

Abstract
Although a robust inflammatory response is needed to combat infection, this response must ultimately be terminated to prevent chronic inflammation. One mechanism that terminates inflammatory signaling is the production of alternative mRNA splice forms in the Toll-like receptor (TLR) signaling pathway. Whereas most genes in the TLR pathway encode positive mediators of inflammatory signaling, several, including that encoding the MyD88 signaling adaptor, also produce alternative spliced mRNA isoforms that encode dominant-negative inhibitors of the response. Production of these negatively acting alternatively spliced isoforms is induced by stimulation with the TLR4 agonist lipopolysaccharide (LPS); thus, this alternative pre-mRNA splicing represents a negative feedback loop that terminates TLR signaling and prevents chronic inflammation. In the current study, we investigated the mechanisms regulating the LPS-induced alternative pre-mRNA splicing of the MyD88 transcript in murine macrophages. We found that 1) the induction of the alternatively spliced MyD88 form is due to alternative pre-mRNA splicing and not caused by another RNA regulatory mechanism, 2) MyD88 splicing is regulated by both the MyD88- and TRIF-dependent arms of the TLR signaling pathway, 3) MyD88 splicing is regulated by the NF-κB transcription factor, and 4) NF-κB likely regulates MyD88 alternative pre-mRNA splicing per se rather than regulating splicing indirectly by altering MyD88 transcription. We conclude that alternative splicing of MyD88 may provide a sensitive mechanism that ensures robust termination of inflammation for tissue repair and restoration of normal tissue homeostasis once an infection is controlled.
Keywords: myeloid differentiation primary response gene 88 (MyD88), Toll-like receptor 4 (TLR4), alternative pre-mRNA splicing, macrophage, inflammation, gene regulation, NF-kappaB (NF-KB), TRIF, lipopolysaccharide
Introduction
The delicate balance between the initiation and termination of inflammation must be tightly regulated. Activation of a robust inflammatory response is needed to fight infection. However, persistent inflammation can damage tissues and contribute to many inflammatory diseases, such as atherosclerosis, chronic obstructive pulmonary disease, and cancer (1–4). Thus, once an infection is cleared, it is critical that inflammation be turned off. Macrophages are sentinel immune cells that represent one of the first lines of immune defense; they engulf and kill invading microorganisms and produce cytokines and chemokines to recruit and stimulate other immune cells (5). In addition to activating inflammatory responses upon infection, at later times during an infection macrophages also play an anti-inflammatory healing role (6, 7). Thus, macrophages mediate both the activation and termination of inflammation.
Toll-like receptors (TLRs)2 are a key receptor family present in macrophages and most other cell types that detect and activate pro-inflammatory signaling pathways (8, 9). For example, TLR4 senses lipopolysaccharide (LPS) from Gram-negative bacteria. When exposed to LPS, TLR4 sequentially recruits two signaling adaptors, MyD88 and TRIF (8, 9). In turn, these adaptor proteins recruit a complex network of proteins that mediate TLR signaling and ultimately activate the transcription factors NF-κB and AP1 to induce a program of pro-inflammatory gene expression.
One key mechanism that terminates TLR signaling and this pro-inflammatory program is the induction of alternative pre-mRNA splicing in the TLR signaling pathway. More than 256 different mRNA isoforms encompassing receptors, adaptors, and downstream effectors have been identified in the TLR signaling pathway; many of these different isoforms encode proteins with differing functions (10). In particular, whereas the canonical mRNAs in this signaling pathway usually encode activators of signaling, many TLR pathway genes also produce alternative mRNA splice forms that encode dominant negative inhibitors of the signaling pathway (11–21). Production of many of these negatively acting alternative splice forms is induced by LPS and/or other TLR agonists (11–21); thus, induction of this alternative splicing constitutes a negative feedback loop that terminates persistent inflammatory signaling.
One of the best-studied examples of this alternative splicing regulatory mechanism is production of an alternate isoform of the MyD88 signaling adaptor. The canonical MyD88 mRNA or long mRNA (MyD88-L) is 5 exons in length and produces a positive regulator of TLR signaling. However, the MyD88 gene also encodes an alternative shorter mRNA (MyD88-S), in which the 135-bp exon 2 is skipped. This shorter isoform contains an in-frame deletion that produces a functional protein that acts as a dominant negative inhibitor of signaling (12, 17, 22, 23). The longer canonical MyD88-L adaptor protein contains an N-terminal Toll-interleukin domain and a C-terminal death domain separated by an intermediate domain (24). MyD88-L bridges the interactions between Toll-interleukin domains on TLRs and the death domain on IL-1 receptor–associated kinases (IRAKs) to form a signaling complex through homotopic protein-protein interactions. The MyD88-S protein lacks the intermediary domain; it can bind to TLRs and the IRAK1 kinase but not the IRAK4 kinase, resulting in dominant-negative inhibition of IRAK1 phosphorylation and NF-κB activation (22).
MyD88-S has been identified in multiple species, including humans and mice, and in multiple cell types, including macrophages, monocytes, T-cells, B-cells, dendritic cells, and epithelial cells (17, 25–30). Thus, production of this negatively acting isoform is likely a universal mechanism for terminating TLR signaling. Production of MyD88-S is induced by LPS and other immune challenges, indicating that production of MyD88-S likely represents a key negative feedback loop to terminate inflammation (12, 17, 26).
Many questions remain about the production of MyD88-S. Whereas it is generally assumed that MyD88-S is produced by an alternative pre-mRNA splicing mechanism, there are other possible explanations for its LPS-mediated induction. Confounding this issue is the fact that most studies reporting on MyD88-S production in different disease contexts only monitor the production of this single isoform (e.g. see Refs. 25, 27, 28, 31, and 32). Moreover, the mechanisms regulating LPS-induced MyD88-S production have not been determined. Here, we establish a controlled macrophage model to monitor LPS-induced production of MyD88-S. We demonstrate that LPS-induced MyD88-S accumulation most likely involves a change in pre-mRNA splicing rather than other possible mechanisms, such as altered mRNA stability. Using genetic and pharmacological manipulation of the TLR signaling pathway, we demonstrate that the LPS-induced production of MyD88-S is mediated by the MyD88 and TRIF signaling adaptors and the downstream signaling components TRAF6 and the pro-inflammatory transcription factor NF-κB. Using a splicing-sensitive MyD88 minigene, we further demonstrate that MyD88 alternative pre-mRNA splicing is not transcriptionally coupled to NF-κB activation, suggesting that NF-κB mediates alternative splicing per se rather than affecting MyD88 transcription. Finally, we provide evidence that MyD88 alternative splicing is a sensitive mechanism that ensures robust termination of inflammation, thereby enabling tissue repair and return to homeostasis once infection is controlled.
Results
LPS induces MyD88-S expression in mouse macrophages
We and others have previously observed increased MyD88-S expression upon LPS stimulation in mouse and human macrophages (12, 17). To develop a system to investigate the mechanisms controlling LPS-induced expression of MyD88-S as well as to better understand the kinetics of MyD88-S expression, we treated the RAW264.7 mouse macrophage cell line with LPS and used qPCR to monitor MyD88-L and MyD88-S expression at multiple time points after LPS stimulation. The expression of both MyD88-L and MyD88-S was determined using isoform-specific qPCR. Other than a small transient increase in MyD88-L expression 6 h after LPS stimulation, the expression of MyD88-L remained largely unchanged at all time points after LPS stimulation (Fig. 1A). In contrast, MyD88-S expression was more dynamic. MyD88-S expression exhibited a moderate transient increase in expression between 1 and 6 h after LPS stimulation (Fig. 1B). By 12 h after LPS stimulation, both MyD88-L and MyD88-S returned to baseline. At later time points, MyD88-S but not MyD88-L levels continued to increase (Fig. 1, A and B).
Figure 1.

LPS induces MyD88-S expression in mouse macrophages. A and B, analysis of MyD88-L and MyD88-S mRNA expression at multiple time points during LPS exposure in RAW264.7 macrophages. Macrophages were treated with 200 ng/ml LPS or were left untreated, and MyD88 isoform levels were monitored at each time point relative to the untreated control using isoform-specific qPCR. C, RAW 264.7 cells were exposed to LPS (200 ng/ml) for 48 h or not exposed to LPS as a control. Total RNAs were then harvested, RT-PCR was performed using primers that bracket MyD88 exon 2, and the resulting PCR products were subjected to agarose gel electrophoresis. This allowed the simultaneous identification of PCR products corresponding to MyD88-L (369 bp) and MyD88-S (234 bp). L, MyD88-L; S, MyD88-S. Each lane represents an independent biological replicate. D and E, analysis of MyD88-L (D) and MyD88-S (E) mRNA expression at multiple LPS doses; RAW264.7 macrophages were treated with the indicated concentrations of LPS for 48 h, and MyD88 isoform expression was monitored by qPCR. F and G, MyD88-L and MyD88-S mRNA expression in LPS pulse–treated macrophages. RAW264.7 macrophages were treated with 200 ng/ml LPS for 2 h, the medium was then changed to medium lacking LPS, and the cells were incubated for an additional 22 h (24 h total) prior to harvesting RNA for isoform-specific qPCR. All qPCR data represent a minimum of three biological replicates. mRNA levels for each isoform are normalized to 1 in the absence of LPS. *, p < 0.05.
To validate the expression of MyD88-L and MyD88-S detected with qPCR, we performed RT-PCR with primers bracketing MyD88 exon 2 to amplify both MyD88-L and MyD88-S simultaneously. The PCR products were then resolved using agarose gel electrophoresis. This also allowed us to determine the relative levels of the two isoforms, as there is substantially more MyD88-L than MyD88-S in unstimulated cells (12, 33). In the absence of LPS, only a single PCR product of 369 bp corresponding to MyD88-L was amplified (Fig. 1C). After stimulation with LPS for 48 h, a 234-bp PCR product corresponding to MyD88-S also was clearly visualized (Fig. 1C). This result further confirms that LPS induces MyD88-S production in RAW264.7 cells.
To determine how LPS dosing affected MyD88-S production, we treated RAW264.7 macrophages with a range of LPS concentrations and monitored MyD88 isoform levels 48 h after challenge. Whereas LPS had only a minor effect on MyD88-L levels, LPS exposure led to a dose-dependent increase in MyD88-S (Fig. 1, D and E). Taken together, these experiments confirm that continuous LPS exposure can induce MyD88-S expression in RAW264.7 macrophages.
Because of the lag observed between the initial transient increase in MyD88-S expression and the second increase in MyD88-S levels, we wondered whether a time-limited LPS exposure could set in motion an early chain of events that would affect MyD88 splicing at later times. To test this hypothesis, RAW264.7 cells were treated with LPS for 2 h, and then the medium was changed to LPS-free medium. After an additional 22 h (24 h total), we monitored MyD88-L and MyD88-S expression. We found that a 2-h pulse exposure of LPS was able to increase MyD88-S levels without substantially changing MyD88-L levels 24 h after challenge (Fig. 1, F and G). These data are consistent with LPS inducing an early signal that can alter MyD88 splicing at later times.
Both MyD88 isoforms have similar half-lives
It has been hypothesized that the LPS-induced increase in MyD88-S expression results from MyD88 alternative pre-mRNA splicing (17). However, changes in the relative levels of MyD88-L and MyD88-S could also be affected by the selective degradation of MyD88-L mRNA if MyD88-L mRNA has a shorter half-life than MyD88-S mRNA. We therefore monitored MyD88-L and MyD88-S mRNA stability in the absence or presence of LPS exposure. We treated RAW264.7 cells with LPS for 24 h (or left some cells untreated as a control) and then treated the cells with actinomycin D to inhibit RNA polymerase II–dependent transcription. We then monitored MyD88 isoform levels by qPCR at multiple times after actinomycin D treatment to assess the rate of MyD88-L and MyD88-S mRNA decay. As confirmation that actinomycin D was inhibiting new mRNA synthesis, we found that actinomycin D treatment ablated LPS-induced TNFα mRNA production (Fig. 2A). In the absence of LPS treatment, the half-lives of MyD88-L and MyD88-S mRNAs were similar, roughly 1.6 h for both (Fig. 2B). LPS exposure increased the stability of both MyD88-L and MyD88-S mRNA, increasing the half-life for both to ∼5.6 h (Fig. 2C). Thus, both isoforms are equivalently stable. Therefore, the differential expression of MyD88-L and MyD88-S isoforms following LPS exposure is not the result of selective degradation of the MyD88-L mRNA and instead is likely the result of synthesis of new alternatively spliced mRNA.
Figure 2.

Both MyD88 isoforms have similar half-lives. A, to verify that actinomycin D was inhibiting transcription, RAW264.7 cells were treated with LPS (200 ng/ml) for 2 h in either the presence or absence of actinomycin D (50 μg/ml). Total RNA was then purified, and TNFα mRNA levels were monitored by qPCR. Data are presented as mean ± S.E. (error bars) with the data normalized so that TNFα expression in the absence of LPS is set to 1. B and C, RAW264.7 macrophages were treated with 200 ng/ml LPS for 24 h (C) or were left untreated (B) as a control. 50 μg/ml actinomycin D was then added to inhibit transcription, and decay of MyD88-L and MyD88-S mRNA was monitored by qPCR at the indicated time points. Time 0 is the time of the addition of actinomycin D; MyD88-L and MyD88-S levels are normalized so that they are both set to 1 at this time point. Note that there is substantially more MyD88-L than MyD88-S present in cells in the absence of LPS treatment; the figure depicts mRNA of each isoform relative to the starting concentration of that isoform. Additionally, MyD88-S levels at time 0 (after 24-h LPS exposure) in C are 2.2 ± 0.2 times greater than MyD88-S levels in the absence of LPS in B. All experiments represent a minimum of three independent biological replicates.
Both MyD88- and TRIF-dependent signaling pathways alter MyD88 splicing
LPS is sensed by TLR4 and its co-receptor MD-2 (9, 34). TLR4, when activated by LPS, in turn uses two adaptor proteins, MyD88 and TRIF, to sequentially transduce downstream signals and activate pro-inflammatory gene expression (Fig. 3A) (9, 34). To determine which adaptor protein(s) regulates MyD88 alternative splicing, we treated RAW264.7 cells with two other TLR agonists, PAM3CSK4 and poly(I:C). PAM3CSK4 stimulates TLR2, which signals through the MyD88 signaling adaptor; in contrast, poly(I:C) is recognized by TLR3, which signals through the TRIF signaling adaptor (Fig. 3A) (9, 34). All three TLR agonists induced robust TNFα production in RAW264.7 cells (Fig. 3B). Treatment of RAW264.7 cells with LPS, PAM3CSK4, or poly(I:C) for 48 h strongly increased MyD88-S expression while having a much more moderate effect on MyD88-L expression (Fig. 3, C and D), indicating that both MyD88- and TRIF-dependent signaling can alter MyD88 pre-mRNA splicing. Moreover, the degree of MyD88-S expression was generally correlated with the extent of TNFα production (Fig. 3, compare B and D), consistent with the observation that activating either the MyD88 or the TRIF signaling pathway can positively induce MyD88-S expression. Finally, we monitored the effect of these TLR agonists on the early induction of MyD88-S expression. All three TLR agonists induced a significant increase in MyD88-S production while having a much more limited effect on MyD88-L production 1 h after challenge (Fig. 3, E and F).
Figure 3.
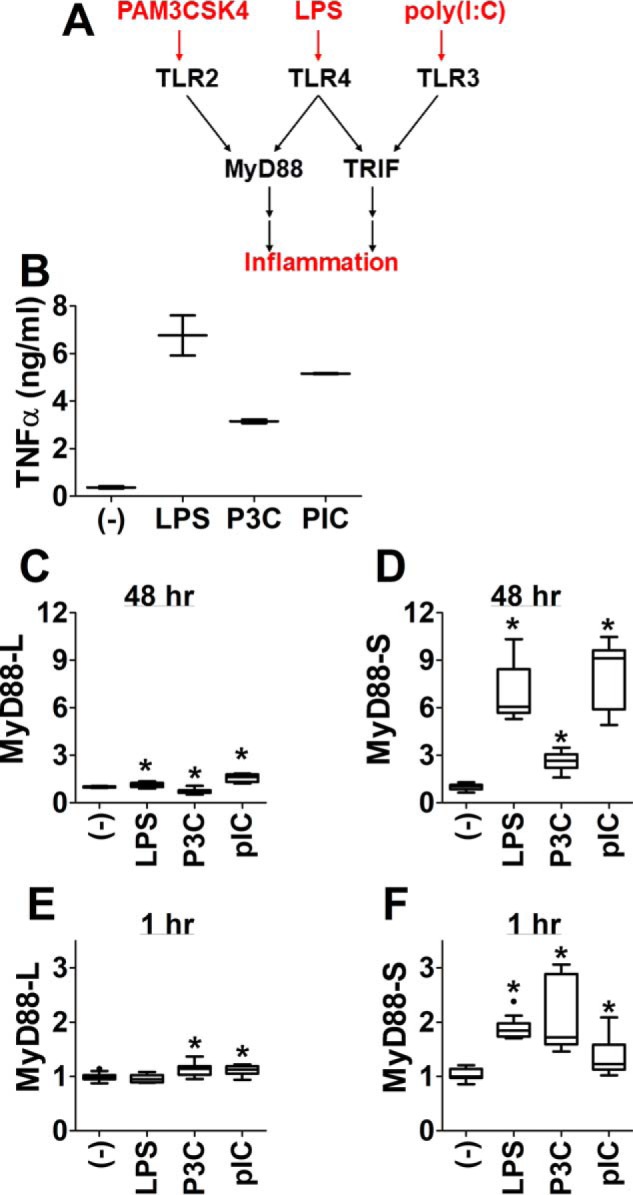
Activation of either MyD88-dependent or TRIF-dependent signaling pathways increases MyD88-S production. A, simplified schematic depicting TLR signaling pathways. LPS is sensed by TLR4, which sequentially uses two adaptor proteins, MyD88 and TRIF, to transduce downstream signals. PAM3CSK4 stimulates TLR2, which uses the MyD88 signaling adaptor to transduce downstream signals. Poly(I:C) stimulates TLR3, which uses the TRIF signaling adaptor to transduce downstream signals. RAW264.7 cells were stimulated for 48 h with either 200 ng/ml LPS, 200 ng/ml PAM3CSK4 (P3C), 10 μg/ml poly(I:C) (pIC), or no agonist, as indicated, and either TNFα production was monitored by ELISA (B) or MyD88-L and MyD88-S expression was monitored by qPCR in conjunction with isoform-specific primers (C and D). In a separate study, RAW264.7 cells were stimulated with the same concentrations of the indicated TLR agonists, and qPCR was used to monitor MyD88-L and MyD88-S production 1 h after challenge (E and F). All experiments represent a minimum of three independent biological replicates. Data are normalized so that MyD88-L or MyD88-S expression in the absence of LPS is set to 1. *, p < 0.05.
To directly investigate the role of MyD88 protein in the regulation of MyD88 alternative pre-mRNA splicing, we overexpressed MyD88 and assessed the effect on production of MyD88-S. We first built a stable RAW264.7 cell line overexpressing MyD88 fused to Escherichia coli DNA Gyrase B. The bacterial gyrase B fusion protein allows MyD88 to be dimerized and activated upon addition of the antibiotic coumermycin A1 (35). As expected, RAW264.7 cells stably overexpressing MyD88-GyrB produced increased levels of MyD88-L compared with cells stably overexpressing the negative control protein chloramphenicol acetyltransferase (CAT), which does not affect the immune response (Fig. 4A). In the absence of stimulation, overexpression of MyD88-GyrB did not affect MyD88-S levels (Fig. 4B). However, the addition of coumermycin A1 increased MyD88-S expression in cells expressing MyD88-GyrB but not cells expressing negative control CAT (Fig. 4B). This indicates that activated MyD88 protein is sufficient to induce MyD88-S production. Because overexpression of MyD88-GyrB interferes with the ability to monitor endogenous MyD88-L (but not MyD88-S), we also monitored the effect of overexpression of a TRAF6-GyrB fusion, which also is activated by coumermycin A1–induced dimerization (35). The E3 ubiquitin ligase TRAF6 functions downstream of MyD88 in the MyD88-dependent arm of the TLR signaling pathway (9, 34). Stable overexpression of TRAF6-GyrB induced MyD88-S but not MyD88-L production in RAW264.7 cells that were treated with coumermycin A1 (Fig. 4, A and B). Thus, both MyD88 and TRAF6 are sufficient to induce MyD88-S expression. As a control, we confirmed that coumermycin A1 was able to induce TNFα protein production in cells expressing MyD88-gyrB or TRAF6-gyrB but not CAT (Fig. 4C).
Figure 4.
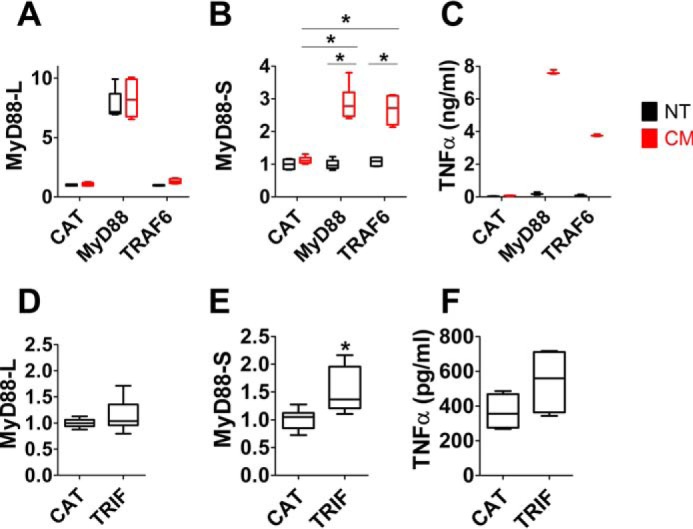
Both MyD88-dependent and TRIF-dependent signaling pathways increase MyD88-S production. A and B, MyD88-L and MyD88-S expression in RAW264.7 cell lines stably expressing either CAT, MyD88-GyrB, or TRAF6-GyrB. The cell lines were either treated with coumermycin A1 (CM), which dimerizes the gyrB fusions, resulting in activation of that protein, or not treated (NT) for 24 h prior to RNA collection for qPCR. C, TNFα protein production in the supernatants (monitored by ELISA) from the studies in A and B. D–F, plasmids overexpressing either negative control protein CAT or TRIF were transiently transfected into RAW264.7 cells; 48 h after transfection, MyD88-L and MyD88-S mRNA levels were assessed by qPCR in conjunction with isoform-specific primers, or TNFα protein production was monitored by ELISA. All experiments represent a minimum of three independent biological replicates. Data are normalized so that MyD88-L or MyD88-S expression in the absence of LPS is set to 1. *, p < 0.05.
To test whether TRIF can also regulate MyD88 alternative splicing, we transiently transfected a plasmid overexpressing WT TRIF into RAW264.7 cells. Overexpression of TRIF led to a moderate increase in MyD88-S expression compared with the cells transfected with a plasmid that expresses negative control CAT; in contrast, MyD88-L levels were not affected when TRIF was overexpressed (Fig. 4, D and E). The relatively modest MyD88-S expression increase induced in cells overexpressing TRIF compared with cells overexpressing MyD88-GyrB might be due to the inefficiency of transfection in RAW264.7 cells (which is only ∼50% or less as assessed by transfection of plasmids expressing GFP; data not shown). The successful but modest activation of signaling by TRIF overexpression was confirmed by monitoring TNFα protein production (Fig. 4F). In summary, MyD88-S expression is induced by both MyD88-dependent and TRIF-dependent signaling as assessed by stimulation with different TLR ligands and by overexpressing signaling pathway components.
The TLR4 signaling pathway regulates MyD88-S production in vivo
Our current study and prior studies indicate that LPS induces MyD88-S expression in cultured cell lines. To verify that LPS induces MyD88-S expression in vivo, we instilled LPS into the lungs of mice via intratracheal instillation and monitored MyD88 isoform levels in resident alveolar macrophages at multiple time points after LPS instillation. We found that LPS treatment led to a moderate increase in MyD88-L levels and a much more substantial increase in MyD88-S levels (Fig. 5, A and B), consistent with the cell line studies.
Figure 5.
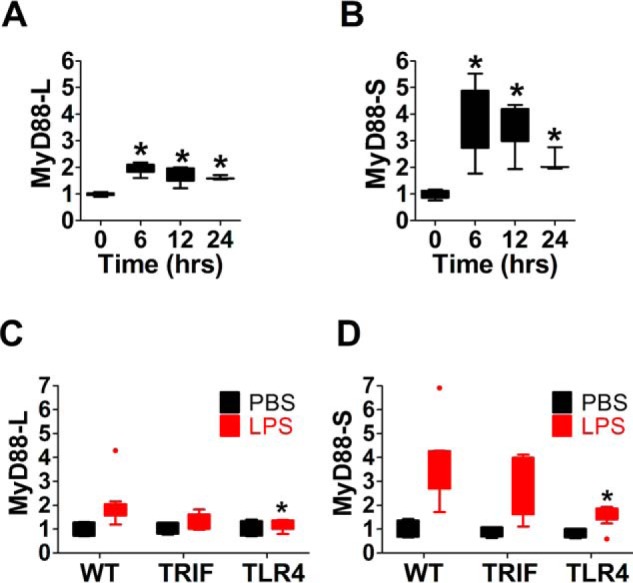
TLR4 signaling mediates LPS-induced MyD88-S production in vivo. A and B, analysis of MyD88-L and MyD88-S mRNA expression in mouse alveolar macrophages treated with LPS. LPS (20 μg/mouse) was instilled intratracheally into C57BL/6 mice, alveolar macrophages were isolated at the indicated times after challenge, and MyD88 isoform mRNA levels were quantitated. MyD88-L and MyD88-S mRNA were quantitated using qPCR in conjunction with isoform-specific primers. n = 7 for 0, 6, and 12 h; n = 3 for 24 h. C and D, analysis of MyD88-L and MyD88-S mRNA expression in mouse alveolar macrophages treated with LPS or treated with PBS control. LPS (20 μg/mouse) or PBS was instilled intratracheally into C57BL/6 mice, alveolar macrophages were isolated 6 h after challenge, and MyD88 isoform levels were quantitated. n = 4 for PBS, and n = 6–10 for LPS. Data are normalized so that MyD88-L or MyD88-S expression in the absence of LPS is set to 1. *, p < 0.05.
Our cell line studies suggested that either MyD88 or TRIF signaling is sufficient to induce MyD88-S production in macrophages. To determine the contributions of these pathways, we monitored the effect of TRIF and TLR4 knockouts on LPS-induced MyD88-S production in alveolar macrophages. The latter block signaling through both the MyD88 and TRIF arms of the signaling pathway and were chosen instead of MyD88 knockouts, because deletion of MyD88 abrogates production of both MyD88-L and MyD88-S. Deletion of TRIF had little effect on LPS-induced MyD88-S production (Fig. 5, C and D), suggesting that MyD88 acts at least partially redundantly in the induction of MyD88-S. Consistent with a requirement for MyD88 in LPS-induced production of MyD88-S, the TLR4 knockout greatly weakened the LPS-induced induction in MyD88-S (Fig. 5, C and D).
NF-κB is required for LPS-induced MyD88 alternative pre-mRNA splicing
Both MyD88 and TRIF signaling ultimately lead to activation of the NF-κB transcription factor (9, 34). To determine whether downstream signaling is involved in the LPS-induced production of MyD88-S, we examined the effect of activation of the NF-κB regulatory kinase IKK2 on MyD88-S production. We transiently overexpressed a constitutively active version of IKK2 (IKK2-S177E-S181E) (36) and found that this led to increased production of MyD88-S without a substantial increase in MyD88-L (Fig. 6, A and B). This result suggests that IKK2 activation is sufficient to positively regulate MyD88-S expression, raising the possibility that NF-κB mediates the effects of LPS on induction of MyD88-S.
Figure 6.

NF-κB regulates MyD88 alternative pre-mRNA splicing. A and B, RAW264.7 macrophages were transiently transfected either with negative control protein CAT or with a plasmid overexpressing constitutively activated (CA) IKK2 (IKK2-S177E-S181E). 48 h after transfection, MyD88-L and MyD88-S mRNA levels were monitored by qPCR. C and D, RAW264.7 cells were pulse-treated with 200 ng/ml LPS for 2 h (or were left untreated as a control) in either the presence or absence of inhibitors of NF-κB (J = JSH23, T = TPCA1, M = MG132). After a further 22-h incubation with or without these inhibitors, RNA was collected, and MyD88-L and MyD88-S mRNA levels were assessed by qPCR. To prevent cell death, the NF-κB inhibition studies were performed in the presence of two apoptosis inhibitors, Z-VAD-fmk and necrostatin. E and F, RAW264.7 cells were treated with 200 ng/ml LPS for 1 h (or were left untreated as a control) in either the presence or absence of the indicated inhibitors of NF-κB. RNA was then collected, and MyD88-L and MyD88-S mRNA levels were assessed by qPCR. All experiments represent a minimum of three independent biological replicates. Data are normalized so that MyD88-L or MyD88-S expression in the absence of LPS is set to 1. *, p < 0.05.
To test whether NF-κB activity was necessary for LPS-induced production of MyD88-S, we conversely inhibited NF-κB using several different pharmacological inhibitors. These inhibitors included the IKK2 kinase activity inhibitor TPCA1 and the NF-κB nuclear translocation inhibitor JSH23 (37, 38). In human macrophages, inhibition of NF-κB in the presence of LPS results in cell apoptosis and necrosis (39, 40), and we observed similar effects in RAW264.7 cells (data not shown). Therefore, we conducted the NF-κB inhibition experiments in the presence of both the caspase inhibitor Z-VAD-fmk and the necrosis inhibitor necrostatin; these two cell death inhibitors were sufficient to prevent RAW264.7 cell death during these studies (data not shown). We treated the RAW264.7 cells with LPS for 2 h in the absence or presence of the NF-κB inhibitors and monitored MyD88 isoform production 24 h after LPS stimulation was started (TPCA1 or JSH23 remained in the medium throughout the experiment). Treatment with TPCA1 alone did not affect MyD88-L or MyD88-S expression (Fig. 6, C and D). JSH23 treatment, however, did lead to a moderate increase in MyD88-S expression (Fig. 6D). LPS treatment, as expected, increased MyD88-S expression significantly without affecting MyD88-L production (Fig. 6, C and D). Inhibition of IKK2 activity with TPCA1 completely abolished the LPS-induced increase in MyD88-S expression, whereas inhibition of NF-κB nuclear translocation with JSH23 brought LPS-induced MyD88-S expression down to the level present when cells were treated with JSH23 alone (Fig. 6D). Thus, both NF-κB inhibitors prevented LPS-induced production of MyD88-S 24 h after challenge. LPS and either TPCA1 or JSH23 co-treatment did not have a substantial effect on MyD88-L expression (Fig. 6C).
To further investigate the role of NF-κB in LPS-induced production of MyD88-S, we also monitored the effects of LPS and these inhibitors at an earlier time point, 1 h after LPS challenge. For these studies, we tested the IKK2 activity inhibitor (TPCA1), the NF-κB nuclear translocation inhibitor (JSH23), and a third pharmacological inhibitor with a distinct mechanism of action, MG132 (we were unable to test MG132 at the later time point as it killed the cells in less than 24 h). MG132 inhibits the proteasome, thereby stabilizing the IκBα inhibitory protein and preventing activation of NF-κB (41). Treatment of RAW264.7 cells with any of the three NF-κB–inhibitory chemicals decreased LPS-induced MyD88-S production 1 h after challenge without substantially altering MyD88-L levels at this time point (Fig. 6, E and F). Thus, NF-κB activity is required for LPS-induced production of MyD88-S both early and late after challenge. Together with the IKK2 activation studies, these data indicate that IKK2 and downstream NF-κB are both necessary and sufficient to regulate MyD88 alternative splicing.
The regulation of MyD88 alternative pre-mRNA splicing by NF-κB is independent of MyD88 transcription
NF-κB could enhance MyD88-S production by altering expression or activity of a component(s) of the pre-mRNA splicing machinery. An alternate, not mutually exclusive, possibility is that NF-κB induces new MyD88 transcription, which would be needed for new MyD88-S production. Consistent with the possibility that TLR signaling might induce MyD88 transcription is our observation that both MyD88-L and MyD88-S exhibit a small transient increase in expression 6 h after LPS challenge (Fig. 1, A and B). To test whether NF-κB–induced MyD88 transcription was needed for the LPS-induced change in MyD88 pre-mRNA splicing, we constructed a MyD88 minigene whose expression was under the control of the NF-κB-independent elongation factor 1α (EF-1α) promoter. We cloned ∼2 kb of MyD88 genomic sequence encompassing all of exons 1–3 and the intervening introns downstream of the EF-1α promoter. Additionally, we engineered a stretch of mutations in exon 3 to allow for the design of minigene-specific qPCR primers that would not amplify the endogenous MyD88 gene. For this mutagenesis, we chose a region of exon 3 that was predicted to not contain critical splicing factor binding information (analysis using the Spliceaid (42) online RNA-binding site prediction program; data not shown). The final minigene plasmid construct is termed pEF1α-MyD88-mini (Fig. 7A).
Figure 7.
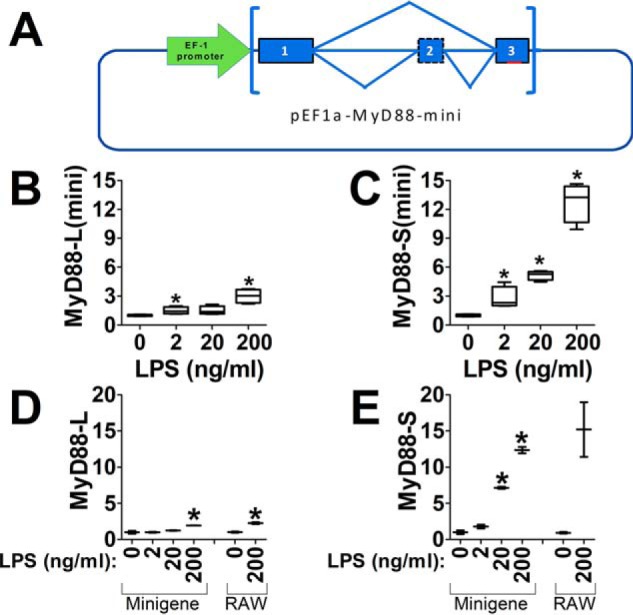
NF-κB regulates MyD88 pre-mRNA splicing independent of transcriptional effects on MyD88. A, schematic of the MyD88 minigene construction. The MyD88 minigene construct contains ∼2 kb of genomic sequence encompassing exon 1–intron 1–exon 2–intron 2–exon 3 of mouse MyD88; this sequence is cloned downstream of the EF-1α promoter. Additionally, 8 bp within exon 3 (indicated in red, not to scale) were mutated to allow for minigene-specific qPCR. B and C, the MyD88 minigene plasmid was transiently transfected into RAW264.7 cells; after 24 h, the cells were stimulated with the indicated LPS concentrations for 48 h, and MyD88-L(minigene) and MyD88-S(minigene) mRNA levels were quantitated by qPCR. D and E, to confirm that the MyD88 minigene does not interfere with splicing of the endogenous locus, the MyD88 minigene construct was transiently transfected into RAW264.7 cells (indicated as minigene) or was not transfected into RAW264.7 cells (indicated as RAW). 24 h after transfection, the cells were stimulated with the indicated concentrations of LPS for 48 h. Production of endogenous MyD88-L and MyD88-S was then assessed by isoform-specific qPCR. Data are normalized so that MyD88-L(mini), MyD88-S(mini), MyD88-L, or MyD88-S expression in the absence of LPS is set to 1. *, p < 0.05.
We transfected the pEF1α-MyD88-mini plasmid into RAW264.7 cells; 24 h later, we stimulated the cells with LPS for an additional 48 h. qPCR using MyD88-L(mini) and MyD88-S(mini) primers that were designed to amplify the minigene mRNA but not the endogenous MyD88 mRNA amplified a product when the minigene was transfected into cells (Fig. 7, B and C). In contrast, when the minigene was not present in RAW264.7 cells, these minigene primers did not amplify a product above background, indicating that the minigene primers were specific to the minigene and not the endogenous MyD88 gene. Increasing doses of LPS led to increased production of MyD88-S(mini) while having only a moderate effect on production of MyD88-L(mini) (Fig. 7, B and C). Thus, the minigene faithfully recapitulated LPS-induced alternative pre-mRNA splicing. Moreover, expression of the minigene did not affect alternative splicing of the endogenous MyD88 gene, as determined using qPCR primers that target the endogenous gene (Fig. 7, D and E).
Alternative pre-mRNA splicing and transcription are known to be functionally coupled (43). Thus, it was possible that MyD88 transcription induced by NF-κB activation could influence MyD88 splicing. However, because transcription of the EF-1α promoter-driven minigene is not controlled by NF-κB, the alteration of MyD88 splicing induced by LPS is likely not the result of functionally coupled transcription and pre-mRNA splicing. It is therefore more likely that NF-κB is regulating MyD88 alternative splicing by altering expression or activity of one or more components of the pre-mRNA splicing machinery.
To further test the possibility that the effect of NF-κB on MyD88-S production involved NF-κB–mediated alteration of splicing as opposed to an effect on MyD88 transcription, we tested the consequence of NF-κB inhibition on LPS-induced production of MyD88-S(mini). For these studies, we built a stable RAW264.7 cell line expressing the EF-1α promoter-driven MyD88 minigene. As expected, a 2-h pulse of LPS stimulation led to increased production of MyD88-S(mini) but not MyD88-L(mini) 24 h after the start of the experiment (Fig. 8, A and B). The addition of the IKK2 inhibitor TPCA1 prevented the LPS-induced increase in MyD88-S mRNA (Fig. 8B). This result mirrors what we observed at the endogenous MyD88 locus and suggests that the effect of NF-κB on MyD88 alternative pre-mRNA splicing occurs independently of a possible role affecting MyD88 transcription and instead likely involves regulation of a splicing machinery component(s).
Figure 8.

NF-κB regulates MyD88 pre-mRNA splicing. A and B, RAW264.7 cells stably overexpressing the MyD88 minigene plasmid were pulse-treated with 200 ng/ml LPS for 2 h (or were left untreated as a control) in either the presence or absence of the IKK2 inhibitor TPCA1. After a further 22-h incubation with or without TPCA1, RNA was collected, and MyD88-L(minigene) and MyD88-S(minigene) mRNA levels were assessed by qPCR. These inhibitor studies were performed in the presence of two apoptosis inhibitors, Z-VAD-fmk and necrostatin (n = 3). Data are normalized so that MyD88-L(mini) or MyD88-S(mini) expression in the absence of LPS is set to 1. *, p < 0.05.
MyD88 is sensitized to undergo alternative splicing to produce MyD88-S
We have previously shown that inhibition of core components of the spliceosome (SF3A1, SF3A2, SF3A3, SF3B1, or U2AF1) weakens the response to LPS in mouse and/or human macrophages (12, 44, 45). These core spliceosome factors all bind to the 3′ end of introns to facilitate pre-mRNA splicing (46–49). Whereas these are essential splicing factors, inhibition at around the 80% level weakened LPS-induced inflammatory cytokine production without affecting cell viability (12, 44, 45). This weakened response to LPS was caused, in part, by increased production of MyD88-S when the spliceosome was inhibited (12, 44, 45). We further showed that moderate inhibition of the spliceosome had relatively specific effects, altering splicing of only a subset of genes (33), including MyD88. This raised the possibility that MyD88 alternative splicing, because of its functional significance in regulating inflammation, may be particularly sensitive to environmental signals such as LPS (12).
To investigate the signals that render MyD88 splicing so sensitive to LPS treatment and perturbation of the spliceosome, we examined the sequences at the 3′ end of MyD88 intron 1 (Fig. 9A). Just upstream of the AG dinucleotide at the end of the intron is the polypyrimidine tract (pY tract), which is bound by the U2AF1 spliceosome component during pre-mRNA splicing (50). Upstream of the pY tract is the branch point sequence, which is recognized by the U2 snRNA and its associated splicing factors, including members of the SF3A and SF3B complexes (50). Our prior observation that moderate inhibition of these spliceosome components weakened exon 2 inclusion (and thus favored production of MyD88-S) suggested that the pY tract and/or the branch point sequence in this intron may be relatively “weak.”
Figure 9.
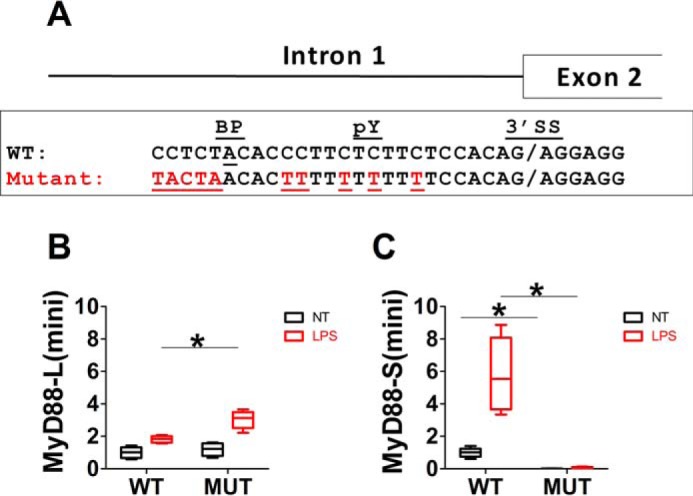
MyD88 is poised to undergo alternative pre-mRNA splicing. A, WT sequence at the 3′ end of intron 1 in the mouse MyD88 gene. Marked is the 3′ splice site (3′SS), the polypyrimidine tract (pY), and the branch point adenosine residue (BP). Also depicted are 10 bp that were mutated (Mutant) highlighted in red to improve the strength of the branch point and pY tract sequences in a mutagenized MyD88 minigene construct. B and C, the MyD88 minigene plasmid (either WT or mutant (MUT)) was transiently transfected into RAW264.7 cells; after 24 h, the cells were stimulated with LPS (red) or were left unstimulated (no treatment (NT), black) for 48 h, and MyD88-L(minigene) and MyD88-S(minigene) mRNA levels were quantitated by qPCR. Data are normalized so that MyD88-L(mini) and MyD88-S(mini) in the WT minigene in the absence of LPS are set to 1. *, p < 0.05.
pY tracts with more thymidine residues than cytidine residues generally correspond to stronger pY tracts (51, 52). Consistent with MyD88 having a relatively weak pY tract, the pY tract in MyD88 contains several cytidine residues (Fig. 9A). Similarly, canonical and thus “stronger” branch point sequences match the inverse complement of the U2 snRNA pre-mRNA binding region (TACTA) (53). The MyD88 branch point has been mapped in human MyD88 (54), and by analogy in the mouse MyD88 sequence, we find that the branch point sequence in MyD88 does not match the canonical sequence (Fig. 9A) and is predicted to be very weak.
To directly test whether these weak splicing regulatory sequences sensitized MyD88 to exon 2 skipping (i.e. MyD88-S production), we mutated 10 residues in the MyD88 minigene construct to “improve” the splicing regulatory sequences. We mutated the branch point sequence to match the canonical sequence, and we converted cytidine to thymidine residues in the pY tract (Fig. 9A). We then transiently transfected either the WT MyD88 minigene or the mutated minigene into RAW264.7 cells and exposed the cells to LPS for 24 h. As observed previously, LPS induced MyD88-S(mini) but not MyD88-L(mini) production for the WT minigene (Fig. 9, B and C). In contrast, MyD88-L(mini) levels were slightly increased, and MyD88-S(mini) levels were all but abolished in the minigene with the mutated “improved” intronic sequences (Fig. 9, B and C). These data are consistent with the splicing regulatory sequences in MyD88 intron 1 being relatively “weak,” contributing to a sensitized LPS-responsive alternative splicing system.
Discussion
The mechanisms regulating the activation of TLR signaling have been studied for some time, and the importance of TLR signaling in combating infection is well-established (55–57). In contrast, whereas terminating this response is necessary to prevent chronic inflammatory disease, the mechanisms that terminate TLR signaling are less well-understood (58). Thus, the LPS-induced production of MyD88-S represents an important mechanism that might be critical to preventing inflammatory disease. Several studies have reported an association of MyD88 isoform levels with inflammatory disease. MyD88-L but not MyD88-S is increased in PBMCs from patients with two different inflammatory lung diseases (acute respiratory distress syndrome and interstitial lung disease with an acute exacerbation) (11). The increase in MyD88-L without a corresponding alteration in MyD88-S could contribute to the inflammatory milieu in these patients. Likewise, MyD88-L but not MyD88-S levels were increased in stimulated PBMCs obtained from HIV-1–exposed seronegative individuals (59), which could be a contributing factor to the stronger immune response in these individuals. Several other studies have monitored MyD88-S without monitoring MyD88-L levels. For example, MyD88-S was increased in monocytes from septic patients and may contribute to the depressed TNFα production in these patient cells (25). MyD88-S levels were decreased in monocytes from patients with major depressive disorder, and the authors suggest that this contributes to chronic low-grade inflammation present in these patients (31). MyD88-S also is reported to be increased in stimulated T-cells obtained from patients with chronic obstructive pulmonary disease (28).
Despite the potential importance of MyD88 alternative splicing in inflammatory disease, there has been very limited investigation of the mechanisms regulating this negative feedback loop. We therefore established a controlled system to investigate LPS-induced production of MyD88-S in mouse macrophages. We found that LPS induced MyD88-S in a time- and dose-dependent fashion. In particular, we observed a change in MyD88 splicing that favored the anti-inflammatory isoform early (1–2 h) and late (>12 h) after LPS challenge. We speculate that the moderate increase in MyD88-S at early times plays a role in attenuating acute inflammation, whereas the increase in MyD88-S at later times may be important in resolving persistent inflammation. Our observation that a short pulse of LPS was sufficient to induce MyD88-S at these late time points suggests that LPS sets in motion an early chain of events that ultimately alters MyD88 splicing. This is consistent with published observations that TLR4 is internalized and down-regulated following initial LPS exposure (60–62).
Whereas it has been assumed that the LPS-induced increase in MyD88-S levels represents a change in MyD88 pre-mRNA splicing, this has not been definitively demonstrated previously, and the changes in MyD88 isoform levels could have involved differential stability of the two MyD88 isoforms. However, in the current study, we found that both MyD88 isoforms have similar half-lives, indicating that selective destruction of one isoform does not contribute to LPS-induced MyD88-S production. Interestingly, we did observe a moderate increase in the half-life of both MyD88 isoforms upon LPS exposure, as has been observed previously for many other LPS-induced genes (63–65).
The TLR signaling pathway mediates NF-κB activation in response to LPS exposure, and our current study likewise indicates that the TLR signaling pathway mediates the effect of LPS on MyD88-S production. We determined that activation of either the MyD88 or TRIF signaling adaptors was sufficient to increase MyD88-S production. It is interesting that TRIF signaling is sufficient to induce MyD88-S production. MyD88-S inhibits IRAK activation and thus inhibits MyD88-dependent signaling. poly(I:C) can induce IL-1β production; because MyD88 signaling mediates the response to IL-1β, it is possible that this increased MyD88-S prevents persistent inflammation mediated by paracrine loops. There is precedent for similar alternative splicing events inhibiting heterologous pathways. For example, a negatively acting isoform of MD-2 (MD-2s) is induced by LPS, IL-6, and IFNγ (14).
Activation and inhibition studies of NF-κB and the NF-κB–regulatory kinase IKK2 demonstrated that the NF-κB transcription factor was both necessary and sufficient to induce MyD88-S production. Our demonstration that IKK2 and NF-κB mediates LPS-induced MyD88-S production in macrophages is consistent with a prior study that indicated that IKK2 was required for H. influenza-induced production of MyD88-S in epithelial cells (26). There are several possibilities for how NF-κB could regulate MyD88 alternative pre-mRNA splicing. Pre-mRNA splicing can be functionally coupled to the activation state of the transcription machinery (43), raising the possibility that NF-κB could indirectly alter MyD88 splicing by affecting MyD88 transcription. To uncouple potential effects of NF-κB on MyD88 transcription from effects on splicing, we built a MyD88 minigene driven by the constitutive EF-1α promoter, which is not significantly affected by LPS exposure (66). The effects of LPS on expression of MyD88-L(mini) and MyD88-S(mini) driven by EF-1α faithfully recapitulate those of LPS on the endogenous MyD88 gene. This result suggests that the effects of LPS on MyD88 splicing are independent of any effect of LPS on MyD88 transcription. Moreover, inhibition of NF-κB abolished LPS-induced MyD88-S(mini) production, indicating that NF-κB is not regulating MyD88 transcription to affect MyD88 splicing. Rather, NF-κB is regulating MyD88 alternative splicing per se. We speculate that NF-κB does so through NF-κB–mediated changes in expression of a component(s) of the pre-mRNA splicing machinery. Precedent for NF-κB regulating the expression of splicing factors includes the NF-κB–mediated expression of CELF2 in T cells to regulate splicing of several genes (67). NF-κB–mediated transcription and subsequent translation of a splicing machinery component would account for the long temporal lag in the effects of LPS on MyD88 alternative pre-mRNA splicing.
Our minigene studies also suggest that the regulatory sequences that control MyD88 exon 2 inclusion, and thus production of MyD88-S, are relatively weak. Thus, MyD88 is poised to undergo signal-induced alternative splicing. It is intriguing to speculate that these intronic sequences have evolved a low threshold to induce MyD88 exon 2 skipping because of the functional significance of MyD88-S production in terminating persistent inflammation and preventing inflammatory disease.
In summary, our current study demonstrates that LPS induces MyD88-S production in murine macrophages both in vitro and in vivo, that it does so in a dose- and time-dependent manner, and that this induction involves a change in MyD88-S pre-mRNA splicing rather than differences in stability of the different MyD88 isoforms. Additionally, we demonstrate that the MyD88, TRIF, TRAF6, and NF-κB components of the TLR signaling pathway regulate MyD88-S production. Moreover, we find that NF-κB regulates MyD88 alternative pre-mRNA splicing directly rather than affecting MyD88 transcription and thus affecting splicing indirectly.
Experimental procedures
Cell line maintenance and transfection
The mouse macrophage cell line RAW264.7 was grown in Dulbecco's modified Eagle's medium (high-glucose with sodium pyruvate; Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific) and 10 units/ml penicillin-streptomycin (Thermo Fisher Scientific). Cells were maintained at 37 °C, 5% CO2. For transfection studies, 200,000 cells were plated in each well of a 24-well tissue culture plate. The next day, test plasmids were transfected (600 ng/well) using Fugene-HD (Roche Applied Science; 3.75 μl/well) as described previously (68). For transient transfections, cells were analyzed 48 h after transfection. To construct stable lines, 400 μg/ml G418 (Thermo Fisher Scientific) was added 2 days after transfection to select for neomycin resistance. Construction of stable cell lines expressing negative control protein CAT was described previously (69).
Source of plasmids
Plasmids expressing MyD88-gyrB and TRAF6-gyrB (35) were a gift from Dr. Hans Haecker. Dimerization and activation of MyD88 or TRAF6 was induced with 100 μm coumermycin A1 (Sigma). Plasmids expressing TRIF (70) or constitutively activated IKK2-S177E-S181E (36) were from Addgene.
Macrophage exposures
RAW264.7 cells were seeded into 24-well plates (200,000 cells/well). The next day, the medium was replaced with medium containing the indicated doses of either Escherichia coli O111:B4 LPS (List Biological Laboratories, Inc.), PAM3CSK4 (Invivogen), or poly(I:C) (Invivogen). Following the exposure, cells were lysed in RLT for RNA preparation. Additionally, in some studies, supernatant was collected for analysis of cytokine protein levels by ELISA. In the studies in which the cells were given a pulse of LPS, after a 2-h LPS exposure, the medium was replaced with fresh medium not containing LPS.
Mouse studies
Mouse studies were approved by the National Jewish Health Animal Care and Use Committee. 8–12-week-old mice of both sexes were obtained from Jackson Laboratories (Bar Harbor, ME). These mice included C57BL/6J (stock no. 000664), TLR4 knockout mice (stock no. 029015), and TRIF knockout mice (stock no. 005037). Mice were anesthetized using isofluorane, and either 20 μg of E. coli O55:B5 LPS (List Biological Laboratories) or PBS buffer control was instilled intratracheally using a modified feeding needle. At the indicated time points, mice were euthanized via intraperitoneal injection of Fatal Plus in accordance with American Veterinary Medical Association guidelines. Bronchoalveolar lavage was performed using PBS containing 5 mm EDTA. The cells were collected by centrifugation and were subsequently resuspended in PBS. Neutrophils were depleted using Ly6G columns (Miltenyi Biotech), and alveolar macrophages were recovered in the flow-through. Macrophages were >99% pure as verified by Wright-Giemsa–stained cytospins and were used for RNA preparation and subsequent qPCR analysis.
qPCR, RT-PCR, and ELISA analysis
RNA was purified from cell lysates using RNAeasy minikits (Qiagen). qPCR was performed on a QuantStudio 7 Flex (Applied Biosystems) using the Quantitect SYBR Green RT-PCR kit (Qiagen). Data were normalized relative to β-actin using the ΔΔCt method (except for the mRNA stability assays, which were normalized relative to 18S rRNA). Oligonucleotides used for qPCR analysis are listed in Table S1. MyD88-L was assessed using a forward primer that annealed to the exon 2–3 boundary and a reverse primer that annealed to exon 3; MyD88-S was assessed using a primer that annealed to the unique exon 1–3 boundary and a reverse primer that annealed exon 3. These MyD88 isoform–specific primers as well as primers used to analyze cytokines and housekeeping genes have been validated extensively by us previously (12, 33, 71). RT-PCR to analyze MyD88-L and MyD88-S simultaneously was performed by first reverse transcribing RNA with the iScript cDNA synthesis kit (Bio-Rad) and subsequently performing PCR using primers that bracket exon 2 (Table S1). RT-PCR products were visualized by agarose gel electrophoresis. ELISAs to measure TNFα in cell supernatants were performed using kits from R&D Biosystems according to the manufacturer's instructions.
NF-κB inhibitor assays
RAW264.7 cells were treated for 2 h with LPS in either the presence or absence of various NF-κB inhibitors. These inhibitors included 10 μm TPCA1 (Tocris Bioscience), 30 μm JSH23 (Sigma), and 10 μm MG132 (Calbiochem). After 2 h, the medium was replaced with medium containing NF-κB inhibitor but lacking LPS. After a further 22 h (24 h total), the indicated readouts were assessed. In other studies, the NF-κB studies were performed for only 1 h. Additionally, all studies were conducted in the additional presence of a 100 μm concentration of the apoptosis inhibitor Z-VAD-fmk (Selleck Chemicals) and an 80 μm concentration of the necrosis inhibitor necrostatin (Selleck Chemicals).
Measuring mRNA stability
RAW264.7 cells were seeded in 24-well plates (200,000 cells/well) and were allowed to adhere overnight. The cells were then either treated for 24 h with 200 ng/ml LPS or left untreated. 50 μg/ml actinomycin D (Gibco) was then added to the growth medium to inhibit RNA polymerase II–dependent RNA synthesis. Total RNAs were collected every 2 h for a total of 6 h after the addition of actinomycin D. The amounts of MyD88-L and MyD88-S were assayed by qPCR at each time point and normalized to 18S rRNAs. The half-lives of the mRNAs were determined using an online half-life calculator.
Construction of the MyD88 minigene
To construct a mouse MyD88 minigene driven by the EF-1α promoter, ∼2 kb of MyD88 genomic sequence extending from the 5′ end of exon 1 to the 3′ end of exon 3 were PCR-amplified using MyD88-outer primers (Table S1) and were cloned into pGem-T (Promega). To allow qPCR primers to distinguish between the minigene and the endogenous gene, an 8-bp mutation (AACGATAT → GGATCCCC) was then introduced into exon 3 using MyD88-mut primers (Table S1) and nested PCR fusion (72); this PCR product was digested with EcoRI and NotI and cloned into pCDA3.1(+) that had been similarly digested. The mutagenized MyD88 minigene was then amplified by PCR with MyD88-inner primers (Table S1) and cloned into the XhoI-NotI sites of pEF-Bos-TRIF-FLAG (Addgene). This replaces TRIF with the MyD88 minigene downstream of the EF-1α promoter. This plasmid was used for transient transfections. Additionally, the EF-1α MyD88 minigene cassette was subcloned into pCDNA3.1(+) using the MluI and NotI restriction sites to generate a plasmid where the EF-1α promoter drives minigene expression in a vector encoding neomycin resistance, allowing for G418-selected construction of stable cell lines. As evidence of the specificity of the minigene-specific qPCR primers, they did not amplify a product until 37 cycles on the PCR machine, which was weaker than amplification from the negative control water.
To generate the mutated MyD88 minigene with altered branch point and pY tract sequence, we applied the same nested PCR fusion strategy with MyD88-outer and MyD88-mut2 primers (Table S1). The PCR product was directly cloned into XhoI-NotI sites of pEF-Bos-TRIF-FLAG plasmid. All minigene constructs were verified by capillary sequencing.
Statistical analysis
All presented data represent a minimum of at least three independent biological replicates. qPCR data were normalized so that the expression of genes in the absence of treatment was averaged to 1. All data were analyzed in GraphPad Prism and are displayed using box and whiskers plots using the Turkey method. Statistical significance was assessed using unpaired t tests; significance was considered p < 0.05.
Data availability
All data are contained within the article.
Author contributions
F. F.-Y. L. and S. A. conceptualization; F. F.-Y. L. and S. A. formal analysis; F. F.-Y. L., K. D., C. H., J. M., W. J. J., and S. A. investigation; F. F.-Y. L. and S. A. writing-original draft; F. F.-Y. L., K. D., C. H., W. J. J., and S. A. writing-review and editing; W. J. J. and S. A. supervision; S. A. funding acquisition.
Supplementary Material
Acknowledgment
We thank H. Haecker for several plasmid reagents.
This study was supported by National Institutes of Health Grants R01ES025161, R01HL148335, R21AI132827, R35HL140039, and T32HL007085 and the Wendy Siegel Fund for Leukemia and Cancer Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
- TLR
- Toll-like receptor
- TPCA1
- 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide
- JSH23
- 4-methyl-N1-(3-phenyl-propyl)-benzene-1,2-diamine
- MG132
- carbobenzoxy-Leu-Leu-leucinal
- LPS
- lipopolysaccharide
- IL
- interleukin
- IRAK
- IL-1 receptor–associated kinase
- qPCR
- quantitative PCR
- CAT
- chloramphenicol acetyltransferase
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- EF-1α
- elongation factor 1α
- pY
- polypyrimidine
- IFN
- interferon.
References
- 1. Cook D. N., Pisetsky D. S., and Schwartz D. A. (2004) Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 5, 975–979 10.1038/ni1116 [DOI] [PubMed] [Google Scholar]
- 2. Grivennikov S. I., Greten F. R., and Karin M. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mohammad Hosseini A., Majidi J., Baradaran B., and Yousefi M. (2015) Toll-like receptors in the pathogenesis of autoimmune diseases. Adv. Pharm. Bull. 5, 605–614 10.15171/apb.2015.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zuo L., Lucas K., Fortuna C. A., Chuang C. C., and Best T. M. (2015) Molecular regulation of Toll-like receptors in asthma and COPD. Front. Physiol. 6, 312 10.3389/fphys.2015.00312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burke B., and Lewis C. E. (2002) The Macrophage, 2nd Ed., Oxford University Press, Oxford [Google Scholar]
- 6. Martinez F. O., and Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 6, 13 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinez F. O., Helming L., and Gordon S. (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483 10.1146/annurev.immunol.021908.132532 [DOI] [PubMed] [Google Scholar]
- 8. Medzhitov R., and Horng T. (2009) Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 10.1038/nri2634 [DOI] [PubMed] [Google Scholar]
- 9. Takeuchi O., and Akira S. (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 10. Wells C. A., Chalk A. M., Forrest A., Taylor D., Waddell N., Schroder K., Himes S. R., Faulkner G., Lo S., Kasukawa T., Kawaji H., Kai C., Kawai J., Katayama S., Carninci P., et al. (2006) Alternate transcription of the Toll-like receptor signaling cascade. Genome Biol. 7, R10 10.1186/gb-2006-7-2-r10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blumhagen R. Z., Hedin B. R., Malcolm K. C., Burnham E. L., Moss M., Abraham E., Huie T. J., Nick J. A., Fingerlin T. E., and Alper S. (2017) Alternative pre-mRNA splicing of Toll-like receptor signaling components in peripheral blood mononuclear cells from ARDS patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L930–L939 10.1152/ajplung.00247.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Arras L., and Alper S. (2013) Limiting of the innate immune response by SF3A-dependent control of MyD88 alternative mRNA splicing. PLoS Genet. 9, e1003855 10.1371/journal.pgen.1003855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng W., Shi M., Han M., Zhong J., Li Z., Li W., Hu Y., Yan L., Wang J., He Y., Tang H., Deubel V., Luo X., Ning Q., and Sun B. (2008) Negative regulation of virus-triggered IFN-beta signaling pathway by alternative splicing of TBK1. J. Biol. Chem. 283, 35590–35597 10.1074/jbc.M805775200 [DOI] [PubMed] [Google Scholar]
- 14. Gray P., Michelsen K. S., Sirois C. M., Lowe E., Shimada K., Crother T. R., Chen S., Brikos C., Bulut Y., Latz E., Underhill D., and Arditi M. (2010) Identification of a novel human MD-2 splice variant that negatively regulates lipopolysaccharide-induced TLR4 signaling. J. Immunol. 184, 6359–6366 10.4049/jimmunol.0903543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hardy M. P., and O'Neill L. A. (2004) The murine IRAK2 gene encodes four alternatively spliced isoforms, two of which are inhibitory. J. Biol. Chem. 279, 27699–27708 10.1074/jbc.M403068200 [DOI] [PubMed] [Google Scholar]
- 16. Iwami K. I., Matsuguchi T., Masuda A., Kikuchi T., Musikacharoen T., and Yoshikai Y. (2000) Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 165, 6682–6686 10.4049/jimmunol.165.12.6682 [DOI] [PubMed] [Google Scholar]
- 17. Janssens S., Burns K., Tschopp J., and Beyaert R. (2002) Regulation of interleukin-1- and lipopolysaccharide-induced NF-κB activation by alternative splicing of MyD88. Curr. Biol. 12, 467–471 10.1016/S0960-9822(02)00712-1 [DOI] [PubMed] [Google Scholar]
- 18. Koop A., Lepenies I., Braum O., Davarnia P., Scherer G., Fickenscher H., Kabelitz D., and Adam-Klages S. (2011) Novel splice variants of human IKKϵ negatively regulate IKKϵ-induced IRF3 and NF-κB activation. Eur. J. Immunol. 41, 224–234 10.1002/eji.201040814 [DOI] [PubMed] [Google Scholar]
- 19. Palsson-McDermott E. M., Doyle S. L., McGettrick A. F., Hardy M., Husebye H., Banahan K., Gong M., Golenbock D., Espevik T., and O'Neill L. A. (2009) TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat. Immunol. 10, 579–586 10.1038/ni.1727 [DOI] [PubMed] [Google Scholar]
- 20. Rao N., Nguyen S., Ngo K., and Fung-Leung W. P. (2005) A novel splice variant of interleukin-1 receptor (IL-1R)-associated kinase 1 plays a negative regulatory role in Toll/IL-1R-induced inflammatory signaling. Mol. Cell. Biol. 25, 6521–6532 10.1128/MCB.25.15.6521-6532.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenstiel P., Huse K., Till A., Hampe J., Hellmig S., Sina C., Billmann S., von Kampen O., Waetzig G. H., Platzer M., Seegert D., and Schreiber S. (2006) A short isoform of NOD2/CARD15, NOD2-S, is an endogenous inhibitor of NOD2/receptor-interacting protein kinase 2-induced signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 103, 3280–3285 10.1073/pnas.0505423103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burns K., Janssens S., Brissoni B., Olivos N., Beyaert R., and Tschopp J. (2003) Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 197, 263–268 10.1084/jem.20021790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janssens S., Burns K., Vercammen E., Tschopp J., and Beyaert R. (2003) MyD88S, a splice variant of MyD88, differentially modulates NF-κB- and AP-1-dependent gene expression. FEBS Lett. 548, 103–107 10.1016/S0014-5793(03)00747-6 [DOI] [PubMed] [Google Scholar]
- 24. Medzhitov R., Preston-Hurlburt P., Kopp E., Stadlen A., Chen C., Ghosh S., and Janeway C. A. Jr. (1998) MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2, 253–258 10.1016/S1097-2765(00)80136-7 [DOI] [PubMed] [Google Scholar]
- 25. Adib-Conquy M., Adrie C., Fitting C., Gattolliat O., Beyaert R., and Cavaillon J. M. (2006) Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit. Care Med. 34, 2377–2385 10.1097/01.CCM.0000233875.93866.88 [DOI] [PubMed] [Google Scholar]
- 26. Andrews C. S., Miyata M., Susuki-Miyata S., Lee B. C., Komatsu K., and Li J. D. (2015) Nontypeable Haemophilus influenzae-induced MyD88 short expression is regulated by positive IKKβ and CREB pathways and negative ERK1/2 pathway. PLoS One 10, e0144840 10.1371/journal.pone.0144840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoogerwerf J. J., de Vos A. F., van't Veer C., Bresser P., de Boer A., Tanck M. W., Draing C., van der Zee J. S., and van der Poll T. (2010) Priming of alveolar macrophages upon instillation of lipopolysaccharide in the human lung. Am. J. Respir. Cell Mol. Biol. 42, 349–356 10.1165/rcmb.2008-0362OC [DOI] [PubMed] [Google Scholar]
- 28. Knobloch J., Schild K., Jungck D., Urban K., Müller K., Schweda E. K., Rupp J., and Koch A. (2011) The T-helper cell type 1 immune response to Gram-negative bacterial infections is impaired in COPD. Am. J. Respir. Crit. Care Med. 183, 204–214 10.1164/rccm.201002-0199OC [DOI] [PubMed] [Google Scholar]
- 29. Liew C. W., Phuong T., Jones C. B., Evans S., Hoot J., Weedling K., Ingram D., Nganga S., and Kurt R. A. (2018) A computational approach to unraveling TLR signaling in murine mammary carcinoma. Comput. Biol. Med. 93, 56–65 10.1016/j.compbiomed.2017.12.013 [DOI] [PubMed] [Google Scholar]
- 30. Nelson A. M., Carew N. T., Smith S. M., and Milcarek C. (2018) RNA splicing in the transition from B cells to antibody-secreting cells: the influences of ELL2, small nuclear RNA, and endoplasmic reticulum stress. J. Immunol. 201, 3073–3083 10.4049/jimmunol.1800557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hung Y. Y. (2018) Antidepressants improve negative regulation of Toll-like receptor signaling in monocytes from patients with major depression. Neuroimmunomodulation 25, 42–48 10.1159/000489562 [DOI] [PubMed] [Google Scholar]
- 32. Wiersinga W. J., van't Veer C., van den Pangaart P. S., Dondorp A. M., Day N. P., Peacock S. J., and van der Poll T. (2009) Immunosuppression associated with interleukin-1R-associated-kinase-M upregulation predicts mortality in Gram-negative sepsis (melioidosis). Crit. Care Med. 37, 569–576 10.1097/CCM.0b013e318194b1bf [DOI] [PubMed] [Google Scholar]
- 33. O'Connor B. P., Danhorn T., De Arras L., Flatley B. R., Marcus R. A., Farias-Hesson E., Leach S. M., and Alper S. (2015) Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet. 11, e1004932 10.1371/journal.pgen.1004932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kawai T., and Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 35. Häcker H., Redecke V., Blagoev B., Kratchmarova I., Hsu L. C., Wang G. G., Kamps M. P., Raz E., Wagner H., Häcker G., Mann M., and Karin M. (2006) Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207 10.1038/nature04369 [DOI] [PubMed] [Google Scholar]
- 36. Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., Young D. B., Barbosa M., Mann M., Manning A., and Rao A. (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278, 860–866 10.1126/science.278.5339.860 [DOI] [PubMed] [Google Scholar]
- 37. Shin H. M., Kim M. H., Kim B. H., Jung S. H., Kim Y. S., Park H. J., Hong J. T., Min K. R., and Kim Y. (2004) Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-κB without affecting IκB degradation. FEBS Lett. 571, 50–54 10.1016/j.febslet.2004.06.056 [DOI] [PubMed] [Google Scholar]
- 38. Podolin P. L., Callahan J. F., Bolognese B. J., Li Y. H., Carlson K., Davis T. G., Mellor G. W., Evans C., and Roshak A. K. (2005) Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IκB kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell proliferation. J. Pharmacol. Exp. Ther. 312, 373–381 10.1124/jpet.104.074484 [DOI] [PubMed] [Google Scholar]
- 39. Ma Y., Temkin V., Liu H., and Pope R. M. (2005) NF-κB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J. Biol. Chem. 280, 41827–41834 10.1074/jbc.M510849200 [DOI] [PubMed] [Google Scholar]
- 40. He S., Liang Y., Shao F., and Wang X. (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059 10.1073/pnas.1116302108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gasparian A. V., Guryanova O. A., Chebotaev D. V., Shishkin A. A., Yemelyanov A. Y., and Budunova I. V. (2009) Targeting transcription factor NFκB: comparative analysis of proteasome and IKK inhibitors. Cell Cycle 8, 1559–1566 10.4161/cc.8.10.8415 [DOI] [PubMed] [Google Scholar]
- 42. Giulietti M., Piva F., D'Antonio M., D'Onorio De Meo P., Paoletti D., Castrignanò T., D'Erchia A. M., Picardi E., Zambelli F., Principato G., Pavesi G., and Pesole G. (2013) SpliceAid-F: a database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 41, D125–D131 10.1093/nar/gks997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saldi T., Cortazar M. A., Sheridan R. M., and Bentley D. L. (2016) Coupling of RNA polymerase II transcription elongation with pre-mRNA splicing. J. Mol. Biol. 428, 2623–2635 10.1016/j.jmb.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Arras L., Seng A., Lackford B., Keikhaee M. R., Bowerman B., Freedman J. H., Schwartz D. A., and Alper S. (2013) An evolutionarily conserved innate immunity protein interaction network. J. Biol. Chem. 288, 1967–1978 10.1074/jbc.M112.407205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pollyea D. A., Harris C., Rabe J. L., Hedin B. R., De Arras L., Katz S., Wheeler E., Bejar R., Walter M. J., Jordan C. T., Pietras E. M., and Alper S. (2019) Myelodysplastic syndrome-associated spliceosome gene mutations enhance innate immune signaling. Haematologica 104, e388–e392 10.3324/haematol.2018.214155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sperling J., Azubel M., and Sperling R. (2008) Structure and function of the pre-mRNA splicing machine. Structure 16, 1605–1615 10.1016/j.str.2008.08.011 [DOI] [PubMed] [Google Scholar]
- 47. Wahl M. C., Will C. L., and Lührmann R. (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 48. Rino J., and Carmo-Fonseca M. (2009) The spliceosome: a self-organized macromolecular machine in the nucleus? Trends Cell Biol. 19, 375–384 10.1016/j.tcb.2009.05.004 [DOI] [PubMed] [Google Scholar]
- 49. Collins L. J., Kurland C. G., Biggs P., and Penny D. (2009) The modern RNP world of eukaryotes. J. Hered. 100, 597–604 10.1093/jhered/esp064 [DOI] [PubMed] [Google Scholar]
- 50. Stamm S., Smith C., and Lührmann R. (eds) (2012) Alternative Pre-mRNA Splicing: Theory and Protocols, Wiley-Blackwell, Weinheim, Germany [Google Scholar]
- 51. Senapathy P., Shapiro M. B., and Harris N. L. (1990) Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 183, 252–278 10.1016/0076-6879(90)83018-5 [DOI] [PubMed] [Google Scholar]
- 52. Roscigno R. F., Weiner M., and Garcia-Blanco M. A. (1993) A mutational analysis of the polypyrimidine tract of introns: effects of sequence differences in pyrimidine tracts on splicing. J. Biol. Chem. 268, 11222–11229 [PubMed] [Google Scholar]
- 53. Taggart A. J., DeSimone A. M., Shih J. S., Filloux M. E., and Fairbrother W. G. (2012) Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol. 19, 719–721 10.1038/nsmb.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pineda J. M. B., and Bradley R. K. (2018) Most human introns are recognized via multiple and tissue-specific branchpoints. Genes Dev. 32, 577–591 10.1101/gad.312058.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frans G., Meyts I., Picard C., Puel A., Zhang S. Y., Moens L., Wuyts G., Van der Werff Ten Bosch J., Casanova J. L., and Bossuyt X. (2014) Addressing diagnostic challenges in primary immunodeficiencies: laboratory evaluation of Toll-like receptor- and NF-κB-mediated immune responses. Crit. Rev. Clin. Lab. Sci. 51, 112–123 10.3109/10408363.2014.881317 [DOI] [PubMed] [Google Scholar]
- 56. Ostuni R., Zanoni I., and Granucci F. (2010) Deciphering the complexity of Toll-like receptor signaling. Cell. Mol. Life Sci. 67, 4109–4134 10.1007/s00018-010-0464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tartey S., and Takeuchi O. (2017) Pathogen recognition and Toll-like receptor targeted therapeutics in innate immune cells. Int. Rev. Immunol. 36, 57–73 10.1080/08830185.2016.1261318 [DOI] [PubMed] [Google Scholar]
- 58. Kondo T., Kawai T., and Akira S. (2012) Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 33, 449–458 10.1016/j.it.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 59. Biasin M., Piacentini L., Lo Caputo S., Naddeo V., Pierotti P., Borelli M., Trabattoni D., Mazzotta F., Shearer G. M., and Clerici M. (2010) TLR activation pathways in HIV-1-exposed seronegative individuals. J. Immunol. 184, 2710–2717 10.4049/jimmunol.0902463 [DOI] [PubMed] [Google Scholar]
- 60. Rosadini C. V., and Kagan J. C. (2017) Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 44, 14–19 10.1016/j.coi.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schappe M. S., and Desai B. N. (2018) Measurement of TLR4 and CD14 receptor endocytosis using flow cytometry. Bio. Protoc. 8, e2926 10.21769/BioProtoc.2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Y., Chen T., Han C., He D., Liu H., An H., Cai Z., and Cao X. (2007) Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in macrophages by promoting lysosomal degradation of TLR4. Blood 110, 962–971 10.1182/blood-2007-01-066027 [DOI] [PubMed] [Google Scholar]
- 63. Hao S., and Baltimore D. (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288 10.1038/ni.1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kontoyiannis D., Pasparakis M., Pizarro T. T., Cominelli F., and Kollias G. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 10.1016/S1074-7613(00)80038-2 [DOI] [PubMed] [Google Scholar]
- 65. Rabani M., Levin J. Z., Fan L., Adiconis X., Raychowdhury R., Garber M., Gnirke A., Nusbaum C., Hacohen N., Friedman N., Amit I., and Regev A. (2011) Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29, 436–442 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lu J., Wu X., Hong M., Tobias P., and Han J. (2013) A potential suppressive effect of natural antisense IL-1β RNA on lipopolysaccharide-induced IL-1β expression. J. Immunol. 190, 6570–6578 10.4049/jimmunol.1102487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mallory M. J., Allon S. J., Qiu J., Gazzara M. R., Tapescu I., Martinez N. M., Fu X. D., and Lynch K. W. (2015) Induced transcription and stability of CELF2 mRNA drives widespread alternative splicing during T-cell signaling. Proc. Natl. Acad. Sci. U.S.A. 112, E2139–E2148 10.1073/pnas.1423695112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee F. F., and Alper S. (2018) Functional Genomics in Murine Macrophages. Methods Mol. Biol. 1809, 289–298 10.1007/978-1-4939-8570-8_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. De Arras L., Yang I. V., Lackford B., Riches D. W., Prekeris R., Freedman J. H., Schwartz D. A., and Alper S. (2012) Spatiotemporal inhibition of innate immunity signaling by the Tbc1d23 RAB-GAP. J. Immunol. 188, 2905–2913 10.4049/jimmunol.1102595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S. M., and Maniatis T. (2003) IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 71. De Arras L., Laws R., Leach S. M., Pontis K., Freedman J. H., Schwartz D. A., and Alper S. (2014) Comparative genomics RNAi screen identifies Eftud2 as a novel regulator of innate immunity. Genetics 197, 485–496 10.1534/genetics.113.160499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hobert O. (2002) PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. BioTechniques 32, 728–730 10.2144/02324bm01 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article.