

Loss of the ClC‐3 chloride/proton exchanger found in intracellular compartments leads to marked neurodegeneration. New genetic work by Weinert et al (2020) now shows that selective impairment of ClC‐3's ion exchange activity is sufficient to elicit this severe phenotype in vivo. ClC‐3 cooperates with the closely related ClC‐4 in protecting endolysosomal chloride balance and neuronal integrity.
Subject Categories: Membrane & Intracellular Transport, Neuroscience
Recent work shows that assembly‐dependent trafficking and chloride exchange by the CLC‐3 and CLC‐4 transporter proteins support neuronal health.
Endosomes and lysosomes are compartments with critical functions in nutrient uptake, hormone signaling, detoxification, and the recycling of metabolites or cellular building blocks. The ion composition of the aqueous phase inside these compartments is critical to the broad spectrum of biochemical reactions that occur in both of them—from uncoupling receptors and their ligands to the activity of many different enzymes. For decades, the concentration of protons, or pH, inside endosomes and lysosomes has received intense attention. While any biochemist would agree that pH is one of the key parameters for optimal enzyme function, it is well known that the correct concentration of other ions can be an equally important factor. In the past decade, it has emerged that the chloride ion is indeed critical for endosomal and lysosomal function (Novarino et al, 2010; Weinert et al, 2010).
The accumulation of protons is an electrogenic (potential‐building) process. Hence, it is more effective when ions of the opposite charge are allowed to accompany the protons inside the vesicular compartment. Simply speaking—if the pH is all that counts (in contrast to the proton electromotive force that is harnessed by mitochondria to make ATP), then send any negatively charged ion in parallel to the protons and you can accumulate them to a higher concentration, i.e., acidify the vesicle more efficiently. This mechanism is called “chloride‐dependent shunt”. For the longest time, Ockham's razor seemed to suggest very plausibly that endosomal or lysosomal chloride was just this, any negatively charged ion compensating for the protons’ positive charge.
It seemed unnecessary to challenge this concept and has, indeed, proven experimentally difficult to do so. The impulse to revisit a seemingly solved problem came from a seminal discovery by Accardi and Miller (2004), their amazing demonstration that prokaryotic CLC homologues act as secondary active transporters and are able to use the energy stored in one ion's gradient to move a second one against its electrochemical potential (secondary active transport). Prior to this discovery, CLC proteins were thought to be chloride channels, passively allowing chloride ions to move in the energetically favored direction.
The unexpected discovery raised many questions regarding the biophysical transport mechanism and the physiological relevance of animal or plant intracellular membrane proteins of the CLC family, such as mammalian ClC‐3, ClC‐4, ClC‐5, ClC‐6, and ClC‐7. Indeed, these proteins were found to function as 2Cl−/H+ exchangers, too. This made the hypothesis that chloride “just” provides negative charge mechanistically more complicated but still tenable since it was observed that losing such transporters did in some cases impair proper acidification of endosomes (Günther et al, 2003) or the osteoclast resorption lacuna, sometimes considered to be an “extracellular lysosome” (Kornak et al, 2001). So, why look any further and wonder about a putative role of chloride beyond being negative? In science, as in real life, the most obvious explanation is not always the correct one or at least not the full truth. To echo George Gershwin, “It ain't necessarily so”.
The normal lysosomal pH of mice lacking lysosomal ClC‐7 or its β‐subunit Ostm1 (Kasper et al, 2005; Lange et al, 2006), which display severe lysosomal storage disease, suggested that CLC exchangers not only serve as chloride‐dependent shunts. Addressing the question if coupled transport activity is per se physiologically relevant, the Jentsch Lab has exploited the discovery of uncoupling CLC mutations (Accardi & Miller, 2004) that convert the exchangers into a passively conducting, chloride‐permeable pore: They embarked on the construction of a series of transgenic mouse models expressing uncoupled intracellular CLC proteins and started a careful and systematic comparison to the corresponding knockout mouse models. This strategy is elegant not only because it segregates the physiological relevance of two biophysically distinct transport modes—passive chloride leak as opposed to proton gradient‐driven chloride accumulation. It also segregates the effects of losing the best‐characterized and presumably central function of a protein from the effect of losing the protein per se, with all its protein–protein interactions, regulation, or unknown functions.
The work of Weinert et al (2020) now reveals just how hard one had to look to tackle the relevance of the exchange mode to the neuroprotective capacity of ClC‐3. They replaced ClC‐3 with the uncoupled variant of the protein ClC‐3unc/unc. Clcn3 −/− knockout mice lose their hippocampus and turn blind due to retinal degeneration (Stobrawa et al, 2001). Surprisingly, and in contrast to the work on ClC‐5 and ClC‐7 where the replacement by the uncoupled transporter phenocopies the knockout (Novarino et al, 2010; Weinert et al, 2010), this was not the case in Clcn3 unc/unc mice, which turned out to be unexpectedly normal (Fig 1).
Figure 1. Assembly‐dependent trafficking and chloride accumulation by the CLC‐3 and CLC‐4 exchanger proteins support neuronal health.
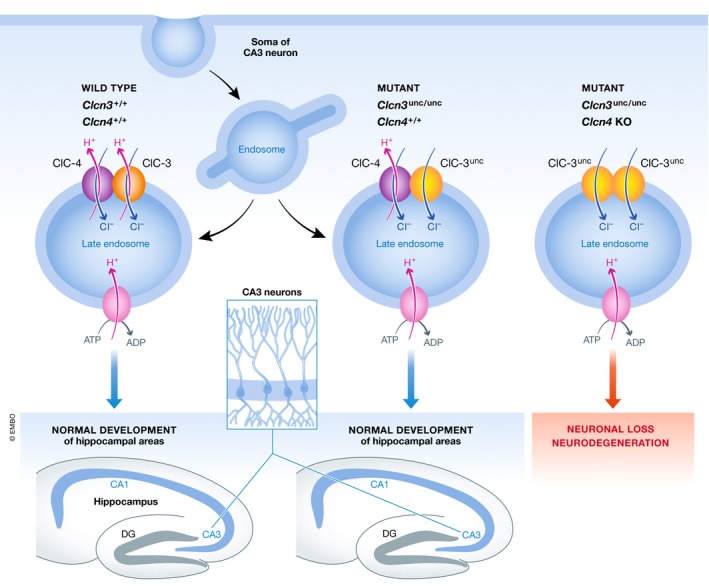
CLC membrane proteins are obligatory dimers with an ion‐conducting pathway in each monomer. The overlapping expression profiles of several CLC proteins raise the possibility of heterodimerization, which has been demonstrated for some combinations but so far not shown to be essential for any CLC transporter's cellular function. Biochemical investigation of the steady‐state levels and glycosylation status of ClC‐4, a close homologue of ClC‐3 also robustly expressed in neurons, enabled Weinert et al (2020) to reveal that ClC‐4 has to co‐assemble with ClC‐3 to leave the early secretory pathway and to provide its ion transport activity to the right cellular compartment in the endolysosomal system. Indeed, mice expressing Clcn3 unc/unc but lacking Clcn4 show severe neurodegeneration (Fig 1). As with the intracellular ClC‐5 and ClC‐7, the exchanger transport mode, and hence the capacity to accumulate chloride inside vesicles, is critical to ClC‐3's physiological function. This is masked by co‐assembly with ClC‐4 and only revealed in a mouse model combining the knock‐in of uncoupled ClC‐3 with a knockout of the Clcn4 gene (which does not give rise to a severe phenotype illustrating that ClC‐3 can fulfill its basic function without ClC‐4).
In conclusion, this work unequivocally shows the central role of CLC‐mediated chloride/proton exchange and hence vesicular chloride accumulation in the neuroprotective function of ClC‐3. It provides a clear example of assembly‐dependent trafficking of a multimeric membrane protein and thereby illustrates that forward protein transport along the secretory pathway is anything but default. Like all intriguing scientific work, the article by Weinert et al (2020) opens new questions: Why is the ClC‐4 protein so strictly attended by ClC‐3? Is there a specific functional or regulatory capacity endowed by ClC‐4 to ClC‐3/ClC‐4 heterodimers? Genetic links to human brain disorders suggest that this may be the case. One can hope that this and related questions around the biochemical function of chloride ions inside endosomal or lysosomal compartments will be pursued by a similarly rich portfolio of cutting‐edge approaches in gene targeting, biochemistry, histology, cell biology, and physiology to elucidate even unexpected facts.
The EMBO Journal (2020) 39: e104812
See also: https://doi.org/10.15252/embj.2019103358 (May 2020)
References
- Accardi A, Miller C (2004) Secondary active transport mediated by a prokaryotic homologue of ClC Cl‐ channels. Nature 427: 803–807 [DOI] [PubMed] [Google Scholar]
- Günther W, Piwon N, Jentsch TJ (2003) The ClC‐5 chloride channel knock‐out mouse – an animal model for Dent's disease. Pflugers Arch 445: 456–462 [DOI] [PubMed] [Google Scholar]
- Kasper D, Planells‐Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poët M, Steinfeld R, Schweizer M et al (2005) Loss of the chloride channel ClC‐7 leads to lysosomal storage disease and neurodegeneration. EMBO J 24: 1079–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ (2001) Loss of the ClC‐7 chloride channel leads to osteopetrosis in mice and man. Cell 104: 205–215 [DOI] [PubMed] [Google Scholar]
- Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC (2006) ClC‐7 requires Ostm1 as a beta‐subunit to support bone resorption and lysosomal function. Nature 440: 220–223 [DOI] [PubMed] [Google Scholar]
- Novarino G, Weinert S, Rickheit G, Jentsch TJ (2010) Endosomal chloride‐proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 328: 1398–1401 [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H, Draguhn A et al (2001) Disruption of ClC‐3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29: 185–196 [DOI] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U, Jentsch TJ (2010) Lysosomal pathology and osteopetrosis upon loss of H+‐driven lysosomal Cl− accumulation. Science 328: 1401–1403 [DOI] [PubMed] [Google Scholar]
- Weinert S, Gimber N, Deuschel D, Stuhlmann T, Puchkov D, Farsi Z, Ludwig CF, Novarino G, López‐Cayuqueo KI, Planells‐Cases R et al (2020) Uncoupling endosomal CLC chloride/proton exchange causes severe neurodegeneration. EMBO J 39: e103358 [DOI] [PMC free article] [PubMed] [Google Scholar]