

Abstract
Background
Breast carcinoma has become a nonnegligible public health problem in China with its increasing incidence and mortality in woman. As a early event regulating tumorigenesis and development, DNA methylation became one of the focuses of current carcinoma researches on potential diagnostic and therapeutic targets.
Methods
In this study, we comprehensively analyzed the gene expression data and DNA methylation data of breast carcinoma and adjacent normal tissues samples in the Gene Expression Omnibus database. Influences of tumor stage, adjuvant therapy, hormone therapy, and chemotherapy on CpG methylation level were explored by linear regression analysis. Correlations between methylation and gene expression levels were determined by spearman rank correlation analysis. Log‐rank test was applied for determining significance of associations between CpG sites methylation level and breast cancer patients' Kaplan–Meier survival.
Results
A total of 229 CpG sites were found to be significantly associated with tumor stage or treatment, and eight of which were potential markers that affect the survival of breast carcinoma and negatively correlated with their genes' expression levels.
Conclusions
We reported eight CpG sites as potential breast cancer prognosis signatures through comprehensively analyzed gene expression and DNA methylation datasets, and excluding influences of tumor stage and treatment. This should be helpful for breast cancer early diagnosis and treatment.
Keywords: breast carcinoma, CpG island, DNA methylation, prognosis
Eight methylated sites that can influence breast carcinoma survival independently of clinical factors such as clinical grade and treatment. Two potential gene markers (ESPL1 and PARPBP) that might affect breast carcinoma survival, which supplemented the existing system of DNA methylation in the regulation of breast carcinoma.

1. INTRODUCTION
Breast carcinoma (BC) is one of the most common reproductive carcinomas. In China, BC is the leading cause of carcinoma death in women younger than 45 years, and also is expected to account for 15% of all new carcinomas in women per year (Chen et al., 2016) In recent decades, the promotion of mammographic screening in physical examination has contributed to improve the early diagnosis of BC (Howell et al., 2014). However, the widespread application of these screening and diagnostic interventions requires significant resources (Eccles et al., 2013). New biomarker development is the basis of BC detection and diagnosis, and personalized treatment (Wagner & Srivastava, 2012). Thus, integrative analysis on high‐throughput data for searching biomarkers and potential therapeutic targets has been extensively studied in recent years (Sethi, Ali, Philip, & Sarkar, 2013). And the good news is that, several promising drugs that target epigenetic alterations are currently available for clinical investigation in solid tumors, including BC (Connolly & Stearns, 2012).
Epigenetic alterations have recently emerged as a common hallmark of multiple tumors (Abdel‐Hafiz & Horwitz, 2015; Chakravarthi, Nepal, & Varambally, 2016; Okugawa, 2015). The occurrence of tumor cells is able to be activated by epigenetic alterations. Furthermore, their cellular behaviors, including proliferation, invasion, metastasis and even escape from chemotherapy and host immune surveillance, are also regulated by epigenetic processes (Klymenko & Nephew, 2018; Marks, Olson, & Fernandez‐Zapico, 2016). The epigenetics refers to changes in gene expression without changes in the DNA, including DNA methylation, histone posttranslational modifications, recruitment of chromatin remodeling factors, and expressions of micro and long noncoding RNA (Baylin & Jones, 2011). Especially, the causal relationships between gene expressions and DNA methylation have received considerable attention, and the epigenetic modification of different gene regions may consequently lead to distinct biological and clinical implications (Dafni, Anna, & Francesc, 2018). In BC, DNA methylation has been proved to be associated with clinicopathological features, such as tumor stage, histological and grade (Fleischer et al., 2017; Holm et al., 2016). Moreover, it can also influence the progression and prognosis of BC patients (Rauscher et al., 2015).
The potential is great for DNA methylation markers to improve carcinoma outcomes across the prevention continuum (Terry, Mcdonald, Wu, Eng, & Santella, 2016). To screen the key methylated sites related to BC prognosis, in this study, we comprehensively analyzed the gene expressions and DNA methylation in BC tumor tissues, and explored the mechanisms and biological processes affecting the occurrence and development. We found that eight methylated sites that can influence breast carcinoma survival independently of clinical factors such as clinical grade and treatment. Finally, we identified two potential gene markers (ESPL1 andPARPBP) that might affect breast carcinoma survival, which supplemented the existing system of DNA methylation in the regulation of breast carcinoma.
2. MATERIALS AND METHODS
2.1. Data source
Datasets on DNA methylation and mRNA expression profiles of BC were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The datasets of DNA methylation (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37754; Platform: Illumina HumanMethylation450 BeadChip [HumanMethylation450 15017482 v.1.1]) and mRNA expression profile (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37751; Platform: Affymetrix Human Gene 1.0 ST Array [HuGene‐1_0‐st]) were selected for further analysis. From http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37754 dataset, 72 samples were obtained, including 10 normal (noncarcinomaous) tissues and 62 tumor (breast carcinoma) tissues. From http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37751 dataset, 108 samples were obtained, including 47 normal tissues and 61 tumor tissues.
2.2. Preprocessing of DNA methylation and mRNA expression data
DNA methylation data were processed via the IMA R Bioconductor package. The following criteria were used: Remove the methylated site if its detection p value (DP) > .05 in >75% samples and the methylated sites on sex chromosomes; remove the sample if >75% methylated sites in it had DP > 10–5. Meanwhile, the mRNA expression data were normalized with Robust Multi‐Chip Averaging with the Affymetrix Expression Console software.
2.3. Screening of differentially methylated genes
The differentially methylated sites (DMGs) between tumors and normal tissues were identified by limma method in IMA package with the thresholds of false discovery rate ≤0.05 (Wang et al., 2012). The Benjamini–Hochberg method was used for statistical corrections.
The correlation of the methylation level of the DMGs with the stage and treatment of the patients was analyzed using limma R Bioconductor package (Ritchie et al., 2015). The correlation was defined as follows:
| (1) |
where a is the methylation level at baseline, and X 1, X 2, X 3, X4 represent for tumor‐node‐metastasis (TNM) carcinoma staging, adjuvant therapy, hormone therapy, and chemotherapy respectively. p ≤ .05 was considered to be significantly correlated.
2.4. Survival analysis
The significantly differentially methylated genes that correlated with prognosis were screened using survival R package. The survival curve analysis was performed by the Kaplan–Meier method and comparison between subgroups were examined by the log‐rank test. The influence of the methylation level of the DMGs on the overall survival of breast carcinoma patients was analyzed using the Cox model.
2.5. Correlation between methylation and mRNA expression
Aberrant DNA methylation is usually closely associated with altered gene expression. Thus, we selected the DMGs that significantly associated with carcinoma staging, treatment and prognosis, and used the spearman rank test to calculate the correlation between their methylation and gene expression levels with the cut‐off of the absolute value of the correlation coefficient >0.75.
3. RESULTS
3.1. Identification of DMGs in BC
The distribution of the mRNA expression profiles of different samples after normalization was suitable for subsequent analysis (Figure 1a). After the preprocessing of DNA methylation, 455,968 methylated sites were retained from the 485,577 methylated sites with a pass rate of 93.9%, and all the samples were retained. The methylation level distribution of remaining methylated sites in tumor and normal tissues after removing the low‐quality methylation site (Figure 1b) indicated that the overall methylation level in tumor was higher than that in normal tissues. After screened by limma method in IMA package, 6,043 significantly differentially methylated genes were identified (Table S1). The heatmap of significantly differentially methylated genes in normal and tumor tissues (Figure 1c), indicating that the gene expression patterns of most tumor samples are consistent.
Figure 1.
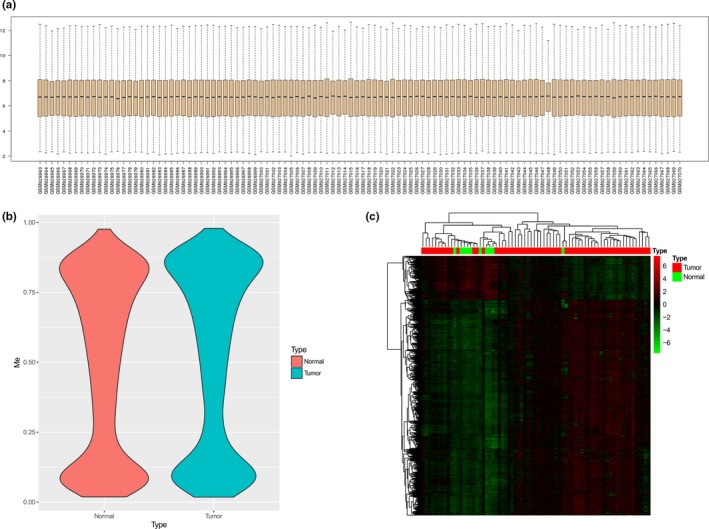
Identification of differentially methylated genes in BC. (a) The distribution of the mRNA expression profiles of normal and tumor tissues after normalization. (b) The methylation level distribution of differentially methylated sites of normal and tumor tissues after preprocessing. (c) Heat map of significantly differentially methylated genes in normal and tumor tissues
3.2. DMGs associated with tumor staging and treatment
As a result, 229 sites were identified out of the 6,043 DMGs with a standard of p ≤ .05 (Table S2). Among the DMGs, 191 were significantly associated with tumor TNM stage, 10 were associated with adjuvant therapy, 13 were associated with hormonal therapy, and 24 were associated with chemotherapy. There were 83.4% of these 229 DMGs associated with tumor stages (Figure 2). Furthermore, the expression pattern of these four DMGs groups were different in their related subgroups. Based on these 229 DMGs, we used principal component analysis to analyze the differences between tumors of different stages, and showed the first three principal components in a three‐dimensional scatter plot (Figure 3).
Figure 2.
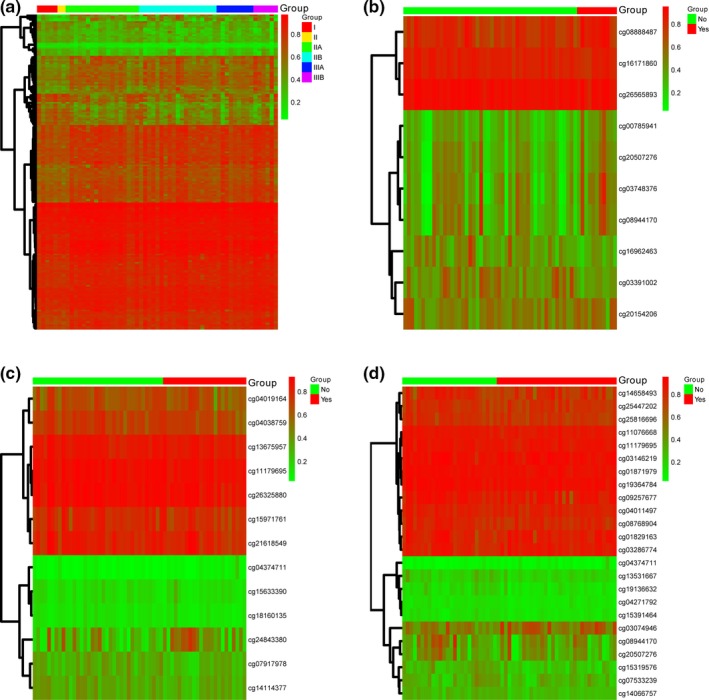
Heat map of differentially methylated genes related to tumor staging and treatments. (a) Methylation level of differentially methylated sites (DMGs) in tissues from patients at different stages. (b) Methylation level of DMGs in tissues from patients with and without adjuvant therapy. (c) Methylation level of DMGs in tissues from patients with and without hormone therapy. (d) Methylation level of DMGs in tissues from patients with and without chemotherapy
Figure 3.
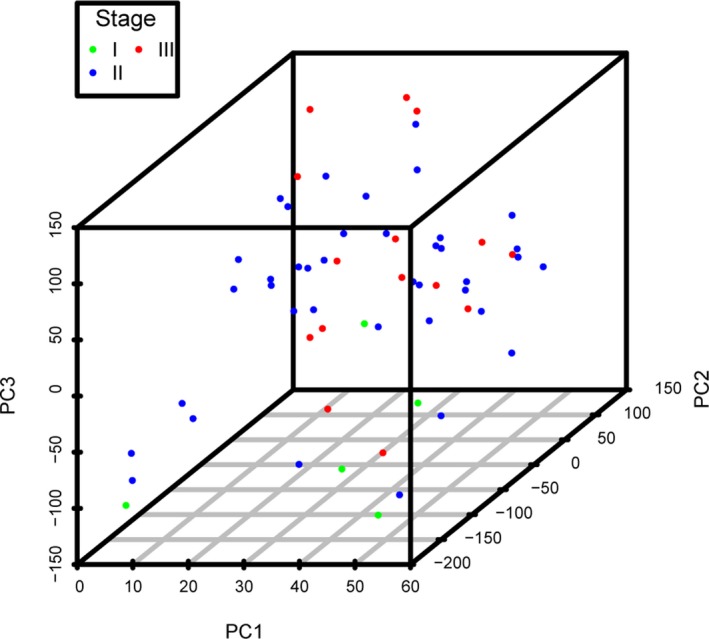
3D scatter plot of principal component analysis
3.3. Analysis of prognosis‐related DNA methylated sites in BC
We performed a survival analysis of the 229 methylated sites significantly associated with tumor staging and treatment. For each methylated site, we divided the tumor samples into two groups using the median of the methylation levels of these sites. The survival curve was plotted using the Kaplan–Meier method. The log‐rank test was used in the judgment of difference in the survival curves. p < .05 was set as the standard. The results showed that the survival curves of 55 methylated sites were significantly different (Table S3). We then performed a cox proportional regression to analyze of the effects of these methylated sites, the effects and relative risks of race, age, tumor stages, and treatments (adjuvant therapy, hormonal therapy and chemotherapy) on survival times (Table S4). The results indicated that among these factors, age, tumor stages, and adjuvant therapy were significantly associated with survival time. Of the 229 DMGs, 87 were significantly associated with carcinoma survival time. To find the methylated sites that associated with survival time independent of age, tumor stages and adjuvant therapy, we analyzed the methylation levels of these 229 DMGs and the factors above in a multivariate cox proportional regression risk model. The results showed that after correcting with these factors, 13 DMGs were significantly associated with survival time (Table S5). In addition, eight of the 13 DMGs were also significant in the single‐factor cox proportional regression, including cg04988216, cg00118989, cg01967564, cg21374754, cg26874872, cg04836851, cg26090534, and cg08783934. The effects of these eight DMGs on BC patients' survival were shown in Figure 4. The red survival curves were the sample with higher methylation level, and the blue survival curves were the sample with lower methylation levels.
Figure 4.
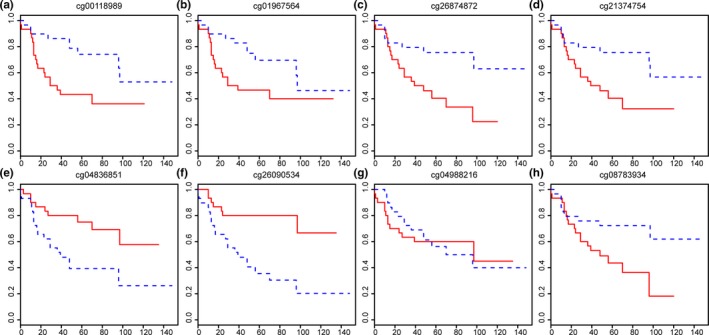
Survival analysis for the most significant differentially methylated sites. (a) Survival analysis stratified by cg00118989. (b) Survival analysis stratified by cg01967564. (c) Survival analysis stratified by cg26874872. (d) Survival analysis stratified by cg21374754. (e) Survival analysis stratified by cg04836851. (f) Survival analysis stratified by cg26090534. (g) Survival analysis stratified by cg04988216. (h) Survival analysis stratified by cg08783934
3.4. Correlation between DNA methylation and gene expression in BC
The level of methylation often affects the level of gene expression. We used the Spearman method to calculate the correlation between the methylation levels of the above 229 methylated sites and their corresponding gene expression levels. Using the absolute value of the correlation coefficient >0.75 as the threshold, a total of 19 methylated sites in the above 229 methylated sites were significantly associated with the expression levels of at least one of the corresponding genes (Table S6).
4. DISCUSSION
DNA hypermethylation is conventionally negatively associated with gene expression, and the DNA methylation in the promoter region of tumor suppressor genes appears to be a key event at early stages of carcinogenesis (Baylin & Jones, 2016). CpG islands are CpG‐rich areas of 200 bp to several kilobases in length, usually located near the promoters of highly expressed genes (Baylin, 2000). The aberrant CpG methylation has been proven to be a nearly universal feature of human carcinoma (Yang & Park, 2012). Damir Herman et al. had shown that CHST11 played an important role in the development of breast carcinoma, and its expression was regulated by DNA methylation (Herman et al., 2015). Studies had also shown that the promoter region of Runx3 gene was methylated, inhibited cell proliferation, apoptosis and differentiation, and promotes the formation of breast carcinoma (Lotem et al., 2017; Rossi, Bagalà, Inzani, Leoncini, & Schinzari, 2017). Liu H et al. revealed that hypermethylation of the RUNX3 gene promoter might play an important role in ER‐positive breast tumor progression (Liu et al., 2018). However, as BC is a heterogeneous disease, which includes several subtypes with different molecular and clinical features, the prognostic value of these aberrantly methylated biomarkers and the complex role of DNA methylation in distinct gene regions are still controversial topics (Győrffy et al., 2016).
In this study, we comprehensively analyzed the gene expressions and DNA methylation in BC tumor tissues, and explored the mechanisms and biological processes affecting the occurrence and development. Comparing with normal tissues, a total of 6,043 significantly differentially methylated genes were identified in BC tumor tissues. Of these, 229 DMGs were significantly associated with tumor staging (83.4%) and treatment (16.6%), and 19 DMGs in the these 229 DMGs were associated with the expression levels of at least one of the corresponding genes. The corresponding genes were CDCA8, KIF2C, ARF1, RGS5, CENPL, TMEM206, EPRS, DHTKD1, CEP55, COL17A1, STIP1, GRIA4, OR5P1P, TUBA1C, ESPL1, PARPBP, RHOJ, PLK1, HERC2P4, HLF, MYO15B, KRT14, L3MBTL4, 8,022,168, CALR, DDX49, MRPS12, SMYD1, C2orf40, TPX2, ADRM1, TP63, PATA18, CENPU, IRX1, CARMN, HIST1H3A, SAMD5, KIAA1456, MTDH, PRDX4, and NAA10.
Among them, the value of 13/42 genes in affecting the tumor cell behavior and BC prognosis has been clarified. CDCA8 is a putative oncogene that is upregulated in multiple types of carcinomas (Ci et al., 2018). In BC, the expression of CDCA8 correlates closely with FOXM1, which might be might be involved in BC progression (Jiao et al., 2015). ARF1 regulates the adhesion and invasion of MDA‐MB‐231 cells (Schlienger, Campbell, & Claing, 2014). EP RS is a critical regulator of cell proliferation and estrogen signaling in ER + BC (Katsyv, Wang, Song, Zhou, & Irie, 2016). CEP55 is a downstream target of FOXM1, which is involved in the proliferation of BC cells (Ye, Tao, Xueming, & Junming, 2016). The aberrant COL17A1 promoter methylation predicts the misexpression and increased invasion in BC (Thangavelu, Krenács, Dray, & Duijf, 2016). ESPL1 is a candidate oncogene of luminal B BC (Finetti et al., 2014). PLK1 is related to the growth and metastasis of Her2+ BC cells (Yao et al., 2012). L3MBTL4 is a potential tumor suppressor gene of chromosome arm 18p regulating the aggressive phenotype of BC (Addou‐Klouche et al., 2010). C2ORF40 suppresses BC cell proliferation and invasion through modulating expression of M phase cell cycle genes (Lu et al., 2013). TPX2 promotes migration and invasion of BC cells (Yang, Li, Shen, et al., 2015). TP63 is involved in the regulation by estrogen receptor‐α and ERK2 that controls BC proliferation and invasiveness properties (Wang et al., 2017). The activation of MTDH can promote chemoresistance and metastasis of poor‐prognosis BC (Hu et al., 2009). Moreover, nine of the other 29 genes have also been named in studies of a variety of carcinomas (Cabagnols, Cayuela, & Vainchenker, 2015; Ho et al., 2012; Jang, Park, Kim, Seong, & Kim, 2014; Jiang et al., 2011; Wang et al., 2010; Wang et al., 2017; Yang, Li, Shen, et al., 2015; Yang, Li, Niu, Li, & Bai, 2015; Zhao et al., 2018).
Breast carcinoma is a complex biological process caused by both hereditary and nonhereditary factors, such as patient age, family heredity, lifestyle habits, estrogen levels, growth factors, cytokines, kinases, and epigenetic regulation (Akram, Iqbal, Daniyal, & Khan, 2017). In this study, we found that age, tumor stage, and adjuvant therapy were significantly associated with the survival time of BC patients. Subsequently, we screened the DMGs that were able to influence survival time independently of these three factors. Eight DMGs showed significant associations, and only one of them (cg26090534) was shown to significantly affect gene expression. These DNA methylation modifications did not directly act on promoter region, but might act on enhancer element to regulate gene expression level to silence tumor suppressor genes. Even though DNA hypermethylation is conventionally negatively associated with gene expression, recently methylation has been demonstrated to positively correlate with gene expression (Fleischer et al., 2014). The genes with the highest positive and negative correlation with cg26090534 were PARPBP and ESPL1. PARPBP, PARP‐1 binding protein, is able to enhance poly (ADP‐ribose) polymerase‐1 (PARP‐1) activity. It has been reported that, PARP‐1 was highly expressed in NSCLC, reducing the effects of CBP in NSCLC. And PARP‐1 as oncogene was found to effect NSCLC cell migration through known oncogene in terms of gene interaction network (Chen, Li, Xu, Zhang, & Wang, 2017). Furthermore, PARP inhibition significantly decreased cell viability, migration, invasion, chromatin loop dimensions, and histone acetylation, and it could play a key role in the compartmentalization of chromatin and in the development of the more aggressive phenotype (Barboro et al., 2015). ESPL1 is located in the 12q13.13 chromosomal region, coding the separase protein (Zhang & Pati, 2017). The separase proteinan is an endopeptidase, which is activated at the onset of anaphase and cleaves the cohesin subunit RAD21, allowing the release of sister chromatid cohesion required for chromosomal disjunction (Xu et al., 2018).
The epigenetic modification of functionally different gene regions may consequently lead to distinct biological and clinical implications. Moreover, specific methylation patterns have been proven to be associated with different BC subtypes. More evidences showed that abnormal methylation of enhancer element, same as gene mutation and abnormal expression played an important role in the occurrence and development of tumor. Vecchione et al. reported that hypermethylation of LZTS1 gene was found in 75% (6/8) of gastric carcinoma cell lines, and the deletion or decrease of this gene expression was more attributed to the enhancer methylation mechanism (Vecchione et al., 2002). In addition, hypermethylation of enhancers was also found in oral squamous cell carcinoma and non‐hodgkin lymphoma (Nakagawa et al., 2006; Rahmatpanah et al., 2006). Thus, the abnormal methylation in enhancer region found in this study might be closely related to the occurrence of BC, and we further study the actual function of these genes in the follow‐up work.
5. CONCLUSION
In this study, we comprehensively analyzed the gene expression data and DNA methylation data of BC and adjacent normal tissues samples in the GEO database, and identified two potential methylated gene markers that may affect the survival of BC patients. This study has reference significance for exploring the molecular mechanism of DNA methylation in the regulation of BC occurrence and development, and would contribute to screening of key methylation markers related to BC prognosis.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
AUTHORS' CONTRIBUTIONS
Jia Li and Xinzheng Li conceived and designed the project, Jia Li acquired the data, analyzed and interpreted the data, and wrote the paper. Xinzheng Li approved the final version.
Supporting information
Li J, Li X. Comprehensive analysis of prognosis‐related methylated sites in breast carcinoma. Mol Genet Genomic Med. 2020;8:e1161 10.1002/mgg3.1161
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article.
REFERENCES
- Abdel‐Hafiz, H. A. , & Horwitz, K. B. (2015). Role of epigenetic modifications in luminal breast cancer. Epigenomics, 7(5), 847–862. 10.2217/epi.15.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addou‐Klouche, L. , Adélaïde, J. , Finetti, P. , Cervera, N. , Ferrari, A. , Bekhouche, I. , … Chaffanet, M. (2010). Loss, mutation and deregulation of L3MBTL4in breast cancers. Molecular Cancer, 9, 213 10.1186/1476-4598-9-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akram, M. , Iqbal, M. , Daniyal, M. , & Khan, A. U. (2017). Awareness and current knowledge of breast cancer. Biological Research, 50, 33 10.1186/s40659-017-0140-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboro, P. , Ferrari, N. , Capaia, M. , Petretto, A. , Salvi, S. , Boccardo, S. , & Balbi, C. (2015). Expression of nuclear matrix proteins binding matrix attachment regions in prostate cancer. PARP‐1: New player in tumor progression. International Journal of Cancer, 137, 1574–1586. 10.1002/ijc.29531 [DOI] [PubMed] [Google Scholar]
- Baylin, S. B. , & Herman, J. G. (2000). DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends in Genetics, 16, 168–174. 10.1016/S0168-9525(99)01971-X [DOI] [PubMed] [Google Scholar]
- Baylin, S. B. , & Jones, P. A. (2011). A decade of exploring the cancer epigenome – Biological and translational implications. Nature Reviews Cancer, 11, 726–734. 10.1038/nrc3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin, S. B. , & Jones, P. A. (2016). Epigenetic determinants of cancer. Cold Spring Harbor Perspectives in Biology, 8, a019505 10.1101/cshperspect.a019505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabagnols, X. , Cayuela, J. M. , & Vainchenker, W. (2015). A CALR mutation preceding BCR‐ABL1 in an atypical myeloproliferative neoplasm. New England Journal of Medicine, 372, 688–690. 10.1056/nejmc1413718 [DOI] [PubMed] [Google Scholar]
- Chakravarthi, B. V. S. K. , Nepal, S. , & Varambally, S. (2016). Genomic and epigenomic alterations in cancer. The American Journal of Pathology, 186, 1724–1735. 10.1016/j.ajpath.2016.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. , Li, Y. , Xu, H. , Zhang, C. , & Wang, B. (2017). An analysis of the gene interaction networks identifying the role of PARP1 in metastasis of non‐small cell lung cancer. Gene, 8, 87263–87275. 10.1016/j.gene.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Zheng, R. , Baade, P. D. , Zhang, S. , Zeng, H. , Bray, F. , … He, J. (2016). Cancer statistics in china, 2015. CA: A Cancer Journal for Clinicians, 66(2), 115–132. 10.3322/caac.21338 [DOI] [PubMed] [Google Scholar]
- Ci, C. , Tang, B. , Lyu, D. , Liu, W. , Qiang, D. , Ji, X. , … Ding, W. (2018). Overexpression of CDCA8 promotes the malignant progression of cutaneous melanoma and leads to poor prognosis. International Journal of Molecular Medicine, 43, 404–412. 10.3892/ijmm.2018.3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, R. , & Stearns, V. (2012). Epigenetics as a therapeutic target in breast cancer. Journal of Mammary Gland Biology and Neoplasia, 17, 191–204. 10.1007/s10911-012-9263-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafni, A. , Anna, E. C. , & Francesc, P. (2018). Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics & Chromatin, 11, 37 10.1186/s13072-018-0205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles, S. A. , Aboagye, E. O. , Ali, S. , Anderson, A. S. , Armes, J. O. , Berditchevski, F. , … Thompson, A. M. (2013). Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Research, 15 10.1186/bcr3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finetti, P. , Guille, A. , Adelaide, J. , Birnbaum, D. , Chaffanet, M. , & Bertucci, F. (2014). ESPL1 is a candidate oncogene of luminal B breast cancers. Breast Cancer Research and Treatment, 147, 51–59. 10.1007/s10549-014-3070-z [DOI] [PubMed] [Google Scholar]
- Fleischer, T. , Edvardsen, H. , Solvang, H. K. , Daviaud, C. , Naume, B. , Børresen‐Dale, A.‐L. , … Tost, J. (2014). Integrated analysis of high‐resolution DNA methylation profiles, gene expression, germline genotypes and clinical end points in breast cancer patients. International Journal of Cancer, 134, 2615–2625. 10.1002/ijc.28606 [DOI] [PubMed] [Google Scholar]
- Fleischer, T. , Tekpli, X. , Mathelier, A. , Wang, S. , Nebdal, D. , Dhakal, H. P. , … Kristensen, V. N. (2017). DNA methylation at enhancers identifies distinct breast cancer lineages. Nature Communications, 8, 1379 10.1038/s41467-017-00510-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Győrffy, B. , Bottai, G. , Fleischer, T. , Munkácsy, G. , Budczies, J. , Paladini, L. , … Santarpia, L. (2016). Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. International Journal of Cancer, 138, 87–97. 10.1002/ijc.29684 [DOI] [PubMed] [Google Scholar]
- Herman, D. , Leakey, T. I. , Behrens, A. , Yao‐borengasser, A. , Cooney, C. A. , Jousheghany, F. , … Monzavi‐karbassi, B. (2015). Chst11 gene expression and DNA methylation in breast cancer. International Journal of Oncology, 46, 1243–1251. 10.3892/ijo.2015.2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, H. , Aruri, J. , Kapadia, R. , Mehr, H. , White, M. A. , & Ganesan, A. K. (2012). RhoJ regulates melanoma chemoresistance by suppressing pathways that sense DNA damage. Cancer Research, 72, 5516–5528. 10.1158/0008-5472.CAN-12-0775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, K. , Staaf, J. , Lauss, M. , Aine, M. , Lindgren, D. , Bendahl, P.‐O. , … Ringnér, M. (2016). An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells. Breast Cancer Research, 18, 27 10.1186/s13058-016-0685-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, A. , Anderson, A. S. , Clarke, R. B. , Duffy, S. W. , Evans, D. G. , Garcia‐Closas, M. , … Harvie, M. N. (2014). Risk determination and prevention of breast cancer. Breast Cancer Research, 16, 446 10.1186/s13058-014-0446-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, G. , Chong, R. A. , Yang, Q. , Wei, Y. , Blanco, M. A. , Li, F. , … Kang, Y. (2009). MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor‐prognosis breast cancer. Cancer Cell, 15, 9–20. 10.1016/j.ccr.2008.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, S. H. , Park, J. W. , Kim, H. R. , Seong, J. K. , & Kim, H. K. (2014). ADRM1 gene amplification is a candidate driver for metastatic gastric cancers. Clinical & Experimental Metastasis, 31, 727–733. 10.1007/s10585-014-9663-4 [DOI] [PubMed] [Google Scholar]
- Jiang, J. , Liu, W. , Guo, X. , Zhang, R. , Zhi, Q. , Ji, J. , … Yu, Y. (2011). IRX1 influences peritoneal spreading and metastasis via inhibiting BDKRB2‐dependent neovascularization on gastric cancer. Oncogene, 30, 4498–4508. 10.1038/onc.2011.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, D. C. , Lu, Z. D. , Qiao, J. H. , Yan, M. , Cui, S. D. , & Liu, Z. Z. (2015). Expression of CDCA8 correlates closely with FOXM1 in breast cancer: Public microarray data analysis and immunohistochemical study. Neoplasma, 62, 464–469. 10.4149/neo_2015_055 [DOI] [PubMed] [Google Scholar]
- Katsyv, I. , Wang, M. , Song, W. M. , Zhou, X. , & Irie, H. Y. (2016). EPRS is a critical regulator of cell proliferation and estrogen signaling in ER+ breast cancer. Oncotarget, 7, 69592–69605. 10.18632/oncotarget.11870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymenko, Y. , & Nephew, K. P. (2018). Epigenetic crosstalk between the tumor microenvironment and ovarian cancer cells: A therapeutic road less traveled. Cancers, 10 295 10.3390/cancers10090295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Yan, Z. , Yin, Q. , Cao, K. , Wei, Y. , Rodriguez‐Canales, J. , … Wu, Y. (2018). RUNX3 epigenetic inactivation is associated with estrogen receptor positive breast cancer. Journal of Histochemistry & Cytochemistry, 66, 709–721. 10.1369/0022155418797315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotem, J. , Levanon, D. , Negreanu, V. , Bauer, O. , Hantisteanu, S. , Dicken, J. , & Groner, Y. (2017). Runx3 in immunity, inflammation and cancer. Advances in Experimental Medicine & Biology, 962, 369–393. 10.1007/978-981-10-3233-2_23 [DOI] [PubMed] [Google Scholar]
- Lu, J. , Wen, M. , Huang, Y. , He, X. , Wang, Y. , Wu, Q. , … Rodriguez, C. A. (2013). C2ORF40 suppresses breast cancer cell proliferation and invasion through modulating expression of m phase cell cycle genes. Epigenetics, 8, 571–583. 10.4161/epi.24626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, D. L. , Olson, R. L. , & Fernandez‐Zapico, M. E. (2016). Epigenetic control of the tumor microenvironment. Epigenomics, 8(12), 1671–1687. 10.2217/epi-2016-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T. , Pimkhaokham, A. , Suzuki, E. , Omura, K. , Inazawa, J. , & Imoto, I. (2006). Genetic or epigenetic silencing of low density lipoprotein receptor‐related protein 1B expression in oral squamous cell carcinoma. Cancer Science, 97, 1070–1074. 10.1111/j.1349-7006.2006.00283.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okugawa, Y. , Grady, W. M. , & Goel, A. (2015). Epigenetic alterations in colorectal cancer: Emerging biomarkers. Gastroenterology, 149, 1204–1225.e12. 10.1053/j.gastro.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmatpanah, F. B. , Carstens, S. , Guo, J. , Sjahputera, O. , Taylor, K. H. , Duff, D. , … Caldwell, C. W. (2006). Differential DNA methylation patterns of small B‐cell lymphoma subclasses with different clinical behavior. Leukemia, 20, 1855–1862. 10.1038/sj.leu.2404345 [DOI] [PubMed] [Google Scholar]
- Rauscher, G. H. , Kresovich, J. K. , Poulin, M. , Yan, L. , Macias, V. , Mahmoud, A. M. , … Ehrlich, M. (2015). Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer, 15, 816 10.1186/s12885-015-1777-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, M. E. , Phipson, B. , Wu, D. I. , Hu, Y. , Law, C. W. , Shi, W. , & Smyth, G. K. (2015). limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Research, 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, E. , Bagalà, C. , Inzani, F. , Leoncini, E. , & Schinzari, G. (2017). RUNX3 as a potential predictor of metastasis in human pancreatic cancer. In Vivo, 31, 833–840. 10.21873/invivo.11136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlienger, S. , Campbell, S. , & Claing, A. (2014). ARF1 regulates the Rho/MLC pathway to control EGF‐dependent breast cancer cell invasion. Molecular Biology of the Cell, 25, 17–29. 10.1091/mbc.e13-06-0335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi, S. , Ali, S. , Philip, P. , & Sarkar, F. (2013). Clinical advances in molecular biomarkers for cancer diagnosis and therapy. International Journal of Molecular Sciences, 14, 14771–14784. 10.3390/ijms140714771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry, M. B. , Mcdonald, J. A. , Wu, H. C. , Eng, S. , & Santella, R. M. (2016). Epigenetic biomarkers of breast cancer risk: Across the breast cancer prevention continuum In Stearns V. (Eds.), Novel biomarkers in the continuum of breast cancer. Advances in Experimental Medicine and Biology (vol 882, pp. 33–68). Cham, Switzerland: Springer International Publishing; 10.1007/978-3-319-22909-6_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavelu, P. U. , Krenács, T. , Dray, E. , & Duijf, P. H. (2016). In epithelial cancers, aberrant COL17A1promoter methylation predicts its misexpression and increased invasion. Clinical Epigenetics, 8 10.1186/s13148-016-0290-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchione, A. , Ishii, H. , Baldassarre, G. , Bassi, P. , Trapasso, F. , Alder, H. , … Baffa, R. (2002). FEZ1/LZTS1 is down‐regulated in high‐grade bladder cancer, and its restoration suppresses tumorigenicity in transitional cell carcinoma cells. American Journal of Pathology, 160, 1345–1352. 10.1016/S0002-9440(10)62561-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, P. D. , & Srivastava, S. (2012). New paradigms in translational science research in cancer biomarkers. Translational Research the Journal of Laboratory & Clinical Medicine, 159, 343–353. 10.1016/j.trsl.2012.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. H. , Huang, W. S. , Hu, C. R. , Guan, X. X. , Zhou, H. B. , & Chen, L. B. (2010). Relationship between RGS5 expression and differentiation and angiogenesis of gastric carcinoma. World Journal of Gastroenterology, 16, 5642 10.3748/wjg.v16.i44.5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Liu, B. , Zhang, J. , Sun, W. , Dai, C. , Sun, W. , & Li, Q. (2017). Centromere protein U is a potential target for gene therapy of human bladder cancer. Oncology Reports, 38(2), 735–744. 10.3892/or.2017.5769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Yan, L. , Hu, Q. , Sucheston, L. E. , Higgins, M. J. , Ambrosone, C. B. , … Liu, S. (2012). IMA: An R package for high‐throughput analysis of illumina's 450K infinium methylation data. Bioinformatics, 28, 729–730. 10.1093/bioinformatics/bts013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Kanai, R. , Nakazawa, N. , Wang, L. , Toyoshima, C. , & Yanagida, M. (2018). Suppressor mutation analysis combined with 3D modeling explains cohesin's capacity to hold and release DNA. Proceedings of the National Academy of Sciences of the United States of America, 115, E4833–E4842. 10.1073/pnas.1803564115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. , & Park, J. Y. (2012). DNA methylation in promoter region as biomarkers in prostate cancer. Cancer Epigenetics, 863, 67–109. 10.1007/978-1-61779-612-8_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Li, Q. , Niu, J. , Li, B. , & Bai, S. (2015). MicroRNA‐342‐5p and mir‐608 inhibit colon cancer tumorigenesis by targeting NAA10. Oncotarget, 7, 2709–2720. 10.18632/oncotarget.6458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Li, D.‐P. , Shen, N. A. , Yu, X.‐C. , Li, J.‐B. , Song, Q. I. , & Zhang, J.‐H. (2015). TPX2 promotes migration and invasion of human breast cancer cells. Asian Pacific Journal of Tropical Medicine, 8, 1064–1070. 10.1016/j.apjtm.2015.11.007 [DOI] [PubMed] [Google Scholar]
- Yao, Y.‐D. , Sun, T.‐M. , Huang, S.‐Y. , Dou, S. , Lin, L. , Chen, J.‐N. , … Song, E. (2012). Targeted delivery of PLK1‐sirna by ScFv suppresses Her2+ breast cancer growth and metastasis. Science Translational Medicine, 4, 130ra48 10.1126/scitranslmed.3003601 [DOI] [PubMed] [Google Scholar]
- Ye, W. , Tao, J. , Xueming, D. , & Junming, X. (2016). Lentivirus‐mediated knockdown of CEP55 suppresses cell proliferation of breast cancer cells. BioScience Trends, 10, 67–73. 10.5582/bst.2016.01010 [DOI] [PubMed] [Google Scholar]
- Zhang, N. , & Pati, D. (2017). Biology and insights into the role of cohesin protease separase in human malignancies. Biological Reviews, 92, 2070–2083. 10.1111/brv.12321 [DOI] [PubMed] [Google Scholar]
- Zhao, J. , Zhu, D. , Zhang, X. , Zhang, Y. , Zhou, J. , & Dong, M. (2018). TMEM206 promotes the malignancy of colorectal cancer cells by interacting with AKT and extracellular signal‐regulated kinase signaling pathways. Journal of Cellular Physiology, 234, 10888–10898. 10.1002/jcp.27751 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.