

Abstract
Background
Panel‐based targeted exome sequencing was used to analyze the genetic and clinical findings of targeted genes in a cohort of northeast Chinese with retinitis pigmentosa.
Methods
A total of 87 subjects, comprising 23 probands and their family members (total patients: 32) with confirmed retinitis pigmentosa were recruited in the study. Panel‐based targeted exome sequencing was used to sequence the patients and family members, all subjects with retinitis pigmentosa underwent a complete ophthalmologic examination.
Results
Of the 23 probands, the clinical manifestations include night blindness, narrowing of vision, secondary cataracts, choroidal atrophy, color blindness, and high myopia, the average age of onset of night blindness is 12.9 ± 14 (range, 0–65; median, 8). Posterior subcapsular opacities is the most common forms of secondary cataracts (nine cases, 39.1%), and peripheral choroidal atrophy is the most common form of secondary choroidal atrophy (12 cases, 52.2%). Of these probands with complication peripheral choroidal atrophy, there were eight probands (66.7%, 8/12) caused by the pathogenic variation in USH2A gene. A total of 17 genes and 45 variants were detected in 23 probands. Among these genes, the commonest genes were USH2A (40%; 18/45), RP1 (15.6%; 7/45), and EYS (8.9%; 4/45), and the top three genes account for 56.5% (13/23) of diagnostic probands. Among these variants, comprising 22 (48.9%) pathogenic variants, 14 (31%) likely pathogenic variants, and nine (20%) uncertain clinical significance variants, and 22 variants was discovered first time. Most of the mutations associated with RP were missense (53.3%, 24/45), and the remaining mutation types include frameshift (35.6%, 16/45), nonsense (6.7%, 3/45), and spliceSite (4.4%, 2/45). Among the probands with mutations detected, compound heterozygous forms was detected in 13 (56.5%, 13/23) probands, and digenic inheritance (DI) forms was detected in five (21.7%, 5/23) probands.
Conclusion
Panel‐based targeted exome sequencing revealed 23 novel mutations, recognized different combinations forms of variants, and extended the mutational spectrum of retinitis pigmentosa and depicted common variants in northeast China.
Keywords: compound heterozygous, digenic inheritance, panel‐based targeted exome sequencing, retinitis pigmentosa
We applied panel‐based targeted exome sequencing to explore the pathogenic variation spectrum and genetic characteristics of retinitis pigmentosa populations in northeast China, and have a deeper understanding of the relationship between clinical manifestations and genotypes.

1. INTRODUCTION
Retinitis pigmentosa (RP, MIM#500004) is the most common group of disorder in retinal diseases, with a common manifestation of progressive photoreceptor cells and loss of retinal pigment epithelial function. Retinitis pigmentosa has a strong clinical heterogeneity (Ayuso & Millan, 2010; Galan et al., 2011). The early typical features mainly start from the peripheral retina and gradually develop into the fovea. The main symptoms include night blindness, progressive reduction of visual field, and ultimately tubular vision and blindness. It is the most common blinding monogenic hereditary fundus disease with a prevalence of approximately 1/3,500–1/5,000 (Galan et al., 2011). The pathogenesis of retinitis pigmentosa is closely related to genetic factors. Different gene mutations have different clinical phenotypes, and the specific pathogenesis research is still unclear (Schuster, 2008).
The clinical and genetic heterogeneity of retinitis pigmentosa is difficult for clinical diagnosis and differential diagnosis. Panel‐based targeted exome sequencing has been widely used in genetic disease screening and clinical diagnosis. With the continuous advancement of this technology and the continuous reduction in the cost of genetic testing, genetic detection‐assisted diagnosis of patients with retinitis pigmentosa and family members has become a reality. Despite the development of targeted sequencing screening strategies for identifying known genes associated with RP, it is estimated that 40% of cases are still not molecularly diagnosed, indicating that there are still many novel mutations in known disease‐causing genes and novel retinitis pigmentosa diseases‐causative gene not found (Ellingford et al., 2016). Here, we reported the mutational spectrum of 23 probands diagnosed with retinitis pigmentosa. The characteristics and clinical manifestations of retinitis pigmentosa in the northeast Chinese population were analyzed, which provided assistance for subsequent clinical diagnosis and genetic counseling.
2. MATERIALS AND METHODS
2.1. Subjects and ethics statement
A total of 87 subjects from 23 families with retinitis pigmentosa diagnosed in Shenyang He Eye Specialist Hospital from January 2017 to July 2018 were recruited in the study. Among these subjects, including 44 males and 43 females, aged 9–88 years old, with a median of 36 years old. Inclusion criteria for patients: (a) night blindness; (b) gradual decline in visual acuity; (c) typical fundus changes, optic disc with waxy yellow atrophy, retinal osteoblast‐like pigmentation, thinning of blood vessels, blue–gray retina; (d) The early peripheral visual field exhibits a circular dark spot, and the late visual field narrows toward the center; (e) After dark adaptation and light adaptation in the early stage of the lesion, the full‐field electroretinogram (ERG) examination showed a decrease in rod function, and decrease in cone function at the same time in the late stage (Hartong, Berson, and Dryja, 2006; Xu, Hu, Ma, Li, and Jonas, 2006). All the examinations and tests involved in this study were approved by the Ethics Committee of Shenyang He Eye Specialist Hospital (approval number: IRB (2016) K001.01), following the Helsinki Declaration, and obtaining informed consent from patients and family members.
2.2. Clinical assessment
Both patients and family members underwent a comprehensive clinical examination to confirm the diagnosis and to exclude ocular diseases caused by nongenetic factors. Clinical examinations include medical history inquiries and physical examinations. Among them, medical history inquiry includes basic personal information (including gender, age, place of origin, ethnicity, etc.), chief complaint, current medical history (age of onset, regularity, treatment status, medication status, type and time of complications, etc.), past history (full body Situation, history of genetic testing, etc.), family history, history of surgery and drug use, history of marriage and childbirth. Physical examination includes vision and corrected visual acuity, non‐contact intraocular pressure, color vision, B‐ultrasound (Tianjin Mida, model ODM‐2200), visual field (Carl Zeiss, model 750i), anterior segment photography, fundus color photography (Topcon, model TRC NW‐300), optical coherence tomography (OCT) (macular, optic nerve layer thickness, Angiography) (Cirrus HD‐OCT 5000), and full‐field electroretinography (ERG). In addition, patients also underwent conventional elbow venous blood collection.
2.3. Panel‐based targeted exome sequencing
All eligible patients and family members routinely collected 5 ml of elbow venous blood, ETDA anticoagulation, and stored at −80°C. The DNA was extracted from whole blood using the FlexiGene DNA Kit (Qiagen) according to the manufacturer's protocols. We designed a Panel‐based high‐throughput targeted enrichment method to capture exon capture regions of 792 genes associated with common Inherited eye diseases (Table S1). The Capture Panel (Target_Eye_792_V2 chip) was custom designed and produced by the Beijing Genomics Institute (BGI) (Gao, Li, et al., 2019; Gao, Qi, Hu, Wang, & Wu, 2019; Hu et al., 2019; Li et al., 2019). To obtain the probe sequence, we obtained the exon sequence of 792 genes and its flanking ±30 bp from the reference human genome (UCSC hg 38). On average, the mean coverage depth was more than 300X, and the coverage of target region was ~99.9% using BGISEQ‐2000 (BGI, Inc.).
2.4. Bioinformatics analysis
We aligned sequence reads to the reference human genome (UCSC hg38) using the Burrows–Wheeler aligner version 0.7.10 (BWA‐MEM). Analysis of the data obtained using previous similar research methods (Gao, Li, et al., 2019). Using the Human Gene Mutation Database, Online Mendelian Inheritance in Man, and the ClinVar database and reference literature reports identified previously reported variations. According to the standards of the American College of Medical Genetics (ACMG), Variants was classified as pathogenic, likely pathogenic, and novel variants of uncertain clinical significance (Richards et al., 2015). Four online function prediction software was used to assess the potential deleterious of the variation, including SIFT (SIFT, http://sift.jcvi.org/), MutationTaster (http://www.mutationtaster.org/), FATHMM (http://fathmm.biocompute.org.uk/), and LRT (http://www.genetics.wustl.edu/jflab/). The obtained candidate variants were first verified by Sanger sequencing or quantitative real time polymerase chain reaction, then reviewed by clinical geneticists and ophthalmologists, and validation of variant segregates with the disease within the family.
3. RESULTS
3.1. Clinical presentation and genetic finding
In this study, there were 23 families (23 probands and their relatives), comprising 32 patients with clinically diagnosed retinitis pigmentosa and 55 subjects with normal visual acuity. Among the 32 patients, six of them had best corrected visual acuity of both eyes greater than 0.1, nine of them had the best corrected visual acuity of both eyes less than finger count (FC), and the remaining patients were between the two level (FC ~0.1). Among the 23 probands, the average age of probands was 48.2 ± 17.7 (range, 21–81; median, 43), and the average age of onset of night blindness was 12.9 ± 14 (range, 0–65; median, 8), of which the majority age of onset of night blindness were under 10 years old, accounts for 65% (15/23) (Figure 1a). The average duration of disease in the 23 probands was 35.3 ± 17.3 (range, 13–72; median, 31). Clinical manifestations of different forms of concurrent lens opacities was found in 17th probands, including nine probands with posterior subcapsular opacities, four probands with anterior subcapsular opacities, two probands with punctate opacities, and two probands with nuclear opacities. Clinical manifestations of different forms concurrent choroidal atrophy was found in 21 probands, including 12 probands with peripheral choroidal atrophy, five probands with total choroidal atrophy, and four probands with posterior choroidal atrophy. Of these probands with peripheral choroidal atrophy, the results showed that eight (66.7%, 8/12) of these probands were caused by the mutation of the USH2A gene. A total of 13 probands with systemic or other ocular manifestations, including five probands with color blindness and two probands with high myopia. Clinical information of the probands with definitive diagnosis is shown in Table 1.
Figure 1.
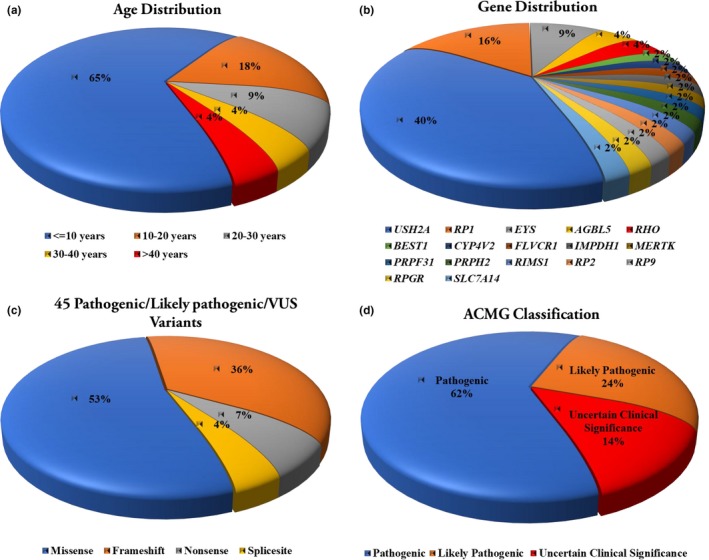
Basic information of clinical presentation and genetic finding of the target exome sequencing in the patients. (a) The age distribution of the total Patients, including age ≤10 years (n = 15), 10–20 years (n = 4), 20–30 years (n = 2), 30–40 years (n = 1), and >40 years (n = 1); (b), Distribution of retinitis pigmentosa (RP)‐causative genes in the 32 patients. Mutations were identified in 17 genes, with 65% of the mutations found in the top three genes (USH2A, RP1, EYS). (c) Forty‐five pathogenic/likely pathogenic/uncertain clinical significance variants were identified, including missense (n = 24), frameshift (n = 16), nonsense (n = 3), and splicing (n = 2) variants. (d) Forty‐five pathogenic/likely pathogenic/uncertain clinical significance variants were identified, including pathogenic (n = 22), likely pathogenic (n = 14), and uncertain clinical significance (n = 9) variants. Of these variants, 23 of them were described for the first time
Table 1.
Basic information of clinical presentation in 23 probands with retinitis pigmentosa
| Family ID | Sex | Age at examination (Yr) | BCVA (OD, OS) | Clinical diagnosis | History | Age of onset (Yr) | Lens opacity | Choroidal atrophy | Other ocular manifestation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 23 | FC/FC | Retinitis pigmentosa | N | 6 | N | N | Retinal detachment |
| 2 | F | 55 | 0.02/0.3 | Retinitis pigmentosa | N | 4 | Posterior subcapsular | Peripheral | N |
| 3 | M | 81 | 0.05/FC | Retinitis pigmentosa | N | 65 | Nuclear | Total choroid | N |
| 4 | F | 62 | 0.01/0.03 | Retinitis pigmentosa | Y | 5 | Nuclear | Peripheral | Red–green color blindness |
| 5 | F | 79 | 0.1/HM | Retinitis pigmentosa | Y | 7 | Anterior subcapsular | Posterior pole | blue color blindness |
| 6 | M | 39 | HM/HM | Retinitis pigmentosa | Y | 5 | Posterior subcapsular | Posterior pole | N |
| 7 | F | 37 | 0.6/0.8 | Retinitis pigmentosa | N | 12 | N | N | Retinal crystal deposit |
| 8 | F | 68 | HM/HM | Retinitis pigmentosa | N | 15 | Posterior subcapsular | Posterior pole | N |
| 9 | F | 33 | 0.6/0.5 | Retinitis pigmentosa | N | 12 | N | Peripheral | Hypotension |
| 10 | M | 45 | 0.1/0.05 | Retinitis pigmentosa | N | 8 | Posterior subcapsular | Peripheral | N |
| 11 | F | 56 | FC/HM | Retinitis pigmentosa | N | 30 | Posterior subcapsular | Peripheral | Hearing loss |
| 12 | M | 72 | HM/HM | Retinitis pigmentosa | N | 7 | Anterior subcapsular | Posterior pole | N |
| 13 | F | 39 | HM/HM | Retinitis pigmentosa | N | 8 | punctate | Total choroid | Macular hole |
| 14 | M | 21 | HM/HM | Retinitis pigmentosa | N | 0 | Posterior subcapsular | Total choroid | Total color blindness |
| 15 | M | 29 | HM/HM | Retinitis pigmentosa | N | 9 | Posterior subcapsular | Total choroid | High myopia |
| 16 | F | 66 | 0.3/0.25 | Retinitis pigmentosa | Y | 8 | Posterior subcapsular | Peripheral | N |
| 17 | F | 38 | 0.15/0.5 | Retinitis pigmentosa | N | 25 | N | Peripheral | N |
| 18 | M | 64 | 0.05/0.05 | Retinitis pigmentosa | Y | 32 | Posterior subcapsular | Peripheral | Ménière disease |
| 19 | M | 43 | 0.1/0.2 | Retinitis pigmentosa | Y | 8 | N | Peripheral | N |
| 20 | F | 31 | 0.1/0.2 | Retinitis pigmentosa | N | 14 | punctate | Peripheral | blue color blindness |
| 21 | M | 36 | 0.05/0.05 | Retinitis pigmentosa | N | 5 | N | Peripheral | Red–green color blindness |
| 22 | M | 36 | 0.05/0.1 | Retinitis pigmentosa | N | 8 | Anterior subcapsular | Peripheral | N |
| 23 | M | 56 | HM/HM | Retinitis pigmentosa | Y | 3 | Anterior subcapsular | Total choroid | High myopia |
Abbreviations: F, female; FC, finger count; HM, high myopia; M, male; N, No; Y, Yes.
A total of 45 pathogenic/likely pathogenic/uncertain clinical significance variants were identified among 17 retinitis pigmentosa (RP)‐causative genes, including pathogenic (n = 22), likely pathogenic (n = 14), and uncertain clinical significance (n = 9) variants, with 65% of the mutations found in the top three genes (USH2A, MIM#608400; RP1, MIM#603937; EYS, MIM#612424), and the top three genes account for 56.5% of diagnostic probands.. Among these variants, including missense (n = 24), frameshift (n = 16), nonsense (n = 3), and splicing (n = 2) mutations, 23 of them were described for the first time (Figure 1b–d). Among the probands with mutations detected, compound heterozygous forms was detected in 13 (56.5%, 13/23) probands, and digenic inheritance forms was detected in five (21.7%, 5/23) probands. Of them, the most common compound heterozygous forms is the mutation of different alleles of the USH2A gene (61.5%, 8/13). Consistent with previous studies, most RP patients carried compound heterozygous variants, but few patients detected digenic gene variants. In addition, five variants of three genes were detected in one proband, and four variants of the RP1 gene were detected in another one proband. Among the first discovered candidate pathogenic mutations, seven unreported mutations were found in the USH2A gene, and five unreported mutations were found in the RP1 gene. Two unreported candidate pathogenic mutations were found for the AGBL5 gene and the EYS gene, respectively. A novel candidate pathogenic variation was detected in the FLVCR1 (MIM#609144), MERTK (MIM#604705), PRPF31 (MIM#606419), RP2 (MIM#300757), RP9 (MIM#607331), and RPGR (MIM#312610) genes, respectively (Table 2).
Table 2.
Genetics finding in the probands with retinitis pigmentosa
| Family ID | Gene | MutName | Amino acid change | Exon Intron ID | Zygous | Chr:por:mut | Functional change | SIFT | Polyphen2 | Mutation taster | Clinical significance | Inheritance mode | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IMPDH1 | c.942_944delGAA | p.Lys314del | EX10/CDS10 | Het | chr7:128398543:GTTC > G | Frameshift | . | . | . | P | AD | Jin, Qu, Qu, Meng, Xu, & Yin, 2014 |
| 2 | SLC7A14 | c.1391G > T | p.Cys464Phe | EX7/CDS6 | Het | chr3:170198680 | Missense | Dam | Neu | . | P | AR | Jin, Huang, et al., 2014 |
| PRPF31 | c.357_358delAA | p.Ser119SerfsX5 | EX5/CDS4 | Het | chr19:54625910.0.54625911 | Frameshift | . | . | . | P | AR | Jianping et al., 2018 | |
| 3 | PRPH2 | c.205delG | p.Val69CysfsX30 | EX1/CDS1 | Het | chr6:42689868 | Frameshift | . | . | . | P | AD | Manes et al., 2015 |
| 4 | RHO | c.403C > T | p.Arg135Trp | EX2/CDS2 | Het | chr3:129530917:C > T | Missense | . | DC | PD | P | AD | Yu et al., 2016 |
| 5 | RHO | c.403C > T | p.Arg135Trp | EX2/CDS2 | Het | chr3:129530917:C > T | Missense | . | DC | PD | P | AD | Yu et al., 2016 |
| 6 | RP2 | c.348_349insT | p.Phe117Phefs7 | EX2/CDS2 | Hemi | chrX:46853721 | Frameshift | . | . | . | LP | XL | This study |
| RP1 | c.1419_1420delTG | p.Thr473Thrfs13 | EX4/CDS3 | Het | chr8:54625300 | Frameshift | . | . | . | LP | XL | This study | |
| 7 | AGBL5 | c.1406C > G | p.Ser469Ter | EX8/CDS7 | Het | chr2:27056663 | Nonsense | . | . | PD | LP | AR | This study |
| AGBL5 | c.1498C > T | p.Arg500Cys | EX8/CDS7 | Het | chr2:27056755 | Missense | Dam | . | PD | VUS | AR | This study | |
| 8 | BEST1 | c.584C > T | p.Ala195Val | EX4/CDS3 | Het | chr11:61956946:C > T | Missense | Dam | PD | P | AD | Gao, Qi, et al., 2019 | |
| 9 | RP9 | c.511_512delGA | p.Glu171ArgfsX2 | EX6/CDS6 | Het | chr7:33135000.0.33135001 | Frameshift | . | . | . | LP | AD | This study |
| CYP4V2 | c.283G > A | p.Gly95Arg | EX2/CDS2 | Het | chr4:187115722 | Missense | . | . | . | P | AR | Aodon et al, 2017 | |
| 10 | EYS | c.8012T > A | p.Leu2671Ter | EX41/CDS38 | Het | chr6:63762520:A > T | Nonsense | . | . | PD | P | AR | Sengillo et al., 2018 |
| EYS | c.6416G > A | p.Cys2139Tyr | EX31/CDS28 | Het | chr6:64230600:C > T | Missense | . | . | Pol | P | AR | Chen et al., 2015 | |
| 11 | FLVCR1 | c.719delC | p.Thr240ThrfsX20 | EX1/CDS1 | Het | chr1:213032513 | Frameshift | . | . | . | LP | AR | This study |
| RIMS1 | c.3136delA | p.Lys1046LysfsX32 | EX20/CDS20 | Het | chr6:72974697 | Frameshift | . | . | . | LP | AR | This study | |
| USH2A | c.2802T > G | p.Cys934Trp | EX13/CDS12 | Het | chr1:216419934 | Missense | Dam | . | PD | P | AR | Lenassi et al., 2015 | |
| USH2A | c.13939G > C | p.Gly4647Arg | EX64/CDS63 | Het | chr1:215844508 | Missense | . | . | . | VUS | AR | This study | |
| USH2A | c.10830G > C | p.Trp3610Cys | EX55/CDS54 | Het | chr1:215953294 | Missense | . | . | . | VUS | AR | This study | |
| 12 | MERTK | c.225delA | p.Thr75Thrfs4 | EX2/CDS2 | Hom | chr2:111929282:CA > C | Frameshift | . | . | . | LP | AR | This study |
| EYS | c.2756G > A | p.Gly919Glu | EX18/CDS15 | Het | chr6:64902203:C > T | Missense | Dam | . | Pol | VUS | AR | This study | |
| EYS | c.6410G > A | p.Arg2137His | EX31/CDS28 | Het | chr6:64230606:C > T | Missense | Dam | . | Pol | VUS | AR | This study | |
| 13 | RP1 | c.2886delA | p.Gly962GlyfsX3 | EX4/CDS3 | Het | chr8:55539328 | Frameshift | . | . | . | LP | AD | This study |
| RP1 | c.4129delG | p.Asp1377ThrfsX20 | EX4/CDS3 | Het | chr8:55540571 | Frameshift | . | . | . | LP | AD | This study | |
| 14 | RP1 | c.4168_4169insT | p.His1390Serfs6 | EX4/CDS3 | Het | chr8:54628050 | Frameshift | . | . | . | LP | AR | This study |
| RP1 | c.4169A > G | p.His1390Arg | EX4/CDS3 | Het | chr8:54628051 | Missense | Dam | . | Pol | VUS | AR | This study | |
| RP1 | c.4196delG | p.Cys1399Leufs5 | EX4/CDS3 | Het | chr8:54628077 | Frameshift | . | . | . | P | AR | Jing et al., 2014 | |
| RP1 | c.6353G > A | p.Ser2118Asn | EX4/CDS3 | Het | chr8:54630235 | Missense | Dam | . | Pol | P | AR | Jing et al., 2014 | |
| 15 | USH2A | c.14285A > G | p.Asn4762Ser | EX65/CDS64 | Hom | chr1:215650650:T > C | Missense | Dam | . | PD | P | AR | Xu et al., 2014 |
| 16 | USH2A | c.8641_8642insTATT | p.Ser2881Tyrfs9 | EX43/CDS42 | Het | chr1:215877797 | Frameshift | . | . | . | LP | AR | This study |
| USH2A | c.13465G > A | p.Gly4489Ser | EX63/CDS62 | Het | chr1:215674446 | Missense | Tol | . | PD | VUS | AR | This study | |
| 17 | USH2A | c.10601A > G | p.Tyr3534Cys | EX54/CDS53 | Het | chr1:215782181:T > C | Missense | Dam | . | PD | VUS | AR | This study |
| USH2A | c.8559‐2A > G | _ | Intron42 | Het | chr1:215877882:T > C | SpliceSite | . | . | PD | P | AR | Lulin et al., 2018 | |
| 18 | USH2A | c.7075_7076delTT | p.Leu2359Asnfs*17 | EX37/CDS36 | Het | chr1:215965360:TAA > T | Frameshift | . | . | . | LP | AR | This study |
| USH2A | c.2802T > G | p.Cys934Trp | EX13/CDS12 | Het | chr1:216246592:A > C | Missense | . | . | PD | P | AR | Lenassi et al., 2015 | |
| 19 | USH2A | c.4021G > C | p.Ala1341Pro | EX18/CDS17 | Het | chr1:216198375:C > G | Missense | Dam | . | PD | VUS | AR | This study |
| USH2A | c.8559‐2A > G | _ | Intron42 | Het | chr1:215877882:T > C | SpliceSite | . | . | PD | P | AR | Lulin et al., 2018 | |
| 20 | USH2A | c.99_100insT | p.Ser33Serfs42 | EX2/CDS1 | Het | chr1:216422237 | Frameshift | . | . | . | P | AR | Dai, Zhang, Zhao, Deng, & Li, 2008 |
| USH2A | c.8254G > A | p.Gly2752Arg | EX42/CDS41 | Het | chr1:215879068 | Missense | Dam | . | PD | P | AR | Perez‐Carro et al., 2016 | |
| 21 | USH2A | c.9469C > T | p.Gln3157Ter | EX48/CDS47 | Het | chr1:215990440 | Nonsense | . | . | . | P | AR | Huang et al., 2013 |
| USH2A | c.11156G > A | p.Arg3719His | EX57/CDS56 | Het | chr1:215933077 | Missense | . | . | . | LP | AR | Lulin et al., 2018 | |
| 22 | USH2A | c.8232G > C | p.Trp2744Cys | EX42/CDS41 | Het | chr1:215879090 | Missense | Dam | . | PD | P | AR | Sodi, Mariottini, Passerini, Murro, & Torricelli, 2014 |
| USH2A | c.2802T > G | p.Cys934Trp | EX13/CDS12 | Het | chr1:216246592 | Missense | . | . | PD | P | AR | Lenassi et al., 2015 | |
| 23 | RPGR | c.905G > A | p.Cys302Tyr | EX8/CDS8 | Hemi | chrX:38304664:C > T | Missense | . | DC | PD | LP | XL | This study |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; Ben, benign; Dam, damaging; DC, disease causing; Del, deleterious; LP, likely pathogenic; Neu, neutral; P, pathogenic; PD, probably damaging; Pol, polymorphism; Tol, tolerated; VUS, uncertain clinical significance; XL, X‐linked inheritance.
3.2. Genetics analysis
A total of 45 variants of retinitis pigmentosa (RP)‐causative genes were identified in the total cohort, including 22 novel variants and 23 known variants. Overall, there were 22 pathogenic variants, 14 likely pathogenic variants, and nine uncertain clinical significance variants, the overall variation detection rate was 80% (36/45). The majority of likely pathogenic variants (n = 13) and all uncertain‐significance (n = 9) were novel variants. Three of the most common known pathogenic variations in two genes (USH2A and RHO) were detected among seven probands, the three commonest variants were USH2A c.2802T > G p.Cys934Trp (13%, 3/23), c.8559‐2A > G (8.7%, 2/23), and RHO c.403C > T c.403C > T (8.7%, 2/23), and all of these were known pathogenic variations.
3.3. Genotype‐phenotype correlations
The proband of the family seven detected a compound heterozygous novel mutation in the AGBL5 gene c.1406C > G (p.Ser469Ter) and c.1498C > T (p.Arg500Cys). After Sanger verification, the patient's father carried the c.1406C > G missense heterozygous mutation with AGBL5 gene, and the patient's mother carried a c.1498C > T missense heterozygous mutation with AGBL5 gene, but the patient's parental eye examination was normal (Figure S1). Through the co‐segregation verification and clinical phenotypic analysis, we consider AGBL5 gene mutations as candidate pathogenic variants in this family of retinitis pigmentosa. The proband had symptoms of night blindness from the age of 12, but the visual acuity was not affected, and the best corrected visual acuity remained at 0.6/0.8. Clinical fundus photography and fundus puzzles show that except for the posterior macular area, a medium amount of bright yellow granule‐like crystals were deposited in the equatorial region and the peripheral retina, with only a small amount of scattered pigmentation was observed, and the retina showed a mottled appearance. Autofluorescence photography showed that the autofluorescence of retinal pigment cells was confined to the central region of the posterior pole, and the peripheral fluorescence was significantly attenuated. This performance was highly consistent with the choroidal atrophy and visual field. Binocular visual field examination showed that the visual field was tubular and the peripheral visual field was completely lost; the flash ERG detection indicated that the Rod–Cone response of both eyes was quenched; The corneal topographic examination suggested that the corneas of both eyes were highly retrograde astigmatism, and the astigmatism was −2.5D (right eye) and −2.1D (left eye), respectively (Figure S2).
In the two families of retinitis pigmentosa in the 13th and 14th, three pairs of compound heterozygous novel mutations of the RP1 gene were detected, and six variants were all in exon 4. Among them, the proband of the 13th family detected a compound heterozygous novel frameshift mutations in the RP1 gene c.2886delA (p.Gly962GlyfsX3), and c.4129delG (p.Asp1377ThrfsX20). Combined with clinical phenotypic analysis, we confirmed that the compound heterozygous mutation was a causative mutation of binocular retinitis pigmentosa. At the age of 8 years, the proband began to experience night blindness. At the age of 20, his vision decreased. At the age of 30, he developed a macular hole. The fundus photography of the eyes showed a small amount of osteoblast‐like pigmentation in the retina, involving the macula. The blood vessels and choroids are slightly atrophied, and autofluorescence indicates that the retinal pigment background fade, and the visual field examination is irregular (Figure S3). The proband of the 14th family detected two pair of compound heterozygous mutations in the RP1 gene: c.4168_4169insT (p.His1390Serfs6), c.4169A > G (p.His1390Arg), c.4196delG (p.Cys1399Leufs5) and c.6353G > A (p.Ser2118Asn). Three of these variants occurred in a gene mutation region ranging from 4,168 to 4,196 in a total of 28 base sequences. By Sanger verification, the c.4168_4169insT and c.4169A > G mutations were detected in the patient's father, while the c.4196delG and c.6353G > A mutations were detected in the patient's mother, but the patient's clinical examination was normal. The patient developed night blindness within 1 year of age. At 12 years old, his vision decreased, with posterior subcapsular cataract and full color blindness. Except for the posterior macular area, the remaining retina had a blue–gray color and did not show obvious osteocyte‐like pigmentation. Fluorescein fundus angiography suggested that the fluorescent background of retinal pigmentation in the fundus was extremely low or absent, showing a mottled appearance. OCT showed that the fovea of the eyes was extremely atrophied, and the thickness was only 37 μm for the right eye and 31 μm for the left eye (Figure S4).
4. DISCUSSION
Of the 23 retinitis pigmentosa families included in the study, 17 probands had clinical manifestations of concurrent lens opacities, and the posterior subcapsular opacities was predominant (nine cases, accounting for 39.13%), suggesting that posterior subcapsular cataracts is the commonest forms of retinitis pigmentosa combined with complication cataract. At the same time, the clinical manifestations of probands with retinitis pigmentosa combined with complication choroidal atrophy were also inconsistent, with peripheral choroidal atrophy as the commonest form (12 cases, accounting for 57.14%). Of these probands with complication peripheral choroidal atrophy, there were eight probands (8/12, 66.7%) caused by the pathogenic variation of USH2A gene. However, there were no significant genotypic differences in probands with concurrent lens opacities. In terms of best corrected visual acuity, peripheral choroidal atrophy has better best corrected visual acuity than patients with posterior choroidal atrophy (the proportion of eyes with visual acuity greater than 0.05 was 79.17% and 12.50%, respectively). It indicated that the best corrected visual acuity may be related to the degree of choroidal atrophy and the degree of pigment epithelium and cone atrophy. Among the probands with mutations detected, compound heterozygous forms was detected in 13 (56.5%, 13/23) probands, and digenic inheritance forms was detected in five (21.7%, 5/23) probands. It suggested that the compound heterozygous forms and digenic inheritance forms are one of the most striking features of the genetic pathogenesis of retinitis pigmentosa (Audo et al., 2012; Corton et al., 2013; Daiger, Sullivan, & Bowne, 2013; Veltel & Wittinghofer, 2009).
The RP1 gene is one of the most common pathogenic genes of retinitis pigmentosa, which encodes oxyregulin 1, involved in the development of photoreceptors and the transport of proteins or the maintenance of cilia between the inner and outer segments, as well as the formation of tissue structures in the outer segments of photoreceptors, and the regulation of photoreceptor microtubules, etc., (Astuti et al., 2016; Liu, Zhou, Daiger, Farber, & Pierce, 2002; Pierce et al., 1999) it's main genetic methods include adRP and arRP (Bowne, 1999; Khaliq, 2005). In the present study, mutations associated with the RP1 gene were detected in probands No.13 and No.14. The difference is that the c.2886delA and c.4129delG mutations of the RP1 gene detected in 13th proband were frameshift mutations, causing a change in the reading frame, resulting in a change in the amino acid sequence of the encoded protein. For the 14th patient, there were four pathogenic mutations in the RP1 gene, c.4168_4169insT and c.4196delG mutations were insertion and deletion frameshift mutations, and c.4169A > G and c.6353G > A mutations were missense mutations. The patient's four heterozygous mutations in the RP1 gene are located at different loci, eventually forming two pairs of compound heterozygous mutations. The effect of two pairs of compound heterozygous mutants may be more severe than that of a pair of compound heterozygous mutations. Corresponding to clinical manifestations, the degree of fundus lesions in proband No. 14 was also much severe than that in patients with No. 13. The age of onset of night blindness in proband No. 14 was less than 1‐year‐old, with full color blindness, severe macular atrophy, disappearance of the outer nuclear layer and cone membrane disc structure of the fovea, and autofluorescence suggesting that the fluorescence of the whole pigment epithelium disappeared, especially obviously of the arterial vascular atrophy. However, the 13th patient began to develop night blindness at the age of 8 years, without color blindness, mild atrophy of blood vessels and choroids, and autofluorescence suggesting that the retinal pigment background fades.
In this study, Proband No. seven is a crystalline retinitis pigmentosa caused by mutation of AGBL5 gene, also known as retinitis pigmentosa 75. Its common genetic pattern is autosomal recessive inheritance. AGBL5 gene encodes protein ATP/GTP binding protein‐like five, which is involved in the regulation of microtubules outside photoreceptors (Branham et al., 2016; Kastner et al., 2015; Patel et al., 2016). The protein is a member of the cytoplasmic carboxypeptidase (CCP) family, which also includes AGTPBP1 (Nna‐1, CCP1, MIM#606830), AGBL1 (CCP4, MIM#615496), and AGBL4 (CCP6, MIM#616476). The main function of these proteins is to remove long‐chain glutamate chains from tubulin, whereas the AGBL5 protein functions in reverse. It mainly localizes the glutamate branching point on tubulin, thereby prolonging the glutamic acid chain (Rogowski et al., 2010). There are few reports of retinitis pigmentosa caused by mutation of AGBL5 gene, and only a few reports indicate that homozygous nonsense mutation of AGBL5 gene can cause non‐syndromic retinitis pigmentosa (Kastner et al., 2015). In turn, it affects the function of tubulin, which eventually leads to atrophy of the optic nerve cell layer and internal and external plexiform layers, causing retinal degenerative diseases. The AGBL5 gene mutation found in this study belongs to a compound heterozygous mutation consisting of p.Ser469Ter (c.1406C > G) and p.Arg500Cys (c.1498C > T), all of which were first discovered novel variations. The proband showed blurred vision at the age of eight and decreased in visual acuity at 12 years of age with posterior subcapsular cataract (PSC). Through the co‐segregation and Sanger verification, it was found that each parent carries a mutation in the gene locus, but the parents have no abnormal clinical manifestations. The crystallized retinitis pigmentosa (BCD) of the probands is different from the BCD manifestation caused by the typical CYP4V2 gene mutation. Crystalline‐like substances are mainly concentrated in the equatorial region and the peripheral retina, but there is no significant change in the posterior polar region. We consider that the tubulin encoded by the AGBL5 gene only affects the cilia activity and metabolism of rod cells, but has less effect on cone cells (Branham et al., 2016; Patel et al., 2016).
In summary, this study provides novel mutations and clinical phenotypes of retinitis pigmentosa, and the result suggest that choroidal atrophy can be used as one of the indicators for best correcting visual acuity and retinitis pigmentosa. Posterior subcapsular opacities is the most common form of secondary cataracts, and peripheral choroidal atrophy is the most common form of secondary choroidal atrophy. At the same time, more than half of the probands with retinitis pigmentosa are caused by compound heterozygous forms, and about one‐fifth of the probands are caused by digenic inheritance forms. Our finding not only extend the existing genotype spectrum, but also provide an effective reference for the design of panel‐based genetic diagnostic testing, genetic counseling, and future gene therapy in northeast Chinese patients with retinitis pigmentosa, and have a deeper understanding of the relationship between clinical manifestations and genotypes.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
WH, FC, WL, and YS conceived and designed this study. YS, Z‐SW, LX, and WH recruited patients, performed clinical examinations and interpretation. WL, YS, BX, and J‐YB collected the clinical samples and clinical data. J‐KL, WY, L‐SW, Z‐WW, and W.L analyzed the sequencing data. WL and YS wrote and revised the manuscript. All authors read and approved the final manuscript.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
The authors thank all of the patients and families who agreed to participate in this study. The authors also thank BGI‐Shenzhen for their technical support and the staff at He Eye Specialist Hospital of He University for their assistance. Finally, we are grateful to Dr WH and Dr FC for their valuable contributions in this work.
Sun Y, Li W, Li J‐K, et al. Genetic and clinical findings of panel‐based targeted exome sequencing in a northeast Chinese cohort with retinitis pigmentosa. Mol Genet Genomic Med. 2020;8:e1184 10.1002/mgg3.1184
Yan Sun and Wei Li are contributed equally as first authors.
Contributor Information
Wei He, Email: hewei@huh.edu.cn.
Fang Chen, Email: fangchen@genomics.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP CNP0000503.
REFERENCES
- Astuti, G. D. , Arno, G. , Hull, S. , Pierrache, L. , Venselaar, H. , Carss, K. , … Webster, A. R. (2016). Mutations in agbl5, encoding a‐tubulin deglutamylase, are associated with autosomal recessive retinitis pigmentosa. Investigative Ophthalmology & Visual Science, 57(14), 6180. [DOI] [PubMed] [Google Scholar]
- Audo, I. , Bujakowska, K. M. , Léveillard, T. , Mohand‐Saïd, S. , Lancelot, M.‐E. , Germain, A. , … Zeitz, C. (2012). Development and application of a next‐generation‐sequencing (ngs) approach to detect known and novel gene defects underlying retinal diseases. Orphanet Journal of Rare Diseases, 7(1), 8 10.1186/1750-1172-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso, C. , & Millan, J. M. (2010). Retinitis pigmentosa and allied conditions today: A paradigm of translational research. Genome Medicine, 2(5), 34 10.1186/gm155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne, S. (1999). Mutations in the rp1 gene causing autosomal dominant retinitis pigmentosa. Human Molecular Genetics, 8(11), 2121–2128. 10.1093/hmg/8.11.2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham, K. , Matsui, H. , Biswas, P. , Guru, A. A. , Hicks, M. , Suk, J. J. , … Nariai, N. (2016). Establishing the involvement of the novel gene agbl5 in retinitis pigmentosa by whole genome sequencing. Physiological Genomics, 48(12), 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Liu, X. , Sheng, X. , Gao, X. , Zhang, X. , Li, Z. , … Zhao, C. (2015). Targeted next‐generation sequencing reveals novel eys mutations in Chinese families with autosomal recessive retinitis pigmentosa. Scientific Reports, 5, 8927 10.1038/srep08927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton, M. , Nishiguchi, K. M. , Avila‐Fernández, A. , Nikopoulos, K. , Riveiro‐Alvarez, R. , Tatu, S. D. , … Rivolta, C. (2013). Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS ONE, 8(6), e65574 10.1371/journal.pone.0065574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, H. , Zhang, X. , Zhao, X. , Deng, T. , & Li, Y. (2008). Identification of five novel mutations in the long isoform of the ush2a gene in Chinese families with usher syndrome type ii. Molecular Vision, 14, 2067–2075. [PMC free article] [PubMed] [Google Scholar]
- Daiger, S. P. , Sullivan, L. S. , & Bowne, S. J. (2013). Genes and mutations causing retinitis pigmentosa. Clinical Genetics, 84(2), 132–141. 10.1111/cge.12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford, J. M. , Barton, S. , Bhaskar, S. , O'Sullivan, J. , Williams, S. G. , Lamb, J. A. , … Black, G. C. M. (2016). Original article: Molecular findings from 537 individuals with inherited retinal disease. Journal of Medical Genetics, 53(11), 761–767. 10.1136/jmedgenet-2016-103837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan, A. , Chizzolini, M. , Milan, E. , Sebastiani, A. , Costagliola, C. , & Parmeggiani, F. (2011). Good epidemiologic practice in retinitis pigmentosa: From phenotyping to biobanking. Current Genomics, 12(4), 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, F.‐J. , Li, J.‐K. , Chen, H. , Hu, F.‐Y. , Zhang, S.‐H. , Qi, Y.‐H. , … Wu, J.‐H. (2019). Genetic and clinical findings in a large cohort of chinese patients with suspected retinitis pigmentosa. Ophthalmology, 126(11), 1549–1556. 10.1016/j.ophtha.2019.04.038 [DOI] [PubMed] [Google Scholar]
- Gao, F. J. , Qi, Y. H. , Hu, F. Y. , Wang, D. D. , & Wu, J. H. (2019). Mutation spectrum of the bestrophin‐1 gene in a large Chinese cohort with bestrophinopathy. British Journal of Ophthalmology, bjophthalmol‐2019‐314679. 10.1136/bjophthalmol-2019-314679 [DOI] [PubMed] [Google Scholar]
- Hartong, D. T. , Berson, E. L. , & Dryja, T. P. (2006). Retinitis pigmentosa. Lancet, 368(9549), 1795–1809. 10.1016/S0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- Hu, F.‐Y. , Li, J.‐K. , Gao, F.‐J. , Qi, Y.‐H. , Xu, P. , Zhang, Y.‐J. , … Wu, J.‐H. (2019). ABCA4 gene screening in a Chinese cohort with stargardt disease: Identification of 37 novel variants. Frontiers in Genetics, 10, 773 10.3389/fgene.2019.00773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X.‐F. , Xiang, P. , Chen, J. , Xing, D.‐J. , Huang, N. A. , Min, Q. , … Jin, Z.‐B. (2013). Targeted exome sequencing identified novel ush2a mutations in usher syndrome families. PLoS ONE, 8(5), e63832 10.1371/journal.pone.0063832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X. , Qu, L. H. , Meng, X. H. , Xu, H. W. , & Yin, Z. Q. (2014). Detecting genetic variations in hereditary retinal dystrophies with next‐generation sequencing technology. Molecular Vision, 20, 553–560. [PMC free article] [PubMed] [Google Scholar]
- Jin, Z. B. , Huang, X. F. , Lv, J. N. , Xiang, L. , Li, D. Q. , Chen, J. , … Qu, J. (2014). Slc7a14 linked to autosomal recessive retinitis pigmentosa. Nature Communications, 5, 3517 10.1038/ncomms4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner, S. , Thiemann, I. J. , Dekomien, G. , Petrasch‐Parwez, E. , Schreiber, S. , Akkad, D. A. , … Bagci, H. (2015). Exome sequencing reveals agbl5 as novel candidate gene and additional variants for retinitis pigmentosa in five Turkish families. Investigative Ophthalmology & Visual Science, 56(13), 8045. [DOI] [PubMed] [Google Scholar]
- Khaliq, S. (2005). Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. Journal of Medical Genetics, 42(5), 436–438. 10.1136/jmg.2004.024281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenassi, E. , Vincent, A. , Li, Z. , Saihan, Z. , Coffey, A. J. , Steele‐Stallard, H. B. , … Webster, A. R. (2015). A detailed clinical and molecular survey of subjects with nonsyndromic ush2a retinopathy reveals an allelic hierarchy of disease‐causing variants. European Journal of Human Genetics, 23(10), 1318–1327. 10.1038/ejhg.2014.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Wang, Z. , Sun, Y. , Wang, Z. , Bai, J. , Xing, B. , … He, W. (2019). A start codon mutation of the TSPAN12 gene in Chinese families causes clinical heterogeneous familial exudative vitreoretinopathy. Molecular Genetics & Genomic Medicine, 7, e00948 10.1002/mgg3.948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Zhou, J. , Daiger, S. P. , Farber, D. B. , & Pierce, E. A. (2002). Identification and subcellular localization of the rp1 protein in human and mouse photoreceptors. Investigative Ophthalmology & Visual Science, 43(1), 22–32. [PMC free article] [PubMed] [Google Scholar]
- Lulin, H. , Yao, M. , Jiyun, Y. , Yuanfeng, L. , Yang, L. , & Zhenglin, Y. (2018). Mutation screening of the ush2a gene in retinitis pigmentosa and usher patients in a han Chinese population. Eye, 32, 1608–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes, G. , Guillaumie, T. , Vos, W. L. , Devos, A. , Audo, I. , Zeitz, C. , … Hamel, C. P. (2015). High prevalence of prph2 in autosomal dominant retinitis pigmentosa in France and characterization of biochemical and clinical features. American Journal of Ophthalmology, 159(2), 302–314. 10.1016/j.ajo.2014.10.033 [DOI] [PubMed] [Google Scholar]
- Patel, N. , Aldahmesh, M. A. , Alkuraya, H. , Anazi, S. , Alsharif, H. , Khan, A. O. , … Alowain, M. (2016). Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genetics in Medicine, 18, 554–562. [DOI] [PubMed] [Google Scholar]
- Perez‐Carro, R. , Corton, M. , Sánchez‐Navarro, I. , Zurita, O. , Sanchez‐Bolivar, N. , Sánchez‐Alcudia, R. , … Ayuso, C. (2016). Panel‐based ngs reveals novel pathogenic mutations in autosomal recessive retinitis pigmentosa. Scientific Reports, 6(1), 19531 10.1038/srep19531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, E. A. , Quinn, T. , Meehan, T. , McGee, T. L. , Berson, E. L. , & Dryja, T. P. (1999). Mutations in a gene encoding a new oxygen‐regulated photoreceptor protein cause dominant retinitis pigmentosa. Nature Genetics, 22(3), 248–254. 10.1038/10305 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogowski, K. , van Dijk, J. , Magiera, M. M. , Bosc, C. , Deloulme, J.‐C. , Bosson, A. , … Janke, C. (2010). A family of protein‐deglutamylating enzymes associated with neurodegeneration. Cell, 143(4), 564–578. 10.1016/j.cell.2010.10.014 [DOI] [PubMed] [Google Scholar]
- Schuster, S. C. (2008). Next‐generation sequencing transforms today's biology. Nature Methods, 5(1), 16–18. 10.1038/nmeth1156 [DOI] [PubMed] [Google Scholar]
- Sengillo, J. D. , Lee, W. , Nagasaki, T. , Schuerch, K. , Yannuzzi, L. A. , Freund, K. B. , … Tsang, S. H. (2018). A distinct phenotype of eyes shut homolog (EYS)‐retinitis pigmentosa is associated with variants near the C‐terminus. American Journal of Ophthalmology, 190, 99–112. 10.1016/j.ajo.2018.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodi, A. , Mariottini, A. , Passerini, I. , Murro, V. , & Torricelli, F. (2014). Myo7a and ush2a gene sequence variants in Italian patients with usher syndrome. Molecular Vision, 20, 1717–1731. [PMC free article] [PubMed] [Google Scholar]
- Veltel, S. , & Wittinghofer, A. (2009). Rpgr and rp2: Targets for the treatment of x‐linked retinitis pigmentosa? Expert Opinion on Therapeutic Targets, 13(10), 1239–1251. 10.1517/14728220903225016 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang, V. W. , Feng, Y. , Tian, X. , Li, F.‐Y. , Truong, C. , … Wong, L.‐J. (2014). Dependable and efficient clinical utility of target capture‐based deep sequencing in molecular diagnosis of retinitis pigmentosa. Investigative Opthalmology & Visual Science, 55(10), 6213 10.1167/iovs.14-14936 [DOI] [PubMed] [Google Scholar]
- Xiao, J. , Guo, X. , Wang, Y. , Shao, M. , Wei, X. , Du, L. , … Yang, Y. (2018). Identification of a disease‐causing mutation in a Chinese patient with retinitis pigmentosa by targeted next‐generation sequencing. European Journal of Ophthalmology, 27(6), 791–796. [DOI] [PubMed] [Google Scholar]
- Xu, L. , Hu, L. , Ma, K. , Li, J. , & Jonas, J. B. (2006). Prevalence of retinitis pigmentosa in urban and rural adult Chinese: The beijing eye study. European Journal of Ophthalmology, 16(6), 865–866. 10.1177/112067210601600614 [DOI] [PubMed] [Google Scholar]
- Xu, Y. , Guan, L. , Shen, T. , Zhang, J. , Xiao, X. , Jiang, H. , … Zhang, Q. (2014). Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Human Genetics, 133(10), 1255–1271. 10.1007/s00439-014-1460-2 [DOI] [PubMed] [Google Scholar]
- Yu, X. , Shi, W. , Cheng, L. , Wang, Y. , Chen, D. , Hu, X. , … Gu, F. (2016). Identification of a rhodopsin gene mutation in a large family with autosomal dominant retinitis pigmentosa. Scientific Reports, 6, 19759 10.1038/srep19759 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP CNP0000503.