

Abstract
Objectives
Triple‐negative breast cancer (TNBC) is well known for its strong invasiveness, rapid recurrence and poor prognosis. Immunotherapy, including chimeric antigen receptor‐modified T (CAR‐T) cells, has emerged as a promising tool to treat TNBC. The identification of a specific target tumor antigen and the design of an effective CAR are among the many challenges of CAR‐T therapy.
Methods
We reported that epidermal growth factor receptor (EGFR) is highly expressed in TNBC and consequently designed an optimal third generation of CAR targeting EGFR. The efficacy of primary T lymphocytes infected with EGFR CAR lentivirus (EGFR CAR‐T) against TNBC was evaluated both in vitro and in vivo. The signalling pathways activated in tumor and EGFR CAR‐T cells were revealed by RNA sequencing analysis.
Results
Third‐generation EGFR CAR‐T cells exerted potent and specific suppression of TNBC cell growth in vitro, whereas limited cytotoxicity was observed towards normal breast epithelial cells or oestrogen receptor‐positive breast cancer cells. This capability was further demonstrated in vivo in a xenograft mouse model, with minimal off‐tumor cytotoxicity. Mechanistically, in vitro stimulation with TNBC cells induced the expansion of naïve‐associated EGFR CAR‐T cells and enhanced their persistence. Furthermore, EGFR CAR‐T cells activated the interferon γ, granzyme–perforin–PARP and Fas–FADD–caspase signalling pathways in TNBC cells.
Conclusion
We demonstrate that EGFR is a relevant immunotherapeutic target in TNBC, and EGFR CAR‐T exhibits potent and specific antitumor activity against TNBC, suggesting the potential of this third‐generation EGFR CAR‐T as an immunotherapy tool to treat TNBC in the clinic.
Keywords: triple‐negative breast cancer, epidermal growth factor receptor, chimeric antigen receptor‐modified T‐cell, immunotherapy
We demonstrated that epidermal growth factor receptor (EGFR) is an immunotherapeutic target in triple‐negative breast cancer (TNBC), and EGFR‐targeted chimeric antigen receptor‐modified T cells (EGFR CAR‐T) exhibited potent and specific antitumor activity towards TNBC in vitro and in vivo, which might serve as a promising immunotherapy tool to treat TNBC in clinic.

Introduction
Breast cancer is the most common malignancy in women worldwide. In 2018, nearly 2.1 million people were diagnosed with breast cancer, and about half a million people died from this disease. 1 There are five main molecular subtypes of breast cancer: luminal A (LumA), luminal B (LumB), human epidermal growth factor receptor 2 (HER2)‐enriched, basal‐like and normal‐like breast cancer. 2 , 3 LumA and LumB together constitute the so‐called oestrogen receptor (ER)‐positive subtype. Triple‐negative breast cancer (TNBC) lacks the expression of ER, progesterone receptor (PR) and HER2, comprising 10–20% of all breast cancers. Molecular profiling studies suggested that about 70% of TNBCs fall into the basal‐like breast cancer subtype. 4 Owing to an inherently aggressive clinical behaviour, TNBC is associated with poor prognosis. 5 Currently, there is no targeted therapy available to treat TNBC. 4 , 6 Although responsive to chemotherapy, TNBCs tend to relapse and metastasize rapidly after treatment. 7 , 8 Therefore, a major effort has been fostered to discover new therapies to treat TNBC patients. 5
Proof‐of‐principle studies with immune checkpoint inhibitors for the treatment of TNBC have yielded promising results, indicating the potential benefits of immunotherapy. 5 , 9 The immune response in tumors mainly relies on the adaptive immunity, usually focusing on T cell‐mediated immunity. 10 Adoptive cell therapy, particularly chimeric antigen receptor‐modified T‐cell (CAR‐T) therapy, has gained much attention in the past decade. 11 In a single fusion molecule, CAR‐T combines the specificity of an antibody to target‐specific antigens on tumor cells with the ability of T cells to kill tumor cells. In fact, CAR‐T immunotherapy led to remarkable tumor regression in patients with haematological malignancies. 12 , 13 Several CAR‐Ts, such as receptor tyrosine kinase‐like orphan receptor CAR‐T, 14 mucin‐1 glycoprotein CAR‐T 15 and natural killer group 2 member D CAR‐T, are currently being tested in clinical trials for the treatment of TNBC. 16 Recent studies have demonstrated that epidermal growth factor receptor (EGFR, also called HER1) is a marker that better distinguishes TNBC from other subtypes of breast cancer. 17 , 18 Additionally, EGFR has been found to be overexpressed in 72% of patients with TNBC. 17 , 19 , 20 , 21 , 22 Kim et al. 23 performed immunohistochemistry (IHC) staining using a tissue microarray enclosing 230 breast cancer tissues and found that EGFR overexpression was observed in 9.1% and 66.7% of all tested samples and TNBCs, respectively (P < 0.001). Similarly, Li et al. 24 demonstrated through IHC staining that EGFR overexpression was observed in 61.2% of TNBCs (n = 60), which was significantly higher than the percentage observed in the non‐TNBC group. In a cohort of TNBC samples (n = 151), 64% were found to overexpress EGFR, which was associated with poor clinical outcome. 25 Small molecule inhibitors or monoclonal antibodies targeting EGFR are being evaluated in clinical trials for treating TNBC. 26 , 27 , 28 All of these studies suggest that EGFR might be a favorable target for the engineering of CAR‐T to treat TNBC. The use of primary T lymphocytes infected with EGFR CAR lentivirus (EGFR CAR‐T) may be a more effective way to generate durable antitumor responses than that of monoclonal antibodies. 29 Indeed, the use of the second generation of EGFR CAR‐T cells for cancer treatment has been reported by multiple groups. 30 , 31 , 32 , 33 , 34 For example, Li et al. 35 produced the second generation of EGFR CAR‐T cells by optimising the non‐viral piggyBac transposon system, which exhibited expansion capability and anticancer efficacy in lung cancer xenografts in vivo. In addition, a phase I clinical study conducted by the Han group using the second generation of EGFR CAR‐T cells showed that they promoted efficient clinical responses in 11 patients with EGFR‐positive, advanced non‐small‐cell lung cancer (NSCLC). 36 The same group later demonstrated that the EGFR CAR‐T cell therapy was a safe and effective strategy for treating EGFR‐positive, advanced biliary tract cancer. 37
In this study, we developed a third‐generation EGFR‐targeted CAR (EGFR CAR) and demonstrated that T lymphocytes infected with the EGFR CAR lentivirus exhibit potent and specific toxicity in TNBC in vitro. After incubation with MDA‐MB‐231, a TNBC model cell line, naïve‐associated EGFR CAR‐T cells expanded, and a gene signature resembling that of naïve T cells (TN) increased, which delayed T‐cell differentiation and exhaustion. The signalling pathways activated by CAR‐T cells in TNBC cells were mainly those of interferon γ (IFN γ), granzyme–perforin–poly adenosine diphosphate (ADP)‐ribose polymerase (PARP), and factor‐associated suicide (Fas)–Fas‐associated death domain (FADD)–caspase. The potential and specificity of our EGFR CAR‐T cells were further demonstrated in a xenograft mouse model.
Results
EGFR is highly expressed in TNBC
Overexpression of EGFR was observed in 15–30% of breast carcinomas and was associated with large tumor size and poor clinical outcomes. 38 We analysed the expression of EGFR in several breast cancer cell lines by immunoblotting and found that EGFR was notably overexpressed in TNBC cells (MDA‐MB‐231, MDA‐MB‐468, HS578T and HCC1860) compared to human breast epithelial cells (MCF10A) and ER‐positive (T47D and MCF7), PR‐positive (BT474) and HER2‐positive (SK‐BR‐3) cells (Figure 1a). Exceptional high levels of EGFR were observed in MDA‐MB‐231 and MDA‐MB‐468 cells (Figure 1a). The expression of EGFR in these two cell lines as well as that in MCF7 and T47D cells was further assessed by flow cytometry analysis, confirming the immunoblotting results (Figure 1b), which was consistent with findings of previous reports. 39 , 40 , 41 , 42 , 43 We then analysed the expression of EGFR in a cohort of breast tumor samples from The Cancer Genome Atlas (TCGA) and found that EGFR was significantly overexpressed in TNBC compared to other breast cancer subtypes (Figure 1c and Supplementary figure 1a). To underscore the clinical significance of highly expressed EGFR, the latter was associated with poor prognosis in TNBC, but not in the LumA, LumB and HER2 subtypes (Supplementary figure 1b–e). Therefore, our results and other previously reported 17 , 23 , 24 , 25 indicate that EGFR is highly expressed in TNBCs.
Figure 1.
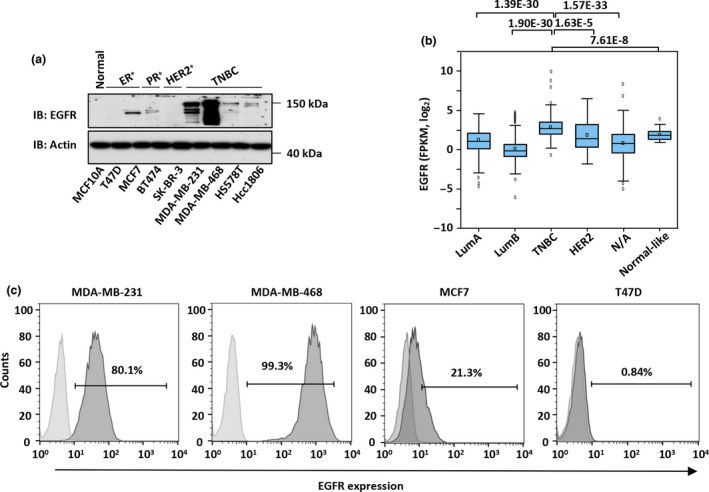
EGFR is overexpressed in TNBC. (a) Cell lysates from different breast cancer cells as well as normal breast epithelial cells as indicated were subjected to IB analysis with an anti‐EGFR‐specific antibody. Actin served as a loading control. Molecular weight is indicated on the right. Experiments were repeated three times, and representative blots are shown. (b) MDA‐MB‐231, MDA‐MB‐468, MCF7 and T47D cells were subjected to flow cytometry analysis by using a PE‐conjugated anti‐EGFR antibody to examine the expression of this receptor. Light grey, blank; dark grey, EGFR. Experiments were repeated three times, and representative histograms are shown. (c) The expression of EGFR (FPKM, log2) in a cohort of clinical breast cancer samples from The Cancer Genome Atlas database is shown in the box plot. LumA, n = 480; LumB, n = 197; TNBC, n = 157; HER2, n = 73; N/A, n = 287; Normal‐like, n = 27. P‐values are shown at the top of the Figure 1c. EGFR, epidermal growth factor receptor; ER+, oestrogen receptor‐positive; FPKM, fragments per kilobase per million; HER2+, human epidermal growth factor receptor 2‐positive; IB, immunoblotting; LumA, luminal A; LumB, luminal B; N/A, not available; PR+, progesterone receptor‐positive; TNBC, triple‐negative breast cancer.
Development of third‐generation EGFR‐targeted CAR‐T cells
The EGFR‐overexpressing TNBC prompted us to engineer a CAR that targets EGFR to suppress this breast cancer subtype. The antigen recognition part of CAR was engineered using variable heavy and light chain sequences from an anti‐EGFR antibody connected by a linker sequence (GlyGlyGlyGlySer GlyGlyGlyGlySer GlyGlyGlyGlySer), producing a recombinant anti‐EGFR single‐chain variable fragment (scFv) antibody (Figure 2a). Moreover, several amino‐terminal signal peptides were compared, and that of interleukin (IL)‐2 receptor (Sp1) was chosen to confer the recombinant anti‐EGFR scFv antibody the ability to target antigen membranes (Figure 2a). The spacer in a typical CAR is the extracellular region that separates the antigen recognition part from the transmembrane domain, providing flexibility to access the targeted antigen. 44 Immunoglobulin (Ig)‐like spacers were proved to be effective in CAR design. 45 Thus, two forms of immunoglobulin G1 (IgG1)‐based spacers, the IgG1 Fc spacer containing IgG1 hinge, constant domain (CH) 2 and CH3, 45 and the IgG1 hinge spacer containing only IgG1 hinge, 45 were used in our third‐generation CAR design (Figure 2a). In addition, the engineered CAR contains a transmembrane domain from CD28 and intracellular signalling domains from CD28, 4‐1BB and CD3ζ (Figure 2a). Flow cytometry analysis showed that both EGFR CARs (Fc‐EGFR CAR and Hinge‐EGFR CAR) were equally well expressed in 293T cells after transfection (Figure 2b), which was further confirmed by immunoblotting analysis using an anti‐CD3ζ antibody (Figure 2c).
Figure 2.
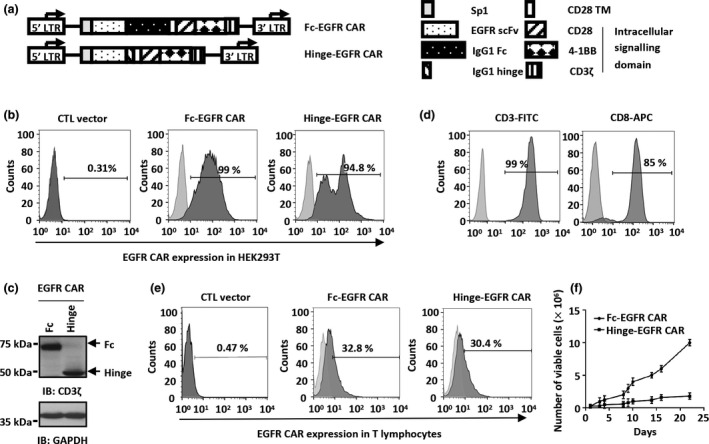
The design and expression of EGFR‐targeted CAR (EGFR CAR). (a) Schematic illustration of the two third‐generation EGFR CAR constructs. The CAR is constituted by a signal peptide of the interleukin (IL)‐2 receptor (Sp1), an anti‐EGFR scFv from cetuximab, a spacer (IgG1 Fc or IgG1 hinge), a CD28 transmembrane domain (CD28 TM) and intracellular signalling domains (CD28, 4‐1BB and CD3ζ). The spacers IgG1 Fc and IgG1 hinge were used in Fc‐EGFR CAR and Hinge‐EGFR CAR constructs, respectively. (b, c) HEK293T cells transfected with a CTL vector or the two EGFR CAR expression vectors as described in a were stained with an APC‐conjugated anti‐human IgG‐Fc antibody or a FITC‐conjugated anti‐human IgG (Fab′)2 antibody followed by flow cytometry analysis (b) or lysed for IB (c) to examine the expression of CD3ζ. GAPDH served as a loading control. Molecular weight is indicated on the left. Light grey, blank; dark grey, CTL vector or EGFR CAR. Experiments were repeated three times, and representative histograms or blots are shown. (d) Primary T lymphocytes from a healthy donor were expanded and stained with an anti‐CD3 antibody conjugated with FITC (CD3‐FITC) (left panel) or an anti‐CD8 antibody conjugated with APC (CD8‐APC) (right panel) followed by flow cytometry analysis. Light grey, blank; dark grey, CD3 or CD8 staining. Experiments were repeated three times, and representative histograms are shown. (e) Primary T lymphocytes were infected with a CTL vector or the two EGFR CAR lentiviruses as described in a and stained with an APC‐conjugated anti‐human IgG‐Fc antibody or a FITC‐conjugated anti‐human IgG (Fab′)2 antibody followed by flow cytometry analysis. Light grey, blank; dark grey, CTL vector or EGFR CAR. Experiments were repeated three times, and representative histograms are shown. (f) Primary T lymphocytes infected with the two EGFR CAR lentiviruses as described in a were maintained in culture medium for the indicated durations, and the number of viable cells was counted at the indicated time points. Initial number, 2.5 × 105. Data were obtained from three replicates and are presented as mean ± s.e.m.. CAR, chimeric antigen receptor; CTL, control; EGFR scFv, single‐chain variable fragment against EGFR; EGFR, epidermal growth factor receptor; Fc, fragment crystallisable; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; IB, immunoblotting; IgG1, immunoglobulin G1; LTR, long terminal repeat; Sp1, signal peptide of the IL‐2 receptor; TM, transmembrane domain.
Primary human T cells from a healthy donor were enriched and expanded in vitro upon treatment with anti‐CD3/CD28 monoclonal antibodies, IL‐2 and IL‐15 for approximately 1 week. The majority of these T cells were found to be a CD3+ CD8+ subpopulation identified by flow cytometry (CD3+, 99%; CD8+, 85%) (Figure 2d), which were then infected with Fc‐EGFR or Hinge‐EGFR CAR lentiviral expression vectors. The infection efficiency of Fc‐EGFR and Hinge‐EGFR CAR was 32.8% and 30.4%, respectively (Figure 2e). However, when expanded under the same protocol, the number of Fc‐EGFR CAR lentivirus‐infected T cells increased at least 40‐fold in 3 weeks, whereas that of Hinge‐EGFR CAR lentivirus‐infected T cells increased only fivefold (Figure 2f). Therefore, the Fc‐EGFR CAR was chosen for cytotoxicity tests towards TNBC.
EGFR CAR‐T cells exhibit potent and specific cytotoxicity against TNBC cells in vitro
It is known that CAR‐T cells can be stimulated to proliferate with tumor‐specific antigens. To examine whether our EGFR CAR‐T can be activated by TNBC cells, EGFR CAR‐T cells were incubated with or without MDA‐MB‐231 or MDA‐MB‐468 cells in culture medium without adding proliferative cytokines after carboxyfluorescein succinimidyl ester (CFSE) labelling. EGFR CAR‐T cells were capable of proliferating for multiple generations within 3 days compared with EGFR CAR‐T cells alone (Figure 3a). Furthermore, control T cells (CTL T, non‐infected) or EGFR CAR‐T cells were incubated with MDA‐MB‐231 or MDA‐MB‐468 cells, followed by flow cytometry analysis to examine the expression of CD69 and CD25, early and late T‐cell activation markers, respectively. Upon co‐incubation with tumor cells, both CD69+/CD8+ and CD25+/CD8+ subpopulations increased more than 32% in EGFR CAR‐T cells, but not in non‐infected T cells, indicating that EGFR CAR‐T cells were activated by tumor cells (Figure 3b and c). Further supporting the activation of EGFR CAR‐T cells after incubation with MDA‐MB‐231 or MDA‐MB‐468 cells, cytokines, such as tumor necrosis factor α (TNFα), IL‐2 and IFNγ, were found to be secreted at a much higher level by EGFR CAR‐T cells than by non‐infected T cells (Figure 3d). In addition, the secretion of cytokines appeared to be dependent on the EGFR CAR‐T dose (Figure 3d). EGFR CAR‐T cells secreted considerable high levels of IFNγ, reaching nearly 30 000 pg mL−1 when the highest number of EGFR CAR‐T cells was tested (Figure 3d).
Figure 3.
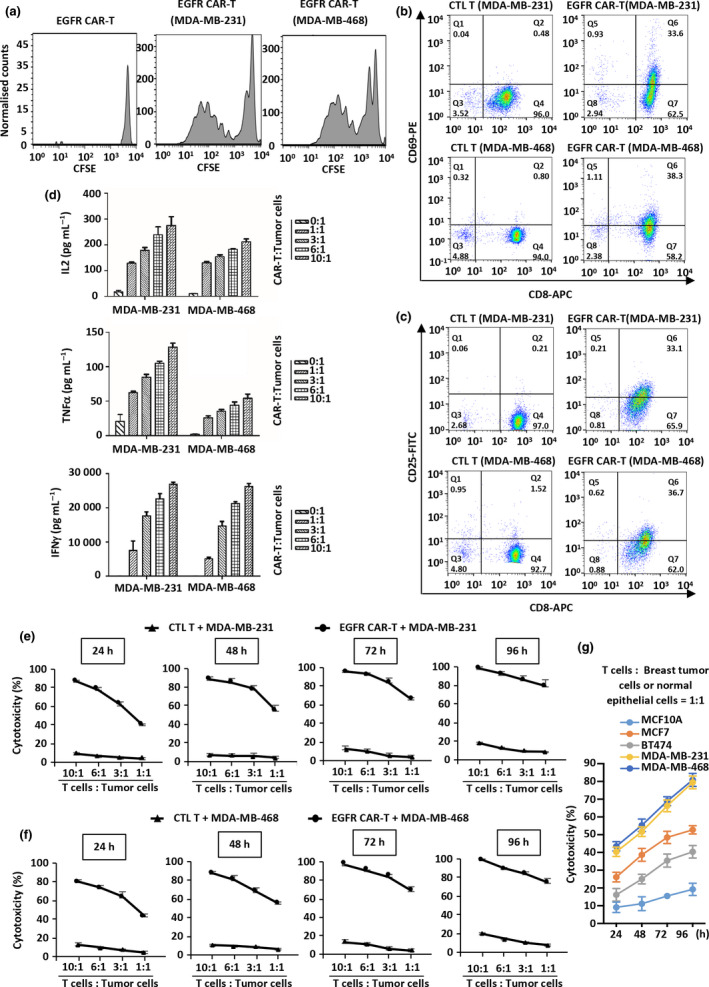
Proliferation, activation and cytotoxicity of EGFR CAR‐T cells. (a) Primary T lymphocytes infected with EGFR CAR lentivirus (EGFR CAR‐T) labelled with CFSE were incubated with or without MDA‐MB‐231 or MDA‐MB‐468 cells in culture medium without adding proliferative cytokines for 3 days and diluted to examine their proliferation. Experiments were repeated three times, and representative histograms are shown. (b, c) CTL T or EGFR CAR‐T cells were incubated with MDA‐MB‐231 or MDA‐MB‐468 cells and stained with CD8‐APC and CD69‐PE (b) or CD25‐FITC (c) followed by flow cytometry analysis. Experiments were repeated three times, and representative histograms are shown. (d) EGFR CAR‐T cells were incubated with MDA‐MB‐231 or MDA‐MB‐468 cells at the indicated ratios for 3 days before measuring the secretion of cytokines, including IL‐2, TNFα and IFNγ. CTL T cells were used as a negative control. Data were obtained from three replicates and are presented as mean ± s.e.m.. (e, f) CTL T or EGFR CAR‐T cells were incubated with MDA‐MB‐231 (e) or MDA‐MB‐468 (f) cells at different ratios for the indicated durations followed by the cytotoxicity assay. Data were obtained from three replicates and are presented as mean ± s.e.m.. (g) EGFR CAR‐T cells were incubated with MCF10A, MCF7, BT474, MDA‐MB‐468 or MDA‐MB‐231 cells at a ratio of 1:1 for the indicated durations followed by the cytotoxicity assay. Data were obtained from three replicates and are presented as mean ± s.e.m.. CAR‐T, chimeric antigen receptor‐modified T cells; CFSE, carboxyfluorescein succinimidyl amino ester; CTL T, control T cells; EGFR, epidermal growth factor receptor; IFNγ, interferon γ; IL‐2, interleukin 2; TNFα, tumor necrosis factor α.
Next, we sought to examine the toxicity of EGFR CAR‐T cells towards TNBC cells. The cytotoxicity assay was performed by incubating MDA‐MB‐231 or MDA‐MB‐468 cells with non‐infected CTL T or EGFR CAR‐T cells at different ratios (T cells:tumor cells = 1:1, 3:1, 6:1 or 10:1) and durations (24–96 h). EGFR CAR‐T cells were found to kill MDA‐MB‐231 and MDA‐MB‐468 cells in a dose‐ and time‐dependent manner (Figure 3e and f). The specificity of EGFR CAR‐T cells was demonstrated by their poor efficiency in killing tumor cells with lower EGFR expression, such as MCF7 and BT474 cells, and nearly no efficiency in killing normal breast epithelial cells (MCF10A cells) (Figure 3g). To further test the efficacy and specificity of EGFR CAR‐T cells in vitro, primary TNBC cell lines, one with high and the other with low EGFR expression, were incubated with EGFR CAR‐T cells, followed by the cytotoxicity assay (Supplementary figure 2a). Our data indicated that EGFR CAR‐T cells displayed higher killing activity towards primary TNBC cells with high expression of EGFR than towards those with low expression of EGFR (Supplementary figure 2b).
EGFR CAR‐T cells exhibit a more naïve T‐cell status upon TNBC stimulation
Upon antigen stimulation, naïve T cells (TN) differentiate into stem cell memory T cells (TSCM), central memory T cells (TCM), effector memory T cells (TEM) and effector T cells (TEFF) (Figure 4a). During this process, the effector function increases, whereas the proliferation, memory and persistence function decrease. 46 TSCM and TCM, naïve‐associated T cells, were shown to enhance the persistence and antitumor effects in murine T‐cell receptor and CAR gene therapy models. 47 Different statuses of T cells are associated with different gene signatures. For instance, naïve‐associated T cells are the least differentiated and therefore present higher expression of naïve‐associated genes than TEM and TEFF. 48 To determine the gene signature of our EGFR CAR‐T cells upon TNBC stimulation, the former were incubated with or without MDA‐MB‐231 cells and then separated from tumor cells before ribonucleic acid (RNA) sequencing (RNA‐seq) analysis. After incubation with tumor cells, 671 and 307 genes were induced and repressed, respectively, in EGFR CAR‐T cells (Figure 4b). The expression profiles of these genes are shown in the heat map (Figure 4c) and box plot (Figure 4d). Human mature T cells are originated from haematopoietic stem cells (HSCs) and haematopoietic progenitor cells. 49 Gene ontology (GO) analysis indicated that the most enriched terms for up‐regulated genes in EGFR CAR‐T cells were associated with the attributes of HSCs, such as cell or tissue morphogenesis and development, which represent characteristics of TN (Figure 4e). In contrast, the enriched GO terms for down‐regulated genes in EGFR CAR‐T cells were mainly associated with T‐cell differentiation, deoxyribonucleic acid (DNA) replication, cell cycle phase transition and among others, which are characteristics of TEFF (Figure 4f). Representative genes with implications in TEFF differentiation, such as eomesodermin (EOMES), 50 T‐box21 (TBX21) 51 and positive regulatory domain containing 1 (PRDM1), 52 genes encoding cytotoxic molecules, such as granzyme A (GZMA) and perforin (PRF1), and genes with implications in T‐cell senescence, such as killer cell lectin‐like receptor subfamily G, member 1 (KLRG1), 51 were down‐regulated in EGFR CAR‐T cells, as shown in the heat map (Figure 4g, top part). The University of California, Santa Cruz (UCSC) Genome Browser views of RNA‐seq for these genes are shown in Figure 4h and Supplementary figure 3a. Conversely, the expression of genes that are associated with persistence, such as forkhead box P1 (FOXP1), 53 actinin alpha 1 (ACTN1), transcription factor 7 (TCF7), C‐C motif chemokine receptor 7 (CCR7), interleukin‐6 signal transducer (IL‐6ST) and selectin L (CD62L), 48 was remarkably increased in EGFR CAR‐T cells upon incubation with MDA‐MB‐231 cells, as shown in the heat map (Figure 4g, bottom part). The UCSC Genome Browser views of RNA‐seq for these up‐regulated genes are shown in Figure 4i and Supplementary figure 3b. The change of expression of representative genes in EGFR CAR‐T cells was confirmed by quantitative reverse transcription polymerase chain reaction (RT‐qPCR) analysis (Figure 4j and k). Our data suggest that, upon antigen stimulation, naïve‐associated CAR‐T cells expanded and contributed to the increased expression of naïve‐associated genes. Meanwhile, a proportion of activated EGFR CAR‐T cells died during co‐culture with tumor cells, which might contribute to the decreased expression of effector‐associated genes.
Figure 4.
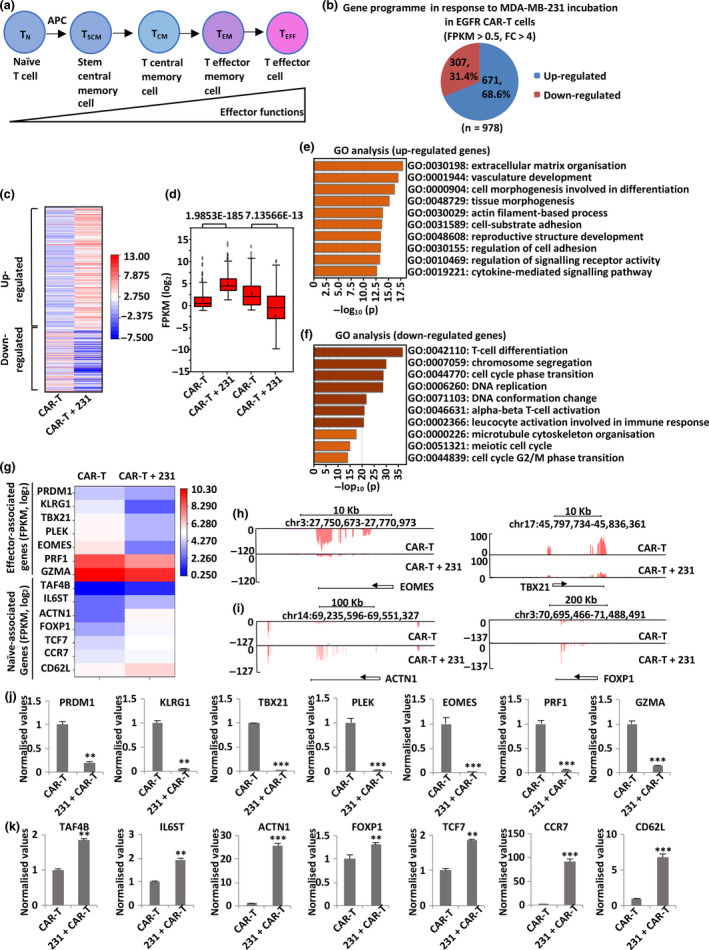
TNBC‐stimulated EGFR CAR‐T cells exhibit an induced naïve‐associated gene signature. (a) Schematic representation of CD8+ T‐cell differentiation. (b) EGFR CAR‐T cells were incubated with or without MDA‐MB‐231 cells (at a ratio of 1:2) for 3 days, and CAR‐T cells in suspension were separated from adherent tumor cells and collected, followed by RNA extraction and RNA sequencing (RNA‐seq) analysis. Up‐ and down‐regulated genes of CAR‐T cells upon incubation with MDA‐MB‐231 cells (FPKM > 0.5, FC > 4) are shown in the pie chart. RNA from three biological replicates was pooled for RNA‐seq. (c, d) Heat map (c) and box plot (d) representation of the expression levels (FPKM, log2) of the up‐ and down‐regulated genes described in b. P‐values are shown at the top (d). (e, f) GO analysis of the up‐ (e) and down‐regulated (f) genes described in b. (g) The expression (FPKM, log2) of representative genes associated with effector and naïve T‐cell function in EGFR CAR‐T cells in response to MDA‐MB‐231 cell co‐incubation as detected by RNA‐seq is shown in the heat map. (h, i) UCSC Genome Browser views of representative genes associated with effector (h) and naïve (i) T‐cell function from RNA‐seq analysis. (j, k) Cells described in b were subjected to RNA extraction and quantitative reverse transcription polymerase chain reaction (RT‐qPCR) analysis to examine the expression of representative genes associated with effector (j) and naïve (k) T‐cell function in EGFR CAR‐T cells. Data were obtained from three replicates and are presented as mean ± s.e.m. (**P < 0.01, ***P < 0.001). 231, MDA‐MB‐231; APC, antigen‐presenting cells; CAR‐T, chimeric antigen receptor‐modified T cells; chr, chromosome; EGFR, epidermal growth factor receptor; FC, fold change; FPKM, fragments per kilobase per million; GO, gene ontology; TCM, central memory T cells; TEFF, effector T cells; TEM, effector memory T cells; TN, naïve T cells; TSCM, T stem central memory cells.
To examine the expansion of naïve‐associated CAR‐T cells, T cells after EGFR CAR infection and ex vivo expansion were incubated with or without MDA‐MB‐231 cells and then separated and coated with CD3–fluorescein isothiocyanate (FITC), CD8–allophycocyanin (APC), CD62L–phycoerythrin (PE) and CCR7–Pacific Blue antibodies, followed by flow cytometry analysis. CD62L and CCR7 served as markers of the naïve‐associated T‐cell population. 46 We found that 29.3% of T cells were a naïve‐associated population (CD3+CD8+CD62L+CCR7+) in the absence of tumor cells (Supplementary figure 4a), increasing to 48.6% upon MDA‐MB‐231 cell stimulation, which supports the expansion of the naïve‐associated T‐cell population (Supplementary figure 4a). Considering the fact that the T‐cell population includes a proportion of non‐transduced cells, which might contribute to the increased number of naïve‐associated T cells, we tested whether EGFR CAR‐transduced T cells expanded after coating them with IgG‐Fc‐APC, CD62L‐PE and CCR7‐Pacific Blue antibodies, with IgG‐Fc serving as a marker of the EGFR CAR‐T cell population. Flow cytometry analysis indicated that naïve‐associated EGFR CAR‐T cells increased significantly during co‐culture with tumor cells (Supplementary figure 4b). Taken together, our results indicate that the induced naïve‐associated gene signature in response to tumor cell co‐culture might result from the expansion of naïve‐associated EGFR CAR‐T cells.
EGFR CAR‐T cells activate multiple signalling pathways in TNBC cells
Chimeric antigen receptor mediates major histocompatibility complex (MHC)‐unrestricted killing by enabling T cells to bind to antigens on the tumor cell surface through a scFv recognition domain. Upon engagement, CAR‐T cells form a non‐classical immune synapse and trigger antitumor effects through the activation of multiple signalling pathways in tumor cells. 54 To determine the signalling pathways activated by EGFR CAR‐T cells in TNBC cells, MDA‐MB‐231 cells were incubated with CTL T or EGFR CAR‐T cells and then separated from T cells, followed by RNA‐seq analysis. Noteworthy, to avoid a large number of dead tumor cells, the latter and T cells were mixed at a ratio of 2:1. Our results show that 1756 and 2392 genes were up‐regulated and down‐regulated, respectively, in MDA‐MB‐231 cells upon EGFR CAR‐T cell co‐culture (Figure 5a). The impact of EGFR CAR‐T on the expression of these genes is shown in the heat map (Figure 5b) and box plot (Figure 5c). GO enrichment analysis revealed that the topmost enriched terms for up‐regulated genes in MDA‐MB‐231 cells were associated with the cytokine‐mediated signalling pathway, cytokine production and apoptotic signalling pathway and among others (Figure 5d). Conversely, the topmost enriched GO terms for down‐regulated genes in MDA‐MB‐231 cells were associated with cell cycle checkpoint, DNA replication, among others, which are critical for tumor growth and proliferation (Figure 5e). The expression of representative genes, both up‐regulated and down‐regulated, is shown in the heat map (Figure 5f and g). The UCSC Genome Browser views of representative genes from RNA‐seq are also shown in Figure 5h and i, and Supplementary figure 5a and b. We also confirmed the change of gene expression in MDA‐MB‐231 cells upon CAR‐T treatment by RT‐qPCR analysis (Figure 5j and k).
Figure 5.
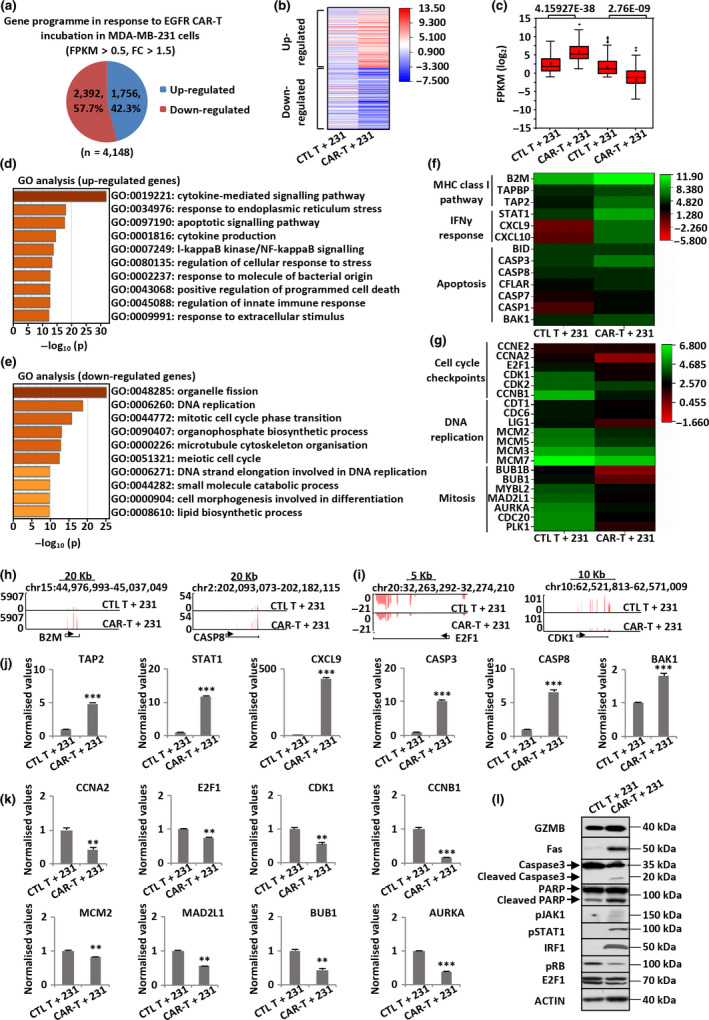
EGFR CAR‐T cells activate multiple signalling pathways in TNBC cells. (a) MDA‐MB‐231 cells were mixed with CTL T or EGFR CAR‐T cells at a ratio of 2:1 for 3 days. T cells in suspension were then removed, and the adherent tumor cells were collected, followed by RNA‐seq analysis. Up‐ and down‐regulated genes of CAR‐T cells upon incubation in MDA‐MB‐231 cells (FPKM > 0.5, FC > 1.5) are shown in the pie chart. RNA from three biological replicates was pooled for RNA‐seq. (b, c) Heat map (b) and box plot (c) representation of the expression levels (FPKM, log2) of the up‐ and down‐regulated genes described in a. P‐values are shown at the top (c). (d, e) GO analysis of up‐ (d) and down‐regulated (e) genes described in a. (f, g) The expression (FPKM, log2) of representative up‐ (f) and down‐regulated (g) genes of MDA‐MB‐231 cells in response to EGFR CAR‐T cell co‐incubation as detected by RNA‐seq is shown in the heat map. (h, i) UCSC Genome Browser views of representative up‐ (h) and down‐regulated (i) genes detected by RNA‐seq. (j, k) Cells described in a were subjected to RNA extraction and RT‐qPCR analysis to examine the expression of representative up‐ (j) and down‐regulated (k) genes of MDA‐MB‐231 cells. Data were obtained from three replicates and are presented as mean ± s.e.m. (**P < 0.01, ***P < 0.001). (l) MDA‐MB‐231 cells treated with CTL T or EGFR CAR‐T cells were subjected to IB analysis with the indicated antibodies. Molecular weight is indicated on the right. Experiments were repeated three times, and representative blots are shown. 231, MDA‐MB‐231; CAR‐T, chimeric antigen receptor‐modified T cells; chr, chromosome; CTL T, control T cells; E2F1, E2F transcription factor 1; EGFR, epidermal growth factor receptor; Fas, factor‐associated suicide; FC, fold change; FPKM, fragments per kilobase per million; GO, gene ontology; GZMB, granzyme B; IB, immunoblotting; IFNγ, interferon γ; IRF1, interferon regulatory factor 1; MHC, major histocompatibility complex; PARP, poly (ADP‐ribose) polymerase; pJAK1, Janus kinase‐1 phosphorylation; pRb, retinoblastoma protein phosphorylation; pSTAT1, signal transducer and activator of transcription 1 phosphorylation.
Examination of the gene programme affected in MDA‐MB‐231 cells revealed that at least three major signalling pathways, namely the granzyme–perforin–PARP, 55 Fas–FADD–caspase 56 and IFNγ signalling pathways, 57 , 58 were altered, which might be at least partially responsible for EGFR CAR‐T‐induced cell killing. At the postsynaptic membrane, perforin forms large transmembrane pores that enable the diffusion of granzymes into the cytosol of target cells. Human granzyme B (GZMB) can then directly cleave key substrates, such as BH3‐interacting domain death agonist (BID), 59 PARP 59 , 60 and caspases, 60 , 61 to activate the mitochondrial and DNA damage pathways. Immunoblotting analysis indicated that GZMB levels and PARP cleavage were induced in EGFR CAR‐T‐treated MDA‐MB‐231 cells (Figure 5l). The Fas/Fas ligand (FasL) signalling pathway results in the activation of caspases, leading to cleavage of substrates, such as PARP, to cause cell death. 62 Similarly, the increase of the Fas protein level and activation of caspase 3 were observed in EGFR CAR‐T‐treated MDA‐MB‐231 cells (Figure 5l). Furthermore, cytokine production by activated CAR‐T cells could further enhance their antitumor capabilities. The exceptionally high IFNγ levels secreted by EGFR CAR‐T cells (Figure 3d) and the associated response to IFNγ from MDA‐MB‐231 cells revealed by RNA‐seq (Figure 5f) prompted us to examine whether the IFNγ‐mediated signalling pathway was activated in MDA‐MB‐231 cells. Immunoblotting analysis demonstrated that IFNγ‐induced Janus kinase‐1 phosphorylation (pJAK1), signal transducer and activator of transcription 1 phosphorylation (pSTAT1) and interferon regulatory factor 1 (IRF1) protein levels were increased in EGFR CAR‐T‐treated MDA‐MB‐231 cells (Figure 5l). Meanwhile, upon EGFR CAR‐T treatment, IFNγ‐induced retinoblastoma protein (Rb) dephosphorylation was observed, leading to inactivation of E2F transcription factor 1 (E2F1) target genes, such as E2F1 itself (Figure 5l). The quantification of band intensity is shown in Supplementary figure 6. Taken together, our results show that multiple signalling pathways were activated in TNBC cells when treated with EGFR CAR‐T cells, including but not limited to the granzyme–perforin–PARP, Fas–FADD–caspase and IFNγ signalling pathways.
EGFR CAR‐T cells suppress TNBC tumorigenesis in a xenograft mouse model
Given the potent cytotoxicity of EGFR CAR‐T cells towards TNBC cells in vitro, we next sought to investigate their antitumor efficacy in mice using xenograft models. Severe combined immunodeficient (SCID) mice were subcutaneously implanted with MDA‐MB‐231 cells stably expressing a luciferase reporter for 5 days, followed by a routinely (once every other day) intravenous injection of CTL T cells (5 × 106 cells per injection) or different doses of EGFR CAR‐T cells (CAR‐T (a), 2.5 × 106 cells per injection; CAR‐T (b), 5 × 106 cells per injection; CAR‐T (c), 1 × 107 cells per injection). Tumor growth/metastasis was then monitored via calliper‐based sizing and bioluminescence imaging for 20 days (Figure 6a–d). EGFR CAR‐T cell‐treated mice showed a decreased tumor growth rate compared to the CTL T cell‐treated group. Interestingly, the medium dose (5 × 106 CAR‐T cells per injection) was the most efficient in suppressing tumor growth (Figure 6a–d). The average body weight of each group did not change significantly throughout the study (data not shown). The persistence of EGFR CAR‐T was evaluated by measuring the CAR gene copy number using genomic DNA (gDNA) extracted from peripheral blood mononuclear cells (PBMCs) of recipient mice. We observed a rapid increase in CAR gene copies 3 days after EGFR CAR‐T cell treatment (> 15 000 copies μg−1 gDNA), which gradually dropped afterwards but maintained a high level even after 20 days (10 000 copies μg−1 gDNA) (Figure 6e). In parallel, we evaluated the survival of mice and observed that in the control group, all six mice died before day 55, whereas in the EGFR CAR‐T cell‐treated group, the following occurred: two mice were cured and presented no signs of tumors, even after 4 months; and two mice were viable, presented tumors and were euthanised because of exceeding the allowable tumor size after 4 months; the last two mice started to show resistance to EGFR CAR‐T cell treatment approximately at 45 days after treatment, but were still viable at day 60, when they were euthanised because of exceeding the allowable tumor size (Figure 6f). All mice were treated with CAR‐T cells for 2 months before treatment withdraw.
Figure 6.
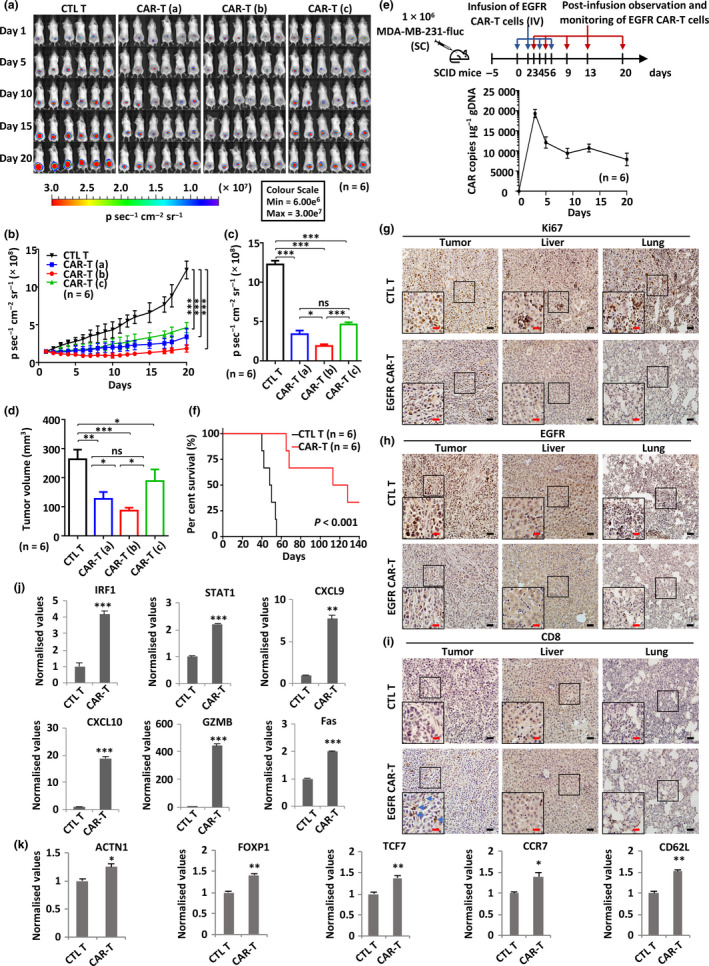
EGFR CAR‐T cells exhibit potent and specific antitumor activities in TNBC xenograft mouse model. (a) SCID mice were injected subcutaneously with MDA‐MB‐231 cells (1.0 × 106) stably expressing a luciferase reporter (MDA‐MB‐231‐fluc). Five days after tumor inoculation, mice were treated intravenously with CTL T or three different dosages of EGFR CAR‐T cells (CAR‐T (a), 2.5 × 106 cells per injection; CAR‐T (b), 5.0 × 106 cells per injection; CAR‐T (c), 1.0 × 107 cells per injection) every other day (n = 6). The day mice received CAR‐T treatment was considered as day 1. Tumor growth was monitored by using bioluminescence imaging for 20 days. Representative data of two independent experiments are shown. (b) Tumor growth curve based on bioluminescence as described in a. Data are presented as mean ± s.e.m.. Statistical significance across multiple comparisons was determined using two‐way ANOVA (***P < 0.001). (c, d) Average bioluminescence (c) and average tumor volume (d) after 20 days of observation as described in a. . Data are presented as mean ± s.e.m. (*P < 0.05, **P < 0.01, ***P < 0.001). (e) gDNA extracted from peripheral blood mononuclear cells (PBMCs) of recipient mice (n = 6) at different time points after injection of EGFR CAR‐T cells was subjected to RT‐qPCR analysis to measure CAR gene copy. The timeline of EGFR CAR‐T injection and blood collection is depicted at the top. Data are presented as mean ± s.e.m.. (f) Survival analysis of mice upon CTL T or EGFR CAR‐T treatment (5 × 106 cells per injection, n = 6). The experiment was terminated at day 140. Representative data of two independent experiments are shown. P‐value was determined using the log‐rank (Mantel–Cox) test. (g–i) Primary tumor, liver and lung sections from CTL T‐ or EGFR CAR‐T‐treated mice (5 × 106 cells per injection) described in a were subjected to IHC staining by anti‐Ki67 (g)‐, EGFR (h)‐ and CD8 (i)‐specific antibodies. Diaminobenzidine (DAB) staining was used for further chromogenic detection (brown). Blue arrows indicate CD8+ staining in i. Representative images are shown. Representative regions are enlarged from 200× (small‐sized black square) to 400× magnification (big‐sized black square) for clarity. Scale bars, 50 µm (black line) and 25 µm (red line). Representative data of three independent experiments are shown. (j, k) Primary tumor tissues from CTL T‐ or EGFR CAR‐T‐treated mice (5 × 106 cells per injection) described in a were subjected to RNA extraction and RT‐qPCR analysis to examine the expression of the indicated IFNγ target genes (j) and T‐cell genes (k). Data were obtained from three replicates and are presented as mean ± s.e.m. (*P < 0.05, **P < 0.01, ***P < 0.001). CAR‐T, chimeric antigen receptor‐modified T cells; CTL T, control T cells; EGFR, epidermal growth factor receptor; gDNA, genomic DNA; IV, intravenous injection; ns, non‐significant; SC, subcutaneous injection; SCID, severe combined immunodeficient.
The effects of EGFR CAR‐T cells on tumor growth/metastasis were also evaluated by IHC staining of the excised tumor tissue or organ sections (Figure 6g–i). Ki67 staining revealed that the expression of this protein decreased dramatically in tumors from the EGFR CAR‐T cell‐treated group (medium dose) compared to the control group (Figure 6g). TNBC has a propensity for visceral metastasis to the lungs, and less to the lymph nodes, bones or liver. 63 , 64 Consistently, in the control group, we observed that remarkable metastasis occurred in the lungs and to a lesser extent in the liver, as revealed by Ki67 staining (Figure 6g). The lung metastasis was confirmed by haematoxylin and eosin (H&E) staining (Supplementary figure 7a) and was consistent with findings of previous reports. 65 , 66 The observed lung metastasis was not seen in EGFR CAR‐T cell‐treated mice (Figure 6g). The suppression of both tumor growth and metastasis was independently confirmed by EGFR IHC staining (Figure 6h). High CD8 staining intensity, which indicates EGFR CAR‐T cells, was only observed in tumors treated with EGFR CAR‐T cells and not in the lungs or liver (Figure 6i), suggesting that there was minimal off‐target toxicity of EGFR CAR‐T cells. Serving as a control, CD8 staining was undetectable in the control group (Figure 6i). H&E‐ and TdT‐mediated dUTP nick‐end labelling (TUNEL) staining were also performed on sections of organs including the liver, lungs and spleen, and no notable toxicity was observed (Supplementary figure 7b and c). Further supporting the specificity of our EGFR CAR‐T, the latter showed to be ineffective in mice harbouring MCF7‐derived xenografts, which present relative lower expression of EGFR (Supplementary figure 8).
Tumor tissues were excised from CTL T or EGFR CAR‐T cell‐treated mice, followed by RNA extraction and RT‐qPCR analysis. Our data showed that representative genes of the IFNγ signalling pathway (IRF1, STAT1, CXCL9 and CXCL10), granzyme–perforin–PARP pathway (GZMB) and Fas–FADD–caspase pathway (Fas) were increased in the EGFR CAR‐T cell‐treated group (Figure 6j). In addition, the expression of T‐cell genes, including ACTN1, FOXP1, TCF7, CCR7 and CD62L, was found to be up‐regulated in tumor samples excised from CAR‐T cell‐treated mice (Figure 6k). These data support the infiltration of EGFR CAR‐T cells in the tumors and the activation of signalling pathways involved in killing MDA‐MB‐231 cells.
Discussion
Immunotherapy, including CAR‐T, has emerged as a promising tool to treat TNBC. The high and specific expression of EGFR in TNBC suggested that it might be an effective target for CAR design. Here, we optimally designed a CAR targeting EGFR and demonstrated that T cells infected with such CAR lentivirus exert potent and specific activity against TNBC cell growth in vitro and tumorigenesis in vivo.
Optimal CAR design is critical for the therapeutic effects of CAR‐T treatment, especially in solid tumors. A typical CAR consists of at least an extracellular antigen recognition domain from the scFv of an antibody, a spacer, a transmembrane domain and intracellular activation domains. 67 The role of a signal peptide is to direct the nascent protein into the endoplasmic reticulum. Here, we screened a number of signal peptides and chose the signal peptide from the IL‐2 receptor, which can augment the secretion of EGFR scFv and enhance the antigen recognition, as already reported. 68 The simplest form of a spacer, the region that connects the antigen binding domain and the transmembrane domain, is the hinge or the Fc region of IgG1, which is sufficient for most scFv‐based constructs. 44 In our study, EGFR CARs carrying a spacer from the hinge or the Fc region of IgG1 showed similar expression, but exhibited a dramatic difference regarding T‐cell proliferation in vitro, suggesting that the inclusion of a longer spacer better stabilises the effects of EGFR CAR. 69
One of the many limiting factors of CAR‐T therapy in clinical trials is poor T‐cell persistence. 70 Several factors can influence the persistence of adoptively transferred T cells, including patient preconditioning, 71 ex vivo culture conditions 72 and development of T‐cell exhaustion. 73 The molecular design of CARs also has a strong influence on T‐cell persistence, and therefore, it deserves attention in CAR‐T therapy research. 74 CARs containing either CD28 or 4‐1BB costimulatory domains have been widely used and yielded dramatic responses in clinical trials. 75 We have compared the efficacy of the second generation of EGFR CAR‐T cells, with only the CD28 stimulatory domain, to that of the third‐generation EGFR CAR‐T cells, with both CD28 and 4‐1BB costimulatory domains (Supplementary figure 9a). Our data indicated that the third generation of EGFR CAR‐T cells expanded more than 15‐fold in 10 days, which was much faster than the expansion of the second generation of CAR‐T cells (Supplementary figure 9b). We also compared the cytotoxicity of these two generations of CAR‐T cells by incubating them with MDA‐MB‐231 or MDA‐MB‐468 cells at different ratios and durations. The cytotoxicity of both generations of CAR‐T cells was quite similar (Supplementary figure 9c). Therefore, we have chosen to investigate the third generation of EGFR CAR‐T cells in the current study. Noteworthy, the second generation of EGFR CAR‐T cell therapy has been investigated in several phase I studies on patients with EGFR‐overexpressing tumors. 36 , 37
The potent killing activity of our EGFR CAR‐T cells towards TNBC cells in vitro prompted us to further evaluate its efficiency in vivo. Consistently with what we observed in vitro, EGFR CAR‐T cell treatment at three different dosages was capable of suppressing TNBC growth/metastasis in xenograft mouse models. A recent study reported that the third generation of EGFR CAR‐T cells effectively suppressed TNBC cell growth in vitro and tumorigenesis in vivo in models including TNBC patient‐derived xenografts. 76 We observed that a medium dose of CAR‐T cell treatment exhibited the best in vivo effect, which might be because of an optimal number of injected CAR‐T cells and amount of released cytokines. Turtle et al. 77 showed that adverse events of cytokine release syndrome (CRS) and neurotoxicity were closely related to a marked increase in the levels of inflammatory cytokines in serum produced directly by a high dosage of CAR‐T cells after encountering the tumor. It was also reported that CRS clinically manifests when large numbers of lymphocytes (B cells, T cells and/or natural killer cells) and/or myeloid cells (macrophages, dendritic cells and monocytes) become activated and release inflammatory cytokines that are directly or indirectly involved in tumor immunosuppression. 78 A recent study suggested that tumor inflammation and immunosuppressive cells can reduce the efficacy of CD19 CAR‐T cell therapy in lymphoma. 79 Furthermore, Ying et al. evaluated the differences between T cells with the variant CAR constructs and found that treatment with CD19 CAR‐T cells, which cause severe CRS, induces weight loss and eventually mortality in a substantial portion of treated mice. This study further demonstrated that the improved version of CD19 CAR‐T cells, which produce lower levels of cytokines, caused no neurological toxicity or CRS and was much more effective in treating refractory B‐cell lymphoma. 80 Based on these previous findings, we propose that a high dosage of CAR‐T cell infusion is more likely to lead to high levels of secreted cytokines and cause CRS, resulting in a lower antitumor efficacy.
Further investigation on the molecular mechanisms underlying the potent activity of EGFR CAR‐T cells revealed that the naïve‐associated EGFR CAR‐T cell population and naïve‐associated gene expression increased upon co‐culture with tumor cells, suggesting that, after stimulation, naïve‐associated T cells undergo robust expansion and persist, as previously reported. 81 , 82 On one hand, the expansion of naïve‐associated CAR‐T cells after tumor exposure might be because of the 4‐1BB CAR signalling, which can reduce T‐cell exhaustion and differentiation. 83 It has also been reported that the population of TCM is progressively enriched after stimulation through 4‐1BB CAR signalling. 84 Moreover, in our study, some of the activated CAR‐T cells died during co‐culture, which might also contribute to an increased expression of naïve‐associated genes. On the other hand, the investigation of the signalling pathways activated in TNBC by EGFR CAR‐T cells revealed that our EGFR CAR‐T induced a strong IFNγ response in TNBC cells. IFNγ is a critical cytokine for antitumor immunity under both physiological and pathological conditions. A previous study reported that IFNγ‐dependent apoptosis and cell cycle arrest are key contributors to tumor growth suppression, which warrant future blocking or knockdown studies to confirm the causal role of IFNγ signalling. 85
Epidermal growth factor receptor is expressed on the cell surface of most normal epithelial tissues, and the off‐tumor toxicity related to EGFR CAR‐T cell therapy is indeed concerning. 86 In our study, CD8 staining showed that the infiltration of EGFR CAR‐T cells was only observed in primary tumors and not in the lungs and liver. H&E and TUNEL staining also showed no notable toxicity of EGFR CAR‐T cells in mice. In contrast to the potent activity exerted against TNBC xenografts, EGFR CAR‐T treatment showed no efficacy against MCF7‐derived xenografts, which present relatively lower EGFR expression. All these data suggest that EGFR CAR‐T‐mediated cytotoxicity can be differentiated between EGFR‐overexpressing TNBC cells and breast cancer cells or normal cells with relatively lower EGFR expression. Of note, the anti‐EGFR scFv was derived from cetuximab, a monoclonal antibody used in the clinic that can recognise the domain III of human EGFR. 87 Sequence alignment between human and mouse EGFR revealed that there is 87% similarity for this region, indicating that cetuximab might cross‐react with mouse EGFR. Thus, as the possibility of mouse EGFR being recognised by anti‐EGFR scFv cannot be ruled out at this stage, we cannot confidently predict the clinical efficacy of our EGFR CAR‐T cell therapy in TNBC patients. Future investigation using the skin graft toxicity model 88 as well as the cynomolgus monkey model 89 will be essential to verify the off‐tumor effects of EGFR CAR‐T cells. A phase I study conducted by the Han group on NSCLC patients treated with the second generation of EGFR CAR‐T cells showed controllable toxicity, suggesting that EGFR CAR therapy might be feasible for cancer treatment. 36 In an independent phase I study on patients with EGFR‐positive, advanced biliary tract cancer treated with the second generation of EGFR CAR‐T cells, data revealed that the most likely on‐target/off‐tumor adverse events were mucosal/cutaneous toxicity, including oral mucositis, oral ulcer, gastrointestinal haemorrhage, desquamation and pruritus. However, all these toxicity events could be reversed, and no treatment‐related deaths occurred. 37 These studies suggest that our third generation of EGFR CAR‐T cells might also be applicable in the treatment of TNBC patients in the clinic. To avoid on‐target/off‐tumor toxicity, more factors should be taken into consideration, such as the dose of CAR‐T cells used and the optimisation of CAR by incorporating suicide genes and tandem CAR. These safety strategies will be included in future pre‐clinical and clinical studies using EGFR CAR‐T cells to treat TNBC.
In conclusion, our study provides an effective and safe EGFR CAR design, and T cells infected with EGFR CAR lentivirus exerted potent and specific suppression of TNBC both in vitro and in vivo. We envision that EGFR CAR‐T therapy may be an attractive strategy for treating solid tumors with EGFR overexpression.
Methods
Cell lines and cell culture
Breast cancer cell lines were obtained from the Cell Bank (Chinese Academy of Sciences, Shanghai, China). MDA‐MB‐231, MCF7, HS578T and SK‐BR‐3 were maintained in Dulbecco's modified Eagle's medium (DMEM) high glucose (Biological Industries, Kibbutz Beit Haemek, Israel) supplemented with 10% heat‐inactivated foetal bovine serum (FBS; Gibco, Grand Island, NY, USA). BT474, MDA‐MB‐468 and Hcc1806 were maintained in Roswell park memorial institute (RPMI) 1640 (Biological Industries) medium supplemented with 10% heat‐inactivated FBS. T47D cells were maintained in RPMI 1640 medium supplemented with 10% heat‐inactivated FBS and sodium pyruvate (Gibco). MCF10A were maintained in DMEM/F‐12 medium (Gibco) supplemented with 5% heat‐inactivated FBS, hydrocortisone (10 mg mL−1; Sangon Biotech, Shanghai, China), insulin (0.01 mg mL−1; Sino, Beijing, China) and recombinant human epidermal growth factor (5 μg mL−1; Invitrogen, Carlsbad, CA, USA). Cells were cultured in a humidified incubator with 5% CO2 at 37 °C.
Animal experiments
All animal experiments were conducted following a protocol approved by the Animal Care and Use Committee of the Xiamen University. MDA‐MB‐231 and MCF7 cells stably expressing firefly luciferase (MDA‐MB‐231‐fluc and MCF7‐fluc cells, respectively) were established by transducing cells with a lentiviral vector expressing a green fluorescent protein (GFP)–internal ribosome entry site (IRES)–luciferase/puromycin cassette. After selection with puromycin (InvivoGen, San Diego, CA, USA) for 2 weeks, cells were sorted by fluorescence‐activated cell sorting (BD Biosciences, San Jose, CA, USA) and verified by fluorescence microscopy (Olympus, Tokyo, Japan) for GFP expression. To evaluate the antitumor activity of EGFR CAR‐T cells in vivo, either 1.0 × 106 MDA‐MB‐231‐fluc cells or 3.0 × 106 MCF7‐fluc cells suspended in 100 μL phosphate‐buffered saline (PBS; BBI Life Science Corporation, Shanghai, China) were inoculated subcutaneously into female SCID mice aged 6 to 12 weeks (Shanghai SLAC Laboratory Animal Center, Shanghai, China). To support the growth of MCF7‐derived xenografts, mice were brushed with oestrogen (E2, 10−2 Mm; Sigma‐Aldrich, Saint Louis, MO, USA) every 3 days for the duration of the experiments. Five days after inoculation of MDA‐MB‐231 cells, CTL T cells or three different dosages of EGFR CAR‐T cells (2.5 × 106, 5.0 × 106 or 1.0 × 107 cells per injection) were administrated intravenously every other day (n = 6 per group). When mice were inoculated with MCF7‐fluc cells, mice were treated with E2 for 7 days before treatment with CTL T or EGFR CAR‐T cells (5.0 × 106 cells per injection) intravenously every other day (n = 3 per group). The day mice received CAR‐T treatment was considered as day 1. To evaluate the effects of CAR‐T cells in mice, the tumor progression was observed for 20 days in either the MDA‐MB‐231‐fluc or the MCF7‐fluc xenograft model. Tumor progression was monitored by bioluminescence using the Xenogen IVIS Lumina imaging system (Caliper Life Sciences, Hopkinton, MA, USA), and tumor volume was measured using a calliper (Fisher Scientific, Pittsburgh, PA, USA). For bioluminescence, mice were injected intraperitoneally with 75 mg kg−1 beetle luciferin (Promega, Madison, WI, USA) and then imaged 6–8 min later with an exposure time of 3 min. Luminescence images were analysed using the Living Image software (Caliper Life Sciences). Tumor volume (V) was calculated according to the formula V = 0.5 × (length × width2). Mice were weighed every 2 days post infusion. After 20 days of observation, mice were euthanised for histopathology and RT‐qPCR analysis. In parallel to experiments to measure the copy number of EGFR CAR in vivo, mice treated with EGFR CAR‐T cells (5.0 × 106 cells per injection; 6 mice per time point) were sacrificed on days 3, 5, 9, 13 and 20 to obtain peripheral blood for gDNA extraction. In parallel to experiments to evaluate the long‐term survival, mice were administrated with CTL T or EGFR CAR‐T cells (5.0 × 106 cells per injection; six mice per group) every other day for 2 months and observed for as long as 140 days.
Generation of human EGFR CAR‐T lymphocytes
The third generation of EGFR CAR is constituted by a signal peptide (Sp1) of the IL‐2 receptor; an anti‐EGFR scFv from cetuximab (a monoclonal antibody used in the clinic); a spacer (IgG1 Fc or hinge); a CD28 transmembrane domain; and CD28, human 4‐1BB and CD3ζ intracellular signalling domains. The second generation of EGFR CAR is constituted by a signal peptide of the IL‐2 receptor, an anti‐EGFR scFv from cetuximab, a spacer (IgG1 Fc), a CD28 transmembrane domain, and CD28 and CD3ζ intracellular signalling domains. Both EGFR CARs were cloned into the lentiviral vector pCDH (Youbio, Changsha, China) and confirmed by DNA sequencing. The sequence information of anti‐EGFR scFv was the following: QVQLKQSGPGLVQPSQSLSITCTVSGFSLTNYGVHWVR QSPGKGLEWLGVIWSGGNTDYNTPFTSRLSINKDNSKSQVFFKMNSLQSNDTAIYYCARALTYYDYEFAYWGQGTLVTVSAGGGGSGGGGSGGGGSDILLTQSPVILSVSPGERVSFSCRASQSIGTNIHWYQQRTNGSPRLLIKYASESISGIPSRFSGSGSGTDFTLSINSVESEDIADYYCQQNNNWPTTFGAGTKLELK.
HEK293T cells were transfected with lentiviral and packaging vectors (CAR:psPAX:pMD2G = 4:3:1) using polyethylenimine (Mw 40000; Polysciences, Warrington, PA, USA) for 60 h, according to the manufacturer's instructions, before collecting supernatants, which were further concentrated 40‐fold by using Amicon Ultra‐15 Centrifugal Filters (100 kDa; Millipore, Billerica, MA, USA). Concentrated lentiviruses were stored at −80 °C. Titrations were measured by the qPCR Lentivirus Titration (Titer) Kit (Applied Biological Materials, Crestwood Place Richmond, BC, Canada).
Peripheral blood mononuclear cells were isolated from whole blood of healthy volunteer donors using the Ficoll (GE Healthcare, Chicago, IL, USA) density gradient centrifugation method. For T‐cell expansion, PBMCs were cultured and stimulated with anti‐CD3 and anti‐CD28 antibodies (Biolegend, San Diego, CA, USA) in X‐VIVO medium (Lonza, Basel, Switzerland) supplemented with IL‐2 and IL‐15 (Sino) for 5–7 days at 37 °C in a humidified incubator with 5% CO2. Primary T cells were then subjected to lentivirus infection. To improve infection efficiency, T cells were spun at 800 × g for 50 min at 32 °C after adding the lentiviral supernatant. Cells were monitored daily, and the culture medium was replaced every 2–3 days for a period of 11–14 days prior to the use of the infected cells in in vitro and/or in vivo experiments. This study was approved by the Ethics Committee of the Xiamen University, and each donor signed an informed consent before blood collection.
Immunoblotting
Immunoblotting was performed as previously described. 90 The following primary antibodies used were as follows: anti‐EGFR and anti‐β actin (Proteintech, Chicago, IL, USA); anti‐CD3ζ, anti‐pRb (S780) and anti‐E2F1 (Abcam, Cambridge, UK); anti‐granzyme B (D6E9W), anti‐IRF1 (D5E4), anti‐PARP (46D11), anti‐Fas (C18C12), anti‐caspase 3 and anti‐pJAK1 (Tyr1034/1035; D7N4Z; Cell Signaling Technology, Danvers, MA, USA), and anti‐pSTAT1 (pY701.4A; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Three biological replicates were performed, and the intensity of the bands of all replicates was quantified by ImageJ (National Institutes of Health, Bethesda, MD, USA) and normalised to that of the actin loading control. Data are presented as mean ± s.e.m. (standard error of the mean).
Flow cytometry
Expression of EGFR on the surface of TNBC cells was detected using a PE‐conjugated mouse anti‐human EGFR antibody (BD Biosciences). The expression of the T‐cell surface markers CD3, CD8, CD25, CD69, CCR7 and CD62L was detected using a FITC‐conjugated mouse anti‐human CD3 antibody, APC‐conjugated mouse anti‐human CD8 antibody, FITC‐conjugated mouse anti‐human CD25 antibody, PE‐conjugated mouse anti‐human CD69 antibody (all from BD Biosciences), Pacific Blue™‐conjugated anti‐human CCR7 antibody and PE‐conjugated anti‐human CD62L antibody (both from Biolegend), respectively. The infection efficiency of Fc‐EGFR CAR and Hinge‐EGFR CAR in T cells was examined by using an APC‐conjugated goat F(ab′)2 anti‐human IgG‐Fc antibody and a FITC‐conjugated goat anti‐human IgG (Fab′)2 (biotin) antibody (Abcam), respectively. Fluorescence was assessed using an Attune NxT Flow Cytometer (Thermo Fisher Scientific, Waltham, MA, USA), and the data were analysed with FlowJo vX.0.7 (BD Biosciences).
Enzyme‐linked immunosorbent assay (ELISA)
T cells (EGFR CAR‐T cells or non‐infected T cells) were co‐cultured with TNBC cells in a 24‐well plate (Corning Incorporated, Corning, NY, USA) for 72 h, and supernatants were collected to determine the presence of TNFα, IFNγ and IL‐2 with the Human TNFα ELISA Kit, Human IFNγ ELISA Kit and Human IL‐2 ELISA Kit (Dakewe Biotech, Beijing, China), respectively, following the manufacturer's instructions.
Cell proliferation assay
Chimeric antigen receptor‐modified T cells labelled with CFSE by using the CellTrace™ CFSE Cell Proliferation Kit (Invitrogen) were incubated with or without TNBC cells at the ratio of 2:1 in culture medium without adding proliferative cytokines for 72 h. Proliferation was assessed by monitoring CFSE dilution. Absolute cell counts during the expansion of EGFR CAR‐T cells were obtained using an Attune NxT Flow Cytometer. The number of viable CAR‐T cells was counted by using a haemocytometer (Paul Marienfeld, Lauda‐Koenigshofen, Germany).
Cytotoxicity assay
The cytotoxicity assay was performed using an xCELLigence real‐time cell analyzer (RTCA) System (ACEA Biosciences, San Diego, CA, USA). The impedance‐based RTCA was used for label‐free and real‐time monitoring of cytolysis activity. The cell index (CI) based on the detected cell–electrode impedance was used to measure cell viability. The % cytotoxicity value was calculated via the following formula: ((CI (tumor only) − CI (tumor + T cells))/CI tumor only) × 100%. TNBC (MDA‐MB‐231 and MDA‐MB‐468), non‐TNBC (MCF7 and BT474) or normal breast epithelial cells (MCF10A) were seeded at a density of 2–5 × 104 cells per well and grown for 24 h. CTL T or EGFR CAR‐T cells were then added into the RTCA unit at different ratios (T cells:tumor cells = 1:1, 3:1, 6:1 or 10:1). The impedance signals were recorded for the duration of 24–96 h at 5‐min intervals. Primary TNBC cells were obtained from the First Affiliated Hospital of the Xiamen University. Tumor samples were collected according to a protocol approved by the institutional review board of the Xiamen University. Written informed consent was obtained from all donors. For the generation and growth of primary TNBC cells, fresh human breast cancer tissues were obtained from patients immediately after resection, cut into small pieces (10 mm3) and grafted onto the mammary fat pad of BALB/C nude mice (Shanghai SLAC Laboratory Animal Center) as previously described. 91 The mice were daily monitored until primary tumors were resected. Cells were then isolated from the primary tumor and cultured in DMEM high glucose supplemented with 10% heat‐inactivated FBS. The cytotoxicity assay with primary TNBC cells was performed as described above.
Histopathological analysis
Mouse tissue samples were resected, formalin‐fixed and paraffin‐embedded. Four‐micrometre tissue sections were prepared, deparaffinised and rehydrated for further staining. For IHC staining, the EDTA antigen retrieval solution and UltraSensitive™ SP (Mouse/Rabbit) IHC Kit (Maxim Biotechnologies, Fuzhou, China) were used according to the instructions provided by the manufacturer. Primary antibodies against human Ki67, human EGFR (both from Maxim Biotechnologies) or human CD8 (Abcam) were incubated at 4 °C overnight at an optimised concentration. Stained tissue sections were developed with the Diaminobenzidine (DAB) Kit (Maxim Biotechnologies) for 1 min and counterstained with haematoxylin solution (Sigma‐Aldrich) for 10 min. For H&E staining, sections were stained with haematoxylin solution (Sigma‐Aldrich) for 5 min and eosin Y solution (Sigma‐Aldrich) for 1 min. For TUNEL assays, sections were treated with 20 μg mL−1 proteinase K (AMRESCO, Solon) at 37 °C for 20 min and then washed in PBS. The commercially available Colorimetric TUNEL Apoptosis Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China) was used to detect apoptotic cells according to the instructions provided by the manufacturer. The histopathological images were obtained and analysed using the Cellsens Standard software (Olympus).
RNA isolation and RT‐qPCR
Total RNA was isolated using the Eastep Super Total RNA Extraction Kit (Promega) following the manufacturer's protocol. RNA from three biological replicates was pooled together, and first‐strand complementary DNA (cDNA) synthesis from total RNA was carried out using the GoScript Reverse Transcription System (Promega), followed by quantitative PCR (qPCR) using an AriaMx Real‐Time PCR machine (Agilent Technologies, Santa Clara, CA, USA). Three biological replicates were performed, and data are presented as mean ± s.e.m..
To assess the persistence of the CAR‐T cells in recipient mice, PBMCs of SCID mice were collected from the eye balls and separated by Ficoll (Solarbio, Beijing, China) centrifugation, followed by gDNA extraction using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). RT‐qPCR was performed by using a primer spanning the junction of the CD137 domain and the adjacent CD3ζ chain (forward primer, 5′‐GAAGAAGGAGGATGTGAACT‐3′; reverse primer, 5′‐TCCTCTCTTCGTCCTAGATT‐3′). Standard curves were prepared using serial dilutions of the CAR plasmid starting at 106 copies μL−1.
RNA‐seq
Epidermal growth factor receptor CAR‐T and MDA‐MB‐231 cells were separated following co‐culture and subjected to RNA extraction. Three biological replicates were performed, and RNA was pooled for sequencing. Specifically, EGFR CAR‐T cells were incubated with MDA‐MB‐231 cells at a ratio of 1:2 or without MDA‐MB‐231 cells for 3 days, and CAR‐T cells in suspension were then collected, separated from adherent tumor cells and spun down at 1000 rpm for 5 min. Dead CAR‐T cells were removed using the Dead Cell Removal Kit (Miltenyi Biotec, Auburn, CA, USA) before RNA extraction and RNA‐seq analysis. MDA‐MB‐231 cells were treated with CTL T or EGFR CAR‐T cells at a ratio of 2:1 for 3 days. T cells in suspension were then removed, and the adherent tumor cells were collected. Dead tumor cells were also removed using the Dead Cell Removal Kit before RNA extraction and RNA‐seq.
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) following the manufacturer's protocol. DNase I (Sigma‐Aldrich) in the column digestion was included to ensure RNA quality. The RNA library preparation was performed by using the NEBNext® Ultra™ Directional RNA Library Prep Kit (Illumina, San Diego, CA, USA). Paired‐end sequencing was performed with Illumina HiSeq 3000. Sequencing reads were aligned to hg19 RefSeq database by using Tophat (http://ccb.jhu.edu/software/tophat/index.shtml). Cuffdiff was used to quantify the expression of RefSeq annotated genes with the option –M (reads aligned to repetitive regions were masked) and –u (multiple aligned reads were corrected using ‘rescue method’). Coding genes with fragments per kilobase per million (FPKM) mapped reads larger than 0.5 were included in our analysis. Up‐ and down‐regulated genes were determined by fold change of gene FPKM. FPKM of a gene was calculated as mapped reads on exons divided by exonic length and the total number of mapped reads. Box plots and heat maps were generated with the R software, and significance was determined using Student's t‐test.
Statistical analysis
The comparison of two groups or data points was performed by the two‐tailed t‐test. Multiple comparisons were analysed by two‐way analysis of variance (ANOVA). Survival curves were constructed according to the Kaplan–Meier method and compared using the log‐rank (Mantel–Cox) test. P‐values < 0.05 were considered statistically significant.
Data extraction and generation from TCGA database
For the expression data shown in Figure 1c, expression data were downloaded from TCGA portal (http://tumorsurvival.org), and the median of EGFR expression in different subtypes was calculated (LumA, n = 480; LumB, n = 197; TNBC, n = 157; HER2, n = 73; N/A (not available), n = 287; Normal, n = 27). Significance was assessed with the Student's t‐test. For Supplementary figure 1a, the plot was generated directly from Breast Cancer Gene‐Expression Miner 4.4 (http://bcgenex.centregauducheau.fr/BC‐GEM/GEM‐Accueil.php?js=1) under the ‘Analysis section’ by choosing EGFR as target gene and TNBC as population. For Supplementary figure 1b–e, Kaplan–Meier plotter analysis was generated directly from Breast Cancer Gene‐Expression Miner 4.4 under the ‘Analysis section’ by following pathway ‘Prognostic/Intrinsic molecular subtypes/target gene’ and then choosing EGFR as target gene.
Conflict of interest
The authors declare no conflict of interest.
Author Contributions
Lin Xia: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Project administration; Writing‐original draft; Writing‐review & editing. Zao‐zao Zheng: Data curation; Methodology; Validation; Writing‐review & editing. Jun‐yi Liu: Data curation; Methodology; Validation; Writing‐review & editing. Yu‐jie Chen: Data curation; Methodology; Writing‐review & editing. Jian‐cheng Ding: Software. Ning‐shao Xia: Resources. Wen‐xin Luo: Conceptualization; Project administration; Supervision. Wen Liu: Conceptualization; Formal analysis; Funding acquisition; Investigation; Project administration; Supervision; Writing‐original draft; Writing‐review & editing.
Ethical approval and consent to participate
Animal studies were approved by the Animal Care and Use Committee of the Xiamen University. This study was approved by the Ethics Committee of the Xiamen University, and experiments with human specimens were undertaken with the understanding and signed informed consent of each donor. Primary TNBC samples were collected according to a protocol approved by the institutional review board of the Xiamen University.
Supporting information
Supplementary figures 1‐9
Acknowledgments
The work of LX was supported by the National Natural Science Foundation of China (31600748) and Project of XMU Training Program of Innovation and Entrepreneurship for Undergraduates (2019X0959), and the work of Wen Liu was supported by the National Natural Science Foundation of China (81761128015, 81861130370, 31871319 and 91953114), Fujian Province Health Education Joint Research Project (WKJ2016‐2‐09), Xiamen Science and Technology Project (2017S0091), Xiamen Science and Technology major projects (3502Z20171001‐20170302) and Fundamental Research Funds for the Central University (20720190145).
Contributor Information
Wen‐xin Luo, Email: wxluo@xmu.edu.cn.
Wen Liu, Email: w2liu@xmu.edu.cn.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2. Perou CM, Sorlie T, Eisen MB et al Molecular portraits of human breast tumours. Nature 2000; 406: 747–752. [DOI] [PubMed] [Google Scholar]
- 3. Sorlie T, Perou CM, Tibshirani R et al Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001; 98: 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Foulkes WD, Smith Ian E, Reisfilho Jorge S. Triple‐negative breast cancer. N Engl J Med 2010; 363: 1938–1948. [DOI] [PubMed] [Google Scholar]
- 5. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple‐negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 2016; 13: 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malorni L, Shetty PB, De Angelis C et al Clinical and biologic features of triple‐negative breast cancers in a large cohort of patients with long‐term follow‐up. Breast Cancer Res Treat 2012; 136: 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Den Brok WD, Speers C, Gondara L, Baxter E, Tyldesley S, Lohrisch CA. Survival with metastatic breast cancer based on initial presentation, de novo versus relapsed. Breast Cancer Res Treat 2017; 161: 549–556. [DOI] [PubMed] [Google Scholar]
- 8. Kassam F, Enright K, Dent R et al Survival outcomes for patients with metastatic triple‐negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer 2009; 9: 29–33. [DOI] [PubMed] [Google Scholar]
- 9. Schmid P, Adams S, Rugo HS et al Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med 2018; 379: 2108–2121. [DOI] [PubMed] [Google Scholar]
- 10. Hwang I, Nguyen N. Mechanisms of tumor‐induced T cell immune suppression and therapeutics to counter those effects. Arch Pharm Res 2015; 38: 1415–1433. [DOI] [PubMed] [Google Scholar]
- 11. Li Z, Qiu Y, Lu W, Jiang Y, Wang J. Immunotherapeutic interventions of triple negative breast cancer. J Transl Med 2018; 16: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Porter DL, Hwang W, Frey NV et al Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7: 303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grupp SA, Kalos M, Barrett DM et al Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Specht JM, Lee S, Turtle C et al Phase I study of immunotherapy for advanced ROR1+ malignancies with autologous ROR1‐specific chimeric antigen receptor‐modified (CAR)‐T cells. J Clin Oncol 2018; 36: TPS79. [Google Scholar]
- 15. Zhou R, Yazdanifar M, Roy LD et al CAR T cells targeting the tumor MUC1 glycoprotein reduce triple‐negative breast cancer growth. Front Immunol 2019; 10: 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han Y, Xie W, Song D, Powell DJ. Control of triple‐negative breast cancer using ex vivo self‐enriched, costimulated NKG2D CAR T cells. J Hematol Oncol 2018; 11: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nielsen TO, Hsu FD, Jensen K et al Immunohistochemical and clinical characterization of the basal‐like subtype of invasive breast carcinoma. Clin Cancer Res 2004; 10: 5367–5374. [DOI] [PubMed] [Google Scholar]
- 18. Cheang MCU, Voduc D, Bajdik C et al Basal‐like breast cancer defined by five biomarkers has superior prognostic value than triple‐negative phenotype. Clin Cancer Res 2008; 14: 1368–1376. [DOI] [PubMed] [Google Scholar]
- 19. Viale G, Rotmensz N, Maisonneuve P et al Invasive ductal carcinoma of the breast with the “triple‐negative” phenotype: prognostic implications of EGFR immunoreactivity. Breast Cancer Res Treat 2009; 116: 317–328. [DOI] [PubMed] [Google Scholar]
- 20. Shien T, Tashiro T, Omatsu M et al Frequent overexpression of epidermal growth factor receptor (EGFR) in mammary high grade ductal carcinomas with myoepithelial differentiation. J Clin Pathol 2005; 58: 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Livasy CA, Karaca G, Nanda R et al Phenotypic evaluation of the basal‐like subtype of invasive breast carcinoma. Mod Pathol 2006; 19: 264–271. [DOI] [PubMed] [Google Scholar]
- 22. Kim M, Ro JY, Ahn SH, Kim HH, Kim S, Gong G. Clinicopathologic significance of the basal‐like subtype of breast cancer: a comparison with hormone receptor and Her2/neu‐overexpressing phenotypes. Hum Pathol 2006; 37: 1217–1226. [DOI] [PubMed] [Google Scholar]
- 23. Kim D, Jung W, Koo JS. The expression of ERCC1, RRM1, and BRCA1 in breast cancer according to the immunohistochemical phenotypes. J Korean Med Sci 2011; 26: 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li RH, Huang WH, Wu JD, Du CW, Zhang GJ. EGFR expression is associated with cytoplasmic staining of CXCR4 and predicts poor prognosis in triple‐negative breast carcinomas. Oncol Lett 2017; 13: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park HS, Jang MH, Kim EJ et al High EGFR gene copy number predicts poor outcome in triple‐negative breast cancer. Mod Pathol 2014; 27: 1212–1222. [DOI] [PubMed] [Google Scholar]
- 26. Baselga J, Gomez P, Greil R et al Randomized phase II study of the anti–epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple‐negative breast cancer. J Clin Oncol 2013; 31: 2586–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nabholtz JM, Abrial C, Mouretreynier M et al Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane based chemotherapy in operable triple negative breast cancer: identification of biologically‐defined signatures predicting treatment impact. Ann Oncol 2014; 25: 1570–1577. [DOI] [PubMed] [Google Scholar]
- 28. Crozier JA, Advani P, Laplant BR et al N0436 (alliance): a phase II trial of irinotecan with cetuximab in patients with metastatic breast cancer previously exposed to anthracycline and/or taxane‐containing therapy. Clin Breast Cancer 2016; 16: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song D, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ. Effective adoptive immunotherapy of triple‐negative breast cancer by folate receptor‐α redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol 2016; 9: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou X, Li J, Wang Z et al Cellular immunotherapy for carcinoma using genetically modified EGFR‐specific T lymphocytes. Neoplasia 2013; 15: 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X, Jiang S, Fang C et al Affinity‐tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015; 75: 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caruso HG, Hurton LV, Najjar A et al Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 2015; 75: 3505–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feng K, Guo Y, Liu Y et al Cocktail treatment with EGFR‐specific and CD133‐specific chimeric antigen receptor‐modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol 2017; 10: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dong Y, Ding Y, Guo W et al The functional verification of EGFR‐CAR T‐cells targeted to hypopharyngeal squamous cell carcinoma. Onco Targets Ther 2018; 11: 7053–7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H, Huang Y, Jiang D et al Antitumor activity of EGFR‐specific CAR T cells against non‐small‐cell lung cancer cells in vitro and in mice. Cell Death Dis 2018; 9: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feng K, Guo Y, Dai H et al Chimeric antigen receptor‐modified T cells for the immunotherapy of patients with EGFR‐expressing advanced relapsed/refractory non‐small cell lung cancer. Sci China Life Sci 2016; 59: 468–479. [DOI] [PubMed] [Google Scholar]
- 37. Guo Y, Feng K, Liu Y et al Phase I study of chimeric antigen receptor–modified T cells in patients with EGFR‐positive advanced biliary tract cancers. Clin Cancer Res 2018; 24: 1277–1286. [DOI] [PubMed] [Google Scholar]
- 38. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5: 341–354. [DOI] [PubMed] [Google Scholar]
- 39. Rae JM, Scheys JO, Clark KM, Chadwick RB, Kiefer MC, Lippman ME. EGFR and EGFRvIII expression in primary breast cancer and cell lines. Breast Cancer Res Treat 2004; 87: 87–95. [DOI] [PubMed] [Google Scholar]
- 40. Davidson NE, Gelmann EP, Lippman ME, Dickson RB. Epidermal growth factor receptor gene expression in estrogen receptor‐positive and negative human breast cancer cell lines. Mol Endocrinol 1987; 1: 216–223. [DOI] [PubMed] [Google Scholar]
- 41. Tan DSP, Marchio C, Jones RL et al Triple negative breast cancer: molecular profiling and prognostic impact in adjuvant anthracycline‐treated patients. Breast Cancer Res Treat 2008; 111: 27–44. [DOI] [PubMed] [Google Scholar]
- 42. Perez R, Pascual MR, Macias A, Lage A. Epidermal growth factor receptors in human breast cancer. Breast Cancer Res Treat 1984; 4: 189–193. [DOI] [PubMed] [Google Scholar]
- 43. Sainsbury JR, Malcolm AJ, Appleton DR, Farndon JR, Harris AL. Presence of epidermal growth factor receptor as an indicator of poor prognosis in patients with breast cancer. J Clin Pathol 1985; 38: 1225–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srivastava S, Riddell SR. Engineering CAR‐T cells: design concepts. Trends Immunol 2015; 36: 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev 2019; 12: 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR‐T immunotherapy. Cancers (Basel) 2016; 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kagoya Y, Nakatsugawa M, Yamashita Y et al BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest 2016; 126: 3479–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gattinoni L, Lugli E, Ji Y et al A human memory T cell subset with stem cell‐like properties. Nat Med 2011; 17: 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seet CS, He C, Bethune MT et al Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods 2017; 14: 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pearce EL, Mullen AC, Martins GA et al Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science 2003; 302: 1041–1043. [DOI] [PubMed] [Google Scholar]
- 51. Joshi NS, Cui W, Chandele A et al Inflammation directs memory precursor and short‐lived effector CD8+ T cell fates via the graded expression of T‐bet transcription factor. Immunity 2007; 27: 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rutishauser RL, Martins GA, Kalachikov S et al Transcriptional repressor blimp‐1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009; 31: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gattinoni L, Zhong XS, Palmer DC et al Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 2009; 15: 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Benmebarek MR, Karches HC, Cadilha LB, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci 2019; 20: 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015; 15: 388–400. [DOI] [PubMed] [Google Scholar]
- 56. Wang M, Su P. The role of the Fas/FasL signaling pathway in environmental toxicant‐induced testicular cell apoptosis: an update. Syst Biol Reprod Med 2018; 64: 93–102. [DOI] [PubMed] [Google Scholar]
- 57. Fulda S, Debatin KM. IFNγ sensitizes for apoptosis by upregulating caspase‐8 expression through the Stat1 pathway. Oncogene 2002; 21: 2295–2308. [DOI] [PubMed] [Google Scholar]
- 58. Ertosun MG, Hapil FZ, Osman Nidai O. E2F1 transcription factor and its impact on growth factor and cytokine signaling. Cytokine Growth Factor Rev 2016; 31: 17–25. [DOI] [PubMed] [Google Scholar]
- 59. Martinvalet D. Mitochondrial entry of cytotoxic proteases: a new insight into the granzyme B cell death pathway. Oxid Med Cell Longev 2019; 2019: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Andrade F, Roy S, Nicholson DW, Thornberry NA, Rosen A, Casciolarosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL‐induced apoptosis. Immunity 1998; 8: 451–460. [DOI] [PubMed] [Google Scholar]
- 61. Metkar SS, Wang B, Ebbs ML et al Granzyme B activates procaspase‐3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol 2003; 160: 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol 2017; 17: 333–340. [DOI] [PubMed] [Google Scholar]
- 63. Rakha EA, Chan S. Metastatic triple‐negative breast cancer. Clin Oncol 2011; 23: 587–600. [DOI] [PubMed] [Google Scholar]
- 64. Jitariu AA, Cîmpean AM, Ribatti D, Raica M. Triple negative breast cancer: the kiss of death. Oncotarget 2017; 8: 46652–46662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rowan BG, Gimble JM, Sheng M et al Human adipose tissue‐derived stromal/stem cells promote migration and early metastasis of triple negative breast cancer xenografts. PLoS One 2014; 9: e89595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chabottaux V, Sounni NE, Pennington CJ et al Membrane‐type 4 matrix metalloproteinase promotes breast cancer growth and metastases. Cancer Res 2006; 66: 5165–5172. [DOI] [PubMed] [Google Scholar]
- 67. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR‐T cells. Biomark Res 2017; 5: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang L, Leng Q, Mixson AJ. Alteration in the IL‐2 signal peptide affects secretion of proteins in vitro and in vivo . J Gene Med 2005; 7: 354–365. [DOI] [PubMed] [Google Scholar]
- 69. Pule M, Finney HM, Lawson AD. Artificial T‐cell receptors. Cytotherapy 2003; 5: 211–226. [DOI] [PubMed] [Google Scholar]
- 70. Ghorashian S, Kramer AM, Onuoha S et al Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low‐affinity CD19 CAR. Nat Med 2019; 25: 1408–1414. [DOI] [PubMed] [Google Scholar]
- 71. Thistlethwaite FC, Gilham DE, Guest RD et al The clinical efficacy of first‐generation carcinoembryonic antigen (CEACAM5)‐specific CAR T cells is limited by poor persistence and transient pre‐conditioning‐dependent respiratory toxicity. Cancer Immunol Immunother 2017; 66: 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third‐generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy 2015; 17: 487–495. [DOI] [PubMed] [Google Scholar]
- 73. Kasakovski D, Xu L, Li Y. T cell senescence and CAR‐T cell exhaustion in hematological malignancies. J Hematol Oncol 2018; 11: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Guedan S, Posey DA, Shaw CE et al Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI insight 2018; 3: e96976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second‐generation chimeric antigen receptors. Nat Rev Drug Discov 2015; 14: 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu Y, Zhou Y, Huang KH et al EGFR‐specific CAR‐T cells trigger cell lysis in EGFR‐positive TNBC. Aging (Milano) 2019; 11: 11054–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Turtle CJ, Hanafi LA, Berger C et al CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016; 126: 2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee DW, Gardner R, Porter DL et al Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124: 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jain M, Zhao H, Atkins R et al Tumor inflammation and myeloid derived suppressor cells reduce the efficacy of CD19 CAR T cell therapy in lymphoma. Blood 2019; 134: 2885. [Google Scholar]
- 80. Ying Z, Huang XF, Xiang X et al A safe and potent anti‐CD19 CAR T cell therapy. Nat Med 2019; 25: 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Obar JJ, Lefrançois L. Memory CD8+ T cell differentiation. Ann N Y Acad Sci 2010; 1183: 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab 2017; 26: 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Long AH, Haso WM, Shern JF et al 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kawalekar OU, O'Connor RS, Fraietta JA et al Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 2016; 44: 380–390. [DOI] [PubMed] [Google Scholar]
- 85. Matsushita H, Hosoi A, Ueha S et al Cytotoxic T lymphocytes block tumor growth both by lytic activity and IFNγ‐dependent cell‐cycle arrest. Cancer Immunol Res 2015; 3: 26–36. [DOI] [PubMed] [Google Scholar]
- 86. Ke EE, Wu Y. EGFR as a pharmacological target in EGFR‐mutant non‐small‐cell lung cancer: where do we stand now? Trends Pharmacol Sci 2016; 37: 887–903. [DOI] [PubMed] [Google Scholar]
- 87. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7: 301–311. [DOI] [PubMed] [Google Scholar]
- 88. Choi BD, Yu X, Castano AP et al CAR‐T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 2019; 37: 1049–1058. [DOI] [PubMed] [Google Scholar]
- 89. Desnoyers LR, Vasiljeva O, Richardson J et al Tumor‐specific activation of an EGFR‐targeting probody enhances therapeutic index. Sci Transl Med 2013; 5: 207ra144. [DOI] [PubMed] [Google Scholar]
- 90. Gao WW, Xiao RQ, Zhang WJ et al JMJD6 licenses ERα‐dependent enhancer and coding gene activation by modulating the recruitment of the CARM1/MED12 co‐activator complex. Mol Cell 2018; 70: 340–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Paez‐Ribes M, Man S, Xu P, Kerbel RS. Development of patient derived xenograft models of overt spontaneous breast cancer metastasis: a cautionary note. PLoS One 2016; 11: e0158034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1‐9