. Park EH, Koh SS, Srisuttee R, et al. Expression of HBX, an oncoprotein of hepatitis B virus, blocks reoviral oncolysis of hepatocellular carcinoma cells. Cancer Gene Ther. 2009;16(5):453‐461. [DOI] [PubMed] [Google Scholar]
- 97. Samson A, Bentham MJ, Scott K, et al. Oncolytic reovirus as a combined antiviral and anti‐tumour agent for the treatment of liver cancer. Gut. 2018;67(3):562‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Etoh T, Himeno Y, Matsumoto T, et al. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin Cancer Res. 2003;9(3):1218‐1223. [PubMed] [Google Scholar]
- 99. Himeno Y, Etoh T, Matsumoto T, Ohta M, Nishizono A, Kitano S. Efficacy of oncolytic reovirus against liver metastasis from pancreatic cancer in immunocompetent models. Int J Oncol. 2005;27(4):901‐906. [PubMed] [Google Scholar]
- 100. Hirano S, Etoh T, Okunaga R, et al. Reovirus inhibits the peritoneal dissemination of pancreatic cancer cells in an immunocompetent animal model. Oncol Rep. 2009;21(6):1381‐1384. [DOI] [PubMed] [Google Scholar]
- 101. Chakrabarty R, Tran H, Selvaggi G, Hagerman A, Thompson B, Coffey M. The oncolytic virus, pelareorep, as a novel anticancer agent: a review. Invest New Drug. 2015;33(3):761‐774. [DOI] [PubMed] [Google Scholar]
- 102. Mahalingam D, Goel S, Aparo S, et al. A phase II study of pelareorep (REOLYSIN((R))) in combination with gemcitabine for patients with advanced pancreatic adenocarcinoma. Cancers (Basel). 2018;10(6):pii:E160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Noonan AM, Farren MR, Geyer SM, et al. Randomized phase 2 trial of the oncolytic virus pelareorep (Reolysin) in upfront treatment of metastatic Pancreatic adenocarcinoma. Mol Ther. 2016;24(6):1150‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shinozaki K, Ebert O, Kournioti C, Tai YS, Woo SL. Oncolysis of multifocal hepatocellular carcinoma in the rat liver by hepatic artery infusion of vesicular stomatitis virus. Mol Ther. 2004;9(3):368‐376. [DOI] [PubMed] [Google Scholar]
- 105. Abdullahi S, Jakel M, Behrend SJ, et al. A novel chimeric oncolytic virus vector for improved safety and efficacy as a platform for the treatment of hepatocellular carcinoma. J Virol. 2018;92(23):pii: e01386‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ebert O, Shinozaki K, Kournioti C, Park MS, Garcia‐Sastre A, Woo SLC. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Mol Ther. 2004;9:S397. [DOI] [PubMed] [Google Scholar]
- 107. Wu L, Huang TG, Meseck M, et al. rVSV(M Delta 51)‐M3 is an effective and safe oncolytic virus for cancer therapy. Hum Gene Ther. 2008;19(6):635‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Cameron C, Hota‐Mitchell S, Chen L, et al. The complete DNA sequence of myxoma virus. Virology. 1999;264(2):298‐318. [DOI] [PubMed] [Google Scholar]
- 109. Wang F, Ma YY, Barrett JW, et al. Disruption of Erk‐dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5(12):1266‐1274. [DOI] [PubMed] [Google Scholar]
- 110. Kim M, Williamson CT, Prudhomme J, et al. The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene. 2010;29(27):3990‐3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang G, Barrett JW, Stanford M, et al. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin‐repeat host range factor. P Natl Acad Sci USA. 2006;103(12):4640‐4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pal I, Mandal M. PI3K and Akt as molecular targets for cancer therapy: current clinical outcomes. Acta Pharmacol Sin. 2012;33(12):1441‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Stanford MM, Werden SJ, Mc FG. Myxoma virus in the European rabbit: interactions between the virus and its susceptible host. Vet Res. 2007;38(2):299‐318. [DOI] [PubMed] [Google Scholar]
- 114. Woo Y, Kelly KJ, Stanford MM, et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329‐2335. [DOI] [PubMed] [Google Scholar]
- 115. Wennier ST, Liu J, Li SD, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20(4):759‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Weng MZ, Zhang MD, Qin YY, et al. Targeting gallbladder carcinoma: bone marrow‐derived stem cells as therapeutic delivery vehicles of myxoma virus. Chinese Med J‐Peking. 2014;127(12):2350‐2356. [PubMed] [Google Scholar]
- 117. Weng MZ, Gong W, Ma MZ, et al. Targeting gallbladder cancer: oncolytic virotherapy with myxoma virus is enhanced by rapamycin in vitro and further improved by hyaluronan in vivo. Mol Cancer. 2014;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cutts FT, Markowitz LE. Successes and failures in measles control. J Infect Dis. 1994;170(Suppl 1):S32‐41. [DOI] [PubMed] [Google Scholar]
- 119. Galanis E. Therapeutic potential of oncolytic measles virus: promises and challenges. Clin Pharmacol Ther. 2010;88(5):620‐625. [DOI] [PubMed] [Google Scholar]
- 120. Longley DB, Harkin DP, Johnston PG. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330‐338. [DOI] [PubMed] [Google Scholar]
- 121. Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64(14):4919‐4926. [DOI] [PubMed] [Google Scholar]
- 122. Carlson SK, Classic KL, Hadac EM, et al. Quantitative molecular imaging of viral therapy for pancreatic cancer using an engineered measles virus expressing the sodium‐iodide symporter reporter gene. Am J Roentgenol. 2009;192(1):279‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lange S, Lampe J, Bossow S, et al. A novel armed oncolytic measles vaccine virus for the treatment of cholangiocarcinoma. Hum Gene Ther. 2013;24(5):554‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kojima Y, Honda K, Hamada H, Kobayashi N. Oncolytic gene therapy combined with double suicide genes for human bile duct cancer in nude mouse models. J Surg Res. 2009;157(1):E63‐E70. [DOI] [PubMed] [Google Scholar]
- 125. Samudio I, Hofs E, Cho B, et al. UV light‐inactivated HSV‐1 stimulates natural killer cell‐induced killing of prostate cancer cells. J Immunother. 2019;42(5):162‐174. [DOI] [PubMed] [Google Scholar]
- 126. Samudio I, Rezvani K, Shaim H, et al. UV‐inactivated HSV‐1 potently activates NK cell killing of leukemic cells. Blood. 2016;127(21):2575‐2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107(10):1373‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266‐4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Medicine. 2016;95(49):e5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ebert O, Shinozaki K, Huang TG, Savontaus MJ, Garcia‐Sastre A, Woo SL. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune‐competent rats. Cancer Res. 2003;63(13):3605‐3611. [PubMed] [Google Scholar]
- 131. Kasuya H, Nishiyama Y, Nomoto S, et al. Suitability of a US3‐inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther. 2007;14(6):533‐542. [DOI] [PubMed] [Google Scholar]
- 132. Bossow S, Grossardt C, Temme A, et al. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18(8):598‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Stackhouse MA, Pederson LC, Grizzle WE, et al. Fractionated radiation therapy in combination with adenoviral delivery of the cytosine deaminase gene and 5‐fluorocytosine enhances cytotoxic and antitumor effects in human colorectal and cholangiocarcinoma models. Gene Ther. 2000;7(12):1019‐1026. [DOI] [PubMed] [Google Scholar]
- 134. Liu C, Sun B, An N, et al. Inhibitory effect of Survivin promoter‐regulated oncolytic adenovirus carrying P53 gene against gallbladder cancer. Mol Oncol. 2011;5(6):545‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wang JH, Yu Y, Yan Z, et al. Anticancer activity of oncolytic adenoviruses carrying p53 is augmented by 11R in gallbladder cancer cell lines in vitro and in vivo. Oncol Rep. 2013;30(2):833‐841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.