

Abstract
T-type (Cav3) Ca2+ channels are important regulators of excitability and rhythmic activity of excitable cells. Among other voltage-gated Ca2+ channels, Cav3 channels are uniquely sensitive to oxidation and zinc. Using recombinant protein expression in HEK293 cells, patch clamp electrophysiology, site-directed mutagenesis, and homology modeling, we report here that modulation of Cav3.2 by redox agents and zinc is mediated by a unique extracellular module containing a high-affinity metal-binding site formed by the extracellular IS1–IS2 and IS3–IS4 loops of domain I and a cluster of extracellular cysteines in the IS1–IS2 loop. Patch clamp recording of recombinant Cav3.2 currents revealed that two cysteine-modifying agents, sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES) and N-ethylmaleimide, as well as a reactive oxygen species–producing neuropeptide, substance P (SP), inhibit Cav3.2 current to similar degrees and that this inhibition is reversed by a reducing agent and a zinc chelator. Pre-application of MTSES prevented further SP-mediated current inhibition. Substitution of the zinc-binding residue His191 in Cav3.2 reduced the channel's sensitivity to MTSES, and introduction of the corresponding histidine into Cav3.1 sensitized it to MTSES. Removal of extracellular cysteines from the IS1–IS2 loop of Cav3.2 reduced its sensitivity to MTSES and SP. We hypothesize that oxidative modification of IS1–IS2 loop cysteines induces allosteric changes in the zinc-binding site of Cav3.2 so that it becomes sensitive to ambient zinc.
Keywords: calcium channel, oxidation–reduction (redox), zinc, electrophysiology, homology modeling, neuropeptide
Introduction
T-type Ca2+ channels (CACNA1G, CACNA1H, and CACNA1I genes; Cav3.1, Cav3.2, and Cav3.3 channel α subunits, respectively) are a family of voltage-gated Ca2+ channels with very negative activation thresholds (less than −60 mV) and fast inactivation kinetics (1, 2). The channels are widely distributed in the central and peripheral nervous systems, heart, and vasculature as well as in several types of nonexcitable cells (3, 4). In the central nervous system, T-type channels are highly expressed in dendrites of thalamic and hippocampal neurons, where they amplify subthreshold postsynaptic potentials and facilitate the spread of depolarization to the cell body (5). A negative activation threshold and fast recovery from inactivation make T-type Ca2+ channels an important contributor to pacemaker activity in the thalamic, corticothalamic, and other rhythmically active neurons (6, 7). Thalamic T-type currents are enhanced in several rodent models of absence epilepsy; correspondingly, several gain-of-function mutations within CACNA1 genes are associated with human epilepsies, whereas T-type channel blockers have been shown to suppress seizures and are efficacious in treatment of absent seizures in humans (for a review, see Refs. 4, 8).
In the peripheral nervous system, T-type Ca2+ channels (and Cav3.2 in particular) are abundant in small-diameter, capsaicin-sensitive (presumably nociceptive) dorsal root ganglion neurons (9–12), as well as in two distinct types of low-threshold mechanoreceptors innervating skin hair follicles (13). The discovery of a relatively high abundance of T-type Ca2+ channels in nociceptors led to establishment of the prominent role of these channels in peripheral nociceptive transmission. Conditional deletion of Cav3.2 (13) or down-regulation in dorsal root ganglia using intrathecal injection of antisense oligonucleotides produced anti-nociceptive effects in rodent pain models of neuropathic and inflammatory pain (14, 15) and reduced the analgesic efficacy of T-type channel blockers (11). Conversely, multiple reports found T-type Ca2+ currents or Cav3.2 expression to be increased in chronic pain conditions, such as diabetic neuropathy (14, 16), peripheral nerve injury, or inflammation (12, 17–19).
Given their clear role in epilepsy and pain, regulation of T-type Ca2+ channel activity receives intense scrutiny. T-type Ca2+ channels are regulated by multiple phosphorylation (20), glycosylation (21, 22), and ubiquitination (12) mechanisms. In addition to these, the Cav3.2 T-type subunit possesses a unique regulatory mode that is targeted by several endogenous regulatory pathways: sensitivity to oxidation and zinc. Thus, Cav3.2 is uniquely sensitive to submicromolar concentrations of extracellular zinc (11, 23–25). The zinc-sensitive module is located extracellularly, involving interaction of the extracellular loops linking the IS1–IS2 and IS3–IS4 transmembrane regions of domain I (23). His191, which is absent in Cav3.1 and Cav3.3 subunits, is critical for the high sensitivity of Cav3.2 to zinc and nickel (11, 23, 26). In addition, T-type Ca2+ currents recorded in native nociceptive neurons and recombinant Cav3.2 currents are enhanced by reducing agents (DTT or l-cysteine) and inhibited by the oxidizing agent 5,5-dithio-bis(2-nitrobenzoic acid) and hydrogen peroxide (11, 27–30). Moreover, neuropeptide substance P, which induces generation of endogenous reactive oxygen species (ROS)3 (31), inhibits endogenous T-type Ca2+ currents in primary somatosensory neurons and recombinant Cav3.2 current via an oxidative mechanism (11). Intriguingly, redox modulation of Cav3.2 also depends on His191 (11, 24, 32). We have recently shown that oxidative modification of Cav3.2 channels (both recombinant and those endogenously expressed in sensory neurons) induced by substance P enhances channel sensitivity to Zn2+ to such an extent that is becomes tonically inhibited by trace amounts of ambient zinc (11). Cav3 channels contain a number of extra- and intracellular cysteines that can be oxidized or reduced, depending on the local redox environment, which, in turn, could affect the conformation of the zinc-binding site. In this study, we combined electrophysiology, site-directed mutagenesis, and computer modeling to delineate the molecular determinants of the unique redox sensitivity of Cav3.2.
Results
Cysteine-modifying reagents mimic the effect of redox modulation of Cav3.2 by substance P
Neuropeptide substance P (SP) induces production of reactive oxygen species in immune (33) and epithelial (34) cells and sensory neurons (31). In the former cell type, SP inhibits T-type Ca2+ current via oxidative modification of Cav3.2 (11). A mechanism of inhibition was proposed to be via enhancement of Zn2+ sensitivity of Cav3.2, whereby the oxidized channel is inhibited by nanomolar free Zn2+ present in the extracellular milieu (11). However, it is presently unknown how oxidation of Cav3.2 is translated into higher sensitivity to Zn2+. Cav3 channels have multiple extra- and intracellular cysteines accessible to redox modulation (Fig. 1A). To test whether cysteine modification is necessary for oxidative modulation of Cav3.2, we performed patch clamp experiments testing the effects of the cell-impermeable cysteine-modifying reagent sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES; 2 mm) and the cell-permeable cysteine-modifying reagent N-ethylmaleimide (NEM; 200 μm), on the recombinant Cav3.2 overexpressed in HEK293 cells together with the SP receptor NK1 (Fig. 1, B–J). MTSES (Fig. 1, B and C) and NEM (Fig. 1, D and E) inhibited the Cav3.2 current to similar levels, and inhibition was also similar in amplitude to that produced by the NK1-specific agonist [Sar9]-substance P (S9SP; 1 μm; Fig. 1, F–H). MTSES, NEM, and SP inhibited peak Cav3.2 current amplitude by 40.1% ± 3.5% (n = 9, p < 0.001), 36.1% ± 5.4% (n = 7, p < 0.001), and 40.1% ± 12.3% (n = 6, p < 0.05), respectively. The reducing agent DTT (1 mm), applied in the presence of MTSES, NEM, or SP (Fig. 1E), recovered most of the inhibitory action of the agents, although, in the case of NEM, recovery was incomplete (Fig. 1, F–H). Interestingly, when S9SP was applied after MTSES, it produced no further inhibition, suggestive of a common mechanism of action for both agents (Fig. 1, I and J). MTSES, NEM, or S9SP did not significantly affect the activation or inactivation kinetics of recombinant Cav3.2 (Table S1).
Figure 1.

Cysteine-modifying reagents and substance P inhibit recombinant Cav3.2 currents with a similar mechanism. A, schematic of the Cav3.2 channel; intra- and extracellular cysteines are indicated by spheres. Shown in red are cysteines mutated in this study. An orange square indicates zinc-binding His191. The purple area indicates the high-affinity zinc-binding site. B, example time course of the effects of 2 mm MTSES and 1 mm DTT) applied in the presence of MTSES) on the Ca2+ current recorded from HEK293 cells transiently overexpressing Cav3.2 and NK1 receptors using a perforated patch clamp. The selective T-type channel blocker Z944 (1 μm) was applied at the end of the experiment. Plotted are peak Ca2+ current amplitudes. Periods of drug application are indicated by vertical gray bars. The inset shows example current traces. C, summary of the effects recorded in the experiments exemplified in B. Individual data points are represented by colored circles. Paired data points from the same experiment are depicted in the same color. D, example time course of an experiment similar to that shown in A, but NEM (200 μm) was applied instead of MTSES. E, summary of the effects recorded in the experiments exemplified in D. F, example time course of an experiment similar to that shown in B, but the selective NK1 receptor agonist S9SP (1 μm) was applied instead of MTSES. G, summary of these experiments. H, comparison of the Cav3.2 current inhibition produced by MTSES and S9SP. I, Example time course of the effects of 1 μm S9SP applied after (and in the presence of) MTSES on the recombinant Cav3.2 current. J, summary of these experiments. In bar/scatter charts, asterisks denote a significant difference between the groups indicated by the line connectors. NS, not significant; *, p < 0.05; ***, p < 0.001 (paired t test or one-way ANOVA, as appropriate). Error bars represent mean ± S.E. The number of individual recordings is shown within the bars.
Ambient zinc and a high-affinity zinc-binding site are necessary for Cav3 channel sensitivity to MTSES
It has been shown previously that Cav3.2 with the H191Q mutation in the high-affinity zinc-binding site displayed much lower sensitivity to SP (11). To examine whether His191 is important for the effect of MTSES, we tested its effect on the Cav3.2 (H191Q) mutant overexpressed in HEK293 cells together with NK1 receptors (Fig. 2, A and D). MTSES had only a modest inhibitory effect on Cav3.2 (H191Q) (23.1% ± 5.6%, n = 6, p < 0.01); inhibition was significantly reduced compared with WT Cav3.2 (Fig. 2D, p < 0.05). Moreover, MTSES-induced inhibition of Cav3.2 (H191Q) was no longer recoverable with DTT (Fig. 2, A and D).
Figure 2.

MTSES-induced inhibition of Cav3 channels requires an intact high-affinity zinc-binding site. A–C, example time courses showing the effects of MTSES (2 mm), DTT (1 mm, applied in the presence of MTSES), and Z944 (1 μm) on the Ca2+ current recorded from HEK293 cells transiently overexpressing Cav3.2 H191Q (A), WT Cav3.1 (B), or Cav3.1 Q172H (C). Plotted are peak Ca2+ current amplitudes; periods of drug application are indicated by vertical gray bars. The inset shows example current traces. D, summary of the effects recorded in the experiments exemplified in A. Individual data points are represented by colored circles. Paired data points from the same experiment are depicted in the same color. Also shown is the dataset for MTSES-induced inhibition of the WT Cav3.2 (gray bar, circles) taken from Fig. 1C. E, summary of the effects recorded in the experiments exemplified in B and C. In bar charts, asterisks denote a significant difference between the groups indicated by the line connectors. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (paired or unpaired t test or one-way ANOVA, as appropriate). Error bars represent mean ± S.E. The number of individual recordings is shown within the bars.
Cav3.1 subunit has glutamine instead of histidine in the position equivalent to 191 in Cav3.2 (position 172 in Cav3.1) and, hence, is much less sensitive to zinc (11, 23). Thus, we tested the effect of MTSES on WT Cav3.1 and the Cav3.1 (Q172H) mutant. MTSES produced only very modest inhibition of 9.3% ± 2.8% in WT Cav3.1 (n = 5, p < 0.05; Fig. 2, B and E). Strikingly, the MTSES effect on Cav3.1 (Q172H) currents was much stronger (inhibition of 36.9% ± 2.8%, n = 6, p < 0.001; Fig. 2, C and E). MTSES inhibition of Cav3.1 (Q172H) was comparable with that of WT Cav3.2 (cf. Figs. 2, C and E, and 1, B and C). The results further reinforce the hypothesis that oxidative modulation of Cav3 channels depends on the high-affinity zinc-binding site.
To further probe the requirement of zinc for the effect of extracellular cysteine modification on Cav3 channel activity, we applied the zinc chelator N,N,N′, N′-tetrakis(2-pyridylmethy1) ethylenediamine (TPEN, 10 μm) before or after MTSES. The experiments presented in Fig. 3 demonstrate that TPEN could totally recover the inhibition induced by MTSES on WT Cav3.2; thus, 2 mm MTSES inhibited the peak Cav3.2 current to 61.2% ± 8.7% of baseline, and TPEN applied in the presence of MTSES recovered the amplitude to 102.4% ± 5.5% of the baseline (n = 6; Fig. 3, A and C). Consistent with previous results, MTSES inhibition of the Cav3.2 (H191Q) mutant was much smaller (inhibition to 83.4% ± 1.7% of baseline, n = 8; Fig. 3, B and C), and this small inhibition was also recovered by TPEN (Fig. 3, B and C). Application of TPEN on its own (Fig. 3, D–F) did not significantly affect the amplitude of WT Cav3.2 or the H191Q mutant (some run-up of the current amplitude was observed in some recordings, but the effect did not reach significance). Importantly, in the presence of TPEN, MTSES was no longer able to inhibit currents produced by either of the channels. Interestingly, under these conditions, we often observed a small and very transient inhibition by MTSES (as can be seen in the examples presented in Fig. 3, D and E) which then spontaneously recovered. Clearly, binding of zinc to its high-affinity extracellular binding site is necessary for the full inhibitory action of MTSES. We also tested the effect of TPEN on WT Cav3.1 and Cav3.1 (Q172H) mutant currents, and there were no strong effects (Fig. 3, G–I). In WT Cav3.1, TPEN produced small inhibition, which is perhaps a nonspecific effect. In the case of the Cav3.1 (Q172H) mutant, TPEN had no significant effect (Fig. 3, H and I). Importantly, when applied in the presence of TPEN, MTSES had no effect on WT Cav3.1 or the Cav3.1 (Q172H) mutant (Fig. 3, G–I). This result was in stark contrast to that shown in Fig. 2, C and E, where, in the absence of TPEN, MTSES produced strong inhibition of the Cav3.1 (Q172H) mutant, which has its high-affinity zinc-binding site reintroduced. Mutations H191Q (Cav3.2) and Q172H (Cav3.1) did not significantly affect the activation and inactivation kinetics and current densities of the respective channels (Table S2).
Figure 3.

MTSES-induced inhibition of Cav3 channels is reversed by zinc chelation. A and B, example time courses showing the effects of MTSES (2 mm), TPEN (10 μm, applied in the presence of MTSES), and Z944 (1 μm) on the Ca2+ currents recorded from HEK293 cells transiently overexpressing WT Cav3.2 (A) or Cav3.2 H191Q (B). C, summary of the effects recorded in experiments exemplified in A and B. Individual data points are represented by colored circles. Paired data points from the same experiment are depicted in the same color. D and E, experiments similar to those shown in A and B, but MTSES was applied after (and in the presence of) TPEN. F, summary of the effects recorded in the experiments exemplified in D and E. G and H, experiments similar to those shown in D and E, but WT Cav3.1 (G) and Cav3.1 Q172H (H) were investigated. I, summary of the effects recorded in the experiments exemplified in G and H. In bar/scatter charts, asterisks denote a significant difference between the groups indicated by the line connectors. NS, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001 (paired or unpaired t test or one-way ANOVA, as appropriate). Error bars represent mean ± S.E. The number of individual recordings is shown within the bars.
In combination, the results presented in Figs. 1–3 point to the following conclusions: cysteine-modifying agents and oxidative modification have similar inhibitory effects on Cav3 channels; these effects require zinc and depend on the high-affinity extracellular zinc-binding site formed by the extracellular loops of domain I, and because MTSES is cell-impermeable, the cysteines mediating the redox inhibition must be located in the extracellular regions of Cav3 channel proteins.
The cysteines in the IS1–IS2 extracellular loop of T-type Ca2+ channels are necessary and sufficient for redox-mediated inhibition of channel activity
Earlier studies defined critical residues of the high-affinity metal-binding site of Cav3.2, including an Asp189-Gly190-His191 motif in IS3–S4 and an additional Asp residue in IS2 (23), with His191 being a key residue absent in other Cav3 subunits (and any other voltage-gated Ca2+ channel α subunits). The high-resolution cryo-EM structure of human Cav3.1 has been solved recently (35). Because Cav3.1 is not highly sensitive to zinc or redox-mediated modulation as a result of absence of the critical histidine residue in its IS3–S4 loop, we obtained the Cav3.2 structure using homology modeling and analyzed the putative metal-binding site (Fig. 4 and Fig. S1). The “local quality” estimate of the model (Fig. 4A) shows that the scores of all regions except for a few loop regions are higher than 0.6, which indicates that the overall structure of the model is reliable (Fig. 1A).
Figure 4.

Homology modeling of the redox/metal-regulatory module in Cav3.2. The initial structure of the human Cav3.2 channel was obtained by homology modeling (see Fig. S1 for further details) based on the structure of human Cav3.1 (35). A, local quality estimate of the model obtained with QMEANBrane. B–D, the redox/metal-regulatory module shown at various rotation points (the protein is shown as ribbons, and key amino acids are indicated). E, electrostatic surface potential of the redox/metal-regulatory module (−50 kT/e to 50 kT/e, in a vacuum).
The putative metal binding site formed by extracellular IS1–IS2 and IS3–IS4 regions is depicted in Fig. 1, B–E. The electrostatic surface potential of the Cav3.2 model indicates that the lower potential in the extracellular region comprised of the IS1–IS2 and IS3–IS4 loops is favorable for metal binding (Fig. 1E). Moreover, the model suggests that the electrostatic surface potential near Glu127 and Glu137 is very low, and we suspect that Glu127 and Glu137 may also contribute to metal binding. Interestingly, four cysteines are located within or near this extracellular region: Cys114, Cys123, Cys128, and Cys133 (Fig. 4, B–D). Although side chains of several amino acids (e.g. methionines, arginines, and aromatic amino acids) can be modified by oxidizers, sulfhydryl groups of cysteines are by far the most susceptible to oxidation protein moieties (36). Cys123, Cys128, and Cys133 have been suggested to be important for modulation of T-type channels by lipoic acid (37) and nitric oxide (38). Thus, we hypothesized that oxidative modification of some or all of these residues may introduce allosteric changes to the metal-binding site, favoring channel inhibition. To test this hypothesis, we substituted these cysteines with alanines and tested the sensitivity of the mutants to MTSES (Fig. 5). All single mutants (C114A, C123A, C128A, and C133A) showed significantly reduced sensitivity to MTSES (Fig. 5, A–D and F); C123A displayed the least sensitivity (Fig. 5, D and F). Importantly, the quadruple mutant in which all of the abovementioned cysteines were substituted with alanines was largely insensitive to MTSES (Fig. 5, E and F). The quadruple mutant expressed very poorly, with current density reduced more than 10-fold compared with WT Cav3.2 (Table S2); hence, only four recordings were produced.
Figure 5.

Extracellular cysteines in the IS1–IS2 loop are necessary for MTSES-mediated inhibition of Cav3.2. A–E, example time courses showing the effects of MTSES (2 mm) and Z944 (1 μm) on the Ca2+ current recorded from HEK293 cells transiently overexpressing Cav3.2 C114A (A), Cav3.2 C123A (B), Cav3.2 C128A (C), and Cav3.2 C133A (D) or a quadruple mutant with Cys-to-Ala mutations at positions 114, 123, 128, and 133 (4xC-A, E). F, summary of the effects recorded in the experiments exemplified in A–E. Individual data points are represented by circles. Asterisks denote a significant difference between the groups indicated by the line connectors. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (one-way ANOVA). Error bars represent mean ± S.E. The number of individual recordings is given below each bar.
We also tested the sensitivity of individual cysteine mutants to SP (Fig. 6), and again, all four displayed much reduced sensitivity with inhibition by 10 μm S9SP in the range of 10%–20% (compared with 45.1% ± 11.6%, n = 7 in the WT Cav3.2). We also tested one intracellular cysteine in the intracellular IS2–IS3 linker, Cys165, as we reasoned it could allosterically influence the arrangement of IS1–IS2 and IS3–IS4 loops. However, the C165A mutant still displayed obvious inhibition by SP (26.5% ± 3.9%, n = 7), which was not significantly different from SP-induced inhibition of WT Cav3.2 (Fig. 6, A and B). None of the cysteine mutants used in this study displayed significantly different activation or inactivation kinetics. The current densities of single mutants also did not change significantly compared with WT Cav3.2. The only exception was the quadruple Cys-to-Ala mutant, which displayed much reduced current density (but the kinetics were not significantly affected; Table S2). Taken together, the results presented in Figs. 4–6 strongly suggest that oxidative modification of extracellular cysteines in the IS1–IS2 loop increases sensitivity of Cav3.2 to inhibition by trace amounts of extracellular zinc by inducing conformational changes within the high-affinity metal-binding site of the channel.
Figure 6.
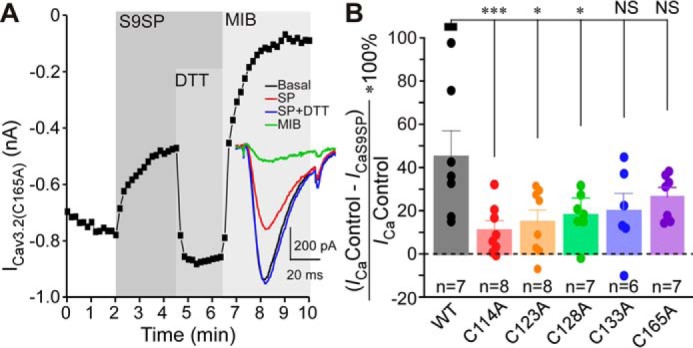
Extracellular cysteines in the IS1–IS2 loop are necessary for SP-mediated inhibition of Cav3.2. A, example time courses showing the effects of S9SP (1 μm), DTT (1 mm, applied in the presence of S9SP), and Z944 (1 μm) on the Ca2+ current recorded from HEK293 cells transiently overexpressing Cav3.2 C165A. B, summary of the effects recorded in experiments exemplified in A for HEK293 cells overexpressing Cav3.2 C114A, Cav3.2 C123A, Cav3.2 C128A, Cav3.2 C133A, or Cav3.2 C165A. Individual data points are represented by circles. Asterisks denote significant difference between the groups indicated by the line connectors. *, p < 0.05; ***, p < 0.001 (one-way ANOVA). Error bars represent mean ± S.E. The number of individual recordings is given below each bar.
Discussion
Here we report that modulation of T-type Ca2+ channels by redox agents and zinc is structurally coupled and depends on the presence of the high-affinity metal-binding site formed by the extracellular IS1–IS2 and IS3–IS4 loops of domain I, as well as on the group of extracellular cysteines present in the IS1–IS2 loop.
A mechanism for convergence of redox- and zinc-dependent modulation of T-type Ca2+ channels has been hypothesized in earlier studies, where it was proposed that His191 could be subject to a metal-catalyzed oxidation reaction (28, 36). Thus, binding of Zn2+ or Zn2+-independent metal-catalyzed oxidation of His191 could result in similar inhibition of channel activity (24, 28, 39). However, the data presented here suggest an alternative mechanism: oxidative modification of extracellular cysteines in the IS1–IS2 loop may allosterically increase the Cav3 channel inhibition induced by binding of zinc to His191. The following observations are in favor of the above hypothesis. The cysteine-modifying reagents MTSES and NEM as well as SP produced similar degrees of Cav3.2 inhibition, which was reversible with the reducing agent DTT (Fig. 1) or the zinc chelator TPEN (Fig. 3); pre-application of MTSES rendered SP ineffective to produce any further current inhibition (Fig. 1, I and J). Removal of His191 from Cav3.2 dramatically reduced inhibition of the channel by MTSES (Fig. 2) or SP (11). Introduction of the corresponding histidine into Cav3.1 induced its sensitivity to MTSES (Fig. 2) and SP (11). Removal of extracellular cysteines from the IS1–IS2 loop of Cav3.2 dramatically reduced the sensitivity of the channel to MTSES (Fig. 5) and SP (Fig. 6); the mutant with the cysteine-less IS1–IS2 loop was found to be resistant to MTSES (Fig. 5, E and F). Thus, we hypothesize that oxidative modification of extracellular cysteines in the IS1–IS2 loop of Cav3.2 induces allosteric changes in its zinc-binding site so that it becomes sensitive to ambient zinc. This effect is unique to Cav3.2 because other Cav3 subunits lack a critical histidine at positions equivalent to 191 in Cav3.2. Indeed, according to our earlier atomic absorption spectroscopy measurements, total zinc levels in nominally zinc-free laboratory solutions are in the range of 5–10 μm (11). A similar or higher range of zinc concentrations has been reported for human plasma (40). Concentrations of free Zn2+ (both in vitro and in vivo) are likely to be much lower compared with total zinc; our estimate suggested a low nanomolar range (11). This would still be sufficient to have a significant effect on channel activity because the high-affinity zinc-binding site in Cav3.2 has a nanomolar zinc affinity (11).
Activation of NK1 receptors has been shown to generate endogenous ROS production (31, 33, 41), which is a necessary step in NK1-mediated modulation of Cav3.2 (11). However, it is presently unclear how intracellularly generated ROS act upon an extracellular site within the Cav3.2 protein. One intriguing possibility is that, in response to endogenous ROS release, cells could release some redox-active molecules, such as thioredoxin (TRX). Indeed, TRX can be secreted (42). Moreover, it is known to inhibit Cav3.2 channels by interfering with their extracellular zinc-binding site (43). A mechanism of TRPC channel regulation through breakdown of the extracellular disulfide bond by secreted TRX has been reported (42), and it is tempting to hypothesize that a related mechanism could be at play in the case of NK1-mediated modulation of T-type Ca2+ channels. However, further investigation is required to decipher this intriguing signaling cascade.
The exact structural consequences of oxidation of IS1–IS2 cysteines have yet to be elucidated. These cysteines may be involved in disulfide bonds or oxidized to cysteine sulfinic (Cys-SO2H) or sulfonic (Cys-SO3H) acids (44). The Cryo-EM structure of Cav3.1 revealed a disulfide bond between Cys104 in IS1–IS2 and Cys889 on the IIS5–IIS6 pore loop, which is unique to Cav3 channels (35). This bond was hypothesized to be important for the unique redox sensitivity of T-type Ca2+ channels (35). Cys104 and Cys889 in Cav3.1 correspond to Cys123 and Cys939 in Cav3.2 (Fig. 1A), and our model of Cav3.2 (Fig. 4 and Fig. S1) also predicts a disulfide bond between these residues in Cav3.2, whereas other cysteines in the IS1–IS2 loop do not form disulfide bonds. Interestingly, the C123A Cav3.2 mutant was the least sensitive to MTSES among all cysteine mutants we tested (Fig. 5). Thus, perhaps this covalent bond, linking the high-affinity zinc-binding site to the pore region of the channel, is indeed important for coupling of zinc binding to channel activity and it could be promoted by oxidation. However, because other IS1–IS2 loop cysteines also affect channel sensitivity to MTSES and SP, the Cys123-Cys939 bond is likely not an exclusive determinant.
Oxidative modification of extracellular cysteines in the IS1–IS2 loop may produce conformational changes within the high-affinity zinc-binding site of the channel that either increases zinc affinity at the binding site or enhances coupling efficiency between zinc binding and channel inhibition. We believe that the latter is more likely to be the case because SP treatment strongly increased the efficacy of zinc-mediated inhibition of Cav3.2 while having no effect on IC50 (11). Resolving Cav3.2 structures in the presence and absence of zinc and at different states of extracellular cysteine oxidation will shed the light on the exact mechanism of coupling between these two modulatory mechanisms. Nevertheless, this study clearly demonstrates that the redox and zinc modulation of Cav3.2 is indeed structurally coupled and requires the metal-coordinating histidine in the IS3–IS4 loop and extracellular cysteines in the IS1–IS2 loop.
T-type Ca2+ channels are important regulators of excitability and rhythmic activity of excitable cells; the activity of these channels is regulated by multiple physiological signaling pathways, many of which act on the channel targeting its redox/zinc-sensitive module. Examples of these modulatory pathways include nitrous oxide (38, 45), carbon monoxide and thioredoxin (43), hydrogen sulfide (46), α-lipoic acid (37), as well as GABAB receptors (47) and substance P (11). Hence, elucidation of structural background of T-type channel modulation via the redox/zinc-sensitive module reported here sheds new light on the physiological regulation of these channels; moreover, it provides valuable insights into the development of future T-type channel modulators for treatment of excitability disorders, such as epilepsy and pain.
Experimental procedures
Cell culture, transfections, cDNA constructs, and chemicals
HEK293 cells were cultured in DMEM supplemented with GlutaMax I, 10% fetal calf serum, penicillin (50 units/ml), and streptomycin (50 μg/ml) in culture flasks in a humidified incubator (37 °C, 5% CO2). Cultures were passaged every 2 days (upon reaching 80%–90% confluency); 1 day before transfection, the cells were passaged on glass coverslips (1.3 × 1.3 cm). All cell culture reagents were purchased from Gibco-BRL unless otherwise stated. Human NK1 receptor (GenBank accession number AY462098) cDNA was purchased from the Missouri Science and Technology cDNA Resource Center. Cav3.1 (GenBank accession number AF027984), Cav3.2 (GenBank accession number AF051946), Cav3.1Q172H, and Cav3.2H191Q were kindly provided by Dr. E. Perez-Reyes (University of Virginia). Cav3.2 C114A, Cav3.2 C123A, Cav3.2 C128A, Cav3.2 C133A, Cav3.2 C165A, and Cav3.2 (C114A, C123A, C128A, and C133A) were custom-made by Sangon Biotech. HEK293 cells were transfected using FuGENE HD (Promega) according to the manufacturer's instructions. MTSES and Z944 were from Toronto Research Chemicals. All other chemicals were from Sigma.
Electrophysiology
Amphotericin B perforated patch clamp recordings were used to record Ca2+ currents from transfected HEK293 cells. Recordings were made using a Multiclamp 700B amplifier in combination with pCLAMP 10.4 software (Axon Instruments, Union City, CA) as described previously (11). Offline analysis was performed using Clampfit 10.4 (Molecular Devices). Voltage clamp recordings were sampled at 4 kHz and performed using the Amphotericin B perforated patch clamp method. The standard bath solution contained 150 mm tetraethylammonium-Cl, 2.5 mm CsCl, 2.5 mm CaCl2, 10 mm HEPES, 0.5 mm MgCl2, and 10 mm glucose (pH 7.4 adjusted with CsOH, 305–310 mosmol/kg). The solutions were applied to the bath chamber using the eight-channel gravity perfusion system VC3–8 (ALA Scientific Instruments) in combination with the local perfusion pencil (04-08-250, AutoMate Scientific; inner diameter, 250 μm) at ∼1 ml/min. The pipette solution contained 155 mm CsCl, 10 mm HEPES, 1 mm EGTA, and 4 mm MgCl2 supplemented with amphotericin B (250 μg/ml), pH 7.4-adjusted with CsOH. Patch electrodes were pulled with a horizontal micropipette puller (P-97, Sutter Instruments) and fire-polished. The access resistance was typically within 6–10 megaohm. Cav3 currents were measured by 50-ms square voltage pulses to −40 mV from a holding potential of −90 mV. Series resistance was compensated online by 50%–80%. All recordings were performed at room temperature (∼22 °C).
Homology modeling
The structural model of the Cav3.2 channel was constructed using the homology modeling server SWISSMODEL (48) and the Cav3.1 channel structure (PDB code 6KZO) (35) as a template. The sequence identity of transmembrane regions of CaV3.1 and CaV3.2 reached 84.27%. The resolution of the Cav3.1 cryo-EM structure is 3.3 Å. The overall root mean square deviation value of atomic positions between the Cav3.2 model and Cav3.1 cryo-EM structure is 0.26 Å (Fig. 1S). QMEANBrane was used for reliable local quality estimation of membrane protein models (49). For evaluation of the overall protein structure, the validation server (SAVES v5.0, Institute of Molecular Biology, University of California; RRID: SCR_018219) was used. All molecular visualization and structural diagrams were made using Open-Source PyMOL (RRID:SCR_000305).
Statistics
All mean data are given as mean ± S.E. Differences between groups were assessed by Student's t test (paired or unpaired, as appropriate) or one-way ANOVA with Dunnett's post hoc test. The differences were considered significant at p ≤ 0.05. Statistical analyses were performed using Origin 8.6 (OriginLab Corp., Northampton, CA).
Data availability
All data are available in the main text or the supporting information.
Author contributions
D. H., C. P., H. Z., and N. G. conceptualization; D. H., S. S., C. L., X. Z., and H. A. investigation; D. H., S. S., C. L., X. Z., H. A., and N. G. methodology; D. H. and N. G. writing-original draft; D. H. and N. G. project administration; S. S. and N. G. visualization; X. D. and N. G. resources; X. D., H. Z., and N. G. supervision; X. D., H. Z., and N. G. funding acquisition; N. G. validation.
Supplementary Material
This work was supported by Biotechnology and Biological Sciences Research Council Grants BB/R003068/1 and BB/R02104X/1 (to N. G.), the 100 Foreign Experts of Hebei Province Award (to N. G.), National Natural Science Foundation of China Grants 81701113 (to D. H.) and 81871075 and 91732108 (to H. Z.), and Excellent Youth Fund of Natural Science Foundation of Hebei Grant C2018206222 and Hebei Province Department of Education Grant BJ2017004 (to D. H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1 and Tables S1 and S2.
- ROS
- reactive oxygen species
- SP
- substance P
- MTSES
- sodium (2-sulfonatoethyl) methanethiosulfonate
- NEM
- N-ethylmaleimide
- S9SP
- [Sar9]-substance P
- TPEN
- N,N,N′,N′-tetrakis(2-pyridylmethy1) ethylenediamine
- TRX
- thioredoxin
- cDNA
- complementary DNA
- ANOVA
- analysis of variance
- Z944
- N-[[1-[2-(tert-butylamino)-2-oxoethyl]piperidin-4-yl]methyl]-3-chloro-5-fluorobenzamide.
References
- 1. Fox A. P., Nowycky M. C., and Tsien R. W. (1987) Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J. Physiol. 394, 149–172 10.1113/jphysiol.1987.sp016864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nowycky M. C., Fox A. P., and Tsien R. W. (1985) Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–443 10.1038/316440a0 [DOI] [PubMed] [Google Scholar]
- 3. Zamponi G. W., Striessnig J., Koschak A., and Dolphin A. C. (2015) The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67, 821–870 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weiss N., and Zamponi G. W. (2020) Genetic T-type calcium channelopathies. J. Med. Genet 57, 1–10 10.1136/jmedgenet-2019-106163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crandall S. R., Govindaiah G., and Cox C. L. (2010) Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J. Neurosci. 30, 15419–15429 10.1523/JNEUROSCI.3636-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huguenard J. R., and Prince D. A. (1994) Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J. Neurosci. 14, 5485–5502 10.1523/JNEUROSCI.14-09-05485.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beurrier C., Congar P., Bioulac B., and Hammond C. (1999) Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J. Neurosci. 19, 599–609 10.1523/JNEUROSCI.19-02-00599.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zamponi G. W. (2016) Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 15, 19–34 10.1038/nrd.2015.5 [DOI] [PubMed] [Google Scholar]
- 9. Jevtovic-Todorovic V., and Todorovic S. M. (2006) The role of peripheral T-type calcium channels in pain transmission. Cell Calcium 40, 197–203 10.1016/j.ceca.2006.04.024 [DOI] [PubMed] [Google Scholar]
- 10. Rose K. E., Lunardi N., Boscolo A., Dong X., Erisir A., Jevtovic-Todorovic V., and Todorovic S. M. (2013) Immunohistological demonstration of CaV3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250, 263–274 10.1016/j.neuroscience.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang D., Huang S., Gao H., Liu Y., Qi J., Chen P., Wang C., Scragg J. L., Vakurov A., Peers C., Du X., Zhang H., and Gamper N. (2016) Redox-dependent modulation of T-type Ca2+ channels in sensory neurons contributes to acute anti-nociceptive effect of substance P. Antioxid. Redox Signal. 25, 233–251 10.1089/ars.2015.6560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. García-Caballero A., Gadotti V. M., Stemkowski P., Weiss N., Souza I. A., Hodgkinson V., Bladen C., Chen L., Hamid J., Pizzoccaro A., Deage M., François A., Bourinet E., and Zamponi G. W. (2014) The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83, 1144–1158 10.1016/j.neuron.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 13. François A., Schüetter N., Laffray S., Sanguesa J., Pizzoccaro A., Dubel S., Mantilleri A., Nargeot J., Noël J., Wood J. N., Moqrich A., Pongs O., and Bourinet E. (2015) The low-threshold calcium channel Cav3.2 determines low-threshold mechanoreceptor function. Cell Rep. 10, 370–382 10.1016/j.celrep.2014.12.042 [DOI] [PubMed] [Google Scholar]
- 14. Messinger R. B., Naik A. K., Jagodic M. M., Nelson M. T., Lee W. Y., Choe W. J., Orestes P., Latham J. R., Todorovic S. M., and Jevtovic-Todorovic V. (2009) In vivo silencing of the CaV3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 145, 184–195 10.1016/j.pain.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bourinet E., Alloui A., Monteil A., Barrère C., Couette B., Poirot O., Pages A., McRory J., Snutch T. P., Eschalier A., and Nargeot J. (2005) Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 24, 315–324 10.1038/sj.emboj.7600515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jagodic M. M., Pathirathna S., Nelson M. T., Mancuso S., Joksovic P. M., Rosenberg E. R., Bayliss D. A., Jevtovic-Todorovic V., and Todorovic S. M. (2007) Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J. Neurosci. 27, 3305–3316 10.1523/JNEUROSCI.4866-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jagodic M. M., Pathirathna S., Joksovic P. M., Lee W., Nelson M. T., Naik A. K., Su P., Jevtovic-Todorovic V., and Todorovic S. M. (2008) Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J. Neurophysiol. 99, 3151–3156 10.1152/jn.01031.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marger F., Gelot A., Alloui A., Matricon J., Ferrer J. F., Barrère C., Pizzoccaro A., Muller E., Nargeot J., Snutch T. P., Eschalier A., Bourinet E., and Ardid D. (2011) T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 11268–11273 10.1073/pnas.1100869108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang D., Liang C., Zhang F., Men H., Du X., Gamper N., and Zhang H. (2016) Inflammatory mediator bradykinin increases population of sensory neurons expressing functional T-type Ca2+ channels. Biochem. Biophys. Res. Commun. 473, 396–402 10.1016/j.bbrc.2016.02.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blesneac I., Chemin J., Bidaud I., Huc-Brandt S., Vandermoere F., and Lory P. (2015) Phosphorylation of the Cav3.2 T-type calcium channel directly regulates its gating properties. Proc. Natl. Acad. Sci. U.S.A. 112, 13705–13710 10.1073/pnas.1511740112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiss N., Black S. A., Bladen C., Chen L., and Zamponi G. W. (2013) Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 465, 1159–1170 10.1007/s00424-013-1259-3 [DOI] [PubMed] [Google Scholar]
- 22. Orestes P., Osuru H. P., McIntire W. E., Jacus M. O., Salajegheh R., Jagodic M. M., Choe W., Lee J., Lee S. S., Rose K. E., Poiro N., Digruccio M. R., Krishnan K., Covey D. F., Lee J. H., et al. (2013) Reversal of neuropathic pain in diabetes by targeting glycosylation of CaV3.2 T-type calcium channels. Diabetes 62, 3828–3838 10.2337/db13-0813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang H. W., Vitko I., Lee S. S., Perez-Reyes E., and Lee J. H. (2010) Structural determinants of the high affinity extracellular zinc binding site on Cav3.2 T-type calcium channels. J. Biol. Chem. 285, 3271–3281 10.1074/jbc.M109.067660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elies J., Scragg J. L., Boyle J. P., Gamper N., and Peers C. (2016) Regulation of the T-type Ca channel Cav3.2 by hydrogen sulfide: emerging controversies concerning the role of H S in nociception. J. Physiol. 594, 4119–4129 10.1113/JP270963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Traboulsie A., Chemin J., Chevalier M., Quignard J. F., Nargeot J., and Lory P. (2007) Subunit-specific modulation of T-type calcium channels by zinc. J. Physiol. 578, 159–171 10.1113/jphysiol.2006.114496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang H. W., Park J. Y., Jeong S. W., Kim J. A., Moon H. J., Perez-Reyes E., and Lee J. H. (2006) A molecular determinant of nickel inhibition in Cav3.2 T-type calcium channels. J. Biol. Chem. 281, 4823–4830 10.1074/jbc.M510197200 [DOI] [PubMed] [Google Scholar]
- 27. Nelson M. T., Joksovic P. M., Perez-Reyes E., and Todorovic S. M. (2005) The endogenous redox agent l-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J. Neurosci. 25, 8766–8775 10.1523/JNEUROSCI.2527-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nelson M. T., Joksovic P. M., Su P., Kang H. W., Van Deusen A., Baumgart J. P., David L. S., Snutch T. P., Barrett P. Q., Lee J. H., Zorumski C. F., Perez-Reyes E., and Todorovic S. M. (2007) Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J. Neurosci. 27, 12577–12583 10.1523/JNEUROSCI.2206-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson M. T., Woo J., Kang H. W., Vitko I., Barrett P. Q., Perez-Reyes E., Lee J. H., Shin H. S., and Todorovic S. M. (2007) Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J. Neurosci. 27, 8250–8260 10.1523/JNEUROSCI.1800-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Todorovic S. M., Jevtovic-Todorovic V., Meyenburg A., Mennerick S., Perez-Reyes E., Romano C., Olney J. W., and Zorumski C. F. (2001) Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron 31, 75–85 10.1016/S0896-6273(01)00338-5 [DOI] [PubMed] [Google Scholar]
- 31. Linley J. E., Ooi L., Pettinger L., Kirton H., Boyle J. P., Peers C., and Gamper N. (2012) Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc. Natl. Acad. Sci. U.S.A. 109, E1578–E1586 10.1073/pnas.1201544109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Voisin T., Bourinet E., and Lory P. (2016) Genetic alteration of the metal/redox modulation of Cav3.2 T-type calcium channel reveals its role in neuronal excitability. J. Physiol. 594, 3561–3574 10.1113/JP271925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanabe T., Otani H., Bao L., Mikami Y., Yasukura T., Ninomiya T., Ogawa R., and Inagaki C. (1996) Intracellular signaling pathway of substance P-induced superoxide production in human neutrophils. Eur. J. Pharmacol 299, 187–195 10.1016/0014-2999(95)00816-0 [DOI] [PubMed] [Google Scholar]
- 34. Springer J., Pleimes D., Scholz F. R., and Fischer A. (2005) Substance P mediates AP-1 induction in A549 cells via reactive oxygen species. Regul. Pept. 124, 99–103 10.1016/j.regpep.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 35. Zhao Y., Huang G., Wu Q., Wu K., Li R., Lei J., Pan X., and Yan N. (2019) Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature 576, 492–497 10.1038/s41586-019-1801-3 [DOI] [PubMed] [Google Scholar]
- 36. Stadtman E. R. (2001) Protein oxidation in aging and age-related diseases. Ann. N.Y. Acad. Sci. 928, 22–38 [DOI] [PubMed] [Google Scholar]
- 37. Lee W. Y., Orestes P., Latham J., Naik A. K., Nelson M. T., Vitko I., Perez-Reyes E., Jevtovic-Todorovic V., and Todorovic S. M. (2009) Molecular mechanisms of lipoic acid modulation of T-type calcium channels in pain pathway. J. Neurosci. 29, 9500–9509 10.1523/JNEUROSCI.5803-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee J., Nelson M. T., Rose K. E., and Todorovic S. M. (2013) Redox mechanism of S-nitrosothiol modulation of neuronal CaV3.2 T-type calcium channels. Mol. Neurobiol. 48, 274–280 10.1007/s12035-013-8493-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gamper N., and Ooi L. (2015) Redox and nitric oxide-mediated regulation of sensory neuron ion channel function. Antioxid. Redox Signal. 22, 486–504 10.1089/ars.2014.5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moran V. H., Stammers A. L., Medina M. W., Patel S., Dykes F., Souverein O. W., Dullemeijer C., Pérez-Rodrigo C., Serra-Majem L., Nissensohn M., and Lowe N. M. (2012) The relationship between zinc intake and serum/plasma zinc concentration in children: a systematic review and dose-response meta-analysis. Nutrients 4, 841–858 10.3390/nu4080841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Springer J., Groneberg D. A., Dinh Q. T., Quarcoo D., Hamelmann E., Braun-Dullaeus R. C., Geppetti P., Anker S. D., and Fischer A. (2007) Neurokinin-1 receptor activation induces reactive oxygen species and epithelial damage in allergic airway inflammation. Clin. Exp. Allergy 37, 1788–1797 10.1111/j.1365-2222.2007.02851.x [DOI] [PubMed] [Google Scholar]
- 42. Xu S. Z., Sukumar P., Zeng F., Li J., Jairaman A., English A., Naylor J., Ciurtin C., Majeed Y., Milligan C. J., Bahnasi Y. M., Al-Shawaf E., Porter K. E., Jiang L. H., Emery P., Sivaprasadarao A., and Beech D. J. (2008) TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72 10.1038/nature06414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boycott H. E., Dallas M. L., Elies J., Pettinger L., Boyle J. P., Scragg J. L., Gamper N., and Peers C. (2013) Carbon monoxide inhibition of Cav3.2 T-type Ca2+ channels reveals tonic modulation by thioredoxin. FASEB J. 27, 3395–3407 10.1096/fj.13-227249 [DOI] [PubMed] [Google Scholar]
- 44. Paget M. S., and Buttner M. J. (2003) Thiol-based regulatory switches. Annu. Rev. Genet. 37, 91–121 10.1146/annurev.genet.37.110801.142538 [DOI] [PubMed] [Google Scholar]
- 45. Orestes P., Bojadzic D., Lee J., Leach E., Salajegheh R., Digruccio M. R., Nelson M. T., and Todorovic S. M. (2011) Free radical signalling underlies inhibition of CaV3.2 T-type calcium channels by nitrous oxide in the pain pathway. J. Physiol. 589, 135–148 10.1113/jphysiol.2010.196220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elies J., Scragg J. L., Huang S., Dallas M. L., Huang D., MacDougall D., Boyle J. P., Gamper N., and Peers C. (2014) Hydrogen sulfide inhibits Cav3.2 T-type Ca2+ channels. FASEB J. 28, 5376–5387 10.1096/fj.14-257113 [DOI] [PubMed] [Google Scholar]
- 47. Huang D., Huang S., Peers C., Du X., Zhang H., and Gamper N. (2015) GABAB receptors inhibit low-voltage activated and high-voltage activated Ca2+ channels in sensory neurons via distinct mechanisms. Biochem. Biophys. Res. Commun. 465, 188–193 10.1016/j.bbrc.2015.07.137 [DOI] [PubMed] [Google Scholar]
- 48. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., and Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benkert P., Biasini M., and Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350 10.1093/bioinformatics/btq662 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supporting information.