

Abstract
Significance: Cytoglobin (Cygb) was discovered as a new addition to the globin superfamily and subsequently identified to have potent nitric oxide (NO) dioxygenase function. Cygb plays a critical role in the oxygen-dependent regulation of NO levels and vascular tone.
Recent Advances: In recent years, the mechanism of the Cygb-mediated NO dioxygenation has been studied in isolated protein, smooth muscle cell, isolated blood vessel, and in vivo animal model systems. Studies in Cygb−/− mice have demonstrated that Cygb plays a critical role in regulating blood pressure and vascular tone. This review summarizes advances in the knowledge of NO dioxygenation/metabolism regulated by Cygb. Advances in measurement of NO diffusion dynamics across blood vessels and kinetic modeling of Cygb-mediated NO dioxygenation are summarized. The oxygen-dependent regulation of NO degradation by Cygb is also reviewed along with how Cygb paradoxically generates NO from nitrite under anaerobic conditions. The important role of Cygb in the regulation of vascular function and disease is reviewed.
Critical Issues: Cygb is a more potent NO dioxygenase (NOD) than previously known globins with structural differences in heme coordination and environment, conferring it with a higher rate of reduction and more rapid process of NO dioxygenation with unique oxygen dependence. Various cellular reducing systems regenerate the catalytic oxyferrous Cygb species, supporting a high rate of NO dioxygenation.
Future Directions: There remains a critical need to further characterize the factors and processes that modulate Cygb-mediated NOD function, and to develop pharmacological or other approaches to modulate Cygb function and expression.
Keywords: globins, cytoglobin, nitric oxide dioxygenation, redox regulation, vascular reactivity, free radicals
Introduction
Vascular function and tone in the body are regulated by many factors that are present in the endothelium and surrounding smooth muscle of the blood vessel wall (32, 74). Nitric oxide (NO) is synthesized by nitric oxide synthase (NOS) enzymes through conversion of substrate l-arginine to products l-citrulline and NO (1, 87). Three isoforms are present and differentially expressed in different tissues, including the calcium/calmodulin-regulated constitutive isoforms, neuronal NOS (nNOS, NOS1) and endothelial NOS (eNOS, NOS3), as well as inducible NOS (iNOS, NOS2) whose activation is calcium independent (1, 87). eNOS is a homodimeric enzyme with monomer molecular weight 134,000 Da (1). Under normal physiological conditions, NO is synthesized in the endothelium and has been identified as the endothelium-derived relaxing factor with its production primarily regulated by eNOS within the endothelium with inward diffusion to the vascular smooth muscle as well as outward diffusion to the vascular lumen (31, 77).
The production of NO by eNOS is highly regulated by a variety of factors, including calcium/calmodulin, substrate and cofactor availability, protein interactions, and posttranslational modifications. The levels and bioavailability of NO ultimately regulate vasomotor tone of blood vessels (32, 74). NO produced in the endothelium causes vasodilation by triggering relaxation of vascular smooth muscle cells (VSMCs) (Fig. 1). Inhibition of or impairment in NO synthesis has been shown to cause hypertension (HTN) with elevated blood pressure noted in both experimental animal models and clinical disease (1). Classically, endothelium-derived NO binds to soluble guanylate cyclase (sGC), which triggers the formation of cyclic guanosine monophosphate (cGMP) (Fig. 1), which then alters cytosolic Ca2+ levels followed by myosin phosphorylation in VSMCs, causing VSMC relaxation (73, 78).
FIG. 1.

Schematic of the mechanism of NO generation in endothelial cells and the process by which it induces smooth muscle relaxation. NO is generated in the endothelium by eNOS via a Ca2+/calmodulin-regulated process utilizing its requisite substrates as depicted. The NO generated diffuses into smooth muscle layer of vessels and binds to sGC to generate cGMP, which in turn induces vascular relaxation. NO that does not bind to sGC is rapidly converted to NO3− by NOD enzymes. cGMP, cyclic guanosine monophosphate; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; NO3−, nitrate; sGC, soluble guanylate cyclase. Color images are available online.
This mechanism of endothelium-dependent regulation of vascular tone through NO production has been extensively studied over the last three decades (73, 74, 78). Its great importance was recognized in 1998 when the Nobel Prize was conferred to three of the pioneering scientists whose observations led to its elucidation (45). NO has also been established to have platelet antiaggregation activity. Through its binding and activation of sGC, NO has been shown to decrease platelet activation, aggregation, and binding with fibrinogen (37). Similarly, NO also exerts other critical regulatory mechanisms, including modulation of smooth cell proliferation (48, 75), endothelial cell proliferation and apoptosis (75), antioxidant/oxidant properties (49, 98), leukocyte adhesion (33, 40), and mitochondrial function (18, 26, 29, 46). Loss of NO production or bioavailability occurs in diseases such as HTN, atherosclerosis, and diabetes (20, 59, 70). In these disease states and others that are accompanied by oxidative stress, it has been shown that eNOS uncoupling can occur with decreased NO production and enhanced superoxide (O2•−) generation. This uncoupling can be triggered by depletion of the critical redox-sensitive cofactor tetrahydrobiopterin (BH4) and by glutathionylation of specific cysteines located at the interface of the reductase and oxygenase domains (19, 24, 99). With superoxide production from eNOS or other sources, NO is scavenged with production of the potent oxidant peroxynitrite (ONOO−).
Apart from the aerobic NOS pathway of NO generation, it has also been demonstrated that NO can be formed independent of NOS, with nitrite, an oxidized product of NO, being reduced back to form NO under hypoxic conditions (3, 22, 57, 58, 103). At low pH, as occurs in ischemic tissues, nitrite-mediated NO production occurs (105). In mammalian cells, nitrite reductase enzymes have been shown to include molybdenum oxoreductases such as xanthine oxidase (XO) and aldehyde oxidase (AO), which have some structural similarity to bacterial and plant nitrite reductases, and also heme proteins and globins in the presence of suitable reducing equivalents or enzymes (57). Thus, while under normoxic conditions NOS enzymes are the major source of NO, under severe hypoxia or acidosis, nitrite can also serve as a potent source of NO (82). In conditions accompanied by severe hypoxia and acidosis, such as occurs in the ischemic heart or following cardiac arrest, nitrite reduction to NO has been shown to be a major source of NO formation (54, 105).
While controlled release and regulated NO bioavailability are beneficial for normal vascular function, excess NO is harmful and needs to be removed to maintain homeostasis of vascular tone. High NO levels, as occurring during septic shock, can cause hypotension resulting in low blood pressure (91, 96). Similarly, if pharmacological treatment with NO donating drugs, such as nitroprusside or nitroglycerin, induces levels of NO that are too high, hypotension will occur. In addition, in pathological conditions with oxidative stress and inflammation along with excess NO production, the reactions of excess NO can be different than when it is present in normal physiological concentrations. Charge neutrality and the high diffusion of NO make it a target for many redox enzymes. Also, high levels of NO have been shown to inhibit mitochondrial respiration through its binding to cytochrome oxidase (13, 72). As noted above, reactive oxygen species such as O2•− rapidly react with NO to form the potent oxidant ONOO− (9, 79, 80). The resulting ONOO− is a strong nitrating agent, causing nitration of proteins and cytotoxicity (79). Thus, for both physiological homeostasis and prevention of disease pathology, it is important that NO levels be precisely regulated. If NO levels are too low, there will be a loss of critical vasodilatory and anti-inflammatory function, while if levels are too high, hypotension with vascular collapse as well as other processes of metabolic inhibition and cellular injury would occur.
While the mechanism of NO synthesis in cells and vessels has been extensively studied and is well understood, until recently the process of NO degradation has been less clear. It has been shown that globins, such as hemoglobin (Hb) and myoglobin (Mb), in the presence of heme-bound O2 metabolize NO to NO3− (23, 35). This NO dioxygenation reaction, where the ferrous oxy-heme complex is oxidized to the ferric state, was well known to occur for Hb and Mb. Indeed, an important role for Hb in NO degradation in blood has been well demonstrated and an important role for Mb in NO degradation in skeletal and cardiac muscle proposed (28, 53). These heme proteins act under severe hypoxic conditions to store NO and possibly deliver it at a later stage, but at higher O2 levels, they degrade the NO via the dioxygenation reaction. While Hb has a major role in NO degradation in red blood cells (RBCs) and Mb in skeletal and cardiac muscle, the mechanism of NO metabolism in VSMCs, which is very critical to understand vasorelaxation and vascular tone, had remained unclear. Notably, smooth muscle cells have little or no expression of Hb or Mb (27, 51).
More recently, two new globins were discovered: neuroglobin (Ngb), primarily in neuronal cells, and cytoglobin (Cygb), which was first identified in liver stellate cells and later found to be expressed in smooth muscle and in many cell types (14, 52, 92). Most recently, Cygb has been shown to have important functions in the oxygen-dependent regulation of NO degradation (63). Interestingly, under anaerobic conditions, it was also shown to be a potent nitrite reductase, generating NO from nitrite (58). Thus, Cygb has been reported to function as an oxygen-dependent regulator of NO levels and related signaling in vessels. Herein, we provide a general review of the NO dioxygenase (NOD) property of globins and then focus on the recent studies showing the unique role of Cygb as a high-affinity NOD providing oxygen-dependent regulation of NO levels and regulating vascular NO homeostasis and tone. The role of various physiological reducing systems, reported to be of importance in regulating this process, is reviewed, and the role of Cygb versus other globins in the regulation of vascular function is also discussed.
NO Diffusion Through the Blood Vessel Wall
As vasodilation by endothelium-generated NO occurs by triggering sGC-mediated signaling in the surrounding smooth muscle, the process of diffusion-controlled transport of NO from the endothelial layer to smooth muscle has received considerable attention. Mathematical models have been developed to predict this process of NO diffusion, as well as its consumption in the smooth muscle of the arterial wall.
NO is freely diffusible outward from the endothelium to the vessel lumen and inward to the medial smooth muscle. Despite the large pool of Hb within the RBCs of luminal blood, endothelium-derived NO reaches the surrounding smooth muscle cells to induce relaxation. To understand how endothelium-derived NO escapes the vast amount of Hb (2.3 mM) (16) in the blood and still diffuses to the muscle cells to stimulate sGC to produce cGMP within these cells, studies have been performed comparing the rate of NO consumption by cell-free oxyHb and the oxyHb enclosed in RBCs. Cell-free oxyHb has been demonstrated to consume NO ∼1000 times faster than identical amounts of RBC-encapsulated Hb (64, 95). The mechanisms that limit NO consumption by RBCs have been debated, with-rate limiting factors evaluated, including diffusion of NO to the RBC surface, through the RBC membrane and inside the RBC (64, 65, 95). As first postulated by Liu et al., extracellular diffusion has been confirmed to be the major rate-limiting factor for NO consumption by RBCs (6, 64, 65).
In considering and modeling NO concentration profiles through the layers of the blood vessel wall, it is important to consider the reaction between NO and O2. The diffusion rates of NO and its coupled reaction with O2 in various media, including solutions (94), cell membranes (65), and intact blood vessels (66), have been reported. NO chemically reacts with O2 via second-order kinetics (17). Therefore, any NO concentration profiling across the blood vessel wall must also consider the transmural O2 variation. In many cases, such as aqueous solutions or cell membranes, NO diffusion is very fast, and its reaction rate is controlled by its diffusion. Despite such challenges, electrochemical methods, especially the Clark electrode-based NO probes, combined with computer modeling, have been extensively used to study the diffusion of NO across blood vessels, and quantitative approaches for comprehensive analyses of O2 diffusion across blood vessels have been developed (62, 66, 68). For example, in the case of one-dimensional NO diffusion through the blood vessel wall, as occurs with the vessel surface in contact with the NO electrode as shown in Figure 2A, the diffusion phenomenon can be described by the following equations:
FIG. 2.

Measurement of O2-dependent NO diffusion through the blood vessel wall. (A) Schematic of NO sensor electrode assembly used to measure NO diffusion across the attached piece of aorta. (B) Section of rat aorta with H&E staining. The rat aortic wall thickness L is ∼140 μm, and the thickness of the TM, LTM, is ∼112 μm. (C) NO diffusion flux across the aortic wall, detected at the electrode assembly (A), in the presence of the O2 concentration indicated. The arrow indicates the time of NO addition. (D) Simulation of NO concentration gradient across the vessel wall, assuming the NO concentration at the endothelial surface is 1 μM [adapted from Liu et al. (61)]. TA, tunica adventitia; TI, tunica intima; TM, tunica media. Color images are available online.


Therefore, the current detected in the electrode is defined as (67),


In a typical experiment, the initial condition defined as [NO] in the aortic wall at t = 0 is zero. When t > 0, and NO is added as bolus to the solution, [NO] is [NO]0. As the electrode perturbation is initiated, an [NO] gradient is created and this NO gradient establishes a steady state, dependent on the O2 levels and gradient across the blood vessel wall (Fig. 2C). From the experimental data, simulated curves of the NO concentration gradient across the vessel wall were obtained as shown in Figure 2D. These demonstrate the O2 dependence of NO diffusion and metabolism in intact vessels. The electrochemical methods used in these studies enable the study of this complex process. NO concentration profiles, calculated using the above expressions, have shown that [NO] falls asymptotically across the vessel wall due to the processes of its diffusion and metabolism (Fig. 2D) (66). These models predicted that, in the smooth muscle of the vessel wall, there is a prominent NOD that rapidly degrades the outward diffusing NO from the endothelium; however, the identity of this NOD remained elusive for over a decade.
Globins as Oxygen-Dependent NO Scavengers
The members of the globin superfamily have unique functions, and also can function differently in different tissues. Globins, primarily Hb or Mb, are classically known as O2 carriers in erythrocytes and muscle, respectively. O2 reversibly binds to the heme iron in the reduced Fe2+ state of globins, and this allows the heme to function as a store and carrier of O2. However, O2 can also be reduced by Fe2+ with the release of the superoxide free radical (O2•−). This potential for redox reactions by the Fe2+ core makes the globin heme a “redox active center.” In the absence of O2, NO binds with high affinity to the Fe2+-heme of deoxyglobins forming mono-nitroso complexes, while with O2 binding to the heme, NO is rapidly degraded through the process of dioxygenation. The balance between these two processes is regulated by the O2 binding affinity of the globin and the concentration of O2 present.
Iron in the globins is present as either five or six coordinates, with proximal or distal histidine residues that influence ligand binding to the Fe-heme core. This difference in structure of the heme coordination and pocket confers different globins with different affinities toward O2 or NO binding. Extensive investigations have been carried out by the Olson group to understand how mutations in these residues can alter the ligand binding and autoxidation properties of the Fe core in globins, especially for Mbs and Hbs (5, 12, 84). Iron is present as hexa-coordinate in Cygb where both fifth and sixth positions are ligated by histidine His81 and His113 (Fig. 3A, B). On the contrary, Mb-like Hb is pentacoordinate, leaving the sixth position free (a coordinated H2O for the met-Mb form as shown in Fig. 3D), while the proximal histidine in the fifth position is coordinated to the Fe (Fig. 3C, D). Therefore, O2 binding efficiency and reactivity at the Fe center vary for each structurally different globin. Moreover, O2 binding to the Fe center in Mb or Hb is stabilized by hydrogen bonding to His (11), and therefore, heme autooxidation to the ferric met form is slowed.
FIG. 3.

Structural comparison of Cygb and Mb. (A) Ferric form of human Cygb (PDB Accession No. 1V5H), (B) and its Fe-heme coordination, (C) aquo-met form of sperm whale Mb (PDB Accession No. 4MBN), (D) and its Fe-heme coordination. The coloring by atom type is: Fe (orange), O (red), N (blue), C (green). Cygb, cytoglobin; Mb, myoglobin. Color images are available online.
The overall association rates of O2, NO, and CO binding to Cygb measured in simple mixing experiments are slow and limited by the rate of dissociation of the internal distal histidine, and on the order of 1 to 2 s−1 at high ligand concentrations (39, 56). However, the association rates for the open conformation measured in laser photolysis experiments before His(E7) rebinding occurs are similar to those of Mb and Hb and on the order of 5 to 30 × 106 M−1 s−1 (39, 56). The NO deoxygenation of CygbO2 is fast and again similar in rate to that observed for MbO2 (63, 83). Thus, after dissociation of His(E7), the accessibility of the heme iron atom in Cygb appears to be similar to that of Mb, which also has a high rate of NO dioxygenation.
The rate of NO reduction by globins and the redox potential of the Fe center are also modulated by other factors. Therefore, the rate of NO dioxygenation also varies. In addition to redox potential-mediated regulation of NO conversion to nitrate, other microenvironmental factors of the heme pocket were recently identified to be of key importance (4). Since the redox potentials of globins do not necessarily correlate with the rate of NO conversion, as recently reported, it appears that local environments such as hydrophilicity/hydrophobicity in the pocket, or accessibility of the pocket, determine the rate of the reaction (4).
Cygb has been shown to have a faster rate of reduction than other globins with simple reducing agents such as ascorbate or with enzymatic reducing systems such as P450 reductase or cytochrome b5/cytochrome b5 reductase (4, 61, 68). In line with this, Cygb has been observed to have the highest rate of NO dioxygenation among these globins.
Cygb Is the Primary NOD in Blood Vessels
Cygb was discovered about two decades ago as the fourth major globin expressed in mammals (100). It was first described by Kawada and colleagues as “stellate cell activation-associated protein” (STEP) in hepatic stellate cells (HSCs) (100). This STEP, later named Cygb, showed higher lipid and peroxidase activity (14, 92, 100). Even though Cygb was discovered in HSCs, later it was found to be expressed in a variety of cells, such as esophageal cells (71) and smooth muscle cells of the vasculature and melanocytes (30, 38). Alterations in Cygb expression have been found to be associated with a variety of disease pathologies, including cancer (76), atherosclerosis (50), HTN (61), and fibrosis (89). Cygb has many similarities to other globins (Hb and Mb), including the classic three over three alpha helical globin fold (Fig. 3A, C); however, its biological and physiological functions are different from other globins, and therefore, the biochemistry of Cygb has been actively investigated. Unlike Hb or Mb, Cygb is six-coordinate in both Fe2+ and Fe3+ states via axial imidazole nitrogen ligands from His81 and His113 in the absence of competing ligands. Therefore, any gasotransmitters such as O2 or NO have to displace the His ligand to bind to the heme Fe. Despite this, O2 binds strongly to Cygb (21, 92). Among the globins, Cygb more efficiently degrades NO due to its higher rate of NO dioxygenation, as detailed below. NO metabolism in VSMCs and NO flux across conduit and resistance vessels, such as aorta and mesenteric arteries, have been studied (60, 61, 63, 67, 68). NO-sensing electrochemical sensors have been widely used to measure the rates of NO degradation and to characterize this process (61, 68, 69, 101).
Recently, Cygb has received increased attention, following the discovery that it is highly expressed in the vascular smooth muscle of vessels and proposed to have a key role in the regulation of vascular tone (38, 58, 61). Halligan et al. first reported that VSMCs from various species, including human, expressed Cygb (38). Knockdown with shRNA successfully reduced Cygb expression and, more importantly, showed decreased NO degradation in these cells. Immunohistology showed expression of Cygb in the smooth muscle medial and advential layers of the aorta. More recently, we reported a quantitative estimation of Cygb and other globins in smooth muscle cells and demonstrated that Cygb is present in the vascular smooth muscle but absent from the endothelium (61). Levels of Cygb and Mb in human aortic smooth muscle were measured to be ∼45 ng/106 cells and only ∼1 ng/106 cells, respectively, suggesting that Cygb is the main NOD in smooth muscle cells. Hb expression was trace or undetectable with levels >200-fold below those of Cygb (Table 1). Therefore, Cygb is the most highly expressed globin in VSMCs, and thus would be expected to have a major role in vascular NO metabolism and secondary regulation of vascular tone (61).
Table 1.
Properties of Relevant Globins
| Globin | Cytoglobin | Myoglobin | Hemoglobin-α |
|---|---|---|---|
| P50(O2), mmHg | 0.7–2.9 | ∼2 | — |
| Sites of expression | Ubiquitous (92) SMCs (61) |
Skeletal muscle Cardiac muscle SMCs |
MEJ of resistance vessels (85), pulmonary endothelial cells (2) |
| Levels of expression | ∼5 μM in SMCs (61) | ∼ 400–500 μM (skeletal muscle) ∼200–330 μM (cardiac muscle) ∼0.13 μM in SMCs (61) |
Present in the MEJ, but not quantitated (85) Not detectible in SMCs (61) |
| Functional role(s) | NOD in SMCs of conduit and resistance vessels O2-dependent regulation of NO metabolism in SMCs (61, 63) |
O2 store NOD in skeletal and cardiac muscle |
NOD in the MEJ of resistance vessels (85) |
| NO dioxygenationa rate, μM−1 s−1 | 22–30 (63) | 34–43 (23, 25, 43) | — |
| Rate of reduction, M−1 s−1 | 1.1–2.7 (Asc) (4, 8, 34) 36.1 (Asc) (61, 63) (1.8–2.9) × 105 (B5R/B5) (61, 88), 0.6 × 105 (B5R/B5)b (55) |
0.09–0.25 (Asc) (34, 61, 68, 88) (0.161–1.27) × 104 (B5R/B5) (61, 88) |
0.32 (Asc) (61) 1.51 × 104 B5R/(B5-system) (61), 0.6 × 104 (B5R/B5)b (55) |
Measured for the reduced ferrous globins.
Derived rate constants from reported rate in s−1.
Asc, ascorbate; MEJ, myoendothelial junction; NO, nitric oxide; NOD, NO dioxygenase; SMC, smooth muscle cell.
Mechanism of O2-Dependent NO Dioxygenation by Cygb
O2-dependent NO metabolism in mammalian cells was reported as early as 2001 by the Gardner group (36), even before Cygb was identified. The metabolism was found to be inhibited by strong Fe-heme ligands such CN− and CO, but neither mitochondrial respiration nor O2•− or H2O2 was required. Therefore, the existence of an efficient mammalian heme- and flavin-dependent NOD was suggested (36). Subsequently, Cygb has been shown to be present in VSMCs with an important role in NO metabolism (38, 61). In the last decade, extensive kinetic modeling and analyses of the process of NO dioxygenation to NO3− by Cygb have been performed by our group and others (34, 63, 68, 101). As pointed out above, in the pocket of the heme core, initial binding of O2 displaces the exchangeable histidine (His81) coordinated to the reduced Fe (Fe2+), and the heme dioxygen complex Fe2+-O2 is formed. This species is the precursor that dioxygenates the NO to NO3− and is oxidized back to met-Cygb (Cygb-Fe3+), which is then reduced back to Fe2+, binding O2 to again form the precursor. This cycle continues as long as O2 and the required reducing equivalents are available (Fig. 4).
FIG. 4.

Reaction mechanism of Cygb and Cygb-mediated NO consumption. Met-cytoglobin Cygb(Fe3+) can be reduced by different reducing systems in SMCs to generate Cygb(Fe2+), and the reduced Cygb establishes an equilibrium with available Fe2+ ligands such as O2, NO, CO, or the His residue. Among these species, Cygb(Fe2+-O2) is the catalytically active species for the dioxygenation of NO to NO3−. SMC, smooth muscle cell. Color images are available online.
We developed a kinetic model of the mechanism of NO dioxygenation (63). This is based on the coupled reactions as below:

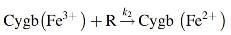
where R represents the total reducing equivalents available to reduce the metCygb. The Cygb(Fe2+) is expected to have an additional equilibrium as described below:

where His81 is the exchangeable histidine. In addition, NO and O2 binding equilibria are considered as follows.


Based on these equations, a kinetic model was developed and experimentally verified in our laboratory (63), which serves to predict the reaction rates that occur. After rearranging the equation as described in Liu et al. (63), the final NO metabolism rate can be related to O2 concentration as follows:

where

where [E] is the total concentration of Cygb.
The above equation is an apparent Michaelis–Menten-type equation where O2 is substrate. The Km defines the O2 concentration at half maximum rate, and it is an indirect measure of substrate (O2) affinity (inversely related) to Cygb(Fe2+) to form the oxy-Cygb (CygbFe2+-O2), which is required for NO dioxygenation. Simulations based on the existing kinetic data have shown that about 98% of the reduced Cygb(Fe2+) exists as the oxyspecies. Validity of this model was tested in vitro with purified proteins (63), and the results were accurately predicted. First, there was a clear decrease in the rate of Cygb-mediated NO consumption with O2 depletion (Fig. 5A). Michaelis–Menten-type curves were obtained for NO consumption rate as a function of O2 concentration. The experimental data were accurately represented by the above equation (Fig. 5C), and relevant parameters could be determined using various reducing systems (63). However, more complex systems such as living cells (where multiple reducing enzyme systems and equivalents are present together) or blood vessels (where, in addition, NO and O2 gradients across the blood vessel exist) await further study and modeling.
FIG. 5.

Comparison of oxygen-dependent NO metabolism by Cygb and Mb. Simultaneous measurements of NO decay and [O2] change for Cygb (A) and Mb (B). (C) Simulated (line) and measured NO consumption rate as function of O2 concentration (VCygb-NO-[O2] and VMb-NO-[O2]) curves at varying O2 concentrations. Experimental data VMb-NO (•) and VCygb-NO (▾) versus [O2] [adapted from Liu et al. (68)].
Differences in the O2-dependent NO dioxygenation properties of each of the mammalian globins may account for their differing expression levels and roles in different cells and tissues (68). For example, the NO dioxygenation reaction rate of Cygb depends on O2 with a sharp decrease under conditions of physiological hypoxia, whereas Mb shows a more gradual, near-linear dependence on O2 concentration (Fig. 5B, C) (68). The higher NOD activity of Cygb and its decline at physiological levels of hypoxia seem to provide a basis for why its expression would be favored for the regulation of NO levels in vascular smooth muscle. In contrast to this, Mb is the major globin expressed in skeletal and cardiac muscle where it is expressed at much higher levels on the order to 100 μM, more than 50-fold higher than the levels of Cygb in smooth muscle (Table 1). In skeletal and cardiac muscle, low O2 levels would occur with mechanical work, and accumulation of NO with hypoxia could be deleterious with inhibition of aerobic metabolism and with enhanced superoxide generation as occurs following ischemia/hypoxia and reperfusion/reoxygenation with formation of NO-derived peroxynitrite or other reactive nitrogen species (97, 102). Thus, the differing NOD properties of each of the globins are an important basis for their pattern and level of cellular expression and their unique functional roles.
Redox Regulation of Cygb O2 Binding and NO Dioxygenation
Redox or chemical modification of Cygb has been shown to modulate its O2 binding affinity and NOD activity (101). The Cygb monomer contains two exposed cysteine residues (Cys38 and Cys83) that enable Cygb to form an intramolecular disulfide bond or intermolecular disulfide bond resulting in a dimeric form (7, 56, 93). The formation of the intramolecular disulfide bond greatly increases the dissociation rate constant of the bound distal histidine, resulting in a greater apparent binding constant of extrinsic ligands (7, 93). This histidine displacement makes Cygb more Mb-like. As an NOD, O2 must bind to Cygb before it metabolizes NO. Under hypoxic conditions, Cygb with intramolecular disulfide bond (Cygb-SS) binds more O2 in the form of Cygb(Fe2+-O2) than Cygb with the free sulfhydryl group (Cygb-SH) or Cygb with cysteine residues blocked by thioether bonds to N-ethylmaleimide (Cygb-SC). However, under ambient oxygen levels, Cygb O2 binding is saturated and its NOD activity is limited and largely controlled by the rate of Cygb reduction (83). Thus, if the rate of Cygb reduction is not altered by modification of the sulfhydryl groups in the Cygb, the NOD activity of Cygb may not be greatly altered. Studies at ambient O2 levels with three different redox forms of Cygb, namely Cygb-SH, Cygb-SC, and Cygb-SS, showed that only Cygb-SS had a perturbed NO dioxygenation rate, decreasing by 25% (101). However, the P50 of O2 binding to Cygb, a measure of oxygen affinity, was shifted to a fourfold lower value with this intramolecular disulfide form. In pathological conditions with oxidative stress, the cysteine of Cygb could be redox modified leading to formation of Cygb S-S. Further studies are needed to explore the role of Cygb thiol modification on its O2-dependent NO dioxygenation rate, and how these thiol modifications affect vessel function.
Cygb Reduces Nitrite (NO2−) to NO Under Anaerobic Conditions
Our early experiments revealed that there is an NOS enzyme-independent process of NO generation in ischemic/hypoxic tissues secondary to the reduction of nitrite (104). While NO2− is ubiquitous in tissues due to NO degradation and dietary sources, with levels typically from 1 to 10 μM, its importance as a source of NO had not been recognized. We observed that in the process of myocardial ischemia, nitrite is reduced to form NO and this was confirmed by electron paramagnetic resonance (EPR) detection of NO and isotope tracer experiments with 15NO2−, proving that nitrite is reduced to form NO. As visualized by EPR imaging in hearts subjected to global ischemia, this process occurs throughout the ischemic myocardium, detectable within 5 min of ischemic onset, and increases with ischemic duration (54, 105). This process progressively increases with the duration of ischemia, and NO2−-derived NO accumulates in the ischemic myocardium bound to Mb. Interestingly, following the onset of cardiac arrest, similar nitrite-derived NO was visualized in ischemic tissues and bound to Hb in the blood stream (54). A role for Mb in the generation of NO during myocardial ischemia was reported and this was confirmed by several groups (42, 81, 90).
Subsequently, experiments were performed to explore the role of nitrite-derived NO in the regulation of vascular function and tone. Our experiments with in vivo hemodynamics in rats and in vitro vasorelaxation in isolated rat aorta under aerobic conditions clearly showed that nitrite-mediated NO formation occurs in vessels and serves to regulate vascular tone independent of NOS (3). As this process of NO generation in isolated vessels was inhibited by heme blocking or oxidation, we speculated about a role of a reduced globin such as Cygb (3). The precise enzyme/pathway in this vascular NO2−-mediated NO formation was uncertain. Work from several investigators suggested a key role of Hb in nitrite-mediated NO generation (22). We observed that NO2− reduction to NO primarily occurs in tissues rather than in blood, and therefore, Hb cannot be the main source (58). XO and AO were identified as other potential mechanisms contributing to this process (57).
While Cygb has been established to metabolize NO through its dioxygenase activity in the presence of O2, we identified that there is an additional process that occurs with severe hypoxia and anoxia, where Cygb can generate NO through the reduction of NO2− (58). As described above, prior studies had reported that Hb can produce NO from NO2− reduction (44). Subsequently, we identified that under anaerobic conditions, Cygb reduces NO2− to NO. Cygb was found to produce much higher levels of free NO than other globins, such as Hb or Mb, which can reduce nitrite to NO, but at their high cellular concentrations tightly bind NO. Thus, Cygb acts as a potent nitrite reductase under anaerobic conditions, while in the presence of O2, it functions as an O2-dependent NOD. This switch with reversal from NO consumption to its synthesis illustrates its biphasic role in sensing O2 level and providing O2-dependent NO regulation. In ischemic syndromes with low or no flow and severe O2 depletion, this mechanism would feed back to enhance NO levels that would serve to increase perfusion and restore O2 levels within the tissue. Thus, one can view Cygb as a critical sensor and regulator of NO in the vessel wall having the function of controlling NO levels to maintain vascular homeostasis and tone over a broad range of O2 tensions.
In anaerobic conditions, nitrite, NO2−, can bind to the reduced Cygb Fe2+ heme and undergoes reduction to regenerate Cygb(Fe3+) and NO as proposed below (58)
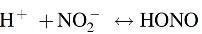

The first evidence of NO generation by Cygb was reported by our group (58). Later, this was confirmed by several groups, and it has also been reported that Ngb can similarly produce NO from NO2− reduction (21, 47, 88). With the use of EPR spin-trapping and also chemiluminescence detection, the generation of Cygb-mediated NO was demonstrated (Fig. 6). This NO generation was found to be proportional to the nitrite concentration up to 1 mM NO2−, with rate of 1 nM s−1 mM−1 NO2−. Furthermore, the NO2−-derived NO was shown to activate sGC with cGMP formation (Fig. 6), proving that Cygb-generated NO signaling in hypoxic conditions can occur and would be predicted to be physiologically relevant.
FIG. 6.

Measurement of Cygb-mediated NO generated from NO2− under anaerobic conditions. (A) EPR measurements of NO at anaerobic conditions in phosphate buffer (pH 7.0) with [NO2−] and [Cygb] as indicated. Reactions were performed at 37°C in the reaction vessel with NO continuously purged with argon from the reaction vessel into a trap vessel containing 1 mL of 2 mM (MGD)2-Fe2+. (B) Measurement of the rate of NO generation using the chemiluminescence NO analyzer under anaerobic conditions in phosphate buffer (pH 7.0) with 10 μM nitrite (a), 200 μM nitrite (b), and 1 mM nitrite (c) with 10 μM Cygb in 5 mL of phosphate buffer at 37°C. (C) Effect of oxygen on NO generation and sGC activation. Oxygen tension was controlled by purging with argon/oxygen gas mixtures. Cygb (10 μM) was incubated with 1 mM nitrite at pH 7.0 in 5 mL of phosphate buffer at 37°C with pO2 values from 0.03 to 0.79 torr. (D) sGC activation was determined from measurements of cGMP formation. Reactions were performed in 1 mL of phosphate buffer with EDTA (5 mM), MgCl2 (2 mM), sGC (10 ng), and GTP (1 mM), incubated with nitrite (10 μM) and Cygb (10 μM) at pH 7.4, and purged with argon/oxygen gas mixtures with a 30-min incubation time [adapted from Li et al. (58)]. EPR, electron paramagnetic resonance.
Overall, these observations support the concept of a central role for Cygb as a key sensor and regulator of NO levels. In the normal well-perfused vessel, it metabolizes excess NO by its dioxygenation function; however, with tissue hypoxia and low arteriolar and capillary O2 levels where increased flow is required by the tissue, this NO degradation is decreased. With more severe hypoxia as occurs in pathologic conditions such as organ ischemia, where eNOS-mediated NO formation in the endothelium would be diminished or even abolished due to lack of O2, Cygb present in the vascular smooth muscle can actually generate NO from NO2−, which will bind to sGC in this smooth muscle and induce vessel relaxation. Hence, Cygb has been shown to be an O2-dependent modulator of NO homeostasis and vascular tone that can serve to regulate tissue perfusion in normal physiology and disease.
Role of Cygb in NO Metabolism in VSMCs
We have recently reported studies in smooth muscle cells and blood vessels that directly demonstrate the important role of Cygb in the regulation of NO metabolism in VSMCs (61). Cygb expression in VSMCs was measured by quantitative immunoblotting and found to be about 45-fold higher than Mb (45 and 1 ng, respectively, in 1 × 106 cells) (61). When Cygb expression was knocked down in VSMCs using specific siRNA, we found that the magnitude of NO dioxygenation was greatly decreased as demonstrated by a much slower rate of NO decay (Fig. 7). The VSMCs from rat aorta or mesenteric artery exhibited similar Cygb dependence, demonstrating that NO metabolism in VSMCs from larger capacitance vessels and smaller resistance vessels is largely Cygb dependent. It was determined that Cygb was responsible for ∼70%–75% of NO metabolism in VSMCs. In parallel with this, NO degradation and the formation of the NO dioxygenation product, NO3−, were demonstrated (61). Furthermore, knockdown of cytochrome b5 reductase decreased NO metabolism by ∼65%, indicating its important role in support of the NO dioxygenation function of Cygb.
FIG. 7.

Knockdown of Cygb in rat aSMC and mSMC. (A) Level of Cygb in rat aSMCs and rat mSMCs was estimated by quantitative immunoblotting. The first four bands are pure Cygb at four different amounts. Bands 5 and 6 are total cellular proteins from rat aSMCs and the related Cygb siRNA-treated cells (Cygb-KD aSMC), respectively; and bands 7 and 8 are rat mSMCs and the related Cygb siRNA-treated cells (Cygb-KD mSMC), respectively. (B) Plots of the rate of NO decay by 7 × 106 SMCs per milliliter (red solid line: aSMC, red dashed line: mSMC) and 7 × 106 Cygb-KD SMCs per milliliter (green solid line: Cygb-KD aSMC, green dashed line: Cygb-KD mSMC) versus time after NO was injected into the solution to achieve an initial concentration of 0.5 μM. Means and standard errors of the rate of NO decay by SMCs and Cygb-KD SMCs from aorta (C) and mesenteric artery (D). Error bars: mean ± SEM, n = 3–5 per group, **p < 0.01 control SMCs versus Cygb-KD SMCs; p-values determined using a two-tailed t-test [adapted from Liu et al. (61)]. aSMCs, aortic smooth muscle cells; mSMCs, mesenteric smooth muscle cells; SEM, standard error of the mean. Color images are available online.
Cygb Reducing Systems in VSMCs
NO dioxygenation requires active regeneration of oxy-Cygb(Fe2+). The process of Cygb(Fe3+) reduction to Cygb(Fe2+) has been extensively studied (4, 63). This reduction rate is on the order of 2–3 × 105 M−1 s−1 for some enzymatic reducing systems, whereas for some chemical reductants it is on the order of 1–36 M−1 s−1 (4). Spectrophotometric assays have been used to study the reduction process, and for fast kinetic analyses, stopped-flow spectroscopy has been used (4). Comparative evaluation of reduction of various globins has shown that Cygb is reduced at a much faster rate than other globins (4, 61, 68). Also, as we have reported, at low O2 concentrations ([O2] < 50 μM), Cygb shows a greatly reduced NO dioxygenation rate, while other globins, for example, Mb, show only a gradual more modest O2-dependent decrease in NO deoxygenation (Table 1). Thus, it has been considered that Cygb is not only a robust NO metabolizing enzyme but also acts as an O2-sensitive NOD, with marked decrease in NO degradation rate under conditions of physiological hypoxia or ischemia. This feature allows the NO to diffuse a longer distance in the blood vessel with an increased lifetime when conditions of low pO2 occur, such as in myocardial ischemia.
Various reducing equivalents and reducing systems have been identified in cells capable of reducing the oxidized form of Cygb (metCygb) to Fe2+ (4). Spectrophotometric studies have demonstrated that ascorbate could function as an effective reducing agent for Cygb in vitro, with electrochemical measurements demonstrating that this leads to rapid NO consumption (63, 68). Interestingly, Cygb exhibits a unique reduction pattern with ascorbate. While other globins, such as Mb, show irreversible reduction with ascorbate, Cygb shows a reversible reduction (i.e., reduced Cygb could be oxidized back to Fe3+ by rising levels of oxidized ascorbate, shifting the equilibrium backward, and thereby slowing down the net Cygb reduction) (63, 68). This unique observation suggests that complex formation between Cygb and ascorbate may occur, as first hypothesized by Gardner et al. (34), although to date there is a lack of direct experimental proof of this complex formation. Various flavin containing reductase enzymes such as cytochrome P450 reductase (CPR) and cytochrome b5 reductase were also found to be highly effective in reduction of Cygb (4). As is the case for other globins, the NADH/cytochrome b5 reductase/cytochrome b5 (b5R/b5) reduction pathway has been proposed to be of particular importance. The b5R/b5 reducing system provides high reduction rates while the rate for ascorbate is much lower; however, the concentration of ascorbate can reach millimolar levels in the cell (10), while expression levels of b5R or b5 would be expected to be 1000-fold lower (4).
The relative role of b5R/b5 versus other reducing enzymes or ascorbate in the process of Cygb reduction within smooth muscle and vessels remains uncertain. Furthermore, it is unclear how this process of reduction is modulated to regulate blood pressure and vascular tone. Resolution of these questions awaits quantitative determination of the levels of each protein as well as ascorbate and other reducing substrates. It is likely in view of the high reduction rate by the b5R/b5 system, that it is the predominant reducing system under normal conditions. The case for a role for ascorbate is that the available concentration of ascorbate is very high (mM in cells) (10), and therefore, it could also play a critical role in reducing metCygb (4, 63). The initial rate of Cygb reduction by ascorbate saturates at higher ascorbate concentrations, following Michaelis–Menten kinetics as follows (63):

which can be reduced to a second-order form

These observations suggest that ascorbate could significantly contribute to Cygb reduction, even though the rate of reduction is much lower than the NADPH/CPR or NADH/b5R/b5 system. Additional work is needed to characterize the role of each of these reducing systems in smooth muscle and vessels.
Cygb in Regulation of Blood Pressure and Vascular Tone
With studies in isolated blood vessels of wild-type (WT) and Cygb−/− mice, it was seen that Cygb gene knockout (Cygb−/−) enhances NO-mediated blood vessel relaxation. These studies confirmed the important functional role of Cygb in regulating NO bioavailability in blood vessels (61). Cygb−/− vessels were much more sensitive to either the endothelium-dependent agonist acetylcholine or the endothelium-independent NO donor nitroprusside. In aortic segments, a marked shift to the left in the endothelial-dependent vasodilation–response curves was observed for Cygb−/− compared with WT, with 39-fold lower levels of the endothelium-dependent relaxation agonist acetylcholine (13 nM vs. 0.33 nM), or 20-fold lower levels of the endothelium-independent NO donor nitroprusside (3.0 nM vs. 0.15 nM) required for 50% relaxation (61). Measurements of the NO diffusion across the aortic wall from Cygb−/− and WT mice were performed using an NO electrode and confirmed that the enhancement of vasodilation in Cygb−/− vessels is due to a lower rate of NO metabolism in the vessel wall. Electrochemical detection of NO flux across aorta isolated from Cygb−/− mice showed higher levels of NO flux across blood vessels due to decreased NO metabolism compared with WT, with the measured peak NO flux more than sixfold higher across the aortic wall of Cygb−/− mice than that of WT (61). Similar results were seen in small resistance arteries such as mesenteric segments (61). Thus, Cygb was shown to regulate endothelium-mediated vasodilation and vascular tone through its metabolism of NO.
In addition to large effects on the function of ex vivo vessels of Cygb−/− mice, we observed large alterations in in vivo vascular tone, blood pressure, and cardiac function compared with matched WT mice (61). Mean arterial blood pressure (MABP) of Cygb−/− mice was 30% lower than the background-matched controls. Echocardiographic evaluations demonstrated that the cardiac output was increased by 68% in these Cygb−/− mice, likely as a compensation for the marked vasodilation present. Systemic vascular resistance (SVR) was 54% lower in the knockout mice. Interestingly, NOS inhibition by L-NAME largely reversed the low MABP and SVR values as well as the elevated cardiac output of the Cygb−/− mice to values close to those in WT, confirming that these alterations were secondary to enhanced NOS-derived NO. From ultrasound measurements, the aorta was also observed to be dilated in Cygb−/− mice compared with WT, and this was also largely reversed by L-NAME. These results suggested that in the absence of Cygb and its potent NOD activity, elevated endothelial-derived NO levels are present in the smooth muscle resulting in enhanced vasodilation. In contrast, in the WT vessels, there was only a slight effect of NOS inhibition suggesting that the normal Cygb levels present were sufficient to metabolize the NO formed in the aorta in situ under basal nonstimulated conditions. Further evidence that Cygb knockout results in increased smooth muscle NO levels with activation of sGC was provided by an assay of cGMP levels, which were observed to be fivefold higher in the aorta of Cygb−/− than matched WT vessels (Fig. 8A, B). A marked increase in tissue perfusion was also observed in the Cygb−/− mice compared with WT (Fig. 8C, D), further demonstrating vasodilation of the small resistance vessels that control tissue perfusion. This increased perfusion was also reversed by NOS inhibition. Together, these observations demonstrated that the NOD function of Cygb is of critical importance for the in vivo regulation of NO levels, in turn controlling vascular tone, blood pressure, cardiac function, and tissue perfusion. Thus, knockout of Cygb enhanced NO bioavailability and secondary vasodilation.
FIG. 8.

Imaging of aortic diameter and perfusion in WT and Cygb−/− mice. (A, B) Inner diameter of aortas in WT mice and Cygb−/− mice before and after treatment with NOS inhibitor L-NAME. ***p < 0.01 WT versus Cygb−/−. Error bars: mean ± SEM; p-values determined using a two-tailed t-test. (C) Perfusion maps show higher baseline perfusion (red) in Cygb−/− mice compared with WT mice as measured on the ventral side of the mouse. Color scale (arbitrary units) is defined as highest perfusion in red and lowest perfusion in blue. (D) Laser speckle intensity comparison of the center panel perfusion maps shown in (C); ∼40% higher tissue perfusion seen in Cygb−/− versus WT, and L-NAME treatment reversed this higher perfusion. N = 4–12 per group. **p < 0.01 WT versus Cygb−/−, +p < 0.05, ++p < 0.01 WT or Cygb−/− versus L-NAME-treated. Error bars: mean ± SEM; p-values determined using a two-tailed t-test [adapted from Liu et al. (61)]. WT, wild type. Color images are available online.
Further experiments evaluated the effect of Cygb knockout in a model of HTN. With angiotensin-II (Ang-II)-induced HTN in rats, we found that knockout of Cygb prevented the Ang-II-associated decrease in NO levels, and prevented the onset of HTN (61). Thus, downregulation of Cygb expression or its NOD function could be a potential approach to ameliorate or prevent HTN.
The Myoendothelial Junction in Vascular Tone Regulation
In recent years, it has also been reported in resistance vessels that the junctional membrane complex of VSMCs with the adjacent endothelial cells functions as a unique subcellular environment with unique localized protein expression and physiological function (41, 86). NO passage from the endothelium to the smooth muscle layer of the thoracodorsal artery was shown to be modulated by Hb-α in the myoendothelial junction. NO was observed to be metabolized by Hb-α present in the endothelial cells of the myoendothelial junction with cytochrome b5 reductase (b5R) serving as the reducing system with this process regulating the flux of NO out to the vascular smooth muscle (55, 85).
In human and mouse arterial endothelial cells, Hb-α was detected at the myoendothelial junction. This function in the myoendothelial junction was observed to be unique to Hb-α as evidenced by abrogation with its genetic depletion. It was proposed that endothelial Hb-α heme iron in the ferric state permits NO signaling, while this signaling is shut off when Hb-α is reduced to the Fe2+ state by endothelial cytochrome b5 reductase 3, b5R3. Genetic and pharmacological inhibition of b5R3 increased NO bioactivity in small arteries (15, 85). Thus, it was proposed that the level of NO available in the smooth muscle of resistance blood vessels is regulated by Hb-α and b5R3 at the myoendothelial junction.
While these results are of great interest, a number of questions remain. It must be considered that b5R also serves as a critical reducing enzyme not only for Hb-α, but also other globins, including Cygb (4, 61). Indeed significant levels of Cygb were also found to be present in the myoendothelial junction, so Cygb could also have some role in this NO decay (85). As such, b5R knockdown alone would not distinguish the role of a given globin. Also, in general, the process and rate of globin reduction by b5R are greatly facilitated by b5, and the role of b5 in this process remains unclear. It is also unclear if gene knock down of Hb-α might have effects on other globins as well. There also is a need to study and compare the process of NO decay in resistance vessels from different locations and of different sizes. Therefore, the relative importance of this process of NO metabolism at the myoendothelial junction compared with that in the smooth muscle has remained unclear, and more study is needed.
Since without Hb-β, the α-globin is unstable and cytotoxic, particularly in its oxidized form, questions arose regarding the mechanisms that regulate α-globin expression in endothelial cells. Recently, it has been shown that the molecular chaperone α-hemoglobin stabilizing protein (AHSP) promotes arteriolar α-globin expression in vivo and facilitates its reduction by cellular reductases, including the reductase domain of eNOS. In AHSP knockout mice, Hb-α levels were decreased by 70% with evidence of increased NO signaling and arteriolar dilation (55).
In an effort to assess the relative role of the myoendothelial junction versus smooth muscle-mediated NO degradation by Cygb in resistance vessels, experiments have been performed in the mouse Cygb knockout model using endothelium-denuded mesenteric artery vessels (61). In these experiments with endothelium-denuded vessels, a slight 6% increase in NO flux was seen in WT vessels with a larger 28% increase in Cygb−/− vessels. In view of the >300% increase in NO flux seen with Cygb knockout, this suggested that the major process of NO consumption regulating NO flux through the wall of resistance vessels is Cygb dependent within the smooth muscle. However, endothelial processes and the myoendothelial junction may also contribute to modulating NO bioavailability and transport to the smooth muscle, with this depending on the relative expression of each globin and the cellular reducing state in the endothelium and smooth muscle. These processes and factors may also vary as a function of vessel location and size. More work is needed to understand the relative importance and interactions between the processes of myoendothelial junction NO metabolism and that which occurs in the smooth muscle cells.
Summary and Conclusions
Until recently, the process of NO metabolism that modulates its bioavailability in the vessel wall was unknown. This had been shown to be highly oxygen dependent, but the underlying molecular mechanism remained elusive. The novel globin, Cygb, has now been shown to have high potency as an NOD with sharp O2-dependent decline in NO consumption with physiological hypoxia. There is clear evidence that the b5R/b5 reductase system has an important role in supporting this NOD function; however, other enzymes and cellular reductants may also have a role in this process. Cygb has also been shown to be expressed at micromolar levels in the vascular smooth muscle wall, but is absent from the endothelium. Cygb has an important role as a key O2-dependent sensor and regulator of NO levels. In the normal well-perfused vessel, it metabolizes excess NO by dioxygenation; however, with tissue hypoxia and low arteriolar O2 levels, this NO degradation is greatly decreased, while with more severe hypoxia, as occurs in organ ischemia, Cygb can generate NO from NO2−, and this NO can bind to sGC in the smooth muscle to induce vessel relaxation. Hence, Cygb is an O2-dependent modulator of NO homeostasis and vascular tone that can serve to regulate tissue perfusion in normal physiology and disease. Recent studies have demonstrated an important role of Cygb as a major regulator of NO bioavailability and decay in the vascular smooth muscle of both resistance and conduit vessels as well as in vivo blood pressure and tissue perfusion. Modulation of Cygb expression can lower blood pressure and reverse HTN. Thus, there remains a critical need to further characterize the factors and processes that modulate and control Cygb-mediated NOD function, and to develop pharmacological or other approaches to modulate Cygb function and expression.
Abbreviations Used
- aSMC
aortic smooth muscle cell
- AHSP
α-hemoglobin stabilizing protein
- Ang-II
angiotensin-II
- AO
aldehyde oxidase
- Asc
ascorbate
- b5R/b5
NADH/cytochrome b5 reductase/cytochrome b5 reducing system
- B5-system
NADH/cytochrome b5 reductase/cytochrome b5 reducing system
- BH4
tetrahydrobiopterin
- cGMP
cyclic guanosine monophosphate
- CPR
cytochrome P450 reductase
- Cygb
cytoglobin
- Cygb−/−
cytoglobin gene knockout
- Cygb-SC
cytoglobin with cysteines blocked by thioether bonds
- Cygb-SH
cytoglobin with cysteines as free sulfhydryls
- Cygb-SS
cytoglobin with an intramolecular disulfide bond
- EPR
electron paramagnetic resonance
- eNOS/NOS3
endothelial nitric oxide synthase
- Hb
hemoglobin
- HSC
hepatic stellate cell
- HTN
hypertension
- iNOS/NOS2
inducible nitric oxide synthase
- MABP
mean arterial pressure
- Mb
myoglobin
- mSMC
mesenteric smooth muscle cell
- Ngb
neuroglobin
- nNOS/NOS1
neuronal nitric oxide synthase
- NO
nitric oxide
- NO3−
nitrate
- NOD
nitric oxide dioxygenase/deoxygenation
- NOS
nitric oxide synthase
- O2•−
superoxide free radical
- ONOO−
peroxynitrite
- SEM
standard error of the mean
- sGC
soluble guanylate cyclase
- SMC
smooth muscle cell
- STEP
stellate cell activation-associated protein
- SVR
systemic vascular resistance
- VSMC
vascular smooth muscle cell
- WT
wild type
- XO
xanthine oxidase
Funding Information
This work was supported by NIH/NHLBI grants R01HL131941 and R01HL135648.
References
- 1. Alderton WK, Cooper CE, and Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 357: 593–615, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alvarez RA, Miller MP, Hahn SA, Galley JC, Bauer E, Bachman T, Hu J, Sembrat J, Goncharov D, Mora AL, Rojas M, Goncharova E, and Straub AC. Targeting pulmonary endothelial hemoglobin alpha improves nitric oxide signaling and reverses pulmonary artery endothelial dysfunction. Am J Respir Cell Mol Biol 57: 733–744, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alzawahra WF, Talukder MA, Liu X, Samouilov A, and Zweier JL. Heme proteins mediate the conversion of nitrite to nitric oxide in the vascular wall. Am J Physiol Heart Circ Physiol 295: H499–H508, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amdahl MB, Sparacino-Watkins CE, Corti P, Gladwin MT, and Tejero J. Efficient reduction of vertebrate cytoglobins by the cytochrome b5/cytochrome b5 reductase/NADH system. Biochemistry 56: 3993–4004, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aranda R, Cai H, Worley CE, Levin EJ, Li R, Olson JS, Phillips GN Jr., and Richards MP. Structural analysis of fish versus mammalian hemoglobins: effect of the heme pocket environment on autooxidation and hemin loss. Proteins 75: 217–230, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azarov I, Liu C, Reynolds H, Tsekouras Z, Lee JS, Gladwin MT, and Kim-Shapiro DB. Mechanisms of slower nitric oxide uptake by red blood cells and other hemoglobin-containing vesicles. J Biol Chem 286: 33567–33579, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beckerson P, Reeder BJ, and Wilson MT. Coupling of disulfide bond and distal histidine dissociation in human ferrous cytoglobin regulates ligand binding. FEBS Lett 589: 507–512, 2015 [DOI] [PubMed] [Google Scholar]
- 8. Beckerson P, Wilson MT, Svistunenko DA, and Reeder BJ. Cytoglobin ligand binding regulated by changing haem-co-ordination in response to intramolecular disulfide bond formation and lipid interaction. Biochem J 465: 127–137, 2015 [DOI] [PubMed] [Google Scholar]
- 9. Beckman JS, Beckman TW, Chen J, Marshall PA, and Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 87: 1620–1624, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG, and Levine M. Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J Biol Chem 265: 2584–2587, 1990 [PubMed] [Google Scholar]
- 11. Birukou I, Schweers RL, and Olson JS. Distal histidine stabilizes bound O2 and acts as a gate for ligand entry in both subunits of adult human hemoglobin. J Biol Chem 285: 8840–8854, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brantley RE, Smerdon SJ, Wilkinson AJ, Singleton EW, and Olson JS. The mechanism of autooxidation of myoglobin. J Biol Chem 268: 6995–7010, 1993 [PubMed] [Google Scholar]
- 13. Brown GC and Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 356: 295–298, 1994 [DOI] [PubMed] [Google Scholar]
- 14. Burmester T, Ebner B, Weich B, and Hankeln T. Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Mol Biol Evol 19: 416–421, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Butcher JT, Johnson T, Beers J, Columbus L, and Isakson BE. Hemoglobin alpha in the blood vessel wall. Free Radic Biol Med 73: 136–142, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cable RG. Hemoglobin determination in blood donors. Transfus Med Rev 9: 131–144, 1995 [DOI] [PubMed] [Google Scholar]
- 17. Caccia S, Denisov I, and Perrella M. The kinetics of the reaction between NO and O2 as studied by a novel approach. Biophys Chem 76: 63–72, 1999 [DOI] [PubMed] [Google Scholar]
- 18. Carreras MC and Poderoso JJ. Mitochondrial nitric oxide in the signaling of cell integrated responses. Am J Physiol Cell Physiol 292: C1569–C1580, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, and Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen JY, Ye ZX, Wang XF, Chang J, Yang MW, Zhong HH, Hong FF, and Yang SL. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed Pharmacother 97: 423–428, 2018 [DOI] [PubMed] [Google Scholar]
- 21. Corti P, Ieraci M, and Tejero J. Characterization of zebrafish neuroglobin and cytoglobins 1 and 2: zebrafish cytoglobins provide insights into the transition from six-coordinate to five-coordinate globins. Nitric Oxide 53: 22–34, 2016 [DOI] [PubMed] [Google Scholar]
- 22. DeMartino AW, Kim-Shapiro DB, Patel RP, and Gladwin MT. Nitrite and nitrate chemical biology and signalling. Br J Pharmacol 176: 228–245, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doyle MP and Hoekstra JW. Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J Inorg Biochem 14: 351–358, 1981 [DOI] [PubMed] [Google Scholar]
- 24. Druhan LJ, Forbes SP, Pope AJ, Chen CA, Zweier JL, and Cardounel AJ. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry 47: 7256–7263, 2008 [DOI] [PubMed] [Google Scholar]
- 25. Eich RF, Li TS, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, and Olson JS. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 35: 6976–6983, 1996 [DOI] [PubMed] [Google Scholar]
- 26. Erusalimsky JD and Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol 27: 2524–2531, 2007 [DOI] [PubMed] [Google Scholar]
- 27. Fasold H, Riedl G, and Jaisle F. Evidence for an absence of myoglobin from human smooth muscle. Eur J Biochem 15: 122–126, 1970 [DOI] [PubMed] [Google Scholar]
- 28. Flogel U, Merx MW, Godecke A, Decking UK, and Schrader J. Myoglobin: a scavenger of bioactive NO. Proc Natl Acad Sci U S A 98: 735–740, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forfia PR, Hintze TH, Wolin MS, and Kaley G. Role of nitric oxide in the control of mitochondrial function. Adv Exp Med Biol 471: 381–388, 1999 [DOI] [PubMed] [Google Scholar]
- 30. Fujita Y, Koinuma S, De Velasco MA, Bolz J, Togashi Y, Terashima M, Hayashi H, Matsuo T, and Nishio K. Melanoma transition is frequently accompanied by a loss of cytoglobin expression in melanocytes: a novel expression site of cytoglobin. PLoS One 9: e94772, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Furchgott RF. Introduction to EDRF research. J Cardiovasc Pharmacol 22(Suppl 7): S1–S2, 1993 [PubMed] [Google Scholar]
- 32. Furchgott RF and Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980 [DOI] [PubMed] [Google Scholar]
- 33. Gao F, Lucke-Wold BP, Li X, Logsdon AF, Xu LC, Xu S, LaPenna KB, Wang H, Talukder MAH, Siedlecki CA, Huber JD, Rosen CL, and He P. Reduction of endothelial nitric oxide increases the adhesiveness of constitutive endothelial membrane ICAM-1 through Src-mediated phosphorylation. Front Physiol 8: 1124, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gardner AM, Cook MR, and Gardner PR. Nitric-oxide dioxygenase function of human cytoglobin with cellular reductants and in rat hepatocytes. J Biol Chem 285: 23850–23857, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gardner PR, Gardner AM, Brashear WT, Suzuki T, Hvitved AN, Setchell KD, and Olson JS. Hemoglobins dioxygenate nitric oxide with high fidelity. J Inorg Biochem 100: 542–550, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Gardner PR, Martin LA, Hall D, and Gardner AM. Dioxygen-dependent metabolism of nitric oxide in mammalian cells. Free Radic Biol Med 31: 191–204, 2001 [DOI] [PubMed] [Google Scholar]
- 37. Gkaliagkousi E, Ritter J, and Ferro A. Platelet-derived nitric oxide signaling and regulation. Circ Res 101: 654–662, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Halligan KE, Jourd'heuil FL, and Jourd'heuil D. Cytoglobin is expressed in the vasculature and regulates cell respiration and proliferation via nitric oxide dioxygenation. J Biol Chem 284: 8539–8547, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamdane D, Kiger L, Dewilde S, Green BN, Pesce A, Uzan J, Burmester T, Hankeln T, Bolognesi M, Moens L, and Marden MC. The redox state of the cell regulates the ligand binding affinity of human neuroglobin and cytoglobin. J Biol Chem 278: 51713–51721, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Hauser B, Matejovic M, and Radermacher P. Nitric oxide, leukocytes and microvascular permeability: causality or bystanders? Crit Care 12: 104, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heberlein KR, Straub AC, and Isakson BE. The myoendothelial junction: breaking through the matrix? Microcirculation 16: 307–322, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, Steinhoff HJ, Goedecke A, Schrader J, Gladwin MT, Kelm M, and Rassaf T. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci U S A 105: 10256–10261, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herold S, Exner M, and Nauser T. Kinetic and mechanistic studies of the NO center dot-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry 40: 3385–3395, 2001 [DOI] [PubMed] [Google Scholar]
- 44. Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, and Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 115: 2099–2107, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ignarro LJ. Nitric oxide is not just blowing in the wind. Br J Pharmacol 176: 131–134, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ilangovan G, Osinbowale S, Bratasz A, Bonar M, Cardounel AJ, Zweier JL, and Kuppusamy P. Heat shock regulates the respiration of cardiac H9c2 cells through upregulation of nitric oxide synthase. Am J Physiol Cell Physiol 287: C1472–C1481, 2004 [DOI] [PubMed] [Google Scholar]
- 47. Jayaraman T, Tejero J, Chen BB, Blood AB, Frizzell S, Shapiro C, Tiso M, Hood BL, Wang X, Zhao X, and Conrads TP, Mallampalli RK, Gladwin MT. 14-3-3 Binding and phosphorylation of neuroglobin during hypoxia modulate six-to-five heme pocket coordination and rate of nitrite reduction to nitric oxide. J Biol Chem 286: 42679–42689, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jeremy JY, Rowe D, Emsley AM, and Newby AC. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovasc Res 43: 580–594, 1999 [DOI] [PubMed] [Google Scholar]
- 49. Joshi MS, Ponthier JL, and Lancaster JR Jr.. Cellular antioxidant and pro-oxidant actions of nitric oxide. Free Radic Biol Med 27: 1357–1366, 1999 [DOI] [PubMed] [Google Scholar]
- 50. Jourd'heuil FL, Xu H, Reilly T, McKellar K, El Alaoui C, Steppich J, Liu YF, Zhao W, Ginnan R, Conti D, Lopez-Soler R, Asif A, Keller RK, Schwarz JJ, Thanh Thuy LT, Kawada N, Long X, Singer HA, and Jourd'heuil D. The hemoglobin homolog cytoglobin in smooth muscle inhibits apoptosis and regulates vascular remodeling. Arterioscler Thromb Vasc Biol 37: 1944–1955, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kagen LJ and Gurevich R. Precipitin reactions of anti-human myoglobin serum with several human and animal muscle extracts. Immunology 12: 667–673, 1967 [PMC free article] [PubMed] [Google Scholar]
- 52. Kawada N, Kristensen DB, Asahina K, Nakatani K, Minamiyama Y, Seki S, and Yoshizato K. Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. J Biol Chem 276: 25318–25323, 2001 [DOI] [PubMed] [Google Scholar]
- 53. Kuhn V, Diederich L, Keller TCS, Kramer CM, Luckstadt W, Panknin C, Suvorava T, Isakson BE, Kelm M, and Cortese-Krott MM. Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism, anemia. Antioxid Redox Signal 26: 718–742, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuppusamy P, Shankar RA, Roubaud VM, and Zweier JL. Whole body detection and imaging of nitric oxide generation in mice following cardiopulmonary arrest: detection of intrinsic nitrosoheme complexes. Magn Reson Med 45: 700–707, 2001 [DOI] [PubMed] [Google Scholar]
- 55. Lechauve C, Butcher JT, Freiwan A, Biwer LA, Keith JM, Good ME, Ackerman H, Tillman HS, Kiger L, Isakson BE, and Weiss MJ. Endothelial cell alpha-globin and its molecular chaperone alpha-hemoglobin-stabilizing protein regulate arteriolar contractility. J Clin Invest 128: 5073–5082, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lechauve C, Chauvierre C, Dewilde S, Moens L, Green BN, Marden MC, Celier C, and Kiger L. Cytoglobin conformations and disulfide bond formation. FEBS J 277: 2696–2704, 2010 [DOI] [PubMed] [Google Scholar]
- 57. Li H, Cui H, Kundu TK, Alzawahra W, and Zweier JL. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem 283: 17855–17863, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li H, Hemann C, Abdelghany TM, El-Mahdy MA, and Zweier JL. Characterization of the mechanism and magnitude of cytoglobin-mediated nitrite reduction and nitric oxide generation under anaerobic conditions. J Biol Chem 287: 36623–36633, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li Q, Youn JY, and Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens 33: 1128–1136, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu X, Cheng C, Zorko N, Cronin S, Chen YR, and Zweier JL. Biphasic modulation of vascular nitric oxide catabolism by oxygen. Am J Physiol Heart Circ Physiol 287: H2421–H2426, 2004 [DOI] [PubMed] [Google Scholar]
- 61. Liu X, El-Mahdy MA, Boslett J, Varadharaj S, Hemann C, Abdelghany TM, Ismail RS, Little SC, Zhou D, Thuy LT, Kawada N, and Zweier JL. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nat Commun 8: 14807, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu X, El-Sherbiny GA, Collard E, Huang X, Follmer D, El-Mahdy M, and Zweier JL. Application of carbon fiber composite minielectrodes for measurement of kinetic constants of nitric oxide decay in solution. Nitric Oxide 23: 311–318, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu X, Follmer D, Zweier JR, Huang X, Hemann C, Liu K, Druhan LJ, and Zweier JL. Characterization of the function of cytoglobin as an oxygen-dependent regulator of nitric oxide concentration. Biochemistry 51: 5072–5082, 2012 [DOI] [PubMed] [Google Scholar]
- 64. Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, and Lancaster JR Jr.. Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem 273: 18709–18713, 1998 [DOI] [PubMed] [Google Scholar]
- 65. Liu X, Samouilov A, Lancaster JR Jr., and Zweier JL. Nitric oxide uptake by erythrocytes is primarily limited by extracellular diffusion not membrane resistance. J Biol Chem 277: 26194–26199, 2002 [DOI] [PubMed] [Google Scholar]
- 66. Liu X, Srinivasan P, Collard E, Grajdeanu P, Lok K, Boyle SE, Friedman A, and Zweier JL. Oxygen regulates the effective diffusion distance of nitric oxide in the aortic wall. Free Radic Biol Med 48: 554–559, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu X, Srinivasan P, Collard E, Grajdeanu P, Zweier JL, and Friedman A. Nitric oxide diffusion rate is reduced in the aortic wall. Biophys J 94: 1880–1889, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu X, Tong J, Zweier JR, Follmer D, Hemann C, Ismail RS, and Zweier JL. Differences in oxygen-dependent nitric oxide metabolism by cytoglobin and myoglobin account for their differing functional roles. FEBS J 280: 3621–3631, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu XP, Srinivasant P, Collard E, Grajdeanu P, Zweier JL, and Friedmant A. Nitric oxide diffusion rate is reduced in the aortic wall. Biophys J 94: 1880–1889, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Masha A, Dinatale S, Allasia S, and Martina V. Role of the decreased nitric oxide bioavailability in the vascular complications of diabetes mellitus. Curr Pharm Biotechnol 12: 1354–1363, 2011 [DOI] [PubMed] [Google Scholar]
- 71. McRonald FE, Risk JM, and Hodges NJ. Protection from intracellular oxidative stress by cytoglobin in normal and cancerous oesophageal cells. PLoS One 7: e30587, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moncada S and Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol 3: 214–220, 2002 [DOI] [PubMed] [Google Scholar]
- 73. Moncada S, Palmer RM, and Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43: 109–142, 1991 [PubMed] [Google Scholar]
- 74. Moncada S, Radomski MW, and Palmer RM. Endothelium-derived relaxing factor. Identification as nitric oxide and role in the control of vascular tone and platelet function. Biochem Pharmacol 37: 2495–2501, 1988 [DOI] [PubMed] [Google Scholar]
- 75. Napoli C, Paolisso G, Casamassimi A, Al-Omran M, Barbieri M, Sommese L, Infante T, and Ignarro LJ. Effects of nitric oxide on cell proliferation: novel insights. J Am Coll Cardiol 62: 89–95, 2013 [DOI] [PubMed] [Google Scholar]
- 76. Oleksiewicz U, Liloglou T, Tasopoulou KM, Daskoulidou N, Bryan J, Gosney JR, Field JK, and Xinarianos G. Cytoglobin has bimodal: tumour suppressor and oncogene functions in lung cancer cell lines. Hum Mol Genet 22: 3207–3217, 2013 [DOI] [PubMed] [Google Scholar]
- 77. Palmer RM, Ferrige AG, and Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526, 1987 [DOI] [PubMed] [Google Scholar]
- 78. Papapetropoulos A, Rudic RD, and Sessa WC. Molecular control of nitric oxide synthases in the cardiovascular system. Cardiovasc Res 43: 509–520, 1999 [DOI] [PubMed] [Google Scholar]
- 79. Radi R. Peroxynitrite, a stealthy biological oxidant. J Biol Chem 288: 26464–26472, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc Natl Acad Sci U S A 115: 5839–5848, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rassaf T, Flogel U, Drexhage C, Hendgen-Cotta U, Kelm M, and Schrader J. Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ Res 100: 1749–1754, 2007 [DOI] [PubMed] [Google Scholar]
- 82. Samouilov A, Kuppusamy P, and Zweier JL. Evaluation of the magnitude and rate of nitric oxide production from nitrite in biological systems. Arch Biochem Biophys 357: 1–7, 1998 [DOI] [PubMed] [Google Scholar]
- 83. Smagghe BJ, Trent JT 3rd, and Hargrove MS. NO dioxygenase activity in hemoglobins is ubiquitous in vitro, but limited by reduction in vivo. PLoS One 3: e2039, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Springer BA, Sligar SG, Olson JS, and Phillips GN. Mechanisms of ligand recognition in myoglobin. Chem Rev 94: 699–714, 1994 [Google Scholar]
- 85. Straub AC, Lohman AW, Billaud M, Johnstone SR, Dwyer ST, Lee MY, Bortz PS, Best AK, Columbus L, Gaston B, and Isakson BE. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature 491: 473–477, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Straub AC, Zeigler AC, and Isakson BE. The myoendothelial junction: connections that deliver the message. Physiology (Bethesda) 29: 242–249, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Stuehr DJ, Santolini J, Wang ZQ, Wei CC, and Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem 279: 36167–36170, 2004 [DOI] [PubMed] [Google Scholar]
- 88. Tejero J, Sparacino-Watkins CE, Ragireddy V, Frizzell S, and Gladwin MT. Exploring the mechanisms of the reductase activity of neuroglobin by site-directed mutagenesis of the heme distal pocket. Biochemistry 54: 722–733, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Thi Thanh Hai N, Thuy LTT, Shiota A, Kadono C, Daikoku A, Hoang DV, Dat NQ, Sato-Matsubara M, Yoshizato K, and Kawada N. Selective overexpression of cytoglobin in stellate cells attenuates thioacetamide-induced liver fibrosis in mice. Sci Rep 8: 17860, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tiravanti E, Samouilov A, and Zweier JL. Nitrosyl-heme complexes are formed in the ischemic heart: evidence of nitrite-derived nitric oxide formation, storage, and signaling in post-ischemic tissues. J Biol Chem 279: 11065–11073, 2004 [DOI] [PubMed] [Google Scholar]
- 91. Titheradge MA. Nitric oxide in septic shock. Biochim Biophys Acta 1411: 437–455, 1999 [DOI] [PubMed] [Google Scholar]
- 92. Trent JT 3rd and Hargrove MS. A ubiquitously expressed human hexacoordinate hemoglobin. J Biol Chem 277: 19538–19545, 2002 [DOI] [PubMed] [Google Scholar]
- 93. Tsujino H, Yamashita T, Nose A, Kukino K, Sawai H, Shiro Y, and Uno T. Disulfide bonds regulate binding of exogenous ligand to human cytoglobin. J Inorg Biochem 135: 20–27, 2014 [DOI] [PubMed] [Google Scholar]
- 94. Vanderkooi JM, Wright WW, and Erecinska M. Nitric oxide diffusion coefficients in solutions, proteins and membranes determined by phosphorescence. Biochim Biophys Acta 1207: 249–254, 1994 [DOI] [PubMed] [Google Scholar]
- 95. Vaughn MW, Huang KT, Kuo L, and Liao JC. Erythrocytes possess an intrinsic barrier to nitric oxide consumption. J Biol Chem 275: 2342–2348, 2000 [DOI] [PubMed] [Google Scholar]
- 96. Vincent JL, Zhang H, Szabo C, and Preiser JC. Effects of nitric oxide in septic shock. Am J Respir Crit Care Med 161: 1781–1785, 2000 [DOI] [PubMed] [Google Scholar]
- 97. Wang P and Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem 271: 29223–29230, 1996 [DOI] [PubMed] [Google Scholar]
- 98. Wink DA, Miranda KM, Espey MG, Pluta RM, Hewett SJ, Colton C, Vitek M, Feelisch M, and Grisham MB. Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal 3: 203–213, 2001 [DOI] [PubMed] [Google Scholar]
- 99. Xia Y, Tsai AL, Berka V, and Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998 [DOI] [PubMed] [Google Scholar]
- 100. Yoshizato K, Thuy le TT, Shiota G, and Kawada N. Discovery of cytoglobin and its roles in physiology and pathology of hepatic stellate cells. Proc Jpn Acad Ser B Phys Biol Sci 92: 77–97, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhou D, Hemann C, Boslett J, Luo A, Zweier JL, and Liu X. Oxygen binding and nitric oxide dioxygenase activity of cytoglobin are altered to different extents by cysteine modification. FEBS Open Bio 7: 845–853, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zweier JL. Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J Biol Chem 263: 1353–1357, 1988 [PubMed] [Google Scholar]
- 103. Zweier JL, Li H, Samouilov A, and Liu X. Mechanisms of nitrite reduction to nitric oxide in the heart and vessel wall. Nitric Oxide 22: 83–90, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zweier JL, Samouilov A, and Kuppusamy P. Non-enzymatic nitric oxide synthesis in biological systems. Biochim Biophys Acta 1411: 250–262, 1999 [DOI] [PubMed] [Google Scholar]
- 105. Zweier JL, Wang P, Samouilov A, and Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med 1: 804–809, 1995 [DOI] [PubMed] [Google Scholar]