

Abstract
Amplification and oncogenic mutations of ERBB2, the gene encoding the HER2 receptor tyrosine kinase, promote receptor hyperactivation and tumor growth. Here we demonstrate that HER2 ubiquitination and internalization, rather than its overexpression, are key mechanisms underlying endocytosis and consequent efficacy of the anti-HER2 antibody-drug conjugates (ADCs) ado-trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-DXd) in lung cancer cell lines and patient-derived xenograft models. These data translated into a 51% response rate in a clinical trial of T-DM1 in 49 patients with ERBB2/HER2-amplified or mutant lung cancers. We show that co-treatment with irreversible pan-HER inhibitors enhances receptor ubiquitination and consequent ADC internalization and efficacy. We also demonstrate that ADC switching to T-DXd, which harbors a different cytotoxic payload, achieves durable responses in a patient with lung cancer and corresponding xenograft model developing resistance to T-DM1. Our findings may help guide future clinical trials and expand the field of ADC as cancer therapy.
Introduction
Each year thousands of cancer patients with amplifications or mutations in the human epidermal growth factor receptor 2 (ERBB2/HER2) are without effective treatments. HER2 is a transmembrane receptor tyrosine kinase (RTK) that is amplified/overexpressed in 15–20% of breast and gastroesophageal cancers, for which targeted anti-HER2 therapy has transformed the standard of care (1–3). In addition to gene amplification, HER2 is also hyperactivated by specific mutations, most of which are located either in its extracellular domain or in its kinase domain. The common denominator of both overexpression and mutation is receptor hyperactivation via an increased homo- or hetero-dimerization and activation of downstream signaling cascades such as the PI3K and MAPK pathways (4). Mutations or amplification of ERBB2 are found also in ~4% of lung tumors (5,6), for which there is currently no approved targeted therapy. HER2-targeted agents investigated in patients with lung cancers over the past two decades include the monoclonal antibodies trastuzumab and pertuzumab, and tyrosine kinase inhibitors such as afatinib, dacomitinib and neratinib (7,8). While clinical activity was seen, neither class of drugs has produced transformative results in clinical trials to date (9–13). Antibody-drug conjugates (ADCs) are emerging antitumoral agents that combine the unique binding capabilities of monoclonal antibodies with the cytotoxic activity of chemotherapy to specifically target and harm cancer cells (14). The mechanism of action of ADCs involve the recognition and binding to the extracellular domain of a cancer-specific transmembrane protein by the monoclonal antibody backbone, the internalization of the conjugated chemotoxin via the endocytic pathway and release of the cytotoxic payload that ultimately kills or arrests the proliferation of the malignant cell (14). To date, there are five FDA-approved ADCs, three are recommended for hematological malignancies, and two, ado-trastuzumab emtansine (T-DM1, Genentech/Roche) and trastuzumab deruxtecan (T-DXd, formerly DS-8201a, Daiichi-Sankyo/AstraZeneca (15)), are indicated for HER2-positive breast cancer (16), with the latter showing also promising results in patients with HER2-positive gastric cancer (17,18).
In this work, we demonstrate that activated HER2, regardless of its oncogenic potential or the addiction status of the cancer cell to its downstream signaling pathways, can serve as a vehicle to funnel potent chemotherapeutic agents into lung tumors. Integrating parallel laboratory and clinical data on the mechanisms of response, we also propose two different strategies to improve the efficacy of anti-HER2 ADCs: co-treatment with irreversible pan-HER inhibitors that enhance receptor ubiquitination and internalization of ADCs or switching to an ADC bearing a different cytotoxic payload.
Results
HER2 mutations increase receptor internalization and T-DM1 activity
We hypothesized that ERBB2 amplified or mutated tumors have exquisite susceptibility to HER2 ADCs on the basis of high rate receptor internalization and trafficking, regardless of their intrinsic dependence on HER2 signaling for cell growth and/or survival. To test whether the presence of an activating mutation influences HER2 internalization rate, affecting in turn the internalization rate of ADC-HER2 complexes, we established isogenic models using a breast non-transformed cell line (MCF10A) and a lung cancer cell line (NCI-H2030) expressing either V5-tagged WT or mutant (S310F or L755S) HER2 or empty vector as control (EV). We used these models to quantitate the internalization rate of T-DM1 linked to a pH-sensitive dye (pHrodo-T-DM1) that becomes fluorescent only at low pH, providing a positive indication of ADC-HER2 endocytosis. Albeit with some variations in the kinetics, we consistently observed that cells expressing either S310F or L755S mutations internalize more T-DM1 than WT cells in both cell models (Figure 1A, B and C, D). In particular, MCF10A cells expressing the HER2 L755S mutant show the highest pHrodo-T-DM1 signal at earlier time points and morphological changes at later time points (Figure 1A, B), compatible to the onset of cell death, as confirmed by increased PARP cleavage (Figure 1E). In both cell lines, expressing comparable WT or mutant HER2, receptor levels were reduced upon T-DM1 treatment, likely due to internalization and subsequent degradation (Figure 1E, F).
Figure 1. Internalization and efficacy of T-DM1 depends on HER2 mutational status.
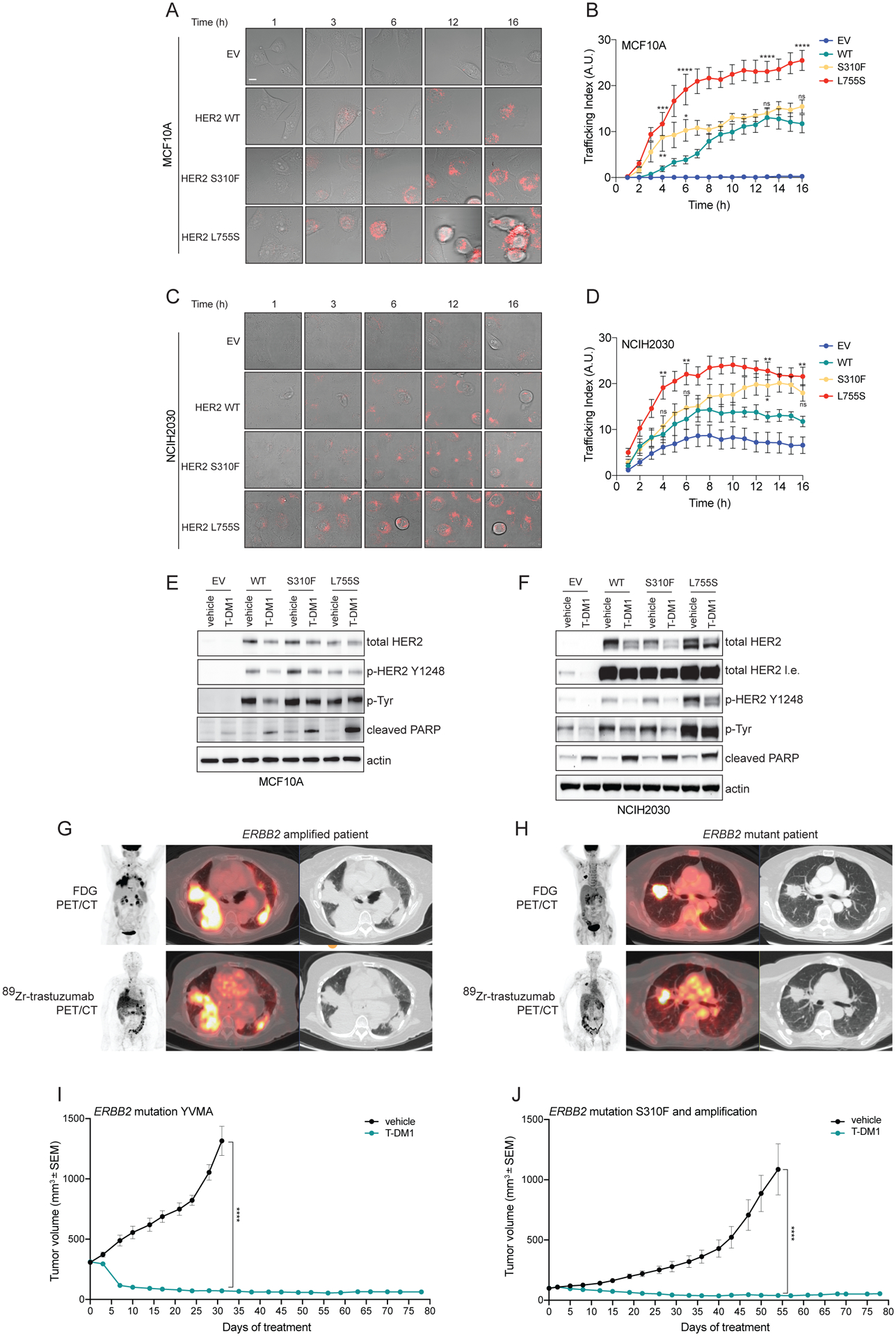
A) Isogenic breast epithelial cells MCF10A ectopically expressing either wild-type (WT) or mutant (S310F or L755S) HER2 or transduced with an empty vector (EV) control were incubated with T-DM1 conjugated to a red fluorescent pH-sensitive dye (pHrodo-T-DM1, 1 μg/mL) for 30 minutes at 4°C. Cells were then released at 37°C and Z-stack imaged every hour over 16 hours on a confocal microscope. Representative images of merged bright-field and Z-projected pHrodo signals depict intracellular red fluorescent dots, corresponding to T-DM1 reaching the endolysosomal compartments. Scale bar, 10 μm. B) Quantification of the experiment described in (A). Data are shown as number of normalized pHrodo dots per cell (Trafficking Index) over time. The error bars indicate SEM. Groups were compared to WT for each time point using 2-way ANOVA test. pValue *=<0.05, **=<0.01, ***=<0.001, ****=<0.0001 at the indicated time point, ns: non-significant. (n = 2 independent experiments, ≥80 cells analyzed in total per condition, per time point). C) Isogenic lung cancer cells NCI-H2030 ectopically expressing either wild-type (WT) or mutant (S310F or L755S) HER2 or transduced with an empty vector (EV) control were treated as in (A). Representative images of merged bright-field and Z-projected pHrodo signals depict intracellular red fluorescent dots, corresponding to T-DM1 reaching the endolysosomal compartments. Scale bar, 10 μm. D) Quantification of the experiment described in (D). Data are shown as number of normalized pHrodo dots per cell (Trafficking Index) over time. The error bars indicate SEM. Groups were compared to WT for each time point using 2-way ANOVA test. pValue *=<0.05, **=<0.01 at the indicated time point, ns: non-significant. (n = 2 independent experiments, ≥80 cells analyzed in total per condition, per time point). E) Western blot analysis of MCF10A isogenic models incubated with T-DM1 (10 μg/mL) or vehicle as control for 24 hours. Total and phospho-HER2, as well as and total phospho-tyrosine were assayed. Cleaved PARP was used to determine the apoptosis induction. Actin was included as loading control. F) Western blot analysis of NCI-H2030 isogenic models incubated with T-DM1 (10 μg/mL) or vehicle as control for 24 hours. Total and phospho-HER2, as well as and total phospho-tyrosine were assayed. Cleaved PARP was used to determine the apoptosis induction. Actin was included as loading control. G, H) 89Zr-Trastuzumab PET/CT scan representative images of ERBB2 amplified (G) and ERBB2 mutant (H) NSCLC patients (from a total of n=4 ERBB2 amplified and n=4 ERBB2 mutant NSCLC patients evaluated by 89Zr-Trastuzumab PET/CT scan). I) In vivo efficacy study of an ERBB2 YVMA mutant lung PDX treated with T-DM1 (15 mg/kg, i.v. once a week). Measurements show average tumor volumes +/− SEM, n=7 animals per group. J) In vivo efficacy study of a ERBB2 S310F mutant and amplified lung PDX treated with T-DM1 (15 mg/kg, i.v. once a week). Measurements show average tumor volumes +/− SEM, n=7 animals per group. Comparisons between the two groups for each time point were performed using 2-way ANOVA test, ****=<0.0001 pValue at the indicated time point.
Based on these data, we posited that T-DM1 can accumulate in both mutant and amplified lung tumors. To evaluate this in the clinical setting, we performed 89Zr-trastuzumab PET/CT functional imaging in lung cancer patients bearing HER2 alterations. We found that 89Zr-trastuzumab can accumulate in both ERBB2 amplified and mutant tumors (Figure 1G, H).
We then tested the antitumoral activity of T-DM1 in different patient-derived xenografts (PDXs) established from lung cancer patients with tumors bearing ERBB2 mutations or amplification. Treatment with T-DM1 resulted in durable responses in both HER2-mutant (YVMA insertion) tumors and in xenografts with co-occurrent amplification (2.8-fold) and mutation (S310F) of ERBB2 (Figure 1I, J). This treatment was well tolerated with no significant change in animal body weight (Supplementary Figure 1A–B).
Clinical activity of T-DM1 in HER2-activated lung cancer patients
The above data provided the rationale for a dedicated analysis of the clinical activity of T-DM1 in patients with ERBB2 amplified or mutant lung cancers within the context of a histology agnostic phase II basket trial (NCT02675829) (Supplementary Figure 2). In total, 49 patients (18/49 patients were part of the previously reported mutation cohort of this clinical trial (19)) with ERBB2 mutant and/or amplified lung cancer (Table 1) were treated with T-DM1 and responses were measured by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and/or modified PET Response Criteria in Solid Tumors (PERCIST). Modified PERCIST was used to capture RECIST non-measurable disease and help assess antitumor activity of new cancer therapies in clinical trial context (13,20,21). The best overall response rate (ORR) by either RECIST or modified PERCIST for ERBB2-amplified/mutant patients was 51% (25/49, 95% CI 36 to 66) (Figure 2a and b), with a median progression-free survival (PFS) of 5 months (95% CI 3.5 to 5.9) (Figure 2c). In the subset of ERBB2 mutant and/or amplified patients with RECIST evaluable disease, the ORR by RECIST was 31% (13/42, 95% CI 18 to 47) (Supplementary Figure 3A–B), while in the subset of ERBB2 mutant and/or amplified patients with PERCIST evaluable disease, the ORR by modified PERCIST was 64% (14/22, 95% CI 41–83) (Supplementary Figure 3C–D). Except for 1 case (2%) of grade 3 febrile neutropenia, treatment was well tolerated (Table 2).
Table 1:
Patient Characteristics.
| Characteristics | N (%) |
|---|---|
| Total patients treated | 49 (100) |
| Age, median | 64 (range 25–84) |
| Sex, Female | 35 (71) |
| Smoking status | |
| Former smoker | 25 (51) |
| Never | 24 (49) |
| Histology | |
| Adenocarcinoma | 47 (96) |
| Large cell neuroendocrine carcinoma | 1 (2) |
| Non-small cell lung cancer not otherwise specified | 1 (2) |
| Median line of therapy for T-DM1 | 2 (range 1–7) |
| 1st line | 5 (10) |
| 2nd line | 23 (47) |
| 3rd line | 12 (24) |
| 4th line | 5 (10) |
| 5th line | 2 (4) |
| 6th line | 1 (2) |
| 7th line | 1 (2) |
| Prior HER2-targeted therapy | 9 (50%) |
| Afatinib | 6 (12) |
| Neratinib | 6 (12) |
| Trastuzumab | 5 (10) |
| Pertuzumab | 1 (2) |
Figure 2. Clinical activity of T-DM1 in NSCLC.
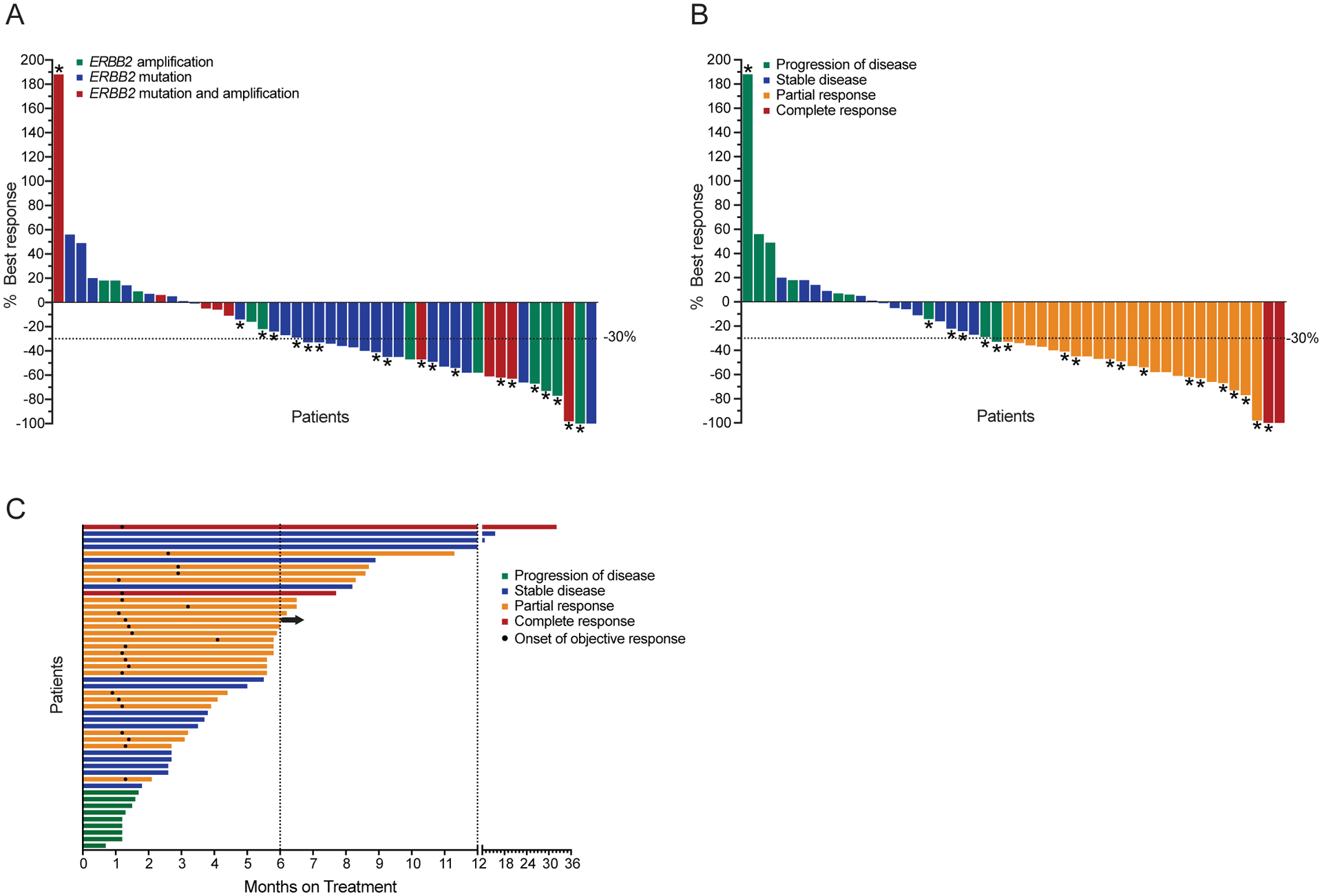
A, B) Waterfall plots showing best overall response to T-DM1 treatment in 48 NSCLC patients, not including one patient who did not have RECIST or PERCIST measurable disease but was still evaluable for progression-free survival study. Colors indicate ERBB2 alteration status (A) or best response (B). Overall Response Rate (ORR) was 51% (25/49, 95% Confidence interval (CI): 36–66). Asterisks indicate responses by PERCIST, otherwise response was assessed by RECISTv1.1. C) Swimmer’s plot showing duration of treatment on T-DM1. Arrow indicates treatment is ongoing at the time of data cut off. Mean PFS: 5.6 months, median PFS: 5.0 months. Mean DOR: 4.2 months, median DOR: 4.4 months.
Table 2. T-DM1-Related Adverse Events.
Treatment related adverse events with total frequencies of greater than 10%, according to CTCAE v4.1. There were no grade 4 or 5 adverse events.
| Adverse Event | Grade 1 N (%) |
Grade 2 N (%) |
Grade 3 N (%) |
Total |
|---|---|---|---|---|
| Elevated AST or ALT | 28 (57) | 3 (6) | - | 31 (63) |
| Thrombocytopenia | 13 (27) | 1 (2) | 1 (2) | 15 (31) |
| Fatigue | 6 (12) | 2 (4) | - | 8 (16) |
| Nausea | 14 (29) | - | - | 14 (29) |
| Infusion reaction | 2 (4) | 5 (10) | - | 7 (14) |
| Anorexia | 3 (6) | 2 (4) | - | 5 (10) |
| Anemia | 1 (1) | 3 (6) | 1 (2) | 5 (10) |
In accordance with our pre-clinical data (Figure 1), we did not observe appreciable differences in response when we stratified patients according to ERBB2 genetic status: the best ORR by either RECIST or modified PERCIST was 55% (6/11, 95% CI 23–83%) for ERBB2-amplified patients, 50% (14/28, 95% CI 31 to 69) for ERBB2-mutant patients and 50% (5/10, 95% CI 19–81) for concurrently ERBB2-mutant and amplified patients (Figure 2A). These data strongly suggest that a response to T-DM1 can be achieved to the same extent in lung cancers with either amplified or mutant ERBB2.
ERBB2 copy number obtained by next-generation sequencing (NGS) using Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) algorithm (22) correlated with HER2 status as assessed by FISH and IHC (Supplementary Figure 3E–G) (Supplementary Table 1). A trend of association between the degree of ERBB2 amplification by FACETS with patient response to T-DM1 therapy was observed, albeit not reaching statistical significance (Supplementary Figure 3H). There were six patients with ERBB2 amplified lung cancer and concurrent EGFR mutation who had progressed on prior EGFR inhibitor, and two patients responded to T-DM1 (Supplementary Table 1).
Irreversible HER kinase inhibitors enhance HER2 internalization and T-DM1 activity
Despite the promising clinical activity observed with T-DM1 in ERBB2 mutant/amplified lung cancers, some patients’ tumors were intrinsically refractory to T-DM1 and some responses were short lived. We then sought strategies to enhance or prolong the duration of the responses to T-DM1 in lung cancers. Irreversible pan-HER kinase inhibitors neratinib and afatinib are approved agents for HER2 overexpressing breast cancer and EGFR-mutant lung cancer, respectively. In addition to their strong activity in inhibiting HER2 phosphorylation, they are thought to induce receptor polyubiquitination and internalization, likely due to hindrance with HSP90 binding (23–25). We thus tested whether these findings could be translated to ERBB2 amplified or mutant lung cancers. Co-treatment of Calu-3 (ERBB2 amplified) and LUAD-10 (patient-derived ERBB2 L755P mutant) lung cancer cells with the irreversible inhibitor neratinib markedly increased T-DM1 internalization, as quantitated by pHrodo-T-DM1 live imaging (Figure 3A, B). In contrast, co-treatment with the reversible HER2 inhibitor lapatinib, known to promote receptor stabilization and accumulation at the plasma membrane (26), decreased the internalization of pHrodo-T-DM1 (Figure 3A, B). Consistently, ubiquitination of immunoprecipitated HER2 was stronger than controls upon treatment with the irreversible inhibitors neratinib or afatinib but not with the reversible inhibitors lapatinib or tucatinib in both cell lines (Figure 3C, D). This increase in HER2 ubiquitination and internalization was recapitulated by HSP90 inhibition (Figure 3C, D and Supplementary Figure 4A, B). The same pattern of HER2 ubiquitination was reproduced also in the NCI-H2030 isogenic models (Figure 3E). Collectively, these data suggest that irreversible inhibitors of HER2 increase poly-ubiquitination and subsequent internalization of the receptor via HSP90 dissociation in ERBB2 amplified or mutant lung cancers. Importantly, this phenomenon seems to be prevented by reversible kinase inhibitors.
Figure 3. pan-HER irreversible inhibitors enhance T-DM1 internalization, ubiquitination and efficacy both in ERBB2 amplified and mutant lung tumors.
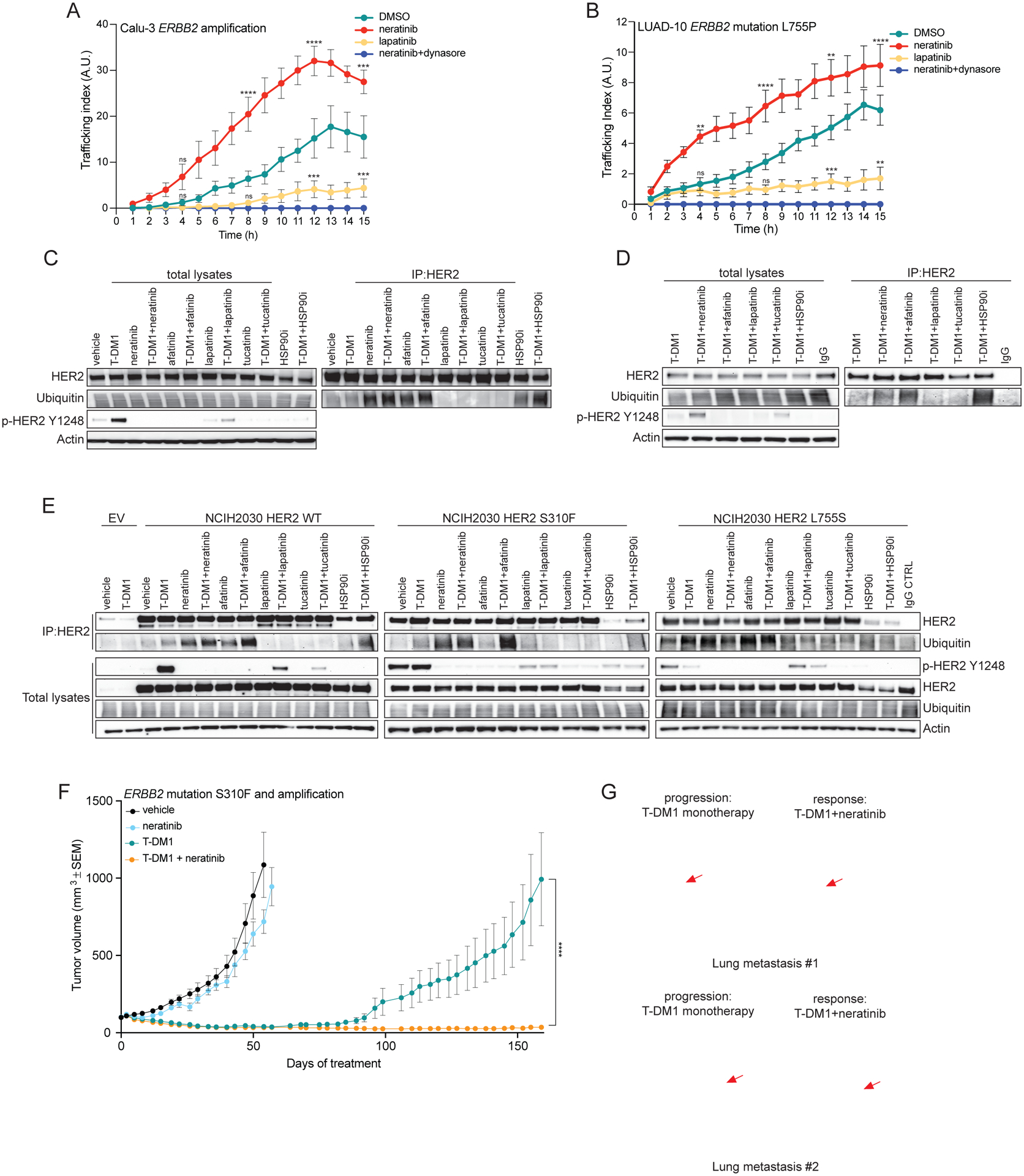
A) Calu-3 ERBB2-amplified NSCLC cells were incubated with pHrodo-T-DM1 (1 μg/mL) together with the pan-HER irreversible inhibitor neratinib (100 nM), the EGFR/HER2 reversible inhibitor lapatinib (100 nM), the inhibitor of endocytosis dynasore (100 μM) or DMSO control for 30 minutes at 4°C and then released at 37°C and Z-stack imaged every hour over 15 hours on a confocal microscope. Data are shown as number of normalized pHrodo dots per cell (Trafficking Index) over time. The error bars indicate SEM. Groups were compared to DMSO for each time point using 2-way ANOVA test. pValue ***=<0.001, ****=<0.0001 at the indicated time point, ns: non-significant. (n = 2 independent experiments, ≥80 cells analyzed in total per condition, per time point). B) LUAD-10 HER2-mutant L755P NSCLC cells were incubated with pHrodo-T-DM1 (10 μg/mL) together with the pan-HER irreversible inhibitor neratinib (10 nM), the pan-HER reversible inhibitor lapatinib (10 nM), the inhibitor of endocytosis dynasore (100 μM) or DMSO control for 30 minutes at 4°C and then released at 37°C and Z-stack imaged every hour over 15 hours on a confocal microscope. Data are shown as number of normalized pHrodo dots per cell (Trafficking Index) over time. The error bars indicate SEM. Groups were compared to DMSO for each time point using 2-way ANOVA test. pValue **=<0.01, ***=<0.001, ****=<0.0001 at the indicated time point, ns: non-significant. (n = 2 independent experiments, ≥80 cells analyzed in total per condition, per time point). C) Calu-3 cells were incubated with T-DM1 (10 μg/mL) or vehicle as control, together with the pan-HER irreversible inhibitors neratinib or afatinib (100 nM), the pan-HER reversible inhibitors lapatinib or tucatinib (100 nM) and HSP90 inhibitor (200 nM) in the presence of the proteasome inhibitor MG-132 (10 μM) for 6 hours at 37°C. HER2 immunoprecipitations (IPs) were performed using either T-DM1 itself or trastuzumab (added only to the protein lysates lacking T-DM1) as primary antibodies. IP or total lysate samples were evaluated by western blot. For the IP, ubiquitin and total HER2 were evaluated, showing a higher HER2 ubiquitination in the samples treated with neratinib, afatinib or HSP90 inhibitor. Ubiquitin and total HER2 were comparable among the total lysates, while phosphorylated HER2 on tyrosine 1248 (p-HER2 Y1248) demonstrated the efficacy of HER2 phosphorylation inhibition. Actin was included as loading control. D) LUAD-10 cells were incubated with T-DM1 (10 μg/mL) or IgG control, together with the irreversible HER2 inhibitors neratinib or afatinib (100 nM), the reversible HER2 inhibitors lapatinib or tucatinib (100 nM) and HSP90 inhibitor (100 nM) in the presence of the proteasome inhibitor MG-132 (10 μM) for 3 hours at 37°C. HER2 immunoprecipitations (IPs) were performed using T-DM1 itself (or IgG control) as primary antibody. IP or total lysate samples were analyzed by western blot. For the IP, ubiquitin and total HER2 were evaluated. Ubiquitin and total HER2 were comparable among the total lysates, while phosphorylated HER2 on tyrosine 1248 (p-HER2 Y1248) demonstrated the efficacy of HER2 phosphorylation inhibition. Actin was included as loading control. E) Isogenic lung cancer cells NCI-H2030 ectopically expressing either wild-type (WT) or mutant (S310F or L755S) HER2 or transduced with an empty vector (EV) control were treated as in (B). IP or total lysate samples were analyzed by western blot. For the IP, ubiquitin and total HER2 were evaluated. Ubiquitin and total HER2 were comparable among the total lysates, while phosphorylated HER2 on tyrosine 1248 (p-HER2 Y1248) demonstrated the efficacy of HER2 phosphorylation inhibition. Actin was included as loading control. F) In vivo efficacy study of the ERBB2 S310F mutant and amplified lung PDX shown in Figure 1e treated with T-DM1 (15 mg/kg, i.v. once a week), neratinib (20 mg/kg, p.o. every day, 5 days a week) and the combination. Measurements show average tumors volumes +/− SEM, n=7 animals per group. Comparisons between the two indicated groups for each time point were performed using 2-way ANOVA test, ****=<0.0001 pValue at the indicated time point. G) CT scan of ERBB2 amplified breast cancer patient who relapsed after T-DM1 single agent treatment and responded to T-DM1+neratinib combination. Arrows point to two different metastatic lesions during T-DM1 monotherapy and after the addition of neratinib.
To determine if the enhanced receptor internalization induced by co-treatment with neratinib and T-DM1 was sufficient to enhance antitumor efficacy, we treated the same lung PDXs bearing ERBB2 amplification and mutation (S310F) shown in Figure 1e with either neratinib, T-DM1, or the combination. While both T-DM1 and the combination of T-DM1 and neratinib induced marked tumor regression, the effect was more durable with combination (despite a negligible activity observed with neratinib monotherapy) (Figure 3F). All treatments were well tolerated by the animals (Supplementary Figure 5).
Since the combination of T-DM1 and neratinib is not given as standard of care at progression to T-DM1, we evaluated the clinical activity of this combination in a 41-year-old patient with ERBB2-amplified breast cancer enrolled in an ongoing clinical trial (NCT01494662). At the time of study entry, the patient had relapsed after multiple lines of anti-HER2 therapy, including T-DM1 (see details in Methods). Immediately upon T-DM1 progression, neratinib was added to T-DM1 and the patient experienced a brisk partial response (−38%) 6 weeks into therapy (Figure 3G) and continued with this treatment until intracranial progression at week 18 (at that time the patient remained in confirmed PR extracranially). This prompt response is consistent with our preclinical data that co-treatment with neratinib improved internalization and efficacy of T-DM1, and with a recently published trial showing encouraging efficacy and tolerability of T-DM1 and neratinib in metastatic breast cancer albeit in a T-DM1 naïve setting (27).
T-DXd shows stronger and more durable responses in lung tumor models compared to T-DM1 and overcomes resistance to T-DM1
Given that T-DXd has demonstrated significant efficacy in T-DM1 resistant ERBB2-amplified breast cancers (18), we next evaluated its antitumor activity in the lung PDXs bearing ERBB2 amplification and mutation (S310F) shown in Figures 1H and 3F, that developed resistance to T-DM1 overtime. Treatment with T-DXd induced striking responses resulting in complete tumor regression in all the mice (Figure 4A), without any sign of toxicity (Supplementary Figure 6A), similarly to the treatment with the combination of T-DM1 and neratinib (Figure 3F). As a preliminary proof-of-concept, we treated the corresponding patient with lung cancer bearing both ERBB2 amplification and mutation (S310F) with T-DXd on a phase 1 trial (NCT02564900) upon progression on T-DM1 after an initial response on 4 months of treatment. Strikingly, the patient achieved a partial response to T-DXd (−70% tumor shrinkage) that lasted for one year (Figure 4B). Moreover, we observed that while lung PDXs harboring a YVMA ERBB2 mutation (different from the PDX shown in Figure 1G) were initially sensitive to all ADC-containing therapeutic regimens, mice receiving T-DM1 or the combination of T-DM1 and neratinib relapsed shortly after treatment discontinuation. In contrast, animals treated with T-DXd monotherapy did not show any sign of tumor regrowth up to 1 month after treatment discontinuation (Figure 4C and Supplementary Figure 6B). To rule out the possibility that the observed superior activity of T-DXd compared to T-DM1 monotherapy was due to a faster internalization kinetic, we performed parallel live imaging experiment with pHrodo-T-DM1 and pHrodo-T-DXd in Calu-3 cells. As expected, we did not observe any significant difference in the internalization rate of the two ADCs (Supplementary Figure 6C).
Figure 4. T-DXd shows increased efficacy in T-DM1 resistant tumors.
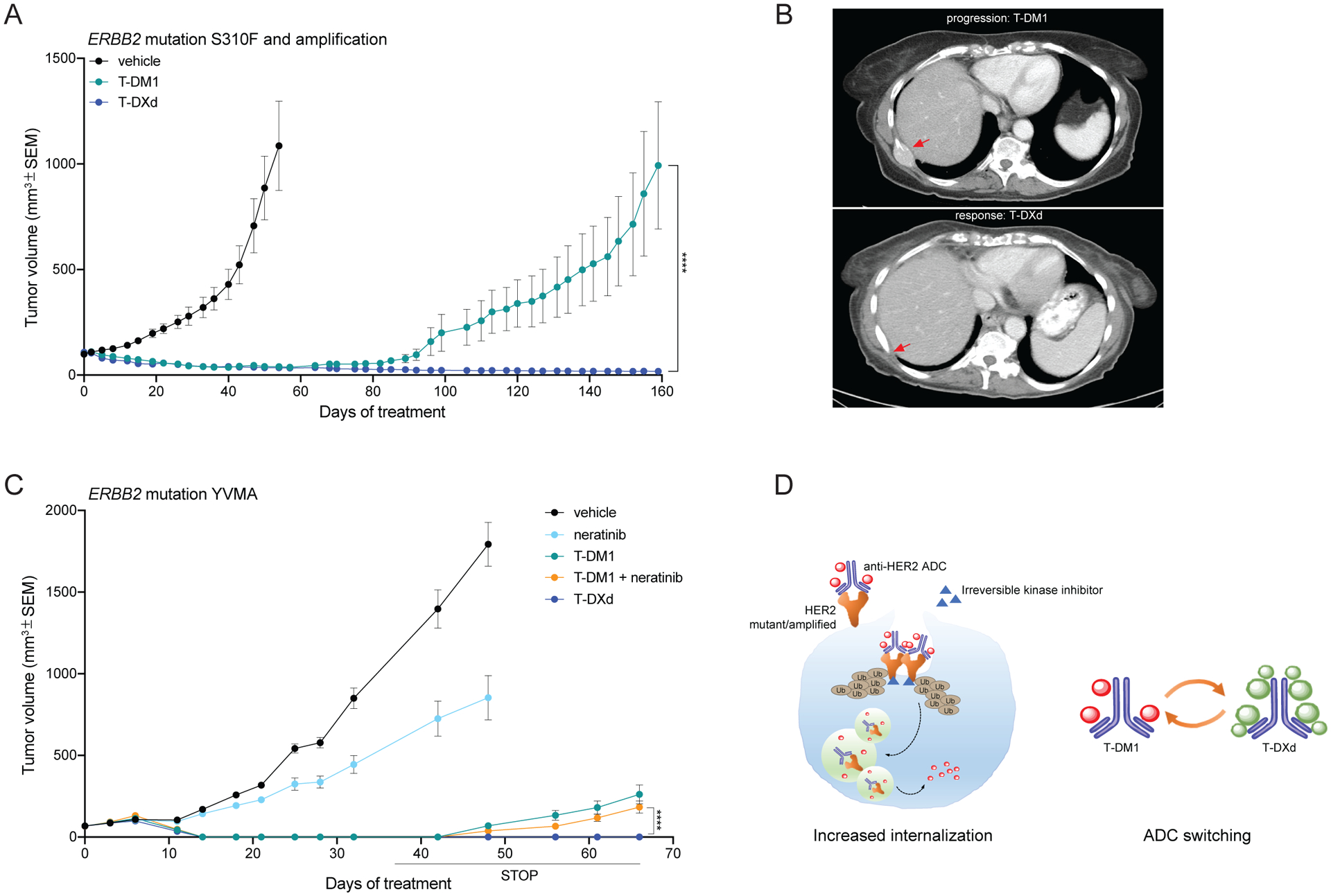
A) In vivo efficacy study of the ERBB2 S310F mutant and amplified lung PDX shown in Figures 1E and 3D treated with T-DM1 (15 mg/kg, i.v. once a week) and T-DXd (10 mg/kg, i.v. once every 3 weeks). Measurements show average tumor volumes +/− SEM, n=7 animals per group. Comparisons between the two indicated groups for each time point were performed using 2-way ANOVA test, ****=<0.0001 pValue at the indicated time point. B) CT-scan of the ERBB2 S310F mutant and amplified lung cancer patient corresponding to the PDX shown in Figure 4c. Arrows point to a bone metastatic lesion at T-DM1 progression and after response to T-DXd. C) In vivo efficacy study of a ERBB2 YVMA mutant lung PDX treated with T-DM1 (15 mg/kg, i.v. once a week), neratinib (20 mg/kg, p.o. 5 days a week), T-DM1+neratinib and T-DXd (10 mg/kg, i.v. once every 3 weeks). Measurements show average tumor volumes +/− SEM, n=6 animals per group. Comparisons between the two indicated groups for each time point were performed using 2-way ANOVA test, ****=<0.0001 pValue at the indicated time point. D) Schematic showing the two strategies proposed in this work to enhance the efficacy of anti-HER2 ADC in lung cancer: increased internalization by pan-HER irreversible inhibitors through increased ubiquitination and consequent endocytosis of the receptor-ADC complex in both ERBB2 mutant or amplified tumors; switching anti-HER2 ADCs from T-DM1 to T-DXd.
Collectively, these data suggest that both the combination of T-DM1 with pan-HER irreversible kinase inhibitors or switching to T-DXd result in superior activity compared to T-DM1 monotherapy (Figure 4D).
Discussion
The activity of T-DM1 shown here represent the first clinical trial evidence that ERBB2/HER2 amplification/overexpression in lung cancers may be therapeutically targeted, which stands in contrast to two decades of disappointing efforts targeting HER2 protein expression in lung cancers (7). This study also confirmed the clinical activity of anti-HER2 ADCs in lung cancers with HER2 activating mutations, regardless of the quantity of protein expression (Supplementary Table 1). These results validate our hypothesis that receptor hyperactivation through gene amplification or mutation facilitates ubiquitination, internalization which may be therapeutically exploited through ADCs. Although two independently conducted clinical trials of T-DM1 targeting HER2 protein expression (IHC 2+/3+) in lung cancers did not reach expected outcomes, a signal of encouraging responses was seen in tumors with ERBB2/HER2 amplification or mutation which is consistent with our findings (28,29). We also observed clinical activity of T-DM1 across all activating HER2 mutation subtypes (Supplementary Table 1), in contrast to the differential activity of tyrosine kinase inhibitors across mutation subtypes seen in previous studies (11,30,31). This finding may be explained by the different mechanism of action of ADCs compared to tyrosine kinase inhibitors, where it acts through the internalization of receptor-ADC complex to deliver the cytotoxic payload rather than inhibiting oncogenic signaling. This also explains the responses to T-DM1 in EGFR-mutant and ERBB2 amplified lung cancer without effective EGFR signal inhibition. Therefore, ADCs represent a fundamentally different targeted therapy for lung cancer, as their efficacy is independent of the drug’s ability to inhibit oncogenic driver signaling.
Our laboratory and clinical data are consistent with the idea that surface-localized and cycling HER2 can serve as a carrier for HER2-specific ADCs. Therefore, strategies that augment the dynamics of internalization may increase the efficacy of these drugs. We observed that co-treatment with a pan-HER irreversible inhibitor such as neratinib enhanced internalization of HER2-ADC complexes in vitro, resulting in a potent antitumor activity in vivo. Importantly, while treatment with reversible inhibitors such as lapatinib or tucatinib decreased HER2 ubiquitination, treatment with irreversible inhibitors such as neratinib or afatinib increased such ubiquitination, similarly to HSP90 inhibition. A superior activity of the combination of the irreversible HER inhibitor poziotinib and T-DM1 compared to single agents has been recently shown in a lung cancer PDX model (32). However, the proposed mechanism of action in that report is HER2 stabilization at the membrane, which is in contrast with our findings. As a matter of fact, we believe that receptor stabilization at the plasma membrane increases antibody-dependent cell cytotoxicity exerted by natural killer cells binding to antibodies such as trastuzumab or cetuximab (26,33).
It has been shown that HSP90 binding regulates the stability of mature membrane-bound form of HER2. Indeed, HSP90 inhibition triggers receptor ubiquitination, destinating HER2 to proteasomal degradation (23,34). Here, we propose that FDA-approved irreversible pan-HER inhibitors, by increasing HER2 ubiquitination, may act as HER2-specific HSP90 inhibitors. Thus, HER2 targeting by irreversible inhibitors would not be used to obtain a stronger inhibition of the kinase and the downstream signaling, but to further enhance internalization of the receptor-ADC improving the antitumor activity of the payload. As a matter of fact, we also speculate that sub-therapeutic doses (or perhaps pulsatile treatment schedules) of neratinib or afatinib may be sufficient to maximize ADC-dependent cell death/tumor shrinkage, thus minimizing the adverse effects associated with daily treatment with these agents (35).
While combined neratinib and T-DM1 elicited an ORR of 63% with manageable toxicity profile in a phase I trial of 19 patients with HER2-positive breast cancer previously treated with trastuzumab- and pertuzumab-based therapies (27), these responses were not compared to T-DM1 monotherapy and no data are available on the efficacy of this combination in T-DM1-refractory breast cancer patients. Furthermore, there are no current clinical trials evaluating the clinical benefit of the combination of T-DM1 and pan-HER irreversible inhibitors in comparison to T-DM1 monotherapy in ERBB2-mutant or amplified lung cancers. Similarly, it is currently unknown whether the antitumor effects of T-DXd, recently FDA-approved for metastatic breast cancer (36) would also be enhanced by the upfront combination with neratinib. The question on potential overlapping gastrointestinal toxicity of the two agents may only be addressed through clinical trial investigation.
The superior efficacy of T-DXd compared to T-DM1 observed in our models is unlikely to be the consequence of differential ADC internalization. Rather, we posit that genetic, epigenetic or transcriptional mechanisms that render tumor cells more refractory to microtubule-directed chemotherapy (37,38) may be at play in this context. The antitumor activity of T-DXd in tumors that are or become resistant to T-DM1 may be the result of a combinations of variables, including the higher drug-to-antibody ratio compared to T-DM1, diversity between stability and cleavability of the respective linkers, a cytotoxic payload with a different mechanism of action, and the higher cell permeability of the payload resulting in bystander cytotoxicity in the neighboring cells (39). The identification of biomarkers of response to the available ADCs (or their payloads) will be critical for a better patient stratification in future trials.
Despite the earlier setbacks, several clinical development strategies targeting HER2 in lung cancers are now underway. T-DXd is currently being evaluated in phase I/II clinical trials as a single agent (NCT03505710) or in combination with immune checkpoint inhibitor pembrolizumab (NCT04042701). Trastuzumab duocarmazine is another anti-HER2 ADC which has demonstrated activity in patients with T-DM1 resistant or HER2-low expressing breast cancers (40). Other anti-HER2 ADCs in early-phase development include ZW-49 (NCT03821233), DHES0815A (NCT03451162), RC48-ADC (NCT03500380) and A166 (NCT03602079). Novel irreversible pan-HER tyrosine kinase inhibitors such as pyrotinib and poziotinib have also shown promising activity in early phase clinical trials targeting HER2-mutant lung cancers (32,41). As the development of brain metastases are common in HER2-mutant lung cancers affecting 47% of patients, central nervous system activity should be further evaluated in future trials as they are in breast cancer trials (42,43). As these trials of HER2-targeted agents bring renewed hope, our findings provide clinical evidence and mechanistic insights to guide the development of ADCs and their combinations for patients with lung cancers and other solid tumors.
In summary, ADC-based therapies are a promising new treatment for patients with ERBB2 amplified or mutant lung cancers. Concurrent treatment with irreversible pan-HER kinase inhibitors or ADC therapy switching can improve the efficacy of these agents. Our findings can potentially be extended to other HER2-activated tumor types, for which pharmacological HER2 signaling blockade is insufficient to elicit strong and durable responses and may help guide future clinical trials and expand the field of ADC as cancer therapy.
Methods
Ethical Compliance
We declare compliance with all relevant ethical regulation.
Patients
A cohort of 49 patients with lung adenocarcinomas carrying either amplification or mutations of ERBB2, was enrolled in a phase II basket trial at MSK #15–335 (NCT02675829) to assess the clinical activity of ado-trastuzumab emtansine (T-DM1). The primary objective was the determination of overall response rate according to RECIST v1.1 or modified PERCIST as assessed by investigator. Secondary objectives included assessment of progression-free survival (PFS) and toxicity according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.1 (NCI CTCAE v4.1). The data cut-off for this analysis was August 1, 2019. All patients received T-DM1 at 3.6 mg/kg by intravenous infusion every 21 days until disease progression or unacceptable toxicity. Physical examination and safety assessments were performed every 3 weeks. Tumor assessments using contrast enhanced CT chest abdomen pelvis and/or PET were performed at baseline, week 6, week 12 then every 12 weeks thereafter until disease progression. One patient had no RECIST or PERCIST measurable disease but was evaluable for progression-free survival. Therefore, the waterfall plot included 48 patients. The 95% exact confidence interval for overall response rate (ORR) was calculated using the Clopper-Pearson method. Progression-free survival time was estimated by the Kaplan-Meier method. ERBB2 mutation and amplification status was evaluated by MSK-IMPACT (see targeted tumor sequencing), another NGS assay in a CLIA-certified laboratory, and/or fluorescence in situ hybridization (FISH). FISH was performed using FDA approved probe sets (PathVysion, Abbott and ERBB2 IQFISH pharmDx™, Dako). HER2 protein by immunohistochemistry (IHC) was assessed using the 4B5 Ventana antibody.
All patients provided written informed consent for genomic sequencing of tumor DNA, and review of medical records for detailed demographic, pathologic, and clinical data and for publication of this information as part of an institutional review board (IRB)-approved investigator sponsored trial (NCT02675829). Research protocols for tumor collection and analysis were approved by the ethical committee of MSK.
The patient shown in Figure 3f has provided written informed consent to DF/HCC protocol #09–204, which is an IRB-approved, prospective cohort study enrolling patients with metastatic breast cancer seen for at least one visit to Dana-Farber Cancer Institute. The consent includes permission to publish deidentified data. In addition, the patient consented to DF/HCC protocol #11–344, a prospective clinical trial testing neratinib-based therapies for patients with breast cancer brain metastases (NCT01494662) and received the combination of neratinib + TDM1 under this protocol. The investigator sponsored trial’s PI (Dr. Rachel Freedman) has provided permission to report on the extracranial response only for this single trial patient ahead of reporting of the primary trial endpoint (CNS objective response rate). Before being enrolled in this clinical trial, the patient was treated with doxorubicin-cyclophosphamide followed by paclitaxel-trastuzumab-pertuzumab, achieving a pathological complete response at surgery. The patient then completed adjuvant trastuzumab and started docetaxel-trastuzumab-pertuzumab, when metastatic relapse (bilateral pulmonary nodules and mediastinal/hilar/supraclavicular adenopathy) occurred 27 months after completion of adjuvant trastuzumab. The patient achieved a clinical response and was transitioned to trastuzumab-pertuzumab maintenance. Seven months after the initial diagnosis of metastatic disease, symptomatic brain metastases were diagnosed, and treated with surgery followed by whole brain radiation therapy. Trastuzumab and pertuzumab were continued for an additional 2 months, however, the patient progressed with pericardial effusion and pulmonary metastases and T-DM1 monotherapy was started. Tumor burden stabilized for 6 months, but resistance to T-DM1 emerged with enlargement of the lung lesions. At this stage, neratinib was added to T-DM1.
The patient shown in Figure 4d is a 73-year-old woman with ERBB2 S310F mutant and amplified metastatic lung adenocarcinoma, who has progressed on prior cisplatin pemetrexed chemotherapy, and ipilimumab nivolumab immunotherapy. She achieved initial partial response to T-DM1 followed by progression of disease after 4 months of treatment. She was then treated with T-DXd and achieved a partial response with 70% tumor shrinkage on RECIST v1.1 with clinical benefit that lasted one year.
Targeted tumor sequencing
DNA from formalin-fixed paraffin-embedded tissue and matched germline DNA underwent targeted next-generation sequencing assay using (MSK-IMPACT). In brief, this assay uses a hybridization-based exon capture design to interrogate all protein-coding exons and select introns of oncogenes, tumor-suppressor genes, and key members of pathways that may be actionable by targeted therapies. In this study, 468 key cancer-associated genes were analyzed. Sequencing data were analyzed as previously described to identify somatic single-nucleotide variants, small insertions and deletions, copy number alterations, and structural rearrangements15.
ERBB2 copy number analysis utilized a computational algorithm termed Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) to adjust for tumor purity, ploidy and variant allele frequency, as previously described (22). The degree of ERBB2 amplification was correlated with clinical response to anti-HER2 therapy.
Compounds
For preclinical studies, T-DM1 was obtained from MSK pharmacy and T-DXd was obtained from Daichii Sankyo, Co., Ltd. The HER2 inhibitors neratinib (provided by Puma Biotechnology), afatinib (Selleckchem, #S1011), lapatinib (Selleckchem, # S1028) and tucatinib (Selleckchem, #S8362) and the HSP90 inhibitor ganetespib (Selleckchem, #S1159) were dissolved in DMSO (10 mM stock), aliquoted and stored at −20℃. The endocytosis inhibitor dynasore (Abcam, #ab120192) was dissolved in DMSO (30 mM stock), aliquoted and stored at −20°C.
Cell lines
MCF10A isogenic cell lines were purchased from ATCC (CRL-10317) and were grown in DMEM/F12 (Invitrogen #11330–032) supplemented with Horse Serum 5% (Invitrogen#16050–122, EGF 20ng/ml (Peprotech #AF-100–15), Hydrocortisone 0.5 mg/ml (Sigma #H-0888), Cholera Toxin 100 ng/ml, (Sigma #C-8052), Insulin 10μg/ml (Sigma #I-1882), Pen/Strep 1% under standard conditions.
NCI-H2030 were purchased from ATCC (CRL-5914) and were grown in RPMI-1640 (Thermo Fisher Scientific, #11875093) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Thermo Fisher Scientific, #16140071), Pen/Strep 1% under standard conditions.
Calu-3 were purchased from ATCC (HTB-55) and were grown in DMEM/F12 supplemented with 10% FBS, Pen/Strep 1% under standard conditions.
Patient-derived LUAD-10 cells were established in our laboratory from a PDX engrafted with the pellet isolated from the pleura effusion of a HER2 L755P mutant lung cancer patient from phase II basket trial at MSK #15–335 (NCT02675829), after T-DM1 progression. Cells were plated on collagen-coated petri dishes and cultured in DMEM/F12 supplemented with 10% FBS, Pen/Strep 1% under standard conditions.
Lentiviral infections
MCF10A and NCI-H2030 isogenic cell lines stably expressing empty vector, ERBB2 WT or ERBB2 mutants were obtained by lentiviral infection as follows. Briefly, 7 × 106 293T cells were seeded into 10cm dishes. Cells were transfected with 1.2 μg pMD2.G, 2.4 μg pCMV-dR8.2, and 3.6 μg pLX302-EV, pLX302- ERBB2 WT, pLX302- ERBB2 S310F or pLX302- ERBB2 L755S using jetPRIME (Polyplus) following the manufacturer’s instructions. Medium was refreshed 6 hours post-transfection. The supernatant was collected 72 hours and filtered with 0.45 μm filters. 3 × 105 MCF10A and NCI-H2030 cells were seeded into 6-well plates and transduced with different dilutions of freshly collected lentiviruses with 8 μg/mL polybrene for 24 hours. Cells were then trasnfered into 10-cm dishes and selected with 2 μg/mL (MCF10A) or 5 μg/mL (NCI-H2030) of puromycin for 2 days. Cells stably expressing comparable levels of WT or mutant HER2 were chosen for subsequent studies.
pHrodo-ADC assay
pHrodo-TDM1 and pHrodo-T-DXd were generated using pHrodo™ iFL Red Microscale Protein Labelling Kit (Invitrogen #P36014) according to manufacturer’s instructions. Briefly, a reaction solution containing 100 μL of the ADC (1 mg/mL) 10 μL of sodium bicarbonate (1 M) and 3.3 μL of pHrodo iFL Red STP ester, amine-reactive dye (2mM in DMSO) was incubate for 30 minutes at room temperature. pHrodo-ADCs were purified by centrifugation using a purification resin, aliquoted and stored at −20°C.
For internalisation assays, 4 × 104 cells were seeded on 8-well chamber slides (Nunc Lab-Tek™ II Chamber Slide, Thermo Scientific #154534). The day after, cells were incubated with serum-free medium containing Hoechst (Thermo Fisher Scientific #62249) for live-cell fluorescent staining of DNA, and pHrodo-ADC (1 μg/mL, unless stated differently in the figure legend) for 30 minutes at 4°C and then released at 37°C in the heated live chamber of the Zeiss LSM880 confocal microscope. Positions were decided using bright-field (at least 4 positions per sample) and 2 μm thick Z-stack images of bright-field and red fluorescence were taken every hour over 16 hours. Images were then analyzed using FiJi Software and results were displayed as average number of cytoplasmic red fluorescent dots per cell (Trafficking Index).
Where indicated, cells were incubated with serum-free medium containing Hoechst, pHrodo-ADC and neratinib (100 nM, unless stated differently in the figure legend) or lapatinib (100 nM, unless stated differently in the figure legend) or HSP90 inhibitor (200 nM) or DMSO as control.
Western Blots
Total protein lysates (20 μg) were extracted using RIPA buffer and separated on SDS-PAGE gels (NuPAGE™ 4–12% Bis-Tris Protein Gels, Invitrogen) according to standard methods.
Membranes were probed using the following antibodies: anti-total HER2 Rabbit mAb (29D8, Cell Signalling #2165), anti-total HER2 Mouse mAb (CB11, Thermo Scientific #MA1–35720), anti-phospho-HER2 (Tyr1248, Cell Signalling #2247), anti-Cleaved PARP Asp214 human specific (Cell Signalling Technology #9541), anti-phospho-tyrosine (Cell Signaling Technology #8954), anti-β-Actin 13E5 (Cell Signalling Technology #4970) and Ubiquitin (P4D1, Cell Signaling Technology #3936S).
HER2 immunoprecipitation
For immunoprecipitation (IP) assays, cells were incubated in 5% FBS medium in the presence of the proteasome inhibitor MG-132 (10 μM), together with 10 μg/mL of T-DM1 (or IgG control, Cell Signalling Technology #2729) and 100 nM of tyrosine kinase inhibitors or HSP90 inhibitor at 37°C for 6 hours (except for LUAD-10, that were incubated for 3 hours). Cells were then washed in cold PBS and lysed using NP-40 buffer (150 mM NaCl, 10 mM Tris pH 8, 1% NP-40, 10% glycerol). 20 μg of proteins were used as total lysates. Protein lysates (350 μg for Calu-3 and NCI-H2030, 1500 μg for LUAD-10) were incubated rotating at 4°C for 4 hours in the presence of 10 μg/mL trastuzumab, which was added only to samples lacking T-DM1 or IgG control. Agarose protein A/G beads (Santa Cruz #sc-2003) were then added and samples were incubated rotating at 4°C for 1 hour. Beads were centrifuged at 3000 rpm for 1 min and supernatant was removed. Similarly, beads were washed twice using NP-40 buffer and once using nuclease-free sterile water. Finally, immunoprecipitates were eluted using 1X sample buffer (NuPAGE™ LDS Sample Buffer, Thermo Fisher Scientific #NP0008 and NuPAGE Sample Reducing Agent, Thermo Fisher Scientific #NP0009) at 95°C for 5 min.
In vivo studies
PDXs were generated as follows: 6-week-old NOD scid gamma (NSG) female mice were implanted subcutaneously with specimens freshly collected from patients at MSK under an MSK-approved IRB biospecimen protocol #06–107. Tumors developed within 2 to 4 months and were expanded into additional mice by serial transplantation. At this point, PDXs were subjected to high coverage next generation sequencing with the MSK-IMPACT assay. For efficacy studies, treatment started when tumor volumes reached approximately 100 mm3. Xenografts were randomized and dosed with T-DM1 (15 mg/kg, intravenously once a week), neratinib (20 mg/kg, orally 5 days a week), a combination of the two agents, T-DXd (10 mg/kg, intravenously once every 3 weeks) or vehicle as control (saline, orally 5 days a week). Mice were observed daily throughout the treatment period for signs of toxicity. Tumors were measured twice weekly using calipers, and tumor volume was calculated using the formula length × width2 × 0.52. Body weight was also assessed twice weekly. At the end of each treatment, animals were sacrificed, and tumors were collected for biochemistry and histology analysis. Mice were cared for in accordance with guidelines approved by the MSK Institutional Animal Care and Use Committee and Research Animal Resource Center. 6 to 7 mice per group were included in each experiment.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 8 (GraphPad Software Inc. San Diego, CA). Two-way ANOVA test was used to evaluate significant differences in tumor volumes in in vivo efficacy studies. Data are presented as mean ± SEM. pValue *=<0.05, **=<0.01, ***=<0.001, ****=<0.0001. Median progression-free survival and 95% confidence interval was calculated using the Kaplan-Meier method. Association of ERBB2 copy number with response status was evaluated using the Wilcoxon rank-sum test. Spearman correlation was used for correlating copy number assessments by FISH, IHC, and NGS.
Data availability
All genomic results and associated clinical data for all patients in this study are publicly available in the cBioPortal for Cancer Genomics at the following URL: http://cbioportal.org/msk-impact. All other relevant data are included in the paper and/or Extended Data files.
Supplementary Material
Statement of significance.
T-DM1 is clinically effective in lung cancers with amplification or mutations of ERBB2. This activity is enhanced by co-treatment with irreversible pan-HER inhibitors, or ADC switching to T-DXd. These results may help address unmet needs of patients with HER2-activated tumors and no approved targeted therapy.
Acknowledgments
We thank the MSKCC imaging unit for their help and all the members of the Scaltriti group for useful discussion. This study was funded by the National Cancer Institute (NCI) under the MSK Cancer Center Support Grant/Core Grant (P30 CA008748), NIH Grants U54 OD020355‑01 (EdS) and P01 CA-129243, the Conquer Cancer Foundation of ASCO-AACR Young Investigator Translational Cancer Research Award, the Carol Lowenstein Fund and a kind gift from Mr. and Mrs. Peter Pritchard. This work was also partially funded by an unrestricted grant from MORE Health, Genentech, Puma Biotechnology and Daiichi Sankyo. E.C. is a recipient of an MSK society scholar prize. P.R.B., T.N. were funded by the CRUK UCL Centre (C416/A25145), CRUK City of London Centre (C7893/A26233), and CRUK KCL-UCL Comprehensive Cancer Imaging Centre (CRUK & EPSRC) in association with the MRC and DoH (C1519/A16463 and C1519/A10331).
Footnotes
Competing interests
M.S. has received research funds from AstraZeneca, Puma Biotechnology, Daiichi Sankyo, Immunomedics, Targimmune and Menarini Ricerche. He is in the scientific advisory board (SAB) of Menarini Ricerche and Bioscience Institute and is a cofounder of Medendi.org. D.B.S. has served as consulted/received honoraria from Pfizer, Loxo Oncology, Illumina, Lilly Oncology and Vivideon Therapeutics. S.M. consulted for Boehringer-Ingelheim. M.S. and B.T.L. have two pending institutional patents at Memorial Sloan Kettering Cancer Center (US62/685,057, US62/514,661). B.T.L. has served as a consultant/advisory board member for Roche/Genentech, Biosceptre International, Thermo Fisher Scientific, Mersana Therapeutics, Hengrui Therapeutics, Guardant Health and has received research funds to his institution from Roche/Genentech, Daiichi Sankyo, Hengrui Therapeutics, Illumina, Guardant Health, BioMed Valley Discoveries, AstraZeneca, GRAIL, MORE Health, Amgen and Lilly. C.M.R. is a consultant/advisory board member for AbbVie, Amgen, Ascentage, AstraZeneca, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Loxo, PharmaMar, Bridge Medicine, and Harpoon. R.A.F. receives research funds from Puma Biotechnology and Eisai Pharmaceuticals. N.U.L. has received research support (to institution) from Pfizer, Genentech, Seattle Genetics, and Merck, and has been a consultant for Puma, Daichii, and Seattle Genetics. D.M.H. reports receiving commercial research grants from Loxo, PUMA Biotechnology, AstraZeneca, and Bayer Pharmaceuticals, and is a consultant/advisory board member for Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer Pharmaceuticals, and Genentech/Roche. G.A.U. is a consultant for Sanofi and receives research support for Sanofi, Novartis, Genentech, and Puma Biotechnology. M.G.K. has received consulting fees from AstraZeneca, Pfizer, and Regeneron. He has received research funding from The National Cancer Institute (USA), The Lung Cancer Research Foundation, Genentech Roche, and PUMA Biotechnology and honoraria He has received honoraria for participation in educational programs from WebMD, OncLive, Physicians Education Resources, Prime Oncology, Intellisphere, Creative Educational Concepts, Peerview, i3 Health, Paradigm Medical Communications, AXIS, Carvive Systems, AstraZeneca, and Research to Practice. N.R. is on the SAB and receives research funding from Chugai, on the SAB and owns equity in Beigene, and Fortress. N.R. is also on the SAB of Daiichi-Sankyo, Astra-Zeneca-MedImmune, and F-Prime, and is a past SAB member of Millenium-Takeda, Kadmon, Kura, and Araxes. N.R. is a consultant to Novartis, Boehringer Ingelheim, Tarveda, and Foresight and consulted in the last three years with Eli Lilly, Merrimack, Kura Oncology, Araxes, and Kadman. N.R. owns equity in ZaiLab, Kura Oncology, Araxes, and Kadman. N.R. also collaborates with Plexxikon. M.F.B. declares consulting/advisory board activities for Roche. J.S.R.-F. is a consultant of Goldman Sachs Merchant Bank and REPARE Therapeutics, a member of the SAB of Volition RX and Paige.AI, and an ad hoc member of the SABs of Roche Tissue Diagnostics, Ventana, InVicro, Novartis and Genentech. D.R.J. is a senior consultant for Diffusions Pharmaceuticals, Consultant for AstraZeneca, and Merck. M.O. is a consultant/advisory board member for PharmaMar, Novartis and Targeted Oncology. He received research funds from Bristol-Myers Squibb (Inst) and Merck Sharp & Dohme (Inst). J.M.I. owns equity in LumaCyte, LLC. F.C. is an AstraZeneca employee. S.T., W-L.L. and A.B. are mProbe Inc. employees.
Memorial Sloan Kettering has an institutional agreement with IBM for Watson For Oncology and receives royalties from IBM.
No potential conflicts of interests were disclosed by the other authors.
References
- 1.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014;25(3):282–303 doi 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cocco E, Javier Carmona F, Razavi P, Won HH, Cai Y, Rossi V, et al. Neratinib is effective in breast tumors bearing both amplification and mutation of ERBB2 (HER2). Sci Signal 2018;11(551) doi 10.1126/scisignal.aat9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montemurro F, Scaltriti M. Biomarkers of drugs targeting HER-family signalling in cancer. J Pathol 2014;232(2):219–29 doi 10.1002/path.4269. [DOI] [PubMed] [Google Scholar]
- 4.Cocco E, Lopez S, Santin AD, Scaltriti M. Prevalence and role of HER2 mutations in cancer. Pharmacol Ther 2019;199:188–96 doi 10.1016/j.pharmthera.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov 2017;7(6):596–609 doi 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ricciardi GR, Russo A, Franchina T, Ferraro G, Zanghi M, Picone A, et al. NSCLC and HER2: between lights and shadows. J Thorac Oncol 2014;9(12):1750–62 doi 10.1097/JTO.0000000000000379. [DOI] [PubMed] [Google Scholar]
- 8.Baraibar I, Mezquita L, Gil-Bazo I, Planchard D. Novel drugs targeting EGFR and HER2 exon 20 mutations in metastatic NSCLC. Crit Rev Oncol Hematol 2020;148:102906 doi 10.1016/j.critrevonc.2020.102906. [DOI] [PubMed] [Google Scholar]
- 9.Gatzemeier U, Groth G, Butts C, Van Zandwijk N, Shepherd F, Ardizzoni A, et al. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann Oncol 2004;15(1):19–27 doi 10.1093/annonc/mdh031. [DOI] [PubMed] [Google Scholar]
- 10.Herbst RS, Davies AM, Natale RB, Dang TP, Schiller JH, Garland LL, et al. Efficacy and safety of single-agent pertuzumab, a human epidermal receptor dimerization inhibitor, in patients with non small cell lung cancer. Clin Cancer Res 2007;13(20):6175–81 doi 10.1158/1078-0432.CCR-07-0460. [DOI] [PubMed] [Google Scholar]
- 11.Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O’Connell J, et al. Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol 2015;26(7):1421–7 doi 10.1093/annonc/mdv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dziadziuszko R, Smit EF, Dafni U, Wolf J, Wasag B, Biernat W, et al. Afatinib in NSCLC With HER2 Mutations: Results of the Prospective, Open-Label Phase II NICHE Trial of European Thoracic Oncology Platform (ETOP). J Thorac Oncol 2019;14(6):1086–94 doi 10.1016/j.jtho.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554(7691):189–94 doi 10.1038/nature25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coats S, Williams M, Kebble B, Dixit R, Tseng L, Yao NS, et al. Antibody-Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin Cancer Res 2019;25(18):5441–8 doi 10.1158/1078-0432.CCR-19-0272. [DOI] [PubMed] [Google Scholar]
- 15.Nakada T, Masuda T, Naito H, Yoshida M, Ashida S, Morita K, et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Bioorg Med Chem Lett 2016;26(6):1542–5 doi 10.1016/j.bmcl.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 16.von Minckwitz G, Huang CS, Mano MS, Loibl S, Mamounas EP, Untch M, et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N Engl J Med 2019;380(7):617–28 doi 10.1056/NEJMoa1814017. [DOI] [PubMed] [Google Scholar]
- 17.Shitara K, Iwata H, Takahashi S, Tamura K, Park H, Modi S, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive gastric cancer: a dose-expansion, phase 1 study. Lancet Oncol 2019;20(6):827–36 doi 10.1016/S1470-2045(19)30088-9. [DOI] [PubMed] [Google Scholar]
- 18.Tamura K, Tsurutani J, Takahashi S, Iwata H, Krop IE, Redfern C, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol 2019;20(6):816–26 doi 10.1016/S1470-2045(19)30097-X. [DOI] [PubMed] [Google Scholar]
- 19.Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J Clin Oncol 2018;36(24):2532–7 doi 10.1200/JCO.2018.77.9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J Nucl Med 2009;50 Suppl 1:122S–50S doi 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ulaner GA, Saura C, Piha-Paul SA, Mayer I, Quinn D, Jhaveri K, et al. Impact of FDG PET Imaging for Expanding Patient Eligibility and Measuring Treatment Response in a Genome-Driven Basket Trial of the Pan-HER Kinase Inhibitor, Neratinib. Clin Cancer Res 2019;25(24):7381–7 doi 10.1158/1078-0432.CCR-19-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, et al. Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J 2002;21(10):2407–17 doi 10.1093/emboj/21.10.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Zhang J, Liu C, Du S, Feng L, Luan X, et al. Neratinib induces ErbB2 ubiquitylation and endocytic degradation via HSP90 dissociation in breast cancer cells. Cancer Lett 2016;382(2):176–85 doi 10.1016/j.canlet.2016.08.026. [DOI] [PubMed] [Google Scholar]
- 25.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem 1996;271(37):22796–801 doi 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 26.Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2009;28(6):803–14 doi 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- 27.Abraham J, Montero AJ, Jankowitz RC, Salkeni MA, Beumer JH, Kiesel BF, et al. Safety and Efficacy of T-DM1 Plus Neratinib in Patients With Metastatic HER2-Positive Breast Cancer: NSABP Foundation Trial FB-10. J Clin Oncol 2019;37(29):2601–9 doi 10.1200/JCO.19.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peters S, Stahel R, Bubendorf L, Bonomi P, Villegas A, Kowalski DM, et al. Trastuzumab Emtansine (T-DM1) in Patients with Previously Treated HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Efficacy, Safety, and Biomarkers. Clin Cancer Res 2019;25(1):64–72 doi 10.1158/1078-0432.CCR-18-1590. [DOI] [PubMed] [Google Scholar]
- 29.Hotta K, Aoe K, Kozuki T, Ohashi K, Ninomiya K, Ichihara E, et al. A Phase II Study of Trastuzumab Emtansine in HER2-Positive Non-Small Cell Lung Cancer. J Thorac Oncol 2018;13(2):273–9 doi 10.1016/j.jtho.2017.10.032. [DOI] [PubMed] [Google Scholar]
- 30.Peters S, Curioni-Fontecedro A, Nechushtan H, Shih JY, Liao WY, Gautschi O, et al. Activity of Afatinib in Heavily Pretreated Patients With ERBB2 Mutation-Positive Advanced NSCLC: Findings From a Global Named Patient Use Program. J Thorac Oncol 2018;13(12):1897–905 doi 10.1016/j.jtho.2018.07.093. [DOI] [PubMed] [Google Scholar]
- 31.Lai WV, Lebas L, Barnes TA, Milia J, Ni A, Gautschi O, et al. Afatinib in patients with metastatic or recurrent HER2-mutant lung cancers: a retrospective international multicentre study. Eur J Cancer 2019;109:28–35 doi 10.1016/j.ejca.2018.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robichaux JP, Elamin YY, Vijayan RSK, Nilsson MB, Hu L, He J, et al. Pan-Cancer Landscape and Analysis of ERBB2 Mutations Identifies Poziotinib as a Clinically Active Inhibitor and Enhancer of T-DM1 Activity. Cancer Cell 2019;36(4):444–57 e7 doi 10.1016/j.ccell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chew HY, De Lima PO, Gonzalez Cruz JL, Banushi B, Echejoh G, Hu L, et al. Endocytosis Inhibition in Humans to Improve Responses to ADCC-Mediating Antibodies. Cell 2020;180(5):895–914 e27 doi 10.1016/j.cell.2020.02.019. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A 2002;99(20):12847–52 doi 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17(3):367–77 doi 10.1016/S1470-2045(15)00551-3. [DOI] [PubMed] [Google Scholar]
- 36.Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med 2020;382(7):610–21 doi 10.1056/NEJMoa1914510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kavallaris M Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 2010;10(3):194–204 doi 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 38.VanderWeele DJ, Zhou R, Rudin CM. Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther 2004;3(12):1605–13. [PubMed] [Google Scholar]
- 39.Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin Cancer Res 2016;22(20):5097–108 doi 10.1158/1078-0432.CCR-15-2822. [DOI] [PubMed] [Google Scholar]
- 40.Banerji U, van Herpen CML, Saura C, Thistlethwaite F, Lord S, Moreno V, et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol 2019;20(8):1124–35 doi 10.1016/S1470-2045(19)30328-6. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Jiang T, Qin Z, Jiang J, Wang Q, Yang S, et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann Oncol 2019;30(3):447–55 doi 10.1093/annonc/mdy542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Offin M, Feldman D, Ni A, Myers ML, Lai WV, Pentsova E, et al. Frequency and outcomes of brain metastases in patients with HER2-mutant lung cancers. Cancer 2019;125(24):4380–7 doi 10.1002/cncr.32461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N Engl J Med 2020;382(7):597–609 doi 10.1056/NEJMoa1914609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic results and associated clinical data for all patients in this study are publicly available in the cBioPortal for Cancer Genomics at the following URL: http://cbioportal.org/msk-impact. All other relevant data are included in the paper and/or Extended Data files.