

Abstract
Background
Agnathia‐otocephaly is a rare and lethal anomaly affecting craniofacial structures derived from the first pharyngeal arch. It is characterized by agnathia, microstomia, aglossia, and abnormally positioned auricles with or without associated anomalies. Variants affecting function of OTX2 and PRRX1, which together regulate the neural crest cells and the patterning of the first pharyngeal arch as well as skeletal and limb development, were identified to be causal for the anomaly in a few patients.
Methods
Family‐based exome sequencing (ES) on a fetus with severe agnathia‐otocephaly, cheilognathopalatoschisis, laryngeal hypoplasia, fused lung lobes and other organ abnormalities and mRNA expression analysis were performed.
Results
Exome sequencing detected a de novo SMAD3 missense variant in exon 6 (c.860G>A) associated with decreased mRNA expression. Variants in SMAD3 cause Loeys–Dietz syndrome 3 presenting with craniofacial anomalies such as mandibular hypoplasia, micro‐ or retro‐gnathia, bifid uvula and cleft palate as well as skeletal anomalies and arterial tortuosity.
The SMAD3 protein acts as a transcriptional regulator in the transforming growth factor β (TGFB) and bone morphogenetic (BMP) signaling pathways, which play a key role in the development of craniofacial structures originating from the pharyngeal arches.
Conclusion
Agnathia‐otocephaly with or without associated anomalies may represent the severe end of a phenotypic spectrum related to variants in genes in the interacting SMAD/TGFB/BMP/SHH/FGF developmental pathways.
Keywords: agnathia‐otocephaly, exome sequencing, prenatal, SMAD3
Variants affecting function of OTX2 and PRRX1, which together regulate neural crest cells and the patterning of the first pharyngeal arch as well as skeletal and limb development, were identified to be causal for aganthia‐otocephaly in a few patients. Using family‐based exome sequencing we found a pathogenic variant in SMAD3. We expand the clinical spectrum of SMAD3 variants from Loeys‐Dietz syndrome 3 and suggest that agnathia‐otocephaly with or without associated anomalies may represent the severe end of a phenotypic spectrum related to variants in genes in the interacting SMAD/TGFB/BMP/SHH/FGF developmental pathways.

1. INTRODUCTION
Agnathia‐otocephaly (OMIM 202650) is a rare congenital anomaly pattern with an estimated incidence of less than 1 in 70,000 births. The anomaly is characterized by mandibular hypoplasia, severe micrognathia or agnathia and variable ventromedial auricular malposition and/or fusion and microstomia with microglossia or aglossia. Holoprosencephaly is the most commonly identified associated anomaly, but cyclopia, uni‐/bilateral microphthalmia/anophthalmia, cleft palate, pharyngeal and laryngeal hypoplasia, skeletal, genitourinary, and cardiovascular anomalies, and situs inversus have been reported. The disorder is almost always lethal. It is considered a defect of blastogenesis, resulting in the variable dysmorphogenesis of structures developing from the first pharyngeal arch derivatives or a failure of neural crest cells to migrate into the first and second pharyngeal arches (Faye‐Petersen et al., 2006; Opitz, Zanni, Reynolds, & Gilbert‐Barness, 2002).
Craniofacial development during embryo‐ and feto‐genesis requires complex interactions of several developmental molecular pathways. Among the pertinent pathways probably involved in the etiology of agnathia‐otocephaly, Sonic Hedgehog (SHH) signaling, bone morphogenetic (BMP) and fibroblast growth factor (FGF) signaling, WNT and glycosylphosphatidylinositol‐anchoring pathways have been suggested (Gekas, Li, & Kamnasaran, 2010). However, pathogenic variants in only two genes, PRRX1 (OMIM 167420) and OTX2 (OMIM 600037) were reported to be causal for the agnathia‐otocephaly phenotype in a few patients so far, suggesting further genetic heterogeneity. PRRX1 encodes a homeobox gene, which functions as transcriptional regulator (Kern, Argao, Birkenmeier, Rowe, & Potter, 1994), is expressed in undifferentiated human embryonic cranial neural crest cells and most abundantly in cardiac, skeletal, and smooth muscle tissues in adults (Çelik et al., 2012). It functions together with other proteins such as PRRX2 and also OTX2, encoded by a homeobox family gene expressed in the developing head, to regulate neural crest cells and the patterning of the first pharyngeal arch as well as skeletal and limb development.
Here we report on non‐consanguineous healthy parents of Albanian descent with a reproductive history of one early miscarriage but an otherwise unremarkable family history. Written informed consent for participation and publication of clinical data and pictures was obtained from the participants. The family was part of a larger study (Meier et al., 2019) with approval of the Ethics Commission Northwest Switzerland (EKNZ 2014‐174).
The family came to our attention during the second pregnancy at 19 + 2 weeks of gestation when severe micrognathia was detected by ultrasound and otocephaly suspected (Figure 1a,b). Fetal postmortem autopsy revealed an agnathia‐otocephaly complex with severe hypoplasia of the mandible and maxilla, ventral median position of the auricles (Figure 1c–d), cheilognathopalatoschisis, microglossia, larynx hypoplasia and esophageal hypoplasia. In addition, the fetus had an aortic isthmus stenosis, fused left lung lobes, a right unilateral rudimental 4th thoracic rib and a cake kidney positioned in the left pelvis with tubular ectasias and megalocytosis of the adrenal cortices. There was no additional brain or eye anomaly. We performed Sanger sequencing for all exons and intron boundaries for both PRRX1 and OTX2 but detected no variants.
Figure 1.
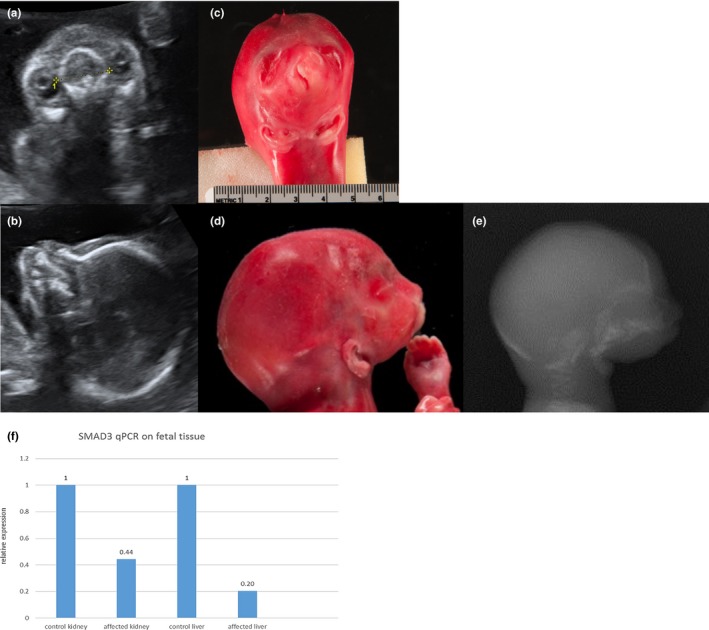
(a–e) The affected fetus at 19 + 2 weeks of gestation in prenatal ultrasound (a and b) and post mortem (c–e) showing cheilognathopalatoschisis, microstomia, absent mandible and ventral median positioned auricles. (f) Relative SMAD3 mRNA expression in kidney and liver tissue of an age‐matched control fetus and the affected fetus (f). RNA was extracted from FFPE tissue, the expression of SMAD3 was normalized to GUSB expression. FFPE, formalin fixed paraffin embedded
In order to further delineate the etiology of this phenotype we performed family‐based exome sequencing using the trio of fetal DNA extracted from formalin fixed paraffin embedded (FFPE) tissue and parental DNA extracted from blood samples. Library preparation was performed with the Nextera© Rapid Capture Kit for the TruSight One panel (Illumina). Paired‐end read sequencing (2 × 100 bp read length) was accomplished on an MiSeq platform (Illumina). Quality estimation of the sequence reads was performed by generating quality control statistics with FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). The Genome Analysis Toolkit (GATK, http://www.broadinstitute.org/gatk) was used to perform variant calling. Variants with a coverage ≤10X and not supported by at least four reads (20%) were discarded. Familial segregation of variants was performed to identify de novo, autosomal recessive and X‐linked recessive inheritance. Hypothesizing a rare Mendelian disorder, we annotated variants as known or novel according to presence or absence in curated databases. Variants were filtered on a heterozygous population frequency (GMAF) of <5% in control databases (dbSNP150, ExAC, 1000G, gnomAD) and absence of homozygosity in healthy individuals (ExAC, gnomAD). We prioritized variants according to their potential to disrupt protein function including the use of prediction tools (SIFT, Provean, Polyphen2, Mutationtaster, Human Splicing Finder v.3.0), the amino acid conservation (PhyloP, PhastCons) and the American College of Medical Genetics (ACMG) variant classification guidelines (Richards et al., 2015). Prioritized variants were inspected for phenotype–genotype correlations reported in humans and other species, based on the medical literature and public databases including functional and expression data (Pubmed, ClinVar, OMIM, HGMD, Uniprot, UniGene, http://www.zfin.org; http://www.informatics.jax.org; http://www.mousephenotype.org). We also focused on genes within the craniofacial developmental pathways including interaction partners of OTX2 and PRRX1.
In the list of de novo variants we prioritized a SMAD3 (OMIM 603109) missense variant in exon 6 (c.860G>A, (p.R287Q), LOVD #0000405920) out of a total of four candidate variants (including variants in KRT86, PRICKLE1, MYH3). This variant was previously described as likely pathogenic in a patient with Aneurysms‐osteoarthritis syndrome (AOS) and a patient with Loeys–Dietz‐Syndrome type 3 (LDS3) (Aubart et al., 2014; Schepers et al., 2018).
Sanger sequencing confirmed the variant in the fetus and its absence in the parents. In silico prediction tools rated the variant as truncating (SIFT, PolyPhen2, Mutationtaster), the HOPE protein prediction (http://www.cmbi.ru.nl/hope/) predicted a loss of hydrogen bonds and salt bridges in the protein, due to the amino acid exchange. According to the ACMG guidelines (Richards et al., 2015) the variant was classified as likely pathogenic (PM1, PM2, PP3, PP5). SMAD3 is a direct mediator of transcriptional activation by the TGF‐beta receptor. SMAD3 consists of nine exons that code for an MH1 and MH2 domain, to interact with other SMAD proteins and signal transducing receptors, that are connected through a linker. The variant is located in the MH2 domain of the protein that is highly conserved across species.
To confirm the functional consequence of the variant, mRNA was extracted following the protocol of the RecoverAll™ Kit (Ambion) from fetal FFPE liver and kidney tissue of the affected fetus and an age‐matched control fetus without structural anomalies. Kidney and liver were chosen based on tissue availability and the expression data in the Unigene database. Reverse transcription was performed with the High‐Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). SYBR™ Green PCR Master Mix (Applied Biosystems) was used for qPCR. Expression levels were normalized to GUSB expression. The results showed a significant decrease of SMAD3 mRNA (Figure 1f) up to 44% in kidney and 20% in liver tissue, confirming that the SMAD3 variant affects the expression of mRNA.
The variant c.860G>A (p.R287Q), we detected in SMAD3 in the fetus presenting with agnathia‐otocephaly, was previously reported to cause LDS3 (Aubart et al., 2014; Schepers et al., 2018), an autosomal dominant connective tissue disorder initially described as AOS (van de Laar et al., 2011). These patients present with generalized arterial tortuosity, arterial aneurysms and an increased risk for dissections, in particular for the ascending aorta, craniofacial anomalies including hypertelorism, micro‐/retro‐gnathia, bifid uvula and cleft palate, skeletal and cutaneous anomalies, and early‐onset osteoarthritis (van de Laar et al., 2012).
Because of the many overlapping clinical features of LDS type 1 and 2 patients, including the craniofacial anomalies, arterial tortuosity and widespread aneurysms, and harboring autosomal dominant variants in the disease‐responsible genes TGFBR1/2 and TGFB2/3 in the same TGF‐β signaling pathway, AOS is now usually classified as LDS type 3 (MacCarrick et al., 2014). Schepers and colleagues extensively reviewed known and novel variants in TGFB2/3 ad SMAD2/3 causing a broad phenotypical LDS spectrum (Schepers et al., 2018). We suggest, to extend the phenotypic spectrum to agnathia‐otocephaly which may represent the lethal or severe end of the TGFBR/SMAD‐pathway associated phenotypes.
The SMAD family member three gene (SMAD3; OMIM 603109) encodes the SMAD3 protein which belongs to the receptor‐activated (R)‐SMAD family. These proteins are intracellular effectors of the canonical transforming growth factor‐β (TGF‐β) signaling pathway including bone morphogenetic proteins (BMPs) that belong to the TGF‐β superfamily (Hata & Chen, 2016; Massagué, 2012). The SMAD3 protein responds to TGF‐β and regulates TGF‐β‐mediated chondrocyte and osteoblast differentiation (Wu, Chen, & Li, 2016). Several studies have shown that SMAD3 deficiency leads also to enhanced BMP signaling and accelerated chondrocyte differentiation which causes osteoarthritis (Li et al., 2006).
Furthermore, the SMAD3 protein acts in the same developmental pathway as OTX2 and PRRX1 as a transcriptional regulator. This pathway is involved in the development of the first pharyngeal arch in the lower jaw (Gekas et al., 2010). Knockout mice show the typical signs of an AOS but also micrognathia (Yang, 1999). A knockdown of SMAD3 in zebrafish resulted in a truncated anterior head and neuronal maldevelopment. However, absence of the lower jaw was not specifically mentioned (Casari et al., 2014).
SMAD3 acts in both the TGF‐β and BMP pathway that are crucial for embryonic development. Figure 2 depicts the complexity of the SMAD3‐related downstream processes. The BMP pathway, where SMAD3 acts as an intracellular effector, is also involved in bone formation (Rahman, Akhtar, Jamil, Banik, & Asaduzzaman, 2015). A knockout mouse model of Alk2, a BMP receptor, resulted in mice with a hypoplastic mandible, missing jugal bones and enlarged fontanels due to lack of ossification (Dudas, Sridurongrit, Nagy, Okazaki, & Kaartinen, 2004). In humans, variants in BMP4 cause anophthalmia, microphthalmia with brain and digit anomalies (Bakrania et al., 2008).
Figure 2.
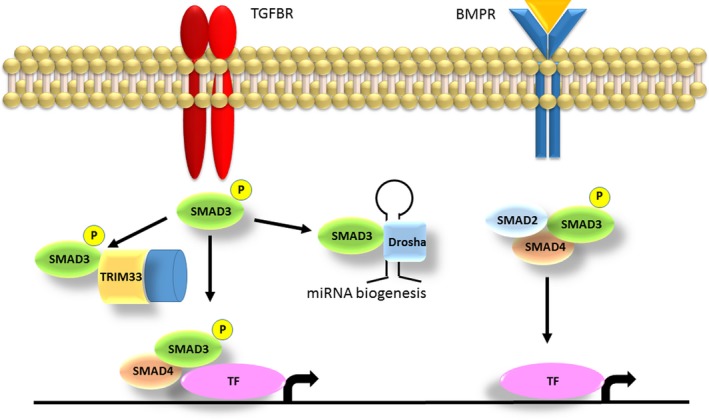
Roles of SMAD3 in the TGF‐β and the BMP pathway. The SMAD3 protein gets activated through TGF‐β signaling and can act in different downstream pathways. If SMAD3 binds to TRIM33, a chromatin reading and remodeling protein, the complex opens the histone to allow other TFs to enter the DNA. Complexes of SMAD3 and SMAD4 get recruited to different genes, and the transcription of a specific gene is determined by varying partner TFs. In those effector pathways SMAD3 is involved in chondrocyte and osteoblast maturation (Massagué, 2012). If SMAD3 binds to DROSHA it regulates the processing of different miRNA precursors. SMAD3 forming a complex with other SMAD proteins driven by BMP signaling also activates transcription via other TFs, regulating various bone formation processes (Rahman et al., 2015). TGF‐β, transforming growth factor‐β; TFs, transcription factors
Interestingly, a loss of function variant in SMAD3 was recently described as a possible candidate for a patient with holoprosencephaly (Roessler et al., 2018). This study concluded that most genes in which variants cause a holoprosencephaly phenotype act in the TGF‐β, hedgehog and FGF signaling pathways. This further supports our hypothesis of agnathia‐otocephaly being a SMAD3‐related phenotype, as agnathia‐otocephaly and holoprosencephaly can occur as a combined phenotype (Faye‐Petersen et al., 2006; Ozden, Fiçicioğlu, Kara, Oral, & Bilgiç, 2000; Puvabanditsin et al., 2006; Rodriguez et al., 2019; Wai & Chandran, 2017).
Agnathia‐otocephaly with or without associated anomalies is clinically and genetically heterogeneous and likely represents the lethal or severe end of a phenotypic spectrum related to variants in genes in the interacting SMAD/TGFB/BMP/SHH/FGF developmental pathways. In order to understand the disease mechanisms and the phenotypic variability, it will be necessary to further study genotype–phenotype correlations as well as the function of genes and their variants in these pathways.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Meier N, Bruder E, Miny P, Tercanli S, Filges I. Expanding the spectrum of SMAD3‐related phenotypes to agnathia‐otocephaly. Mol Genet Genomic Med. 2020;8:e1178 10.1002/mgg3.1178
Funding information
This work was supported by the Swiss National Science Foundation (SNSF), Project Grant to Isabel Filges (320030_160200) and the Freie Akademische Gesellschaft Basel (FAG) to Nicole Meier.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in LOVD at ://databases.lovd.nl, reference number #0000405920.
REFERENCES
- Aubart, M. , Gobert, D. , Aubart‐Cohen, F. , Detaint, D. , Hanna, N. , d’Indya, H. , … Jondeau, G. (2014). Early‐onset osteoarthritis, Charcot‐Marie‐Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: An unrecognized and life threatening condition. PLoS ONE, 9(5), e96387 10.1371/journal.pone.0096387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania, P. , Efthymiou, M. , Klein, J. C. , Salt, A. , Bunyan, D. J. , Wyatt, A. , … Ragge, N. K. (2008). Mutations in BMP4 cause eye, brain, and digit developmental anomalies: Overlap between the BMP4 and hedgehog signaling pathways. The American Journal of Human Genetics, 82(2), 304–319. 10.1016/J.AJHG.2007.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari, A. , Schiavone, M. , Facchinello, N. , Vettori, A. , Meyer, D. , Tiso, N. , … Argenton, F. (2014). A Smad3 transgenic reporter reveals TGF‐beta control of zebrafish spinal cord development. Developmental Biology, 396(1), 81–93. 10.1016/j.ydbio.2014.09.025 [DOI] [PubMed] [Google Scholar]
- Çelik, T. , Simsek, P. O. , Sozen, T. , Ozyuncu, O. , Utine, G. E. , Talim, B. , … Kamnasaran, D. (2012). PRRX1 is mutated in an otocephalic newborn infant conceived by consanguineous parents. Clinical Genetics, 81(3), 294–297. 10.1111/j.1399-0004.2011.01730.x [DOI] [PubMed] [Google Scholar]
- Dudas, M. , Sridurongrit, S. , Nagy, A. , Okazaki, K. , & Kaartinen, V. (2004). Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mechanisms of Development, 121(2), 173–182. 10.1016/j.mod.2003.12.003 [DOI] [PubMed] [Google Scholar]
- Faye‐Petersen, O. , David, E. , Rangwala, N. , Seaman, J. P. , Hua, Z. , & Heller, D. S. (2006). Otocephaly: Report of five new cases and a literature review. Fetal and Pediatric Pathology, 25(5), 277–296. 10.1080/15513810601123417 [DOI] [PubMed] [Google Scholar]
- Gekas, J. , Li, B. , & Kamnasaran, D. (2010). Current perspectives on the etiology of agnathia‐otocephaly. European Journal of Medical Genetics, 53(6), 358–366. 10.1016/j.ejmg.2010.09.002 [DOI] [PubMed] [Google Scholar]
- Hata, A. , & Chen, Y.‐G. (2016). TGF‐β signaling from receptors to Smads. Cold Spring Harbor Perspectives in Biology, 8(9), a022061 10.1101/cshperspect.a022061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, M. J. , Argao, E. A. , Birkenmeier, E. H. , Rowe, L. B. , & Potter, S. S. (1994). Genomic organization and chromosome localization of the murine homeobox gene Pmx. Genomics, 19(2), 334–340. 10.1006/geno.1994.1066 [DOI] [PubMed] [Google Scholar]
- Li, T.‐F. , Darowish, M. , Zuscik, M. J. , Chen, D. I. , Schwarz, E. M. , Rosier, R. N. , … O'Keefe, R. J. (2006). Smad3‐deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. Journal of Bone and Mineral Research, 21(1), 4–16. 10.1359/JBMR.050911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCarrick, G. , Black, J. H. , Bowdin, S. , El‐Hamamsy, I. , Frischmeyer‐Guerrerio, P. A. , Guerrerio, A. L. , … Dietz, H. C. (2014). Loeys‐Dietz syndrome: A primer for diagnosis and management. Genetics in Medicine, 16(8), 576–587. 10.1038/gim.2014.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué, J. (2012). TGFβ signalling in context. Nature Reviews. Molecular Cell Biology, 13(10), 616–630. 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, N. , Bruder, E. , Lapaire, O. , Hoesli, I. , Kang, A. , Hench, J. , … Filges, I. (2019). Exome sequencing of fetal anomaly syndromes: Novel phenotype-genotype discoveries. European Journal of Human Genetics, 27(5), 730–737. 10.1038/s41431-018-0324-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz, J. M. , Zanni, G. , Reynolds, J. F. Jr , & Gilbert-Barness, E. (2002). Defects of blastogenesis. American Journal of Medical Genetics, 115(4), 269–286. [DOI] [PubMed] [Google Scholar]
- Ozden, S. , Fiçicioğlu, C. , Kara, M. , Oral, O. , & Bilgiç, R. (2000). Agnathia‐holoprosencephaly‐situs inversus. American Journal of Medical Genetics, 91(3), 235–236. [DOI] [PubMed] [Google Scholar]
- Puvabanditsin, S. , Garrow, E. , Umaru, S. , Padilla, J. , Chowdawarapu, S. , & Biswas, A. (2006). Otocephaly, and pulmonary malformation association: Two case reports. Genetic Counseling (Geneva, Switzerland), 17(2), 167–171. [PubMed] [Google Scholar]
- Rahman, M. S. , Akhtar, N. , Jamil, H. M. , Banik, R. S. , & Asaduzzaman, S. M. (2015). TGF‐β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Research, 3, 15005 10.1038/boneres.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, N. , Casasbuenas, A. , Andreeva, E. , Odegova, N. , Wong, A. E. , & Sepulveda, W. (2019). First‐trimester diagnosis of agnathia‐otocephaly complex: A series of 4 cases and review of the literature. Journal of Ultrasound in Medicine, 38(3), 805–809. 10.1002/jum.14759 [DOI] [PubMed] [Google Scholar]
- Roessler, E. , Hu, P. , Marino, J. , Hong, S. , Hart, R. , Berger, S. , … Muenke, M. (2018). Common genetic causes of holoprosencephaly are limited to a small set of evolutionarily conserved driver genes of midline development coordinated by TGF‐β, hedgehog, and FGF signaling. Human Mutation, 39(10), 1416–1427. 10.1002/humu.23590 [DOI] [PubMed] [Google Scholar]
- Schepers, D. , Tortora, G. , Morisaki, H. , MacCarrick, G. , Lindsay, M. , Liang, D. , … Loeys, B. (2018). A mutation update on the LDS‐associated genes TGFB2/3 and SMAD2/3 . Human Mutation, 39(5), 621–634. 10.1002/humu.23407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Laar, I. M. B. H. , Oldenburg, R. A. , Pals, G. , Roos‐Hesselink, J. W. , de Graaf, B. M. , Verhagen, J. M. A. , … Bertoli‐Avella, A. M. (2011). Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early‐onset osteoarthritis. Nature Genetics, 43(2), 121–126. 10.1038/ng.744 [DOI] [PubMed] [Google Scholar]
- van de Laar, I. M. B. H. , van der Linde, D. , Oei, E. H. G. , Bos, P. K. , Bessems, J. H. , Bierma‐Zeinstra, S. M. , … Wessels, M. W. (2012). Phenotypic spectrum of the SMAD3‐related aneurysms‐osteoarthritis syndrome. Journal of Medical Genetics, 49(1), 47–57. 10.1136/jmedgenet-2011-100382 [DOI] [PubMed] [Google Scholar]
- Wai, L. T. , & Chandran, S. (2017). Cyclopia: Isolated and with agnathia–otocephaly complex. BMJ Case Reports, 2017, bcr‐2017‐220159 10.1136/bcr-2017-220159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M. , Chen, G. , & Li, Y. P. (2016). TGF‐β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Research, 4(1), 16009 10.1038/boneres.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. (1999). Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF‐beta. The EMBO Journal, 18(5), 1280–1291. 10.1093/emboj/18.5.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available in LOVD at ://databases.lovd.nl, reference number #0000405920.