

Phosphotyrosine (pY)-specific phosphoproteome profiles have been determined for 16 AML cell lines through a pY immunoprecipitation-based protocol. Kinase activity inference using dedicated ranking analysis, combining kinome-, activation loop-, and substrate-based analyses, identified potential drivers for all cell lines and driver function was confirmed through drug experiments. Results for two patient samples show potential of phosphoproteomics in a clinical setting.
Keywords: Tyrosine kinases, personalized medicine, mass spectrometry, molecular biology, phosphoproteome, drug targets, receptor tyrosine kinases, acute myeloid leukemia, cell signaling, kinase inhibitors, phosphoproteomics
Graphical Abstract

Highlights
pY phosphoproteomes and dedicated ranking analyses for 16 AML cell lines.
RTK drivers, 6 mutant cell lines confirmed, identification for 4 more cell lines.
MAPK1/3 phosphorylation for cell lines without TK driver, indicating RAS mutation.
Drug target space phosphorylation correlates with drug IC50s in specific cell lines.
Abstract
Acute myeloid leukemia (AML) is a clonal disorder arising from hematopoietic myeloid progenitors. Aberrantly activated tyrosine kinases (TK) are involved in leukemogenesis and are associated with poor treatment outcome. Kinase inhibitor (KI) treatment has shown promise in improving patient outcome in AML. However, inhibitor selection for patients is suboptimal.
In a preclinical effort to address KI selection, we analyzed a panel of 16 AML cell lines using phosphotyrosine (pY) enrichment-based, label-free phosphoproteomics. The Integrative Inferred Kinase Activity (INKA) algorithm was used to identify hyperphosphorylated, active kinases as candidates for KI treatment, and efficacy of selected KIs was tested.
Heterogeneous signaling was observed with between 241 and 2764 phosphopeptides detected per cell line. Of 4853 identified phosphopeptides with 4229 phosphosites, 4459 phosphopeptides (4430 pY) were linked to 3605 class I sites (3525 pY). INKA analysis in single cell lines successfully pinpointed driver kinases (PDGFRA, JAK2, KIT and FLT3) corresponding with activating mutations present in these cell lines. Furthermore, potential receptor tyrosine kinase (RTK) drivers, undetected by standard molecular analyses, were identified in four cell lines (FGFR1 in KG-1 and KG-1a, PDGFRA in Kasumi-3, and FLT3 in MM6). These cell lines proved highly sensitive to specific KIs. Six AML cell lines without a clear RTK driver showed evidence of MAPK1/3 activation, indicative of the presence of activating upstream RAS mutations. Importantly, FLT3 phosphorylation was demonstrated in two clinical AML samples with a FLT3 internal tandem duplication (ITD) mutation.
Our data show the potential of pY-phosphoproteomics and INKA analysis to provide insight in AML TK signaling and identify hyperactive kinases as potential targets for treatment in AML cell lines. These results warrant future investigation of clinical samples to further our understanding of TK phosphorylation in relation to clinical response in the individual patient.
Acute myeloid leukemia (AML)1 is a clonal hematopoietic stem cell disorder, characterized by expansion of immature leukemic blasts in the bone marrow, resulting in suppression of normal hematopoiesis. In AML, protein kinase mutations are associated with proliferative and survival advantages (1, 2) and treatment of AML with kinase inhibitors is therefore gaining much interest (3).
For example, the FMS-like receptor tyrosine kinase 3 (FLT3) gene is frequently mutated in AML, either by internal tandem duplication of the juxtamembrane domain (∼20–30% of cases) (4–8) or a point mutation of the tyrosine kinase domain (TKD) (∼7%) (5, 7, 8). These mutations lead to constitutive FLT3 signaling and are associated with high peripheral blast counts and a poor treatment outcome (4, 6, 8–10). Recently, addition of the (rather non-selective) FLT3 inhibitor midostaurin to standard chemotherapy was shown to increase overall survival compared with chemotherapy alone in FLT3-ITD/TKD-positive AML patients (11). Clinical studies, using more selective inhibitors, have also shown promising, but variable, results and suggested that patient selection based solely on mutational status is suboptimal for predicting patient response and/or that additional activated pathways may be missed by mutational analysis (12–15). Clearly there is a need for predictive markers that reflect the functional state of the cancer cells. In order to advance KI-based precision medicine for the individual patient, knowledge of individual AML kinase activity profiles could improve treatment decisions.
The emergence of mass spectrometry-based phosphoproteomics has enabled dissection of cellular signaling in cancer at a global scale. Previous global phosphoproteomic studies of AML have focused on unraveling AML signaling pathways (16–21) and assessing the effect of kinase inhibitors in the context of therapeutic response (17, 20, 22–26). Global phosphopeptide patterns have been found to be discriminative between AML cell lines on the one hand, and cell lines derived from other hematological malignancies on the other (23). Heterogeneity was further found among different AML cell lines, underlining the heterogeneous nature of the disease (22, 23).
Global phosphoproteomic analyses, involving all major types of phosphorylation (serine, threonine, and tyrosine), are heavily dominated by serine- and threonine-specific modifications that are much more prevalent than pY modifications. In contrast, pY enrichment-based phosphoproteomics allows for a thorough study of TK signaling that is frequently disturbed in cancer and enables identification of driver kinases (27). In the context of AML, pY-phosphoproteomics has contributed to increased understanding of aberrant TK signaling (16, 18, 19, 27), and identified JAK2 and FGFR1 as drivers in AML cell lines (28, 29). Though limited in their depth and sample size, these studies have shown the ability of phosphoproteomics to detect kinase hyperactivity in AML.
We have recently developed an analysis strategy (called INKA) to infer kinase activity in a single biological sample, combining knowledge on the phosphorylation of both kinases and their substrates (30). To further evaluate the potential of phosphoproteomics for identifying kinase hyperactivity as a possible target in AML, we have performed pY enrichment-based phosphoproteomics of a panel of 16 AML cell lines, coupled to dedicated INKA analysis and functional testing. Additionally, we were able to detect the phosphorylation profile associated with activated FLT3 in two clinical samples. Altogether, our study provides a comprehensive overview of AML signaling and TK activity on an individual cell line basis and shows potential for target selection for effective treatment with kinase inhibitors.
EXPERIMENTAL PROCEDURES
Cell Culture
The HEL, HL-60, Kasumi-3, Kasumi-6, MV4–11, and THP-1 cell lines were obtained from ATCC (Wesel, Germany) and the EoL-1, KG-1, KG-1a, Kasumi-1, ME-1, ML-2, MOLM-13, MM-6, NB4, and OCI-AML3 cell lines were purchased from DSMZ (Braunschweig, Germany). Kasumi-1, Kasumi-3, Kasumi-6, KG-1a, MOLM-13, ML-2, and ME-1 were grown in RPMI 1640 medium with l-glutamine (Life Technologies, Paisley, UK) supplemented with 20% fetal calf serum (FCS). MV4–11, NB4, EoL-1, HL-60, KG-1, MonoMac-6 (MM6), THP-1, and HEL were grown in RPMI 1640 with l-glutamine supplemented with 10% FCS. OCI-AML3 was grown in MEM-α, no nucleosides medium (Life Technologies) supplemented with 20% FCS. Cell lines were cultured at concentrations between 0.3–0.5 × 106 cells/ml and were harvested during the logarithmic growth phase. To obtain ∼10–12 mg protein, 1–1.5 × 108 cells were centrifuged at room temperature and washed twice with cold (4 °C) PBS (Invitrogen, Carlsbad, CA) before further processing.
Clinical Samples
Periferal blood samples containing 93–97% of blasts were obtained from two FLT3-ITD AML patients at the time of diagnosis at Amsterdam UMC, location VUmc, Cancer Center Amsterdam. These patients were included in the HOVON 102 trial (Netherlands Trial Register number NL2070). All study protocols were performed in accordance with the Declaration of Helsinki and approved by the central medical ethical committee (METC-2009–293). Written informed consent was obtained from each patient prior to study entry.
The start of the isolation procedure was within 30–60 min after sample collection. Mononuclear cells (MNC) were enriched using Ficoll-Paque Plus (GE Healthcare, Chicago, IL). Erythrocytes were lysed using lysis buffer (155 mm NH4Cl, 10 mm KHCO3, 0.1 mm Na2EDTA). Cell pellets were snap frozen and stored at −80 °C. Isolation was completed within ∼2.5–3 h after collection.
Sample Processing and pY Immunoprecipitation
Cell lines and clinical samples were lysed in urea lysis buffer for phosphoproteomics (8 m urea, 1 mm orthovanadate, 2.5 mm pyrophosphate, 1 mm β-glycerophosphate in MilliQ water) followed by 1 min. of vortexing and subsequent sonication. Sonicated lysates were cleared by spinning at 5400 × g for 15 min. at 13 °C. Protein content was determined using the DCTM Protein Assay (BioRad, Hercules, CA). Sample quality was examined by SDS-PAGE and Coomassie Blue staining.
Ten miligrams protein input was used as starting material for each cell line. Starting material for the two clinical samples consisted of two 5-mg workflow replicates. Lysates were brought to equal volumes at a concentration of 2 mg/ml protein. Sample preparation and phosphotyrosine immunoprecipitation (IP) procedures were performed as previously reported (31, 32). IP was performed using PTMScan pTyr antibody beads (p-Tyr-1000) (Cell Signaling Technology, Danvers, IL) at a ratio of 4 μl bead slurry per mg protein. Lysate aliquots were taken before the pTyr IP step, and were diluted to 0.1 μg/μl in 0.1% TFA for proteomic analysis.
Phosphopeptide Identification and Quantification
Peptides were separated by an Ultimate 3000 nanoLC system (Dionex LC-Packings, Amsterdam, The Netherlands) coupled online to a Q Exactive mass spectrometer (Thermo Fisher, Bremen, Germany) and equipped with a 40 cm × 75 μm (ID) fused silica column custom packed with 2-μm, 120-Å-pore ReproSil Pur C18 aqua (Dr Maisch GMBH, Ammerbuch-Entringen, Germany). After injection, peptides were trapped at 6 μl/min on a 10 mm × 100 μm (ID) trap column packed with 5-μm, 120-Å-pore ReproSil Pur C18 aqua at 2% buffer B (buffer A: 0.5% acetic acid, buffer B: 80% ACN, 0.5% acetic acid) and separated at 300 nl/min in a 10–40% buffer B gradient in 90 min (120 min. inject-to-inject). Eluting peptides were ionized at a potential of +2 kV and introduced into the mass spectrometer. Intact masses were measured at a resolution of 70,000 (at m/z 200) in the orbitrap using an AGC target value of 3E6 charges. The top 10 peptide signals (charge states 2+ and higher) were submitted to the higher-energy collision (HCD) cell for MS/MS (1.6 amu isolation width, 25% normalized collision energy). MS/MS spectra were acquired at a resolution of 17,500 (at m/z 200) in the orbitrap using an AGC target value of 2E5 charges, a maximum inject time of 80 ms, and an underfill ratio of 0.1%. Dynamic exclusion was applied with a repeat count of 1 and an exclusion time of 30 s. MS/MS spectra for the cell line samples were searched against a Uniprot human FASTA file (release January 2014, no fragments; 42104 entries) using MaxQuant version 1.4.1.2 (33). Clinical samples were searched against the Swissprot human FASTA file (release September 2015, canonical and isoforms; 42122 entries) using MaxQuant version 1.5.2.8. Enzyme specificity was set to trypsin, and up to two missed cleavages were allowed. Cysteine carboxamidomethylation (+57.021464 Da) was treated as a fixed modification, and serine, threonine and tyrosine phosphorylation (+79.966330 Da), methionine oxidation (+15.994915 Da), and N-terminal acetylation (+42.010565 Da) as variable modifications. Peptide precursor ions were searched with a maximum mass deviation of 4.5 ppm, and fragment ions with a maximum mass deviation of 20 ppm. Peptide, protein and site identifications were filtered at a false discovery rate of 1% using the decoy database strategy. The minimal peptide length was set at 7 amino acids, the minimum Andromeda score for modified peptides was 40, and the corresponding minimum delta score was 17 for the cell lines, and 6 for clinical samples (default settings for the respective MaxQuant software versions). Proteins that could not be differentiated based on MS/MS spectra alone were clustered into protein groups (default MaxQuant settings). For the clinical samples, the “match between runs” option was selected in MaxQuant (not for the cell line samples). Phosphopeptide intensities derived from the integrated MS1 signal (isotope cluster) of the eluting peak for each precursor mass were obtained from the modificationSpecificPeptides.txt table from MaxQuant. Intensities were normalized on the summed intensity of all (phospho and non-phospho) peptides in a profiling run of the corresponding tryptic lysate digest and multiplied by the mean intensity across profiled cell line lysates. Phosphopeptide MS/MS spectral counts (34) were calculated from the MaxQuant evidence.txt file using R (http://www.r-project.org) (35). For subsequent analysis, values for clinical sample replicates were averaged. Information on identified phosphopeptides, phosphosites, and proteins can be found in supplemental Table S1. For all phosphopeptide identifications, annotated, mass-labeled MS/MS spectra are provided in supplemental Figs. S8 and S9.
INKA Analysis
Kinase activity was predicted using the INKA analysis tool as described (30). For the INKA analysis, phosphopeptide abundance measures were obtained from the individual cell line measurements. Label-free spectral counts were used as a proxy for abundancy and used in a standard INKA analysis based on four in-silico metrics calculated from phosphopeptide spectral count data. Two kinase-centric metrics focus on phosphopeptides derived from protein kinases themselves: a “kinome” metric, for all phosphopeptides derived from a given kinase, and an “activation loop” metric, which is restricted to phosphopeptides derived from the kinase activation loop segment. Two substrate-centric metrics focus on phosphopeptides derived from proteins that are deemed substrates for a given kinase: a “NetworKIN” (NWK) metric, for phosphopeptides from proteins that are predicted to be a substrate for a specific kinase by the motif-based NetworKIN algorithm (36), and a “PhosphoSitePlus” (PSP) metric, for phosphopeptides from proteins that have been experimentally observed to be a substrate for a particular kinase, as documented in the PhosphoSitePlus catalogue (37). These four metrics, all related to kinase activity directly (kinase-centric) or indirectly (substrate-centric), are integrated into a single INKA score with a requirement for kinases to be implicated by both the kinase- and the substrate-centric side of the analysis. The INKA tool provides bar graphs for all individual analyses, and final INKA scores for top 20 kinases.
Analysis of Kinase Inhibitor Targeting of (Inferred) Kinases
The (kinase) target space of kinase inhibitors quizartinib, ponatinib, ibrutinib, tofacitinib, and GDC0994 was compiled from literature (38–43), selecting kinases inhibited at concentrations below 500 nm. Inhibition data was supplemented with data on drug-kinase binding affinity (500 nm) for kinases for which no inhibition data was available. For each cell line, for each of its kinome, activation-loop, NWK, PSP and INKA metric analyses, kinase activity data were filtered for the kinases included in the pertinent target space (supplemental Table S2). For a given combination of cell line, metric analysis, and kinase inhibitor, the percentage potential kinase inhibition was calculated as the sum of activity values (ia, inferred activity) for kinases in the drug target space, divided by the activity values for all kinases in the data for a cell line (equation 1) (supplemental Table S2A, G, M, S and Y).
| (1) |
Log EC50 values for drugs were compared with the percentage potential kinase inhibition of these drugs for the AML cell lines using Pearson correlation. The EC50 values for ponatinib were supplemented with data from the Genome of Drug Sensitivity in Cancer resource (GDSC), http://www.cancerrxgene.org, consulted Nov.2018) (44). These are indicated in red in supplemental Table S2.
In Silico Analysis of Cell Line KI Sensitivity
Thirty KIs were selected from the dose dependent protein-drug interaction analysis (40) from the proteomicsDB analytics toolbox for which drug sensitivity data was available in the GDSC database or from our own experiments. For each of the 30 KIs the (kinase) target space was determined from literature (38–43, 45–59). Target spaces include kinases inhibited with an IC50 below 500 nm, supplemented with evidence of drug-kinase binding at a Kd below 500 nm as described above (supplemental Table S4). Drug response data were found in the GDSC database for 13 out of 16 cell lines in our data set. For independently tested drugs in this manuscript that were selected based on the INKA profile (see above), the corresponding EC50s were used instead of those in the GDSC database. Per cell line, drug EC50s were divided into two groups based on the presence of the INKA-implicated driver kinase in the target space of the drug. Drug EC50s were visualized in a scatterplot and distribution of the EC50s for each group is shown with a Tukey box plot. The difference in response between the two groups was evaluated using the Mann-Whitney U test.
Molecular Analysis
AML cell lines (n = 16) and two primary patient AML mononuclear fractions were analyzed for common AML-associated molecular aberrancies according to standard diagnostic protocols. DNA and/or RNA was extracted from cell pellets made at the time of lysate preparation as described previously (60, 61). Samples were analyzed for the presence of molecular aberrations in FLT3 (ITD), c-KIT (exon8/17), NPM1 (exon12), JAK2 (exon 14), and CEBPA, FIP1L1-PDGFRA, BCR-ABL, PML-RARA, RUNX1-RUNX1T1 (t(8;21)(q22;q22); AML-ETO), and CBFB-MYH11 (inv(16)) fusions, and 11q23 (MLL) rearrangements according to standard procedures (http://www.modhem.nl). When FLT3-ITD was present, the allelic ratio (AR) was determined by dividing the area under the curve (AUC) of the ITD peak by the total AUC of the ITD and wild-type peaks as found in fragment analysis.
Pathway Analysis
Exploration of downstream pathway activation was done by matching detected phosphoprotein data to an AML scaffold pathway, consisting of the PI3K, MAPK, and JAK-STAT pathways. The AML scaffold pathway was constructed by combining pathway information from four sources, KEGG (62), PANTHER (63), REACTOME (64), and Cell Signaling Technology pathways (https://www.cellsignal.com/contents/science/cst-pathways/science-pathways). Gene symbols were converted to HGNC nomenclature for each source list, and combined lists were made for each pathway. Pathway lists were further integrated to create a list with pathway-associated proteins. The final list was matched to the phosphoprotein list of our data set, and pathway-associated proteins detected in our data set were included in an AML signaling list (n = 199). For each sample, phosphoprotein-level spectral counts were matched to members of this list to create a per sample overview of pathway activation.
Pathway Visualization
To provide a per sample view of pathway activation, we built a simplified pathway scaffold in Cytoscape (65) (version 3.3.0), based on the kinases and phosphatases present in the AML signaling list that were identified in our data set, supplemented with a few other pathway proteins identified in our data set. Protein relations were extracted from the STRING database (https://string-db.org) (66). Phosphoprotein counts per sample were matched to this scaffold pathway to create a per sample visualization of pathway activation. The color key range was set to 0 to 50 for all samples, with proteins undetected in a sample visualized in green.
Signaling Profiles
The spectral count-based AML pathway activation lists (supplemental Table S3) were used to calculate z-scores with median normalization of phosphoprotein counts within each sample column, using the Perseus software package (49) (version 1.5.5.0). Hierarchical clustering (distance measure: Euclidean, linkage method: average) was done for all cell lines and clinical samples using the hclust() function in R.
Western Blot Analysis
Per sample, 20 μg of lysate was loaded on a pre-cast 4–12% gradient gel (Bio-Rad), and proteins were transferred onto an Immobulon P PVDF membrane (Merck Millipore, Billerica, MA). Blocking buffer consisted of 5% BSA in PBS/0.1% Tween20 (PBST). The blot was incubated overnight at 4 °C with anti-phosphotyrosine antibody 4G10 Platinum (Merck Millipore) diluted 1:1000 in PBST containing 1% BSA.
Drug Sensitivity Assay (MTT)
Quizartinib (AC220), imatinib, ponatinib, masitinib, tofacitinib, GDC-0994, and binimetinib were acquired from Selleckchem (Munich, Germany). Ibrutinib was purchased from MedChem Express (Sollentuna, Sweden). Optimal cell amounts for plating were determined with growth curves for each cell line to ensure logarithmic growth (between 3000 and 13,000 cells/well). Proliferation was determined at 96 h to ensure a minimum of three doublings for each cell line. Cell proliferation was assessed after a 4-hour incubation with 10% MTT (3-(4,5-dimethylthiazolyl-2)-2–5-diophenyl tetrazolium bromide) (Sigma, Taufkirchen, Germany) at 37 °C following a previously described protocol (67). The EC50 value was defined as the drug concentration needed to inhibit 50% of cell growth compared with growth of untreated control cells. All experiments were done in quadruplicate.
Experimental Design and Statistical Rationale
Phosphoproteomics was performed for 16 AML cell lines with different genetic profiles and mutations. Workflow reproducibility of the pY phosphoproteomics workflow was benchmarked previously (31). Samples were measured in a single-shot experiment. For cell lines, single biological samples (n = 1) were used for analysis. For clinical samples, biological material was split at the start of sample processing and measured as two technical replicates. For further analysis, values for phosphopeptides identified in both replicates were averaged into one value per clinical sample. For phosphopeptides identified in only one replicate the value of the identification was used. The INKA score and its component metrics are within-sample values, and statistical relevance of the former has been addressed (35). For drug response analyses, fourteen out of 16 cell lines were tested for multiple KIs in quadruplicate, and drug response curves show mean viability with the standard deviation indicated. For correlation between reduction of INKA scores for a drug's target space and drug EC50s in Fig. 6, Pearson correlation in R was used.
Fig. 6.

Systematic analysis of ranked inferred kinases and their INKA metrics in relation to drug EC50 values. Percentage potential kinase inhibition for quizartinib, ponatinib, ibrutinib, tofaitinib and GDC0994 was separately calculated for “Kinome”-, “Activation loop”-, “PhosphoSitePlus” (PSP)- and “NetworKIN′ (NWK)-based metrics of kinase activity as well as for the aggregate INKA score. This involves, for a given metric/score, taking the ratio of the summed values of kinases in the target space of a kinase inhibitor and the summed values of all kinases within a cell line. Log EC50 values for quizartinib, ponatinib, ibrutinib, tofaitinib and GDC0994 were compared with the percentage potential kinase inhibition of these drugs for the AML cell lines using Pearson correlation.
RESULTS
Phosphoproteomic Profiling of AML Cell Lines Shows Heterogeneity in Phosphorylation Patterns
Label-free phosphoproteomics has been shown to be a valuable tool to investigate kinase signaling (57–59), and, unlike isotopic labeling approaches, is feasible for application to large clinical multi-group cohorts that we envisage to screen in future. To investigate kinase hyperactivity and intracellular signaling in AML, we used our robust and reproducible workflow for label-free quantitative pY-based phosphoproteomics (31) combined with a dedicated analysis on a panel of 16 AML cell lines (Fig. 1). In total, 4853 phosphopeptides from 2279 phosphoproteins were identified, carrying 4229 phosphosites (3605 class I, localization probability > 0.75). The bulk of the class I phosphosites (3525) were phosphotyrosine sites on 4430 phosphopeptides (supplemental Tables S1 and S5). The number of identified phosphopeptides per sample was highly variable, ranging from 241 to 2764 phosphopeptides in individual cell lines (Fig. 1; supplemental Table S5). This high variability in phosphorylation patterns was confirmed by anti-pY Western blot analysis using aliquots of cell line lysates (supplemental Fig. S1) and underlines the need for analysis of individual samples.
Fig. 1.

Experimental outline. A panel of AML cell lines with known and unidentified kinase drivers was subjected to phosphoproteomics. To this end, immunoprecipitated phosphotyrosine peptides of 16 AML cell lines were analyzed by nanoLC-MS/MS. Combining phosphopeptides per protein allowed for quantification of overall protein phosphorylation state. Kinase activity in the cell lines was assessed by four different analyses. Kinase activation was inferred from total kinase phosphorylation and activation-loop phosphorylation and substrate driven analysis was done using known kinase-substrate relations and the motif-based kinase-substrate associations. Alongside, downstream pathway analysis was done, focused on AML associated pathways (PI3K, JAK-STAT, and MAPK), to gain insight in downstream network activation. Based on the results, candidate drivers were selected for functional validation using kinase inhibitors.
Unsupervised clustering using all normalized phosphopeptide intensities separated cell lines with high phosphorylation levels and clusters displaying intermediate and low levels of phosphorylation (Fig. 2A). Cell lines did not cluster based on French-American-British (FAB) classification of hematologic diseases (Fig. 2A) or growth conditions such as doubling time or medium (data not shown). Importantly, the closely related KG-1 and KG-1a cell lines, where KG-1a is a less mature subline of KG-1, showed comparable phosphorylation patterns and clustered together.
Fig. 2.

Unsupervised clustering of the panel of 16 AML cell lines. Unsupervised clustering of phosphopeptide intensities indicates that cell lines cluster independent of mutation status or FAB classification.
Out of 2279 identified phosphorylated proteins, 138 were classified as protein kinases (Fig. 1, supplemental Table S5). The heterogeneous levels of phosphorylation per cell line observed on Western blotting and at the phosphopeptide level were also reflected in the unsupervised clustering using kinase counts (supplemental Fig. S2A).
Molecular characterization of the cell line panel following standard molecular diagnostic procedures identified common genomic aberrations in only 6 out of 16 cell lines (supplemental Table S6). These included genomic aberrations in the AML-linked kinase genes FLT3 (MV4–11, MOLM-13, Kasumi-6), PDGFRA (EoL-1), KIT (Kasumi-1), and JAK2 (HEL). Interestingly, analysis of Kasumi-6 revealed two distinct FLT3-ITDs, one of which was only detected at a low AR. No kinase aberrations were detected in any of the other 10 cell lines. Phosphopeptide clustering did not group the MV4–11, MOLM-13 and Kasumi-6 cell lines based on their FLT3-ITD mutation, indicating that phosphoproteomics adds a complementary layer of information.
INKA Ranking Pinpoints Hyperactive Kinases in Accordance with Corresponding Molecular Aberrations, Confirming Their Potential as Drug Targets
Several fusions and gain-of-function mutations in kinases are known driving factors in AML. An initial exploration into phosphorylation of the known AML TK drivers revealed that cell lines with an identified kinase mutation showed unusually high phosphorylation at the protein level when compared with the other cell lines in the panel (supplemental Fig. S2B). In addition, four cell lines without identified mutations according to standard genetic testing (KG-1, KG-1a, MM6 and Kasumi-3) also showed high phosphorylation of FGFR1, FLT3 or PDGFRA, indicating potential kinase driver activity in these cell lines (see outliers in supplemental Fig. S2B, Fig. 3, green panel).
Fig. 3.

Top 20 ranking phosphokinases in the 16 AML cell lines. Kinases were ranked based on their INKA score. Potential drivers are indicated by an arrow and bars are colored (PDGFRA = pink, FGFR1 = purple, KIT = orange, FLT3 = blue, JAK2 = green). Six cell lines showed high phosphorylation for known AML driver kinases that harbored mutations confirmed by standard diagnostic tests (supplemental Table S1). Furthermore, phosphoproteomics uncovered potential driver kinases in 10 other cell lines (green and blue shaded cell lines) that were missed by the standard diagnostic tests.
To gain insight in (hyper)activity of kinases in a single cell line and pinpoint hyperactivity indicative of driver activity, we performed INKA analysis (35). The INKA approach is based on a 4-component analysis, for each given kinase, of phosphorylation of the kinase itself (“kinome” metric) and its activation loop segment (“activation loop” metric) on the one hand, and phosphorylation of all of its possible substrates as deduced from NetworKIN predictions and the PhosphoSitePlus catalogue of experimentally observed substrates (“NWK” and “PSP” metrics, respectively) on the other. Only kinases with both kinase-centric and substrate-centric evidence get a non-zero INKA score.
Bar graph visualizations of the INKA scores for the six cell lines with an identified TK mutation indicated high activity of these kinases, with ranks between position 1 and 6 of the highest INKA scores (Fig. 3). The high ranking of these mutant kinases was also apparent in the four individual INKA component analyses (supplemental Fig. S3A–S3F). INKA scores of mutant kinases were especially pronounced for the FIP1L1-PDGFRA fusion cell line EoL-1 and the Kasumi-1 cell line carrying an N822K KIT point mutation (Fig. 3). INKA ranking of the FLT3-ITD mutant cell lines MV4–11, MOLM-13, and Kasumi-6, and the V617F JAK2 cell line HEL showed a top 2–6 ranking of FLT3 and JAK2, next to several other kinases. Interestingly, other high-ranking kinases in these cell lines were generally located downstream in the FLT3 and JAK2 cellular signaling hierarchy, thereby still implicating FLT3 and JAK2 as primary suspects of driver activity. KIT activity was also indicated for Kasumi-6, indicating this kinase might also contribute to signaling in this cell line.
Exposure of EoL-1, Kasumi-1, MV4–11 and MOLM-13 cells to quizartinib (a PDGFRA, c-KIT and FLT3 inhibitor) confirmed dependence on their drivers, with EC50 values below 10 nm (supplemental Fig. S3A–S3D). The JAK2 mutant HEL was only moderately sensitive to JAK2 inhibition with an EC50 for tofacitinib at 5.5 μm (supplemental Fig. S3F). This result was comparable to sensitivities found with the JAK inhibitors ruxolitinib (0.66 μm) and fedratinib (1.02 μm) for this cell line in the GDSC web resource. To check whether these results were specific to the cell lines with activity of a kinase targeted by the inhibitor, a few of the other cell lines without the intended target driver were taken along as negative controls. As expected, these cell lines were much less sensitive to the KIs tested compared with those with an indicated driver targeted by the inhibitor (supplemental Fig. S4). The KI ibrutinib was also tested, as several relevant kinases (both the major target, BTK, and multiple other kinases to which the drug binds in the nanomolar range) showed high INKA rankings. In correspondence with the general presence of these kinases in all cell lines, EC50s for ibrutinib were moderate with EC50s generally above 1 μm (supplemental Fig. S4). Overall, EoL-1, Kasumi-1, MV4–11, MOLM-13, and HEL cell lines were much less sensitive to ibrutinib than to the driver targeting inhibitor (supplemental Fig. S3A–S3F).
Phosphoproteomics Reveals Unexpected Hyperphosphorylated Kinases and INKA Analysis Indicates Their Potential as Drug Targets in Four Cell Lines
Outlier analysis of phosphorylated kinases in our data set indicated four additional cell lines in our AML panel that potentially contain a kinase driver (supplemental Fig. S2B) although not indicated as such by a standard diagnostic mutation screen. These included FGFR1 (KG1 and KG1a), FLT3 (MM6), and PDGFRA (Kasumi-3), and kinome ranking for these cell lines showed high phosphorylation for each kinase (rank 1–4) (Fig. 3, green highlighted area; Fig. 4A). Kinase activity in these four cell lines was further supported by phosphorylation of the respective activation loops (rank 1–2) and by one or both substrate-centric analyses (supplemental Fig. S3B–S3C and S3I–S3J), resulting in a top 2 INKA ranking and thereby indicating high activity of these kinases in these cell lines.
Fig. 4.

Validation of potential kinase drivers in four AML cell lines without identified driver mutations. A, INKA score ranking for the MM6 and Kasumi-3 cell lines indicates high activity of the FLT3 and PDGFRA kinases while ranking in KG-1 and KG-1a cell lines uncovers FGFR1 (arrows). B, List of experimentally established drug targets of ponatinib (green panel), quizartinib (pink panel) and ibrutinib (blue panel) with an affinity below 500 nm. Targets indicated by circles were discovered through a chemical proteomics approach (40), and those indicated by triangles used cell-free essays for target identification (39, 43, 72). Bold font indicates kinases that are intended targets of the drug, whereas plain font indicates additional target kinases. Kinases with a top 20 INKA score in the KG-1, KG-1a, MM-6 and Kasumi-3 cell lines are indicated with the corresponding colors as in panel A. C, Targeting of suspected driver kinases in the above cell lines with a KI effective against FGFR1 (ponatinib), or FLT3 and PDGFRA (quizartinib) shows effective inhibition of cell viability in the nanomolar range. Ibrutinib is used as a control.
Targeting of FGFR1 in the KG-1 and KG-1a cell lines with ponatinib further supports driver function of FGFR1 in these cell lines with drug EC50 values of 2.8 nm and 1.0 nm, respectively (Fig. 4C, supplemental Fig. S3B–S3C, supplemental Fig. S4). Inhibitor selection based on indicated FLT3 and PDGFRA hyperactivity in the MM6 and Kasumi-3 cell lines was equally successful, with an EC50 of 0,068 nm in MM6 and 1.8 nm in Kasumi-3 for quizartinib (Fig. 4C, supplemental Fig. S3I–S3J, supplemental Fig. S4). As expected, the related KG-1 and KG-1a cell lines showed comparable responses to ponatinib. Ibrutinib had a comparatively minimal effect in all four cell lines with EC50s above 1 μm. A subsequent literature search for these cell lines confirmed mutations in the respective driver kinases identified (29, 68, 69). Together, the results support driver activity of the respective kinases as predicted by our phosphoproteomics-based INKA analysis.
MAPK Signaling is Relatively High in Cell Lines Without a Clear TK Driver
In six of the sixteen cell lines in our panel, HL-60, ML-2, OCI-AML3, NB4, THP-1 and ME-1, we did not identify a clearly hyperactive TK driver, as assessed by group-based outlier analysis and INKA analysis (Fig. 3, blue highlighted area; Fig. 5A; supplemental Fig. S3K–S3P). Although a subset of these cell lines (ML-2, OCI-AML3, THP-1, and ME-1) showed some phosphorylation of known AML drivers such as FLT3 and KIT, phosphorylation levels detected in these cell lines were much lower than in the cell lines carrying a FLT3 or KIT mutation. Similarly, limited substrate-derived evidence for activation of these kinases was found in these cell lines, and INKA analysis only indicated potential activity of KIT (rank 5) in ME-1.
Fig. 5.

Ranking of phosphorylated kinases in six cell lines without an identified mutation or TK hyperphosphorylation. A, INKA scores of MAPK1 and MAPK3 (arrow) rank high in four out of six cell lines without a clear hyperactive TK driver. B, List of experimentally established drug targets of GDC-0994 (orange panel), binimetinib (purple panel) and ibrutinib (blue panel) with an affinity below 500 nm. As in Fig. 4, the target space (40, 42, 43), is indicated in the phosphokinase profiles. Bold font indicates kinases that are intended targets, whereas plain font indicates additional targets of the drug. Kinases with a top 20 INKA score are indicated with corresponding colors as in panel A. C, Targeting of MAPK1 and MAPK3 with GDC-0994 shows effective inhibition of cell viability in the nanomolar range compared with treatment with ibrutinib (control) (p < 0.0001, two-way ANOVA). Targeting the MAPK pathway by upstream MAP2K1 and MAP2K2 inhibition using binimetinib was even more effective (p < 0.0001 and p < 0.0001, respectively). Colors as in panel A.
Inspection of the INKA ranking in these cell lines revealed relatively high INKA ranking for MAPK3 (rank 5–8) in four cell lines, i.e. ML-2, HL-60, OCI-AML3 and THP-1, as compared with cell lines with a TK driver (Fig. 3; Fig. 5A). Importantly, activity of MAPK 3 and of MAPK1 was supported by both the kinase-centric and substrate-centric analyses, in which the latter indicated activation of upstream activators MAP2K1 and MAP2K2 in these cell lines (supplemental Fig. S3K–S3N). An inspection of the Cancer Cell Line Encyclopedia (CCLE) database (70) (https://portals.broadinstitute.org/ccle) and the Cosmic Cell Lines Project (CCLP) database (71) (https://cancer.sanger.ac.uk/cell_lines) indicated the presence of upstream RAS mutations in cell lines displaying MAPK1/3 activation, offering an explanation for the observed activity.
Incubation of ML-2, HL-60, OCI-AML3 and THP-1 with the MAPK1/3 inhibitor GDC-0994 showed a relatively better inhibitory response (EC50 = 0.5 μm–2.7 μm) when compared with ibrutinib (p < 0.0001) (Fig. 5C, supplemental Fig. S3K–S3O). Additionally, consistent with upstream RAS activation, inhibition with the upstream MEK inhibitor binimetinib proved even more successful in these cell lines (EC50 = 8 nm–144 nm). Despite the relatively low kinase rank of MAPK3 in the NB4 cell line, just outside the top 20 (rank 21), pathway visualization of NB4 signaling still indicated involvement of MAPK in NB4 cellular signaling (supplemental Fig. S3O). Indeed, MEK-ERK inhibition proved successful for this cell line (EC50 of 0.5 μm for MAPK1/3 and 28 nm for MAP2K1/2; Fig. 5C, supplemental Fig. S3O), and these results are in line with the KRAS mutation present in this cell line (CCLE and CCLP data).
Systematic Analysis of Correlation Between Kinase Ranking and Drug EC50 Values for Individual Cell Lines
We explored to what extent INKA scores and the metric values of the four underlying analyses could be linked to drug EC50 values for individual cell lines. To this end, a target space was specified for each drug, selecting experimentally proven targets inhibited by the drug at concentrations below 500 nm, supplemented with targets for which no inhibition data were available but with a binding affinity below 500 nm. For each cell line in each specific analysis, the percentage potential kinase inhibition was formulated as the ratio between the summed metric values of kinases in the target space of a KI and the summed metric values of all kinases. The resulting % potential kinase inhibition was then compared with the drug-specific log EC50 values for a cell line. In case of predictive value, an inverse correlation between % potential inhibition and log EC50 would be expected.
Overall, the kinase-centric data (phosphorylated kinase and phosphorylated activation loop) showed more consistent inverse correlations as compared with substrate-centric kinase analyses (Fig. 6). This was especially true for the drugs targeting an RTK driver, whereas the relation was less clear for the drugs targeting downstream kinases like tofacitinib (JAK2), GDC-0994 (MAPK1/3) and ibrutinib (BTK, LYN). This is most likely because of the broad involvement of these kinases in downstream cell signaling, thereby not limiting their involvement to those cell lines carrying a mutation. Though INKA scores are based on all four underlying analyses, correlation between % potential kinase inhibition and log EC50 showed similar results to the kinase-centric analyses, with correlation clearest for those inhibitors targeting RTKs.
In Silico Validation of INKA-guided Drug Selection
To further validate our INKA-guided drug selection in a wider panel of drugs, we performed an in silico analysis of cell line sensitivity for a panel of 30 KIs for which kinase inhibition data was available in the proteomicsDB analytics toolkit, and for which data on drug sensitivity was available in the GDSC (supplemental Table S4). Comparison of drug EC50s for KIs targeting the INKA-identified driver kinase of an RTK-driven cell line versus selected non-profile KIs showed that EoL-1 (p = 0.0001), Kasumi-1 (p = 0.038), MV4–11 (p = 0.0015), MOLM-13 (p = 0.0006), KG-1 (p = 0.0075), and MM-6 (p = 0.0005) were significantly more sensitive to KIs targeting their respective drivers (supplemental Fig. S5).
Though sensitivity of HEL to other JAK2-inhibiting KIs was comparable to its response to tofacitinib, the JAK2-driven cell line HEL did not respond significantly better to JAK2-inhibiting drugs compared with other KIs (p = 0.2217), indicating that targeting of JAK2 alone might not be enough in this cell line. Analysis of RAS-mutant cell lines also showed variable results with p values ranging between 0.2245 and 0.0248. Overall, response to driver inhibition was less distinct for cell lines without an RTK driver.
Phosphoproteomics Demonstrates FLT3 Activation in FLT3-ITD-positive Patient Samples
To assess if identification of hyperactive phosphorylated kinases is also feasible in AML clinical samples that undergo a ∼1.5-hour purification protocol, we analyzed AML cells of two FLT3-ITD-positive AML patients. The first sample (Pt.1) was collected from a 66-year-old female AML patient carrying two FLT3-ITD variants, with an AR of 46% for the dominant variant, and an NPM1 mutation (supplemental Table S7). Blast percentage before MNC isolation using Ficoll-Paque was 97% as determined by immunophenotyping. The second sample (Pt.2) was acquired from a 45-year-old female AML patient with 93% blast cells before isolation. This sample also carried both an FLT3-ITD (AR of 95%) and an NPM1 mutation.
Phosphoproteomic analysis of the two clinical samples yielded 1089 phosphopeptides, with 880 phosphosites (768 class I) on 553 phosphoproteins, including 55 phosphorylated kinases (supplemental Table S8). Despite a lower sample input (5 mg versus 10 mg), phosphorylation of FLT3 was detected at a similar order of magnitude as observed for the FLT3-ITD mutant cell lines (supplemental Fig. S6, kinome analysis). Additionally, a higher extent of FLT3 phosphorylation was detected for the patient with the highest FLT3-ITD AR (Pt.2). INKA analysis of the two clinical samples indicated activity of FLT3 in both samples (rank 6), which was supported by both kinase-centric and substrate-centric analyses (Fig. 7A, supplemental Fig. S6), which is similar to the ranking found in ITD-FLT3 AML cell lines (Fig. 3, supplemental Fig. 3C–S3E).
Fig. 7.
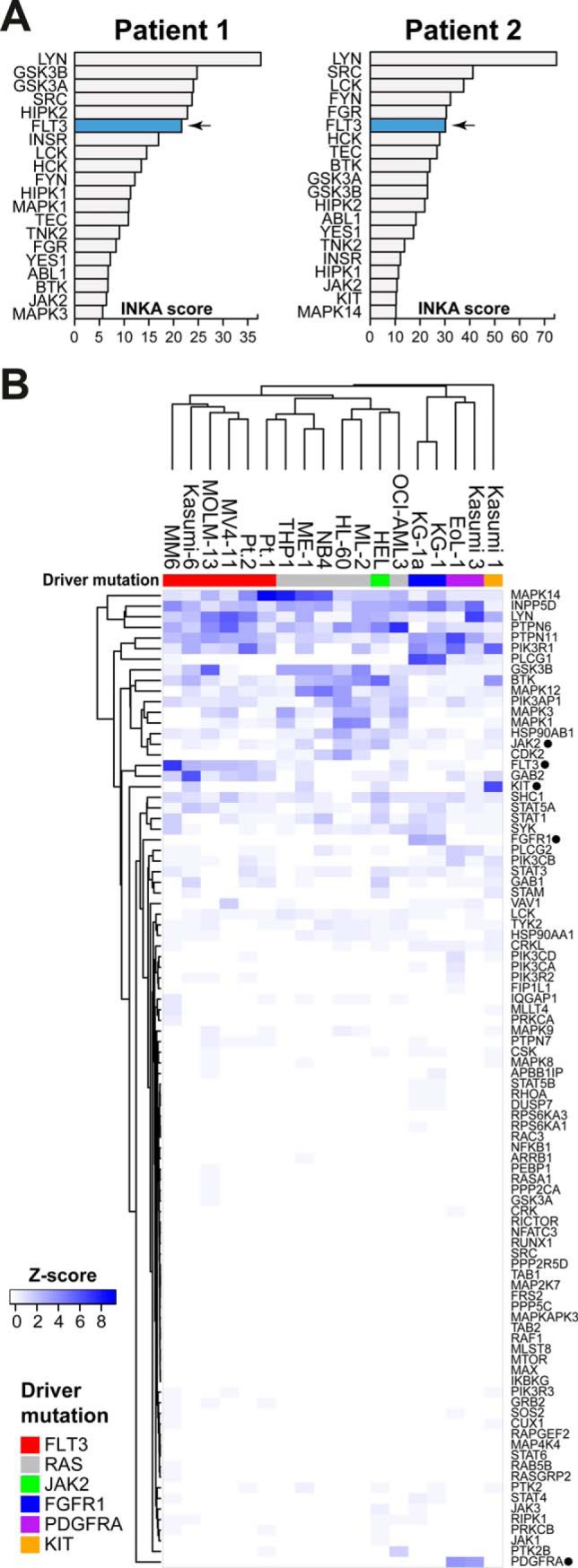
Phosphoproteome analysis of two FLT3-ITD positive patients. A, INKA score ranking of kinases for each patient sample identified FLT3 as highly active in both patients. Ranking of FLT3 was comparable to that in FLT3-ITD-positive cell lines. FLT3 is indicated in blue. B, Hierarchical clustering of all samples shows the similarity in pathway phosphorylation between cell lines and clinical samples. Samples clustered predominantly by their identified AML drivers. Clustering is based on within-sample z-scores of summed phosphopeptide counts for proteins that are part of the defined AML signaling network (supplemental Table S3).
Overlapping phosphopeptide data of the patient samples with that of the three FLT3-ITD-positive cell lines MV4–11, MOLM-13 and Kasumi-6 revealed that nearly all phosphopeptides measured in the cell lines could also be identified in the clinical samples (supplemental Fig. S7A). Furthermore, despite the lower protein input, an additional 251 phosphopeptides were detected in the clinical samples. In total, 71 kinases were identified in the combination of both data sets, and 42 of these were detected in both cell lines and clinical samples (supplemental Fig. S7B).
To further investigate the potential effect of sample processing on phosphorylation in clinical samples, we clustered cell lines and patient samples together based on their AML signaling pathway components (Fig. 7B, supplemental Table S3). This analysis showed that most samples clustered by their identified RTK drivers. Although the Pt.2 patient sample clustered together with the FLT3 aberrant cell lines, the Pt.1 patient sample clustered in an adjacent sub-cluster together with RAS-driven cell lines. These results underscore the similarity in signaling patterns of cell lines and patient samples and suggest that sample processing has minimal impact on AML signaling.
DISCUSSION
In view of the discovery of frequent molecular aberrations in RTKs in AML, and the limitations of mutation-based treatment selection, there is a clinical need to predict drug sensitivity of AML patients for kinase inhibitors. Phosphoproteomic approaches may provide an additional layer of functionally relevant molecular information that is advantageous for effective treatment selection. Here, as a first preclinical step, we have explored application of phosphoproteomics coupled to INKA analyses for KI selection for individual “AML “cases” in an in vitro setting. To our knowledge, we provide the largest AML pY-phosphoproteome data set to date (4853 phosphopeptides from 2279 phosphoproteins), thereby significantly extending previous findings of limited depth (∼48–450 phosphopeptides) in 13 AML cell lines (16, 19, 29, 30), five of which are included in our data set.
We have analyzed cellular kinome states by ranking kinase phosphorylation levels in single samples and by cohort outlier analysis. Most notably, our study revealed that INKA analysis, based on quantification of kinase phosphorylation, both at the level of the entire molecule and at the level of the activation loop specifically, in combination with phosphosubstrate analysis, can serve to deduce aberrant kinase activation. Application of single-sample INKA ranking to a panel of AML cell lines enabled us to explore how different genomic kinase aberrations translate into differences in the phosphoproteome of six AML cell lines, and whether phosphoproteomics-based INKA analysis could reveal their RTK hyperactivity. Furthermore, we were able to identify unexpected FLT3, PDGFR, FGFR1, and MAPK pathway activity in ten more cell lines. Analysis of two patient samples produced comparable results to those of the cell lines, indicating that the elaborate sample processing needed to purify AML blast cells from blood has no major influence on clinical sample phosphorylation state and that our method might be translatable to clinical samples. Our data show that phosphoproteomics in combination with INKA analysis can be used as a global readout of kinase activation that can be harnessed to select KIs for inhibition of identified targets. Potentially, this could also include elucidation of kinase-driven resistance mechanisms as well as stimulatory influences from the microenvironment that are missed by mutation-based analyses.
Evaluation of INKA-guided drug selection in a panel of 30 KIs from publicly available data showed that RTK-driven cell lines were significantly more sensitive to KIs targeting the INKA-identified RTK drivers, confirming the ability of our method to identify driver RTKs and the success of KI-based treatment of RTK-driven cell lines. However, though INKA was able to indicate activity of the mutant driver JAK2 and RAS pathway-related kinases MAPK1 and MAPK3 in non-RTK-driven cell lines, response to single KI inhibition of these downstream hyperactive kinases was much less distinct from selected non-profile kinase inhibitors. Possibly, monotherapy is not enough for these cell lines. Interestingly, despite evidence for only three kinases in its target space, the MEK inhibitor trametinib caused the largest response by far in HL-60, ML-2, and OCI-AML3 compared with other MEK inhibitors. Also, as the insulin receptor is ranked high (position 3–4) in ML-2, OCI-AML3, and THP-1, this receptor might be an interesting co-target in these cell lines. Further research is needed to reveal the mechanisms behind this variable response and the success of combination treatment in these cell lines.
Identification of kinase activity was not limited to the identification of driver kinases, as several other kinases showed high INKA score ranking. These kinases were generally involved downstream of RTKs such as SRC-family kinases, and kinases functioning further downstream such as MAPK14, GSK3A/B and CDKs. Though some components, such as adaptor proteins, were more specific to cell lines with a hyperactive RTK, kinases such as BTK, LYN, MAPK14, GSK3A/B and CDKs were generally phosphorylated in all cell lines and clinical samples, indicating a possible “house-hold” function.
In the FLT3-ITD mutant cell lines, FLT3 generally scored lower than or similar to several downstream components in the INKA analysis. However, these cell lines still proved highly responsive to FLT3 inhibition, indicating that pathway hierarchy is equally important in selecting potential driver kinases for treatment. Indeed, despite the less striking difference in INKA scores in the FLT3-ITD mutant cell lines, sensitivity to a FLT3-targeting inhibitor was similar to that of the FLT3 point mutant cell line MM6, where FLT3 scored exceptionally high. The importance of targeting RTKs over other kinases was also evident from our systematic analysis of kinase activity in relation to drug EC50 values, reflecting the involvement of RTKs as known drivers in AML. Co-activated kinases could potentially be interesting for use in combination treatment.
Previous efforts in AML mainly involved global phosphoproteomics in conjunction with phosphosubstrate-based analyses for kinase inference (17, 54, 55, 57, 58). For the phosphotyrosine-specific data set of AML phosphoproteomes presented here, we showed the superior correlation of kinase-centric ranking with drug EC50 values as compared with substrate-centric ranking. Substrate-centric metrics do significantly contribute to the performance of INKA scoring (35), however, and their low correlation with drug EC50 values did not compromise the correlation of the aggregate INKA scores with EC50. Whether these results may be extrapolated to global phosphoproteomics data dominated by serine and threonine phosphorylation remains to be determined. On a further note, most KIs demonstrate at least some degree of polypharmacology (see Figs. 4 and 5, and supplemental Fig. S3). This may be considered an added benefit, with one drug inhibiting multiple active kinases, and is therefore taken into account in the analysis shown in Fig. 6, where the target space of a drug is used to assess the relation between drug EC50 and the magnitude of individual kinase activity metrics for separate cell lines.
In summary, our study provides an in-depth global analysis of the phosphotyrosine-based phosphoproteome in AML. We demonstrated the potential of the INKA ranking strategy based on kinase phosphorylation, supported by evidence of substrate phosphorylation, to provide a readout for kinase activation in single AML samples. Underscoring our approach, we functionally verified target potential of hyperactive kinases in ten cell lines for which standard diagnostic mutational analysis did not reveal any drivers. Our analysis of two clinical samples illustrates, in principle, the feasibility of kinase activity analysis in patient material. A more elaborate assessment of the phosphoproteome-guided drug matching strategy in de novo and relapsed patient samples will shed light on its potential for future clinical application in guiding specific kinase inhibitor selection for individual AML patients.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the set identifiers PXD007237 (cell line data), PXD015639 (clinical sample phosphoproteomics data), and PXD015662 (clinical sample proteomics data) (https://proteomecentral.proteomexchange.org).
Supplementary Material
Acknowledgments
We thank the molecular diagnostics team of the Department of Hematology for performing the molecular characterization of the cell lines and clinical samples used in this study. Furthermore, we would like to thank Peter Sminia for providing us with binimetinib, and Anneloes van Duijn for performing several of the cell viability assays.
Footnotes
* This work was funded by the VUmc-Cancer Center Amsterdam through grant CCA2014-1-15 and grant CCA2012-5-08. The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- AML
- acute myeloid leukemia
- TK
- tyrosine kinase
- KI
- kinase inhibitor
- pY
- phosphotyrosine
- INKA
- integrative inferred kinase activity
- ITD
- internal tandem duplication
- FLT3
- FMS-like receptor tyrosine kinase 3
- TKD
- tyrosine kinase domain
- PB
- peripheral blood
- MNC
- mononuclear cell
- IP
- immunoaffinity precipitation
- PDGFRA
- platelet-derived growth factor receptor alpha
- FIP1L1
- pre-mRNA 3′-end-processing factor FIP1
- KIT
- Mast/Stem cell growth factor receptor KIT
- JAK2
- janus kinase 2
- FGFR1
- fibroblast growth factor receptor-1
- RTK
- receptor tyrosine kinase
- AR
- allelic ratio.
REFERENCES
- 1. Kelly L. M., and Gilliland D. G. (2002) Genetics of myeloid leukemias. Annu. Rev. Genomics Hum. Genet. 3, 179–198 [DOI] [PubMed] [Google Scholar]
- 2. Cools J., and Schwaller J. (2004) Molecular mechanisms of myeloid malignancies: on the hunt for new therapeutic targets. Drug Discov. Today Dis. Mech. 1, 259–266 [Google Scholar]
- 3. Smith C. C., and Shah N. P. (2013) The role of kinase inhibitors in the treatment of patients with acute myeloid leukemia. Am. Soc. Clin. Oncol. Educ. book. 2013, 313–318. doi: 10.1200/EdBook_AM.2013.33.313 [DOI] [PubMed] [Google Scholar]
- 4. Kottaridis P. D., Gale R. E., Frew M. E., Harrison G., Langabeer S. E., Belton A. A., Walker H., Wheatley K., Bowen D. T., Burnett A. K., Goldstone A. H., and Linch D. C. (2001) The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United King. Blood 98, 1752–1759 [DOI] [PubMed] [Google Scholar]
- 5. Yamamoto Y., Kiyoi H., Nakano Y., Suzuki R., Kodera Y., Miyawaki S., Asou N., Kuriyama K., Yagasaki F., Shimazaki C., Akiyama H., Saito K., Nishimura M., Motoji T., Shinagawa K., Takeshita A., Saito H., Ueda R., Ohno R., and Naoe T. (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97, 2434–2439 [DOI] [PubMed] [Google Scholar]
- 6. Schnittger S., Schoch C., Dugas M., Kern W., Staib P., Wuchter C., Lö H., Sauerland C. M., Serve H., Bü T., Haferlach T., and Hiddemann W. (2002) Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100, 59–66 [DOI] [PubMed] [Google Scholar]
- 7. Thiede C., Steudel C., Mohr B., Schaich M., Schä U., Platzbecker U., Wermke M., Bornhä M., Ritter M., Neubauer A., Ehninger G., and Illmer T. (2002) Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99, 4326–4335 [DOI] [PubMed] [Google Scholar]
- 8. Lo-Coco F., Cuneo A., Pane F., Cilloni D., Diverio D., Mancini M., Testoni N., Bardi A., Izzo B., Bolli N., La Starza R., Fazi P., Iacobelli S., Piciocchi A., Vignetti M., Amadori S., Mandelli F., Pelicci P. G., Mecucci C., Falini B., Saglio G., and Acute Leukemia Working Party of the GIMEMA group. (2008) Prognostic impact of genetic characterization in the GIMEMA LAM99P multicenter study for newly diagnosed acute myeloid leukemia. Haematologica 93, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 9. Fröhling S., Schlenk R. F., Breitruck J., Benner A., Kreitmeier S., Tobis K., Döhner H., and Döhner K. (2002) Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML study group Ulm. Blood 100, 4372–4380 [DOI] [PubMed] [Google Scholar]
- 10. Abu-Duhier F. M., Goodeve A. C., Wilson G. A., Gari M. A., Peake I. R., Rees D. C., Vandenberghe E. A., Winship P. R., and Reilly J. T. (2000) FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br. J. Haematol. 111, 190–195 [DOI] [PubMed] [Google Scholar]
- 11. Stone R. M., Mandrekar S. J., Sanford B. L., Laumann K., Geyer S., Bloomfield C. D., Thiede C., Prior T. W., Döhner K., Marcucci G., Lo-Coco F., Klisovic R. B., Wei A., Sierra J., Sanz M. A., Brandwein J. M., de Witte T., Niederwieser D., Appelbaum F. R., Medeiros B. C., Tallman M. S., Krauter J., Schlenk R. F., Ganser A., Serve H., Ehninger G., Amadori S., Larson R. A., and Döhner H. (2017) Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 377, 454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wander Sa. Levis M. J., and Fathi A. T. (2014) The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther. Adv. Hematol. 5, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kadia T. M., Ravandi F., Cortes J., and Kantarjian H. (2016) New drugs in acute myeloid leukemia. Ann. Oncol. mdw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knapper S., Burnett A. K., Littlewood T., Kell W. J., Agrawal S., Chopra R., Clark R., Levis M. J., and Small D. (2006) A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 108, 3262–3270 [DOI] [PubMed] [Google Scholar]
- 15. Levis M. J., Perl A. E., Dombret H., Döhner H., Steffen B., Rousselot P., Martinelli G., Estey E. H., Burnett A. K., Gammon G., Trone D., Leo E., and Cortes J. E. (2012) Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. Blood 120, 48 [Google Scholar]
- 16. Buet D., Gallais I., Lauret E., Denis N., Lombard B., Guillonneau F., Kosmider O., Loew D., Dusanter-Fourt I., Guillouf C., Mayeux P., and Moreau-Gachelin F. (2012) Cotargeting signaling pathways driving survival and cell cycle circumvents resistance to Kit inhibitors in leukemia. Blood 119, 4228–4241 [DOI] [PubMed] [Google Scholar]
- 17. Casado P., Rodriguez-Prados J.-C., Cosulich S. C., Guichard S., Vanhaesebroeck B., Joel S., and Cutillas P. R. (2013) Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 6, rs6. [DOI] [PubMed] [Google Scholar]
- 18. Choudhary C., Olsen J. V., Brandts C., Cox J., Reddy P. N. G., Böhmer F. D., Gerke V., Schmidt-Arras D.-E., Berdel W. E., Müller-Tidow C., Mann M., and Serve H. (2009) Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 36, 326–339 [DOI] [PubMed] [Google Scholar]
- 19. Gu T., Nardone J., Wang Y., Loriaux M., Villén J., Beausoleil S., Tucker M., Kornhauser J., Ren J., MacNeill J., Gygi S. P., Druker B. J., Heinrich M. C., Rush J., and Polakiewicz R. D. (2011) Survey of activated FLT3 signaling in leukemia. PLoS ONE 6, e19169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roolf C., Dybowski N., Sekora A., Mueller S., Knuebel G., Tebbe A., Murua Escobar H., Godl K., Junghanss C., and Schaab C. (2017) Phosphoproteome analysis reveals differential mode of action of sorafenib in wildtype and mutated FLT3 AML Cells. Mol. Cell. Proteomics 16, 1365–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Askenazi M., Jiang J., Luckey C. J., Griffin J. D., and Marto J. a (2010) A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia. Mol. Cell. Proteomics 9, 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alcolea M. P., Casado P., Rodríguez-Prados J.-C., Vanhaesebroeck B., and Cutillas P. R. (2012) Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol. Cell. Proteomics 11, 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casado P., Alcolea M. P., Iorio F., Rodríguez-Prados J.-C., Vanhaesebroeck B., Saez-Rodriguez J., Joel S., and Cutillas P. R. (2013) Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 14, R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hahn C. K., Berchuck J. E., Ross K. N., Kakoza R. M., Clauser K., Schinzel A. C., Ross L., Galinsky I., Davis T. N., Silver S. J., Root D. E., Stone R. M., DeAngelo D. J., Carroll M., Hahn W. C., Carr S. a Golub T. R., Kung A. L., and Stegmaier K. (2009) Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell 16, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schaab C., Oppermann F. S., Klammer M., Pfeifer H., Tebbe A., Oellerich T., Krauter J., Levis M., Perl AE, Daub H., Steffen B., Godl K., and Serve H. (2014) Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia 28, 716–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weber C., Schreiber T. B., and Daub H. (2012) Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J. Proteomics 75, 1343–1356 [DOI] [PubMed] [Google Scholar]
- 27. Tong J., Helmy M., Cavalli F. M. G., Jin L., St-Germain J., Karisch R., Taylor P., Minden M. D., Taylor M. D., Neel B. G., Bader G. D., and Moran M. F. (2017) Integrated analysis of proteome, phosphotyrosine-proteome, tyrosine-kinome and tyrosine-phosphatome in acute myeloid leukemia. Proteomics 17, doi: 10.1002/pmic.201600361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walters D. K., Goss V. L., Stoffregen E. P., Gu T.-L., Lee K., Nardone J., McGreevey L., Heinrich M. C., Deininger M. W., Polakiewicz R., and Druker B. J. (2006) Phosphoproteomic analysis of AML cell lines identifies leukemic oncogenes. Leuk. Res. 30, 1097–1104 [DOI] [PubMed] [Google Scholar]
- 29. Gu T. L., Goss V. L., Reeves C., Popova L., Nardone J., MacNeill J., Walters D. K., Wang Y., Rush J., Comb M. J., Druker B. J., and Polakiewicz R. D. (2006) Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 108, 4202–4204 [DOI] [PubMed] [Google Scholar]
- 30. Beekhof R., Alphen C., Henneman A. A., Knol J. C., Pham T. V., Rolfs F., Labots M., Henneberry E., Le Large T. Y., Haas R. R., Piersma S. R., Vurchio V., Bertotti A., Trusolino L., Verheul H. M., and Jimenez C. R. (2019) INKA, an integrative data analysis pipeline for phosphoproteomic inference of active kinases. Mol. Syst. Biol. 15, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Mijn J. C., Labots M., Piersma S. R., Pham T. V., Knol J. C., Broxterman H. J., Verheul H. M., and Jim??nez CR. (2015) Evaluation of different phospho-tyrosine antibodies for label-free phosphoproteomics. J. Proteomics 127, 259–263 [DOI] [PubMed] [Google Scholar]
- 32. Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X.-M., Polakiewicz R. D., and Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 33. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 34. Liu H., Sadygov R. G., and Yates J. R. (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 35. RDevelopment Core Team. (2016) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 36. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A. T. M., Jørgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., Samson L. D., Woodgett J. R., Russell R. B., Bork P., Yaffe M. B., and Pawson T. (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M., and Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 [DOI] [PubMed] [Google Scholar]
- 39. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., and Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 [DOI] [PubMed] [Google Scholar]
- 40. Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P. A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., Zecha J., Reiter K., Qiao H., Helm D., Koch H., Schoof M., Canevari G., Casale E., Re Depaolini S., Feuchtinger A., Wu Z., Schmidt T., Rueckert L., Becker W., Huenges J., Garz A. K., Gohlke B. O., Zolg D. P., Kayser G., Vooder T., Preissner R., Hahne H., Tõnisson N., Kramer K., Götze K., Bassermann F., Schlegl J., Ehrlich H. C., Aiche S., Walch A., Greif P. A., Schneider S., Felder E. R., Ruland J., Médard G., Jeremias I., Spiekermann K., and Kuster B. (2017) The target landscape of clinical kinase drugs. Science 358, pii: eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Changelian P. S., Flanagan M. E., Ball D. J., Kent C. R., Magnuson K. S., Martin W. H., Rizzuti B. J., Sawyer P. S., Perry B. D., Brissette W. H., Mccurdy S. P., Kudlacz E. M., Conklyn M. J., Elliott E. A., Koslov E. R., Fisher M. B., Strelevitz T. J., Yoon K., Whipple D. A., Sun J., Munchhof M. J., Doty J. L., Casavant J. M., Blumenkopf T. A., Hines M., Brown M. F., Lillie B. M., Subramanyam C., Shang-Poa C., Milici A. J., Beckius G. E., Moyer J. D., Su C., Woodworth T. G., Gaweco A. S., Beals C. R., Littman B. H., Fisher D. A., Smith J. F., Zagouras P., Magna H. A., Saltarelli M. J., Johnson K. S., Nelms L. F., Des Etages S. G., Hayes L. S., Kawabata T. T., Finco-Kent D., Baker D. L., Larson M., Si M.-S., Paniagua R., Higgins J., Holm B., Reitz B., Zhou Y.-J., Morris R. E., O'shea J. J., and Borie D. C. (2003) Prevention of Organ Allograft Rejection by a Specific Janus Kinase 3 Inhibitor. Science 302, 875–878 [DOI] [PubMed] [Google Scholar]
- 42. Blake J. F., Burkard M., Chan J., Chen H., Chou K.-J., Diaz D., Dudley D. A., Gaudino J. J., Gould S. E., Grina J., Hunsaker T., Liu L., Martinson M., Moreno D., Mueller L., Orr C., Pacheco P., Qin A., Rasor K., Ren L., Robarge K., Shahidi-Latham S., Stults J., Sullivan F., Wang W., Yin J., Zhou A., Belvin M., Merchant M., Moffat J., and Schwarz J. B. (2016) Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in early clinical development. J. Med. Chem. 59, 5650–5660 [DOI] [PubMed] [Google Scholar]
- 43. Honigberg L. a. Smith A. M., Sirisawad M., Verner E., Loury D., Chang B., Li S., Pan Z., Thamm D. H., Miller R. a and Buggy J. J. (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U.S.A. 107, 13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang W., Soares J., Greninger P., Edelman E. J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J. A., Thompson I. R., Ramaswamy S., Futreal P. A., Haber D. A., Stratton M. R., Benes C., McDermott U., and Garnett M. J. (2013) Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, 955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harrington E. A., Bebbington D., Moore J., Rasmussen R. K., Ajose-Adeogun A. O., Nakayama T., Graham J. A., Demur C., Hercend T., Diu-Hercend A., Su M., Golec J. M. C., and Miller K. M. (2004) VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 10, 262–267 [DOI] [PubMed] [Google Scholar]
- 46. Harris P. A., Boloor A., Cheung M., Kumar R., Crosby R. M., Davis-Ward R. G., Epperly A. H., Hinkle K. W., Hunter R. N., Johnson J. H., Knick V. B., Laudeman C. P., Luttrell D. K., Mook R. A., Nolte R. T., Rudolph S. K., Szewczyk J. R., Truesdale A. T., Veal J. M., Wang L., and Stafford J. A. (2008) Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J. Med. Chem. 51, 4632–4640 [DOI] [PubMed] [Google Scholar]
- 47. Hickinson D. M., Klinowska T., Speake G., Vincent J., Trigwell C., Anderton J., Beck S., Marshall G., Davenport S., Callis R., Mills E., Grosios K., Smith P., Barlaam B., Wilkinson R. W., and Ogilvie D. (2010) AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin. Cancer Res. 16, 1159–1169 [DOI] [PubMed] [Google Scholar]
- 48. Iverson C., Larson G., Lai C., Yeh L.-T., Dadson C., Weingarten P., Appleby T., Vo T., Maderna A., Vernier J.-M., Hamatake R., Miner J. N., and Quart B. (2009) RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 69, 6839–6847 [DOI] [PubMed] [Google Scholar]
- 49. Meijer L., Borgne A., Mulner O., Chong J. P., Blow J. J., Inagaki N., Inagaki M., Delcros J. G., and Moulinoux J. P. (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243, 527–536 [DOI] [PubMed] [Google Scholar]
- 50. Nakamura K., Taguchi E., Miura T., Yamamoto A., Takahashi K., Bichat F., Guilbaud N., Hasegawa K., Kubo K., Fujiwara Y., Suzuki R., Kubo K., Shibuya M., and Isae T. (2006) KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 66, 9134–9142 [DOI] [PubMed] [Google Scholar]
- 51. Pachl F., Plattner P., Ruprecht B., Médard G., Sewald N., and Kuster B. (2013) Characterization of a chemical affinity probe targeting Akt kinases. J. Proteome Res. 12, 3792–3800 [DOI] [PubMed] [Google Scholar]
- 52. Quintás-Cardama A., Vaddi K., Liu P., Manshouri T., Li J., Scherle P. A., Caulder E., Wen X., Li Y., Waeltz P., Rupar M., Burn T., Lo Y., Kelley J., Covington M., Shepard S., Rodgers J. D., Haley P., Kantarjian H., Fridman J. S., and Verstovsek S. (2010) Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115, 3109–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tokushige H., Waki M., Takayama Y., and Tanihara H. (2011) Effects of Y-39983, a selective Rho-associated protein kinase inhibitor, on blood flow in optic nerve head in rabbits and axonal regeneration of retinal ganglion cells in rats. Curr. Eye Res. 36, 964–970 [DOI] [PubMed] [Google Scholar]
- 54. Torrance C. J., Jackson P. E., Montgomery E., Kinzler K. W., Vogelstein B., Wissner A., Nunes M., Frost P., and Discafani C. M. (2000) Combinatorial chemoprevention of intestinal neoplasia. Nat. Med. 6, 1024–1028 [DOI] [PubMed] [Google Scholar]
- 55. Wilhelm S. M., Carter C., Tang L., Wilkie D., McNabola A., Rong H., Chen C., Zhang X., Vincent P., McHugh M., Cao Y., Shujath J., Gawlak S., Eveleigh D., Rowley B., Liu L., Adnane L., Lynch M., Auclair D., Taylor I., Gedrich R., Voznesensky A., Riedl B., Post L. E., Bollag G., and Trail P. A. (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109 [DOI] [PubMed] [Google Scholar]
- 56. Brasca M. G., Albanese C., Alzani R., Amici R., Avanzi N., Ballinari D., Bischoff J., Borghi D., Casale E., Croci V., Fiorentini F., Isacchi A., Mercurio C., Nesi M., Orsini P., Pastori W., Pesenti E., Pevarello P., Roussel P., Varasi M., Volpi D., Vulpetti A., and Ciomei M. (2010) Optimization of 6,6-dimethyl pyrrolo[3,4-c]pyrazoles: Identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing. Bioorg. Med. Chem. 18, 1844–1853 [DOI] [PubMed] [Google Scholar]
- 57. Zou H. Y., Li Q., Lee J. H., Arango M. E., McDonnell S. R., Yamazaki S., Koudriakova T. B., Alton G., Cui J. J., Kung P.-P., Nambu M. D., Los G., Bender S. L., Mroczkowski B., and Christensen J. G. (2007) An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 67, 4408–4417 [DOI] [PubMed] [Google Scholar]
- 58. Folkes A. J., Ahmadi K., Alderton W. K., Alix S., Baker S. J., Box G., Chuckowree I. S., Clarke P. A., Depledge P., Eccles S. A., Friedman L. S., Hayes A., Hancox T. C., Kugendradas A., Lensun L., Moore P., Olivero A. G., Pang J., Patel S., Pergl-Wilson G. H., Raynaud F. I., Robson A., Saghir N., Salphati L., Sohal S., Ultsch M. H., Valenti M., Wallweber H. J. A., Wan N. C., Wiesmann C., Workman P., Zhyvoloup A., Zvelebil M. J., and Shuttleworth S. J. (2008) The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51, 5522–5532 [DOI] [PubMed] [Google Scholar]
- 59. Green T. P., Fennell M., Whittaker R., Curwen J., Jacobs V., Allen J., Logie A., Hargreaves J., Hickinson D. M., Wilkinson R. W., Elvin P., Boyer B., Carragher N., Plé P. A., Bermingham A., Holdgate G. A., Ward W. H. J., Hennequin L. F., Davies B. R., and Costello G. F. (2009) Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol. Oncol. 3, 248–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bachas C., Schuurhuis G. J., Hollink I. H. I. M., Kwidama Z. J., Goemans B. F., Zwaan C. M., Van Den Heuvel-Eibrink M. M., De Bont E. S. J. M., Reinhardt D., Creutzig U., De Haas V., Assaraf Y. G., Kaspers G. J. L., and Cloos J. (2010) High-frequency type I/II mutational shifts between diagnosis and relapse are associated with outcome in pediatric AML: Implications for personalized medicine. Blood 116, 2752–2758 [DOI] [PubMed] [Google Scholar]
- 61. Bachas C., Schuurhuis G. J., Zwaan C. M., Van Den Heuvel-Eibrink M. M., Boer M. L. D., De Bont E. S. J. M., Kwidama Z. J., Reinhardt D., Creutzig U., De Haas V., Kaspers G. J. L., and Cloos J. (2015) Gene expression profiles associated with pediatric relapsed AML. PLoS ONE 10, 30–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goto S., Bono H., Ogata H., Fujibuchi W., Nishioka T., Sato K., and Kanehisa M. (1997) Organizing and computing metabolic pathway data in terms of binary relations. Pac. Symp. Biocomput. 1997, 175–186 [PubMed] [Google Scholar]
- 63. Mi H., and Thomas P. (2009) PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 563, 123–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., and D'Eustachio P. (2016) The reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van Meerloo J., Kaspers G. J. L., and Cloos J. (2011) Cell Sensitivity Assays: The MTT Assay. Methods Mol. Biol. 731, 237–245 [DOI] [PubMed] [Google Scholar]
- 68. Spiekermann K., Dirschinger R. J., Schwab R., Bagrintseva K., Faber F., Buske C., Schnittger S., Kelly L. M., Gilliland D. G., and Hiddemann W. (2003) The protein tyrosine kinase inhibitor SU5614 inhibits FLT3 and induces growth arrest and apoptosis in AML-derived cell lines expressing a constitutively activated FLT3. Blood 101, 1494–1504 [DOI] [PubMed] [Google Scholar]
- 69. Asou H., Imagawa J., Harada K., Harada H., and Harada Y. (2003) Acute myeloid leukemia with either mutant KIT or PDGFRA-ITD is a novel therapeutic target of imatinib mesylate. Blood 102, 374a [Google Scholar]
- 70. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. a Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. a Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., and Garraway L. a (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Forbes S. A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S., Kok C. Y., Jia M., De T., Teague J. W., Stratton M. R., McDermott U., and Campbell P. J. (2015) COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. O'Hare T., Shakespeare W. C., Zhu X., Eide C. A., Rivera V. M., Wang F., Adrian L. T., Zhou T., Huang W. S., Xu Q., Metcalf C. A., Tyner J. W., Loriaux M. M., Corbin A. S., Wardwell S., Ning Y., Keats J. A., Wang Y., Sundaramoorthi R., Thomas M., Zhou D., Snodgrass J., Commodore L., Sawyer T. K., Dalgarno D. C., Deininger M. W. N., Druker B. J., and Clackson T. (2009) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16, 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the set identifiers PXD007237 (cell line data), PXD015639 (clinical sample phosphoproteomics data), and PXD015662 (clinical sample proteomics data) (https://proteomecentral.proteomexchange.org).