

Abstract
Incorporation of ribonucleotides into DNA can severely diminish genome integrity. However, how ribonucleotides instigate DNA damage is poorly understood. In DNA, they can promote replication stress and genomic instability and have been implicated in several diseases. We report here the impact of the ribonucleotide rATP and of its naturally occurring damaged analog 1,N6-ethenoadenosine (1,N6-ϵrA) on translesion synthesis (TLS), mediated by human DNA polymerase η (hpol η), and on RNase H2–mediated incision. Mass spectral analysis revealed that 1,N6-ϵrA in DNA generates extensive frameshifts during TLS, which can lead to genomic instability. Moreover, steady-state kinetic analysis of the TLS process indicated that deoxypurines (i.e. dATP and dGTP) are inserted predominantly opposite 1,N6-ϵrA. We also show that hpol η acts as a reverse transcriptase in the presence of damaged ribonucleotide 1,N6-ϵrA but has poor RNA primer extension activities. Steady-state kinetic analysis of reverse transcription and RNA primer extension showed that hpol η favors the addition of dATP and dGTP opposite 1,N6-ϵrA. We also found that RNase H2 recognizes 1,N6-ϵrA but has limited incision activity across from this lesion, which can lead to the persistence of this detrimental DNA adduct. We conclude that the damaged and unrepaired ribonucleotide 1,N6-ϵrA in DNA exhibits mutagenic potential and can also alter the reading frame in an mRNA transcript because 1,N6-ϵrA is incompletely incised by RNase H2.
Keywords: DNA damage; DNA polymerase; DNA enzyme; RNA modification; RNA processing; reverse transcription; 1,N6-ethenoadenosine (1,N6-εrA); DNA adducts; genome integrity; polymerase eta; translesion synthesis (TLS); DNA alkylation
Introduction
High-fidelity DNA polymerase-mediated replication is a central tenet of biology. It is important to ensure that the genome is copied completely and accurately during replication without any incorrect insertions to avoid further consequences. On the basis of several studies done during the past decade, ribonucleotide incorporation in DNA is considered a major threat to genome stability (1, 2). Mitochondrial (pol γ)2 as well as nuclear DNA polymerases (pols α, β, and ϵ) (3, 4) incorporate ribonucleotides into the genome because the cellular concentrations of ribonucleoside triphosphates (rNTPs) are much higher than the dNTPs (5–7). Discrimination between deoxyribo- and ribosugars varies among DNA polymerases, mainly due to the active site “steric gate” residue that plays an important role in limiting rNTP insertions (8). Other sources of ribonucleotide incorporation include HIV-1 reverse transcriptase (RT)-mediated reverse transcription and incomplete removal of Okazaki fragment primers (1, 9). Embedded ribonucleotides in the genome present an enigma in the DNA replication process due to the presence of a 2′-OH group, which renders the backbone susceptible to cleavage and various sugar puckered conformations (10–12). Cells repair such ribonucleotides (in DNA) primarily by the ribonucleotide excision repair (RER) process (13–15) and a mechanism that involves RNase H2 enzyme-mediated recognition and incision of ribonucleotides (15–17). The loss of RNase H2 activity is linked to Aicardi–Goutieres syndrome in humans (18). If RNase H2 is unable to remove these ribonucleotides (19), they can have both favorable (20, 21) and lethal biological consequences (22). Recent investigations have shown that ribonucleotides are deleterious lesions that can lead to mutations, large deletions, replication stress, strand breaks (topoisomerase-1–mediated repair), chromosomal rearrangements, loss of hereditary information, and disruption of transcription processes (1, 23–26).
In cells, reactive oxygen species and lipid peroxidation can cause damage of the nucleotide pool (damaged rNTPs/dNTPs) (7, 27) and can be a threat to the synthesis of genetic material (28). Thus, the coding potential of genetic material can be compromised if damaged rNTPs/dNTPs are incorporated into the genome during replication. Oxidation of a deoxyribo- to a ribosugar can also occur (29). In the cellular nucleotide pool, free dGTP is more vulnerable to oxidation than the G present in DNA and leads primarily to the formation of 8-oxo-dGTP (30), which can be inserted into the nascent DNA (31–35). Among the rNTPs, the cellular concentration of rATP is very high, and it may therefore be inserted into DNA (33). In this connection, Clausen and co-workers also (36) reported the levels of rATP in mitochondrial DNA. The possibility of insertion of rATP and damaged rATPs into the DNA during replication cannot be excluded (33). The biological consequences of rATP and damaged rATPs in DNA remain elusive. Our own work has shown that low-fidelity human DNA polymerase η (hpol η) incorporates rATP opposite a cyclobutane pyrimidine dimer as well as 8-oxo-dG lesions (37, 38).
DNA and RNA lesions are ubiquitous to the genome. If DNA damage is left unrepaired, cells utilize low-fidelity DNA polymerases to continue replication across damaged sites (to rescue the stalled replication forks) for cell survival, a process termed translesion synthesis (TLS) (39, 40). The TLS process has garnered considerable attention due to the tolerance of damage in DNA in an error-free or error-prone manner (39, 40). TLS also plays an important role in tolerance of ribonucleotides in DNA, and the main key player is hpol η (especially in dealing with lagging strands derived from Okazaki fragments) (16, 24, 38, 41–46). Studies with 8-oxo-rG in DNA indicate that hpol η is required for the tolerance of this adduct (24). The versatile hpol η is now also known for its reverse transcriptase and transcription-associated roles (38, 47–50). Damage in RNA has not been considered to disturb the cellular processes, possibly due to general lack of replication of RNA and its innately lower stability as well as the more rapid turnover (51–54). However, RNA is more vulnerable to oxidative damage due to its abundance and single-stranded nature (55, 56), and this damage can be detrimental for the cell (57). RNA damage is also associated with the pathogenesis of some neurodegenerative diseases (58–60). RNA is not exempt from damaging events from reactive oxygen species and lipid peroxidation.
2-Chloroethylene oxide, an oxidation product of the known human liver carcinogen vinyl chloride (VC), reacts with nucleophilic sites in DNA and RNA and leads to the formation of etheno (ϵ) lesions (61). Exposure of animals to VC or vinyl bromide (VB) resulted in chemical modification of RNA, and a common product was 1,N6-ethenoadenosine (1,N6-ϵrA) (62). Several studies have shown that 1,N6-ϵrA can be formed in the presence of metabolites of VC or other compounds that generate reactive epoxides (63–65). Bolt and co-workers (66, 67) measured in vivo alkylation of RNA by VC or VB and its effect on protein synthesis; they reported the synthesis of two unusual proteins from mRNA of VC-exposed animals as compared with control rats (67). Exposure of rats to VB resulted in the synthesis of one unusual protein (67). Schulman and Pelka (68) reported that a total of five adenosine structures were altered after reaction of 2-chloroacetaldehyde (a rearrangement product of the VC metabolite 2-chlorooxirane, also known to generate ϵ adducts) with Escherichia coli fMet tRNA and also suggested that adenosine residues were modified more rapidly than the cytosine in tRNA (69). Biernat et al. (70) also reported 1,N6-ϵrA formation in tRNA after treatment with 2-chloroacetaldehyde. Transcription errors were also observed in the presence of 1,N6-ϵrA (71). ϵ-Lesions can also be generated indirectly from oxidative stress through lipid peroxidation (72). Leumann and co-workers (73) studied various damaged bases in RNA in the context of translation, but there is only limited knowledge of the biochemistry of damage in stable RNA. It is important to understand the possible consequences of 1,N6-ϵrA in RNA, especially for RT and translation processes.
We now report TLS, RT, and RNA primer extension events and incision of the most abundant nucleotide, rATP, and its naturally occurring damaged analog 1,N6-ϵrA. We utilized hpol η for the TLS, RT, and RNA primer extension studies due to its recently discovered novel functions (37, 38, 44, 47, 49, 74, 75) and RNase H2 for the incision studies. Our laboratory has recently shown that hpol η can act as a reverse transcriptase in human cells (38) and also that it utilizes RNA as a primer in in vitro replication assays, apart from its role in TLS (74). We examined the roles of hpol η as a TLS polymerase and reverse transcriptase and in RNA primer extension events in the presence of a damaged ribonucleotide in DNA as well as in RNA under physiological conditions (Fig. 1).
Figure 1.

Human pol η–mediated translesion synthesis, reverse transcription, and RNA primer extension activities across adenosine and 1,N6-ϵrA.
Results
hpol η tolerates the damaged ribonucleotide 1,N6-ϵrA
To gain detailed insights into the biological consequences of 1,N6-ϵrA and rATP, multiple modified oligonucleotides were used for the TLS, RT, and RNA primer extension studies (Table S1). A primary concern was feasible side reactions related to the presence of the ribo-backbone in the DNA and RNA. Recent studies have shown that strand cleavage, as well as transesterification reactions (isomerization from 3′–5′ to 2′–5′), at ribonucleotide sites are drastically suppressed in nucleosome core particles (76). In contradiction, it is also known that a ribo-backbone in DNA or RNA is temperature–, pH–, and metal ion–sensitive and can lead to transesterification as well as strand cleavage (77). Accordingly, we excluded the possibility of side reactions during our experiments by maintaining physiological conditions, as well as a desired metal ion concentration (Mg2+), low temperature, and pH.
To investigate TLS across a ribonucleotide embedded in DNA, full-length extension and single nucleotide incorporation reactions were carried out using physiological concentrations of dNTPs as well as rNTPs (78, 79) and hpol η (Fig. 2 and Fig. S1 (A and B); see “Experimental procedures” for details). Full-length extension reactions were carried out using higher concentrations of hpol η, whereas single-nucleotide incorporation reactions were carried out using lower concentrations of hpol η. With an unmodified DNA template (DNA-dA), hpol η rapidly extended the DNA primer using dNTPs as well as rNTPs (Fig. S1A, lanes 1–8). When an rA-embedded DNA template was used (DNA-rA), hpol η fully extended the DNA primer using dNTPs (Fig. 2A, lanes 1–4). The DNA primer was also elongated to the full-length product in the presence of rNTPs, but with less processivity (Fig. 2A, lanes 5–8). These results show that generation of a ribo-ribo pair at an rA site in a DNA template is an unusual behavior of hpol η. Changing the coding base from dA to rA had only a minor effect on rNTP incorporation. TLS activities were examined in the presence of the modified ribonucleotide 1,N6-ϵrA, and hpol η was less efficient in bypassing 1,N6-ϵrA in a DNA template (Fig. 2C, lanes 1–4) as compared with an rA template. However, extension reactions were strongly blocked when rNTPs were used (Fig. 2C, lanes 5–8). As expected, the TLS process was attenuated in the presence of 1,N6-ϵrA (Fig. 2 (A and C), compare lane 1).
Figure 2.

TLS by hpol η. PAGE (20%, 7 m urea) was conducted. A and C, hpol η (200 nm) elongated DNA primer opposite rA-containing (A) and ϵrA-containing (C) DNA templates in the presence of a mixture of dNTPs or rNTPs (physiological concentrations). All reactions were done at 37 °C for 5, 30, 60, and 180 min (time gradients indicated with wedges). Lanes 1–4, dNTPs; lanes 5–8, rNTPs. B and D, hpol η (50 nm) was incubated with DNA/DNA-rA; DNA/DNA-1,N6-ϵrA with individual dNTPs and rNTPs (physiological concentrations). Lanes 1–3, dTTP; lanes 4–6, dATP; lanes 7–9, dCTP; lanes 10–12, dGTP; lanes 13–15, rUTP; lanes 16–18, rATP; lanes 19–21, rCTP; lanes 22–24, rGTP. All reactions were done at 37 °C for 5, 30, and 60 min. See “Experimental procedures” and Table S1 for the oligonucleotide sequences used. P, FAM-labeled DNA primer.
In the single-nucleotide incorporation assays, hpol η preferentially added dTTP opposite A in DNA-dA and DNA-rA templates (Fig. S1B and Fig. 2B (lanes 1–3)). With the DNA-rA template, the incorporation preference was dTTP > dATP > dGTP > dCTP > rUTP > rATP > rGTP (Fig. 2B, lanes 1–24). These results showed that hpol η can generate an rA-rU pair during TLS by inserting rUTP opposite rA in DNA. In the case of a 1,N6-ϵrA–modified DNA template, hpol η preferably added dATP and dGTP, as compared with dTTP and dCTP (Fig. 2D, lanes 1–24). Additionally, no rNTP incorporation was observed opposite 1,N6-ϵrA in DNA (with a lower concentration of hpol η), indicating that hpol η prefers to add deoxypurines instead of ribopurines opposite 1,N6-ϵrA.
Our laboratory has previously shown that hpol η can act as a reverse transcriptase in human cell extracts (38), and here we examined this novel function of hpol η using modified RNA as a template, employing physiological concentrations of dNTPs and rNTPs (Fig. 3) under in vitro conditions. With an unmodified RNA template, hpol η elongated a DNA primer in the presence of a mixture of dNTPs (Fig. 3A, lanes 1–4), but only one or two rNTPs were added even after 3 h (Fig. 3A, lanes 5–8). In the case of a 1,N6-ϵrA–modified RNA template, the DNA primer was elongated with a mixture of dNTPs with low processivity (Fig. 3C, lanes 1–4), whereas extension reactions were strongly blocked in the assays with rNTPs (Fig. 3C, lanes 5–8). These results indicate that hpol η acts as a specific reverse transcriptase in the presence of 1,N6-ϵrA and prefers dNTPs under physiological conditions.
Figure 3.

Reverse transcription by hpol η. PAGE (20%, 7 m urea) was conducted. A and C, hpol η (200 nm) elongated DNA primer opposite rA-containing (A) and ϵrA-containing (C) RNA templates in the presence of a mixture of dNTPs or rNTPs (physiological concentrations). All reactions were done at 37 °C for 5, 30, 60, and 180 min (time gradients indicated with wedges). Lanes 1–4, dNTPs; lanes 5–8, rNTPs. B and D, hpol η (50 nm) was incubated with DNA/RNA-rA; DNA/RNA-1,N6-ϵrA with individual dNTPs and rNTPs (physiological concentrations). Lanes 1–3, dTTP; lanes 4–6, dATP; lanes 7–9, dCTP; lanes 10–12 dGTP; lanes 13–15, rUTP; lanes 16–18, rATP; lanes 19–21, rCTP; lanes 22–24, rGTP. All reactions were done at 37 °C for 5, 30, and 60 min. See “Experimental procedures” and Table S1 for the oligonucleotide sequences used. P, FAM-labeled DNA primer.
As a reverse transcriptase, hpol η preferentially added dTTP opposite an unmodified RNA template (Fig. 3B, lanes 1–3), and the incorporation preference was dTTP > dATP > dGTP > dCTP > rUTP (Fig. 3B, lanes 1–24). These results also suggest that hpol η can generate rA-rU pairs during reverse transcription. In the case of a 1,N6-ϵrA–modified template, hpol η preferably added dATP and dGTP (Fig. 3D, lanes 1–24), following similar base selectivity as in the TLS process. Overall, hpol η catalyzes reverse transcription with preference for incorporation of dNTPs over rNTPs.
We previously reported that hpol η can extend an RNA primer (74), and we next examined the ability of hpol η to extend an RNA primer in the presence of an unmodified as well as a modified ribonucleotide in DNA using physiological concentrations of dNTPs and rNTPs. With the unmodified DNA template (DNA-dA), hpol η readily extended the RNA primer using dNTPs, but the process was slow with rNTPs (Fig. S1C, lanes 1–8). Surprisingly, fully extended as well as stalled products were observed for a DNA-rA template (Fig. 4A, lanes 1–8). The RNA primer was partially extended in the presence of a 1,N6-ϵrA–modified ribonucleotide. Single-base addition was predominant with the use of dNTPs (Fig. 4C, lanes 1–4), and RNA primer extension was completely retarded opposite 1,N6-ϵrA when rNTPs were used (Fig. 4C, lanes 5–8).
Figure 4.

RNA primer extension activities of hpol η. PAGE (20%, 7 m urea) was performed. A and C, hpol η (200 nm) elongated RNA primer opposite rA-containing (A) and ϵrA-containing (C) DNA templates in the presence of a mixture of dNTPs or rNTPs (physiological concentrations). All reactions were done at 37 °C for 5, 30, 60, and 180 min (time gradients indicated with wedges). Lanes 1–4, dNTPs; lanes 5–8, rNTPs. B and D, hpol η (50 nm) was incubated with RNA/DNA-rA; RNA/DNA-1,N6-ϵrA with individual dNTPs and rNTPs (physiological concentrations). Lanes 1–3, dTTP; lanes 4–6, dATP; lanes 7–9, dCTP; lanes 10–12, dGTP, lanes 13–15, rUTP; lanes 16–18, rATP; lanes 19–21, rCTP; lanes 22–24, rGTP. All reactions were done at 37 °C for 5, 30, and 60 min. See “Experimental procedures” and Table S1 for the oligonucleotide sequences used. P, FAM-labeled RNA primer.
For RNA primer extension, hpol η preferentially added dTTP opposite DNA-dA and DNA-rA templates (Fig. S1D and Fig. 4B, lanes 1–3). For the DNA-rA template, the incorporation preference was dTTP/dGTP/dATP > dCTP (Fig. 4B, lanes 1–24). In the case of a 1,N6-ϵrA–modified template, hpol η preferentially added dGTP compared with dATP (Fig. 4D, lanes 1–24). No rNTP incorporation was observed opposite 1,N6-ϵrA.
Miscoding potential of the damaged ribonucleotide 1,N6-ϵrA
To further measure the efficiency and fidelity of hpol η during TLS, RT, and RNA primer extension processes, steady-state kinetic analysis (80, 81) was done (Table 1, Figs. 5 and 6, and Figs. S2–S16; see “Experimental procedures” for details). In the TLS assays, steady-state kinetic analysis showed that incorporation of dTTP opposite 1,N6-ϵrA was highly unfavorable. The specificity constant (ksp; i.e. kcat/Km) for dTTP insertion (0.001 μm−1 min−1, Table 1) was dramatically reduced, 4200-fold compared with DNA-dA (4.2 μm−1 min−1; Table 1) and 1600-fold compared with DNA-rA templates (1.7 μm−1 min−1, Table 1 and Fig. S2), respectively. For insertion of dATP opposite 1,N6-ϵrA, the specificity constant decreased 4.5-fold compared with DNA-dA and 6.5-fold compared with a DNA-rA template (Table 1 and Fig. 5). The insertion of dGTP opposite 1,N6-ϵrA was also decreased 5-fold compared with a DNA-dA and 8-fold compared with a DNA-rA template (Table 1 and Fig. S3). In the case of dCTP insertion opposite to 1,N6-ϵrA, the specificity constant was decreased 50-fold compared with a DNA-dA and 80-fold compared with a DNA-rA template (Table 1 and Fig. S4). As discussed earlier, rNTP insertion was only observed for the DNA-dA and DNA-rA templates. Surprisingly, the specificity constant for rUTP insertion opposite DNA-rA was only 1.5-fold lower compared with the DNA-dA template (Table 1 and Fig. S5). For rATP insertion, the same specificity constants were observed for both templates (Table 1 and Fig. S6), and rGTP incorporation was observed only with the DNA-rA template (Table 1 and Fig. S7).
Table 1.
Steady-state kinetic analysis of hpol η-catalyzed translesion synthesis, reverse transcription, and RNA primer extension
ϵrA, 1, N6-ethenoadenosine; rA, adenosine; dA, 2′-deoxyadenosine. The complete data set is presented in Figs. 5 and 6 and Figs. S2–S16.
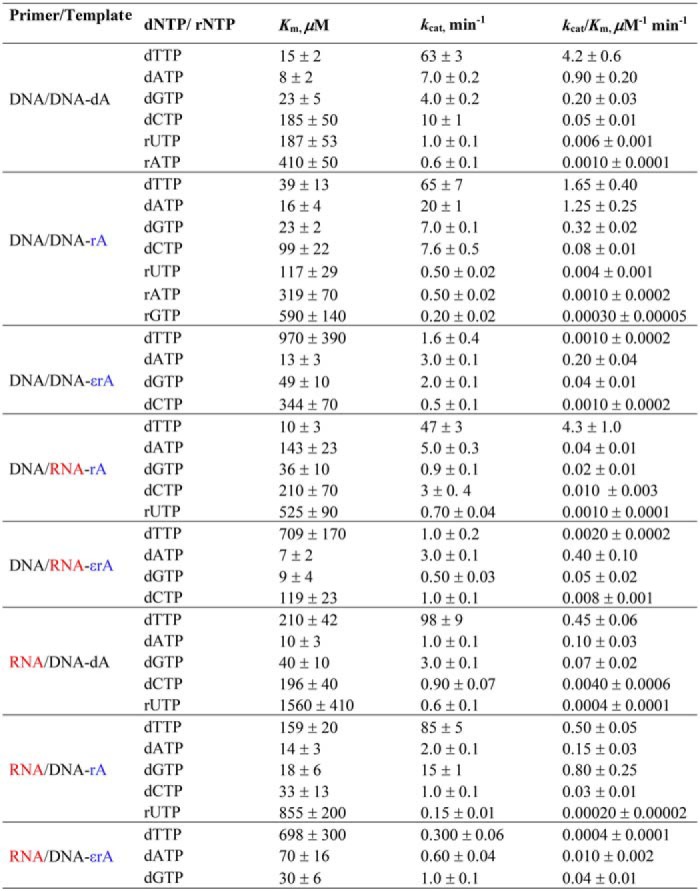
Figure 5.

Steady-state kinetic analysis of dATP insertion by hpol η during translesion synthesis. PAGE (20%, 7 m urea) of steady-state kinetic analysis using hpol η (1–2.3 nm) with varying concentrations of dATP (0–800 μm, indicated with wedges) was conducted for DNA/DNA-dA (A), DNA/DNA-rA (B), and DNA/DNA-1,N6-ϵrA (C). All reactions were carried out at 37 °C for 5 min in duplicate. The indicated data points are shown as means ± S.D. (error bars); see Table 1 for kcat and Km values (estimated using fits to a hyperbolic equation in Prism (GraphPad)); see “Experimental procedures” and Table S1 for the oligonucleotide sequences. P, FAM-labeled DNA primer.
Figure 6.

Steady-state kinetic analysis of dATP insertion by hpol η during reverse transcription. PAGE (20%, 7 m urea) of steady-state kinetic analysis using hpol η (2–2.5 nm) was conducted with varying concentrations of dATP (0–800 μm, indicated with wedges). A, DNA/RNA-rA; B, DNA/RNA-1,N6-ϵrA. All reactions were carried out at 37 °C for 5 min in duplicate. The indicated data points are shown as means ± S.D. (error bars); see Table 1 for kcat and Km values (estimated using fits to a hyperbolic equation in Prism (GraphPad)); see “Experimental procedures” and Table S1 for the oligonucleotide sequences. P, FAM-labeled DNA primer.
In comparing the specificity constants for insertion of individual dNTPs opposite 1,N6-ϵrA, hpol η incorporated dATP 200-fold and dGTP 40-fold more efficiently than dTTP and dCTP, a result driven by the Km values (Table 1). Incorporation of dCTP showed the same low-specificity constant as dTTP (0.001 μm−1 min−1; Table 1). We conclude that hpol η catalyzes translesion synthesis across the damaged ribonucleotide 1,N6-ϵrA in a highly error-prone manner, with preference for insertion of deoxypurines, which can affect the reading frame of mRNA. This observation is in line with the previous bypass studies with the 1,N6-ϵdA adduct (82).
We next focused on the efficiency of hpol η as a reverse transcriptase (Table 1, Fig. 6, and Figs. S8–S11). Steady-state kinetic analysis revealed that the incorporation of dTTP across 1,N6-ϵrA was quite unfavorable, as seen in the TLS process. The specificity constant for insertion of dTTP (0.002 μm−1 min−1; Table 1 and Fig. S8) decreased 2100-fold compared with the RNA-rA template (4.3 μm−1 min−1; Table 1), driven by the much larger Km value for 1,N6-ϵrA template (Table 1) as compared with the RNA-rA template. For insertion of dATP opposite 1,N6-ϵrA, the specificity constant was 10-fold higher than with the RNA-rA template (Table 1 and Fig. 6), again driven by the Km difference (Table 1). Thus, hpol η prefers to insert dATP opposite 1,N6-ϵrA in an RNA template, similar to HIV-1 RT (83). In the case of dGTP insertion across 1,N6-ϵrA, the specificity constant was 2.5-fold higher compared with the RNA-rA template (Table 1 and Fig. S9). For dCTP insertion, the specificity constant was 1.25-fold lower compared with the RNA-rA template (Table 1 and Fig. S10). rUTP incorporation was only observed opposite the RNA-rA template (Table 1 and Fig. S11).
With 1,N6-ϵrA in the RNA template, hpol η incorporated dATP 200-fold, dGTP 25-fold, and dCTP 4-fold more efficiently than dTTP (Table 1), indicating that hpol η catalyzes reverse transcription across the damaged ribonucleotide 1,N6-ϵrA by the preferential addition of dNTPs instead of rNTPs. With the use of an RT strategy, the position of 1,N6-ϵrA in the RNA could be mapped concerning distinctive mutations in the DNA (84).
The efficiency of hpol η in RNA primer extension was examined (Table 1 and Figs. S12–S16). Steady-state kinetic analysis showed that insertion of dTTP opposite 1,N6-ϵrA was highly unfavorable, as observed for both TLS and RT processes (Table 1). The specificity constant (0.0004 μm−1 min−1) was 1100-fold lower compared with DNA-dA (0.45 μm−1 min−1) and 1200-fold lower than for a DNA-rA template (0.50 μm−1 min−1; Table 1 and Fig. S12). For dATP insertion opposite 1,N6-ϵrA, the specificity constant was 10-fold lower compared with a DNA-dA template and 15-fold lower compared with a DNA-rA template (Table 1 and Fig. S13). These results indicate a preference for dATP insertion opposite to the DNA-rA template (1.5-fold higher specificity constant) compared with a DNA-dA template. For dGTP insertion opposite 1,N6-ϵrA, the specificity constant was 1.7-fold lower compared with a DNA-dA template and 20-fold lower compared with a DNA-rA template (Table 1 and Fig. S14). These results indicate that dGTP is preferentially added opposite a DNA-rA template compared with DNA-dA, as judged by an 11-fold higher specificity constant. The dCTP incorporation was observed only with the DNA-dA and DNA-rA templates, not with 1,N6-ϵrA. The dCTP was preferentially added opposite the DNA-rA (7.5-fold higher specificity constant; Table 1 and Fig. S15). Among the four rNTPs, the insertion of rUTP was observed for DNA-dA and DNA-rA templates but not for the 1,N6-ϵrA template. The specificity constant for rUTP insertion opposite DNA-rA was 2-fold lower than with the DNA-dA template (0.0004 μm−1 min−1; Table 1 and Fig. S16).
The specificity constants for insertion of three dNTPs across 1,N6-ϵrA were compared. Interestingly, hpol η incorporated dGTP 100-fold and dATP 25-fold more efficiently compared with dTTP (Table 1), suggesting that hpol η extends an RNA primer opposite the damaged ribonucleotide 1,N6-ϵrA–containing DNA template by the preferential addition of dGTP.
hpol η generates frameshifts opposite the damaged ribonucleotide 1,N6-ϵrA
Steady-state kinetic analysis of hpol η–mediated TLS showed a preference for dATP and dGTP insertion opposite 1,N6-ϵrA. Other possible miscoding events during the lesion bypass were also considered, and semiquantitative LC-ESI-MS/MS analysis was performed using a previously developed protocol (85, 86) for TLS processing (see “Experimental procedures” and Table S1 for the oligonucleotide sequences used) because both the RT and RNA primer extension processes were slower. A 2′-deoxyuridine (dU)–containing primer and physiological concentrations of dNTPs were used for the full-length extension reactions (see “Experimental procedures” for details). The fully extended products were treated sequentially with uracil-DNA glycosylase (UDG) and piperidine to obtain short pieces of extended products (Fig. S17) (85). Sequences of full-length extended products and their relative yields were determined by LC-ESI-MS/MS analysis and are summarized in Table 2 (see Figs. S18–S35 and Tables S2–S12 for fragmentation patterns).
Table 2.
LC-ESI-MS/MS sequencing: Summary of products of extension of template-primer complexes by hpol η
X, 1, N6-ethenoadenosine (1,N6-ϵrA), adenosine (rA), or 2′-deoxyadenosine (dA). LC-ESI-MS/MS data used to summarize these products are presented in Figs. S18–S35 and Tables S2–S12. Δ1, a one-base deletion. T, DNA template; P, FAM-labeled dU-containing DNA primer.
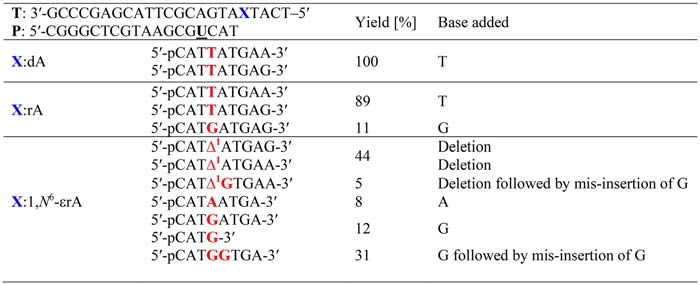
As expected, hpol η yielded error-free products for the DNA/DNA-dA complex, with blunt end additions of G and A, respectively (m/z 938.3 (−3) and 933.0 (−3); Figs. S18–S20 and Tables S2 and S3). hpol η is well-known to catalyze the blunt end addition of A and G to oligonucleotides (86–88). For the DNA/DNA-rA complex, hpol η yielded ∼89% extended products having T incorporated opposite rA (Table 2) with blunt end additions of G and A (m/z 938.3 (−3) and 933.0 (−3); see Figs. S21–S23 and Tables S4 and S5 for fragmentation patterns). Apart from the insertion of T, incorporation of G was also observed across rA, accounting for 11% of the products (m/z 946.7 (−3); Table 2 Fig. S24, and Table S6).
hpol η–mediated bypass of the DNA/DNA-1,N6-ϵrA complex was analyzed (Figs. S25–S35 and Tables S7–S12), yielding multiple extended products of the same m/z (837.0 (−3) and 831.9 (−3)) and eluted together (Figs. S26, S30, S31, and S34). Mass spectral analysis revealed that frameshift products (one base deletion) contributed 44% of the total, with blunt end additions of G and A, respectively (Table 2, m/z 837.0 (−3) and 831.9 (−3); Figs. S27 and S32 and Tables S7 and S10). Extended products with frameshift (one base deletion) followed by misinsertion of G were also observed (5%), with a blunt end addition of A (Table 2, m/z 837.0 (−3); Fig. S28 and Table S8). Overall, the frameshift products accounted for 49% of the total products derived using physiological concentrations of dNTPs. Apart from frameshift products, small amounts of products with insertion of G (12%) and A (8%) opposite 1,N6-ϵrA were also observed in the MS/MS analysis (Table 2, m/z 837.0 (−3) and 831.9 (−3); Figs. S29 and S33 and Tables S9 and S11). Interestingly, we did not detect insertion of T opposite 1,N6-ϵrA, indicating that hpol η is not reading this lesion as a deoxyadenosine residue. Surprisingly, LC-ESI-MS/MS analysis also revealed an unexpected extended product containing a G opposite 1,N6-ϵrA and the subsequent misinsertion of another G (insertion of two Gs; Table 2, m/z 842.0 (−3); Fig. S35 and Table S12). We included this product in a different category (Table 2) of base substitutions. Such base substitutions have been observed for 8-oxo-rG in human cells (24). LC-ESI-MS/MS provides insight regarding the frameshift products, and the steady-state kinetic and LC-ESI-MS/MS analyses both showed the insertion of A and G opposite 1,N6-ϵrA during TLS.
Human RNase H2–mediated recognition but partial incision of the damaged ribonucleotide 1,N6-ϵrA
RNase H2–mediated RER plays an important role in removing rNTPs in DNA (89), and we analyzed the endoribonuclease activity of RNase H2 enzyme opposite rA and 1,N6-ϵrA in the DNA template. On the basis of our steady-state kinetic and LC-ESI-MS/MS results, both rA and 1,N6-ϵrA in the modified DNA templates were paired with the sequences containing T, A, and G complementary to the modification site (CS_1 to CS_3; see “Experimental procedures” for details; Table S1). We performed the incision assays employing FAM-labeled DNA-dA-, DNA-rA-, and DNA-1,N6-ϵrA-modified templates paired with T, A, and G in their complementary sequences (Fig. 7). DNA-dA dsDNAs were used as negative controls, and, as expected, no cleaved product for the DNA-dA strand was observed (Fig. 7, lanes 1–6, 19–24, and 37–42). With DNA-rA, RNase H2 recognized and incised rA very efficiently within 5 min (96% cleaved product, 4 nucleotides) irrespective of its complementary base in dsDNAs (Fig. 7, lanes 7–12, 25–30, and 43–48). The endoribonuclease activity of human RNase H2 was drastically reduced when 1,N6-ϵrA–modified dsDNAs were paired with T, A, and G (Fig. 7, lanes 13–18, 31–36, and 49–54). These results indicate that RNase H2 recognizes the modified ribonucleotide 1,N6-ϵrA but exhibited only partial incision activity. For 1,N6-ϵrA dsDNAs under all pairing conditions, ∼30% cleaved product (4 nt) was observed after 1 h. RNase H2 could cleave a DNA strand containing 1,N6-ϵrA, whether positioned opposite dT, dG, or dA. To determine the rate of 5′-cleavage by RNase H2, we measured initial burst rates (Fig. S36) employing pre-steady-state kinetic assays using an established protocol (90). The burst amplitude was 38 ± 8 nm for DNA-rA and 1.8 ± 0.3 nm for DNA-1,N6-ϵrA. The burst rate demonstrated cleavage at rates of 47 ± 7 s−1 for DNA-rA and 78 ± 24 s−1 for DNA-1,N6-ϵrA (Fig. S36, inset), and the steady-state rate was 0.15 ± 0.02 s−1 for DNA-rA and 0.025 ± 0.001 s−1 for DNA-1,N6-ϵrA.
Figure 7.

Human RNase H2–mediated incision of ribonucleotide embedded in DNA. PAGE (20%, 7 m urea) was conducted of incision assays employing human RNase H2 (10 nm) with dsDNAs and different pairing conditions. A, DNA-X/DNA-dT; B, DNA-X/DNA-dA; C, DNA-X/DNA-dG (where X represents dA, rA, or 1,N6-ϵrA). All reactions were done in duplicate at 37 °C for 5, 15, 30, 45, and 60 min (time gradients indicated with wedges). S, substrate position (23 nt); P, product position (4 nt). See “Experimental procedures” and Table S1 for the DNA sequences used (FAM-labeled templates and biotin-labeled complementary strands (CS) were used).
E. coli RNase HII is reported to be very efficient in removing a wide range of damaged ribonucleotides (91), and we examined the endoribonuclease activity of E. coli RNase HII employing the same dsDNAs (Fig. S37). As expected, E. coli RNase HII recognized and incised rA very efficiently irrespective of its complementary base in dsDNAs (Fig. S37, lanes 7–12, 25–30, and 43–48), and there was no effect on the DNA-dA strand (Fig. S37, lanes 1–6, 19–24, and 37–42). Endoribonuclease cleavage was ∼50% (after 1 h) for 1,N6-ϵrA–modified DNA templates paired with T, A, and G (Fig. S37, lanes 13–18, 31–36, and 49–54). Our results indicate that E. coli RNase HII (91) also recognized 1,N6-ϵrA, but incision was slow.
Discussion
The DNA replication process is continuously challenged by various obstacles, such as non-B-form structures of DNA (e.g. G-quartets), fragile sites, transcription factors, and numerous DNA lesions (92). In addition to this, rNTP insertion during DNA replication is a major concern because more than one million rNTPs are incorporated per cell cycle (in human cells) due to their cellular abundance (93). rNTPs and dNTPs are the main precursors of RNAs and DNAs and can be damaged endogenously in the nucleotide pool (e.g. formation 8-oxo-dGTP). Recent studies have shown that 8-oxo-rG, an oxidized form of rGTP, is incorporated into DNAs and that nucleotide excision repair and RER mechanisms are not able to remove this adduct efficiently (24). 1,N6-ϵrA, a well-known oxidized adduct derived from rATP, is generated from VC (and its reactive metabolites) as well as from lipid peroxidation. 1,N6-ϵrA is known to affect protein translation, as discussed earlier. In the present study, we investigated the impact of rATP and the damaged ribonucleotide 1,N6-ϵrA on TLS, reverse transcription, RNA primer extension, and ribonucleotide incision processes catalyzed by hpol η and human RNase H2 employing steady-state kinetics, MS, and incision assays. Interestingly, the TLS process was strongly affected due to the presence of 1,N6-ϵrA in DNA as compared with rA (Fig. 2). It has previously been reported that hpol η bypasses the major groove DNA adduct 1,N6-ϵdA poorly (82, 94). Steady-state kinetic analysis revealed the error-prone bypass of 1,N6-ϵrA with the preferable addition of deoxypurines (Table 1). These outcomes are congruent with the results of our previous studies on hpol η–catalyzed TLS past a 1,N6-ϵdA DNA lesion (i.e. more efficient insertion of dATP (3.7-fold) and dGTP (2.5-fold) than dTTP opposite 1,N6-ϵdA) (82).
Among the dNTPs, the dGTP insertion opposite DNA-dA and DNA-rA templates suggests that hpol η preferably adds dGTP opposite DNA-rA, with a specificity constant 1.6-fold higher than for the DNA-dA template (Table 1). In comparing the specificity constants for DNA-dA and DNA-rA templates, the insertion of dATP was 1.4-fold more efficient for DNA-rA compared with the DNA-dA template. dCTP insertion opposite a DNA-rA template yielded a higher specificity constant (1.6-fold) than for the DNA-dA template, with the basis being in the Km values (Table 1).
When we compare 1,N6-ϵdA and 1,N6-ϵrA lesions, the impact of ribo-backbone on the TLS process can be seen. These studies led to a new direction in understanding whether slow TLS across 1,N6-ϵrA is due to the ribo-backbone or the damaged ribonucleotide. Recent investigations with 8-oxo-rG in DNA indicate that the ribonucleotide backbone has a strong influence on biological processes (24, 35).
Further, the specificity constants for insertion of dNTP versus rNTP opposite the DNA-dA and DNA-rA templates were compared. Interestingly, the incorporation of dTTP was 700-fold more efficient compared with rUTP for the DNA-dA template, and incorporation of dTTP was 410-fold more efficient compared with rUTP for the DNA-rA template (Table 1). The specificity constants for dATP and rATP insertion opposite the DNA-dA and DNA-rA templates indicated that incorporation of dATP was favored 900-fold compared with rATP for the DNA-dA template, and incorporation of dATP was 1200-fold more efficient than rATP for the DNA-rA template. It is important to note that the sugar selectivity was well-maintained during the TLS as reflected in the kinetic parameters (Table 1).
LC-ESI-MS/MS analysis showed that the presence of 1,N6-ϵrA in DNA produces extensive frameshift (one-base deletion) products (Table 2). On the basis of earlier results, we hypothesize that TLS may occur in two different ways. First, the incoming A or G can be stacked across from 1,N6-ϵrA in cross-strand fashion and result in frameshifts (one base deletion), with hpol η skipping the lesion (82, 95). This process could contribute to the misinsertion opposite the next base T, positioned 5′ to the lesion on the template strand. These results are consonant with our previous LC-ESI-MS/MS analysis of a 1,N6-ϵdA DNA lesion, which yielded 37% frameshifts (82). Structural studies revealed that incoming dATP and dGTP molecules are present in a staggered configuration across the anti-oriented 1,N6-ϵdA lesion (82). Subsequent analyses of these studies for both of the lesions indicates that insertion of A and G was reduced drastically across 1,N6-ϵrA as compared with 1,N6-ϵdA. We assume that the ribo-backbone of 1,N6-ϵrA may play an important role in this case. The addition of two Gs by hpol η, possibly due to the tight binding of hpol η with dGTP during the extension process, leads to the apparent misincorporation of G opposite the T positioned 5′ to the lesion (base substitution) or due to the misalignment of the template strand (95, 96). Overall, we have observed extensive frameshifts (one deletion) followed by misinsertion of G as well as another unusual product that indicates the insertion of two Gs (Table 2). The preference for insertion of purines over pyrimidines is presumably due to better-stacking interactions to achieve a proper orientation of the lesion and incoming nucleotide in the active site of hpol η for further replication. Previous TLS studies with 1,N6-ϵdA and an abasic site also showed that hpol η predominantly follows the purine rule (82, 97). The in vitro ribonucleotide incision assays indicate that recognition of unmodified rA and 1,N6-ϵrA by human RNase H2 is completely different, apparently due to the presence of an etheno group on the adenosine (Fig. 7). It is important to note that ribonucleotide excision repair involves RNase H2–mediated incision of a 5′-ribonucleotide followed by strand displacement, flap cleavage by the flap endonuclease FEN1, and ligation (13–15). Our results indicate that E. coli RNase HII (91) recognizes and incises 1,N6-ϵrA slowly. Overall, RNase H2–mediated incomplete incision of 1,N6-ϵrA from DNA poses a threat to genome stability. Alternative repair pathways such as base excision repair may have potential roles in removing this adduct from DNA employing the alkyladenine DNA glycosylase AAG. If this adduct persists in the DNA, the versatile human TLS pol η may tolerate this adduct but in an error-prone way. These studies provide new insight into the consequences of 1,N6-ϵrA when TLS is the only means of coping with damaged ribonucleotides in DNA (outlined in Fig. 8).
Figure 8.

Processing of 1,N6-ϵrA in DNA.
Damage to DNA has been studied extensively, as compared with RNA. RNA is an innately reactive biomolecule, reacting with both endogenous and exogenous genotoxic agents to yield a plethora of lesions and changes in structural integrity. Leumann and co-workers (98) studied various damaged bases in RNA (8-oxo-rA, 8-oxo-rG, ribo-abasic site, 5-hydroxycytidine, and uridine) in the context of protein translation, as discussed earlier. The ϵ-adducts in RNA were also detected in various tissues of mice after exposure to [14C]VC as discussed earlier (63). In liver, RNA accumulated 1,N6-ϵrA, and in vivo levels of ϵ-adducts in RNA were higher than in DNA (63). 1,N6-ϵrA was also detected in mouse liver RNA after treatment with ethyl carbamate (64).
To this end, we explored the 1,N6-ϵrA modification to understand the RT employing hpol η. As a result, hpol η was able to synthesize DNA utilizing 1,N6-ϵrA–modified RNA as a template with the preferred addition of dNTPs (Fig. 3). These studies suggested that hpol η may utilize RNA as a template for double-strand break repair (99, 100). However, hpol η showed reduced reverse transcriptase activities as compared with HIV-1 RT. The preferable addition of deoxypurines across 1,N6-ϵrA by hpol η (Table 1) was observed. Previous RT studies showed a similar trend with HIV-1 RT, which preferentially added dATP and dGTP across 1,N6-ϵrA (83). Overall, 1,N6-ϵrA was bypassed by hpol η, and yielded full-length DNA molecules in this study may be due to the lack of a more effective polar filter, as proposed by Nair and co-workers (75).
RNA primer extension activities indicate that hpol η may play a role in the extension of RNA primer across the templates containing unmodified ribonucleotides using only dNTPs (Fig. 4). The incorporation pattern showed that the hpol η prefers to add deoxypurines (preferably dGTP) across 1,N6-ϵrA. This result differs from the TLS and RT processes but the basis is unclear for the G preference over A with the RNA primer.
RNase H2 activity toward an oligonucleotide duplex was strongly attenuated when rA (opposite the DNA strand) was replaced by 1,N6-ϵrA (Fig. S36), as seen in both the steady-state and pre-steady-state kinetics. The steady-state kinetic rate was reduced 6-fold. RNase H2 shows burst kinetics, indicative of a rate-limiting step following cleavage (38, 101). The amplitude of the burst (Fig. S36) was reduced from 38 to 1.8 nm (i.e. 20-fold). Although the calculated pre-steady-state burst rate was still 78 s−1, the error in this measurement is high due to the very low amplitude. We previously reported that the presence of a cyclobutane dimer (T∧T) opposite the DNA strand only reduced the rate of RNase H2 cleavage ∼2-fold relative to two Ts (T-T) (38). The effect of the presence of 1,N6-ϵrA was much more dramatic than a cyclobutane dimer. There is precedent for the presence of a DNA adduct to change the burst amplitude of an enzyme, specifically a DNA polymerase (102, 103). One kinetic explanation, which has been supported over the years, is that the presence of an adduct facilitates the equilibrium of the enzyme with nonproductive forms (103, 104). This may well be the case here, although other kinetic possibilities could be involved and cannot be excluded without more extensive experiments. The main point is that the adduct 1,N6-ϵrA is highly disruptive of RNase H2 catalysis, as judged by both steady-state and pre-steady-state kinetics (Fig. S36).
In summary, our findings indicate the unique roles of hpol η in dealing with an oxidatively damaged ribonucleotide. hpol η tolerates the damaged ribonucleotide 1,N6-ϵrA during the TLS process with lower efficiency and also generates extensive frameshifts. RNase H2–mediated partial incision of 1,N6-ϵrA in DNA was supported by the observation of a (low) burst amplitude. hpol η acts as specific reverse transcriptase in the presence of damaged ribonucleotide 1,N6-ϵrA in RNA, showing preference for dNTPs. In addition, poor RNA primer extension activities were observed by hpol η.
Experimental procedures
Reagents
The catalytic core of human DNA pol η (amino acids 1–432) was expressed in E. coli and purified as reported previously (105, 106). Human RNase H2 was expressed in E. coli and purified as reported previously (107, 108). Unlabeled dNTPs, rNTPs, UDG, and E. coli RNase HII were purchased from New England Biolabs (Ipswich, MA). C18 Sep-Pak columns were purchased from Waters Corp. (Milford, MA). Micro Biospin-6 columns were purchased from Bio-Rad. Piperidine was from Sigma–Aldrich. Unmodified oligonucleotides and FAM-labeled DNA and RNA primers (twice HPLC-purified) were purchased from Integrated DNA Technologies (Coralville, IA). 1,N6-ϵrA phosphoramidite and FAM-labeled modified oligonucleotides were purchased from ChemGenes Corp. (Wilmington, MA). The tert-butyldimethylsilyl–protected ribo-phosphoramidites were purchased from Glen Research (Sterling, VA).
Oligonucleotide synthesis, purification, and characterization
The oligonucleotide sequences were designed on the basis of our previous in vitro and cell-based studies (38, 74). Solid-phase synthesis of the 1,N6-ϵrA–modified DNAs and RNAs was done on a PerSeptive Biosystems model 8909 DNA synthesizer (see Table S1 for the in-house oligonucleotide sequences). tert-Butyldimethylsilyl–protected ribo-phosphoramidites were used, and the modified ribo- and deoxyribooligonucleotides were synthesized on a 1-μmol scale using the appropriate controlled pore glass as solid support. 5-Ethylthio-1H-tetrazole was used as an activator, and the coupling time for the modified phosphoramidites was 10 min. The deprotection of modified ribo- and deoxyribooligonucleotides was carried out using a reported procedure (83). The crude modified ribo- and deoxyribooligonucleotides were purified by denaturing PAGE (20% PAGE, 7 m urea) at 55 W for 2.5 h using 1× TBE buffer (89 mm Tris-HCl buffer containing boric acid (89 mm) and 2 mm EDTA, pH 8.0). The gel was visualized under a UV lamp at 260 nm, and the desired DNA/RNA bands were isolated from the gel and extracted using TEN buffer (10 mm Tris-HCl buffer containing 1 mm EDTA and 300 mm NaCl, pH 8.0) overnight at room temperature. The modified ribo- and deoxyribooligonucleotides were desalted using C18 Sep-Pak columns, and their integrity was confirmed by MALDI-TOF in each case using a matrix of 3-hydroxypicolinic acid and ammonium citrate (9:1 molar ratio) (109) and represented as an average mass (positive linear mode; Figs. S38–S39).
Physiological concentrations of dNTPs and rNTPs (78, 79)
All polymerase reactions were carried out using physiological concentrations of dNTPs/rNTPs. Physiological concentrations of dNTPs used in these studies were 25 μm dATP, 30 μm dCTP, 90 μm dGTP, and 40 μm dTTP. Physiological concentrations of rNTPs used in these studies were 2 mm rATP, 0.25 mm rCTP, 0.5 mm rGTP, and 0.5 mm rUTP. The desired stocks of dNTPs/rNTPs were prepared in RNase-free water.
Full-length extension reactions for translesion synthesis, reverse transcription, and RNA primer extension studies (Figs. 2 (A and C), 3 (A and C), and 4 (A and C) and Fig. S1 (A and C)).
A primer-template complex containing a FAM-labeled DNA or RNA primer and an unmodified or modified template were annealed (1:1 molar ratio) at 75 °C for 5 min followed by slow cooling overnight. Full-length extension reactions were carried out using 40 mm Tris-HCl buffer (pH 7.5) containing 100 mm KCl, 5 mm MgCl2, 5% glycerol (v/v), 10 mm DTT, and 0.1 mg/ml BSA at 37 °C. The final concentration of primer-template complex was 0.5 μm, and 200 nm hpol η was used to obtain fully extended primers. Reactions were initiated by adding a 1-μl mixture of dNTPs/rNTPs (physiological concentrations) to a total volume of 10 μl. Aliquots (1.5 μl) of reaction mixtures were taken at different time points (0, 5, 30, 60, and 180 min) and quenched with 8.5 μl of 10 mm EDTA (pH 8.0) in 95% deionized formamide (v/v). Products were separated using 20% PAGE (7 m urea), and results were visualized using a Typhoon scanner (GE Healthcare) and analyzed by ImageJ software (80).
Single nucleotide incorporation reactions (Figs. 2 (B and D), 3 (B and D), and 4 (B and D) and Fig. S1 (B and D))
A primer-template DNA complex containing a FAM-labeled DNA or RNA primer and an unmodified or modified template were annealed (1:1 molar ratio) at 75 °C for 5 min, followed by slow cooling overnight. All single-nucleotide insertion reactions were performed using 40 mm Tris-HCl buffer (pH 7.5) containing 100 mm KCl, 5 mm MgCl2, 5% glycerol (v/v), 10 mm DTT, and 0.1 mg/ml BSA at 37 °C. The final concentration of primer-template complex was 0.5 μm, and 50 nm hpol η was used, followed by the addition of 1 μl of an individual dNTP/rNTP (physiological concentrations) to a total volume of 10 μl. Aliquots (1.5 μl) of reaction mixtures were taken at each time point (0, 5, 30, and 60 min) and quenched with 8.5 μl of 10 mm EDTA (pH 8.0) in 95% deionized formamide (v/v). Products were separated using 20% PAGE (7 m urea), and results were visualized using a Typhoon scanner (GE Healthcare) and analyzed by ImageJ software.
Steady-state kinetics (Table 1, Figs. 5 and 6, and Figs. S2–S16)
A primer-template DNA complex containing a FAM-labeled DNA or RNA primer and an unmodified or modified template were annealed (1:1 molar ratio) at 75 °C for 5 min, followed by slow cooling overnight. All steady-state kinetic reactions were performed using 40 mm Tris-HCl buffer (pH 7.5) containing 100 mm KCl, 5 mm MgCl2, 5% glycerol (v/v), 10 mm DTT, and 0.1 mg/ml BSA at 37 °C for 5 min. The final concentration of primer-template complex was 0.5 μm, and hpol η was added, ranging from 0.25 to 38 nm to achieve steady-state kinetics of dNTP/rNTP insertion. Reactions were initiated by adding 1 μl of an individual dNTP/rNTP stock solution, at each of 10 different concentrations, to a total volume of 5 μl. A general rule of ≤20% (P + 1) product formation was maintained (80). Aliquots (1.5 μl) of reaction mixtures were taken at 5-min time points and quenched with 8.5 μl of 10 mm EDTA (pH 8.0) in 95% deionized formamide (v/v). Products were separated using 20% PAGE (7 m urea), and results were visualized using a Typhoon scanner (GE Healthcare) and analyzed by ImageJ software. Data points are shown as means ± S.D. from two independent experiments (see Table 1 for kcat and Km values) and estimated using fit to a hyperbolic equation in Prism software (GraphPad, San Diego, CA) to obtain kcat/Km (ksp) and kcat directly (81).
LC-ESI-MS/MS analysis: Full-length extension followed by UDG and piperidine treatment (Fig. S17)
A 2′-deoxyuridine (dU)-containing FAM-labeled 18-mer primer and an unmodified or modified template were annealed (1:1 molar ratio) at 75 °C for 5 min followed by slow cooling overnight. The full-length extension reactions were carried out under the same reaction conditions as described for the full-length extension assays, except that the final concentrations were as follows: primer-template complex was 2.5 μm; hpol η was 1.2 μm for DNA/DNA-dA and DNA/DNA-rA complexes and 2 μm for DNA/DNA-1,N6-ϵrA complex, in a total reaction volume of 85 μl. Reactions were carried out in the presence of mixture of dNTPs (physiological concentrations) at 37 °C for 4 h for DNA/DNA-dA and DNA/DNA-rA complexes and overnight for DNA/DNA-1,N6-ϵrA complex. Reactions were terminated using Micro Biospin-6 column separations to extract Mg2+ and dNTPs. The resulting products were treated with 25 units of UDG at 37 °C for 4 h, followed by 0.25 m piperidine at 95 °C for 1 h (85). The reaction mixture was evaporated to dryness by lyophilization. The dried pellet was resuspended in 500 μl of nuclease-free water and taken to dryness by lyophilization. Finally, this dried pellet was resuspended in 25 μl of nuclease-free water for LC-ESI-MS/MS analysis.
LC-ESI-MS/MS analysis (Figs. S18–S35 and Tables S2–S12)
LC-ESI-MS/MS analyses were performed on a Finnigan LTQ mass spectrometer (Thermo Scientific Corp., San Jose, CA) connected to an Acquity ultraperformance LC (UPLC) system (Waters). The data were acquired in the ESI negative ion mode using an C18 column (Acquity UPLC BEH, 1.7 μm, 2.1 × 100 mm). UPLC conditions were as follows. Buffer A contained 10 mm NH4CH3CO2, 2% CH3CN, and 98% H2O (v/v), pH 7.0, and buffer B contained 10 mm NH4CH3CO2, 95% CH3CN, 5% H2O (v/v), pH 7.0. The column temperature was 50 °C. The following gradient program was used with a flow rate of 0.3 ml/min: 0–30% B over 8 min, 30–90% B over 1 min, held at 90% B for 1 min, 90–0% B over 1 min, held at 0% B for 4 min (all v/v). A 15-μl sample aliquot was injected with an autosampler system. ESI settings were as follows: source voltage 4.5 kV, sweep gas flow rate 5, auxiliary gas flow rate 30, capillary voltage −48 V, sheath gas flow 60, capillary temperature 270 °C, and tube lens voltage −143 V. MS/MS conditions were as follows: activation Q 0.25, activation time 30 ms. Product spectra were acquired over the range m/z 300–2000. The most abundant species were fragmented in the ion trap with a normalized collision energy of 35% (data-dependent mode). The extended product sequences were identified (Figs. S18–S35 and Tables S2–S12) by comparing the observed CID fragments with the theoretical values using the Mongo Oligo Mass Calculator version 2.08 (State University of New York at Albany). The relative yields of extended product sequences were calculated based on the peak areas of extracted ion chromatograms. For the co-eluted products, relative peak areas were calculated on the basis of the intensity of representative fragments (as shown in Fig. S30 for a6-B(−2) and Fig. S34 for W3(−1) fragments). The fragmentation patterns of the observed products were confirmed by comparing with the fragmentation patterns of commercial standards (IDT, Coralville, IA).
RNase H2–mediated incision assays (Fig. 7 and Fig. S37)
The complementary sequences (CS_1 to CS_3; Table S1) were annealed with FAM-labeled unmodified and modified template (1:1 molar ratio) at 75 °C for 5 min followed by slow cooling overnight (see Table S1 for the oligonucleotide sequences). The final concentration of dsDNA was 0.5 μm. The assays were done at 37 °C in the incision buffer: 40 mm Tris-HCl buffer (pH 7.5) containing 50 mm NaCl, 10 mm MgCl2, 1 mm DTT, 5% glycerol (v/v), and 0.1 mg/ml BSA. The reactions were initiated by adding 10 nm RNase H2. Aliquots (1.5 μl) of reaction mixtures were taken at different time points (0, 5, 15, 30, 45, and 60 min) and quenched with 8.5 μl of slow-moving dye (10 mm EDTA (pH 8.0) in 95% deionized formamide (v/v) and xylene cyanol FF, only slow moving dye). Products were separated using 20% PAGE (7 m urea), and the results were visualized using a Typhoon scanner (GE Healthcare) and analyzed by ImageJ software. The E. coli RNase HII-mediated incision assays were carried out in 1× ThermoPol reaction buffer (New England Biolabs) using 1 unit of enzyme at 37 °C. The same time points were taken as described above.
Pre-steady-state kinetics (Fig. S36)
The complementary sequence (CS_1; Table S1) was annealed with a FAM-labeled unmodified as well as a modified ribonucleotide-containing DNA template (1:1 molar ratio) at 75 °C for 5 min, followed by slow cooling overnight (see Table S1 for oligonucleotide sequences). The final concentration of dsDNA was 0.25 μm, and concentration of RNase H2 was 203 nm (for DNA-rA) and 263 nm (for DNA-1,N6-ϵrA), respectively. Pre-steady-state kinetic experiments were carried out at 37 °C in the incision buffer (40 mm Tris-HCl buffer (pH 7.5) containing 50 mm NaCl, 10 mm MgCl2, 1 mm DTT, 5% glycerol (v/v), and 0.1 mg/ml BSA. The dsDNA and RNase H2 were mixed in an RQF-3 rapid quench-flow instrument (KinTek Corp., Snow Shoe, PA), allowing the reaction to proceed for time ranges from 0.005 to 2 s. Aliquots (6 μl) of each reaction mixture were taken with 24 μl of slow-moving dye (0.5 mm EDTA (pH 8.0) in 90% deionized formamide (v/v) and xylene cyanol FF (0.01%, w/v)). Part of each reaction mixture (7 μl) was loaded on the 20% PAGE (7 m urea), and the results were visualized using a Typhoon scanner (GE Healthcare) and analyzed by ImageJ software, and the data were fit to a burst equation, y = A(1 − e−kpt) + kssE0t, using GraphPad Prism software (GraphPad), where A denotes the apparent concentration of the active form of the enzyme, kp denotes the burst rate, kss represents the steady-state rate, E0 is the total enzyme concentration, and t is the time. The initial time points (0–0.08 s) were analyzed by non-linear regression of an exponential decay equation fits in Prism software and presented in Fig. S36 (inset).
Supporting information
The supporting information contains a list of oligonucleotide sequences used for the studies; PAGE analyses of full-length extension and single-nucleotide incorporation using physiological concentrations of dNTPs and rNTPs; PAGE analyses of steady-state kinetic analysis and the respective Prism graphs; PAGE analyses of full-length extension assays for LC-ESI-MS/MS analysis using physiological concentrations of dNTPs; LC-ESI-MS/MS sequencing analysis of full-length extension reactions for DNA/DNA-dA, DNA/DNA-rA, and DNA/DNA-1,N6-ϵrA complexes; respective extracted ion chromatograms and CID fragmentation spectrums; Prism graphs of pre-steady-state kinetic analysis; PAGE of E. coli RNase HII-mediated incision assays; and the MALDI spectrum for DNA-1,N6-ϵrA and RNA-1,N6-ϵrA.
Data availability
All data are included in the article and supporting information.
Author contributions
P. P. G. formal analysis; P. P. G. investigation; P. P. G. methodology; P. P. G. and F. P. G. writing-original draft; P. P. G. and F. P. G. writing-review and editing; F. P. G. conceptualization; F. P. G. resources; F. P. G. supervision; F. P. G. funding acquisition; F. P. G. project administration.
Supplementary Material
Acknowledgments
We thank Prof. Carmelo J. Rizzo for providing access to the DNA synthesizer, Dr. Yan Su for help with expression and purification of hpol η and RNase H2, and Prof. Martin Egli for valuable suggestions. We also thank K. A. Trisler for assistance in the preparation of the manuscript.
This work was supported by National Institutes of Health Grant R01 ES026955 (to F. P. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S12 and Figs. S1–S39.
- pol
- polymerase
- hpol
- human polymerase
- ESI
- electrospray ionization
- RER
- ribonucleotide excision repair
- TLS
- translesion synthesis
- VC
- vinyl chloride
- VB
- vinyl bromide
- 1,N6-ϵrA
- 1,N6-ethenoadenosine
- ϵ
- etheno
- FAM
- 6-carboxyfluorescein
- UDG
- uracil-DNA glycosylase
- UPLC
- ultraperformance liquid chromatography
- CID
- collision-induced dissociation
- RT
- reverse transcriptase
- nt
- nucleotides.
References
- 1. Caldecott K. W. (2014) Ribose—an internal threat to DNA. Science 343, 260–261 10.1126/science.1248234 [DOI] [PubMed] [Google Scholar]
- 2. Forslund J. M. E., Pfeiffer A., Stojko5 vič5 G., Wanrooij P. H., and Wanrooij S. (2018) The presence of rNTPs decreases the speed of mitochondrial DNA replication. PLoS Genet. 14, e1007315 10.1371/journal.pgen.1007315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams J. S., and Kunkel T. A. (2014) Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst.) 19, 27–37 10.1016/j.dnarep.2014.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wanrooij P. H., and Chabes A. (2019) Ribonucleotides in mitochondrial DNA. FEBS Lett. 593, 1554–1565 10.1002/1873-3468.13440 [DOI] [PubMed] [Google Scholar]
- 5. Nick McElhinny S. A., Watts B. E., Kumar D., Watt D. L., Lundström E.-B., Burgers P. M. J., Johansson E., Chabes A., and Kunkel T. A. (2010) Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. U.S.A. 107, 4949–4954 10.1073/pnas.0914857107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yao N. Y., Schroeder J. W., Yurieva O., Simmons L. A., and O'Donnell M. E. (2013) Cost of rNTP/dNTP pool imbalance at the replication fork. Proc. Natl. Acad. Sci. U.S.A. 110, 12942–12947 10.1073/pnas.1309506110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pai C.-C., and Kearsey S. E. (2017) A critical balance: dNTPs and the maintenance of genome stability. Genes (Basel) 8, E57 10.3390/genes8020057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vaisman A., and Woodgate R. (2018) Ribonucleotide discrimination by translesion synthesis DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 53, 382–402 10.1080/10409238.2018.1483889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kennedy E. M., Amie S. M., Bambara R. A., and Kim B. (2012) Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J. Biol. Chem. 287, 14280–14288 10.1074/jbc.M112.348482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeRose E. F., Perera L., Murray M. S., Kunkel T. A., and London R. E. (2012) Solution structure of the Dickerson DNA dodecamer containing a single ribonucleotide. Biochemistry 51, 2407–2416 10.1021/bi201710q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Evich M., Spring-Connell A. M., Storici F., and Germann M. W. (2016) Structural impact of single ribonucleotide residues in DNA. ChemBioChem 17, 1968–1977 10.1002/cbic.201600385 [DOI] [PubMed] [Google Scholar]
- 12. Fu I., Smith D. J., and Broyde S. (2019) Rotational and translational positions determine the structural and dynamic impact of a single ribonucleotide incorporated in the nucleosome. DNA Repair (Amst.) 73, 155–163 10.1016/j.dnarep.2018.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reijns M. A. M., Rabe B., Rigby R. E., Mill P., Astell K. R., Lettice L. A., Boyle S., Leitch A., Keighren M., Kilanowski F., Devenney P. S., Sexton D., Grimes G., Holt I. J., Hill R. E., et al. (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022 10.1016/j.cell.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sparks J. L., Chon H., Cerritelli S. M., Kunkel T. A., Johansson E., Crouch R. J., and Burgers P. M. (2012) RNase H2-initiated ribonucleotide excision repair. Mol. Cell 47, 980–986 10.1016/j.molcel.2012.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerritelli S. M., and Crouch R. J. (2016) The balancing act of ribonucleotides in DNA. Trends Biochem. Sci. 41, 434–445 10.1016/j.tibs.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cerritelli S. M., and Crouch R. J. (2019) RNase H2-RED carpets the path to eukaryotic RNase H2 functions. DNA Repair (Amst.) 84, 102736 10.1016/j.dnarep.2019.102736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cerritelli S. M., and Crouch R. J. (2009) Ribonuclease H: the enzymes in eukaryotes. FEBS J. 276, 1494–1505 10.1111/j.1742-4658.2009.06908.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uehara R., Cerritelli S. M., Hasin N., Sakhuja K., London M., Iranzo J., Chon H., Grinberg A., and Crouch R. J. (2018) Two RNase H2 mutants with differential rNMP processing activity reveal a threshold of ribonucleotide tolerance for embryonic development. Cell Rep. 25, 1135–1145.e5 10.1016/j.celrep.2018.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kellner V., and Luke B. (2020) Molecular and physiological consequences of faulty eukaryotic ribonucleotide excision repair. EMBO J. 39, e102309 10.15252/embj.2019102309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghodgaonkar M. M., Lazzaro F., Olivera-Pimentel M., Artola-Borán M., Cejka P., Reijns M. A., Jackson A. P., Plevani P., Muzi-Falconi M., and Jiricny J. (2013) Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol. Cell 50, 323–332 10.1016/j.molcel.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pryor J. M., Conlin M. P., Carvajal-Garcia J., Luedeman M. E., Luthman A. J., Small G. W., and Ramsden D. A. (2018) Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 361, 1126–1129 10.1126/science.aat2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Potenski C. J., and Klein H. L. (2014) How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res. 42, 10226–10234 10.1093/nar/gku773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cronan G. E., Kouzminova E. A., and Kuzminov A. (2019) Near-continuously synthesized leading strands in Escherichia coli are broken by ribonucleotide excision. Proc. Natl. Acad. Sci. U.S.A. 116, 1251–1260 10.1073/pnas.1814512116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sassa A., Tada H., Takeishi A., Harada K., Suzuki M., Tsuda M., Sasanuma H., Takeda S., Sugasawa K., Yasui M., Honma M., and Ura K. (2019) Processing of a single ribonucleotide embedded into DNA by human nucleotide excision repair and DNA polymerase η. Sci. Rep. 9, 13910 10.1038/s41598-019-50421-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conover H. N., Lujan S. A., Chapman M. J., Cornelio D. A., Sharif R., Williams J. S., Clark A. B., Camilo F., Kunkel T. A., and Argueso J. L. (2015) Stimulation of chromosomal rearrangements by ribonucleotides. Genetics 201, 951–961 10.1534/genetics.115.181149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moss C. F., Dalla Rosa I., Hunt L. E., Yasukawa T., Young R., Jones A. W. E., Reddy K., Desai R., Virtue S., Elgar G., Voshol P., Taylor M. S., Holt I. J., Reijns M. A. M., and Spinazzola A. (2017) Aberrant ribonucleotide incorporation and multiple deletions in mitochondrial DNA of the murine MPV17 disease model. Nucleic Acids Res. 45, 12808–12815 10.1093/nar/gkx1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Colussi C., Parlanti E., Degan P., Aquilina G., Barnes D., Macpherson P., Karran P., Crescenzi M., Dogliotti E., and Bignami M. (2002) The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 12, 912–918 10.1016/S0960-9822(02)00863-1 [DOI] [PubMed] [Google Scholar]
- 28. Foti J. J., Devadoss B., Winkler J. A., Collins J. J., and Walker G. C. (2012) Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336, 315–319 10.1126/science.1219192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Randerath K., Reddy R., Danna T. F., Watson W. P., Crane A. E., and Randerath E. (1992) Formation of ribonucleotides in DNA modified by oxidative damage in vitro and in vivo: characterization by 32P-postlabeling. Mutat. Res. 275, 355–366 10.1016/0921-8734(92)90038-Q [DOI] [PubMed] [Google Scholar]
- 30. Markkanen E. (2017) Not breathing is not an option: how to deal with oxidative DNA damage. DNA Repair 59, 82–105 10.1016/j.dnarep.2017.09.007 [DOI] [PubMed] [Google Scholar]
- 31. Shimizu M., Gruz P., Kamiya H., Masutani C., Xu Y., Usui Y., Sugiyama H., Harashima H., Hanaoka F., and Nohmi T. (2007) Efficient and erroneous incorporation of oxidized DNA precursors by human DNA polymerase η. Biochemistry 46, 5515–5522 10.1021/bi062238r [DOI] [PubMed] [Google Scholar]
- 32. Pursell Z. F., McDonald J. T., Mathews C. K., and Kunkel T. A. (2008) Trace amounts of 8-oxo-dGTP in mitochondrial dNTP pools reduce DNA polymerase γ replication fidelity. Nucleic Acids Res. 36, 2174–2181 10.1093/nar/gkn062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sastre-Moreno G., Sánchez A., Esteban V., and Blanco L. (2014) ATP insertion opposite 8-oxo-deoxyguanosine by Pol4 mediates error-free tolerance in Schizosaccharomyces pombe. Nucleic Acids Res. 42, 9821–9837 10.1093/nar/gku711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katafuchi A., Sassa A., Niimi N., Grúz P., Fujimoto H., Masutani C., Hanaoka F., Ohta T., and Nohmi T. (2010) Critical amino acids in human DNA polymerases η and κ involved in erroneous incorporation of oxidized nucleotides. Nucleic Acids Res. 38, 859–867 10.1093/nar/gkp1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sassa A., Yasui M., and Honma M. (2019) Current perspectives on mechanisms of ribonucleotide incorporation and processing in mammalian DNA. Genes Environ. 41, 3 10.1186/s41021-019-0118-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berglund A.-K., Navarrete C., Engqvist M. K. M., Hoberg E., Szilagyi Z., Taylor R. W., Gustafsson C. M., Falkenberg M., and Clausen A. R. (2017) Nucleotide pools dictate the identity and frequency of ribonucleotide incorporation in mitochondrial DNA. PLoS Genet. 13, e1006628 10.1371/journal.pgen.1006628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Su Y., Egli M., and Guengerich F. P. (2016) Mechanism of ribonucleotide incorporation by human DNA polymerase η. J. Biol. Chem. 291, 3747–3756 10.1074/jbc.M115.706226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su Y., Ghodke P. P., Egli M., Li L., Wang Y., and Guengerich F. P. (2019) Human DNA polymerase η has reverse transcriptase activity in cellular environments. J. Biol. Chem. 294, 6073–6081 10.1074/jbc.RA119.007925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vaisman A., and Woodgate R. (2017) Translesion DNA polymerases in eukaryotes: what makes them tick? Crit. Rev. Biochem. Mol. Biol. 52, 274–303 10.1080/10409238.2017.1291576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang W., and Gao Y. (2018) Translesion and repair DNA polymerases: diverse structure and mechanism. Annu. Rev. Biochem. 87, 239–261 10.1146/annurev-biochem-062917-012405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meroni A., Nava G. M., Bianco E., Grasso L., Galati E., Bosio M. C., Delmastro D., Muzi-Falconi M., and Lazzaro F. (2019) RNase H activities counteract a toxic effect of polymerase η in cells replicating with depleted dNTP pools. Nucleic Acids Res. 47, 4612–4623 10.1093/nar/gkz165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sassa A., Çaǧlayan M., layan M., Rodriguez Y., Beard W. A., Wilson S. H., Nohmi T., Honma M., and Yasui M. (2016) Impact of ribonucleotide backbone on translesion synthesis and repair of 7,8-dihydro-8-oxoguanine. J. Biol. Chem. 291, 24314–24323 10.1074/jbc.M116.738732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Acharya N., Manohar K., Peroumal D., Khandagale P., Patel S. K., Sahu S. R., and Kumari P. (2019) Multifaceted activities of DNA polymerase η: beyond translesion DNA synthesis. Curr. Genet. 65, 649–656 10.1007/s00294-018-0918-5 [DOI] [PubMed] [Google Scholar]
- 44. Kreisel K., Engqvist M. K. M., Kalm J., Thompson L. J., Boström M., Navarrete C., McDonald J. P., Larsson E., Woodgate R., and Clausen A. R. (2019) DNA polymerase η contributes to genome-wide lagging strand synthesis. Nucleic Acids Res. 47, 2425–2435 10.1093/nar/gky1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garcia-Exposito L., Bournique E., Bergoglio V., Bose A., Barroso-Gonzalez J., Zhang S., Roncaioli J. L., Lee M., Wallace C. T., Watkins S. C., Opresko P. L., Hoffmann J. S., and O'Sullivan R. J. (2016) Proteomic profiling reveals a specific role for translesion DNA polymerase η in the alternative lengthening of telomeres. Cell Rep. 17, 1858–1871 10.1016/j.celrep.2016.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barnes R. P., Hile S. E., Lee M. Y., and Eckert K. A. (2017) DNA polymerases η and κ exchange with the polymerase δ holoenzyme to complete common fragile site synthesis. DNA Repair 57, 1–11 10.1016/j.dnarep.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Franklin A., Milburn P. J., Blanden R. V., and Steele E. J. (2004) Human DNA polymerase-η, an A-T mutator in somatic hypermutation of rearranged immunoglobulin genes, is a reverse transcriptase. Immunol. Cell Biol. 82, 219–225 10.1046/j.0818-9641.2004.01221.x [DOI] [PubMed] [Google Scholar]
- 48. Mentegari E., Crespan E., Bavagnoli L., Kissova M., Bertoletti F., Sabbioneda S., Imhof R., Sturla S. J., Nilforoushan A., Hübscher U., van Loon B., and Maga G. (2017) Ribonucleotide incorporation by human DNA polymerase η impacts translesion synthesis and RNase H2 activity. Nucleic Acids Res. 45, 2600–2614 10.1093/nar/gkw1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gali V. K., Balint E., Serbyn N., Frittmann O., Stutz F., and Unk I. (2017) Translesion synthesis DNA polymerase η exhibits a specific RNA extension activity and a transcription-associated function. Sci. Rep. 7, 13055 10.1038/s41598-017-12915-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rey L., Sidorova J. M., Puget N., Boudsocq F., Biard D. S. F., Monnat R. J. Jr., Cazaux C., and Hoffmann J.-S. (2009) Human DNA polymerase η Is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 29, 3344–3354 10.1128/MCB.00115-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Z., Wu J., and DeLeo C. J. (2006) RNA damage and surveillance under oxidative stress. IUBMB Life 58, 581–588 10.1080/15216540600946456 [DOI] [PubMed] [Google Scholar]
- 52. Bernstein J. A., Khodursky A. B., Lin P.-H., Lin-Chao S., and Cohen S. N. (2002) Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 99, 9697–9702 10.1073/pnas.112318199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wurtmann E. J., and Wolin S. L. (2009) RNA under attack: cellular handling of RNA damage. Crit. Rev. Biochem. Mol. Biol. 44, 34–49 10.1080/10409230802594043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Defoiche J., Zhang Y., Lagneaux L., Pettengell R., Hegedus A., Willems L., and Macallan D. C. (2009) Measurement of ribosomal RNA turnover in vivo by use of deuterium-labeled glucose. Clin. Chem. 55, 1824–1833 10.1373/clinchem.2008.119446 [DOI] [PubMed] [Google Scholar]
- 55. Hofer T., Badouard C., Bajak E., Ravanat J.-L., Mattsson Å., Cotgreave I. A. (2005) Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol. Chem. 386, 333–337 10.1515/BC.2005.040 [DOI] [PubMed] [Google Scholar]
- 56. Liu M., Gong X., Alluri R. K., Wu J., Sablo T., and Li Z. (2012) Characterization of RNA damage under oxidative stress in Escherichia coli. Biol. Chem. 393, 123–132 10.1515/hsz-2011-0247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thapar R., Bacolla A., Oyeniran C., Brickner J. R., Chinnam N. B., Mosammaparast N., and Tainer J. A. (2019) RNA modifications: reversal mechanisms and cancer. Biochemistry 58, 312–329 10.1021/acs.biochem.8b00949 [DOI] [PubMed] [Google Scholar]
- 58. Nawrot B., Sochacka E., and Düchler M. (2011) tRNA structural and functional changes induced by oxidative stress. Cell Mol. Life Sci. 68, 4023–4032 10.1007/s00018-011-0773-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Poulsen H. E., Specht E., Broedbaek K., Henriksen T., Ellervik C., Mandrup-Poulsen T., Tonnesen M., Nielsen P. E., Andersen H. U., and Weimann A. (2012) RNA modifications by oxidation: a novel disease mechanism? Free Radic. Biol. Med. 52, 1353–1361 10.1016/j.freeradbiomed.2012.01.009 [DOI] [PubMed] [Google Scholar]
- 60. Che Y., Wang J.-F., Shao L., and Young T. (2010) Oxidative damage to RNA but not DNA in the hippocampus of patients with major mental illness. J. Psychiatry Neurosci. 35, 296–302 10.1503/jpn.090083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guengerich F. P., and Watanabe P. G. (1979) Metabolism of [14C]- and [36Cl]-labeled vinyl chloride in vivo and in vitro. Biochem. Pharmacol. 28, 589–596 10.1016/0006-2952(79)90140-0 [DOI] [PubMed] [Google Scholar]
- 62. Ottenwälder H., Laib R. J., and Bolt H. M. (1979) Alkylation of RNA by vinyl bromide metabolites in vitro and in vivo. Arch. Toxicol. 41, 279–286 10.1007/BF00296897 [DOI] [PubMed] [Google Scholar]
- 63. Bergman K. (1982) Reactions of vinyl chloride with RNA and DNA of various mouse tissues in vivo. Arch. Toxicol. 49, 117–129 10.1007/BF00332359 [DOI] [PubMed] [Google Scholar]
- 64. Ribovich M. L., Miller J. A., Miller E. C., and Timmins L. G. (1982) Labeled 1,N6-ethenoadenosine and 3,N4-ethenocytidine in hepatic RNA of mice given [ethyl-1,2-3H or ethyl-1-14C]ethyl carbamate (urethan). Carcinogenesis 3, 539–546 10.1093/carcin/3.5.539 [DOI] [PubMed] [Google Scholar]
- 65. Guengerich F. P., and Raney V. D. (1992) Formation of etheno adducts of adenosine and cytidine from 1-halooxiranes: evidence for a mechanism involving initial reaction with the endocyclic nitrogen atoms. J. Am. Chem. Soc. 114, 1074–1080 10.1021/ja00029a041 [DOI] [Google Scholar]
- 66. Laib R. J., and Bolt H. M. (1977) Alkylation of RNA by vinyl chloride metabolites in vitro and in vivo: formation of 1,N6-ethenoadenosine. Toxicology 8, 185–195 10.1016/0300-483X(77)90007-5 [DOI] [PubMed] [Google Scholar]
- 67. Laib R. J., Ottenwälder H., and Bolt H. M. (1981) Alkylation of RNA by vinyl chloride and vinyl bromide metabolites in vivo: effect on protein biosynthesis. In Industrial and Environmental Xenobiotics (Gut I., Cikrt M., and Plaa G. L., eds) pp. 319–322, Springer, Berlin [Google Scholar]
- 68. Secrist J. A. 3rd, Barrio J. R., Leonard N. J., and Weber G. (1972) Fluorescent modification of adenosine-containing coenzymes: biological activities and spectroscopic properties. Biochemistry 11, 3499–3506 10.1021/bi00769a001 [DOI] [PubMed] [Google Scholar]
- 69. Schulman L. H., and Pelka H. (1976) Location of accessible bases in Escherichia coli formylmethionine transfer RNA as determined by chemical modification. Biochemistry 15, 5769–5775 10.1021/bi00671a013 [DOI] [PubMed] [Google Scholar]
- 70. Biernat J., Ciesiołka J., Górnicki P., Adamiak R. W., Kryzosiak W. J., and Wiewió5 rowski M. (1978) New observations concerning the chloroacetaldehyde reaction with some tRNA constituents: stable intermediates, kinetics and selectivity of the reaction. Nucleic Acids Res. 5, 789–804 10.1093/nar/5.3.789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Spengler S., and Singer B. (1981) Transcriptional errors and ambiguity resulting from the presence of 1,N6-ethenoadenosine or 3,N4-ethenocytidine in polyribonucleotides. Nucleic Acids Res. 9, 365–373 10.1093/nar/9.2.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. el Ghissassi F., Barbin A., Nair J., and Bartsch H. (1995) Formation of 1,N6-ethenoadenine and 3,N4-ethenocytosine by lipid peroxidation products and nucleic acid bases. Chem. Res. Toxicol. 8, 278–283 10.1021/tx00044a013 [DOI] [PubMed] [Google Scholar]
- 73. Yan L. L., and Zaher H. S. (2019) How do cells cope with RNA damage and its consequences? J. Biol. Chem. 294, 15158–15171 10.1074/jbc.REV119.006513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Su Y., Egli M., and Guengerich F. P. (2017) Human DNA polymerase η accommodates RNA for strand extension. J. Biol. Chem. 292, 18044–18051 10.1074/jbc.M117.809723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Johnson M. K., Kottur J., and Nair D. T. (2019) A polar filter in DNA polymerases prevents ribonucleotide incorporation. Nucleic Acids Res. 47, 10693–10705 10.1093/nar/gkz792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ren M., Cheng Y., Duan Q., and Zhou C. (2019) Transesterification reaction and the repair of embedded ribonucleotides in DNA are suppressed upon the assembly of DNA into nucleosome core particles. Chem. Res. Toxicol. 32, 926–934 10.1021/acs.chemrestox.9b00059 [DOI] [PubMed] [Google Scholar]
- 77. Seiffert S., Debelak H., Hadwiger P., Jahn-Hofmann K., Roehl I., Vornlocher H.-P., and Noll B. (2011) Characterization of side reactions during the annealing of small interfering RNAs. Anal. Biochem. 414, 47–57 10.1016/j.ab.2011.02.040 [DOI] [PubMed] [Google Scholar]
- 78. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 10.1007/BF00928361 [DOI] [PubMed] [Google Scholar]
- 79. Crespan E., Furrer A., Rösinger M., Bertoletti F., Mentegari E., Chiapparini G., Imhof R., Ziegler N., Sturla S. J., Hübscher U., van Loon B., and Maga G. (2016) Impact of ribonucleotide incorporation by DNA polymerases β and λ on oxidative base excision repair. Nat. Commun. 7, 10805 10.1038/ncomms10805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. O'Flaherty D. K., and Guengerich F. P. (2014) Steady-state kinetic analysis of DNA polymerase single-nucleotide incorporation products. Curr. Protoc. Nucleic Acid Chem. 59, 7.21.21–27.21.13 10.1002/0471142700.nc0721s59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Johnson K. A. (2019) New standards for collecting and fitting steady state kinetic data. Beilstein J. Org. Chem. 15, 16–29 10.3762/bjoc.15.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Patra A., Su Y., Zhang Q., Johnson K. M., Guengerich F. P., and Egli M. (2016) Structural and kinetic analysis of miscoding opposite the DNA adduct 1,N6-ethenodeoxyadenosine by human translesion DNA polymerase η. J. Biol. Chem. 291, 14134–14145 10.1074/jbc.M116.732487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Calabretta A., and Leumann C. J. (2013) Base pairing and miscoding properties of 1,N6-ethenoadenine- and 3,N4-ethenocytosine-containing RNA oligonucleotides. Biochemistry 52, 1990–1997 10.1021/bi400116y [DOI] [PubMed] [Google Scholar]
- 84. Alenko A., Fleming A. M., and Burrows C. J. (2017) Reverse transcription past products of guanine oxidation in RNA leads to insertion of A and C opposite 8-oxo-7,8-dihydroguanine and A and G opposite 5-guanidinohydantoin and spiroiminodihydantoin diastereomers. Biochemistry 56, 5053–5064 10.1021/acs.biochem.7b00730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chowdhury G., and Guengerich F. P. (2011) Liquid chromatography-mass spectrometry analysis of DNA polymerase reaction products. Curr. Protoc. Nucleic Acid Chem. 47, 7.16.11–17.16.11 10.1002/0471142700.nc0716s47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zang H., Goodenough A. K., Choi J.-Y., Irimia A., Loukachevitch L. V., Kozekov I. D., Angel K. C., Rizzo C. J., Egli M., and Guengerich F. P. (2005) DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem. 280, 29750–29764 10.1074/jbc.M504756200 [DOI] [PubMed] [Google Scholar]
- 87. Hwang H., and Taylor J.-S. (2004) Role of base stacking and sequence context in the inhibition of yeast DNA polymerase η by pyrene nucleotide. Biochemistry 43, 14612–14623 10.1021/bi0489558 [DOI] [PubMed] [Google Scholar]
- 88. Fiala K. A., Brown J. A., Ling H., Kshetry A. K., Zhang J., Taylor J.-S., Yang W., and Suo Z. (2007) Mechanism of template-independent nucleotide incorporation catalyzed by a template-dependent DNA polymerase. J. Mol. Biol. 365, 590–602 10.1016/j.jmb.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hyjek M., Figiel M., and Nowotny M. (2019) RNases H: structure and mechanism. DNA Repair (Amst.) 84, 102672 10.1016/j.dnarep.2019.102672 [DOI] [PubMed] [Google Scholar]
- 90. Su Y., and Guengerich F. P. (2016) Pre-steady-state kinetic analysis of single-nucleotide incorporation by DNA polymerases. Curr. Protoc. Nucleic Acid Chem. 65, 7.23.21–27.23.10 10.1002/cpnc.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Malfatti M. C., Henneke G., Balachander S., Koh K. D., Newnam G., Uehara R., Crouch R. J., Storici F., and Tell G. (2019) Unlike the Escherichia coli counterpart, archaeal RNase HII cannot process ribose monophosphate abasic sites and oxidized ribonucleotides embedded in DNA. J. Biol. Chem. 294, 13061–13072 10.1074/jbc.RA119.009493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bétous R., Rey L., Wang G., Pillaire M.-J., Puget N., Selves J., Biard D. S. F., Shin-ya K., Vasquez K. M., Cazaux C., and Hoffmann J.-S. (2009) Role of TLS DNA polymerases η and κ in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 48, 369–378 10.1002/mc.20509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Clausen A. R., Zhang S., Burgers P. M., Lee M. Y., and Kunkel T. A. (2013) Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair 12, 121–127 10.1016/j.dnarep.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Levine R. L., Miller H., Grollman A., Ohashi E., Ohmori H., Masutani C., Hanaoka F., and Moriya M. (2001) Translesion DNA synthesis catalyzed by human pol η and pol κ across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 276, 18717–18721 10.1074/jbc.M102158200 [DOI] [PubMed] [Google Scholar]
- 95. Ling H., Boudsocq F., Woodgate R., and Yang W. (2004) Snapshots of replication through an abasic lesion: structural basis for base substitutions and frameshifts. Mol. Cell 13, 751–762 10.1016/S1097-2765(04)00101-7 [DOI] [PubMed] [Google Scholar]
- 96. Zhao Y., Gregory M. T., Biertümpfel C., Hua Y.-J., Hanaoka F., and Yang W. (2013) Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc. Natl. Acad. Sci. U.S.A. 110, 8146–8151 10.1073/pnas.1303126110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Patra A., Zhang Q., Lei L., Su Y., Egli M., and Guengerich F. P. (2015) Structural and kinetic analysis of nucleoside triphosphate incorporation opposite an abasic site by human translesion DNA polymerase η. J. Biol. Chem. 290, 8028–8038 10.1074/jbc.M115.637561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Willi J., Küpfer P., Evéquoz D., Fernandez G., Katz A., Leumann C., and Polacek N. (2018) Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 46, 1945–1957 10.1093/nar/gkx1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kim N., Cho J.-E., Li Y. C., and Jinks-Robertson S. (2013) RNA:DNA hybrids initiate quasi-palindrome-associated mutations in highly transcribed yeast DNA. PLOS Genet. 9, e1003924 10.1371/journal.pgen.1003924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Storici F., Bebenek K., Kunkel T. A., Gordenin D. A., and Resnick M. A. (2007) RNA-templated DNA repair. Nature 447, 338–341 10.1038/nature05720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Heider M. R., Burkhart B. W., Santangelo T. J., and Gardner A. F. (2017) Defining the RNaseH2 enzyme-initiated ribonucleotide excision repair pathway in Archaea. J. Biol. Chem. 292, 8835–8845 10.1074/jbc.M117.783472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Suo Z., and Johnson K. A. (1998) DNA secondary structure effects on DNA synthesis catalyzed by HIV-1 reverse transcriptase. J. Biol. Chem. 273, 27259–27267 10.1074/jbc.273.42.27259 [DOI] [PubMed] [Google Scholar]
- 103. Furge L. L., and Guengerich F. P. (1999) Explanation of pre-steady-state kinetics and decreased burst amplitude of HIV-1 reverse transcriptase at sites of modified DNA bases with an additional, nonproductive enzyme-DNA-nucleotide complex. Biochemistry 38, 4818–4825 10.1021/bi982163u [DOI] [PubMed] [Google Scholar]
- 104. Beckman J. W., Wang Q., and Guengerich F. P. (2008) Kinetic analysis of correct nucleotide insertion by a Y-family DNA polymerase reveals conformational changes both prior to and following phosphodiester bond formation as detected by tryptophan fluorescence. J. Biol. Chem. 283, 36711–36723 10.1074/jbc.M806785200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Su Y., Patra A., Harp J. M., Egli M., and Guengerich F. P. (2015) Roles of residues Arg-61 and Gln-38 of human DNA polymerase η in bypass of deoxyguanosine and 7,8-dihydro-8-oxo-2′-deoxyguanosine. J. Biol. Chem. 290, 15921–15933 10.1074/jbc.M115.653691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Biertümpfel C., Zhao Y., Kondo Y., Ramón-Maiques S., Gregory M., Lee J. Y., Masutani C., Lehmann A. R., Hanaoka F., and Yang W. (2010) Structure and mechanism of human DNA polymerase η. Nature 465, 1044–1048 10.1038/nature09196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Figiel M., Chon H., Cerritelli S. M., Cybulska M., Crouch R. J., and Nowotny M. (2011) The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi-Goutières syndrome defects. J. Biol. Chem. 286, 10540–10550 10.1074/jbc.M110.181974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chon H., Vassilev A., DePamphilis M. L., Zhao Y., Zhang J., Burgers P. M., Crouch R. J., and Cerritelli S. M. (2009) Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 37, 96–110 10.1093/nar/gkn913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shah S., and Friedman S. H. (2008) An ESI-MS method for characterization of native and modified oligonucleotides used for RNA interference and other biological applications. Nat. Protoc. 3, 351–356 10.1038/nprot.2007.535 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the article and supporting information.