

Abstract
Background
Ebstein's anomaly (EA) is a rare congenital heart disease of the tricuspid valve and right ventricle. Patients with EA often manifest with left ventricular noncompaction (LVNC), a cardiomyopathy. Despite implication of cardiac sarcomere genes in some cases, very little is understood regarding the genetic etiology of EA/LVNC. Our study describes a multigenerational family with at least 10 of 17 members affected by EA/LVNC.
Methods
We performed echocardiography on all family members and conducted exome sequencing of six individuals. After identifying candidate variants using two different bioinformatic strategies, we confirmed segregation with phenotype using Sanger sequencing. We investigated structural implications of candidate variants using protein prediction models.
Results
Exome sequencing analysis of four affected and two unaffected members identified a novel, rare, and damaging coding variant in the Kelch‐like family member 26 (KLHL26) gene located on chromosome 19 at position 237 of the protein (GRCh37). This variant region was confirmed by Sanger sequencing in the remaining family members. KLHL26 (c.709C > T p.R237C) segregates only with EA/LVNC‐affected individuals (FBAT p < .05). Investigating structural implications of the candidate variant using protein prediction models suggested that the KLHL26 variant disrupts electrostatic interactions when binding to part of the ubiquitin proteasome, specifically Cullin3 (CUL3), a component of E3 ubiquitin ligase.
Conclusion
In this familial case of EA/LVNC, we have identified a candidate gene variant, KLHL26 (p.R237C), which may have an important role in ubiquitin‐mediated protein degradation during cardiac development.
Keywords: congenital heart defects, Ebstein's anomaly, Kelch‐like family member 26, left‐ventricular noncompaction, ubiquitin proteasome
Patient's Ebstein's anomaly (EA), a rare congenital heart disease of the tricuspid valve and right ventricle often manifest with left ventricular noncompaction (LVNC), a cardiomyopathy. Our study identifies a novel, rare, and damaging coding variant c.709C > T (p.R237C) in the Kelch‐like family member 26 (KLHL26) gene with 10 affected out of 17 affected members using Sanger sequencing. Through structural modeling, we show that the KLHL26 variant may affect protein binding to Cullin3 (CUL3), a component of E3 ubiquitin ligase in the ubiquitin‐mediated protein degradation.

1. INTRODUCTION
Congenital heart defects (CHDs) are malformations of cardiac development and function, ranging from chamber to valve formation. CHDs account for a high burden of neonatal morbidity and mortality in the developed world (Attenhofer Jost, Connolly, Dearani, Edwards, & Danielson, 2007). Despite tremendous advances in genomic technologies, diagnostic variants cannot be identified for most patients. Therefore, additional approaches are required to elucidate the etiology of CHDs. Ebstein's anomaly (EA) is a rare CHD, comprising less than 1% of all cases with a prevalence of 1 in 200,000 live births (Attenhofer Jost et al., 2007). EA is characterized by the failure of posterior and septal tricuspid valve leaflets to delaminate, resulting in apical displacement of the valve, “atrialization” of the right ventricle above the valve, redundancy and fenestration of the anterior leaflet, and dilation of the atrioventricular junction (Attenhofer Jost et al., 2007). EA commonly manifests with left ventricular noncompaction (LVNC), a cardiomyopathy theorized to be due to hyper‐trabeculation or abnormal compaction of the ventricular trabeculations during normal development, causing sponge‐like myocardium (Pignatelli et al., 2014; Zhang, Chen, Qu, Chang, & Shou, 2013). In one study, 10 of 61 patients with EA (16.4%) also had LVNC (Pignatelli et al., 2014). Clinically, patients with EA can present at any age due to the varying degrees of anatomic and physiologic presentations of the disease. If degree of tricuspid valve insufficiency is mild, presentation can be delayed. On the other hand, presentation with severe tricuspid valve insufficiency can cause hydrops fetalis and death (Voges, Al‐Mallah, Scognamiglio, & Di Salvo, 2018). Signs and symptoms include cyanosis, dyspnea, right‐sided heart failure, hepatomegaly, cardiomegaly, and arrhythmias (Galea, Ellul, Schembri, Schembri‐Wismayer, & Calleja‐Agius, 2014). As such, management is individualized for each patient, in severe cases including surgical treatment such as tricuspid valve repair or replacement (Galea et al., 2014).
Both sporadic and familial EA cases have been associated with sarcomeric genes including myosin heavy chain 7 (MYH7) (Bettinelli et al., 2013; Postma et al., 2011) and filamin A (FLNA) (Mercer et al., 2017), transcription factor genes including NKX2.5 (Benson et al., 1999; Gioli‐Pereira et al., 2010) and GATA4 (Digilio et al., 2011), and channel genes such as sodium channel voltage gated type V (SCN5A) (Neu et al., 2010), all of which have a role in myocardial development (Sicko et al., 2016). Likewise, isolated and familial LVNC has been associated with various genes including tafazzin (G4.5) (Bione et al., 1996), Z‐band alternatively spliced PDZ motif‐containing protein (ZASP) (Vatta et al., 2003), transcription factor TBX20 (Kodo et al., 2016), MYH7 (Budde et al., 2007), myosin‐binding protein C (MYBPC3) (van Waning et al., 2018), titin (TTN) (van Waning et al., 2018), alpha‐dystrobrevin (DTNA) (Kenton et al., 2004), and FK‐binding protein‐12(FKBP12) (Kenton et al., 2004). Although genes such as MYH7 (Vermeer et al., 2013), α‐tropomyosin (TPM1) (Kelle, Bentley, Rohena, Cabalka, & Olson, 2016; Nijak et al., 2018), and NKX2.5 (Benson et al., 1999) have been implicated in both EA and LVNC, little is understood regarding the genetic etiology of this disease phenotype. Genetic heterogeneity, low recurrence, and variable clinical phenotypes indicate the interaction of multiple pathobiological mechanisms yet to be clearly defined in patients with EA and LVNC.
In this study we report a rare, novel variant of KLHL26 in a family with autosomal dominant inheritance of EA/LVNC. This gene has not been previously associated with Mendelian disease. We describe multiple in vitro and computational approaches designed to characterize this variant and its potential physiologic functions.
2. METHODS
2.1. Ethics Statement
This study is in accordance with institutionally approved research (IRB) protocols by the Children's Hospital of Wisconsin (CHW) and conforms to the US Federal Policy for the Protection of Human Subjects. Subjects were consented through the IRB‐approved CHW ‐ CHD Tissue Bank (IRB #CHW 06/229, GC 300). This biorepository provided all DNA samples and cardiac tissue of patients and family members.
2.2. Family Pedigree
We identified this family after the proband (VIII:5) presented with severe EA/LVNC to the Children's Hospital of Wisconsin (CHW) (Figure 1). Historical information provided by the family and clinical information from medical records was used to create the pedigree. We performed all measurements of the four chambers, apical displacement of tricuspid valve, presence of LVNC, and degree of tricuspid insufficiency (Table 1 and Table S1). We recorded characteristics of EA according to diagnostic criteria including 1) tethering of tricuspid valve, 2) apical displacement of valve >8mm/m2 body surface area, and 3) combined area of right ventricle plus right atrium larger than combined area of right ventricle, left ventricle, and left atrium (ratio >1.0) (Attenhofer Jost et al., 2007). We extracted DNA (gDNA) from peripheral blood samples of family members and performed exome sequencing of VI:9, VII:7, VII:8, VIII:1, VIII:5, and VIII:7 (encircled blue in Figure 1). Methods of exome sequencing, variant filtering, and in silico modeling are described in the Data Supplement.
Figure 1.
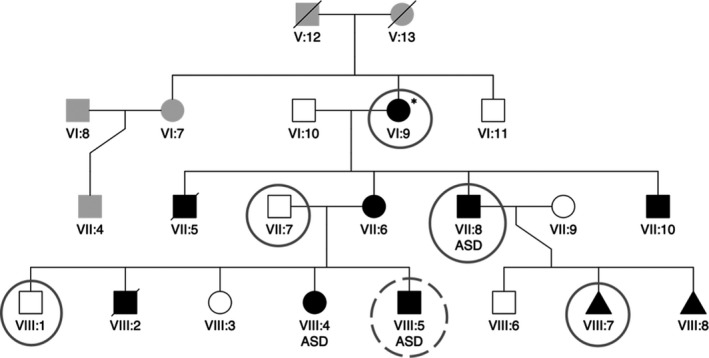
Pedigree of family with members affected by EA/LVNC. EA/LVNC‐affected members are denoted black. VII:5 was a stillborn male, VIII:2 was a premature birth, and VIII:7 was an IUFD; all were affected with cardiac malformations. VIII:8 was an IUFD at 8 weeks, also believed to be of cardiac origin. VI:9 does not fit the complete criteria for EA but is within the spectrum of the phenotype. The proband (VIII:5) is denoted by a dotted circle. Exome sequencing was performed on all encircled family members (VI:9, VII:7, VII:8, VIII:1, VIII:5, and VIII:7). EA/LVNC, Ebstein's anomaly with left ventricular noncompaction; IUFD, intrauterine fetal death
Table 1.
Echocardiography results
| Pedigree ID | Relationship | Age (yr) | LVNC? | TVd/BSA (mm/m2) | (RA + RV)/(RV + LV +LA) | EA? |
|---|---|---|---|---|---|---|
| VIII:5 | Proband | Day 0 | Yes | 25.5 | 1.25 | Yes |
| VII:7 | Father | 37 | No | 3.88 | 0.44 | No |
| VII:6 | Mother | 37 | Yes | 14.94 | 0.98 | Yes |
| VIII:4 | Sister | 2 | Yes | 20.69 | 1.05 | Yes |
| VIII:1 | Brother | 12 | No | 5.23 | 0.73 | No |
| VIII:3 | Sister | 10 | No | 6.58 | 0.63 | No |
| VII:8 | Maternal uncle | 34 | Yes | 8.06 | 1.26 | Yes |
| VI:9 | Maternal grandmothera | — | No | 4.31 | — | No |
| VIII:6 | Maternal first cousin | — | No | 7.32 | — | No |
| VI:11 | Maternal great uncle | 60 | No | 6.21 | 0.54 | No |
Abbreviations: BSA, body surface area; EA, Ebstein's anomaly; LA, left atrium; LV, left ventricle; LVNC, left ventricular noncompaction; RA, right atrium; RV, right ventricle; TVd, tricuspid valve displacement.
Echocardiographic diagnostic criteria for Ebstein's anomaly include TVd/BSA of greater than 8 mm/m2 and/or combined right atrium RA and RV areas to be greater than areas of LV, RV, and LA combined (Attenhofer Jost et al., 2007). Maternal grandmother does not satisfy these criteria, but her echocardiogram did exhibit tricuspid anomaly and mild LVNC, which is within the spectrum of disease. Individual measurements can be found in Table S1.
3. RESULTS
3.1. Family Phenotyping
This family's CHDs pedigree (Figure 1) was revealed when prenatal ultrasound discovered severe EA/LVNC in utero for the proband (VIII:5), which was the mother's (VII:6) fifth pregnancy. This prompted our inquiry into the family's history of heart disease, which indicated the presence of cardiac anomalies in 10 of 17 family members. The mother's third living child (VIII:4) had previously undergone surgical repair for an atrial septal defect (ASD); it was also during this pregnancy that the mother herself (VII:6) was diagnosed with EA. Evaluation for CHDs confirmed an autosomal dominant inheritance pattern (Figure 1) for EA/LVNC. Echocardiography performed on her two other children (VIII:1 and VIII:3) indicated normal cardiac anatomy. The maternal grandmother (VI:9) had four pregnancies resulting in a stillborn male with cardiac defects (VII:5) and three living children with EA/LVNC (VII:6, VII:8, and VII:10).
The diagnostic criteria for EA/LVNC are defined by tricuspid valve displacement to body surface ratio greater than 8 mm/m2, and combined right atrial and right ventricular volume greater than combined right ventricular, left atrial, and left ventricular volume (Table 1) (Attenhofer Jost et al., 2007). Echocardiography and chart reviews revealed six family members with EA/LVNC (VI:9, VII:6, VII:8, VII:10, VIII:4, and VIII:5), and three with ASD (VII:8, VIII:4, and VIII:5). VIII:2 was a premature birth and VIII:7 was an intrauterine fetal death (IUFD) at 20 weeks gestational age; both demises were due to cardiac disease. VIII:8 was an IUFD at 8 weeks gestational age also theorized to be due to a cardiac defect. While the maternal grandmother (VI:9) did not qualify for EA according to the strict criteria, echocardiography indicated tricuspid valve anomaly and mild LVNC, which is within the spectrum of EA/LVNC. All affected individuals demonstrated some degree of tricuspid valve insufficiency (Table S1).
3.2. Exome Sequencing and Analysis
Exome sequencing of subjects VIII:5 and VII:8, followed by filtering with reference to a published differential mRNA expression pattern during murine cardiogenesis (Li et al., 2014), was employed to generate a gene candidate list. Although sequencing of several candidate genes did not reveal any variants segregating with EA/LVNC‐affected family members, additional exome sequencing of VI:9, VIII:1, and VIII:7 narrowed the list to six candidate genes, among which five were missense variants (Table S2). Two of the six candidate genes were also identified via a separate filtering method using Golden Helix VarSeq (described in the Data Supplement). Both filtering strategies resulted in two candidate variants—RP1 (c.3532G > T p.D1178Y) and KLHL26 (c.709C > T p.R237C). Sanger sequencing of the exon containing each candidate variant revealed that the RP1 variant did not segregate with disease. However, the nonsynonymous variant on chromosome 19 (GRCh37) in the region of KLHL26 (NM_001345981.1:c.709C > T p.R237C) segregated with EA/LVNC in all genotyped subjects (n = 14, eight affected and six unaffected; Figure 1a) (Family‐Based Association Test p < .05) and was fully penetrant. KLHL26 (p.R237C), like other KLHL family members is involved in ubiquitin‐mediated protein degradation, but the specific functions of all family members have not been elucidated. KLHL26 is conserved across many species including chimpanzee, dog, rat, chicken, zebrafish, and frog.
3.3. Structural Modeling
KLHL26 belongs to the Kelch‐like (KLHL) gene family and is a multidomain protein consisting of three types of domains (Dhanoa, Cogliati, Satish, Bruford, & Friedman, 2013) depicted in Figure 2a. The first is a homodimerization type domain known by two names: BTB (Broad‐Complex, Tramtrack and Bric a brac) and POZ (POxvirus and Zinc finger) (Stogios, Downs, Jauhal, Nandra, & Prive, 2005). The second domain is termed a BACK (BTB and C‐terminal Kelch) domain, named for its conserved presence in genes with BTB and Kelch domains. Third, KLHL26 has six Kelch repeats that form a single topologic fold (Stogios & Prive, 2004).
Figure 2.

Structural Model of KLHL26‐CUL3 complex. (a) Our structural model is colored according to domain, depicting the predicted interaction with CUL3 mediated by the BACK and BTB domains. Residues outside of established domains by sequence comparison are colored white. Residues in the BTB domain that flank a site of additional sequence due to alternative splicing are circled. (b) Magnification of the BACK domain defines the location of R237 and its proximity to the CUL3 binding surface. (c). The electrostatic surface of KLHL26‐CUL3 interaction, wherein R237 is circled. (d) The analogous image of 2C for C237, wherein the electrostatic surface is more neutral and partially negative. (e) A 180° rotated view of the electrostatic surface of KLHL26‐CUL3 interaction; the gold circle marks the location of R237. (f) The positive patch on CUL3 is seen across from R237 (parts of KLHL26 that occlude the CUL3 surface are hidden). This patch is created by an HKH sequence whose sidechains fan out radially, contributing to a positive crescent‐shaped region, leaving the backbone oxygen atoms to make a negative surface patch
There are no published data describing the structure of KLHL26 protein. As an initial approach, we employed homology‐based modeling to assess KLHL26 structure. The BioPlex database (Huttlin et al., 2015) identifies with 99% probability that KLHL26 interacts with Cullin‐3 (CUL3), an important subunit of E3 ubiquitin ligase. Query of other genes in the KLHL gene family with prior data revealed that KLHL11 forms a complex with CUL3 (Canning et al., 2013). Therefore, we used this experimentally validated structure of KLHL11 to build a model of KLHL26 (Figure 2a,b). Although the R237C variant is located in the BACK domain of KLHL26, it is predicted to have little effect on stability (ΔΔGfold = 0.09 kcal/mol) based on FoldX (Parra et al., 2016). Amino acids are considered to be frustrated when favorable and unfavorable interactions are simultaneously experienced. Local frustration analysis identified R237 as a neutral amino acid, while C237 is minimally frustrated (energetically more stable), displaying a significant shift in frustration index, ΔFR237C = FC237 − FR237 = 1.67 − (−0.41) = 2.08. Electrostatic profiling of the interface between proteins KLHL26 and CUL3 showed that whereas R237 forms a positive surface patch complementary to the negative CUL3 patch, the C237 variant forms a more neutral patch (Figure 2c‐f). This suggests that the C237 variant could alter KLHL26‐CUL3 binding affinity, perhaps decoupling the Cullin system in a fashion that disrupts the ubiquitin‐mediated pathway of protein degradation.
4. DISCUSSION
We have discovered that a rare, novel variant in KLHL26 segregates with disease in a family with a highly penetrant form of EA/LVNC. In order to suggest an underlying pathogenic mechanism, we obtained a predicted molecular structure by comparing protein domains across the KLHL26 gene family.
There is high probability that KLHL26 is a component of the ubiquitin‒proteasome system (UPS) (Hershko, Heller, Elias, & Ciechanover, 1983), wherein protein ubiquitylation occurs through a sequence of reactions catalyzed by E1, E2, and E3 enzymes, via its interaction with CUL3. The largest family of multisubunit E3 ubiquitin ligases is the cullin‐RING ligases (CRLs) (Petroski & Deshaies, 2005; Zimmerman, Schulman, & Zheng, 2010). KLHL family members are substrate adaptors for CUL3, which is part of the CRL3 subclass and is highly enriched in muscle tissues (Hori et al., 1999). The N‐terminal domain of CRLs binds to a substrate adaptor that recruits target proteins for ubiquitination, while the C‐terminal domain binds a RING protein that recruits E2. The target protein and E2 are brought into correct conformation through neddylation (NEDD8) of the C‐terminal domain. Both the BTB and BACK domains permit assembly with CUL3 (Canning et al., 2013). Other such domains commonly within the same protein as a BTB domain include C‐terminal Kelch, PHR, or zinc finger domains, with the Kelch β‐propeller domain being the most common for target recognition (Prag & Adams, 2003; Stogios et al., 2005). Thus, within KLHL26, the BTB and BACK domains likely interact with CUL3, while the Kelch domain recruit targets for ubiquitination (Figure 3). This model suggests that although R237C has little direct effect on KLHL26, it may significantly affect proteolytic function by altering protein:protein electrostatic interactions; as such, decoupling the Cullin system.
Figure 3.

Proposed mechanism of KLHL26 interaction with CUL3. CUL3 is a core subunit of E3 ubiquitin ligase. Its N‐terminal domain binds to a receptor protein that confers substrate specificity – in this case KLHL26. The C‐ terminal domain binds a RING protein that recruits E2. NEDD8 brings substrate and E2 to the correct conformation for ubiquitination. In KLHL26, most likely the BTB domain interacts with CUL3 and the Kelch domain acts as the substrate‐recognition module
Considering the substantial physical intracellular forces exerted during the continual contraction of cardiac and skeletal muscle, it is not surprising that these cells have evolved mechanisms, including the UPS, to efficiently turn‐over sarcomeric proteins. Consequences of disrupting the CRL3‐KLHL pathway in striated muscle have been documented (Dhanoa et al., 2013; Papizan, Vidal, Bezprozvannaya, Bassel‐Duby, & Olson, 2018). KLHL40 (Bowlin, Embree, Garry, Garry, & Shi, 2013), KLHL41 (du Puy et al., 2012), KLHDC1 (Chin, Xu, Ching, & Jin, 2007; Sekine et al., 2012), and KLHDC2 (Chin et al., 2007; Sekine et al., 2012) are associated with myoblast differentiation. KLHL31 (Abou‐Elhamd, Cooper, & Munsterberg, 2009; Yu et al., 2008) and KLHL41 (Greenberg, Connelly, Daniels, & Horowits, 2008) have roles in muscle maturation. KLHL41 is also associated with myoblast proliferation (Paxton et al., 2011). KLHL19 (Miller et al., 2012), KLHL20 (Hara et al., 2004), ND1‐L (Sasagawa et al., 2002), and KLHL27 (I. F. Kim, Mohammadi, & Huang, 1999) either bind to actin or are associated with actin, playing critical roles in cytoskeletal arrangement. KLHL9 (Cirak et al., 2010), KBTBD13 (Sambuughin et al., 2010), KLHL40 (Ravenscroft et al., 2013), and KLHL41 (Gupta et al., 2013) have been implicated in distal and nemaline myopathy. CUL3 is enriched in muscle tissues, essential for myoblast differentiation, and conditional CM‐KO of CUL3 is neonatal lethal in mice (Blondelle, Shapiro, Domenighetti, & Lange, 2017; Papizan et al., 2018). CM deletion of NAE1, a regulatory subunit of neddylation, in mice CMs caused heart failure and perinatal lethality (Zou et al., 2018). In summary, substantial data exist implicating KLHL proteins in muscular function, in processes ranging from myoblast migration to cytoskeletal arrangement.
Evidence suggests that the EA/LVNC phenotype may result from sarcomeric disarray, as indicated by electron microscopic images revealing disrupted Z‐bands in the right atrium and atrialized part of the right ventricle of EA patients (Egorova, Penyaeva, & Bockeria, 2015). The authors suggested that the similarity between electron dense deposits observed in cardiomyocytes of EA patients and skeletal muscle myopathies occurring in patients harboring mutations in genes encoding α‐actin, nebulin, and titin might link EA pathogenicity to a sarcomeric protein, or perhaps to a transcription factor required to maintain the expression of sarcomeric genes. In this regard, RNA‐seq analysis performed on H1‐ESCs (Kim et al., 2015) showed that KLHL26 is expressed during cardiomyocyte differentiation. As noted earlier, previously identified genetic risk factors for EA/LVNC are mainly comprised of sarcomeric proteins. We accordingly hypothesize that KLHL26 (p.R237C) dysregulates the degradation of a sarcomeric protein, causing altered cardiomyocyte proliferation and differentiation, and ultimately results in presentation of the familial EA/LVNC phenotype.
In summary, while the specific function of KLHL26 in the UPS remains unknown, growing evidence that mutated KLHL proteins disrupt striated muscle homeostasis, combined with the high disease‐associated penetrance of this specific KLHL26 variant, warrants investigation to validate the predictions suggested by the modeling described in Figure 3. In order to determine possible effects of disrupted UPS processing on the developing heart, we are modeling this familial EA/LVNC using induced pluripotent stem cells derived from the family depicted in Figure 1 to identify KLHL26‐dependent changes in cardiomyocyte proliferation and differentiation.
DISCLOSURES
A. Tomita‐Mitchell and M. E. Mitchell are cofounders of TAI Diagnostics (Milwaukee, WI), a biotechnology company involved in transplant diagnostics, and members of its scientific advisory board.
Supporting information
ACKNOWLEDGMENTS
We are grateful to the families and clinicians of the Herma Heart Institute from CHW. We thank Mary Krolikowski for IRB support. We gratefully acknowledge Mary Goetsch, MS for making this study possible. We also thank Dr. Min‐Su Kim, PhD and Dr. John Lough, PhD for their careful review to this manuscript.
Samudrala SSK, North LM, Stamm KD, et al. Novel KLHL26 variant associated with a familial case of Ebstein’s anomaly and left ventricular noncompaction. Mol Genet Genomic Med. 2020;8:e1152 10.1002/mgg3.1152
Funding information
This work was supported by National Heart, Lung, and Blood Institute 5T35HL072483‐32 (L.N) and 5T35HL072483‐34 (S.S.), the Wolfe Family Foundation, the Little Hearts for Life Foundation, the Medical College of Wisconsin's Department of Surgery, and the Children's Hospital of Wisconsin Research Institute. S.S. is a member of the Medical Scientist Training Program at MCW, which is partially supported by a training grant from National Institute of General Medical Sciences T32‐GM080202.
DATA AVAILABILITY STATEMENT
The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Abou‐Elhamd, A. , Cooper, O. , & Munsterberg, A. (2009). Klhl31 is associated with skeletal myogenesis and its expression is regulated by myogenic signals and Myf‐5. Mechanisms of Development, 126(10), 852–862. 10.1016/j.mod.2009.07.006 [DOI] [PubMed] [Google Scholar]
- Attenhofer Jost, C. H. , Connolly, H. M. , Dearani, J. A. , Edwards, W. D. , & Danielson, G. K. (2007). Ebstein's anomaly. Circulation, 115(2), 277–285. 10.1161/CIRCULATIONAHA.106.619338 [DOI] [PubMed] [Google Scholar]
- Benson, D. W. , Silberbach, G. M. , Kavanaugh‐McHugh, A. , Cottrill, C. , Zhang, Y. , Riggs, S. , … Kugler, J. D. (1999). Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. Journal of Clinical Investigation, 104(11), 1567–1573. 10.1172/JCI8154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettinelli, A. L. , Mulder, T. J. , Funke, B. H. , Lafferty, K. A. , Longo, S. A. , & Niyazov, D. M. (2013). Familial ebstein anomaly, left ventricular hypertrabeculation, and ventricular septal defect associated with a MYH7 mutation. American Journal of Medical Genetics. Part A, 161A(12), 3187–3190. 10.1002/ajmg.a.36182 [DOI] [PubMed] [Google Scholar]
- Bione, S. , D'Adamo, P. , Maestrini, E. , Gedeon, A. K. , Bolhuis, P. A. , & Toniolo, D. (1996). A novel X‐linked gene, G4.5. is responsible for Barth syndrome. Nature Genetics, 12(4), 385–389. 10.1038/ng0496-385 [DOI] [PubMed] [Google Scholar]
- Blondelle, J. , Shapiro, P. , Domenighetti, A. A. , & Lange, S. (2017). Cullin E3 ligase activity is required for myoblast differentiation. Journal of Molecular Biology, 429(7), 1045–1066. 10.1016/j.jmb.2017.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowlin, K. M. , Embree, L. J. , Garry, M. G. , Garry, D. J. , & Shi, X. (2013). Kbtbd5 is regulated by MyoD and restricted to the myogenic lineage. Differentiation, 86(4–5), 184–191. 10.1016/j.diff.2013.08.002 [DOI] [PubMed] [Google Scholar]
- Budde, B. S. , Binner, P. , Waldmuller, S. , Hohne, W. , Blankenfeldt, W. , Hassfeld, S. , … Scheffold, T. (2007). Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta‐myosin heavy chain gene. PLoS ONE, 2(12), e1362 10.1371/journal.pone.0001362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning, P. , Cooper, C. D. O. , Krojer, T. , Murray, J. W. , Pike, A. C. W. , Chaikuad, A. , … Bullock, A. N. (2013). Structural basis for Cul3 protein assembly with the BTB‐Kelch family of E3 ubiquitin ligases. Journal of Biological Chemistry, 288(11), 7803–7814. 10.1074/jbc.M112.437996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, K. T. , Xu, H. T. , Ching, Y. P. , & Jin, D. Y. (2007). Differential subcellular localization and activity of kelch repeat proteins KLHDC1 and KLHDC2. Molecular and Cellular Biochemistry, 296(1–2), 109–119. 10.1007/s11010-006-9304-6 [DOI] [PubMed] [Google Scholar]
- Cirak, S. , von Deimling, F. , Sachdev, S. , Errington, W. J. , Herrmann, R. , Bönnemann, C. , … Voit, T. (2010). Kelch‐like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain, 133(Pt 7), 2123–2135. 10.1093/brain/awq108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanoa, B. S. , Cogliati, T. , Satish, A. G. , Bruford, E. A. , & Friedman, J. S. (2013). Update on the Kelch‐like (KLHL) gene family. Hum Genomics, 7, 13 10.1186/1479-7364-7-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digilio, M. C. , Bernardini, L. , Lepri, F. , Giuffrida, M. G. , Guida, V. , Baban, A. , … Dallapiccola, B. (2011). Ebstein anomaly: Genetic heterogeneity and association with microdeletions 1p36 and 8p23.1. American Journal of Medical Genetics. Part A, 155A(9), 2196–2202. 10.1002/ajmg.a.34131 [DOI] [PubMed] [Google Scholar]
- du Puy, L. , Beqqali, A. , van Tol, H. T. , Monshouwer‐Kloots, J. , Passier, R. , Haagsman, H. P. , & Roelen, B. A. (2012). Sarcosin (Krp1) in skeletal muscle differentiation: Gene expression profiling and knockdown experiments. International Journal of Developmental Biology, 56(4), 301–309. 10.1387/ijdb.113327lp [DOI] [PubMed] [Google Scholar]
- Egorova, I. F. , Penyaeva, E. V. , & Bockeria, L. A. (2015). Altered Z‐disks of myofibrils in the cardiomyocytes from patients with Ebstein's anomaly. Arkhiv Patologii, 77(6), 3–8. 10.17116/patol20157763-8 [DOI] [PubMed] [Google Scholar]
- Galea, J. , Ellul, S. , Schembri, A. , Schembri‐Wismayer, P. , & Calleja‐Agius, J. (2014). Ebstein anomaly: A review. Neonatal Network, 33(5), 268–274. 10.1891/0730-0832.33.5.268 [DOI] [PubMed] [Google Scholar]
- Gioli‐Pereira, L. , Pereira, A. C. , Mesquita, S. M. , Xavier‐Neto, J. , Lopes, A. A. , & Krieger, J. E. (2010). NKX2.5 mutations in patients with non‐syndromic congenital heart disease. International Journal of Cardiology, 138(3), 261–265. 10.1016/j.ijcard.2008.08.035 [DOI] [PubMed] [Google Scholar]
- Greenberg, C. C. , Connelly, P. S. , Daniels, M. P. , & Horowits, R. (2008). Krp1 (Sarcosin) promotes lateral fusion of myofibril assembly intermediates in cultured mouse cardiomyocytes. Experimental Cell Research, 314(5), 1177–1191. 10.1016/j.yexcr.2007.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, V. A. , Ravenscroft, G. , Shaheen, R. , Todd, E. J. , Swanson, L. C. , Shiina, M. , … Beggs, A. H. (2013). Identification of KLHL41 mutations implicates BTB‐Kelch‐mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. American Journal of Human Genetics, 93(6), 1108–1117. 10.1016/j.ajhg.2013.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, T. , Ishida, H. , Raziuddin, R. , Dorkhom, S. , Kamijo, K. , & Miki, T. (2004). Novel kelch‐like protein, KLEIP, is involved in actin assembly at cell‐cell contact sites of Madin‐Darby canine kidney cells. Molecular Biology of the Cell, 15(3), 1172–1184. 10.1091/mbc.e03-07-0531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, A. , Heller, H. , Elias, S. , & Ciechanover, A. (1983). Components of ubiquitin‐protein ligase system. Resolution, affinity purification, and role in protein breakdown. Journal of Biological Chemistry, 258(13), 8206–8214 https://www.ncbi.nlm.nih.gov/pubmed/6305978 [PubMed] [Google Scholar]
- Hori, T. , Osaka, F. , Chiba, T. , Miyamoto, C. , Okabayashi, K. , Shimbara, N. , … Tanaka, K. (1999). Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene, 18(48), 6829–6834. 10.1038/sj.onc.1203093 [DOI] [PubMed] [Google Scholar]
- Huttlin, E. L. , Ting, L. , Bruckner, R. J. , Gebreab, F. , Gygi, M. P. , Szpyt, J. , … Gygi, S. P. (2015). The BioPlex Network: A Systematic exploration of the human interactome. Cell, 162(2), 425–440. 10.1016/j.cell.2015.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelle, A. M. , Bentley, S. J. , Rohena, L. O. , Cabalka, A. K. , & Olson, T. M. (2016). Ebstein anomaly, left ventricular non‐compaction, and early onset heart failure associated with a de novo alpha‐tropomyosin gene mutation. American Journal of Medical Genetics. Part A, 170(8), 2186–2190. 10.1002/ajmg.a.37745 [DOI] [PubMed] [Google Scholar]
- Kenton, A. B. , Sanchez, X. , Coveler, K. J. , Makar, K. A. , Jimenez, S. , Ichida, F. , … Bowles, K. R. (2004). Isolated left ventricular noncompaction is rarely caused by mutations in G4.5, alpha‐dystrobrevin and FK Binding Protein‐12. Molecular Genetics and Metabolism, 82(2), 162–166. 10.1016/j.ymgme.2004.02.009 [DOI] [PubMed] [Google Scholar]
- Kim, I. F. , Mohammadi, E. , & Huang, R. C. (1999). Isolation and characterization of IPP, a novel human gene encoding an actin‐binding, kelch‐like protein. Gene, 228(1–2), 73–83. 10.1016/s0378-1119(99)00006-2 [DOI] [PubMed] [Google Scholar]
- Kim, M.‐S. , Horst, A. , Blinka, S. , Stamm, K. , Mahnke, D. , Schuman, J. , … Lough, J. (2015). Activin‐A and Bmp4 levels modulate cell type specification during CHIR‐induced cardiomyogenesis. PLoS ONE, 10(2), e0118670 10.1371/journal.pone.0118670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodo, K. , Ong, S. G. , Jahanbani, F. , Termglinchan, V. , Hirono, K. , InanlooRahatloo, K. , … Wu, J. C. (2016). iPSC‐derived cardiomyocytes reveal abnormal TGF‐beta signalling in left ventricular non‐compaction cardiomyopathy. Nature Cell Biology, 18(10), 1031–1042. 10.1038/ncb3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Martinez‐Fernandez, A. , Hartjes, K. A. , Kocher, J. P. , Olson, T. M. , Terzic, A. , & Nelson, T. J. (2014). Transcriptional atlas of cardiogenesis maps congenital heart disease interactome. Physiological Genomics, 46(13), 482–495. 10.1152/physiolgenomics.00015.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer, C. L. , Andreoletti, G. , Carroll, A. , Salmon, A. P. , Temple, I. K. , & Ennis, S. (2017). Familial Ebstein anomaly: Whole exome sequencing identifies novel phenotype associated with FLNA. Circulation: Cardiovascular Genetics, 10(6), 10.1161/CIRCGENETICS.116.001683 [DOI] [PubMed] [Google Scholar]
- Miller, C. J. , Gounder, S. S. , Kannan, S. , Goutam, K. , Muthusamy, V. R. , Firpo, M. A. , … Rajasekaran, N. S. (2012). Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochimica Et Biophysica Acta, 1822(6), 1038–1050. 10.1016/j.bbadis.2012.02.007 [DOI] [PubMed] [Google Scholar]
- Neu, A. , Eiselt, M. , Paul, M. , Sauter, K. , Stallmeyer, B. , Isbrandt, D. , & Schulze‐Bahr, E. (2010). A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Human Mutation, 31(8), E1609–1621. 10.1002/humu.21302 [DOI] [PubMed] [Google Scholar]
- Nijak, A. , Alaerts, M. , Kuiperi, C. , Corveleyn, A. , Suys, B. , Paelinck, B. , … Verstraeten, A. (2018). Left ventricular non‐compaction with Ebstein anomaly attributed to a TPM1 mutation. European Journal of Medical Genetics, 61(1), 8–10. 10.1016/j.ejmg.2017.10.003 [DOI] [PubMed] [Google Scholar]
- Papizan, J. B. , Vidal, A. H. , Bezprozvannaya, S. , Bassel‐Duby, R. , & Olson, E. N. (2018). Cullin‐3‐RING ubiquitin ligase activity is required for striated muscle function in mice. Journal of Biological Chemistry, 293(23), 8802–8811. 10.1074/jbc.RA118.002104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, R. G. , Schafer, N. P. , Radusky, L. G. , Tsai, M. Y. , Guzovsky, A. B. , Wolynes, P. G. , & Ferreiro, D. U. (2016). Protein Frustratometer 2: A tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Research, 44(W1), W356–360. 10.1093/nar/gkw304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxton, C. W. , Cosgrove, R. A. , Drozd, A. C. , Wiggins, E. L. , Woodhouse, S. , Watson, R. A. , … Pell, J. M. (2011). BTB‐Kelch protein Krp1 regulates proliferation and differentiation of myoblasts. American Journal of Physiology. Cell Physiology, 300(6), C1345–1355. 10.1152/ajpcell.00321.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski, M. D. , & Deshaies, R. J. (2005). Function and regulation of cullin‐RING ubiquitin ligases. Nature Reviews Molecular Cell Biology, 6(1), 9–20. 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- Pignatelli, R. H. , Texter, K. M. , Denfield, S. W. , Grenier, M. A. , Altman, C. A. , Ayres, N. A. , & Chandra‐Bose Reddy, S. (2014). LV Noncompaction in Ebstein's anomaly in infants and outcomes. JACC: Cardiovascular Imaging, 7(2), 207–209. 10.1016/j.jcmg.2013.05.021 [DOI] [PubMed] [Google Scholar]
- Postma, A. V. , van Engelen, K. , van de Meerakker, J. , Rahman, T. , Probst, S. , Baars, M. J. , … Klaassen, S. (2011). Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circulation: Cardiovascular Genetics, 4(1), 43–50. 10.1161/CIRCGENETICS.110.957985 [DOI] [PubMed] [Google Scholar]
- Prag, S. , & Adams, J. C. (2003). Molecular phylogeny of the kelch‐repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics, 4, 42 10.1186/1471-2105-4-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft, G. , Miyatake, S. , Lehtokari, V.‐L. , Todd, E. J. , Vornanen, P. , Yau, K. S. , … Laing, N. G. (2013). Mutations in KLHL40 are a frequent cause of severe autosomal‐recessive nemaline myopathy. American Journal of Human Genetics, 93(1), 6–18. 10.1016/j.ajhg.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambuughin, N. , Yau, K. S. , Olivé, M. , Duff, R. M. , Bayarsaikhan, M. , Lu, S. , … Goldfarb, L. G. (2010). Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. American Journal of Human Genetics, 87(6), 842–847. 10.1016/j.ajhg.2010.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa, K. , Matsudo, Y. , Kang, M. , Fujimura, L. , Iitsuka, Y. , Okada, S. , … Hatano, M. (2002). Identification of Nd1, a novel murine kelch family protein, involved in stabilization of actin filaments. Journal of Biological Chemistry, 277(46), 44140–44146. 10.1074/jbc.M202596200 [DOI] [PubMed] [Google Scholar]
- Sekine, Y. , Hatanaka, R. , Watanabe, T. , Sono, N. , Iemura, S.‐I. , Natsume, T. , … Ichijo, H. (2012). The Kelch repeat protein KLHDC10 regulates oxidative stress‐induced ASK1 activation by suppressing PP5. Molecular Cell, 48(5), 692–704. 10.1016/j.molcel.2012.09.018 [DOI] [PubMed] [Google Scholar]
- Sicko, R. J. , Browne, M. L. , Rigler, S. L. , Druschel, C. M. , Liu, G. , Fan, R. , … Mills, J. L. (2016). Genetic variants in isolated Ebstein anomaly implicated in myocardial development pathways. PLoS ONE, 11(10), e0165174 10.1371/journal.pone.0165174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogios, P. J. , Downs, G. S. , Jauhal, J. J. , Nandra, S. K. , & Prive, G. G. (2005). Sequence and structural analysis of BTB domain proteins. Genome Biology, 6(10), R82 10.1186/gb-2005-6-10-r82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogios, P. J. , & Prive, G. G. (2004). The BACK domain in BTB‐kelch proteins. Trends in Biochemical Sciences, 29(12), 634–637. 10.1016/j.tibs.2004.10.003 [DOI] [PubMed] [Google Scholar]
- van Waning, J. I. , Caliskan, K. , Hoedemaekers, Y. M. , van Spaendonck‐Zwarts, K. Y. , Baas, A. F. , Boekholdt, S. M. , … Majoor‐Krakauer, D. (2018). Genetics, clinical features, and long‐term outcome of noncompaction cardiomyopathy. Journal of the American College of Cardiology, 71(7), 711–722. 10.1016/j.jacc.2017.12.019 [DOI] [PubMed] [Google Scholar]
- Vatta, M. , Mohapatra, B. , Jimenez, S. , Sanchez, X. , Faulkner, G. , Perles, Z. , … Towbin, J. A. (2003). Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non‐compaction. Journal of the American College of Cardiology, 42(11), 2014–2027. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/14662268 [DOI] [PubMed] [Google Scholar]
- Vermeer, A. M. , van Engelen, K. , Postma, A. V. , Baars, M. J. , Christiaans, I. , De Haij, S. , … Keavney, B. (2013). Ebstein anomaly associated with left ventricular noncompaction: An autosomal dominant condition that can be caused by mutations in MYH7. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 163C(3), 178–184. 10.1002/ajmg.c.31365 [DOI] [PubMed] [Google Scholar]
- Voges, I. , Al‐Mallah, M. H. , Scognamiglio, G. , & Di Salvo, G. (2018). Right heart‐pulmonary circulation unit in congenital heart diseases. Heart Failure Clinics, 14(3), 283–295. 10.1016/j.hfc.2018.02.005 [DOI] [PubMed] [Google Scholar]
- Yu, W. , Li, Y. , Zhou, X. , Deng, Y. , Wang, Z. , Yuan, W. , … Wu, X. (2008). A novel human BTB‐kelch protein KLHL31, strongly expressed in muscle and heart, inhibits transcriptional activities of TRE and SRE. Molecules and Cells, 26(5), 443–453. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/18719355 [PubMed] [Google Scholar]
- Zhang, W. , Chen, H. , Qu, X. , Chang, C. P. , & Shou, W. (2013). Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 163C(3), 144–156. 10.1002/ajmg.c.31369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, E. S. , Schulman, B. A. , & Zheng, N. (2010). Structural assembly of cullin‐RING ubiquitin ligase complexes. Current Opinion in Structural Biology, 20(6), 714–721. 10.1016/j.sbi.2010.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, J. , Ma, W. , Li, J. , Littlejohn, R. , Zhou, H. , Kim, I.‐M. , … Su, H. (2018). Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proceedings of the National Academy of Sciences of the United States of America, 115(17), E4101–E4110. 10.1073/pnas.1719309115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are not publicly available due to privacy or ethical restrictions.