

Abstract
The ribosome biogenesis factor Las1 is an essential endoribonuclease that is well-conserved across eukaryotes and a newly established member of the higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domain-containing nuclease family. HEPN nucleases participate in diverse RNA cleavage pathways and share a short HEPN nuclease motif (RφXXXH) important for RNA cleavage. Most HEPN nucleases participate in stress-activated RNA cleavage pathways; Las1 plays a fundamental role in processing pre-rRNA. Underscoring the significance of Las1 function in the cell, mutations in the human LAS1L (LAS1-like) gene have been associated with neurological dysfunction. Two juxtaposed HEPN nuclease motifs create Las1's composite nuclease active site, but the roles of the individual HEPN motif residues are poorly defined. Here using a combination of in vivo experiments in Saccharomyces cerevisiae and in vitro assays, we show that both HEPN nuclease motifs are required for Las1 nuclease activity and fidelity. Through in-depth sequence analysis and systematic mutagenesis, we determined the consensus HEPN motif in the Las1 subfamily and uncovered its canonical and specialized elements. Using reconstituted Las1 HEPN-HEPN′ chimeras, we defined the molecular requirements for RNA cleavage. Intriguingly, both copies of the Las1 HEPN motif were important for nuclease function, revealing that both HEPN motifs participate in coordinating the RNA within the Las1 active site. We also established that conformational flexibility of the two HEPN domains is important for proper nuclease function. The results of our work reveal critical information about how dual HEPN domains come together to drive Las1-mediated RNA cleavage.
Keywords: endoribonuclease, ribosome assembly, RNA degradation, RNA processing, ribosomal RNA processing (rRNA processing), higher eukaryotes and prokaryotes nucleotide-binding (HEPN), Las1, Las1L, nuclease specificity, RNA cleavage, ribosome biogenesis factor, LAS1L, RNA processing
Introduction
Endoribonucleases play critical roles in the processing, maturation, and destruction of diverse RNAs by catalyzing the hydrolysis of phosphodiester bonds (1). The HEPN (higher eukaryotes and prokaryotes nucleotide-binding) superfamily is an emerging group of endoribonucleases spanning across all walks of life. The HEPN domain is characterized by a small α-helical domain originally suggested to be important for nucleotide binding (2). Subsequent bioinformatic analysis transformed the field through the widespread identification of HEPN-containing proteins and the classification of a catalytic subset that encode for a short, but defined, RφXXXH (where φ is often N, D, or H, and X is any amino acid that can vary from three to five residues) endoribonuclease motif (3) (Fig. 1A). The expanding list of HEPN nucleases highlights their profound role in numerous biological processes, most notably host-defense and stress response pathways. For example, the eukaryotic HEPN nuclease Ire1 triggers the unfolded protein response and the bacterial HEPN nuclease SO_3166 is the toxic component of the dueling type II toxin-antitoxin system (4, 5). In contrast to these defense systems, the HEPN nuclease Las1 ensures the translational capacity of the cell through its role in ribosome assembly (6). Furthermore, the bacterial Cas13 CRISPR effectors represent a set of programmable HEPN-containing nucleases that are gaining broad attention for their potential therapeutic applications (7–11). Despite the strong prevalence of the HEPN domain in critical RNA-targeting nucleases, the molecular basis for how HEPN nucleases catalyze RNA cleavage has remained elusive.
Figure 1.

Members of the HEPN superfamily encode a canonical RφXXXH motif responsible for RNA cleavage. A, amino acid sequence alignment of the HEPN motif from several different HEPN nucleases. The HEPN canonical motif RφXXXH (where φ is commonly N, H, or D and X is any residue) is responsible for RNA cleavage and highlighted in purple. Abbreviations are as follows: C. thermophilum (Ct), S. cerevisiae (Sc), Sus Scrofa (Ss), Leptotrichia buccalis (Lb), Eubacterium siraeum (Es), Ruminococcus species (Rs), Staphylococcus epidermidis (Se), Pyrococcus furiosus (Pf), and Shewanella oneidensis (So). B, diagram of ITS2 pre-rRNA processing by RNase PNK. RNase PNK is composed of the Las1 RNase and the Grc3 PNK. Las1 cleaves the C2 site leaving a 2′,3′-cyclic phosphate (cP) and a 5′-hydroxyl (OH) (14). Subsequently, Grc3 phosphorylates (P) the 5′-hydroxyl end marking the ITS2 for decay by a series of exoribonucleases. C, orthogonal views of C. thermophilum RNase PNK model (PDB ID 6OF3). Grc3 promoters are shown in gray and Las1 protomers are shown in orange and yellow. Las1 RφXXXH motifs are highlighted in purple and ATP-γS are shown as sticks. Las1 RNase and Grc3 PNK active sites are boxed. D, cartoon schematic of metal-independent RNA cleavage by members of the HEPN superfamily. HEPN members dimerize (orange/yellow) to form an active nuclease, which cleaves the phosphodiester backbone through an unclear mechanism resulting in a 2′,3′-cyclic phosphate and 5′-hydroxyl RNA ends.
The Las1 HEPN nuclease harbors unique characteristics that distinguish it from other members of the HEPN superfamily. Defects in Las1 (Las1L in humans) are linked to motor neuron diseases and X-linked intellectual disability (12, 13); however, its molecular function in ribosome production was only elucidated following its recent classification as a HEPN nuclease (3, 14). Unlike most HEPN nucleases that are activated following cellular stress (3), Las1 is essential for cell growth and proliferation (6, 15). Las1 plays a fundamental role in processing precursor rRNA (pre-rRNA)4 (6, 14–17). Ribosome production involves a complex and highly coordinated pre-rRNA processing cascade tasked with removing four spacer sequences from the pre-rRNA (14, 18–21). Removal of the ITS2 (internal transcribed spacer 2), which lies between the 5.8S and 25S rRNAs, is initiated by Las1 pre-rRNA cleavage at the defined C2 site (Fig. 1B) (14). Following endoribonucleolytic cleavage, the ITS2 is phosphorylated by the Grc3 RNA kinase, which in turn triggers the recruitment of 5′ and 3′ exoribonucleases that sequentially degrade the ITS2 spacer (14, 16, 22). Further distinguishing Las1 from other HEPN nucleases is Las1's dependence on its binding partner, the Grc3 polynucleotide kinase for nuclease activation (23). Although no other HEPN nuclease is known to require an auxiliary protein for HEPN activation, Las1 and Grc3 are reliant on one another for protein stability and enzyme activation (15, 23, 24). Together the ribonuclease (RNase) Las1 and polynucleotide kinase (PNK) Grc3 assemble into a tetrameric complex, known as RNase PNK. Through this higher-order assembly, RNase PNK coordinates its dual enzymatic functions through a poorly understood mechanism of molecular crosstalk that ensures efficient processing of the pre-rRNA (23–25).
Las1 adopts a canonical HEPN active site, suggesting it shares a common mechanism for RNA cleavage with other HEPN nucleases. We recently solved a series of cryo-EM structures of RNase PNK, which unveiled its butterfly-like architecture (26). Two Las1 protomers homodimerize at the axis of symmetry where the HEPN domains form the “body” of the butterfly, and a Grc3 protomer makes up each “wing” and holds the HEPN domains together (Fig. 1C) (26). This is reminiscent to all characterized HEPN nucleases, which require dimerization of two HEPN domains to activate nuclease activity (4, 5, 7, 26–28). Furthermore, the composite Las1 HEPN active site resembles that of other HEPN nucleases, which are formed by the juxtaposition of two RφXXXH HEPN motifs (Fig. 1C). As with all members of this superfamily, the Las1 HEPN nuclease catalyzes metal-independent RNA cleavage, resulting in the production of a terminal 2′,3′-cyclic phosphate and 5′-hydroxyl (Fig. 1D). Despite the extensive structural and biochemical characterization of numerous HEPN nucleases, the role of the individual residues composing HEPN RφXXXH motifs remains unclear for this superfamily. With the widespread prevalence and functional significance of HEPN nucleases in diverse biological processes, including the use of HEPN-associated CRISPR-Cas nucleases for in vivo RNA-targeting applications (7, 11, 29–31), it is paramount to define the molecular mechanism of HEPN domains in RNA cleavage.
To gain a better understanding of the molecular features driving RNase PNK nuclease activity, we generated a series of Las1 RφXXXH HEPN variants to determine the functional significance of each individual residue. Through a combination of in vivo studies in Saccharomyces cerevisiae and in vitro activity assays, we determined that the flanking invariant arginine and histidine residues along with several intervening residues of the RφXXXH motif are critical for nuclease activity. Unexpectedly, we also discovered that conformational flexibility of the two HEPN domains contributes to nuclease fidelity. Restriction of the conformational flexibility of the two HEPN domains and alteration of the HEPN motif leads to off-target RNA cleavage products.
Results
The Las1 HEPN domain encodes a conserved RHXhTH motif
Las1 is conserved across eukaryotes and encodes a six–amino acid RφXXXH motif within its HEPN domain. Previous work has established that the invariant arginine and histidine residues of RφXXXH are critical to support S. cerevisiae Las1 RNA cleavage in vitro and in vivo (14, 23, 24), yet little is known about the intervening residues composing this motif. To determine the significance of the entire Las1 RφXXXH HEPN motif, we curated over 300 unique Las1 orthologs from species spanning fungi to vertebrates and carried out a comprehensive sequence alignment. Analysis of the alignment encompassing the Las1 HEPN domain revealed a single well-conserved RφXXXH motif previously associated with Las1 nuclease activity (Fig. 2A). This led to the discovery that beyond the first arginine (Arg-1) and last histidine (His-6) residues found in all HEPN family nucleases, the intervening residues within the Las1 RφXXXH HEPN motif are also well-conserved. The second position within this motif is an invariant histidine (His-2), which fits with the general trend that HEPN nucleases often encode for a polar amino acid at this position (3). On the other hand, the third position is a poorly conserved residue (X3) with a subtle preference for alanine and the fourth position has a strong preference for a hydrophobic amino acid (h4). Although a subset of plant Las1 homologs encode for a serine residue at the fifth position, the majority of Las1 homologs harbor a threonine (Thr-5). Based on this high sequence conservation, we define the consensus HEPN motif of the Las1 subfamily as RHXhTH (where X is any residue and h is a hydrophobic residue) (Fig. 2A).
Figure 2.

The Las1 HEPN nuclease encodes a consensus RHXhTH catalytic motif. A, sequence logo for the Las1 HEPN motif generated from over 101 vertebrates and 219 fungi orthologs. The height of each letter is correlated with its conservation within the motif. The logo defines the consensus Las1 HEPN motif RHXhTH (where X is any residue and h is commonly hydrophobic). B, Las1 HEPN nuclease active site from C. thermophilum RNase PNK in state 1 (PDB ID 6OF3) and state 2 (PDB ID 6OF2). Grc3 protomers are colored in gray and the two Las1 HEPN domains are shown in orange and yellow. The second HEPN protomer is denoted by prime. Residues of the Las1 HEPN motifs RHXhTH are shown in purple. Conserved residues Arg-1, His-2, Thr-5 and His-6 all face the catalytic center whereas X3 (CtLas1 Q3) points toward the cleft formed between Las1 and Grc3 and h4 (CtLas1 A4) contributes to the hydrophobic core. Black arrows highlight the rearrangement of invariant residue His-2 between state 1 (light purple) and state 2 (dark purple). Dotted line between residues Thr-5 and His-6 represents a putative hydrogen bond. Residues 108–140 of the proximal Las1HEPN′ protomer (orange) was removed for display purposes. C, table of the equivalent RHXhTH consensus residues in C. thermophilum Las1 (CtLas1) and S. cerevisiae Las1 (ScLas1).
The structural architecture surrounding the Las1 RHXhTH motif reveals the molecular basis for its residue preference at each position. Our recent cryo-EM reconstructions of Chaetomium thermophilum RNase PNK revealed the juxtaposed Las1 RHXhTH motifs within the composite nuclease active site (Fig. 2B) (26). Each Las1 RHXhTH motif is embedded within an α-helix that lies at the interface of the Las1 HEPN homodimer. By capturing multiple conformational states of RNase PNK, we revealed that His-2 is an important active site switch that toggles between nuclease active and inactive conformations (26). In the active state, His-2 is pointed toward the center of the active site whereas His-2 is pointing away in the inactive state (Fig. 2B). Although His-2 undergoes a distinct conformational rearrangement, the remaining residues of the Las1 RHXhTH motif remain largely unchanged. In the nuclease active conformation, the four well-conserved residues of the RHXhTH motif (Arg-1, His-2, Thr-5, His-6) all point toward the nuclease active site, suggesting they each play an important role in catalysis (Fig. 2, B and C). The well-conserved Thr-5 lies at the base of the nuclease active site where it is within hydrogen bonding distance of His-6 and could contribute to the spatial positioning of His-6. The other two residues, which lie within the middle of the motif, are positioned away from the active site. The variable residue at the third position (CtLas1 Gln-3) points toward the broad cleft formed between the nuclease and kinase active sites of RNase PNK, explaining the lack of conservation at this position. Conversely, the hydrophobic residue at the fourth position (CtLas1 Ala-4) is buried in the Las1 HEPN–HEPN′ core and explains its strong preference for a nonpolar residue to preserve its native fold (Fig. 2, B and C). Together, the identification of the Las1 HEPN consensus RHXhTH motif, along with the recent structural characterization of RNase PNK, uncovers the molecular requirements for the Las1 composite HEPN nuclease active site.
The Las1 RHXhTH motif is essential for S. cerevisiae cell proliferation
To investigate the functional significance of the Las1 RHXhTH motif, we disrupted the motif and monitored its functional effects in S. cerevisiae (15, 23). Analogous to all Las1 homologs, ScLas1 is composed of a well-conserved N-terminal HEPN nuclease domain followed by a poorly conserved coiled-coil domain and a conserved C-terminal tail, herein called LCT for Las1 C-terminal tail (Figs. 1C and 3A) (6, 17). The ScLas1 HEPN domain harbors the HEPN nuclease motif 129RHWGTH, which emulates the consensus RHXhTH motif found in all Las1 homologs. Although previous work has established that ScLas1 variants harboring R1A, H2A, or H6A mutations are nonviable in S. cerevisiae and unable to cleave the ITS2 pre-rRNA in vitro (14, 26); it is unknown if any other amino acid substitutions are tolerated within this motif. To determine the requirements of the RHXhTH consensus motif, we utilized a strain of S. cerevisiae containing a tetracycline-inducible promoter (tetO7) upstream of endogenous LAS1 (tet-LAS1) (26). This strain was modified to include a 3×Myc tag on the N terminus of endogenous GRC3 (tet-LAS1/Myc-GRC3) so that we could monitor Grc3 expression. Addition of doxycycline (DOX) to the growth medium represses the expression of endogenous Las1 and inhibits cell growth and proliferation (Fig. 3B and Fig. S1) (15). Repression of endogenous LAS1 was confirmed by RT-PCR (Fig. S2A). The tet-LAS1 and tet-LAS1/Myc-GRC3 strains were transformed with a plasmid harboring a 3×-FLAG tagged WT Las1 construct (FLAG-Las1) or an empty ARS1-CEN4 YCplac vector (32). We tested the complementation of FLAG-Las1 by repressing endogenous LAS1 expression with doxycycline at 30 °C. Unlike the transformed yeast strain harboring the empty vector, yeast expressing FLAG-Las1 rescues S. cerevisiae growth in the presence of doxycycline both on solid media and in liquid culture (Fig. 3, B and C).
Figure 3.

The Las1 RHXhTH motif is essential for S. cerevisiae proliferation. A, cartoon of S. cerevisiae (Sc) Las1 with the numbers defining the amino acid residue domain boundaries. The Las1 HEPN domain is shown as an orange box, the coiled-coil (CC) domain is a white box and the Las1 C-terminal tail (LCT: residues 469–502) is shown in brown. Individual residues of the ScLas1 RHXhTH motif (129RHWGTH) are highlighted in red, orange, yellow, green, blue, and purple, respectively. B, S. cerevisiae tet-LAS1 and tet-LAS1/Myc-GRC3 strains were either left untransformed (no plasmid) or transformed with plasmids encoding WT Las1 with an N-terminal 3× FLAG tag (FLAG-Las1) and empty YCplac 33 vector (vector). Serial dilutions of each strain were grown on YPD agar in the absence (−DOX) and presence (+DOX) of 40 μg/ml doxycycline at 30 °C for 3 days. Doxycycline represses transcription of endogenous LAS1 (Fig. S2A). C, growth curves of tet-LAS1/Myc-GRC3 strains transformed with control plasmids encoding WT FLAG-Las1 (WT) and empty YCplac 33 (vector). Strains were incubated at 30 °C in the absence and presence of doxycycline and absorbance was recorded over 25 h at 595 nm. Each curve is an average of three independent replicates and error bars define the S.D. D, growth curves as seen in panel C with the tet-LAS1/Myc-GRC3 strain transformed with FLAG-Las1 RHXhTH variants (Table S2). Color scheme is the same as seen in panel A. The doubling time was calculated using the +DOX curve and shown in the lower right corner. The average doubling time and S.D. were calculated from three biological replicates. E, -fold change in the doubling time for the different yeast strains normalized to the WT strain.
To assess the requirements of the Las1 RHXhTH motif, we engineered a series of single missense mutations to each residue (Fig. 3A and Table S1). Twelve individual Las1 RHXhTH HEPN motif variants (R1E, R1K, H2N, H2D, H2R, W3F, W3L, G4A, T5S, T5A, H6A, and H6N) were transformed into the tet-LAS1/Myc-GRC3 strain (Table S2). The complementation of these variants was tested by repressing endogenous LAS1 expression with doxycycline at 30 °C and growth curves were recorded over a 25-h time period by measuring the absorbance at 595 nm (Fig. 3D). We determined the doubling time and -fold change in doubling time for each variant as compared with the WT control (Fig. 3, D and E). Disruption of the first arginine (R1E, R1K) and last histidine (H6A, H6N) residues prevents cell proliferation underscoring the significance of these residues for Las1 function. Mutations to the invariant His-2 residue (H2N, H2D, H2R) yielded variable results. The H2D and H2R variants cause severe growth defects; however the H2N variant only causes a minor growth defect. Although the role of H2 in Las1 function is unknown, the observation that either a histidine or asparagine can support Las1 function in vivo is suggestive of a role in RNA engagement and/or positioning in the nuclease active site. Disruption of either the third or fourth residue from the Las1 RHXhTH HEPN motif (W3F, W3L, G4A) only causes mild growth defects, which is not unexpected given the poor sequence conservation and position within the RNase PNK structure. Mutagenesis of the threonine at the fifth position to a serine (T5S) causes a mild growth defect, whereas an alanine (T5A) causes a moderate growth defect. This corresponds well with a subset of plant Las1 homologs that encode a serine at this position and suggests these homologs are functional. A serine residue at the fifth position may be tolerated because it could still maintain a hydrogen bond to the adjacent catalytic histidine (His-6), as seen with Thr-5 in the RNase PNK structure (Fig. 2B). Conversely, an alanine substitution (T5A) would prevent hydrogen bonding to His-6, thus potentially disrupting Las1 function and explaining its growth defect. Interestingly, the catalytic histidine found in the nuclease active site of RNase A also forms a similar hydrogen bond with a nearby aspartic acid residue. Although disruption of this hydrogen bond leads to a modest defect in RNA cleavage, this interaction is thought to promote proper orientation of the catalytic histidine for RNA hydrolysis and enhance the conformational stability of the active site (33, 34). Therefore, these results suggest a threonine or serine at the fifth position is important for Las1 function. We also tested temperature sensitivity by plating serial dilutions of these strains on solid media and monitored their growth at five different temperatures in the presence and absence of doxycycline (Fig. S1). With the exception of W3L, T5S, and T5A, the majority of the HEPN variants do not display temperature sensitivity. Temperatures higher or lower than 30 °C result in more significant growth defects for the W3L, T5S, and T5A variants. Taken together these results highlight the importance of the entire Las1 RHXhTH HEPN motif for cell viability in S. cerevisiae.
Las1 RHXhTH variants do not disrupt protein stability or Grc3 association
We performed control experiments to determine that the growth defects observed with the Las1 HEPN variants are not the result of Las1 protein instability. To confirm that the HEPN variants do not compromise Las1 protein stability in vivo, we grew tet-LAS1/Myc-GRC3 strains expressing the Las1 HEPN variants to mid-log phase in the presence of doxycycline at 30 °C and then analyzed the whole cell lysate by Western blotting. Because of the co-dependence of Las1 and Grc3 for protein stability (15), we also analyzed endogenous levels of 3×Myc-tagged Grc3 (15). FLAG-tagged Las1 was detected with an anti-FLAG antibody, Grc3 was detected with an anti-Myc antibody and tubulin was used as a loading control. Expression of the Las1 HEPN variants does not substantially alter the endogenous levels of Las1 or Grc3 in vivo (Fig. S2B). Therefore, the growth defects caused by the Las1 HEPN variants are not because of a loss in Las1 protein stability.
The Las1 HEPN variants also retain their association with Grc3 and support higher-order assembly of the RNase PNK complex. The Las1 HEPN nuclease directly depends on its binding partner Grc3 to execute its ITS2 pre-rRNA processing activity (23). To ensure that the defects observed in cell growth and proliferation are not the result of an inability to associate with Grc3, we monitored assembly of RNase PNK using the Las1 HEPN variants and WT Grc3. To reconstitute recombinant RNase PNK complexes, we generated a series of Escherichia coli co-expression vectors encoding WT Grc3 along with poly-histidine–tagged Las1 HEPN variants (Table S3). Using affinity chromatography, we immobilized Las1 HEPN variants and assessed their association with Grc3. SDS-PAGE analysis of the purified samples revealed that all of the Las1 HEPN variants stably expressed and co-purified with Grc3 (Fig. S2C). Because the higher-order assembly of the RNase PNK complex is also crucial for ITS2 pre-rRNA processing (23), we monitored RNase PNK heterotetrameric assembly by gel filtration. All of the RNase PNK variants show similar retention volumes to WT RNase PNK, suggesting they all maintain their heterotetrameric organization (Fig. S2D). Thus, this confirms that the Las1 HEPN variants do not hinder Grc3 association or oligomerization of RNase PNK.
Las1 RHXhTH motif is required for C2 pre-rRNA cleavage
The Las1 HEPN variants reveal the importance of the RHXhTH motif for C2 cleavage in vitro and in vivo. We speculated that the yeast growth defects observed in the presence of the Las1 HEPN variants were because of a defect in C2 pre-rRNA cleavage. To determine the effects of the Las1 variants on C2 pre-rRNA cleavage, we performed in vitro C2 cleavage assays using a 3′ fluorescently labeled ITS2 RNA mimic (C2 RNA substrate) (Fig. 4A). These reactions were carried out under enzyme excess, where RNase PNK variants were in molar excess over the C2 RNA substrate. Incubation of the C2 RNA substrate with WT RNase PNK results in a specific 5′-hydroxyl RNA fragment that can be resolved on a denaturing urea gel and was previously mapped to the C2 site (Fig. 4B) (23). In contrast, incubation of the C2 RNA substrate with the RNase PNK Las1 variants led to the observation that many of the variants are deficient at C2 cleavage. A representative gel summarizing C2 cleavage reactions of all the HEPN variants is shown in Fig. 4B. The variants that cause severe growth defects in S. cerevisiae (R1E, R1K, H2D, H2R, T5A, H6A, H6N) are unable to cleave the C2 site indicating that the observed growth defects are the result of an inability to cut the pre-rRNA (Fig. 4, B and C). In contrast, the Las1 HEPN variants that cause minor growth defects (H2N, W3L, G4A, T5S) are able to cleave the ITS2 at the C2 site, albeit less efficiently than WT (Fig. 3D and 4C).
Figure 4.

Las1 RHXhTH is critical for efficient C2 cleavage. A, C2 RNA substrate used for in vitro C2 processing assays. This RNA substrate mimics the S. cerevisiae ITS2 C2 site (23). Black asterisk marks the 3′-fluorescein label (3′-FAM). B, representative C2 RNA cleavage by recombinant S. cerevisiae Las1 RHXhTH variants bound to WT Grc3. Excess ScRNase PNK variants (4.5 μm) were incubated with C2 RNA substrate (50 nm) for 1 h at 37 °C and resolved using denaturing urea gels. RNA marks the reaction set without protein. C, quantification of C2 RNA cleavage product shown in panel B. The mean and S.D. were calculated from three independent replicates. n.s. not significant, *, p < 6 × 10−3; **, p < 8 × 10−4; and ***, p < 6 × 10−6 were calculated by two-tailed Student's t tests. D, representative C2 RNA cleavage and phosphorylation by recombinant ScRNase PNK variants. Protein variants (4.5 μm) were incubated with C2 RNA substrate (50 nm) in the absence (−) and presence (+) of 2 mm ATP for 1 h at 37 °C. RNA marks the reaction set in the absence of protein. Dots define C2 RNA cleavage products and triangles label C2 RNA phosphorylation products.
Beyond its role in nuclease activity, Las1 is also important for supporting nuclease-directed kinase activity, raising the possibility that the Las1 HEPN variants may also hinder Grc3 kinase activity. Previously we showed that the Las1R1E,H6A variant hinders Grc3 phosphotransferase activity in vitro (23). To determine whether the individual Las1 variants impact Grc3 kinase activity, we added ATP to the C2 cleavage reactions and visualized RNA phosphorylation through altered RNA mobility in a denaturing urea gel. All of the Las1 HEPN variants are able to phosphorylate either the 5′-hydroxyl end of the unprocessed C2 RNA substrate and/or the 5′-hydroxyl end of the C2 cleavage product (Fig. 4D). The observation of multiple phosphorylation events confirms our earlier work that showed the Grc3 kinase component of RNase PNK has broad RNA specificity (24). Taken together, these results indicate that the Las1 HEPN variants do not prevent Grc3 kinase activity on the ITS2 substrate, but are crucial for supporting Las1 nuclease activity.
The Las1 HEPN variants also disrupt pre-rRNA processing in vivo. Previous work has shown that knockdown of endogenous Las1 and expression of R1A, H2A, or H6A variants blocks ITS2 processing (14, 15, 26). To determine the effects of the Las1 variants on pre-rRNA processing, we extracted RNA from cells expressing the Las1 variants in the presence of doxycycline and then analyzed the total RNA with a bioanalyzer (Fig. S2E). We compared the ratio of the 25S/18S mature rRNA and total rRNA (25S + 18S) for all the variants against the WT control. We see a decrease in the ratio of 25S/18S and total rRNA with all the variants, with a more pronounced effect with the R1E, R1K, H2D, H2R, H6A, and H6N variants (Fig. S2F). These results are consistent with a defect in maturation of pre-rRNA processing.
Las1 HEPN–HEPN′ chimeras alter nuclease fidelity
Next, we sought to determine the significance of each Las1 RHXhTH motif for ITS2 pre-rRNA recognition and hydrolysis. Composite HEPN nuclease active sites are formed by two RφXXXH motifs through either trans or cis assembly. Trans-homodimerization of HEPN domains is typically observed in HEPN nucleases, including RNase PNK, Ire1, RNase L, and Csm6 (5, 23, 27, 28, 35, 36). Cis-heterodimerization of tandem HEPN domains encoded within a single protomer has thus far only been observed in Cas13 nucleases (7, 11, 29, 31). Individual mutagenesis of the Ala-1 or His-6 residues from a single copy of the tandem HEPN heterodimer of the Cas13 subfamily impairs cleavage in vitro (11, 31). In contrast, trans complementation assays with HEPN homodimers of Ire1 and RNase L do not impair cleavage (28, 36). Collectively these results suggest that the requirement for juxtaposed Arg-1 and His-6 residues varies among HEPN family nucleases.
To determine whether RNase PNK requires both copies of its Las1 HEPN motifs for C2 cleavage, we engineered Las1 constructs that harbored mutations to a single copy of the HEPN homodimer. The constitutive dimerization of the Las1 HEPN domains poses a challenge for examining the significance of residues from the individual RHXhTH motifs. To overcome this problem, we used the RNase PNK structure to design a chimera of ScLas1 in which we fused two Las1 HEPN domains (HEPN–HEPN′) together with a flexible linker (Fig. 5A). The rational for the Las1 chimera was based off our previous work demonstrating that with the exception of the LCT, which is critical for Grc3 stability, the coiled-coil domain of Las1 is not required for RNase PNK nuclease and kinase activity in vitro (23). We reconstituted the chimeric ScRNase PNK complex using an E. coli co-expression vector encoding the Las1HEPN-HEPN′ chimera, a poly-histidine–tagged Las1LCT and full-length Grc3 (Fig. 5A). First, we purified the chimeric RNase PNK complex and confirmed Las1HEPN-HEPN′ retains its association with Grc3 and maintains its higher-order assembly using SDS-PAGE and gel filtration, respectively (Fig. S3A). Furthermore, we confirmed that the chimeric RNase PNK complex (WT–WT′) retains nuclease and kinase activities along with specificity for the C2 site (Fig. 5B, full-length versus WT–WT′, and Fig. S3B). Chimeric RNase PNK titrations revealed that the Las1WT-WT′ chimera cleaves the C2 RNA substrate at the appropriate site but is not as active as full-length Las1. The double mutant Las1H6A-H6A′ chimera is severely deficient in C2 cleavage (Fig. 5B and Fig. S3B).
Figure 5.

Las1 HEPN-HEPN′ chimeras have altered cleavage fidelity. A, cartoon schematic of the chimeric ScRNase PNK complex comprised of the Las1 HEPN-HEPN′ dimer (orange-yellow) connected by a flexible linker, the Las1 LCT (brown) and WT Grc3 (gray). The black line represents the linker tethering the Las1 HEPN-HEPN′ dimer. The Las1 coiled-coil domain is dispensable for C2 RNA processing in vitro (23) and was removed to create the chimeric ScLas1HEPN-HEPN′+LCT-Grc3 complex. B, denaturing urea gels of RNA cleavage and phosphorylation reactions with full-length RNase PNK (FL) and chimeric RNase PNK HEPN variants (HEPN-HEPN′). The lanes are marked as follows: RNA (100 nm RNA alone), X (0.8 μm RNase PNK variant and 100 nm RNA), ATP (0.8 μm RNase PNK variant, 100 nm RNA, and 10 mm ATP), ADPnP (0.8 μm RNase PNK variant, 100 nm RNA, and 10 mm ADPnP). Circles mark RNA cleavage products and triangles identify phosphorylated RNA. Black symbols specify C2 RNA processing products and red symbols are off-target RNA processing products.
After confirming that the WT–WT′ chimeric RNase PNK cleaves the RNA at the correct site in vitro, we generated a series of chimeric RNase PNK variants that encode missense mutations to a single RHXhTH motif. We carried out both nuclease and kinase assays under enzyme excess with double the amount of C2 RNA substrate to enhance the detection of low abundance RNA cleavage and phosphorylation products (Fig. 5B). Chimeric RNase PNK complexes harboring Las1WT-R1E′, Las1WT-R1K′, Las1WT-H6A′, or Las1WT-H6N′ all retained nuclease and kinase activity in vitro. We also generated chimeric RNase PNK complexes harboring two mutations to the RHXhTH motif either in cis or in trans. Both the trans mutants (Las1H6A-H6A′ and Las1R1E-H6A′) and the cis mutant (Las1R1E,H6A-WT′) dramatically hinder Las1 RNA cleavage activity (Fig. 5B). In accordance with our earlier work that shows the Las1 nuclease domain is important for supporting Grc3 kinase activity (23), we also observe a detectable RNA phosphorylation defect of the unprocessed RNA substrate for chimeric RNase PNK harboring Las1R1E,H6A-WT′.
Unexpectedly, several chimeric variants (Las1WT-R1E′, Las1WT-H6N′, Las1H6A-H6A′, Las1R1E,H6A-WT′, and Las1R1E-H6A′) produced additional cleavage products (Fig. 5B, bottom gel). These additional products are all phosphorylated by Grc3 in the presence of ATP, but not a nonhydrolysable ATP analog, confirming that they must arise from Las1 cleavage. For instance, the chimeric RNase PNK comprising Las1WT-R1E′ produces two cleavage products, one that coincides with canonical C2 cleavage and another that is the result of an off-target cleavage event which lies at least one nucleotide away from the C2 site. The chimeric RNase PNK complex harboring Las1WT-H6N′ also produces the same two cleavage products, but the majority is the off-target product. To determine the identity of the off-target product, we performed LC electrospray ionization MS (LC-ESI-MS) analysis on the uncleaved C2 RNA substrate and the C2 RNA substrate following an incubation with chimeric RNase PNK harboring Las1WT-H6N′. The unprocessed C2 RNA substrate gave rise to a single peak above background that corresponds with the theoretical mass of the uncleaved C2 RNA substrate containing 5′- and 3′-hydroxyl ends (Fig. S4A). The mutant chimeric RNase PNK cleavage reaction gave rise to three RNA peaks above background including the unprocessed C2 RNA substrate and two product peaks (Fig. S4B). One product peak corresponds to an 8-nucleotide RNA fragment containing a 5′-hydroxyl and 2′,3′-cyclic phosphate; the second product is a 19-nucleotide RNA fragment containing 5′- and 3′-hydroxyl ends. This unambiguously maps the Las1-mediated off-target cleavage event to the phosphodiester bond 5′ to the canonical C2 scissile phosphate (off-target site: U139-A140 of ScITS2). Thus, we confirm that the mutant chimeric RNase PNK complex has altered specificity and we denote the resulting off-target cleavage event as C2(−1) RNA cleavage.
Conformational flexibility is important for nuclease fidelity
To ensure that the off-target cleavage observed with the Las1 chimeric variants was not a result of the presence of the glycine-serine linker between the HEPN domains we added a tobacco etch virus (TEV) protease cleavage site within the linker (Fig. 6A). We repeated our nuclease assay with RNase PNK harboring Las1WT-WT′ before (WT-WT′) and after (WT|WT′) TEV cleavage (Fig. S5A). We optimized the TEV reaction conditions to achieve as much cleavage of the linker as possible, because it is not feasible to separate TEV cleaved from uncleaved RNase PNK chimeras. TEV cleavage also facilitates the removal of the hexahistidine tag on the LCT peptide. We quantified the specific activity of the Las1 nuclease and found that the presence of the linker restricts nuclease activity by about 50% (Fig. 6B). Cleavage of the linker with TEV protease almost completely restores the activity to that of WT RNase PNK (WT+WT′) lacking the coiled-coil domain of Las1 (Fig. 6B and Fig. S5B). We presume that we cannot fully restore the activity because the TEV cleavage reaction was not 100%. These results suggest that conformational flexibility of the two HEPN domains is important for Las1 nuclease activity.
Figure 6.
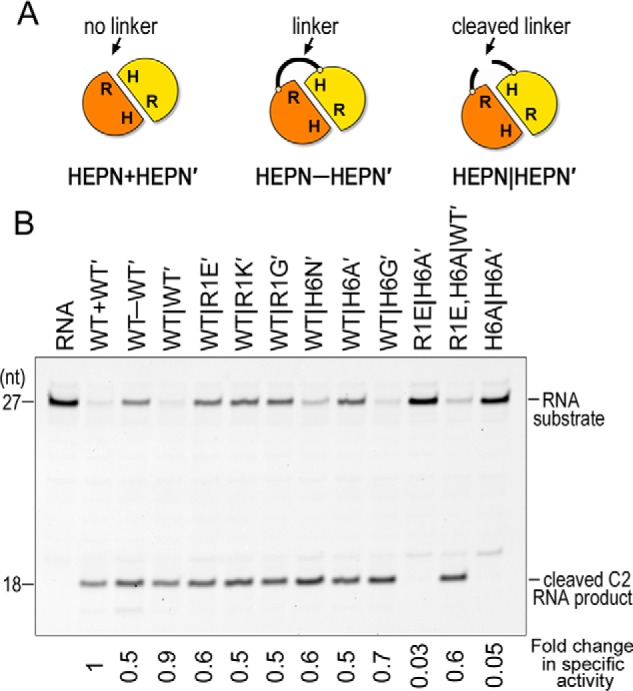
The chimera linker restricts nuclease activity. A, cartoon schematic of the different constructs used for nuclease assays. HEPN+HEPN′ represents a construct with no linker between the Las1 HEPN domains. HEPN-HEPN′ is the construct with the linker containing a TEV site. HEPN|HEPN′ represents TEV cleaved linkers. B, denaturing urea gels of RNA cleavage with chimeric RNase PNK HEPN variants. The -fold change in specific activity is reported below the gel and calculated from three independent replicates of enzyme titration experiments shown in Fig. S5B.
We then made a series of chimeric RNase PNK variants with a TEV site in the HEPN–HEPN′ linker region and carried out nuclease assays following cleavage of the linker (HEPN–HEPN′). Surprisingly, once the linker holding together the HEPN domains is cleaved we no longer observe off-target cleavage with any of the RNase PNK variants (Fig. 6B). This suggests that it is a combination of conformational flexibility and active site variants that give rise to the observed mis-cleavage. Taken together these results confirm that beyond the Las1 RHXhTH motif, conformational flexibility between the two HEPN domains is important for Las1 function. This is further supported by recent cryo-EM structures that revealed hingelike conformational changes between the Las1 HEPN domains when comparing distinct conformational states of RNase PNK (26).
To determine the contribution of each individual Arg-1 and His-6 residue from the Las1 RHXhTH motif, we carried out nuclease assays with titration gradients of the TEV-cleaved Las1 variants. The TEV cleavage reactions were not 100% complete, but the amount of cleavage was fairly consistent among the variants (Fig. S5A). From these titrations we calculated the specific activity of the Las1 nuclease. Individual mutations of R1E, R1K, R1G, H6N, H6A, and H6G cause a 0.5- to 0.7-fold change in specific nuclease activity (Fig. 6B). We also made double mutations to the HEPN motif in cis and trans. The trans H6A,H6A′ and R1E,H6A′ double mutants do not have nuclease activity. In contrast the cis R1E,H6A WT′ mutant results in a 0.6-fold change in specific nuclease activity, similar to the single active site mutants. Collectively these results highlight the individual significance of each Arg-1 and His-6 active site residue and establishes that there must be a minimum of one fully intact Las1 RHXhTH motif for nuclease activity.
Discussion
In this study we report a comprehensive molecular characterization of the Las1 HEPN nuclease motif, which plays a critical role in pre-rRNA processing. HEPN nucleases participate in a wide spectrum of RNA processing pathways and the recent identification of many new HEPN nucleases (3) has led to a surge in atomic-resolution structures of different HEPN nuclease family members (Fig. 7) (4, 5, 26, 28–30, 35, 37–44). The overall architecture of HEPN nucleases varies dramatically from the butterfly-shaped RNase PNK to bi-lobed Cas13 nucleases and intertwined RNase L. These diverse assemblies are important contributors to the activation and specialized functions of individual HEPN nucleases. Yet, each of these nucleases contains a common homo or heterodimeric HEPN core that is responsible for nuclease activity. The presence of this common core suggests that HEPN nucleases cleave RNA following a similar mechanism. Each HEPN core contains a composite nuclease active site at its center and is defined by the juxtaposition of two RφXXXH motifs (Fig. 7). Despite numerous recent advances in HEPN nuclease biology, the molecular mechanism of RNA cleavage remains unresolved. By implementing an in-depth sequence analysis, yeast genetics, and in vitro activity assays we determined that the first, second, fifth, and sixth residues from this motif are absolutely essential for supporting Las1 function. Because of the strong parallels among all HEPN nucleases, our work has broad implications for the HEPN nuclease field.
Figure 7.

Structural comparison of HEPN nucleases. A–D, ribbon diagram of (A) C. thermophilum RNase PNK (PDB ID 6OF3), (B) Staphylococcus epidermis Csm6 (PDB ID 5YJC), (C) Eubacterium siraeum Cas13d (PDB ID 6E9F) and (D) Sus scrofa RNase L (PDB ID 4O1P). HEPN-HEPN′ dimers are colored in yellow and orange along with their juxtaposed RφXXXH motifs colored in purple. Enzyme-specific insertions of RNase PNK, Csm6, Cas13d, and RNase L are colored in gray, light blue, brown, and light green, respectively. Black boxes mark the HEPN nuclease active sites formed by well-conserved HEPN nuclease motifs (purple). The inset is a zoom of the juxtaposed RφXXXH motifs forming the catalytic site. Conserved residues of the RφXXXH motifs are shown and the second copy of the RφXXXH motif is designated by prime.
Significance of the individual residues from the HEPN motif
Although it is well-established that the canonical Arg-1 and His-6 residues from HEPN nucleases are important for catalysis (3, 7), this work highlights the significance of His-2 and Thr-5 from the Las1 HEPN motif. The invariant His-6 residue is thought to be important for triggering 2′-OH nucleophilic attack, and Arg-1 has been proposed to stabilize the transition state and/or the RNA substrate (3). Our work expands this working model by uncovering the functional significance of the intervening Las1 HEPN residues. Amino acid composition of the second residue from the HEPN motif is most often a polar residue. Indeed our complementation and in vitro cleavage assays revealed that Las1 can function with a conservative histidine to asparagine mutation at this position, but other substitutions are not tolerated. Similarly, HEPN nucleases Ire1, Csm6, and Cas13 encode an asparagine at its second position and previous work has shown that N2A mutants are functionally inactive (5, 27, 30). Together, this signifies a universal importance of the second residue from the RφXXXH motif. Considering recent structural characterization of RNase PNK revealed that Las1 His-2 is a functional molecular switch that undergoes conformational rearrangements within the HEPN active site, we compared the active sites from several recent HEPN nuclease structures. Intriguingly, within these structures the His-2/Asn-2 residue either points toward the center of the composite active site, as seen in the nuclease active state of RNase PNK where it could coordinate RNA, or it points away from the catalytic center, as seen in the nuclease inactive state of RNase PNK. This raises the question whether the second amino acid residue of the HEPN motif is a universal molecular switch regulating the nuclease activity of the HEPN superfamily. What then is the role of this residue? One possibility is that the His-2 residue could participate in the general acid-base mechanism that has been proposed for HEPN family nucleases (3, 7, 36). However, this seems unlikely because we determined that H2N mutants of Las1 are functionally active, and H6N mutants are inactive. Furthermore, both asparagine and histidine can form hydrogen bonds with RNA, but only histidine is able to donate a proton because the pKa of asparagine is too high (45). Because the second position of the RφXXXH motif is consistently a proton acceptor, we do not anticipate that His-2 interacts with a phosphate group in the RNA backbone, but more likely interacts with a hydroxyl group on the pentose ring. Thus, we hypothesize that the second position residue of the HEPN motif is critical for proper positioning of the RNA substrate within the active site, and His-6 participates in acid-base catalysis.
Our work also establishes the significance of Thr-5 within the Las1 nuclease motif. Among HEPN nucleases there is a prevalence for a small amino acid at this position (2). This is observed in the recent structures of both Csm6 (Ala-5/Ala-5′) and Cas13d (Val-5/Cys-5′), which harbor small amino acids at the fifth position (Fig. 7). Within composite HEPN nuclease active sites, the fifth-position residue is located along the base of the active site. Although the location of this residue suggests that it does not directly participate in catalysis, it likely plays a supportive role in maintaining the structural integrity of the active site. Here we show that Las1 depends upon having either a threonine or serine at this position. This dependence is supported by the sequence conversation among Las1 homologs and the presence of a hydrogen bond between Thr-5 and His-6 that was observed in the cryo-EM reconstruction of RNase PNK. Taken together, our work indicates that beyond the invariant Arg-1 and His-6 residues from the RφXXXH HEPN nuclease motif, the intervening residues play important supporting roles in substrate engagement and active site architecture.
It takes two (RφXXXH domains) to cut the RNA right
A fundamental outstanding question is why are HEPN nucleases reliant on the juxtaposition of two RφXXXH motifs to cleave the RNA backbone. A single RφXXXH motif presumably contains the residues that are sufficient for cleavage; however all HEPN nucleases require dimerization to be active. There are several possibilities as to why dimerization could be important. For example, one RφXXXH motif could be important for RNA cleavage whereas the other copy could participate in positioning the RNA. Another possibility is that the HEPN active site could be reliant on Arg-1 from one motif and His-6′ from the other copy for catalysis. We suggest it is a combination of both because our work reveals that all four Las1 Arg-1, Arg-1′, His-6 and His-6′ residues are needed for full nuclease activity. Moreover our biochemical analysis with the Las1 HEPN chimeras establishes that the minimal requirement for nuclease activity is an intact RφXXXH from one of the HEPN domains.
We also uncovered an unexpected finding through the creation of the RNase PNK chimeras. Conformational flexibility between the HEPN domains is important for both nuclease activity and fidelity. Having an intact linker between the two HEPN domains leads to a 50% reduction in specific activity. The combination of the intact linker and specific active site variants also leads to altered cleavage specificity. The most striking alteration in specificity was observed with the WT-H6N′ variant which cleaves the ITS2 predominantly at the C2(−1) position. Cleavage of the linker between the two HEPN domains prevents the altered specificity, strongly implying that conformational dynamics are critical for proper Las1 nuclease fidelity. This is further supported by recent cryo-EM reconstructions of RNase PNK that revealed a hingelike motion between different conformational states of the Las1 HEPN domains (26).
Defining the precise mechanism of HEPN-directed RNA cleavage will require a series of high-resolution structures of HEPN domains engaged to their endogenous RNA substrates. However, to date, capturing RNA-associated HEPN states has proven challenging because of their transient nature. Our work lays the foundation for understanding the contributions of the individual residues from the Las1 RHXhTH motif. Beyond advancing our understanding of pre-rRNA processing, the observation that Las1 can be re-wired to cleave RNA at different positions has far reaching implications in the rapidly developing field of RNA targeting applications. The Cas13 CRISPR effectors, which contain HEPN nuclease domains linked within the same polypeptide chain, are currently being adapted for a range of applications including RNA knockdown, RNA editing, and RNA detection/diagnostics (7–11, 46). Once activated by CRISPR RNA, the Cas13 HEPN domains orchestrate nonspecific RNA cleavage. The ability to re-wire the Cas13 HEPN domains with altered specificity could open the door for new RNA-targeting applications.
Materials and methods
Las1 sequence analysis
Homo sapiens Las1L (NP_112483.1) was used as a query to extract 101 Las1 orthologs for vertebrate genomes using Ensembl (RRID: SCR_002344, release 95) (47) and S. cerevisiae Las1 (NP_012989.3) was used as the query to extract 219 Las1 orthologs from Ensembl Fungi (release 42). Sequence alignments were performed with MAFFT 7 (RRID: SCR_011811) (48) using default settings. CLC Genomics version 8 was used for visualization of multiple sequence alignments and WebLogo (49) was used to generate the Las1 HEPN sequence logo.
Generation of Las1 yeast strains
Generation of the S. cerevisiae LAS1 tetracycline-titratable promoter (tetO7) strain was described previously (26). This strain was modified to include a 3×Myc-tag upstream of endogenous GRC3 for detection purposes (yMP125). N-terminal 3×FLAG-tagged ScLas1 (pMP 580) was amplified along with 300 nucleotides of flanking endogenous DNA sequence and inserted into YCplac33 (32) using KpnI and SacI. Las1 HEPN variants were generated by overlap PCR, inserted into YCplac33 and verified by DNA sequencing (Genewiz). The yMP125 strain was transformed with plasmids encoding the N-terminal 3×FLAG tagged Las1 WT (pMP 580), Las1 HEPN variants (see Table S1), or empty YCplac33 vector. All yeast strains used in this study are listed in Table S2.
Yeast spotting assays and growth curves
Yeast spotting assays and growth curves were performed as described previously (24) with minor modifications. Transformed LAS1 yeast strains were pre-incubated in YPD that was supplemented with 40 μg/ml doxycycline for 24 h at 22 °C prior to performing the assays. For proliferation assays, transformed tet-LAS1 and tet-LAS1/Myc-GRC3 strains (Table S2) were spotted on YPD plates in the absence and presence of doxycycline (40 μg/ml) and incubated at 30 °C for 2–3 days. Temperature sensitivity was tested by carrying out additional proliferation assays at 16 °C, 25 °C, 34 °C, and 37 °C for 2–6 days. Growth curves were generated using transformed tet-LAS1/Myc-GRC3 strains by measuring the absorbance at 595 nm of 100 μl yeast cultures inoculated at an A600 of 0.05 and incubated at 30 °C in YPD and YPD with doxycycline (40 μg/ml). A600 measurements were recorded every 15 min over a 25-h time period with an Infinite F200 Pro (Tecan) and i-control 1.11 software. The mean and S.D. of each growth curve were calculated from three independent replicates.
Bioanalyzer analysis
Tet-LAS1/Myc-GRC3 strains were grown in YPD supplemented with 40 μg/ml doxycycline for 24 h at 22 °C. Strains were then growing at 30 °C to mid-log phase before total RNA was extracted. RNA was quantified using the Qubit RNA HS Assay Kit (Thermo Fischer Scientific), and 250 ng total RNA was analyzed on the bioanalyzer. Using 2100 Expert Software (Agilent), electropherograms were analyzed to calculate the area under the peaks corresponding to the 25S and 18S rRNA. The ratio of mature 25S to 18S rRNA and total rRNA (sum of 25S and 18S) were calculated from three technical replicates.
RT-PCR
Three independent cultures of the CML476 parental strain and three independent cultures of tet-LAS1/Myc-GRC3 strain (Table S2) were grown at 22 °C in YPD in the absence and presence of doxycycline (40 μg/ml) for ∼24 h. Total RNA was isolated using the RiboPure Yeast RNA Purification kit (Life Technologies) following the manufacturer's protocol. The RNA was quantitated with a NanoDrop 2000C, and 1 μg of each sample was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad), following the manufacturer's protocol. Real-time PCR analysis was performed using the ABI Quant Studio 7 Flex system. All cDNAs were diluted and then subjected to real-time PCR using the TaqMan SYBR green PCR Master Mix (Life Technologies) with transcript-specific primers (Table S4), according to the manufacturer's protocol. Relative abundance was determined by normalizing to an internal control, TFC1 (50).
Western blotting
Western blotting was performed as described previously (24) with minor modifications. Transformed tet-LAS1/Myc-GRC3 strains were grown in YPD in the presence of doxycycline (40 μg/ml) to mid-log phase. The whole cell lysate was prepared by lysing the cells with glass beads followed by TCA precipitation. Proteins were resolved by SDS-PAGE and analyzed by Western blotting using anti-Myc (Grc3; EMD Millipore), anti-FLAG (Las1; Sigma), and anti-α-tubulin (Abcam).
Recombinant Las1-Grc3 complex purification
S. cerevisiae RNase PNK variants were generated from the original Las1-Grc3 co-expression vector (pMP001) (23) by Q5 site-directed mutagenesis (New England Biolabs) and verified by DNA sequencing (Genewiz). ScLas1 HEPN-HEPN′ fusion (Las1 residues 1–179 amino acids followed by a 2× Gly-Gly-Gly-Gly-Ser linker, NotI restriction site, and Las1 residues 1–185) was inserted into the expression plasmid ScLas1LCT-Grc3 (pMP072) (23) using HindIII and SacI to generate ScLas1HEPN-HEPN′+LCT-Grc3 (pMP673). Chimeric ScLas1HEPN-HEPN′+LCT-Grc3 was generated by amplifying ScLas1 HEPN′ (residues 1–185) harboring mutations R129E (R1E; pMP683), R129K (R1K; pMP684), H134A (H6A; pMP681), and H134N (H6N; pMP682) were amplified by PCR and inserted into pMP673 using NotI and SacI. Additional chimeric ScLas1HEPN-HEPN′+LCT-Grc3 constructs were generated by amplifying ScLas1 HEPN (residues 1–179) harboring mutations R129E, H134A (R1E,H6A; pMP697), H134A (H6A; pMP695), and R129E (R1E; pMP691) and inserted into pMP673 or pMP681 using HindIII and NotI. All Las1-Grc3 co-expression plasmids used and generated in this study were sequenced (Genewiz) and are listed in Table S3.
ScLas1-Grc3 variant proteins were expressed and purified as described previously (23) with minor modifications. Las1-Grc3 variants were overexpressed in E. coli LOBSTR (Kerafast) cells following cold shock and induction with 0.5 mm IPTG. Harvested cells were resuspended in lysis buffer (50 mm Tris, pH 8.0, 500 mm NaCl, 5 mm MgCl2, 1% (v/v) Triton X-100, 10% (v/v) glycerol) and lysed by sonication. Clarified lysate was applied to a gravity flow column with His60 Ni Superflow Resin (Clontech) that was pre-equilibrated in lysis buffer. After washing with 200 ml of wash buffer (50 mm Tris, pH 8.0, 500 mm NaCl, 5 mm MgCl2, 30 mm imidazole, 10% glycerol) the complex was eluted with imidazole (50 mm Tris, pH 8.0, 500 mm NaCl, 5 mm MgCl2, 200 mm imidazole, 10% glycerol). To cleave the HEPN-HEPN′ TEV encoded linker, recombinant TEV protease (48 μg/ml) was incubated with 1 μm of HEPN-HEPN′ chimera overnight at 4 °C (51). Las1-Grc3 variants were further resolved by gel filtration (HiLoad 16/600 Superdex 200 Prep Grade; GE Healthcare) with storage buffer (20 mm Tris, pH 8.0, 200 mm NaCl, 5 mm MgCl2, 5% glycerol).
C2 pre-rRNA cleavage and phosphorylation assays
C2 RNA cleavage assays were performed as described previously (23, 24) with minor changes. Full-length ScLas1-Grc3 and chimeric ScLas1HEPN-HEPN′+LCT-Grc3 variants (0–3.2 μm) were incubated with 1 mm EDTA and 50 or 100 nm 3′ fluorescein-labeled C2 RNA substrate (5′-GUCGUUUUAGGUUUUACCAACUGCGGC/36-FAM/-3′) in the absence and presence of 2 mm nucleotide (ATP or ADPnP). Cleavage reactions were incubated for 1 h at 37 °C and then quenched with urea-loading dye (20 mm Tris, pH 8.0, 8 m urea, 0.05% bromphenol blue, 1 mm EDTA). Samples were resolved on 15% polyacrylamide (8 m urea) gels in 1× Tris-borate-EDTA buffer and visualized using a Typhoon FLA 9500. Representative gels are shown from three independent replicates.
Mass spectrometric sample preparation and analysis of oligonucleotides
10 μl of RNA sample (5 μm) was injected onto the column for LC MS (LC-MS) analysis. Data were acquired on a Q Exactive Plus mass spectrometer (QE-MS, Thermo Fisher Scientific) interfaced with a Vanquish (Thermo Fisher Scientific) UHPLC system. Reverse-phase chromatography was performed using a CORTECS C18 column (100 × 2.1 mm diameter, 1.6 μm particle size; Waters Corporation) and a CORTECS C18 VanGuard precolumn (5 × 2.1 mm diameter, 1.6 μm particle size; Waters Corporation) with solvent A being 5 mm ammonium formate in water (pH 6.5) and solvent B being methanol and a flow rate of 150 μl per minute. The LC gradient included a ramp from 0 to 42% B from 0 to 6 min followed by a ramp from 42 to 95% B over 1 min, and then a 3-min hold at 95% B. The run was completed with a ramp of 95% to 0% B for 0.5 min followed by a 4.5 min recondition at 0% B. The QE-MS was equipped with a HESI source used in the negative ion mode and performing only MS1 scans. Mass calibration was performed before data acquisition using the Pierce ESI Negative Ion Calibration Mixture (Pierce). Data were processed and deconvoluted using the Intact Protein Analysis function of BioPharma Finder (Thermo Fisher Scientific). Mass predictions of the oligonucleotides were performed using the web version of Mongo Oligo Mass Calculator v2.08 maintained by the RNA Institute.
Data availability
All data is included in the manuscript figures and supporting information. All corresponding raw data files will be made available upon request to the corresponding author, Robin E. Stanley (robin.stanley@nih.gov).
Author contributions
M. C. P. and R. E. S. conceptualization; M. C. P., K. H. G., J. G., and M. L. W. data curation; M. C. P., K. H. G., J. G., M. L. W., and J. G. W. formal analysis; M. C. P. and R. E. S. writing-original draft; M. C. P., K. H. G., J. G., M. L. W., J. G. W., and R. E. S. writing-review and editing; J. G. and R. E. S. investigation; J. G. W. methodology; R. E. S. supervision; R. E. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank Drs. Lars Pedersen and Chen Qiu, as well as all the members of the Stanley Lab for their critical reading of this manuscript. We are grateful to Dr. Thomas Randall from the NIEHS Integrative Bioinformatics Support Group for help with Las1 sequence alignments.
This work was supported by the National Institutes of Health Intramural Research Program and NIEHS, National Institutes of Health Grant ZIA ES103247 (to R. E. S.) and by the Canadian Institutes of Health Research (CIHR) Grant 146626 (to M. C. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1–S4.
- pre-rRNA
- precursor rRNA
- PNK
- polynucleotide kinase
- DOX
- doxycycline
- TEV
- tobacco etch virus
- YPD
- yeast extract peptone dextrose.
References
- 1. Yang W. (2011) Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 44, 1–93 10.1017/S0033583510000181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grynberg M., Erlandsen H., and Godzik A. (2003) HEPN: A common domain in bacterial drug resistance and human neurodegenerative proteins. Trends Biochem. Sci. 28, 224–226 10.1016/S0968-0004(03)00060-4 [DOI] [PubMed] [Google Scholar]
- 3. Anantharaman V., Makarova K. S., Burroughs A. M., Koonin E. V., and Aravind L. (2013) Comprehensive analysis of the HEPN superfamily: Identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct. 8, 15 10.1186/1745-6150-8-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jia X., Yao J., Gao Z., Liu G., Dong Y. H., Wang X., and Zhang H. (2018) Structure-function analyses reveal the molecular architecture and neutralization mechanism of a bacterial HEPN-MNT toxin-antitoxin system. J. Biol. Chem. 293, 6812–6823 10.1074/jbc.RA118.002421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee K. P., Dey M., Neculai D., Cao C., Dever T. E., and Sicheri F. (2008) Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100 10.1016/j.cell.2007.10.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castle C. D., Cassimere E. K., Lee J., and Denicourt C. (2010) Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol. Cell Biol. 30, 4404–4414 10.1128/MCB.00358-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Connell M. R. (2019) Molecular mechanisms of RNA targeting by Cas13-containing type VI CRISPR-Cas systems. J. Mol. Biol. 431, 66–87 10.1016/j.jmb.2018.06.029 [DOI] [PubMed] [Google Scholar]
- 8. Konermann S., Lotfy P., Brideau N. J., Oki J., Shokhirev M. N., and Hsu P. D. (2018) Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676.e614 10.1016/j.cell.2018.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gootenberg J. S., Abudayyeh O. O., Kellner M. J., Joung J., Collins J. J., and Zhang F. (2018) Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 10.1126/science.aaq0179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox D. B. T., Gootenberg J. S., Abudayyeh O. O., Franklin B., Kellner M. J., Joung J., and Zhang F. (2017) RNA editing with CRISPR-Cas13. Science 358, 1019–1027 10.1126/science.aaq0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. East-Seletsky A., O'Connell M. R., Knight S. C., Burstein D., Cate J. H., Tjian R., and Doudna J. A. (2016) Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 10.1038/nature19802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu H., Haas S. A., Chelly J., Van Esch H., Raynaud M., de Brouwer A. P., Weinert S., Froyen G., Frints S. G., Laumonnier F., Zemojtel T., Love M. I., Richard H., Emde A. K., Bienek M., et al. (2016) X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 21, 133–148 10.1038/mp.2014.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Butterfield R. J., Stevenson T. J., Xing L., Newcomb T. M., Nelson B., Zeng W., Li X., Lu H. M., Lu H., Farwell Gonzalez K. D., Wei J. P., Chao E. C., Prior T. W., Snyder P. J., Bonkowsky J. L., and Swoboda K. J. (2014) Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurology 82, 1322–1330 10.1212/WNL.0000000000000305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gasse L., Flemming D., and Hurt E. (2015) Coordinated ribosomal ITS2 RNA processing by the Las1 complex integrating endonuclease, polynucleotide kinase, and exonuclease activities. Mol. Cell 60, 808–815 10.1016/j.molcel.2015.10.021 [DOI] [PubMed] [Google Scholar]
- 15. Castle C. D., Sardana R., Dandekar V., Borgianini V., Johnson A. W., and Denicourt C. (2013) Las1 interacts with Grc3 polynucleotide kinase and is required for ribosome synthesis in Saccharomyces cerevisiae. Nucleic Acids Res. 41, 1135–1150 10.1093/nar/gks1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fromm L., Falk S., Flemming D., Schuller J. M., Thoms M., Conti E., and Hurt E. (2017) Reconstitution of the complete pathway of ITS2 processing at the pre-ribosome. Nat. Commun. 8, 1787 10.1038/s41467-017-01786-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schillewaert S., Wacheul L., Lhomme F., and Lafontaine D. L. (2012) The evolutionarily conserved protein Las1 is required for pre-rRNA processing at both ends of ITS2. Mol. Cell Biol. 32, 430–444 10.1128/MCB.06019-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woolford J. L. Jr., and Baserga S. J. (2013) Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195, 643–681 10.1534/genetics.113.153197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klinge S., and Woolford J. L. Jr. (2019) Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 20, 116–131 10.1038/s41580-018-0078-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bassler J., and Hurt E. (2019) Eukaryotic ribosome assembly. Annu. Rev. Biochem. 88, 281–306 10.1146/annurev-biochem-013118-110817 [DOI] [PubMed] [Google Scholar]
- 21. Tomecki R., Sikorski P. J., and Zakrzewska-Placzek M. (2017) Comparison of preribosomal RNA processing pathways in yeast, plant and human cells—focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 591, 1801–1850 10.1002/1873-3468.12682 [DOI] [PubMed] [Google Scholar]
- 22. Heindl K., and Martinez J. (2010) Nol9 is a novel polynucleotide 5′-kinase involved in ribosomal RNA processing. EMBO J. 29, 4161–4171 10.1038/emboj.2010.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pillon M. C., Sobhany M., Borgnia M. J., Williams J. G., and Stanley R. E. (2017) Grc3 programs the essential endoribonuclease Las1 for specific RNA cleavage. Proc. Natl. Acad. Sci. U.S.A. 114, E5530–E5538 10.1073/pnas.1703133114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pillon M. C., Sobhany M., and Stanley R. E. (2018) Characterization of the molecular crosstalk within the essential Grc3/Las1 pre-rRNA processing complex. RNA 24, 721–738 10.1261/rna.065037.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pillon M. C., and Stanley R. E. (2018) Nuclease integrated kinase super assemblies (NiKs) and their role in RNA processing. Curr. Genet. 64, 183–190 10.1007/s00294-017-0749-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pillon M. C., Hsu A. L., Krahn J. M., Williams J. G., Goslen K. H., Sobhany M., Borgnia M. J., and Stanley R. E. (2019) Cryo-EM reveals active site coordination within a multienzyme pre-rRNA processing complex. Nat. Struct. Mol. Biol. 26, 830–839 10.1038/s41594-019-0289-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niewoehner O., and Jinek M. (2016) Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA 22, 318–329 10.1261/rna.054098.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han Y., Donovan J., Rath S., Whitney G., Chitrakar A., and Korennykh A. (2014) Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343, 1244–1248 10.1126/science.1249845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang C., Konermann S., Brideau N. J., Lotfy P., Wu X., Novick S. J., Strutzenberg T., Griffin P. R., Hsu P. D., and Lyumkis D. (2018) Structural basis for the RNA-guided ribonuclease activity of CRISPR-Cas13d. Cell 175, 212–223.e17 10.1016/j.cell.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu L., Li X., Wang J., Wang M., Chen P., Yin M., Li J., Sheng G., and Wang Y. (2017) Two distant catalytic sites are responsible for C2c2 RNase activities. Cell 168, 121–134.e12 10.1016/j.cell.2016.12.031 [DOI] [PubMed] [Google Scholar]
- 31. Abudayyeh O. O., Gootenberg J. S., Konermann S., Joung J., Slaymaker I. M., Cox D. B., Shmakov S., Makarova K. S., Semenova E., Minakhin L., Severinov K., Regev A., Lander E. S., Koonin E. V., and Zhang F. (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 10.1126/science.aaf5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gietz R. D., and Sugino A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 10.1016/0378-1119(88)90185-0 [DOI] [PubMed] [Google Scholar]
- 33. Quirk D. J., and Raines R. T. (1999) His … Asp catalytic dyad of ribonuclease A: Histidine pKa values in the wild-type, D121N, and D121A enzymes. Biophys. J. 76, 1571–1579 10.1016/S0006-3495(99)77316-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quirk D. J., Park C., Thompson J. E., and Raines R. T. (1998) His … Asp catalytic dyad of ribonuclease A: Conformational stability of the wild-type, D121N, D121A, and H119A enzymes. Biochemistry 37, 17958–17964 10.1021/bi981688j [DOI] [PubMed] [Google Scholar]
- 35. Huang H., Zeqiraj E., Dong B., Jha B. K., Duffy N. M., Orlicky S., Thevakumaran N., Talukdar M., Pillon M. C., Ceccarelli D. F., Wan L. C., Juang Y. C., Mao D. Y., Gaughan C., Brinton M. A., et al. (2014) Dimeric structure of pseudokinase RNase L bound to 2–5A reveals a basis for interferon-induced antiviral activity. Mol. Cell 53, 221–234 10.1016/j.molcel.2013.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korennykh A. V., Korostelev A. A., Egea P. F., Finer-Moore J., Stroud R. M., Zhang C., Shokat K. M., and Walter P. (2011) Structural and functional basis for RNA cleavage by Ire1. BMC Biol. 9, 47 10.1186/1741-7007-9-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feldman H. C., Tong M., Wang L., Meza-Acevedo R., Gobillot T. A., Lebedev I., Gliedt M. J., Hari S. B., Mitra A. K., Backes B. J., Papa F. R., Seeliger M. A., and Maly D. J. (2016) Structural and functional analysis of the allosteric inhibition of IRE1α with ATP-competitive ligands. ACS Chem. Biol. 11, 2195–2205 10.1021/acschembio.5b00940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang B., Ye Y., Ye W., Perčulija V., Jiang H., Chen Y., Li Y., Chen J., Lin J., Wang S., Chen Q., Han Y. S., and Ouyang S. (2019) Two HEPN domains dictate CRISPR RNA maturation and target cleavage in Cas13d. Nat. Commun. 10, 2544 10.1038/s41467-019-10507-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knott G. J., East-Seletsky A., Cofsky J. C., Holton J. M., Charles E., O'Connell M. R., and Doudna J. A. (2017) Guide-bound structures of an RNA-targeting A-cleaving CRISPR-Cas13a enzyme. Nat. Struct. Mol. Biol. 24, 825–833 10.1038/nsmb.3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu L., Li X., Ma J., Li Z., You L., Wang J., Wang M., Zhang X., and Wang Y. (2017) The molecular architecture for RNA-guided RNA cleavage by Cas13a. Cell 170, 714–726.e10 10.1016/j.cell.2017.06.050 [DOI] [PubMed] [Google Scholar]
- 41. Slaymaker I. M., Mesa P., Kellner M. J., Kannan S., Brignole E., Koob J., Feliciano P. R., Stella S., Abudayyeh O. O., Gootenberg J. S., Strecker J., Montoya G., and Zhang F. (2019) High-resolution structure of Cas13b and biochemical characterization of RNA targeting and cleavage. Cell Rep. 26, 3741–3751.e5 10.1016/j.celrep.2019.02.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang B., Ye W., Ye Y., Zhou H., Saeed A. F. U. H., Chen J., Lin J., Perčulija V., Chen Q., Chen C. J., Chang M. X., Choudhary M. I., and Ouyang S. (2018) Structural insights into Cas13b-guided CRISPR RNA maturation and recognition. Cell Res. 28, 1198–1201 10.1038/s41422-018-0109-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niewoehner O., Garcia-Doval C., Rostøl J. T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L. A., and Jinek M. (2017) Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548 10.1038/nature23467 [DOI] [PubMed] [Google Scholar]
- 44. Kim Y. K., Kim Y. G., and Oh B. H. (2013) Crystal structure and nucleic acid-binding activity of the CRISPR-associated protein Csx1 of Pyrococcus furiosus. Proteins 81, 261–270 10.1002/prot.24183 [DOI] [PubMed] [Google Scholar]
- 45. Panov K. I., Kolbanovskaya E. Y., Okorokov A. L., Panova T. B., Terwisscha van Scheltinga A. C., Karpeisky M. Ya., and Beintema J. J. (1996) Ribonuclease A mutant His119 Asn: The role of histidine in catalysis. FEBS Lett. 398, 57–60 10.1016/S0014-5793(96)01173-8 [DOI] [PubMed] [Google Scholar]
- 46. Terns M. P. (2018) CRISPR-based technologies: Impact of RNA-targeting systems. Mol. Cell 72, 404–412 10.1016/j.molcel.2018.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., et al. (2018) Ensembl 2018. Nucleic Acids Res. 46, D754–D761 10.1093/nar/gkx1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Katoh K., Rozewicki J., and Yamada K. D. (2019) MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166 10.1093/bib/bbx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Teste M. A., Duquenne M., François J. M., and Parrou J. L. (2009) Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol. Biol. 10, 99 10.1186/1471-2199-10-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tropea J. E., Cherry S., and Waugh D. S. (2009) Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol. Biol. 498, 297–307 10.1007/978-1-59745-196-3_19 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is included in the manuscript figures and supporting information. All corresponding raw data files will be made available upon request to the corresponding author, Robin E. Stanley (robin.stanley@nih.gov).