

Proximity-dependent biotinylation approaches such as BioID and APEX overcome classical limitations of biochemical purification and have gained widespread use in recent years for revealing cellular neighborhoods. Here we focus on the structural diversity and mechanisms of the two classes of enzymes, biotin protein ligases and peroxidases, and discuss current and emerging applications of these enzymes for proximity dependent biotinylation. We provide guidelines for enzyme selection and experimental design for performing and interpreting proximity-dependent biotinylation experiments.
Keywords: Protein-protein interactions, cellular organelles, enzymes, mass spectrometry, molecular biology, protein engineering, APEX, BioID, biotin ligase, peroxidase, proximity-dependent biotinylation
Graphical Abstract

Highlights
Proximity-dependent biotinylation (PDB) approaches involve fusion of a bait with an enzyme.
BioID (biotin protein ligase) and APEX (peroxidase) are distinct enzymes used in PDB.
Past, present and future development and applications of PDB are discussed.
We review labeling mechanisms and kinetics to provide guidance for experimental design.
We discuss controls and considerations for data interpretation.
Abstract
The study of protein subcellular distribution, their assembly into complexes and the set of proteins with which they interact with is essential to our understanding of fundamental biological processes. Complementary to traditional assays, proximity-dependent biotinylation (PDB) approaches coupled with mass spectrometry (such as BioID or APEX) have emerged as powerful techniques to study proximal protein interactions and the subcellular proteome in the context of living cells and organisms. Since their introduction in 2012, PDB approaches have been used in an increasing number of studies and the enzymes themselves have been subjected to intensive optimization. How these enzymes have been optimized and considerations for their use in proteomics experiments are important questions. Here, we review the structural diversity and mechanisms of the two main classes of PDB enzymes: the biotin protein ligases (BioID) and the peroxidases (APEX). We describe the engineering of these enzymes for PDB and review emerging applications, including the development of PDB for coincidence detection (split-PDB). Lastly, we briefly review enzyme selection and experimental design guidelines and reflect on the labeling chemistries and their implication for data interpretation.
In eukaryotic cells, most processes and reactions are compartmentalized into organelles and other subcellular structures and often effected through the concerted action of molecular machines. To elucidate the composition of protein complexes or organelles, biochemical fractionation followed by mass spectrometric identification - now most often through systematic quantitation of chromatographic elution profiles (1–9) - can be performed. Alternatively, a widely-used approach to define protein-protein interactions is to perform affinity purification of a protein of interest followed by the identification of its direct and indirect interaction partners by mass spectrometry (a technique commonly referred to as AP-MS; reviewed in (10, 11)). Importantly, however, these methods all rely on the principle that organelles or interactions must be preserved during cell lysis and purification of complexes or organelles. However, this can be challenging when recovering structures or molecules that are difficult to solubilize or easily lose integrity through purification (12, 13). Strategies that attempt to overcome these limitations have been introduced in the past decade. Optimization of lysis and purification conditions has enabled the definition of interactomes for membrane proteins (14) and a more complete characterization of a variety of other macromolecular complexes (15). Additionally, use of low concentrations of chemical cross-linkers in live cells or immediately after cell lysis may also alleviate some of the challenges associated with interaction preservation in AP-MS and fractionation experiments (16, 17), though this may also increase the number of false positive interactors in some cases.
In the past 8 years, alternative approaches have been introduced that instead bypass the requirement to maintain protein-protein interactions or organellar integrity during sample purification. Collectively, these are referred to as proximity-dependent biotinylation (PDB)1 approaches and consist of directing an enzyme capable of catalyzing covalent transfer of biotin (or other derivatives) to endogenous proteins that are located within a certain distance of the enzyme. By fusing the enzyme to specific proteins (referred to as “baits”), the enzyme can be localized to distinct areas of the cell, for example to a protein complex or an organelle (Fig. 1A). Addition of the enzyme substrate leads to the covalent biotinylation of proteins located near the bait (these are referred to as “preys”). Importantly, the labeling can be performed in live cells (or whole organisms), on fixed samples, or even in lysates or semi-purified structures. The primary advantage of PDB is that protein-protein interactions or the integrity of organelles do not need to be maintained post-labeling as the covalently biotinylated preys can be captured using an affinity matrix, most often streptavidin. This principle has enabled purification of preys under harsh lysis and wash conditions because of the high affinity of the biotin-streptavidin interaction (Kd ∼10−14 m), which is also resistant to many conditions (detergents, salt, or denaturing agents) that typically disrupt protein-protein interactions or organellar integrity. Subsequently, streptavidin-purified proteins can be identified and quantified by mass spectrometry (Fig. 1B). When appropriate controls and mass spectrometric quantification are employed (as discussed elsewhere - (18–20)), PDB-MS can report on specific proximity relationships. It is important to keep in mind that what PDB-MS provides is a qualitative metric of the relative proximity between bait and prey and cannot explicitly determine whether the detected proteins are physically interacting (either directly or indirectly), or whether they are simply localized to the same area. Additionally, these PDB-MS approaches will reveal the proximal relationships of a bait throughout the entire life cycle of the protein from its synthesis until the end point of the assay. This is discussed below (in Proximal Labeling Chemistry and Labeling Propensity) and elsewhere (19).
Fig. 1.

Principles of Proximity-Dependent Biotinylation. A, A protein of interest (bait) is fused in-frame to a PDB enzyme from one of two families, biotin protein ligases or peroxidases, that have distinct substrate requirements and modify different amino acids. B, Schematic workflow for a PDB experiment identifying proximal proteins. Proteins are labeled inside living cells prior to a harsh lysis and a protein-level capture on streptavidin beads. After stringent washing, elution is most often effected by proteolysis with trypsin, and the non-biotinylated peptides are released and identified by mass spectrometry. A variation of this approach consists of performing an elution with high concentrations of acid such as trifluoroacetic acid; in this case information about the site of biotinylation may be obtained. C, Alternative workflow for peptide-level capture and identification of the biotinylated peptides: an antibody against biotin is used to capture biotinylated peptides directly. Alternatively, other biotin affinity capture strategies can be employed.
Since the introduction of the first PDB-MS approach (reviewed in (18, 19, 21, 22)), a growing number of enzymes - that largely fall into two groups: the biotin protein ligases and the peroxidases - as well as additional tools and experimental designs have made the strategy a flexible mainstay of interaction and organellar proteomics. Here, we will focus on the molecular basis for the main proximity-dependent biotinylation approaches and on the development of distinct toolsets for the application of proximity dependent biotinylation to different biological questions.
Overview of PDB Enzymes
In this section, we will describe the two major classes of enzymes currently used for PDB-MS: biotin protein ligases and peroxidases, with an emphasis on their structural characteristics, their natural enzymatic reactions, and the modifications that have made BioID and APEX possible.
Biotin Protein Ligases and BioID
Biotin is an essential vitamin for all organisms that is produced by plants, fungi and most prokaryotes, but not mammalian cells (23). In mammalian cells, biotin uptake is primarily mediated by the sodium multivitamin transporter SMVT (encoded by the SLC5A6 gene in humans) (24). Intracellular biotin serves as a covalently-attached cofactor for the biotin-dependent carboxylase enzymes (four enzyme families are present in humans: PC, PCCA/PCCB, MCCC1/MCCC2, ACACA/ACACB) that have crucial roles in amino acid, fatty acid, carbohydrate and energy metabolism. Carboxylases transfer carboxyl groups to small molecule substrates, most of which are coenzyme A (CoA) esters of organic acids (though other compounds including urea and pyruvate can also serve as substrates). This transfer is enabled by biotin, which first becomes enzymatically carboxylated (bicarbonate serves as the CO2 donor), and in a second enzymatic step, releases the carboxyl group to the substrate (for a review of carboxylases and their mechanisms, please refer to (23)).
Biotin protein ligases (here referred to as BPLs), also known as holocarboxylase synthetases in eukaryotes, are responsible for the covalent attachment of biotin to the carboxylases (25), and are present in all living species. They exhibit a high substrate specificity for the carboxylases and this has been evolutionarily conserved as specific biotinylation can still occur when the BPL and carboxylase come from divergent species (26). This high specificity for a very small number of substrates (largely localized in the mitochondrial matrix in eukaryotes) is important for the use of BPLs in several biotechnology applications, including BioID.
BPL enzymes (PFAM: PF03099) can be grouped into three classes based on their structural architecture (27) (Fig. 2A). All three classes have a conserved central catalytic domain responsible for the protein biotinylation reaction and a C-terminal domain of unknown function that is essential for its enzymatic activity (28). However, they differ in their N termini. Class II enzymes possess a winged helix-turn-helix DNA-binding domain that functions as a biotin-controlled repressor of the biotin biosynthesis operon (29), whereas class III enzymes have a large N terminus without DNA binding activity. Class I enzymes lack this N-terminal domain altogether. Class I and II BPLs are found in Archaea, prokaryotes and plants whereas class III BPLs are found in yeast, insects and mammals and include the human holocarboxylase synthetase HLCS (Fig. 2A). As discussed below, class II BPLs (such as that found in E. coli) have been extensively used in PDB-MS and in biotechnology in general, whereas class I enzymes (such as that found in Aquifex aeolicus, a thermophilic bacteria) have been more recently introduced for use in PDB-MS and have unique properties (30).
Fig. 2.
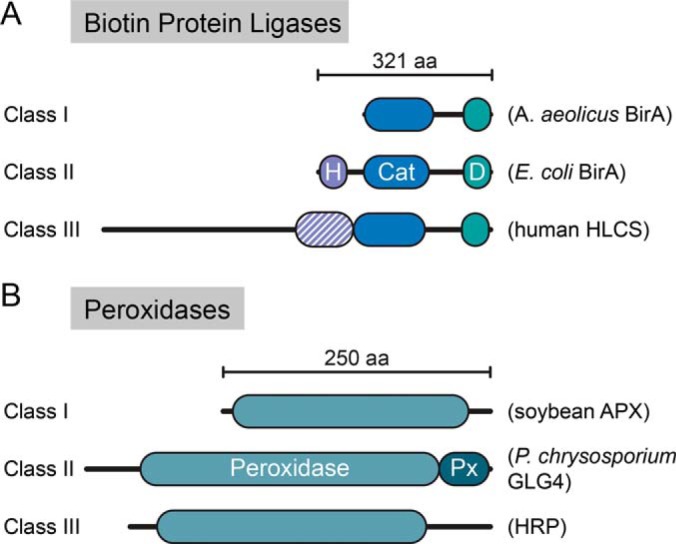
Domain structure of the different classes of PDB enzymes. A, Biotin protein ligases. H: helix-turn-helix DNA binding domain; Cat: central catalytic domain (in blue); D: domain of unknown function. Examples of proteins for each of the three structural classes are shown. B, Peroxidases. APX: Ascorbate peroxidase; GLG4: P. chrysosporium Ligninase H2; HRP: Horseradish peroxidase. Px: peroxidase extension region, peroxidase catalytic domain (in teal).
The structure and activity of several BPLs from different bacterial species have been described, providing insight into their reaction mechanism (27, 31). The E. coli BPL, also known as Bifunctional ligase/repressor (BirA), is an archetypal type-II enzyme and is one of the best-studied enzymes of this class. Upon binding of biotin to BirA, the biotin-binding loop undergoes a conformational change that allows for subsequent binding of ATP, leading to a structural rearrangement of the adenylate-binding loop, stabilizing the bound ATP (Fig. 3). Subsequently, a nucleophilic substitution mediated by K183 of BirA catalyzes the attack of the biotin carboxylate on the alpha phosphate of ATP, producing biotinyl-5′-AMP. Biotinyl-5′-AMP remains stably associated with the enzyme in a mixed anhydride form through hydrogen bonding with the R118 backbone (27, 32, 33). This is stabilized through a salt-bridge interaction between R118 and D176, another highly conserved residue (32). The second step of the transfer reaction involves the nucleophilic attack of this mixed anhydride by the epsilon amine of lysine from the substrate (K122 on the biotin-carboxylase cargo protein, BCCP, a subunit of the acetyl-CoA carboxylase), resulting in covalent biotinylation of BCCP on the attacking lysine (25, 34, 35).
Fig. 3.

Mechanism of labeling by BPLs. Side chains on the BPL enzyme involved in the reaction are highlighted in pink, whereas the lysine side chain of the substrate (here BCCP) is shown in blue. See text for details.
By exploring the specificity of BirA for its substrate (36), a minimum short biotin-acceptor peptide (referred to as AviTag) was identified that could be biotinylated by BirA in the absence of the full length BCCP (37). AviTag sequences have been valuable reagents for various applications. For example, by fusing an AviTag sequence and BirA to two respective proteins, the interaction between the two proteins can be monitored through biotinylation of the AviTag (38, 39). Alternatively, the biotinylation of the AviTag-tagged protein can be harnessed for affinity purification on streptavidin-conjugated resin (40–42), or protein visualization using fluorophores conjugated to streptavidin (43). Other applications include the tagging of ribosomes localized to different parts of the cell to elucidate which transcripts they translate (44), and the selective purification of structures, e.g. the nucleus, to assist in downstream assay design (e.g. (45)). Importantly, however, this application of BirA requires the expression of two proteins, one fused to BirA and one fused to the AviTag, which limits discovery proteomics assays.
Although the wildtype BirA remains widely used, many BirA mutants have been described over the years, presenting opportunities for new applications (Fig. 4). The study of mutants that affect the biotin operon activity in E. coli resulted in identification of the BirA91 mutant allele in 1980 (46), with the specific mutation (R118G) identified in 1986 (47). Relative to wildtype BirA, this mutant was found to have 100-fold greater Kd for biotin and a 400-fold higher dissociation rate for biotinyl-5′-AMP (48–50), consistent with the role of R118 in stabilizing the biotinyl-5′-AMP intermediate. Later, the R118G mutant was demonstrated to act as a non-sequence specific biotinylation reagent by Choi-Rhee et al., who first described the potential use of this mutant for the “recovery of interacting proteins by existing avidin/streptavidin technology” (51). However, it was nearly a decade before Roux et al. would bring this idea into fruition in the technique now known as BioID (52).
Fig. 4.

Structures of the main enzymes employed in PDB research. PDB files were downloaded from the RCSB Protein Data Bank and visualized in Pymol. Mutations over the wild type enzymes are indicated. A, Original enzymes used in BioID. Structures of the wild type E. coli BirA with biotinyl-5′-AMP (source PDB 2EWN) and A. aeolicus BPL with biotin (source PDB 3EFS) with position of the mutation decreases the affinity for biotinyl-5′-AMP intermediate. B, Molecularly evolved TurboID and miniTurbo displayed on the E. coli wild type BirA structure. C, Peroxidases. HRP with heme and calcium ions (source 1HCH) and APEX/APEX2 mutations displayed on the structure of soybean APX with heme (source PDB 1OAG) are indicated (note that APEX2 contains the additional A134P mutation compared with APEX).
The BioID technique takes advantage of the reduced affinity of the BirA enzyme (R118G mutation, denoted as BirA*) for biotinyl-5′-AMP. This is thought to result in the generation of a reactive biotinyl-5′-AMP cloud (estimated in one study to be in the range of 10 nm (53)) around the enzyme, which can react with primary amines (such as epsilon-amines on lysines) of proximal proteins, resulting in their covalent biotinylation. The reaction can take place inside living cells and over time leads to a build-up of biotinylated proteins in the vicinity of the bait. As discussed in the introduction, this in situ covalent labeling allows for lysis of cells under harsh conditions to maximize protein solubility without the need for preserving protein interactions or subcellular organization. The biotinylated proteins can then be captured on streptavidin-conjugated resin (or alternatively through anti-biotin antibodies; see below) and identified by mass spectrometry (19, 52, 54) or detected through different means, such as immunoblotting (Fig. 1).
BioID was first used to explore the proximal associations within the nuclear envelope, leading to the assignment of a previously uncharacterized protein, SLAP75, as a novel nuclear envelope protein (52). This led to the realization that the approach was a powerful method to detect proximity partners for typically insoluble cellular compartments. This study was followed by many others, including an investigation of the Hippo signaling pathway in which phosphorylation-specific interactions could be detected in the absence of sustained signal, revealing a propensity for signal amplification in BioID experiments (19, 55). Since its initial development, BioID has been widely used to explore proximal associations in both large and small-scale experimentations in various experimental models (reviewed in (20)) that importantly now include the characterization of the proteome composition and organization of membraneless organelles, including focal adhesions (56) and cell junctions (57–60), the centrosome (61, 62), P-bodies and stress granules (63), and the generation of a draft proximity map of a human cell (64). As we discuss in the later sections, continued development of this technology has enabled the identification of more active enzymes and permitted the implementation of new assay designs.
Peroxidases and APEX
Peroxidases (like other oxidoreductases) generally catalyze redox reactions, such as reduction of hydrogen peroxide through oxidizing various substrates. Peroxidases are first classified on the basis of their prosthetic group into heme and non-heme peroxidases (refer to (65) for an interactive database of oxidoreductases and (66) for an evolutionary view of the heme-containing peroxidases). Heme peroxidases (PFAM PF00141) can be divided into four superfamilies of which the peroxidase-catalase family is the largest, consisting of over 8100 members (66). All heme peroxidases are characterized by the presence of an iron-protoporphyrin IX prosthetic group (the heme group) in their active site that is used as a reduction-oxidation reaction cofactor, and a catalytic histidine. The principal reaction catalyzes hydrogen peroxide (H2O2)-mediated oxidation of a variety of molecules (including aromatic molecules such as tyrosine) acting as electron donors (AH2). This results in the formation of active radicals (●AH) and the conversion of hydrogen peroxide to water (H2O). Some of the enzymes in this class can also perform other catalytic reactions, including peroxygenation (66).
There exists a large sequence diversity in the heme peroxidase enzymes, which have arisen four times through evolution (66), are present across all kingdoms, and catalyze one- and two-electron mediated oxidation of multiple organic and inorganic substrates (66). The peroxidase-catalase superfamily is characterized by a structurally-conserved globular fold of 12 alpha-helices and the presence of a sequence signature. This superfamily can be further separated in three distinct classes, which exhibit different primary functions (67) (Fig. 2B). Class I is the most divergent across these three classes, consisting of more than 1800 annotated members and notably includes cytochrome c and ascorbate peroxidases (including the ascorbate peroxidase APX) that primarily function in the scavenging of excess H2O2. Class II is a smaller group that contains fungal manganese and lignin peroxidases and are involved in the degradation of lignin-containing soil debris. Lastly, Class III contains the highest number of reported sequences, and includes plant secretory peroxidases (such as horseradish peroxidase that will be described in depth below) that are implicated in oxidation of lignin, auxin and secondary metabolites. The evolutionary divergence of these enzyme classes confers different biochemical characteristics, which have been leveraged for their application in biotechnology.
Horseradish peroxidase C (commonly known as HRP) is the best-studied peroxidase and has demonstrated immense biotechnological value. HRP is a class III secreted glycoprotein that binds a heme-group and calcium, both of which are essential to its function (68) (Figs. 2B, 4). HRP has been a workhorse of many molecular biology techniques, such as oxidation of chromogenic or chemiluminescent substrates for signal detection and amplification or deposition of molecules including diaminobenzidine for electron microscopy (69). Because of the versatility in the oxidation of various phenolic substrates (e.g. biotin-phenol, a.k.a. biotin-tyramide; Fig. 5), HRP has also been utilized for protein labeling. In this context, a tyramide molecule that has been coupled to either a fluorophore or a small molecule such as biotin, can be activated by a peroxidase (in the presence of H2O2), to produce a short-lived radical that can react with electron-rich amino acids such as tyrosine (70) (Fig. 1A).
Fig. 5.

Substrates used in PDB approaches. Different related substrates for PDB have been developed specifically for different variations on the PDB theme. See text and references for details.
Conjugation of HRP to an antibody allows for targeting of the peroxidase activity to specific cellular compartments such as the cell surface of live cells or intracellular compartments of permeabilized cells. This has been demonstrated by the EMARS (Enzyme-Mediated Activation of Radical Sources) technique using arylazide-biotin (a.k.a. phenyl azide-biotin) as the substrate (71, 72) and the SPPLAT (Selective Proteomic Proximity LAbeling using Tyramide) technique using biotinyl-tyramide (73), respectively. These proximal protein-labeling approaches increase sensitivity in signal detection in immunoassays (69, 74, 75), and SPPLAT has been coupled to mass spectrometry for quantification of proximity partners of a B-cell receptor (73).
An important variation on this concept, which is particularly useful when a high-specificity antibody directed against a target is not available, is through expression of a genetically encoded enzyme in model cells or organisms, allowing for protein labeling in live cells under physiological conditions, similar to the development of BioID described above. This was first demonstrated by directing expression of HRP to the endoplasmic reticulum (ER) through fusion with a signal peptide and an ER retention motif to label the secretory compartments for visualization by electron microscopy (76). Similarly, genetically encoded HRP was targeted to the cell surface (by making fusions to four synaptic cleft-resident proteins) for proteomic profiling of lipid raft domains (77) and neuronal synaptic clefts (78). Intriguingly, although HRP retains its catalytic activity in the oxidizing environments of endomembrane system lumens (ER or Golgi) or extracellular spaces, it is inactive when expressed in the reducing cytosol environment (79, 80). It has been hypothesized that this may be because HRP is natively a secreted glycoprotein, resulting in its incompatibility with expression within the cytosol.
To overcome the limitations of HRP in these intracellular contexts, a novel enzyme was engineered based on the ascorbate peroxidase (APX) (80). APX is a Class I enzyme (Fig. 2B, 4) that regulates intracellular H2O2 levels in chloroplasts and the cytosol of higher plants by oxidizing l-ascorbate (81–83). Similar to HRP, APX can convert its substrate into a radical through reduction of H2O2. However, unlike HRP, APX does not contain disulfide bonds, is not a glycoprotein and it does not require calcium for its activity (82, 84–86) (Fig. 4). Martell et al. postulated that the structural differences between APX and HRP could allow for retention of its activity in reducing cellular environments such as the cytosol (80). Through mutagenesis of the pea APX protein, they developed a novel enzyme they called APEX that, when expressed in mammalian cells, could allow for localized deposition of diaminobenzidine for electron microscopy (80). The mutagenesis strategy was targeted at reducing the propensity of APX to dimerize (monomeric APX: K14D, E112K). Although these mutations also decrease catalytic activity, this was restored by an additional W41F mutation, which is part of the catalytic triad in the active site (81) (Fig. 4). Intriguingly, HRP (which has a high catalytic activity) also harbors a phenylalanine at this corresponding position and its mutation to a tryptophan hinders its substrate binding ability (87). The pea APX mutations described above were also generated in the soybean APX, which resulted in an enzyme with a further reduction of unwanted dimerization, leading to the first generation of the APEX recombinant enzyme (88).
APEX can oxidize phenol derivatives (such as biotin-phenol, a.k.a. biotinyl tyramide) to phenoxyl radicals, like the use of HRP in the SPPLAT technique. This reactive radical, which has an estimated half-life of <1 ms (74), can then covalently react with a number of electron-rich amino acids (predominantly tyrosine), resulting in proximal protein labeling in living cells (Fig. 1A) (88, 89). Like BioID, this labeling in live cells allows for the lysis of cells under harsh conditions to maximize protein solubility. The labeled proteins can then be captured with streptavidin-conjugated resin and identified using mass spectrometry (19, 52). The first proteomic implementations of APEX were to map the proteome of the mitochondrial matrix (88) and then the mitochondrial intermembrane space (89). APEX was also applied to isolated tissues from Drosophila to study their mitochondrial proteomes (90). Subsequent applications have included the profiling of a stress granule marker in the presence or absence of stress treatment (91), as well as in the definition of the proteome composition of another inducible organelle, the lipid droplet (92).
A key difference in the labeling approaches of APEX and BioID is that the cells expressing the bait protein fused to APEX are first incubated with biotinyl tyramide and subsequently treated with H2O2 to initiate proximal labeling. This labeling can be performed for as little as 1 min before quenching the reaction, providing a relatively short snapshot of the proximal proteins for the bait when compared with the hours time-scale needed for obtaining enough signal to noise with the original BioID approach. This significant increase in temporal resolution offered by APEX serves as a powerful tool in the PDB arsenal to study proximal associations. It has most notably been exploited for profiling the proximity interactomes of G-protein coupled receptors following signal induction (93, 94). Like BioID, these original APEX studies proved to only be the beginning of advanced technological developments that further expand the application of PDB-based techniques.
Improvement of PDB Enzyme Characteristics
As a testament to the utility of PDB-MS techniques and their quick uptake by the scientific community, both biotin ligases and peroxidases have seen rapid development and improvement. This has been accomplished through a combination of exploring enzyme homologues, structure-guided protein engineering and unbiased molecular evolution.
Improving BPLs
Although BPLs from different classes display high degrees of structural conservation (especially in their catalytic domains) (95), they show little sequence homology, with the exception of a few key short motifs essential for catalytic activity (96–100). The sequence differences between different BPLs likely result in structural differences, which in turn can affect their thermal stability, affinity for substrates or reaction products (biotinyl-5′-AMP), or even alter their catalytic activity (98, 100, 101). Therefore, exploring BPLs from different organisms can yield enzymes with unique characteristics.
For example, Roux et al. found that the BPL from the thermophilic bacteria Aquifex aeolicus (30, 100, 102) could be used in PDB (they refer to it as BioID2 (30)). The R40G mutation, orthologous to the R118G mutation in BirA*, lowers the affinity of the A. aeolicus BPL for the reaction intermediate biotinyl-5′-AMP (100), resulting in an abortive enzyme that, similar to BirA*, is capable of generating a cloud of activated biotin (52). Because BioID2 also displays a higher affinity for biotin compared with BirA*, this allows the use of lower biotin concentrations for protein labeling. [We note however, that this also allows the BioID2-enzyme to utilize the biotin in the serum (low nm range) or some media (such as RPMI) to carry out protein biotinylation (101), and that removing biotin from the serum may be important to establish a clean baseline for differential proximity proteomics. This can be done by charcoal stripped FBS, dialyzed FBS or streptavidin-depletion of FBS - See section Enzyme Selection]
An interesting characteristic of BioID2 is that it is a Type-I BPL, and hence it lacks the N-terminal DNA-binding domain found in Type-II BPLs, such as the E. coli BirA (Fig. 2A). This domain structurally resembles linker histone H5 (103) and could potentially mediate unwanted proximity labeling of chromatin-associated proteins. Therefore, the absence of the N-terminal domain from BPL Type-II enzymes like BioID2 could also result in decreased background of proteins associated with DNA/chromatin. Although N-terminal truncation has not been successfully done with wild type E. coli BirA because of impaired catalytic activity (28, 29, 98, 104–106), the N-terminal domain of the Type-II BPL from Bacillus subtilis could be removed without deleterious effect on its enzymatic activity (104). By generating an R124G mutation in the B. subtilis BPL (orthologous to the R118G in E. coli BirA*), along with two mutations in the C-terminal domain (E323S, G325R) an alternative abortive biotin ligase, BaSu, was engineered (107). We note however that although this enzyme exhibited improved activity compared with BirA* and BioID2 in the original study, follow-up comparison by Branon et al. shows little difference between these enzymes (108), suggesting that further development of BaSu may be necessary.
In addition to exploring natural biodiversity or structure-guided protein engineering to obtain enzymes with unique properties, directed evolution can also evolve proteins with desired characteristics (109). Recently, Branon et al. used error-prone PCR, in combination with yeast-display, to generate two new E. coli BirA variants that they referred to as TurboID and miniTurbo (108). By immunoblotting with Streptavidin-HRP, they determined that TurboID and miniTurbo exhibited a 3∼6-fold increase in activity compared with BirA* over a short period of labeling and up to 15∼23-fold increase over longer durations (6–18 h of biotin treatment). These enzymes contain 14 and 12 mutations (plus a N-terminal deletion of the first 63 amino acids for miniTurbo), respectively (Fig. 4). TurboID has the same 12 mutations as miniTurbo, with an additional two mutations (S263P and M241T). Interestingly, these mutations are for the most part distal to the catalytic pocket and distributed throughout the entire structure with no obvious mechanistic explanation for increased enzymatic activity. It is plausible that these mutations (or a subset of them) alter the structure or rigidity of the catalytic pocket, which could result in increased catalytic activity. It is also intriguing that although the deletion of the N-terminal domain of wild type BirA drastically impairs its activity, miniTurbo tolerates the removal of its N-terminal domain. Though the structural and mechanistic implications of these mutations have not been systematically investigated, these new enzymes provide an opportunity to better understand the activity of BPLs and aid in the design of the next generation of enzymes.
Importantly, the increased activity in the resulting TurboID and miniTurbo enzymes enables shorter incubation times in cell culture to deliver specific biotinylation enrichment in minutes, compared with the hours needed for the original BirA*. We note however that although the initial and several subsequent BioID studies employed a labeling time of 24 h, this is not a requirement for BioID, and shorter labeling times - as little as 3 h (63) - enable efficient biotinylation and recovery of proximal interactors. Yet, this is quite far from the 5–10 min labeling times reported sufficient for miniTurbo and TurboID (108), and it is expected that these newer enzymes will enable temporal studies in the minute(s) time scale. Importantly, TurboID seems to also have an increased affinity for biotin, much like BioID2, allowing it to use the biotin present in media serum to carry out proximal labeling (30, 108); this should be considered in the assay designs when short labeling times are desired. Furthermore, this increase in affinity may result in biotin scavenging when TurboID is constitutively expressed, which may have detrimental repercussions when generating stable cells lines or transgenic animals.
As further research and development gives rise to newer enzymes with varying characteristics, it would be beneficial to have a thorough direct comparison of the enzymatic properties of all the emerging BPL enzymes using the same bait and matched purification and analysis by mass spectrometry. This will be of significant value in helping researchers to select the most appropriate enzyme for PDB experiments.
Improving Peroxidases
Similar to the BPLs, heme peroxidases are a large family of enzymes expressed in a diverse array of organisms, and many organisms express multiple different enzymes with distinct patterns of expression and preferred substrates ((68); e.g. Arabidopsis thaliana encodes 73 different heme peroxidases (65)). Although to date only APX and HRP have been used for PDB assays, continued research into these enzymes may reveal novel properties or substrate specificities that can have biotechnological use.
Like the workflow used to generate TurboID and miniTurbo, the Ting group used directed evolution to improve upon the soybean APEX enzyme (88). They noted that although APEX was suitable for electron microscopy, it lacked sensitivity for proteomics (110). Through a combination of error-prone PCR and yeast-display, they improved the activity of APEX to generate a new enzyme they named APEX2 (110). Interestingly, unlike the evolution of TurboID/miniTurbo, evolution of APEX2 only added a single new mutation to APEX, an A134P substitution (110) (Fig. 4). The authors note that the majority of sequenced Class II and III peroxidases, including HRP, already possess a proline at the corresponding position that may promote a higher tolerance against inactivation by H2O2 and/or enhancing oxidation of phenolic substrates (110, 111). Yet, despite the improvements made to the enzyme, APEX2 remains less active than HRP and more sensitive to H2O2 mediated inhibition (110), suggesting that further improvements may be possible. Recently, site directed mutagenesis also yielded a non-glycosylated recombinant HRP (rHRP) with 8-fold improved catalytic activity and 2-fold improved thermal stability over non-mutated HRP (112), but its application to intracellular labeling has yet to be determined.
With the pace of development of new reagents and strategies in the PDB field, it is difficult to speculate where the next breakthrough will come from. Although exploring only a few enzymes and their mutants from different species had led to the invaluable tools we have today, we have likely only scratched the surface. A more systematic exploration of enzymes across the large super-families could for example yield PDB enzymes with unique properties, such as varying temperature and pH dependences, which may allow for broader application. In addition to exploring enzymes across species, examination of the substrate repertoire of current and next generation enzymes could provide their own unique applications to PDB. For example, E. coli BirA has also been evolved to utilize desthiobiotin as a substrate (113), offering easier elution from streptavidin beads and an easier identification of the modified sites, similar to the use of desthiobiotin-phenol in APEX applications (114). BirA also accepts a ketone isostere of biotin as a cofactor, which can then be specifically conjugated to hydrazide- or hydroxylamine-functionalized molecules (43). Alternatively, Pyrococcus horikoshii BPL can accept azide derivatives of biotin (115), which can be functionalized using Staudinger ligation (116), triarylphosphine-FLAG epitope or Fluorogenic phosphines (116). More systematic exploration of directed evolution strategies with multiple enzymatic templates could also expand the substrate repertoire of these enzymes, paving the way for new applications.
Development of New PDB Modalities
As with any successful new technique, the development of PDB has prompted a growing number of modifications that further expand its scope beyond the original design. Here we list some of the most promising developments.
Protein-Nucleic Acid Contacts
Beyond the development of PDB-MS for detecting proximity among proteins, a logical application of the approach was to use it as a tool to detect proteins recruited to nucleic acids. There are now several publications that have fused PDB enzymes to an inactive Cas9 to direct the protein fusion to specific genomic loci (117–119), or to nucleic acid binding moieties (e.g. MS2-coat protein) that can recognize engineered hairpins in RNAs (120). These approaches can in theory identify proteins that are recruited to specific RNA or DNA sequences, though in these experimental designs, this task is rendered more difficult by the high background noise because of the presence of the active enzyme throughout the cell in addition to the fraction that is targeted to the desired site. This has made well-controlled experimental design and strong quantification techniques paramount to the success of this approach, which remains difficult for most laboratories, especially for the discovery of proteins associating with a single locus (118, 121).
Excitingly, the labeling of RNA by APEX enzymes is enabling the parallel probing of RNA and protein proximity interactomes, as recently reported by the Ting and Ingolia groups (122–124). Furthermore, aromatic substrate exploration identified that biotin-aniline and biotin-naphthylamine are preferentially transferred to RNA and DNA compared with biotin-phenol (125), providing new avenues of exploration for the APEX-seq modalities described below. This type of approach should enable a better understanding of the protein-nucleic acid interactions.
Context Dependent PDB
Most of the PDB studies to date have fused a protein of interest to a full-length PDB enzyme, which can be active from biogenesis onwards. Therefore, the final PDB profile is the lifetime footprint of proximal labeling by the PDB-fusion. Additionally, when a protein can occupy different locations inside the cell, the standard PDB approaches will reveal a convoluted signal that results from activity at each of these localizations. Although computational approaches for signal deconvolution have been developed (reviewed in (19)), more direct approaches that report on the proximity interactome of a bait protein in very specific contexts (e.g. when in a given organelle or in association with a certain protein complex) are needed.
A successful approach to capture context-dependent interactions/associations includes using protein-fragment complementation assays (PCAs) (126, 127). PCA entails splitting a “reporter” molecule (for example an enzyme or a fluorescent protein) into two fragments that are each inactive. Each of these fragments is then fused to a different protein of interest and simultaneously expressed (e.g. in a cell system). If the two proteins interact, the two fragments of the reporter molecule can reconstitute its activity. This approach has been widely used as a binary protein interaction method using colorimetric (e.g. β-galactosidase (128–130)) or fluorescent (e.g. bimolecular fluorescence complementation, BiFC, a.k.a. split-EGFP (131, 132)) reporters, as well as proteins or enzymes mediating drug resistance (e.g. DHFR-methotrexate (133)). This activity dependent PCA strategy was clearly applicable to PDB enzymes as well, and parallel efforts by several groups have already generated functional splits.
The first PDB-PCA consisted of a split HRP selected through structure-guided enzyme splitting followed by molecular evolution that was used to investigate cell-cell interactions in the extracellular compartment (134). However, as was discussed above, HRP is not suitable for intracellular PDB-PCA and this split has not been coupled to mass spectrometric identification of proximal proteins. Subsequent fragmentation of APEX between amino acids 201 and 202 generated the first split APEX (135). More recently, structure-guided fragment scanning followed by directed evolution was applied to APEX2 to generate a more active split enzyme, which harbors a further 9-mutations relative to APEX2 (136). The evolution of this split enzyme utilized both positive and negative selection steps to reduce interaction-independent reactivation of the fragments, while maintaining high peroxidase activity when reconstituted. This was implemented as a coincidence detector for protein-RNA interactions, and for labeling ER-mitochondria contact sites (136). In principle, using a split-enzyme strategy should improve the signal-to-noise in proximal labeling, and could be of particular use for defining proteins proximal to specific loci in the next generation of nucleic-acid tethered system. For fast-acting enzymes such as the split APEX2, this should also facilitate signal transduction studies and enable the identification of proximity interactomes for a specific subset of a protein pool (e.g. that are defined by distinct binding partners).
As with peroxidases, BPLs were also adapted to a PCA system. The first split BioID truncated BirA* at the hinge between E140/Q141, and was applied to define the proximity interactome of protein phosphatase 1, PP1, in complex with one of its substrate-targeting subunits (137). Another pair of splits with apparent higher activity truncated BirA* between its central catalytic domain and its C-terminal domain (E256/G257) and was used to study miRNA-mediated translation repression (138). Although this was an elegant demonstration of PCA using BirA*, it is important to note that the slow kinetics of the BirA* enzyme as well as the lower activity of the reconstituted enzymes compared with the full-length protein has limited the application of this approach for the study of dynamically-regulated systems. This is likely to change when split versions of the more active enzymes, such as TurboID, are generated.
Although PDB-PCA can be very useful for certain biological applications, it is by no means trivial to setup and does come with the potential risk for false-negative results. Like other PCA approaches, it may be necessary to optimize various bait-enzyme linker lengths, the protein termini tagged, and the stoichiometry of the two proteins expressed. These are a few of the factors that can affect the ability of the two enzyme halves to be able to assemble and reconstitute its PDB activity. As such, versatile PCA toolbox development is likely to remain an active area of research.
Other Emerging Strategies
There are a number of other noteworthy techniques that have been developed based on the principles of PDB. Although at their core they use similar labeling chemistry and protein identification, they provide an alternative means to capture a more restricted set of proximal interactors.
A technique that deserves attention is the 2C-BioID technique, developed by the Burke group (139). When tagging of a protein of interest with the relatively large PDB enzymes is problematic with regards to protein localization and function (such as for the lamina associated protein LAP2 beta), the PDB enzyme can be directed to the desirable bait through inducible dimerization. In this case, a generic fusion of the prolyl isomerase FK506-binding protein (FKBP) to BioID (or, more recently, GFP-APEX2 (140)) is co-expressed with a fusion of the protein of interest with the FKBP-rapamycin binding (FRB) domain of the mTOR kinase (∼100 amino acids). These two fusion proteins will only interact, recruiting the BioID enzyme to the protein of interest, in the presence of a dimerizer (a rapamycin derivative (141)). This inducible recruitment of the enzyme to the “bait” may be helpful to both prevent trafficking issues of a directly tagged PDB bait and provide a convenient control (the same cells in the absence of the rapamycin analog). This induced dimerization system was also applied to VAPB-APEX2 to further increase the spatial resolution of labeling (140). Although two-component systems are by definition more complex to optimize and are associated with their own issues (e.g. the relative stoichiometries of the PDB-FKBP and bait-FRB fusions), it should be expected that stable cell lines (and perhaps even animal models) constitutively expressing the generic PDB-FKBP fusion (ideally under the control of a regulatable promoter) would facilitate the broader adoption of this elegant design.
Although a clear strength of PDB approaches is the capability of performing experiments in living cells and organisms, several groups have explored in vitro approaches to label proteins located in proximity to a bait of interest. This includes antibody-HRP fusions that extends PDB approaches to fixed cells and tissues (e.g. (73) or the more recently developed Biotinylation by Antibody Recognition (BAR) technique (142). Similarly, Remnant et al., developed an in vitro BioID (ivBioID) technique (143) to improve the kinetic parameters of the BioID reaction and specifically focus on the proximal interactome of CENP-A at mitotic centromeres by permeabilizing the cells and performing a shorter labeling reaction in the presence of ATP and biotin. The full applicability of these approaches is not yet clear: for antibody-based approaches, issues of antibody cross-reactivity will need to be addressed (they may actually be amplified by coupling to a PDB method) whereas the ivBioID technique is likely to be more useful for proteins (like CENP-A) that remain tightly anchored to a structure following permeabilization.
In summary, both BPLs and peroxidases have been subjected to protein engineering to target them to nucleic acids or specific subcellular compartments to explore their use as coincidence detectors. Although it is still early days, future developments in these areas should generate robust approaches for studying the proximal associations between proteins and other molecules in the context of living cells or ex vivo.
Considerations for Experimental Design
With so many variations on the initial successful BioID and APEX approaches already available, it is important to reflect on the considerations for the selection of a specific enzyme and experimental design. In the following sections, we briefly comment on some of these aspects.
Enzyme Selection
The list of enzymes for PDB is still growing, but because most studies use a single enzyme, systematic comparisons are still largely lacking. A notable exception, that performed both APEX (APEX2) and BioID (BirA*) on baits within the ribosome quality control pathway, revealed little overlap between the approaches (and a better recall of known interactors with BioID) (144). Although it is not clear whether this is a widespread phenomenon, it raises interesting questions regarding the degree of complementarity between these different methods. In our hands (unpublished), proximity interactomes of the same bait (e.g. Lamin A) obtained with alternative biotin ligases are remarkably similar after filtering the background, so it remains to be seen whether the different classes of enzymes are responsible for the differences noted in the ribosomal quality control pathways.
There are conditions where selection of a given enzyme should be dictated by its properties. For instance, if proximity interactomes need to be established under short labeling times (for example to follow a time course of intracellular signaling), the ability to acquire proximity interactomes in seconds or minutes may be the dominant factor to consider. After optimization of the expression and biotinylation conditions, the original BirA* and BioID2 enzymes could generate enough signal-to-noise labeling with several tested proteins in as little as 3 h (63) and 1 h (unpublished), respectively. By contrast, second- to minute-scale labeling, such as would be required to study signaling from G-protein coupled receptors, could until recently only be accomplished with peroxidases (93, 94). However, the recent introduction of more active biotin ligases such as miniTurbo and TurboID permits labeling within 5–15 min (108), enabling PDB to be performed with either class of enzymes for all but the shortest labeling times. However, faster approaches may not always be the best choice. In particular, the longer incubation with biotin ligases may be helpful to “de-noise” the system by amplifying the signal at the predominant locations of the fusion protein, though this may also result in increasing complexity of the data [see our recent review (19) for an extended discussion].
Another property that may help guide the selection of a specific enzyme is the type of background generated by a given PDB enzyme (we have noticed that each of the biotin ligases generate a specific background pattern). Therefore, avoiding an enzyme that generates high backgrounds for proteins or cellular structures of interest can be a major advantage. For example, the lack of a DNA binding domain (e.g. in BioID2) appears to decrease chromatin-specific background as discussed above and may be beneficial for projects exploring the nuclear proximity proteome.
Other enzymatic properties should also be considered, especially when establishing the approach in different organisms, including the temperature at which the enzyme is functional (e.g. BioID2, from a thermophile which grows at temperatures up to 85–90 °C, may be more compatible with heat-shock treatment experiments). Though scant comparative information is available, properties such as tolerance over a broader range of pH, salt, etc., may likewise be useful. For instance, whether some enzymes work better than others inside acidic organelles remains to be defined. Furthermore, biotin has a well-characterized role in metabolism but extends to other processes such as the cell cycle, transcription and DNA damage, and high concentrations of biotin may alter these processes (reviewed elsewhere (145, 146)). Therefore, the ability for some BPL enzymes to work with minute concentrations of biotin can help minimize the need for high concentrations of biotin. However, this may necessitate the use of biotin-depleted media to establish a clean baseline in kinetic studies (note, however that prolonged biotin depletion can also alter metabolic processes, transcription and cell cycle). Anecdotal reports that the TurboID enzyme is capable of scavenging endogenous biotin and thus may be toxic to some cell lines or model systems (108) may be a further concern when using enzymes with enhanced affinity for biotin. Therefore, selection of the optimal system and protocols requires careful consideration.
Selection of more complex experimental designs such as 2C-BioID or split enzymes over simpler ones also requires careful consideration. Although split-BioID was first introduced as a way to identify proteins that are in proximity to a pair of baits in a complex, it is still unclear how the proximity interactome generated by the pair of baits using a split approach would have differed from performing standard BioID experiments with each bait separately. Similarly, whether the successes of 2C-BioID have more to do with the specific mislocalization of the LAP beta protein rather than a general improvement in the technology will remain to be systematically tested. What seems clear with the split systems in particular is that they lead to a more focused view of proximal interactomes, and concomitantly a lower overall background, which may be particularly desirable for proteins expressed at a low level, or for detecting rare association events between the selected baits (whether they are proteins, nucleic acids or other molecules). Yet, these advantages need to be considered in the context of increased burden with assay design for split enzymes that tend to be affected by binding geometries, as well as the need to express both recombinant proteins at optimal levels.
Other Experimental Considerations
Although this manuscript does not include a step-by-step “how-to” guide for PDB (please refer instead to (22, 147, 148)), there are certain guidelines regarding experimental set-up and control systems that are relevant to whichever enzymatic system is selected. Because every PDB experiment will lead to the identification of hundreds, if not thousands of proteins, how to design the experiment and implement relevant controls is fundamental.
In some cases, the tagging and expression of the bait may result in its aberrant function and/or mislocalization (149, 150), which can yield misleading results. This can be mitigated to some extent by maintaining expression of the bait at near endogenous levels (e.g. through regulated promoters or via CRISPR-tagging (151)), but this does not offer any guarantee that the fusion will behave the same way as the endogenous protein. Whenever possible, localization and functional assays should therefore be employed to ensure that the bait is—at least grossly—behaving as expected. Detection of “unexpected” proteins, or high enrichment of chaperones or components of folding and trafficking machineries by mass spectrometry (or lack of recall of previous interactors) may also be an indication that the bait is not working as expected, in which case revisiting expression levels, location of the tag, functional assays, etc., should be performed.
Even when the bait behaves as expected, negative controls are critical to a PDB experiment and some kind of quantitative mass spectrometry approach should be employed to discriminate between specific proximity interactors and contaminants (reviewed in (19)). Although we have not focused here on the quantitative mass spectrometry aspects of PDB studies, how one designs and executes these experiments is critical. When the background biotinylation is consistent and the signal-to-noise ratio is strong, it is often feasible to use robust semi-quantitative approaches such as spectral counting to identify proximity-dependent biotinylation. Yet, when the profiles are more complex and discriminating between signal and noise is challenging (a good example here is the exemplary work of Myers et al. to identify proteins proximal to a single locus with dCas9-APEX (118)), approaches that provide more accurate quantitative assessment are beneficial (and sometimes essential). In principle, any quantitative proteomics approach can be used for this purpose. For example, the Ting group used Stable Isotope Labeling by Amino acids in Culture (SILAC) for ratiometric assessment of high-confidence proximity interactors in mitochondrial subcompartments (89). Different groups have instead used labeling with isobaric reagents (such as Tandem Mass Tags, TMT) to discriminate between signal and noise in G-protein complex signaling and in identification of proteins associated with single DNA loci (93, 118). A different GPCR study instead used label-free Selected Reaction Monitoring (SRM) to follow temporal profiles of their candidate specific proximity partners with high sensitivity (94). Although it may be daunting to decide on a specific quantitative proteomics technique to use for PDB experiments, it may be easier to start with a few pilot experiments (perhaps using shotgun proteomics) to explore the signal-to-noise characteristics of an experimental set-up before deciding on more complex quantitative proteomics design. However, a constant for all PDB experiments, regardless of the quantitative approach ultimately selected, is that proper controls must be included, and performed in parallel to the true experiment.
In a PDB experiment, there are several categories of controls that could help with data analysis and the identification of true versus false-positive proximal associations. The first category of controls is the specific cell line without the enzyme present (or in the case of peroxidases - omission of H2O2, or inclusion of H2O2 but omission of biotin-phenol) which can help identify endogenously biotinylated proteins or proteins that non-specifically bind to the affinity matrix used for purification. The second category of controls is the expression of the BPL or peroxidase enzyme either by itself or fused to a ubiquitously distributed protein such as GFP. Because all proteins associate with the protein biogenesis machinery, using such a control can help remove some of these frequent flyer proteins. For many projects, this control may be sufficient to also account for nonspecific proximal labeling, provided that the “control” is expressed to at least the same level as the most expressed bait in the dataset. However, for proteins that are localized to specific subcellular compartments, it may also be useful to have additional controls. In this third category of controls, the fusion of a signal sequence to the enzyme of choice (or alternatively, a different bait that localizes to the same general area) can direct its activity to a specific compartment, allowing for a more refined subtraction of nonspecific proximal labeling (152, 153). Selection of such “compartment-specific” controls for BioID could be facilitated by the humancellmap.org resource (64). However, these compartment controls need to be tested to balance gains in specificity without over-penalizing the true-positive proximal partners, which we have found may happen when the controls are expressed at much higher levels than the baits. Lastly, whenever possible, inclusion of additional controls such as mutants of the protein under study may also be important for fully interpreting experimental results.
It is also worth reiterating that based on our experience with the various enzymes described here, each enzyme has a distinct nonspecific labeling signature. Therefore, we recommend using controls performed with the same enzyme and cell lines whenever possible.
Proximal Labeling Chemistry and Labeling Propensity
It is unclear how changes in bait protein structure or interactions (as well as post-translational modifications) affect proximal labeling in PDB experiments. However, this is certainly something to keep in mind when interpreting experimental results where a proteomic change is observed upon some treatment or experimental perturbation. Measured differences in proximal proteomes can very well be because of changes in the abundance and localization of proteins but could also be a result of alterations in biochemical or stereochemistry of proteins and their side chains.
PDB experiments are chemically-driven attacks that result in biotin transfer to lysines or tyrosines in BioID and APEX, respectively, and that a single biotinylated amino acid can enable recovery of a prey protein. Although there may be multiple modifiable residues among proteins in the vicinity of a bait of interest, only a small fraction of them need to become modified in order to enable capturing and detection. Which residues of a protein that have the highest propensity to become modified likely results from a combination of factors, including the distance to the PDB enzyme, but also the solvent accessibility of amino acid side chains competent to mediate the chemical reactions. Hydrophobic residues tend to be buried within or between proteins and hence less solvent exposed, whereas polar or charged residues (such as lysine) tend to be solvent accessible (154). In the case of BioID, this involves free (i.e. surface exposed and not engaged in a protein-protein interaction), unmodified lysine residue because the epsilon amine is responsible for the attack on biotinyl-5′-AMP. Lysines are also preferentially enriched in intrinsically disordered regions (IDRs), which have high solvent accessibility (155, 156). Consistent with this, IDR content correlates with protein detection in BioID experiments, though a more thorough investigation will be warranted (157).
In addition to solvent accessibility, the context-dependent biochemical properties of a side chain can also affect its labeling. This was highlighted using labeling reagents such as N-hydroxysuccinimide-biotin (NHS-biotin), which also involves a nucleophilic attack by primary amines on the NHS group. Here, biotinylation was significantly correlated with four factors relevant to the local environment of lysine residues; the solvent accessibility, the electrostatic energy, the number of hydrogen bonds, and the estimated pKa value of the sidechain (158). In agreement with this, nucleophilic attack by primary amines on biotinyl-5′-AMP is dependent on the side chain pKa, which can vary based on the pH of the local environment as well as the biochemical properties of amino acids in the vicinity of the amine group (159, 160). This has been demonstrated through non-enzymatic in vitro biotinylation of BCCP using biotinyl-5′-AMP, where only one of the five solvent accessible lysines is biotinylated (35), highlighting the complex role local side chain environments have on proximal labeling by PDB. As additional studies that monitor the biotinylated peptides directly are used (161–164), the availability of peptide-level data may shed better light on these still poorly understood aspects of PDB-MS.
Like lysine biotinylation, the reaction of biotin-phenoxyl radicals with tyrosine in peroxidase-based PDB is also dependent on solvent accessibility of the amino acid side chain and the local environment. A unique and lesser mentioned caveat to the use of peroxidase based reactions is that they can also catalyze crosslinking reactions between tyrosines in the presence of H2O2, inducing the formation of dityrosine linkages that are not labeled with biotin (68, 88, 165, 166). The abundance of this side reaction and its implication for protein labeling and identification is not currently known and requires further investigation.
In summary, the detection of prey proteins in a PDB-MS experiment can result from actual changes in the proximal proteome or may reflect changes in side chain reactivity or accessibility from conformational states following cellular state perturbations, which could alter proximal protein labeling in a less direct manner. In addition, if the perturbation modulates the abundance of some of the proteins in the proteome, changes in the detected PDB-MS profiles may simply reflect that. Therefore, besides the inclusion of proper controls in the experimental design, validation of results using complementary approaches is essential to distinguish the reasons for the changes observed.
CONCLUSION
PDB approaches are increasingly used to investigate a variety of different biological questions. As such, it is becoming progressively more important to understand the mechanisms, kinetics and considerations for each enzyme and experimental design. With this in mind, we have attempted to provide users with some background regarding the enzymes currently used for PDB-MS alongside brief considerations regarding the selection of the enzyme and experimental design that can be used. We hope that by reviewing the development and application of latest PDB tools, we will further stimulate their expansion for use in various biological contexts.
Acknowledgments
We thank members of the Gingras lab, and in particular Christopher Go, Geoff Hesketh, James Knight and Ji-Young Youn for discussions and critical comments.
Footnotes
* This work was supported by a Canadian Institutes of Health Research (CIHR) Foundation Grant (FDN 143301) to A.-C.G. Proteomics work in the Gingras lab is performed at the Network Biology Collaborative Centre at the Lunenfeld-Tanenbaum Research Institute, a facility supported by Canada Foundation for Innovation funding, by the Ontarian Government and by Genome Canada and Ontario Genomics (OGI-097, OGI-139). Anne-Claude Gingras is the Canada Research Chair (Tier 1) in Functional Proteomics. The authors declare that they have no conflicts of interest with the contents of this article.
1 The abbreviations used are:
- PDB
- proximity-dependent biotinylation
- AP-MS
- affinity purification coupled to mass spectrometry
- APX
- ascorbate peroxidase
- BAR
- biotinylation by antibody recognition
- BirA
- bifunctional ligase/repressor
- BCCP
- biotin-carboxylase cargo protein
- BPL
- biotin protein ligase
- EMARS
- enzyme-mediated activation of radical sources
- ER
- endoplasmic reticulum
- IDR
- intrinsically disordered region
- ivBioID
- in vitro BioID
- FKBP
- FK506-binding protein
- FRB
- FKBP-rapamycin binding domain
- HRP
- horseradish peroxidase C
- NHS-biotin
- N-hydroxysuccinimide-biotin
- PCA
- protein-fragment complementation assay
- PDB-MS
- proximity-dependent biotinylation coupled to mass spectrometry
- SPPLAT
- selective proteomic proximity labeling using tyramide
- SILAC
- stable isotope labeling by amino acids in cell culture
- TMT
- tandem mass tag
- iTRAQ
- isobaric tag for relative and absolute quantification.
REFERENCES
- 1. Yates J. R. 3rd, Gilchrist A., Howell K. E., and Bergeron J. J. (2005) Proteomics of organelles and large cellular structures. Nat. Rev. 6, 702–714 [DOI] [PubMed] [Google Scholar]
- 2. Itzhak D. N., Tyanova S., Cox J., and Borner G. H. (2016) Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mulvey C. M., Breckels L. M., Geladaki A., Britovsek N. K., Nightingale D. J. H., Christoforou A., Elzek M., Deery M. J., Gatto L., and Lilley K. S. (2017) Using hyperLOPIT to perform high-resolution mapping of the spatial proteome. Nat. Protocols 12, 1110–1135 [DOI] [PubMed] [Google Scholar]
- 4. Geladaki A., Kocevar Britovsek N., Breckels L. M., Smith T. S., Vennard O. L., Mulvey C. M., Crook O. M., Gatto L., and Lilley K. S. (2019) Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun. 10, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kristensen A. R., Gsponer J., and Foster L. J. (2012) A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods 9, 907–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wan C., Borgeson B., Phanse S., Tu F., Drew K., Clark G., Xiong X., Kagan O., Kwan J., Bezginov A., Chessman K., Pal S., Cromar G., Papoulas O., Ni Z., Boutz D. R., Stoilova S., Havugimana P. C., Guo X., Malty R. H., Sarov M., Greenblatt J., Babu M., Derry W. B., Tillier E. R., Wallingford J. B., Parkinson J., Marcotte E. M., and Emili A. (2015) Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Havugimana P. C., Hart G. T., Nepusz T., Yang H., Turinsky A. L., Li Z., Wang P. I., Boutz D. R., Fong V., Phanse S., Babu M., Craig S. A., Hu P., Wan C., Vlasblom J., Dar V. U., Bezginov A., Clark G. W., Wu G. C., Wodak S. J., Tillier E. R., Paccanaro A., Marcotte E. M., and Emili A. (2012) A census of human soluble protein complexes. Cell 150, 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heusel M., Bludau I., Rosenberger G., Hafen R., Frank M., Banaei-Esfahani A., van Drogen A., Collins B. C., Gstaiger M., and Aebersold R. (2019) Complex-centric proteome profiling by SEC-SWATH-MS. Mol. Syst. Biol. 15, e8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salas D., Stacey R. G., Akinlaja M., and Foster L. J. (2019) Next-generation interactomics: considerations for the use of co-elution to measure protein interaction networks. Mol. Cell Proteomics 19, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gingras A. C., and Raught B. (2012) Beyond hairballs: The use of quantitative mass spectrometry data to understand protein-protein interactions. FEBS letters 586, 2723–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mehta V., and Trinkle-Mulcahy L. (2016) Recent advances in large-scale protein interactome mapping. F1000Res 5, pii: F1000 Faculty Rev-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mousson F., Kolkman A., Pijnappel W. W., Timmers H. T., and Heck A. J. (2008) Quantitative proteomics reveals regulation of dynamic components within TATA-binding protein (TBP) transcription complexes. Mol. Cell. Proteomics 7, 845–852 [DOI] [PubMed] [Google Scholar]
- 13. Wang X., and Huang L. (2008) Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell. Proteomics 7, 46–57 [DOI] [PubMed] [Google Scholar]
- 14. Babu M., Vlasblom J., Pu S., Guo X., Graham C., Bean B. D., Burston H. E., Vizeacoumar F. J., Snider J., Phanse S., Fong V., Tam Y. Y., Davey M., Hnatshak O., Bajaj N., Chandran S., Punna T., Christopolous C., Wong V., Yu A., Zhong G., Li J., Stagljar I., Conibear E., Wodak S. J., Emili A., and Greenblatt J. F. (2012) Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 489, 585–589 [DOI] [PubMed] [Google Scholar]
- 15. Hakhverdyan Z., Domanski M., Hough L. E., Oroskar A. A., Oroskar A. R., Keegan S., Dilworth D. J., Molloy K. R., Sherman V., Aitchison J. D., Fenyo D., Chait B. T., Jensen T. H., Rout M. P., and LaCava J. (2015) Rapid, optimized interactomic screening. Nat. Methods 12, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subbotin R. I., and Chait B. T. (2014) A pipeline for determining protein-protein interactions and proximities in the cellular milieu. Mol. Cell Proteomics 13, 2824–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu C., Yang Y., Wang X., Guan S., Fang L., Liu F., Walters K. J., Kaiser P., and Huang L. (2016) Characterization of Dynamic UbR-proteasome subcomplexes by in vivo cross-linking (X) assisted bimolecular tandem affinity purification (XBAP) and label-free quantitation. Mol. Cell Proteomics 15, 2279–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trinkle-Mulcahy L. (2019) Recent advances in proximity-based labeling methods for interactome mapping. F1000Research 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gingras A. C., Abe K. T., and Raught B. (2019) Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opinion Chem. Biol. 48, 44–54 [DOI] [PubMed] [Google Scholar]
- 20. Varnaite R., and MacNeill S. A. (2016) Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 16, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rees J. S., Li X. W., Perrett S., Lilley K. S., and Jackson A. P. (2015) Protein Neighbors and Proximity Proteomics. Mol. Cell. Proteomics 14, 2848–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim D. I., and Roux K. J. (2016) Filling the void: proximity-based labeling of proteins in living cells. Trends Cell Biol. 26, 804–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tong L. (2013) Structure and function of biotin-dependent carboxylases. Cell Mol. Life Sci. 70, 863–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Azhar A., Booker G., and Polyak S. (2015) Mechanisms of biotin transport. Biochem. Anal. Biochem. 4, 1000210-1000211-1000218 [Google Scholar]
- 25. Chapman-Smith A., and Cronan J. E. Jr. (1999) Molecular biology of biotin attachment to proteins. J. Nutrition 129, 477S–484S [DOI] [PubMed] [Google Scholar]
- 26. McAllister H. C., and Coon M. J. (1966) Further studies on the properties of liver propionyl coenzyme A holocarboxylase synthetase and the specificity of holocarboxylase formation. J. Biol. Chem. 241, 2855–2861 [PubMed] [Google Scholar]
- 27. Sternicki L. M., Wegener K. L., Bruning J. B., Booker G. W., and Polyak S. W. (2017) Mechanisms Governing Precise Protein Biotinylation. Trends biochemical sciences 42, 383–394 [DOI] [PubMed] [Google Scholar]
- 28. Chapman-Smith A., Mulhern T. D., Whelan F., Cronan J. E. Jr, and Wallace J. C. (2001) The C-terminal domain of biotin protein ligase from E. coli is required for catalytic activity. Protein Sci. 10, 2608–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu Y., and Beckett D. (1996) Evidence for interdomain interaction in the Escherichia coli repressor of biotin biosynthesis from studies of an N-terminal domain deletion mutant. Biochemistry 35, 1783–1792 [DOI] [PubMed] [Google Scholar]
- 30. Kim D. I., Jensen S. C., Noble K. A., Kc B., Roux K. H., Motamedchaboki K., and Roux K. J. (2016) An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng J., Paparella A. S., Booker G. W., Polyak S. W., and Abell A. D. (2016) Biotin protein ligase is a target for new antibacterials. Antibiotics 5, pii: E26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weaver L. H., Kwon K., Beckett D., and Matthews B. W. (2001) Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator. Proc. Natl. Acad. Sci. U.S.A. 98, 6045–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu Y., and Beckett D. (1994) Kinetics of biotinyl-5′-adenylate synthesis catalyzed by the Escherichia coli repressor of biotin biosynthesis and the stability of the enzyme-product complex. Biochemistry 33, 7354–7360 [DOI] [PubMed] [Google Scholar]
- 34. Yao X., Wei D., Soden C. Jr, Summers M. F., and Beckett D. (1997) Structure of the carboxy-terminal fragment of the apo-biotin carboxyl carrier subunit of Escherichia coli acetyl-CoA carboxylase. Biochemistry 36, 15089–15100 [DOI] [PubMed] [Google Scholar]
- 35. Streaker E. D., and Beckett D. (2006) Nonenzymatic biotinylation of a biotin carboxyl carrier protein: unusual reactivity of the physiological target lysine. Protein Sci. 15, 1928–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schatz P. J. (1993) Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Bio/technology 11, 1138–1143 [DOI] [PubMed] [Google Scholar]
- 37. Beckett D., Kovaleva E., and Schatz P. J. (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8, 921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kulyyassov A., Shoaib M., Pichugin A., Kannouche P., Ramanculov E., Lipinski M., and Ogryzko V. (2011) PUB-MS: a mass spectrometry-based method to monitor protein-protein proximity in vivo. J. Proteome Res. 10, 4416–4427 [DOI] [PubMed] [Google Scholar]
- 39. Fernandez-Suarez M., Chen T. S., and Ting A. Y. (2008) Protein-protein interaction detection in vitro and in cells by proximity biotinylation. J. Am. Chem. Soc. 130, 9251–9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duffy S., Tsao K. L., and Waugh D. S. (1998) Site-specific, enzymatic biotinylation of recombinant proteins in Spodoptera frugiperda cells using biotin acceptor peptides. Anal. Biochem. 262, 122–128 [DOI] [PubMed] [Google Scholar]
- 41. Viens A., Mechold U., Lehrmann H., Harel-Bellan A., and Ogryzko V. (2004) Use of protein biotinylation in vivo for chromatin immunoprecipitation. Anal. Biochem. 325, 68–76 [DOI] [PubMed] [Google Scholar]
- 42. Cronan J. E. Jr., and Reed K. E. (2000) Biotinylation of proteins in vivo: a useful posttranslational modification for protein analysis. Methods Enzymol. 326, 440–458 [DOI] [PubMed] [Google Scholar]
- 43. Chen I., Howarth M., Lin W., and Ting A. Y. (2005) Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104 [DOI] [PubMed] [Google Scholar]
- 44. Jan C. H., Williams C. C., and Weissman J. S. (2014) Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deal R. B., and Henikoff S. (2011) The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 6, 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barker D. F., and Campbell A. M. (1980) Use of bio-lac fusion strains to study regulation of biotin biosynthesis in Escherichia coli. J. Bacteriol. 143, 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Buoncristiani M. R., Howard P. K., and Otsuka A. J. (1986) DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia coli. Gene 44, 255–261 [DOI] [PubMed] [Google Scholar]
- 48. Kwon K., and Beckett D. (2000) Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 9, 1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kwon K., Streaker E. D., Ruparelia S., and Beckett D. (2000) Multiple disordered loops function in corepressor-induced dimerization of the biotin repressor. J. Mol. Biol. 304, 821–833 [DOI] [PubMed] [Google Scholar]
- 50. Cronan J. E. (2005) Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase. J. Nutritional Biochem. 16, 416–418 [DOI] [PubMed] [Google Scholar]
- 51. Choi-Rhee E., Schulman H., and Cronan J. E. (2004) Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci. 13, 3043–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim D. I., Birendra K. C., Zhu W., Motamedchaboki K., Doye V., and Roux K. J. (2014) Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U.S.A. 111, E2453–E2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sears R. M., May D. G., and Roux K. J. (2019) BioID as a tool for protein-proximity labeling in living cells. Methods Mol. Biol. 2012, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Couzens A. L., Knight J. D., Kean M. J., Teo G., Weiss A., Dunham W. H., Lin Z. Y., Bagshaw R. D., Sicheri F., Pawson T., Wrana J. L., Choi H., and Gingras A. C. (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal 6, rs15. [DOI] [PubMed] [Google Scholar]
- 56. Dong J. M., Tay F. P., Swa H. L., Gunaratne J., Leung T., Burke B., and Manser E. (2016) Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci. Signal 9, rs4. [DOI] [PubMed] [Google Scholar]
- 57. Guo Z., Neilson L. J., Zhong H., Murray P. S., Zanivan S., and Zaidel-Bar R. (2014) E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal 7, rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Van Itallie C. M., Tietgens A. J., Aponte A., Fredriksson K., Fanning A. S., Gucek M., and Anderson J. M. (2014) Biotin ligase tagging identifies proteins proximal to E-cadherin, including lipoma preferred partner, a regulator of epithelial cell-cell and cell-substrate adhesion. J. Cell Sci. 127, 885–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hennigan R. F., Fletcher J. S., Guard S., and Ratner N. (2019) Proximity biotinylation identifies a set of conformation-specific interactions between Merlin and cell junction proteins. Sci. Signal 12, pii: eaau8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Y., Merkel C. D., Zeng X., Heier J. A., Cantrell P. S., Sun M., Stolz D. B., Watkins S. C., Yates N. A., and Kwiatkowski A. V. (2019) The N-cadherin interactome in primary cardiomyocytes as defined using quantitative proximity proteomics. J. Cell Sci. 132, pii: jcs221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Firat-Karalar E. N., Rauniyar N., Yates J. R. 3rd, and Stearns T. (2014) Proximity interactions among centrosome components identify regulators of centriole duplication. Curr. Biol. 24, 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gupta G. D., Coyaud E., Goncalves J., Mojarad B. A., Liu Y., Wu Q., Gheiratmand L., Comartin D., Tkach J. M., Cheung S. W., Bashkurov M., Hasegan M., Knight J. D., Lin Z. Y., Schueler M., Hildebrandt F., Moffat J., Gingras A. C., Raught B., and Pelletier L. (2015) A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 163, 1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Youn J. Y., Dunham W. H., Hong S. J., Knight J. D. R., Bashkurov M., Chen G. I., Bagci H., Rathod B., MacLeod G., Eng S. W. M., Angers S., Morris Q., Fabian M., Cote J. F., and Gingras A. C. (2018) High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532.e511 [DOI] [PubMed] [Google Scholar]
- 64. Go C. D., Knight J. D. R., Rajasekharan A., Rathod B., Hesketh G. G., Abe K., Youn J.-Y., Samavarchi-Tehrani P., Zhang H., Zhu L. Y., Popiel E., Lambert J. P., Coyaud E., Cheung S. W. T., Rajendran D., Wong C. J., Antonicka H., Pelletier L., Raught B., Palazzo A. F., Shoubridge E. A., and Gingras A. C. (2019) A proximity map of a human cell. bioRxiv 796391 [Google Scholar]
- 65. Savelli B., Li Q., Webber M., Jemmat A. M., Robitaille A., Zamocky M., Mathe C., and Dunand C. (2019) RedoxiBase: A database for ROS homeostasis regulated proteins. Redox. Biol. 26, 101247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zamocky M., Hofbauer S., Schaffner I., Gasselhuber B., Nicolussi A., Soudi M., Pirker K. F., Furtmuller P. G., and Obinger C. (2015) Independent evolution of four heme peroxidase superfamilies. Arch. Biochem. Biophys. 574, 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Welinder K. G. (1992) Superfamily of plant, fungal and bacterial peroxidases. Curr. Opin. Chem. Biol. 2, 338–393 [Google Scholar]
- 68. Veitch N. C. (2004) Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 65, 249–259 [DOI] [PubMed] [Google Scholar]
- 69. Mayer G., and Bendayan M. (1997) Biotinyl-tyramide: a novel approach for electron microscopic immunocytochemistry. J. Histochem. Cytochem. 45, 1449–1454 [DOI] [PubMed] [Google Scholar]
- 70. Bobrow M. N., Litt G. J., Shaughnessy K. J., Mayer P. C., and Conlon J. (1992) The use of catalyzed reporter deposition as a means of signal amplification in a variety of formats. J. Immunol. Methods 150, 145–149 [DOI] [PubMed] [Google Scholar]
- 71. Kotani N., Gu J., Isaji T., Udaka K., Taniguchi N., and Honke K. (2008) Biochemical visualization of cell surface molecular clustering in living cells. Proc. Natl. Acad. Sci. U.S.A. 105, 7405–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Honke K., Miyagawa-Yamaguchi A., and Kotani N. (2019) The EMARS reaction for proximity labeling. Methods Mol. Biol. 2008, 1–12 [DOI] [PubMed] [Google Scholar]
- 73. Li X. W., Rees J. S., Xue P., Zhang H., Hamaia S. W., Sanderson B., Funk P. E., Farndale R. W., Lilley K. S., Perrett S., and Jackson A. P. (2014) New insights into the DT40 B cell receptor cluster using a proteomic proximity labeling assay. J. Biol. Chem. 289, 14434–14447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bobrow M. N., Harris T. D., Shaughnessy K. J., and Litt G. J. (1989) Catalyzed reporter deposition, a novel method of signal amplification. Application to immunoassays. J. Immunol. Methods 125, 279–285 [DOI] [PubMed] [Google Scholar]
- 75. Adams J. C. (1992) Biotin amplification of biotin and horseradish peroxidase signals in histochemical stains. J. Histochem. Cytochem. 40, 1457–1463 [DOI] [PubMed] [Google Scholar]
- 76. Connolly C. N., Futter C. E., Gibson A., Hopkins C. R., and Cutler D. F. (1994) Transport into and out of the Golgi complex studied by transfecting cells with cDNAs encoding horseradish peroxidase. J. Cell Biol. 127, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miyagawa-Yamaguchi A., Kotani N., and Honke K. (2014) Expressed glycosylphosphatidylinositol-anchored horseradish peroxidase identifies co-clustering molecules in individual lipid raft domains. PloS One 9, e93054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Loh K. H., Stawski P. S., Draycott A. S., Udeshi N. D., Lehrman E. K., Wilton D. K., Svinkina T., Deerinck T. J., Ellisman M. H., Stevens B., Carr S. A., and Ting A. Y. (2016) Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166, 1295–1307.e1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hopkins C., Gibson A., Stinchcombe J., and Futter C. (2000) Chimeric molecules employing horseradish peroxidase as reporter enzyme for protein localization in the electron microscope. Methods Enzymol. 327, 35–45 [DOI] [PubMed] [Google Scholar]
- 80. Martell J. D., Deerinck T. J., Sancak Y., Poulos T. L., Mootha V. K., Sosinsky G. E., Ellisman M. H., and Ting A. Y. (2012) Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 30, 1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Patterson W. R., and Poulos T. L. (1995) Crystal structure of recombinant pea cytosolic ascorbate peroxidase. Biochemistry 34, 4331–4341 [DOI] [PubMed] [Google Scholar]
- 82. Zamocky M., Furtmuller P. G., and Obinger C. (2010) Evolution of structure and function of Class I peroxidases. Arch. Biochem. Biophys. 500, 45–57 [DOI] [PubMed] [Google Scholar]
- 83. Maruta T., Sawa Y., Shigeoka S., and Ishikawa T. (2016) Diversity and evolution of ascorbate peroxidase functions in chloroplasts: more than just a classical antioxidant enzyme? Plant Cell Physiol. 57, 1377–1386 [DOI] [PubMed] [Google Scholar]
- 84. Pipirou Z., Bottrill A. R., Metcalfe C. M., Mistry S. C., Badyal S. K., Rawlings B. J., and Raven E. L. (2007) Autocatalytic formation of a covalent link between tryptophan 41 and the heme in ascorbate peroxidase. Biochemistry 46, 2174–2180 [DOI] [PubMed] [Google Scholar]
- 85. Badyal S. K., Joyce M. G., Sharp K. H., Seward H. E., Mewies M., Basran J., Macdonald I. K., Moody P. C., and Raven E. L. (2006) Conformational mobility in the active site of a heme peroxidase. J. Biol. Chem. 281, 24512–24520 [DOI] [PubMed] [Google Scholar]
- 86. Shigeoka S., Ishikawa T., Tamoi M., Miyagawa Y., Takeda T., Yabuta Y., and Yoshimura K. (2002) Regulation and function of ascorbate peroxidase isoenzymes. J. Exp. Botany 53, 1305–1319 [PubMed] [Google Scholar]
- 87. Smulevich G., Paoli M., Burke J. F., Sanders S. A., Thorneley R. N., and Smith A. T. (1994) Characterization of recombinant horseradish peroxidase C and three site-directed mutants, F41V, F41W, and R38K, by resonance Raman spectroscopy. Biochemistry 33, 7398–7407 [DOI] [PubMed] [Google Scholar]
- 88. Rhee H. W., Zou P., Udeshi N. D., Martell J. D., Mootha V. K., Carr S. A., and Ting A. Y. (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hung V., Zou P., Rhee H. W., Udeshi N. D., Cracan V., Svinkina T., Carr S. A., Mootha V. K., and Ting A. Y. (2014) Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen C. L., Hu Y., Udeshi N. D., Lau T. Y., Wirtz-Peitz F., He L., Ting A. Y., Carr S. A., and Perrimon N. (2015) Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc. Natl. Acad. Sci. U.S.A. 112, 12093–12098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Markmiller S., Soltanieh S., Server K. L., Mak R., Jin W., Fang M. Y., Luo E. C., Krach F., Yang D., Sen A., Fulzele A., Wozniak J. M., Gonzalez D. J., Kankel M. W., Gao F. B., Bennett E. J., Lecuyer E., and Yeo G. W. (2018) Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604.e513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bersuker K., Peterson C. W. H., To M., Sahl S. J., Savikhin V., Grossman E. A., Nomura D. K., and Olzmann J. A. (2018) A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell 44, 97–112.e117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Paek J., Kalocsay M., Staus D. P., Wingler L., Pascolutti R., Paulo J. A., Gygi S. P., and Kruse A. C. (2017) Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell 169, 338–349.e311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lobingier B. T., Huttenhain R., Eichel K., Miller K. B., Ting A. Y., von Zastrow M., and Krogan N. J. (2017) An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 169, 350–360.e312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Reche P. A. (2000) Lipoylating and biotinylating enzymes contain a homologous catalytic module. Protein Sci. 9, 1922–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jitrapakdee S., and Wallace J. C. (2003) The biotin enzyme family: conserved structural motifs and domain rearrangements. Current Protein Peptide Sci. 4, 217–229 [DOI] [PubMed] [Google Scholar]
- 97. Lombard J., and Moreira D. (2011) Early evolution of the biotin-dependent carboxylase family. BMC Evolutionary Biol. 11, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gupta V., Gupta R. K., Khare G., Salunke D. M., Surolia A., and Tyagi A. K. (2010) Structural ordering of disordered ligand-binding loops of biotin protein ligase into active conformations as a consequence of dehydration. PloS One 5, e9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang J., and Beckett D. (2017) A conserved regulatory mechanism in bifunctional biotin protein ligases. Protein Sci. 26, 1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tron C. M., McNae I. W., Nutley M., Clarke D. J., Cooper A., Walkinshaw M. D., Baxter R. L., and Campopiano D. J. (2009) Structural and functional studies of the biotin protein ligase from Aquifex aeolicus reveal a critical role for a conserved residue in target specificity. J. Mol. Biol. 387, 129–146 [DOI] [PubMed] [Google Scholar]
- 101. Purushothaman S., Gupta G., Srivastava R., Ramu V. G., and Surolia A. (2008) Ligand specificity of group I biotin protein ligase of Mycobacterium tuberculosis. PloS One 3, e2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wilson K. P., Shewchuk L. M., Brennan R. G., Otsuka A. J., and Matthews B. W. (1992) Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc. Natl. Acad. Sci. U.S.A. 89, 9257–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Groft C. M., Uljon S. N., Wang R., and Werner M. H. (1998) Structural homology between the Rap30 DNA-binding domain and linker histone H5: implications for preinitiation complex assembly. Proc. Natl. Acad. Sci. U.S.A. 95, 9117–9122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Henke S. K., and Cronan J. E. (2014) Successful conversion of the Bacillus subtilis BirA Group II biotin protein ligase into a Group I ligase. PloS One 9, e96757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chakravartty V., and Cronan J. E. (2013) The wing of a winged helix-turn-helix transcription factor organizes the active site of BirA, a bifunctional repressor/ligase. J. Biol. Chem. 288, 36029–36039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Naganathan S., and Beckett D. (2007) Nucleation of an allosteric response via ligand-induced loop folding. J. Mol. Biol. 373, 96–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ramanathan M., Majzoub K., Rao D. S., Neela P. H., Zarnegar B. J., Mondal S., Roth J. G., Gai H., Kovalski J. R., Siprashvili Z., Palmer T. D., Carette J. E., and Khavari P. A. (2018) RNA-protein interaction detection in living cells. Nat. Methods 15, 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Branon T. C., Bosch J. A., Sanchez A. D., Udeshi N. D., Svinkina T., Carr S. A., Feldman J. L., Perrimon N., and Ting A. Y. (2018) Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Packer M. S., and Liu D. R. (2015) Methods for the directed evolution of proteins. Nature Reviews. Genetics 16, 379–394 [DOI] [PubMed] [Google Scholar]
- 110. Lam S. S., Martell J. D., Kamer K. J., Deerinck T. J., Ellisman M. H., Mootha V. K., and Ting A. Y. (2015) Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Turner D. D., Lad L., Kwon H., Basran J., Carr K. H., Moody P. C. E., and Raven E. L. (2018) The role of Ala134 in controlling substrate binding and reactivity in ascorbate peroxidase. J. Inorganic Biochem. 180, 230–234 [DOI] [PubMed] [Google Scholar]
- 112. Humer D., and Spadiut O. (2019) Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis. Int. J. Mol. Sci. 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lu W. C., Levy M., Kincaid R., and Ellington A. D. (2014) Directed evolution of the substrate specificity of biotin ligase. Biotechnol. Bioeng. 111, 1071–1081 [DOI] [PubMed] [Google Scholar]
- 114. Lee S. Y., Kang M. G., Shin S., Kwak C., Kwon T., Seo J. K., Kim J. S., and Rhee H. W. (2017) Architecture mapping of the inner mitochondrial membrane proteome by chemical tools in live cells. J. Am. Chem. Soc. 139, 3651–3662 [DOI] [PubMed] [Google Scholar]
- 115. Slavoff S. A., Chen I., Choi Y. A., and Ting A. Y. (2008) Expanding the substrate tolerance of biotin ligase through exploration of enzymes from diverse species. J. Am. Chem. Soc. 130, 1160–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kiick K. L., Saxon E., Tirrell D. A., and Bertozzi C. R. (2002) Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. U.S.A. 99, 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gao X. D., Tu L. C., Mir A., Rodriguez T., Ding Y., Leszyk J., Dekker J., Shaffer S. A., Zhu L. J., Wolfe S. A., and Sontheimer E. J. (2018) C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods 15, 433–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Myers S. A., Wright J., Peckner R., Kalish B. T., Zhang F., and Carr S. A. (2018) Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat. Methods 15, 437–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu X., Zhang Y., Chen Y., Li M., Shao Z., Zhang M. Q., and Xu J. (2018) CAPTURE: In situ analysis of chromatin composition of endogenous genomic loci by biotinylated dCas9. Curr. Protoc. Mol. Biol. 123, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mukherjee J., Hermesh O., Eliscovich C., Nalpas N., Franz-Wachtel M., Macek B., and Jansen R. P. (2019) beta-Actin mRNA interactome mapping by proximity biotinylation. Proc. Natl. Acad. Sci. U.S.A. 116, 12863–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schmidtmann E., Anton T., Rombaut P., Herzog F., and Leonhardt H. (2016) Determination of local chromatin composition by CasID. Nucleus 7, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Padron A., Iwasaki S., and Ingolia N. T. (2019) Proximity RNA labeling by APEX-Seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol. Cell 75, 875–887.e875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Fazal F. M., Han S., Parker K. R., Kaewsapsak P., Xu J., Boettiger A. N., Chang H. Y., and Ting A. Y. (2019) Atlas of subcellular RNA localization revealed by APEX-Seq. Cell 178, 473–490.e426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kaewsapsak P., Shechner D. M., Mallard W., Rinn J. L., and Ting A. Y. (2017) Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. Elife 6, pii: e29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhou Y., Wang G., Wang P., Li Z., Yue T., Wang J., and Zou P. (2019) Expanding APEX2 substrates for proximity-dependent labeling of nucleic acids and proteins in living cells. Angew Chem. Int. Ed. Engl. 58, 11763–11767 [DOI] [PubMed] [Google Scholar]
- 126. Remy I., and Michnick S. W. (2015) Mapping biochemical networks with protein fragment complementation assays. Methods Mol. Biol. 1278, 467–481 [DOI] [PubMed] [Google Scholar]
- 127. Shekhawat S. S., and Ghosh I. (2011) Split-protein systems: beyond binary protein-protein interactions. Curr. Opin. Chem. Biol. 15, 789–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Galarneau A., Primeau M., Trudeau L. E., and Michnick S. W. (2002) Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat. Biotechnol. 20, 619–622 [DOI] [PubMed] [Google Scholar]
- 129. Mohler W. A., and Blau H. M. (1996) Gene expression and cell fusion analyzed by lacZ complementation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 93, 12423–12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rossi F., Charlton C. A., and Blau H. M. (1997) Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc. Natl. Acad. Sci. U.S.A. 94, 8405–8410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Paulmurugan R., and Gambhir S. S. (2005) Novel fusion protein approach for efficient high-throughput screening of small molecule-mediating protein-protein interactions in cells and living animals. Cancer Res. 65, 7413–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hu C. D., and Kerppola T. K. (2003) Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pelletier J. N., Campbell-Valois F. X., and Michnick S. W. (1998) Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. U.S.A. 95, 12141–12146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Martell J. D., Yamagata M., Deerinck T. J., Phan S., Kwa C. G., Ellisman M. H., Sanes J. R., and Ting A. Y. (2016) A split horseradish peroxidase for the detection of intercellular protein-protein interactions and sensitive visualization of synapses. Nat. Biotechnol. 34, 774–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Xue M., Hou J., Wang L., Cheng D., Lu J., Zheng L., and Xu T. (2017) Optimizing the fragment complementation of APEX2 for detection of specific protein-protein interactions in live cells. Sci. Reports 7, 12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Han Y., Branon T. C., Martell J. D., Boassa D., Shechner D., Ellisman M. H., and Ting A. (2019) Directed evolution of split APEX2 peroxidase. ACS Chem. Biol. 14, 619–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. De Munter S., Gornemann J., Derua R., Lesage B., Qian J., Heroes E., Waelkens E., Van Eynde A., Beullens M., and Bollen M. (2017) Split-BioID: a proximity biotinylation assay for dimerization-dependent protein interactions. FEBS Letters 591, 415–424 [DOI] [PubMed] [Google Scholar]
- 138. Schopp I. M., Amaya Ramirez C. C., Debeljak J., Kreibich E., Skribbe M., Wild K., and Bethune J. (2017) Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat. Commun. 8, 15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chojnowski A., Sobota R. M., Ong P. F., Xie W., Wong X., Dreesen O., Burke B., and Stewart C. L. (2018) 2C-BioID: an advanced two component BioID system for precision mapping of protein interactomes. iScience 10, 40–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. James C., Muller M., Goldberg M. W., Lenz C., Urlaub H., and Kehlenbach R. H. (2019) Proteomic mapping by rapamycin-dependent targeting of APEX2 identifies binding partners of VAPB at the inner nuclear membrane. J. Biol. Chem. 294, 16241–16254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Liberles S. D., Diver S. T., Austin D. J., and Schreiber S. L. (1997) Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc. Natl. Acad. Sci. U.S.A. 94, 7825–7830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bar D. Z., Atkatsh K., Tavarez U., Erdos M. R., Gruenbaum Y., and Collins F. S. (2018) Biotinylation by antibody recognition-a method for proximity labeling. Nat. Methods 15, 127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Remnant L., Booth D. G., Vargiu G., Spanos C., Kerr A. R. W., and Earnshaw W. C. (2019) In vitro BioID: mapping the CENP-A microenvironment with high temporal and spatial resolution. Molecular biology of the cell 30, 1314–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zuzow N., Ghosh A., Leonard M., Liao J., Yang B., and Bennett E. J. (2018) Mapping the mammalian ribosome quality control complex interactome using proximity labeling approaches. Mol. Biol. Cell 29, 1258–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Riveron-Negrete L., and Fernandez-Mejia C. (2017) Pharmacological effects of biotin in animals. Mini. Rev. Med. Chem. 17, 529–540 [DOI] [PubMed] [Google Scholar]
- 146. Leon-Del-Rio A. (2019) Biotin in metabolism, gene expression, and human disease. J. Inherit. Metab. Dis. 42, 647–654 [DOI] [PubMed] [Google Scholar]
- 147. Hesketh G. G., Youn J. Y., Samavarchi-Tehrani P., Raught B., and Gingras A. C. (2017) Parallel exploration of interaction space by BioID and affinity purification coupled to mass spectrometry. Methods Mol. Biol. 1550, 115–136 [DOI] [PubMed] [Google Scholar]
- 148. Hung V., Udeshi N. D., Lam S. S., Loh K. H., Cox K. J., Pedram K., Carr S. A., and Ting A. Y. (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protocols 11, 456–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Weill U., Krieger G., Avihou Z., Milo R., Schuldiner M., and Davidi D. (2019) Assessment of GFP tag position on protein localization and growth fitness in yeast. J. Mol. Biol. 431, 636–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Stadler C., Rexhepaj E., Singan V. R., Murphy R. F., Pepperkok R., Uhlen M., Simpson J. C., and Lundberg E. (2013) Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 10, 315–323 [DOI] [PubMed] [Google Scholar]
- 151. Long S., Brown K. M., and Sibley L. D. (2018) CRISPR-mediated tagging with BirA allows proximity labeling in Toxoplasma gondii. Bio. Protoc. 8, pii: e2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lambert J. P., Tucholska M., Go C., Knight J. D., and Gingras A. C. (2015) Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteomics 118, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Bagci H., Sriskandarajah N., Robert A., Boulais J., Elkholi I. E., Tran V., Lin Z. Y., Thibault M. P., Dube N., Faubert D., Hipfner D. R., Gingras A. C., and Cote J. F. (2019) Mapping the proximity interaction network of the Rho-family GTPases reveals signalling pathways and regulatory mechanisms. Nat. Cell Biol. 22, 120–134 [DOI] [PubMed] [Google Scholar]
- 154. Lins L., Thomas A., and Brasseur R. (2003) Analysis of accessible surface of residues in proteins. Protein Sci. 12, 1406–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Uversky V. N. (2016) Paradoxes and wonders of intrinsic disorder: Complexity of simplicity. Intrinsically Disordered Proteins 4, e1135015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Campen A., Williams R. M., Brown C. J., Meng J., Uversky V. N., and Dunker A. K. (2008) TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Peptide Lett. 15, 956–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Minde D. P., Ramakrishna M., and Lilley K. S. (2018) Cellular labelling favours unfolded proteins. bioRxiv 274761 [Google Scholar]
- 158. Muraoka A., Matsuura Y., Naitow H., Ihara M., and Kunishima N. (2018) Availability of NHS-biotin labeling to identify free protein lysine revealed by experiment and MD simulation. Anal. Biochem. 557, 46–58 [DOI] [PubMed] [Google Scholar]
- 159. Isom D. G., Castaneda C. A., Cannon B. R., and Garcia-Moreno B. (2011) Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U.S.A. 108, 5260–5265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Gutteridge A., and Thornton J. M. (2005) Understanding nature's catalytic toolkit. Trends Biochem. Sci. 30, 622–629 [DOI] [PubMed] [Google Scholar]
- 161. Mackmull M. T., Klaus B., Heinze I., Chokkalingam M., Beyer A., Russell R. B., Ori A., and Beck M. (2017) Landscape of nuclear transport receptor cargo specificity. Mol. Syst. Biol. 13, 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kim D. I., Cutler J. A., Na C. H., Reckel S., Renuse S., Madugundu A. K., Tahir R., Goldschmidt H. L., Reddy K. L., Huganir R. L., Wu X., Zachara N. E., Hantschel O., and Pandey A. (2018) BioSITe: A Method for Direct Detection and Quantitation of Site-Specific Biotinylation. J. Proteome Res. 17, 759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Udeshi N. D., Pedram K., Svinkina T., Fereshetian S., Myers S. A., Aygun O., Krug K., Clauser K., Ryan D., Ast T., Mootha V. K., Ting A. Y., and Carr S. A. (2017) Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat. Methods 14, 1167–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Lee S. Y., Lee H., Lee H. K., Lee S. W., Ha S. C., Kwon T., Seo J. K., Lee C., and Rhee H. W. (2016) Proximity-directed labeling reveals a new rapamycin-induced heterodimer of FKBP25 and FRB in live cells. ACS Cent Sci. 2, 506–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gross A. J., and Sizer I. W. (1959) The oxidation of tyramine, tyrosine, and related compounds by peroxidase. J. Biol. Chem. 234, 1611–1614 [PubMed] [Google Scholar]
- 166. Minamihata K., Goto M., and Kamiya N. (2011) Site-specific protein cross-linking by peroxidase-catalyzed activation of a tyrosine-containing peptide tag. Bioconjugate Chem. 22, 74–81 [DOI] [PubMed] [Google Scholar]