

Abstract
RNA–protein interactions are the crucial basis for many steps of bacterial gene expression, including post‐transcriptional control by small regulatory RNAs (sRNAs). In stark contrast to recent progress in the analysis of Gram‐negative bacteria, knowledge about RNA–protein complexes in Gram‐positive species remains scarce. Here, we used the Grad‐seq approach to draft a comprehensive landscape of such complexes in Streptococcus pneumoniae, in total determining the sedimentation profiles of ~ 88% of the transcripts and ~ 62% of the proteins of this important human pathogen. Analysis of in‐gradient distributions and subsequent tag‐based protein capture identified interactions of the exoribonuclease Cbf1/YhaM with sRNAs that control bacterial competence for DNA uptake. Unexpectedly, the nucleolytic activity of Cbf1 stabilizes these sRNAs, thereby promoting their function as repressors of competence. Overall, these results provide the first RNA/protein complexome resource of a Gram‐positive species and illustrate how this can be utilized to identify new molecular factors with functions in RNA‐based regulation of virulence‐relevant pathways.
Keywords: Cbf1, competence, Grad‐seq, RNA–protein complex, Streptococcus pneumoniae
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; RNA Biology
Comprehensive assessment of protein‐RNA complexes in Streptococcus pneumoniae via Grad‐seq uncovers an unexpected role for the exoribonuclease Cbf1 in stabilizing sRNAs that control bacterial competence for DNA uptake.
Introduction
Within the past two decades, our view of bacterial gene regulation has dramatically changed. Once considered as organisms with a protein output that is a straight function of transcription initiation, we know now that bacteria amply use noncoding RNAs (ncRNAs) and RNA‐binding proteins (RBPs) in post‐transcriptional control networks that impact almost every aspect of physiology (Storz & Papenfort, 2019). Interactions with cellular proteins crucially underlie many of these RNA functions, running the gamut of universal ribonucleoprotein particles (RNPs) with housekeeping functions, 6S RNA‐mediated modulation of RNA polymerase (RNAP), CRISPR/Cas complexes for genome defense, and an increasing number of small regulatory RNAs (sRNAs) that associate with global RNA‐binding proteins (RBPs) (Holmqvist & Vogel, 2018).
Much of this recent progress has come from studies in Gram‐negative bacteria, especially Escherichia coli and Salmonella enterica (Hör et al, 2018, 2020). These model bacteria express at least three global RBPs—CsrA, Hfq, and ProQ—that form complexes with hundreds of different sRNAs to facilitate extensive post‐transcriptional control of mRNAs (Olejniczak & Storz, 2017; Holmqvist & Vogel, 2018; Kavita et al, 2018; Babitzke et al, 2019). The situation is very different with Gram‐positive bacteria: Despite much evidence that sRNAs also play important roles in these organisms (Brantl & Brückner, 2014; Wagner & Romby, 2015; Quereda & Cossart, 2017; Wassarman, 2018; Desgranges et al, 2019), many aspects of their sRNA biology seem to differ from Gram‐negative species. For example, CsrA circuits work without antagonistic sRNAs, Hfq seems to play a minor role, ProQ is generally absent, and a major sRNA‐related RBP yet awaits to be discovered. Major ribonucleases, and hence RNA metabolism, also differ from Gram‐negative bacteria (Durand & Condon, 2018), and in more general terms, it is fair to say that our knowledge on RNA–protein interactions and molecular complexes is lagging behind in Gram‐positive species.
We have recently developed gradient profiling by sequencing (Grad‐seq) to accelerate the discovery of the major functional RNAs and RBPs in a bacterium of interest (Smirnov et al, 2016). This new type of complexomics approach predicts native cellular complexes by high‐throughput RNA and protein analysis of biochemically fractionated cellular lysates. In its pioneering application to Salmonella (Smirnov et al, 2016), Grad‐seq brought to light ProQ as a previously overlooked major RBP in enteric bacteria (Smirnov et al, 2017b; Holmqvist et al, 2018; Westermann et al, 2019; Melamed et al, 2020). Given the generic nature of the method, we reasoned that Grad‐seq would lend itself to guide the discovery of new RNA‐based mechanisms in Gram‐positive bacteria as well.
In the present work, we have applied Grad‐seq to Streptococcus pneumoniae to provide the first census of potential RNA and protein complexes in a Gram‐positive bacterium. The pneumococcus is a leading human pathogen, causing diverse infectious diseases such as otitis media, sinusitis, sepsis, meningitis, and pneumonia, and is responsible for more than one million deaths annually (O'Brien et al, 2009; Henriques‐Normark & Tuomanen, 2013). Unsurprisingly, pneumococcal transcriptomes, including condition‐specific sRNA expression, have been extensively characterized (Acebo et al, 2012; Mann et al, 2012; Aprianto et al, 2018; Slager et al, 2018, 2019; Warrier et al, 2018; Sinha et al, 2019) and potential binary protein–protein interactions have also been predicted (Wuchty et al, 2017). Although these studies yielded invaluable new insight into the molecular underpinnings of pneumococcal virulence, they did not cover functional RNA–protein interactions, especially for sRNAs with known roles in these processes. Here, the application of Grad‐seq allowed us to identify the sedimentation profiles of thousands of S. pneumoniae transcripts and proteins simultaneously in a single experiment. We establish a use case for this complexome resource by uncovering an unexpected exonucleolytic stabilization of sRNAs in the competence regulon, a pathway essential for DNA uptake and important for pneumococcal virulence (Lin et al, 2016; Muschiol et al, 2019; Salvadori et al, 2019).
Results
Grad‐seq captures major RNA–protein complexes of the pneumococcus
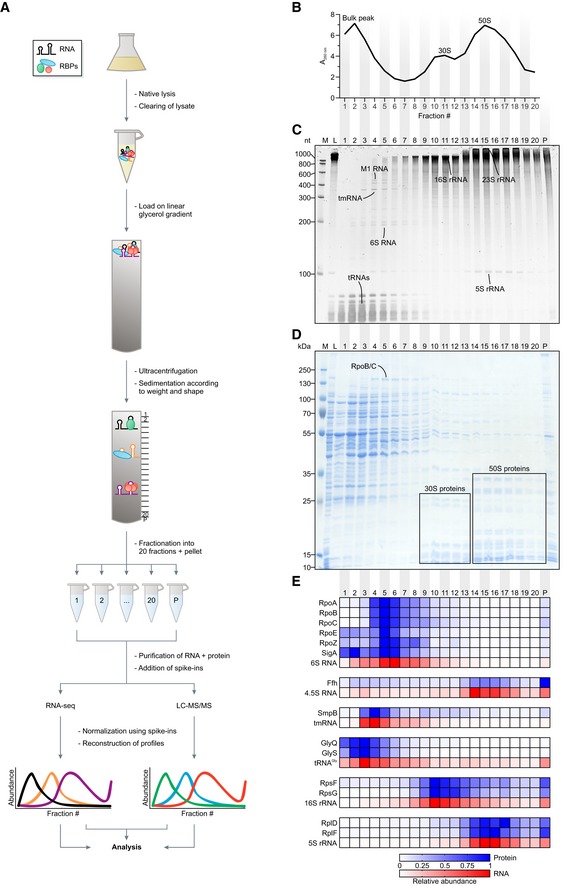
To draft a digital map of soluble cellular complexes formed by pneumococcal RNA and proteins, a lysate of the model strain TIGR4 grown to mid‐exponential phase was fractionated on a linear glycerol gradient. Each of 20 fractions was then analyzed by RNA‐seq and mass spectrometry (Fig 1A). Quality control by A260 nm measurements showed the expected sedimentation profile (Smirnov et al, 2016): one bulk peak in the low‐molecular‐weight (LMW) fractions and two additional peaks for the 30S and 50S ribosomal subunits (Fig 1B). Importantly, these three peaks corresponded well with ribosome‐free tRNAs, 16S rRNA, and 5S/23S rRNAs, respectively, visible in a stained RNA gel (Fig 1C). Additional abundant transcripts known to form stable complexes with proteins, e.g., 6S RNA, M1 RNA of RNase P, and tmRNA, were readily visible (Fig 1C), thus confirming the quality of the gradient. Complementary SDS gel analysis offered a coarse‐grained view of the in‐gradient distribution of some of the corresponding proteins of, e.g., RNAP [target of 6S RNA (Wassarman & Storz, 2000)] and the 30S and 50S ribosomal subunits (Fig 1D).
Figure 1. Grad‐seq reveals the RNA/protein complexome of Streptococcus pneumoniae .
-
AGrad‐seq workflow.
-
BA260 nm profile of the gradient with highlighted low‐molecular‐weight complexes (bulk peak) and ribosomal subunits (30S, 50S). Particles larger than the 50S subunit were pelleted.
-
CEthidium bromide‐stained RNA gel. Bands corresponding to abundant housekeeping RNAs are indicated.
-
DCoomassie‐stained SDS–PAGE. Bands corresponding to abundant housekeeping proteins are indicated.
-
EHeat map of digital in‐gradient distributions of known RNA–protein complexes derived from RNA‐seq and LC‐MS/MS data. The profiles are normalized to the range from 0 to 1. RBP, RNA‐binding protein. LC‐MS/MS, liquid chromatography‐tandem mass spectrometry. M, size marker. L, lysate (input control). P, pellet fraction.
Source data are available online for this figure.
RNA‐seq of the 20 fractions detected 2,240 transcripts, accounting for ~ 88% of the S. pneumoniae transcriptome (Dataset EV1). As seen previously with Gram‐negative Salmonella (Smirnov et al, 2016), mRNAs populated the whole gradient but peaked in the 30S and pellet (containing 70S ribosomes) fractions (Fig EV1A). Notable differences from Salmonella Grad‐seq are a slight shift of mRNAs toward the soluble fractions (Fig EV1A), indicative of partial ribosome dissociation, and a broader average distribution of ncRNAs as compared to the mRNAs (Fig EV1B). For the ncRNAs, this broader distribution may be due to the inclusion of various different classes such as riboswitches and sRNAs in the annotation used for the study here. Importantly, the sRNAs that are transcribed from independent genes (i.e., do not result from processing of longer transcripts) tend to be found in small complexes, as suggested by their accumulation in low‐molecular‐weight fractions (Fig EV1C).
Figure EV1. Average Grad‐seq transcript profiles.
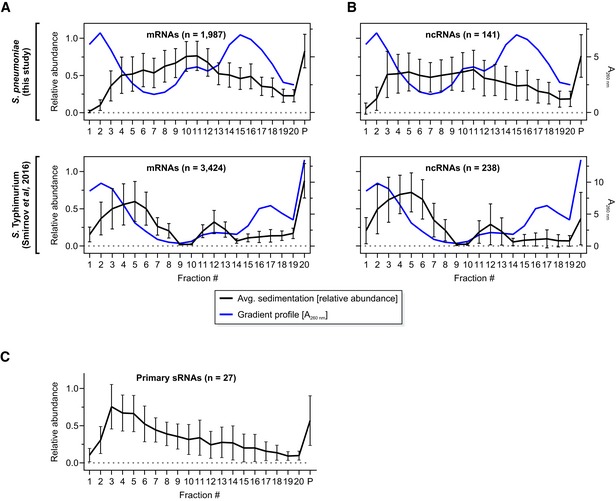
- A, B
-
CAverage profile of high confidence, independently transcribed sRNAs.
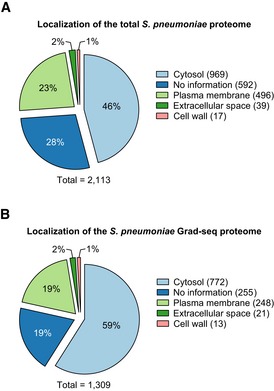
Complementary mass spectrometry analysis of the gradient detected 1,301 proteins (Dataset EV2), most of which were cytosolic and in sum represented ~ 62% of the annotated proteome (Fig EV2A and B). Of known stable RNPs, we observed excellent correlations in the digital RNA and protein data for 30S and 50S proteins with their respective rRNAs, as well as for the 6S RNA‐RNAP complex (Wassarman, 2018), the signal recognition particle (SRP; complex of Ffh protein and 4.5S RNA) (Akopian et al, 2013), and the tmRNA‐based ribosome rescue system (Keiler, 2015; (Fig 1E). Interestingly, different from Salmonella, where the SRP peaks in fraction 3 (Smirnov et al, 2016), the pneumococcal SRP sediments in the 50S region, suggesting that its quaternary configuration is more stable in lysed pneumococcal cells. Lastly, as observed previously (Smirnov et al, 2016), reads of major tRNAs such as those decoding glycine coincided with peptides of their corresponding tRNA synthetases (Fig 1E).
Figure EV2. Localization of detected proteins.
-
A, BPredicted localization of the total (A) and the Grad‐seq (B) proteome of Streptococcus pneumoniae. Note that the numbers for (B) do not fit with Dataset EV2 because some proteins have more than one localization assigned. Predicted localizations were downloaded from BioCyc (Karp et al, 2019).
Grad‐seq assists in the characterization of protein functions
Our focus on RNA–protein complexes notwithstanding, the digital in‐gradient distributions obtained by Grad‐seq proteomics should also aid the prediction of other macromolecular assemblies of proteins. For example, the nearly congruent profiles of the RpoA, RpoB, and RpoC proteins readily predict that they form a large complex, i.e., RNAP (Fig 1E).
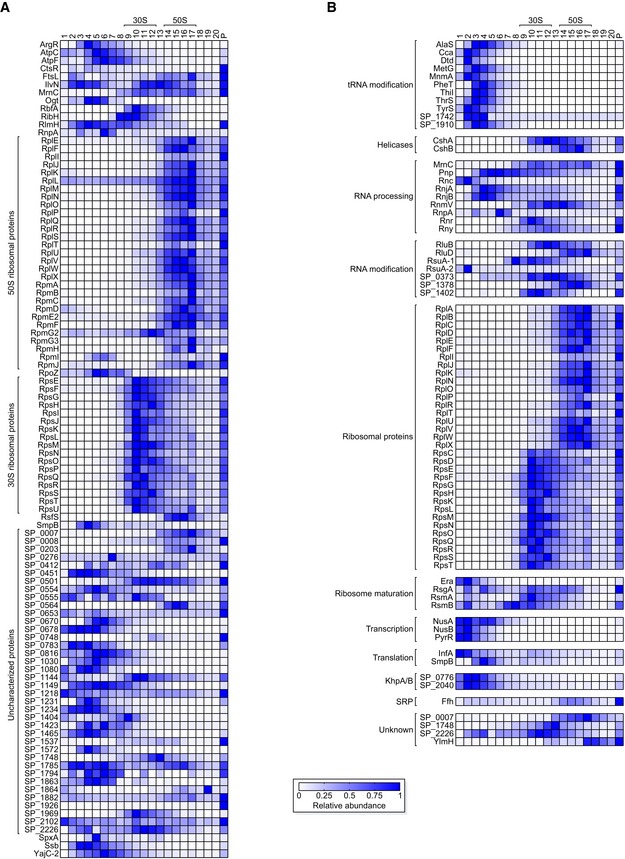
To test this assumption, we focused our analysis on proteins < 20 kDa. Considering a moderately elongated shape, these proteins should sediment at < 3S (fractions ~ 1–3) unless in a complex (Erickson, 2009). Of 284 such proteins in total in our data, 102 showed top peaks in fraction 4 or higher (Dataset EV3), indicating complex formation (Fig 2A). This latter set contained many expected proteins, such as 46 known ribosomal proteins, RpoZ, SmpB, and RnpA. Further, the peaks of RbfA (ribosome‐binding factor A) and RsfS (ribosomal silencing factor) coincided with the 30S and 50S peaks, respectively, which agrees with their activities on disassembled subunits only (Shajani et al, 2011; Häuser et al, 2012).
Figure 2. Grad‐seq predicts complexes.
-
AProfiles of 102 proteins < 20 kDa that show fast sedimentation, indicating involvement in complexes.
-
BHeat map of migration profiles of 77 proteins with the UniProt keyword “RNA‐binding”. For clarity, they were assigned to functionally related classes of RBPs. Note that SP_2040 was added due to its interaction with SP_0776 in the KhpA/B heterodimer (Zheng et al, 2017; Winther et al, 2019), even though it does not have the keyword on UniProt.
Grad‐seq data also add confidence to domain‐based functional assignment of uncharacterized proteins. For example, SP_1969 is annotated as a member of the InterPro family IPR004398 (Mitchell et al, 2019) that also contains the 16S rRNA methyltransferase RsmD of E. coli (Lesnyak et al, 2007; Sergeeva et al, 2012; Appendix Fig S1). Grad‐seq showed SP_1969 peaking with the S. pneumoniae 30S proteins, suggesting that it has an RsmD‐like function in 30S subunit maturation. Another example is SP_0007, which, like its putative E. coli homolog Hsp15, contains an RNA‐binding S4 domain. Interestingly, both SP_0007 and its Gram‐positive homologs lack the disordered ~ 40 aa C‐terminus of E. coli Hsp15 (Appendix Fig S2) which increases the affinity of the latter to translationally inactive 50S subunits (Jiang et al, 2009). Nonetheless, S. pneumoniae SP_0007 peaked with the 50S proteins, supporting the prediction of a conserved role in 50S recycling. On the contrary, S4 domain protein SP_2226, whose Bacillus subtilis homolog YaaA was proposed to be involved in 50S assembly (Suzuki et al, 2014), co‐migrated primarily with the 30S proteins, therefore suggesting that it is involved in a different part of ribosomal biology than originally predicted. C‐termini of different lengths in Streptococcus, Bacillus, and Staphylococcus species (Appendix Fig S3) may alter the interactomes of these homologous proteins. Taken together, these examples illustrate how digital Grad‐seq data can, on the protein part, support functional predictions of uncharacterized pneumococcal proteins.
Next, we interrogated the Grad‐seq proteomics data for co‐migrating proteins from 388 predicted operons of S. pneumoniae TIGR4 (Warrier et al, 2018; Dataset EV4), which often encode for proteins involved in the same complex (Wells et al, 2016). For the first time in the pneumococcus, this analysis proves higher‐order macromolecular assemblies such as glutamyl‐tRNAGln amidotransferase (GatBAC), ribonucleoside‐diphosphate reductase (NrdEF), galactose‐6‐phosphate isomerase (LacBA), and an intact 9‐subunit cytosolic ATP synthase F 1 complex (AtpCDGAH) (Fig EV3). Surprisingly, although these proteins had been gradient‐fractionated from a cleared lysate, we observed co‐migrating proteins with predicted membrane localization. For example, the membrane‐associated, virulence‐related protein UppP (a.k.a. BacA) (Chalker et al, 2000) co‐migrated with SP_0454, a protein of unknown function with several trans‐membrane domains that are encoded in the same operon. While we cannot assess whether these putative membrane proteins remain properly folded, this illustrates the potential for predicting in vivo complexes beyond those formed by cytosolic proteins.
Figure EV3. Grad‐seq profiles of proteins deriving from operons.
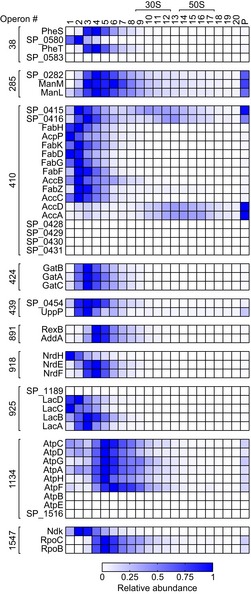
Exemplary operons [numbers according to Warrier et al (2018)] that provide evidence for predicted complexes.
UniProt lists 78 S. pneumoniae TIGR4 proteins with the keyword “RNA‐binding”, 77 of which were detected in our gradient (Fig 2B). While tRNA‐modifying enzymes expectedly migrated in early fractions, others such as RNases showed no common profile. The RNA‐binding heterodimer KhpA/B, known as SP_0776 and SP_2040 in the strain used here, was recently reported to bind a diverse set of cellular transcripts, including tRNAs and sRNAs (Zheng et al, 2017). Here, we detected a stable KhpA/B complex with a peak in fractions 2–3, which coincided with tRNAs and primary sRNAs (i.e., those transcribed from independent genes) (Fig EV1C). Taken together, these profiles provide the first global information about which RBPs in Gram‐positive bacteria may exist within macromolecular assemblies.
Identification of biochemically distinct classes of ncRNAs
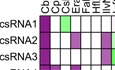
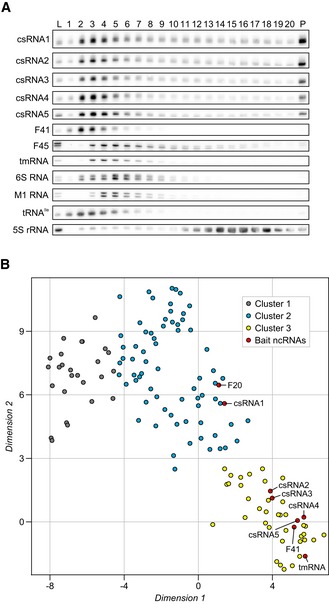
ncRNAs are particularly promising with respect to RBP discovery because the well‐studied ones usually require helper proteins for cellular function or stability (Gorski et al, 2017; Holmqvist & Vogel, 2018). Importantly, we observed migration patterns of many pneumococcal ncRNAs that were indicative of RNPs different from the major RNPs described above. Northern blot analysis of several such ncRNAs (Fig 3A) showed strong correlation of their in‐gradient migration with the digital RNA‐seq data (Fig EV4A). For example, the five competence‐regulating cia‐dependent small RNAs (csRNAs) (Halfmann et al, 2007) all showed profiles very different from the ubiquitous tmRNA, 6S RNA, and M1 RNA (Fig 3A).
Figure 3. Grad‐seq identifies clusters of biochemically distinct ncRNAs.
-
ANorthern blots of ncRNAs. The different classes of ncRNAs show unique profiles on the gradient. Of note, the functionally related csRNAs co‐migrate. L, lysate (input control). P, pellet fraction.
-
Bt‐SNE analysis of all 141 ncRNAs detected by Grad‐seq. Cluster 3 was of particular interest since it contained mostly ncRNAs present in low‐molecular‐weight fractions (Fig EV4F). ncRNAs selected for downstream pull‐down experiments are highlighted in red.
Source data are available online for this figure.
Figure EV4. Grad‐seq profiles of ncRNAs.
-
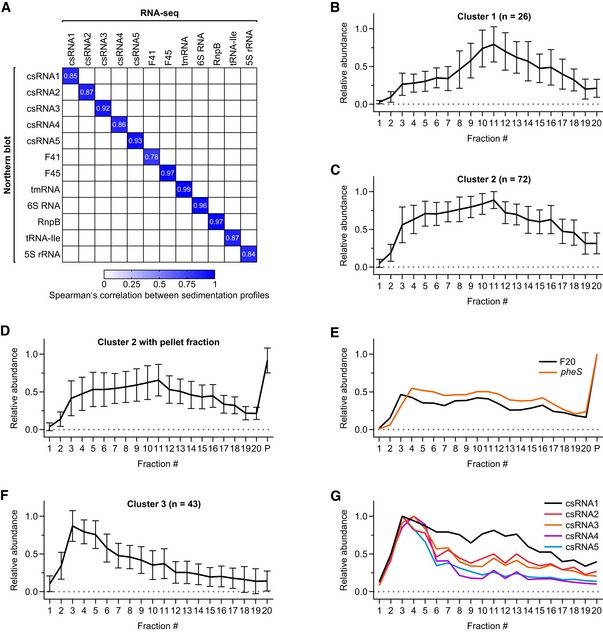
AHeat map of Spearman's correlation between northern blot profiles (Fig 3A) and RNA‐seq profiles for selected ncRNAs. Two‐tailed P‐values for all correlations are 2.77 × 10−17 < P < 2.60 × 10−5.
-
B–FAverage ncRNA profiles for cluster 1 (B), cluster 2 (C), cluster 2 including the pellet fraction (D), and cluster 3 (F) show distinct sedimentation, separating them from each other (Fig 3B). Cluster 2 includes the riboswitch RNA F20, which has similar sedimentation to its downstream CDS pheS (E).
-
GRNA‐seq profiles of the five csRNAs. Note that csRNA1 has a clear peak in fraction 3, albeit not as pronounced as the other csRNAs.
A general assumption in Grad‐seq is that transcripts with similar in‐gradient distributions share interacting RBPs (Smirnov et al, 2017a). To define biochemically similar ncRNAs as potential baits for subsequent RBP recovery, we performed a t‐SNE analysis on the RNA‐seq data of gradient fractions 1–20 (Fig 3B, Appendix Fig S4). This yielded three distinct clusters of which cluster 1 contained 26 ncRNAs that generally showed low abundance in the LMW fractions and peaks around the ribosomal subunits (Fig EV4B). As expected, this cluster contained the 4.5S RNA component of the ribosome‐associated SRP, but also several ncRNAs with previously demonstrated roles in virulence, e.g., F48, F62, F65, and R8 (Mann et al, 2012). Of note, many of these latter ncRNAs originate from repetitive elements and show poor sequence conservation in closely related species, hampering the evaluation of potential overlooked ORFs that would explain their sedimentation in the ribosomal fraction. Cluster 2 contained 72 ncRNAs that generally exhibited broad distribution in the gradient (Fig EV4C), and most (~ 69%) of these were also much more abundant in the pellet than in the gradient fractions (Fig EV4D). A main peak in the pellet indicates 70S ribosome association, which in the exemplary case of the F20 ncRNA (a T‐box riboswitch) may be driven by the parental pheS mRNA (Fig EV4E).
Cluster 3 containing 43 ncRNAs with a tendency to peak before the 30S subunit (Fig EV4F) was the most interesting. This cluster contained not only ncRNAs of well‐characterized stable RBPs but also 21 primary sRNAs (Fig EV1C), including four of the five csRNAs. The csRNAs are post‐transcriptional repressors of comC mRNA encoding the precursor protein of the pneumococcal competence‐stimulating peptide (CSP). Without synthesis of CSP, competence and therefore DNA uptake from the environment cannot be induced (Schnorpfeil et al, 2013; Laux et al, 2015). Molecular details of csRNA‐mediated control and the involvement of RBPs are yet to be determined. To us, their co‐migration in the gradient indicated that they shared associated protein(s).
Cbf1 is an sRNA‐interacting protein
To identify csRNA‐interacting proteins, we sought to co‐purify them from lysates using in vitro‐transcribed MS2‐tagged csRNAs as baits, i.e., using the same strategy that recently enabled us to identify a new global RBP (ProQ) in enteric bacteria (Smirnov et al, 2016). Several additional ncRNAs were included for comparison or controls: the functionally related csRNA1, which ended up in cluster 2, probably because its peak in fractions 3–4 was weaker than for the other csRNAs (Fig EV4G); riboswitch‐derived F20 from cluster 2; F41 sRNA from cluster 3, which also clearly peaks in the LMW fractions; and tmRNA, which is known to bind the SmpB protein (Fig 3B). For unclear reasons, attempts to use the MS2 aptamer, which successfully captured RBPs and RNA–RNA interactions from Salmonella, E. coli, and Staphylococcus aureus lysates (Said et al, 2009; Corcoran et al, 2012; Lalaouna et al, 2015, 2019; Smirnov et al, 2016; Tomasini et al, 2017), failed. While we did recover the MS2‐tagged RNAs after incubation with lysates, no protein co‐purified at all.
Therefore, we adopted a protocol developed for eukaryotic RBPs in which the bait RNA is in vitro transcribed with a 5′ located, 14 nt capture tag (Treiber et al, 2017). Using this alternative procedure, we successfully enriched proteins for each of the tagged pneumococcal ncRNAs (Fig 4A). LC‐MS/MS‐based identification of these proteins (Appendix Fig S5A–H) followed by comparison of their in‐gradient distributions with those of the respective ncRNAs used as baits (Fig 4B) indicated strong enrichment of several degradosome components. Of these, RNases J1 and J2, as well as PNPase to some extent, correlated well with the respective ncRNA baits. Another recurring protein was the essential RNase Z, as pulled down by F41 and all csRNAs except csRNA3. However, the highest correlation of all enriched proteins was observed for Cbf1: This protein stably associated with all csRNAs and F41 (Fig 4C) but did not elute with tmRNA or riboswitch RNA F20 (Appendix Fig S5G and H).
Figure 4. Identification of Cbf1 as a ncRNA‐binding protein.
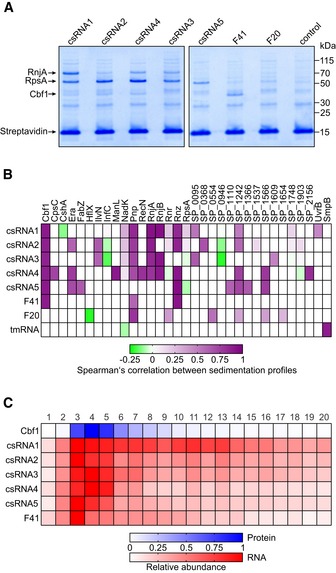
-
ARepresentative gel images of pull‐downs performed with ncRNAs as baits (compare to Fig 3B). Cbf1 was identified as a specific binder of all csRNAs and F41, but not of riboswitch RNA F20.
-
BHeat map showing Spearman's correlation coefficients between the sedimentation profiles of bait ncRNAs and proteins enriched in the pull‐downs. Ribosomal proteins except for S1 (RpsA) and proteins only co‐purified with tmRNA (except for SmpB) are omitted for clarity. Cbf1 is a recurring binding partner of the bait ncRNAs that also shows highly correlating sedimentation profiles in the gradient.
-
CHeat map showing gradient profiles of Cbf1, csRNAs, and F41. Cbf1 co‐sediments with these sRNAs.
Source data are available online for this figure.
To validate the proposed Cbf1–RNA interaction, we also performed the reverse experiment: UV CLIP‐seq using a cbf1‐3xFLAG strain. UV crosslinking resulted in a ~ 5–10‐fold overall enrichment of RNA compared to the non‐crosslinked control (Fig 5A). Deep sequencing of the bound RNA fragments yielded 528 peaks that passed our stringent cutoffs, i.e., a log2 fold change > 1 and an adjusted P‐value < 0.01 (Fig 5B and Dataset EV5). 354 (~ 67%) of these peaks derived from CDSs, whereas 94 (~ 18%) peaks could be attributed to intergenic sequences, which originated from unannotated antisense transcripts, intergenic transcripts, and UTRs. 38 (~ 7%) peaks mapped to annotated ncRNAs, 30 of which were unique (Fig 5C). Importantly, F41 was the most enriched ncRNA in the dataset, matching the strong enrichment of Cbf1 in the F41 RNA pull‐down (Fig 4A). As expected, we further found csRNA1, csRNA3, and csRNA5 to be among the most enriched ncRNAs (Fig 5C). csRNA2 and csRNA4 were not enriched, which could be due to an unfavorable constellation of crosslinking‐prone nucleotides in these transcripts. Other detected, mildly enriched ncRNAs included 6S RNA, tmRNA, and M1 RNA, all of which are abundant RNAs that tend to crosslink to RBPs, as already observed previously (Holmqvist et al, 2018). The identified peaks mostly mapped to the 3′ ends of the sRNAs, whereas there was little‐to‐no coverage toward 5′ ends (Fig 5D). The observed binding of Cbf1 at the 3′ ends of its targets supports its predicted role as a 3′→5′ exonuclease (see below). Taken together, Cbf1 was identified as a new ncRNA‐associated protein in the pneumococcus and was therefore selected for deeper investigation.
Figure 5. CLIP‐seq validates ncRNA binding of Cbf1 in vivo .
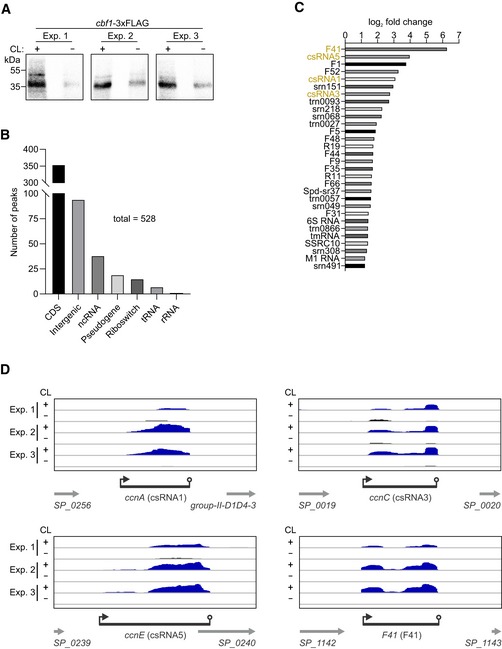
-
AAutoradiographs of radioactively labeled RNA fragments covalently bound to Cbf1 after in vivo UV crosslinking, immunoprecipitation, gel electrophoresis, and membrane transfer. Three independent experiments are shown that were subjected to sequencing. CL, UV crosslinking. Exp., experiment.
-
BOrigins of the peaks found in the Cbf1 CLIP‐seq dataset.
-
CEnrichment of the 30 ncRNAs found in the CLIP‐seq dataset. F41, csRNA1, csRNA3, and csRNA5 (highlighted in orange) are strongly enriched and were previously used to pull‐down Cbf1 (Fig 4A and B). If more than one peak was identified for a single ncRNA in the dataset, only the highest fold change is shown.
-
DCLIP‐seq coverage plots of csRNA1 (ccnA), csRNA3 (ccnC), csRNA5 (ccnE), and F41 (F41) showing crosslinking‐dependent enrichment of peaks at the 3′ ends of the transcripts.
Source data are available online for this figure.
Cbf1 processes and stabilizes csRNAs
Cbf1 (cmp‐binding factor 1) was first described as a host factor for replication of plasmid pT181 in S. aureus (Zhang et al, 1997). Subsequently, both S. aureus Cbf1 and its B. subtilis homolog YhaM were shown to be manganese‐dependent 3′→5′ exonucleases that are present in a wide range of Gram positives (Oussenko et al, 2002, 2005; Fang et al, 2009; Redko & Condon, 2010). Most recently, an RNA‐seq study in Streptococcus pyogenes concluded that YhaM acted to trim transcripts by a few nucleotides until it reaches a stem‐loop structure such as a Rho‐independent terminator (Lécrivain et al, 2018). However, specific physiological functions of Cbf1/YhaM proteins remain unknown.
We validated the predicted 3′→5′ RNase activity by incubating recombinant S. pneumoniae Cbf1 protein with in vitro‐transcribed ncRNAs (Fig 6A). Matching the in vivo observations in S. pyogenes (Lécrivain et al, 2018), Cbf1 trimmed the RNA substrates by several nucleotides, as judged by their faster migration in a gel. No shortening was observed with 5S rRNA (Fig 6A), which is known to be processed by RNase M5 (Bechhofer & Deutscher, 2019).
Figure 6. Cbf1 acts as an sRNA‐stabilizing 3′→5′ exonuclease.
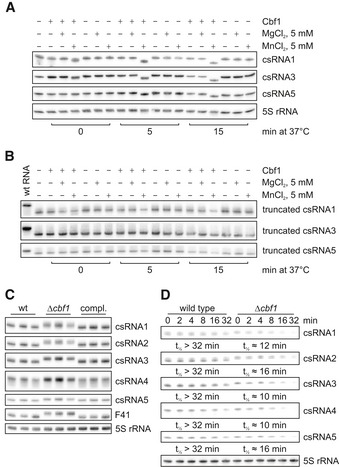
-
AIn vitro RNase assay. In vitro‐transcribed RNAs were incubated in the presence or absence of Cbf1, MgCl2, and MnCl2. Reactions were stopped at the indicated time points and the RNA run on denaturing 10% PAGE followed by northern blotting. All csRNAs, but not 5S rRNA, are cleaved in the presence of Cbf1 and MnCl2.
-
BIn vitro RNase assay with truncated RNAs. The setup of the experiment was the same as in A, the difference being that the in vitro‐transcribed RNAs were missing the single‐stranded uridine overhang 3′ of the Rho‐independent terminator. Wild‐type (wt) RNA used in A was loaded as reference. No cleavage can be observed in any of the conditions.
-
CNorthern blots of denaturing 10% PAGE of total RNA extracted from wild type (wt), Δcbf1 and the complementation (compl.) strain. Upon deletion of cbf1, an upshift of the tested ncRNAs, but not of 5S rRNA is visible.
-
DIn vivo RNA stability assays. After blocking transcription with rifampicin, RNA was collected at the indicated time points and visualized using northern blotting of 6% denaturing PAGE. Interestingly, knockout of cbf1 increases stability of the tested RNAs. 5S rRNA was used as loading control. RNA half‐lives were calculated based on at least two independent experiments (Fig EV5B–F).
Source data are available online for this figure.
Cbf1‐mediated trimming required the presence of Mn2+ ions, thus recapitulating the manganese dependence of B. subtilis YhaM (Oussenko et al, 2002; Fang et al, 2009). At the concentration used, the trimming occurred fast, as evident from a slight shift immediately after Cbf1 was added (time point 0 min). After 5 and 15 min of incubation, the RNAs migrated as single, shorter bands, suggesting complete digestion. Importantly, Cbf1 did not process 3′‐truncated RNAs lacking the uridine stretch of their Rho‐independent terminators, confirming these single‐stranded 3′ overhangs as its targets (Fig 6B). We note that truncated csRNA1 showed reduced RNA levels after incubation with Cbf1. Probing of the 5′ end of this RNA revealed a ~ 45 nt RNA species specific for manganese‐dependent cleavage by Cbf1, which is not visible when probing for the 3′ end (Appendix Fig S6A and B). Subsequent 3′ RACE determined the 3′ nucleotide of this short species to be at the C at position 45 of the truncated csRNA1, which lies directly upstream of a uridine stretch (Appendix Fig S6C–E). The predicted secondary structure of this 45 nt species contains a weak stem‐loop upstream of its 3′ end, with a short single‐stranded overhang (Appendix Fig S6F). We speculate that removal of the uridine stretch from the full‐length RNA probably leaves a less protective 3′ end structure, which then allows Cbf1 to digest more of the RNA.
Next, we studied Cbf1‐dependent processing in vivo, probing total RNA from wild‐type and Δcbf1 strains on northern blots (Fig 6C). As expected, both the csRNAs and F41 showed slower migration in the knockout mutant, with their lengths being restored by chromosomal complementation of cbf1. Note that, in rich medium, the cbf1 deletion does not affect bacterial growth (Fig EV5A). To address the consequence of trimming by Cbf1, we monitored the decay of target transcripts after shutting off transcription with rifampicin (Fig 6D). Intriguingly, the half‐lives of all csRNAs were diminished from > 32 min in the wild‐type strain to approximately 10–16 min in the cbf1 knockout, suggesting a protective function of Cbf1 (Fig EV5B–F). Therefore, we interpret the Cbf1–RNA complexes detected by Grad‐seq as stable intermediates that persist after 3′ trimming and stabilize the associated sRNAs by protecting them from further degradation.
Figure EV5. Growth and csRNA stability in the absence of cbf1 .
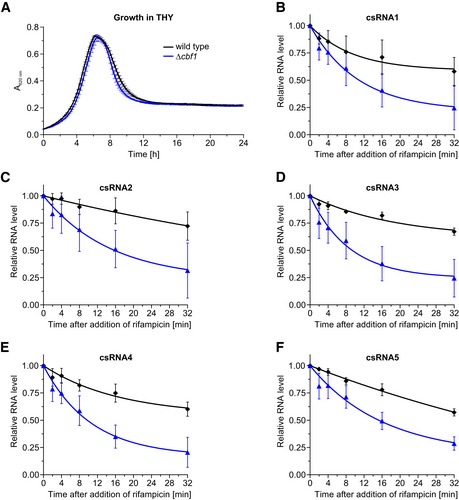
-
AGrowth curves in rich medium. Deletion of cbf1 has no influence on growth when compared to the wild type. The average cell density is based on three independent biological replicates.
-
B–FDetermination of the stability of csRNAs based on rifampicin assays shown in Fig 6D. In the absence of cbf1, csRNAs become less stable when compared to the wild type. The relative RNA levels were calculated based on at least two independent experiments.
Cbf1 is a negative regulator of competence
The observed stabilization of csRNAs links Cbf1 to the regulation of competence. Intriguingly, others have observed in a global transposon screen that disruption of cbf1 reduces fitness in transformation conditions, compromises the colonization of the murine lung, and terminates the colonization of the murine nasopharynx (van Opijnen & Camilli, 2012). Moreover, a recent gene expression catalogue for S. pneumoniae strain D39 grown in many different infection‐relevant conditions predicts a specific upregulation of cbf1 when competence is stimulated by CSP (Aprianto et al, 2018). To test this prediction, we measured cbf1 expression after stimulating S. pneumoniae strain TIGR4 with CSP. We observed higher levels of both the Cbf1 protein (Fig 7A and Appendix Fig S7A; dependence on CSP stimulation shown in Appendix Fig S7B) and the cbf1 mRNA (Appendix Fig S7C) under this condition, thus validating cbf1 as a member of the competence regulon.
Figure 7. Cbf1 negatively regulates competence.
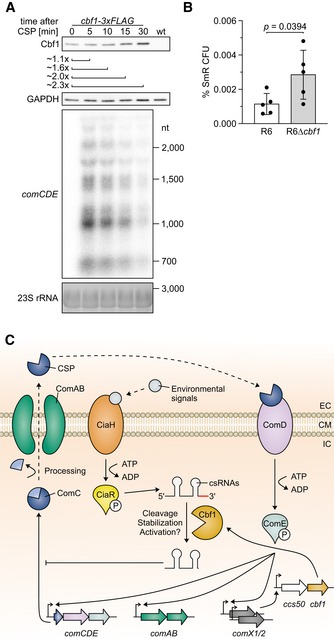
-
AWestern blot of Cbf1‐3xFLAG levels after CSP stimulation. After 15 min, ˜ 2‐fold higher Cbf1‐3xFLAG levels could be detected, confirming cbf1 as a late‐competence gene as predicted by others (Peterson et al, 2004; Slager et al, 2019). CSP‐specific fold changes in Cbf1‐3xFLAG levels were calculated based on three independent experiments (Appendix Fig S7A). As control for the CSP induction, total RNA was extracted, run on an agarose gel, blotted, and probed for comCDE. A GAPDH‐specific antiserum and ethidium bromide staining were used as loading controls for the Western and northern blots, respectively.
-
BPercentage of transformants using R6 and R6Δcbf1 in a spontaneous transformation assay. Bars represent the mean ± standard deviation percentage SmR CFU from 5 independent experiments. P‐value was calculated using an unpaired, two‐tailed t‐test.
-
CModel of Cbf1‐dependent regulation of competence. The pneumococcal competence regulon is a quorum sensing system induced by activation of ComDE via CSP. Phosphorylated ComE induces transcription of the early competence gene loci comAB, comCDE, comX1, and comX2, leading to a positive feedback loop by production of ComC, which is processed into CSP and exported by ComAB. The paralogous alternative sigma factors ComX1 and ComX2 induce transcription of late‐competence gene loci, one of which is ccs50/cbf1 (Slager et al, 2019). Cbf1 is able to cleave, stabilize, and possibly activate csRNAs, which are induced by CiaRH, which in turn is activated by a variety of factors (Gómez‐Mejia et al, 2018). csRNAs are able to post‐transcriptionally downregulate comC. EC, extracellular. CM, cell membrane. IC, intracellular. CSP, competence‐stimulating peptide.
Source data are available online for this figure.
The established function of the csRNAs is that they repress the comC mRNA and thereby the synthesis of CSP, thus counteracting competence activation. If the Cbf1‐mediated csRNA stabilization were of physiological relevance, the absence of this protein should render S. pneumoniae more competent. We were unable to obtain transformants in the absence of exogenously added CSP with S. pneumoniae strain TIGR4, in contrast to strain R6, which is known to spontaneously develop competence when grown at pH 7.9–8.0 (Moscoso & Claverys, 2004). Hence, we constructed a knockout mutant strain in R6, R6Δcbf1, and tested the predicted role of cbf1 using a spontaneous competence assay. We indeed observed ~ 2.5‐fold higher transformation in the Δcbf1 strain as compared to the wild‐type strain R6 (Fig 7B) with all replicates showing the same positive trend (Appendix Fig S7D). This suggests a model, whereby Cbf1‐mediated csRNA stabilization contributes to a negative feedback loop that controls competence after CSP stimulation (Fig 7C).
Discussion
Our Grad‐seq analysis of S. pneumoniae provides the first global census of major stable complexes formed by RNAs and proteins in a Gram‐positive bacterium. The results not only provide crucial knowledge for this important human pathogen, but should also be a valuable resource for other Gram‐positive species including the model bacteria B. subtilis, Listeria monocytogenes, S. aureus, and S. pyogenes. Establishing a use case for the Grad‐seq data, we identify a novel physiological function of the conserved exonuclease Cbf1 in trimming and thus stabilizing the csRNAs that are central regulators of competence. As discussed below, the data now permit conclusions as to what may be conserved major RNA–protein complexes and global RBPs outside the well‐characterized enterobacterial clade.
Although the pneumococcus expresses a great number of sRNAs with evidence for important physiological roles (Mann et al, 2012; Laux et al, 2015; Wilton et al, 2015), homologs of CsrA, Hfq, or ProQ are not present in this organism (Tettelin et al, 2001). A preliminary prediction from our cluster analysis and subsequent protein co‐purification experiments is that S. pneumoniae perhaps does not possess a general RBP analogous to Hfq or ProQ, the two RBPs that together associate with > 80% of the sRNAs in E. coli and Salmonella (Holmqvist & Vogel, 2018). Therefore, the csRNAs may be the first example of redundant sRNAs that regulate complex behavior without an RNA chaperone, i.e., different from the well‐studied quorum sensing‐related Qrr sRNAs in Vibrio spp. that crucially depend on Hfq (Feng et al, 2015). More generally, the S. pneumoniae sRNAs may rely upon more specialized RBPs such as the KhpA/B complex (Zheng et al, 2017) instead of a common RNA chaperone. Grad‐seq readily captures the KhpA/B complex, detecting nearly congruent peaks of KhpA and KhpB in gradient fractions 2–3 (Fig 2B). These narrow peaks in the LMW fractions differ from the profiles of Hfq and ProQ in Salmonella (Smirnov et al, 2016), suggesting that the KhpA/B complex stably associates with fewer cellular transcripts than those global RBPs.
Of more widely conserved RNA–protein constellations, 6S RNA and RNAP show overlapping peaks in the gradient (Fig 1E), supporting previous reports for such a complex in B. subtilis (Trotochaud & Wassarman, 2005; Cavanagh et al, 2012; Burenina et al, 2014). However, in light of the available copy numbers of the involved RNA and proteins in S. pneumoniae, their Grad‐seq‐based capture suggests that the 6S RNA‐RNAP complex may be the major cellular form of RNAP when it is not transcribing DNA. We propose that the 6S RNA–RNAP complex, two decades after its discovery in E. coli (Wassarman & Storz, 2000), should be considered on par with RNase P, SRP, tmRNA‐SmpB, and the ribosome when it comes to ubiquitous RNPs in bacteria.
We have discovered stable in vivo complexes of the 3′→5′ exonuclease Cbf1 (Figs 4 and 5) and shown that, contrary to expectation, the associated csRNAs are stabilized by Cbf1 (Figs 6D and EV5B–F). This stabilization, however, makes sense as one considers the documented physiological function of the csRNAs that—after their upregulation by CiaRH—act to post‐transcriptionally repress ComC synthesis (Laux et al, 2015; Gómez‐Mejia et al, 2018). We propose a model in which Cbf1 binds and trims the csRNAs by a few nucleotides and, through its continued association, shields them from other decay enzymes. If so, loss of Cbf1 function should decrease the post‐transcriptional inhibition of the competence regulon by the csRNAs. In support of this model, we find that a Δcbf1 strain is more competent (Fig 7B). Importantly, cbf1 was proposed to constitute a ComX‐activated late‐competence gene (Peterson et al, 2004; Slager et al, 2019), which we validated here on both the RNA and protein levels (Fig 7A, Appendix Fig S7A–C). Alternatively, Cbf1‐dependent trimming could increase the activity of csRNAs, similar to microRNA, piRNA, and siRNA trimming in eukaryotes (Han et al, 2011; Liu et al, 2011; Marasovic et al, 2013; Tang et al, 2016). Overall, this finding is relevant for in vivo survival: Disruption of the cbf1 gene renders the pneumococcus unable to infect the murine nasopharynx and strongly attenuates lung colonization (van Opijnen & Camilli, 2012).
Arguably, our model does not exclude possible csRNA‐independent functions of Cbf1 in competence regulation, which remain to be tested by combined inactivation of cbf1 and multiple csRNAs. These regulations may indeed be more complex: Others have reported that competence is reduced (20% down) when cbf1 is deleted in a different S. pneumoniae strain and exogenous CSP is added (Peterson et al, 2004). Further, in the related species S. pyogenes, Cbf1 (YhaM) acts on a large number of transcripts (Lécrivain et al, 2018), which we also observed here (Fig 5B). These observations notwithstanding, the evidence for a direct function of Cbf1 on the csRNAs is compelling. We have reconstituted in vitro the protective target trimming by Cbf1 and confirmed that, as in the original observations with B. subtilis YhaM (Oussenko et al, 2002; Fang et al, 2009), this reaction is Mn2+‐dependent (Fig 6A). Intriguingly, Cbf1 is the first 3′→5′ exonuclease in Gram‐positive bacteria with a protective effect on sRNAs, and the second when PNPase‐mediated stabilization of some Hfq‐dependent sRNAs in E. coli is counted (Cameron et al, 2018, 2019). This protection is also reminiscent of certain eukaryotic ncRNAs, which can be trimmed at their 3′ ends by PARN and TOE1, leading to enhanced stability (Berndt et al, 2012; Tseng et al, 2015; Son et al, 2018).
While our previous Grad‐seq analysis in Salmonella focused on complex formation of cellular RNA species, the depth of the present proteomics analysis for the S. pneumoniae gradient bears great potential for prediction and validation of protein–protein interactions as well. Importantly, Grad‐seq drafts a global landscape of the major protein complexes in vivo in a single experiment, without the massive epitope‐tagging of individual proteins in previous binary protein–protein interactome studies (Hu et al, 2009; Rajagopala et al, 2014). As such, our Grad‐seq proteomics data will help to add confidence to the available pneumococcal protein–protein interactions obtained in a yeast two‐hybrid screen (Wuchty et al, 2017). Illustrating potential, we find that the putative 16S rRNA methyltransferase homolog SP_1969, which might be involved in ribosome maturation and bacterial fitness (Lesnyak et al, 2007; Burakovsky et al, 2012; Sergeeva et al, 2012), co‐migrates with the 30S ribosome (Fig 2A). Analogously, the putative Hsp15 homolog SP_0007 co‐migrates with the 50S ribosome, supporting its possible role as a factor that recycles dead‐end 50S subunits (Korber et al, 2000; Jiang et al, 2009). In contrast, SP_2226 co‐migrated mostly with the 30S ribosome, even though its B. subtilis homolog YaaA was suggested to be a player in 50S assembly (Suzuki et al, 2014), indicating that SP_2226 could have alternative functions in the pneumococcus.
Taken together, our Grad‐seq study adds a new level of information to the increasing number of high‐throughput studies of the pneumococcus. It will provide an important link between predictions of protein–protein interactions (Wuchty et al, 2017) and the wealth of recent transcriptomics‐based studies (Acebo et al, 2012; Mann et al, 2012; Aprianto et al, 2018; Slager et al, 2018, 2019; Warrier et al, 2018; Sinha et al, 2019) in the quest to better understand the molecular underpinnings of pneumococcal physiology and virulence.
Materials and Methods
Bacteria and media
Streptococcus pneumoniae strains TIGR4 and R6 were streaked on tryptic soy agar plates containing 5% sheep blood (Oxoid) and grown overnight at 37°C and 5% CO2. Strains used are listed in Table EV1. The next day, pre‐cultures were grown in THY medium (to the exception of competence assays, see below) [Todd Hewitt broth (BD Bacto) supplemented with 0.5% yeast extract (Carl Roth)] to an OD600 nm of 0.5 at 37°C without shaking. Subsequently, the pre‐culture was refreshed in THY to a starting OD600 nm of 0.05 and grown at 37°C without shaking to a final OD600 nm of 0.5.
Glycerol gradient fractionation
400 ml of S. pneumoniae TIGR4 wild type was grown to an OD600 nm of 0.5, cooled down in an ice‐water bath for 15 min, and then harvested by centrifugation for 20 min at 4°C and 4,000 g. The cells were washed three times in ice‐cold 1× TBS, resuspended in 500 μl ice‐cold 1× lysis buffer A [20 mM Tris–HCl, pH 7.5, 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 0.2% Triton X 100, 20 U/ml DNase I (Thermo Fisher Scientific), 200 U/ml RNase inhibitor]. Mechanical lysis was performed using the FastPrep‐24 instrument (MP Biomedicals) in 2‐ml tubes with lysing matrix E (MP Biomedicals) at 6 m/s for 30 s. To remove insoluble debris and the beads, the lysate was cleared by centrifugation for 30 min at 4°C and 16,100 g. Of the cleared lysate, 10 μl was mixed with 1 ml TRIzol (Thermo Fisher Scientific) for the RNA input control and 20 μl was mixed with 20 μl 5× protein loading buffer for the protein input control.
400 μl of the cleared lysate was then layered on top of a linear 10–40% (w/v) glycerol gradient (in 1× lysis buffer A without DNase I or RNase inhibitor), which was formed in an open‐top polyallomer tube (Seton) using the Gradient Station model 153 (Biocomp). The gradient was centrifuged for 17 h at 4°C and 100,000 g (23,700 rpm) using a Beckman Coulter SW40Ti rotor, followed by manual fractionation into 20 590 μl fractions and measurement of the A260 nm of each fraction. 90 μl of each fraction and 40 μl of the pellet were mixed with 30 μl of 5× protein loading buffer for protein analysis and stored at −20°C.
The remaining 500 μl of each fraction was used for RNA isolation by addition of 50 μl of 10% SDS (25 μl for the pellet) and 600 μl of acidic phenol/chloroform/isoamyl alcohol (P/C/I; 300 μl for the pellet). The fractions were then vortexed for 30 s and let rest at room temperature for 5 min before separating the phases by centrifugation for 15 min at 4°C and 16,100 g. The aqueous phases were collected, and 1 μl of GlycoBlue (Thermo Fisher Scientific) and 1.4 ml of ice‐cold ethanol/3 M NaOAc, pH 6.5 (30:1) were added and precipitated for at least 1 h at −20°C. The RNA was collected by centrifugation for 30 min at 4°C and 16,100 g and washed with 350 μl ice‐cold 70% ethanol, followed by centrifugation for 15 min at 4°C and 16,100 g. The lysate RNA sample stored in TRIzol was purified according to the manufacturer's protocol, except that the precipitation was performed using the mentioned ethanol mix. After drying of the RNA pellet, it was dissolved in 40 μl DEPC‐treated H2O and DNase‐digested by addition of 5 μl DNase I buffer with MgCl2 (Thermo Fisher Scientific), 0.5 μl RNase inhibitor, 4 μl DNase I (Thermo Fisher Scientific), and 0.5 μl DEPC‐treated H2O, followed by incubation for 45 min at 37°C. The DNase‐treated RNA was purified by the addition of 150 μl DEPC‐treated H2O and 200 μl acidic P/C/I as described above. The purified, DNase‐treated RNA was dissolved in 35 μl DEPC‐treated H2O and stored at −80°C.
RNA gel electrophoresis and northern blotting
Equal volumes of the gradient RNA samples were separated by denaturing 6% PAGE in 1× TBE and 7 M urea and stained with ethidium bromide. For northern blotting, unstained gels were transferred onto Hybond+ membranes (GE Healthcare Life Sciences) and probed with RNA‐specific radioactively labeled DNA oligonucleotides.
Protein gel electrophoresis and Western blotting
Equal volumes of the gradient protein samples were separated by 12% SDS–PAGE and stained with Coomassie. For Western blotting of Cbf1‐3xFLAG, unstained gels were transferred to PVDF membranes (GE Healthcare Life Sciences) and probed with an anti‐FLAG primary antibody (Sigma, cat# F1804, clone M2) and an anti‐mouse secondary antibody (Thermo Fisher Scientific, cat# 31430). As loading controls, the membranes were stripped and probed for glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) using a rabbit antiserum. Immunoreactive serum to S. pneumoniae GAPDH (GenBank AAK76079) was raised in rabbit against a synthetic peptide (DPIVSSDIVGMS) corresponding to amino acids 275–286. Synthetic peptide production, immunization, and validation by ELISA were done by Innovagen (Lund, Sweden), following a 41‐day immunization protocol including two booster injections. It was further used in previous studies (Codemo et al, 2018; Pathak et al, 2018). Detection of the GAPDH‐specific serum was performed using an anti‐rabbit secondary antibody (Thermo Fisher Scientific, cat# 31460).
RNA‐seq
For RNA‐seq, 5 μl of the gradient samples was diluted in 45 μl DEPC‐treated H2O. 10 μl of the resulting 1:10 dilution was mixed with 10 μl of a 1:100 dilution of the ERCC spike‐in mix 2 (Thermo Fisher Scientific) and subjected to library preparation for next‐generation sequencing (Vertis Biotechnologie). Briefly, the RNA samples were fragmented using ultrasound (four pulses of 30 s at 4°C) followed by 3′ adapter ligation. Using the 3′ adapter as primer, first‐strand cDNA synthesis was performed using M‐MLV reverse transcriptase. After purification, the 5′ Illumina TruSeq sequencing adapter was ligated to the 3′ end of the antisense cDNA. The resulting cDNA was PCR‐amplified to about 10–20 ng/μl using a high‐fidelity DNA polymerase followed by purification using the Agencourt AMPure XP Kit (Beckman Coulter Genomics). The cDNA samples were pooled with ratios according to the RNA concentrations of the input samples, and a size range of 200–550 bp was eluted from a preparative agarose gel. This size‐selected cDNA pool was finally subjected to sequencing on an Illumina NextSeq 500 system using 75 nt single‐end read length.
RNA‐seq data analysis
Read trimming and clipping were done with cutadapt (Martin, 2011). Further analysis steps (read filtering, read mapping, nucleotide‐wise coverage calculation, and genome feature‐wise counting) were done using READemption (Förstner et al, 2014) (v0.4.3; https://doi.org/10.5281/zenodo.250598) and the short read mapper segemehl (Hoffmann et al, 2014) (v0.2.0‐418). The S. pneumoniae TIGR4 genome version was NC_003028.3. Grad‐seq‐specific data analysis was performed with the tool GRADitude (Di Giorgio S, Hör J, Vogel J, Förstner KU, unpublished, https://foerstner-lab.github.io/GRADitude/). For the downstream analysis, only transcripts with a sum of ≥ 100 reads in all fractions within the gradient were considered. For each fraction, read counts were normalized by calculating size factors following the DESeq2 approach (Anders & Huber, 2010) generated from the ERCC spike‐in read counts added previously (see above). To remove left‐over disturbances in the data, the size factors were then manually adjusted by multiplication based on quantified northern blots: 1.15 (fraction 3), 1.15 (fraction 4), 0.85 (fraction 5), 0.95 (fraction 6), 0.85 (fraction 7), and 1.25 (fraction 15). To allow comparison of all transcript counts, they were scaled to the maximum value.
Based on these normalized values, two analyses were performed: one containing all the detectable transcripts, and one containing only the detectable ncRNAs. For the ncRNAs, t‐SNE dimension reduction (van der Maaten & Hinton, 2008) and k‐means clustering (Lloyd, 1982) were performed using the Python package scikit‐learn (Pedregosa et al, 2011). t‐SNE was performed excluding the pellet fraction using all the default parameters provided by the sklearn.manifold.TSNE class. The only exception was the perplexity parameter, set to 35 for Fig 3B and to 25 for Appendix Fig S4 instead of the default of 30. k‐means clustering was performed using the class sklearn.cluster.KMeans, grouping the ncRNAs into three clusters. Subsequently, the ncRNAs in the t‐SNE plot were colored according to their cluster number. Appendix Fig S4 shows stronger separation of the clusters by adding information of the k‐means analysis to the read count table before the t‐SNE dimensionality reduction. A script representing the analysis workflow, including Unix Shell calls and Python scripts, and documentation have been deposited at Zenodo (https://doi.org/10.5281/zenodo.3475892).
Sample preparation for mass spectrometry
For mass spectrometry (MS), the gradient protein samples (diluted in 1.25× protein loading buffer) were homogenized using ultrasound [five cycles of 30 s on followed by 30 s off, high power at 4°C (Bioruptor Plus, Diagenode)]. Insoluble material was then removed by centrifugation for 15 min at 4°C and 16,100 g. 20 μl of the cleared protein sample was mixed with 10 μl of UPS2 spike‐in (Sigma‐Aldrich) diluted in 250 μl 1.25× protein loading buffer. The samples were subsequently reduced in 50 mM DTT for 10 min at 70°C and alkylated with 120 mM iodoacetamide for 20 min at room temperature in the dark. The proteins were precipitated in four volumes of acetone overnight at −20°C. Pellets were washed four times with acetone at −20°C and dissolved in 50 μl 8 M urea, 100 mM ammonium bicarbonate.
Digestion of the proteins was performed by the addition of 0.25 μg Lys‐C (Wako) for 2 h at 30°C, followed by dilution to 2 M urea by the addition of 150 μl 100 mM ammonium bicarbonate, pH 8 and overnight digestion with 0.25 μg trypsin at 37°C. Peptides were desalted using C‐18 Stage Tips (Rappsilber et al, 2003). Each Stage Tip was prepared with three disks of C‐18 Empore SPE Disks (3M) in a 200‐μl pipette tip. Peptides were eluted with 60% acetonitrile/0.3% formic acid, dried in a laboratory freeze‐dryer (Christ), and stored at −20°C. Prior to nanoLC‐MS/MS, the peptides were dissolved in 2% acetonitrile/0.1% formic acid.
NanoLC‐MS/MS analysis
NanoLC‐MS/MS analysis was performed similarly to Cossa et al (2020) using an Orbitrap Fusion (Thermo Fisher Scientific) equipped with a PicoView Ion Source (New Objective) and coupled to an EASY‐nLC 1000 (Thermo Fisher Scientific). Peptides were loaded on capillary columns (PicoFrit, 30 cm × 150 μm ID, New Objective) self‐packed with ReproSil‐Pur 120 C18‐AQ, 1.9 μm (Dr. Maisch) and separated with a 140‐min linear gradient from 3 to 40% acetonitrile and 0.1% formic acid at a flow rate of 500 nl/min. Both MS and MS/MS scans were acquired in the Orbitrap analyzer with a resolution of 60,000 for MS scans and 15,000 for MS/MS scans. HCD fragmentation with 35% normalized collision energy was applied. A Top Speed data‐dependent MS/MS method with a fixed cycle time of 3 s was used. Dynamic exclusion was applied with a repeat count of 1 and an exclusion duration of 60 s; singly charged precursors were excluded from selection. Minimum signal threshold for precursor selection was set to 50,000. Predictive AGC was used with a target value of 2 × 105 for MS scans and 5 × 104 for MS/MS scans. EASY‐IC was used for internal calibration.
Grad‐seq MS data analysis
Raw MS data files were analyzed with MaxQuant version 1.5.7.4 (Cox & Mann, 2008). Database search was performed using Andromeda (integrated into MaxQuant) against the UniProt database for S. pneumoniae TIGR4 (UP000000585, organism identifier: STRPN), a database containing the UPS2 spike‐in and a database containing common contaminants. The search was performed with tryptic cleavage specificity with three allowed miscleavages. Protein identification was under control of a false discovery rate of 1% on both protein and peptide levels. In addition to the MaxQuant default settings, the search was performed against the following variable modifications: protein N‐terminal acetylation, Gln to pyro‐Glu formation (N‐terminal Gln), and oxidation of Met. For protein quantitation, the LFQ intensities were used (Cox et al, 2014). Proteins with less than 2 identified razor/unique peptides were dismissed.
Normalization of the proteins across the fractions was performed using the UPS2 spike‐in. For this, only spike‐in proteins with detectable intensities in all fractions were used. The spike‐in proteins showing the highest variance (median average deviation of log10 intensities > 1.5× lQR) were eliminated. Following this, for each spike‐in protein, the median log10 intensity was subtracted from the log10 intensities of each fraction. The fraction‐wise median of the resulting values was then subtracted from the log10 intensities for each bacterial protein in the corresponding fractions. Finally, all log10 intensities smaller than the 5% quantile of all intensities in the dataset were replaced by the value of the 5% quantile of all intensities in the dataset.
Protein pull‐down using RNA as bait
To pull down proteins using RNA as bait, a modified version of a published protocol (Treiber et al, 2017, 2018) was used. PCR templates for in vitro transcription with T7 RNAP were generated with a 39 nt 5′ overhang: GTTTTTTTTAATACGACTCACTATA G GG AGACCTAGCCT (italicized nucleotides belong to the T7 promoter, bold nucleotides represent the 14 nt 5′ tag, the underlined nucleotide is the transcription start site). Primers used are listed in Table EV2. In vitro transcription was performed using the TranscriptAid kit (Thermo Fisher Scientific) according to the manufacturer's instructions. The resulting RNA was purified using denaturing 6% PAGE in 1× TBE and 7 M urea. An RNA oligonucleotide representing only the 14 nt tag (GGGAGACCUAGCCU) was used as negative control.
For the pull‐down, 100 μl magnetic streptavidin beads (Dynabeads M‐270, Thermo Fisher Scientific) were washed 3× with 1 ml of lysis buffer B (50 mM Tris–HCl, pH 8, 150 mM KCl, 1 mM MgCl2, 5% glycerol, 0.05% Tween‐20). The rest of the protocol was performed at 4°C. The washed beads were coupled to 4 μg of a 3′‐biotinylated, 2′‐O‐methyl‐modified RNA adaptor complementary to the 14 nt tag of the bait RNAs (AGGCUAGGUCUCCC‐biotin) for 1 h. The adaptor‐coupled beads were washed twice with 1 ml of lysis buffer B, resuspended in 1 ml of lysis buffer B, and split into two tubes with 500 μl each. One tube was used to couple 10 μg per 100 nt of bait RNA overnight, and the other was stored for pre‐clearing.
To prepare the lysate for the pull‐down, 100 OD600 nm of cells were lysed and cleared in 500 μl lysis buffer C (lysis buffer B with 1 mM DTT and 1 mM PMSF) as described above. To pre‐clear the lysate from content that binds the beads unspecifically, the stored beads were incubated with the lysate for 3.5 h. The beads were subsequently removed by centrifugation for 10 min at 16,100 g. The bait RNA‐coupled beads were washed twice with lysis buffer B and incubated with the pre‐cleared lysate supernatant for 2 h to capture interacting proteins of the bait RNAs. To get rid of unspecific binders, the beads were washed with 1 ml each of wash buffer A (lysis buffer C with 300 mM KCl in total), wash buffer B (lysis buffer C with 0.1% Triton X‐100), and lysis buffer C.
The beads were resuspended in 35 μl of 1× LDS sample buffer (Thermo Fisher Scientific) containing 50 mM DTT and boiled for 5 min at 95°C to elute the bound proteins. Following alkylation as described above, the pull‐down samples were run on a 4–12% Bolt Bis‐Tris plus gel (Thermo Fisher Scientific) using MES buffer (Thermo Fisher Scientific). After staining with SimplyBlue Coomassie (Thermo Fisher Scientific), each lane of the gel was either cut into 11 pieces or specific prominent bands were cut. The gel pieces were prepared for LC/MS‐MS by destaining with 30% acetonitrile in 100 mM ammonium bicarbonate, pH 8 followed by shrinking with 100% acetonitrile and drying in a vacuum concentrator (Eppendorf). 0.1 μg trypsin was added per gel piece and digestion performed overnight at 37°C in 100 mM ammonium bicarbonate, pH 8. The supernatant was removed, and the peptides were extracted from the gel pieces with 5% formic acid. Finally, the supernatant was pooled with the extracted peptides.
Pull‐down nanoLC‐MS/MS analysis
NanoLC‐MS/MS analyses were performed similarly to Braun et al (2018) on an LTQ‐Orbitrap Velos Pro (Thermo Fisher Scientific) equipped with a PicoView Ion Source (New Objective) and coupled to an EASY‐nLC 1000 (Thermo Fisher Scientific). Peptides were loaded on capillary columns (PicoFrit, 30 cm × 150 μm ID, New Objective) self‐packed with ReproSil‐Pur 120 C18‐AQ, 1.9 μm (Dr. Maisch) and separated with a 30‐min linear gradient from 3 to 30% acetonitrile and 0.1% formic acid at a flow rate of 500 nl/min. MS scans were acquired in the Orbitrap analyzer with a resolution of 30,000 at m/z 400, and MS/MS scans were acquired in the Orbitrap analyzer with a resolution of 7,500 at m/z 400 using HCD fragmentation with 30% normalized collision energy. A TOP5 data‐dependent MS/MS method was used; dynamic exclusion was applied with a repeat count of 1 and an exclusion duration of 30 s; singly charged precursors were excluded from selection. Minimum signal threshold for precursor selection was set to 50,000. Predictive AGC was used with a target value of 1 × 106 for MS scans and 5 × 104 for MS/MS scans. Lock mass option was applied for internal calibration in all runs using background ions from protonated decamethylcyclopentasiloxane (m/z 371.10124).
Pull‐down MS data analysis
Analysis of the pull‐down MS data was performed as described for the Grad‐seq MS data, except that the search against the UPS2 spike‐in database was skipped.
UV CLIP‐seq sample preparation and sequencing
Sample preparation for UV crosslinking and immunoprecipitation followed by RNA‐seq (CLIP‐seq) followed a previously published protocol (Holmqvist et al, 2018) with a few changes. For each biological replicate, 800 ml of the cbf1‐3xFLAG strain was grown to an OD600 nm of 0.5. Half of the culture was irradiated in 50 ml fractions at 254 nm in a 22 × 22 cm plastic tray at 0.8 J. After collection of the cells by centrifugation at 4°C, the pellets were frozen in liquid nitrogen and stored at −80°C. Cells were thawed on ice, resuspended in 800 μl NP‐T buffer (50 mM NaH2PO4, 300 mM NaCl, 0.05% Tween‐20, pH 8), and transferred to tubes containing 1 ml of glass beads. Following lysis for 10 min at 30 Hz using a mixer mill (Retsch MM400), the lysates were cleared twice by centrifugation for 15 min at 16,100 g and 4°C and mixed with one volume of NP‐T buffer containing 8 M urea. Finally, the lysates were incubated for 5 min at 65°C with shaking at 900 rpm, put on ice for 2 min and diluted 10× in ice‐cold NP‐T buffer, and put back on ice.
To perform the immunoprecipitation, 30 μl of anti‐FLAG magnetic beads was washed 3× with 800 μl NP‐T buffer, added to the lysate, and rotated for 1 h at 4°C. The beads were collected by centrifugation at 1,000 g and 4°C, and washed twice with 2 ml of high‐salt buffer (NP‐T buffer containing 1 M NaCl) and twice with 2 ml of NP‐T buffer. Subsequently, the beads were resuspended in 100 μl NP‐T buffer containing 1 mM MgCl2 and 2.5 U benzonase (Sigma) followed by incubation for 10 min at 37°C with shaking at 800 rpm. Then, the beads were put on ice for 2 min and washed once with 500 μl high‐salt buffer and twice with 500 μl CIP buffer (100 mM NaCl, 50 mM Tris–HCl, pH 7.4, 10 mM MgCl2) followed by resuspension in 100 μl CIP buffer containing 10 U of calf intestinal alkaline phosphatase (NEB) and incubation for 30 min at 37°C with shaking at 800 rpm. The beads were washed once with 500 μl high‐salt buffer and twice with 500 μl PNK buffer (50 mM Tris–HCl, pH 7.4, 10 mM MgCl2, 0.1 mM spermidine) followed by resuspension in 100 μl PNK buffer containing 1 μl T4 polynucleotide kinase (Thermo Fisher Scientific) and 1 μl γ‐32P‐ATP for 30 min at 37°C. Then, 10 μl 1 mM ATP was added and the beads were incubated for 5 min at 37°C, followed by two washes with 1 ml of NP‐T buffer and resuspension in 10 μl protein loading buffer. The labeled RNA–protein complexes were eluted from the beads by incubation for 5 min at 95°C, which was repeated once.
15 μl of the eluted samples was separated using 12% SDS–PAGE followed by transfer to a nitrocellulose membrane (GE Healthcare Life Sciences). Marker sizes of the protein ladder were highlighted using a radioactive marker, and the autoradiogram of the blot was subsequently used as a template to cut out the labeled complexes and their corresponding controls. The membrane pieces were then cut into smaller pieces, transferred to LoBind tubes (Eppendorf), and incubated with 200 μl PK buffer [50 mM Tris–HCl, pH 7.4, 75 mM NaCl, 6 mM EDTA, 1% SDS, 10 U SUPERaseIN (Thermo Fisher Scientific), and 0.4 mg proteinase K (Thermo Fisher Scientific)] for 1 h at 37°C with shaking at 800 rpm. Then, 100 μl PK buffer containing 9 M urea was added and the incubation continued for 1 h. The membrane pieces were briefly centrifuged and the supernatant extracted with one volume of P/C/I. The purified RNA fragments were resuspended in 10 μl DEPC‐treated H2O.
Library preparation for next‐generation sequencing was performed by the CoreUnit SysMed at the University of Würzburg, Germany. The protocol used is the same as published before (Holmqvist et al, 2018). Sequencing was also performed by the CoreUnit SysMed on an Illumina NextSeq 500 system using 2 × 75 nt paired‐end sequencing.
CLIP‐seq data analysis
CLIP‐seq data analysis was performed as described previously (Chihara et al, 2019). Briefly, read 1 and read 2 files containing the paired‐end reads were quality and adapter trimmed via Cutadapt (Martin, 2011), version 1.17, using a cutoff Phred score of 20 in NextSeq mode, and reads without any remaining bases were discarded. Putative PCR duplicates were discarded by collapsing the reads using FastUniq (Xu et al, 2012). After trimming, all reads longer than 11 nt were aligned to the S. pneumoniae TIGR4 genome (NC_003028.3) using READemption (Förstner et al, 2014) (version 0.4.5) and segemehl (Hoffmann et al, 2014) (version 0.2.0) with an accuracy cutoff of 80%. Only reads uniquely mapping to the genome were considered for all subsequent analyses.
Size factors for normalization of the CLIP‐seq data were calculated based on positional coverage files generated via READemption. In brief, positions present in both crosslinked and non‐crosslinked library pairs were isolated, as described previously (Holmqvist et al, 2018). Positions with read counts less than 6 standard deviations from 0 were filtered from both crosslinked and non‐crosslinked libraries. The distribution of ratios between the paired crosslinked and non‐crosslinked samples presented a clear bimodal distribution, with modes corresponding to background (high counts in both libraries) and crosslink‐enriched positions, which were separated by k‐means clustering. Size factors were then calculated from the background positions present across all replicates against a geometric mean pseudo‐reference, as introduced in DESeq (Anders & Huber, 2010).
We applied PEAKachu (https://github.com/tbischler/PEAKachu) (version 0.1.0) for peak calling in a similar way as described previously (Holmqvist et al, 2018). The tool was run in paired‐end and paired‐replicates mode using BAM files for the respective pairs of crosslinked and control libraries as input. The maximum fragment size was set to 50, and the S. pneumoniae TIGR4 annotation (Warrier et al, 2018) was used to map overlapping features to called peaks. For normalization, “manual” mode was selected together with previously calculated size factors (see above). Peak calling via the adaptive approach is performed in two consecutive steps. In the first step, initial peaks are defined via heuristic decomposition of read clusters computed by the blockbuster algorithm (Langenberger et al, 2009) based on pooled read alignments from all crosslinked libraries. In this step, PEAKachu applies a set of parameters for which default values were used. In the second step, PEAKachu runs DESeq2 (Love et al, 2014) to test each peak for significant enrichment of normalized read counts in the crosslinked compared to the control libraries. Initial peaks were tested for significance using the following parameter values: mad‐multiplier 1.0, fold change 1.0, and adjusted P‐value 0.01. Normalized coverage plots representing the numbers of mapped reads per nt were generated by PEAKachu for each strand to facilitate data visualization in a genome browser.
Purification of recombinant Cbf1
The purification of recombinant Cbf1 was performed by the Recombinant Protein Expression core unit at the Rudolf Virchow Center in Würzburg, Germany. Briefly, to purify recombinantly expressed Cbf1, the CDS was cloned into pet21b+, which adds a C‐terminal His‐tag to the sequence (N‐terminal His‐tagging did not allow purification of Cbf1). After transformation into E. coli BL21, cells were grown to an OD600 nm of 0.5 and induced with 0.5 mM IPTG for 3 h at 37°C. After centrifugation, the cell pellet was dissolved in 20 mM HEPES, pH 8, 250 mM NaCl, 0.4 mM PMSF, and 1.5 U/ml DNase I and subjected to affinity purification using Protino Ni‐NTA Agarose (Macherey Nagel). Following elution, the purity was tested on a Superdex 16/600 column (GE Healthcare Life Sciences). Purified Cbf1 was stored in 100 μl aliquots of 20 mM HEPES, pH 8, 250 mM NaCl, and 10% glycerol at −80°C (~ 1.35 mg/ml).
In vitro RNase assay
To test the activity of Cbf1 on target RNAs, 400 ng in vitro‐transcribed RNAs (~ 9–14 pmol, depending on the RNA) were digested with 3.5 μg Cbf1 (~ 100 pmol) in a total volume of 20 μl [50 mM Tris–HCl, pH 8, 100 mM KCl; according to Fang et al (2009)]. Primers used are listed in Table EV2. Reactions contained either no divalent cations or 5 mM of MgCl2 or MnCl2. Samples (10% of the reaction volume) were taken immediately after addition of Cbf1 (time point 0 min) and then, after shifting the reaction to 37°C, after 5 and 15 min. Collection was performed on ice in RNA loading buffer and immediately frozen in order to stop the reaction. The samples were analyzed by northern blotting using denaturing 10% PAGE.
3′ RACE
Identification of 3′ ends via 3′ rapid amplification of cDNA ends (RACE) was performed as described before (Fröhlich et al, 2012) with few modifications. First, a Cbf1 in vitro RNase assay with truncated csRNA1 was performed as described above, except that the total reaction volume was scaled up to 200 μl containing 4 μg of RNA. 50 μl of the reaction (1 μg of RNA) was taken at the indicated time points and stopped by addition of 150 μl acidic P/C/I and 200 μl of H2O. As negative control, 1 μg of untreated RNA was used. After P/C/I extraction, the RNA was dephosphorylated using 10 U of calf intestinal alkaline phosphatase (NEB) in 1× NEB buffer 3 in 25 μl for 1 h at 37°C. The RNA was then P/C/I‐extracted and precipitated in the presence of 1 μl GlycoBlue (Thermo Fisher Scientific) and 250 pmol of RNA adapter E1. Ligation of the adapter was performed in a 20 μl reaction containing 20 U T4 RNA ligase (NEB), 1× T4 RNA ligase buffer, and 10% DMSO overnight at 16°C. After ligation, the RNA was P/C/I‐extracted and reverse transcribed for 5 min at 50°C followed by 60 min at 55°C using 200 U SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and 39 pmol of adapter E1‐specific oligo E3RACE in a 20 μl reaction containing 1× FS buffer, 2 mM dNTPs, and 5 mM DTT. The template RNA was digested by addition of 1 μl RNase H (NEB) and incubation at 37°C for 30 min. 1 μl of the cDNA was then used as template for a Taq PCR using E3RACE and JVO‐15297 as primers. The PCR products were run on a 3.5% agarose gel, the bands cut from the gel and cloned using the StrataClone PCR cloning kit (Agilent Technologies) following the manufacturer's instructions. For the 5‐ and 15‐min time points, the inserts of 20 positive clones each were amplified by PCR using the primers M13 and M13rev and analyzed by Sanger sequencing. Primers used are listed in Table EV2.
Rifampicin RNA stability assays
The in vivo stability of RNAs was determined by growing bacterial cultures in a water bath until they reached an OD600 nm of 0.5. Four OD600 nm were collected as the non‐treated 0‐min sample. Then, 500 μg/ml rifampicin was added to stop transcription and four OD600 nm samples were collected after 2, 4, 8, 16, and 32 min. Each sample was immediately mixed with 0.2 volumes of stop‐mix (95% ethanol, 5% phenol) and snap‐frozen in liquid nitrogen. Following RNA isolation using the hot phenol method and DNase treatment, 5 μg of RNA was analyzed by northern blotting and quantified using ImageJ (Schneider et al, 2012). 5S rRNA was used as loading control.
CSP induction assay
To determine the influence of CSP induction, the cbf1‐3xFLAG strain was grown in a water bath until it reached an OD600 nm of 0.5. For determination of protein levels, 1 ml of culture was collected as the non‐treated 0‐min sample and immediately snap‐frozen in liquid nitrogen. For RNA purification, instead 4 ml of culture were mixed with 0.2 volumes of stop‐mix and snap‐frozen in liquid nitrogen. Stimulation was then performed by addition of CSP‐2 (AnaSpec) to a final concentration of 100 ng/ml. For the control, the same volume of sterile H2O was added instead. After 5, 10, 15 and 30 min of stimulation, samples were collected the same way as for the 0 min time point. Protein samples were thawed on ice and diluted in 200 μl protein loading buffer, and equal volumes (10 μl) were subjected to PAGE and Western blotting. RNA isolation was performed using the hot phenol method followed by DNase treatment. 15 μg of RNA was loaded on a 1.2% agarose gel, run, and stained with ethidium bromide to visualize 16S and 23S rRNAs as loading control. Finally, capillary blotting on a Hybond+ membrane was performed and the membranes probed with RNA‐specific radioactively labeled DNA oligonucleotides.
RT–qPCR
For the determination of mRNA levels via RT–qPCR, RNA was extracted as described for the rifampicin RNA stability assays. Following PCR‐based analysis of potential gDNA contamination, RT–qPCR was carried out using the Power SYBR Green RNA‐to‐CT 1‐Step kit (Thermo Fisher Scientific) and a CFX96 system (Bio‐Rad). gyrA was used as the control gene. Data were analyzed using the comparative ΔΔC T method (Livak & Schmittgen, 2001). Primers used are listed in Table EV2.
Strain construction
Primers used are listed in Table EV2. All PCRs were carried out using Phusion Flash High‐Fidelity PCR Master Mix (Thermo Fisher Scientific) following manufacturer's instructions. Strains TIGR4Δcbf1::Sp, R6Δcbf1::Sp, and TIGR4 cbf1‐3xFLAG‐Sp were constructed by homologous recombination in the genome of a cassette composed of a spectinomycin (Sp) resistance gene flanked with ~ 1,000 bp homologous to the regions bordering the region of interest. Cassettes were assembled by PCR assembly via overlapping regions in the primers used for amplifying individual fragments. Briefly, the upstream (up) and downstream (dwn) of TIGR4/R6 cbf1 were amplified using primers cbf1DF/cbf1DJ1R (up) and cbf1DJ2F/cbf1DR (dwn), and TIGR4 or R6 gDNA as the template. The spectinomycin resistance (Sp) gene, along with its promoter and terminator, was amplified from plasmid pSP72::Sp (courtesy of P. Mellroth) using primers cbf1DJ1F and cbf1DJ2R. The final up‐Sp‐dwn cassette was amplified using primers cbf1DF/cbf1DR (TIGR4) and cbf1DF2/cbf1DR2 (R6) and transformed into competent TIGR4/R6 cells. The cbf1‐3xFLAG strain was constructed by assembling cbf1 from TIGR4 gDNA, amplified with primers cbf1VF/cbf13xFLAGR, with the Sp‐dwn region of TIGR4Δcbf1::Sp strain amplified with primers AbRFLAGF/cbf1DR. The final product was amplified using primers cbf1_nestedF/cbf1_nestedR and transformed into competent TIGR4 cells.
The cbf1 deletion was complemented in TIGR4Δcbf1::Sp using plasmid pJWV25 which integrates into the chromosome at the bga locus by homologous recombination between sequences flanking the insert (Eberhardt et al, 2009). Briefly, cbf1 was cloned into pJWV25 under control of the zinc‐inducible promoter PczcD using ligase‐independent cloning (Li et al, 2011). cbf1 was amplified from TIGR4 gDNA with primers cbf1_pGG8F/cbf1_pGG7R, and pJWV25 was amplified using primers pGG7F/pGG8R. Products were digested with DpnI and co‐transformed into E. coli XL‐GOLD Ultracompetent cells (Stratagene) following manufacturer's instructions and selected on LB plates supplemented with 100 μg/ml ampicillin. The resulting vector, pGG8A, was purified using the QIAprep Spin Miniprep Kit (QIAGEN), and 300 ng of plasmid DNA was transformed into competent TIGR4Δcbf1::Sp. All PCR products described above were verified on agarose gel and, when necessary, excised, and purified using the Wizard SV Gel and PCR Clean‐Up System (Promega). Transformations were performed as described below for induced competence using 100 ng/ml CSP‐1 (R6) or 100 ng/ml CSP‐2 (TIGR4). Transformant colonies were isolated on blood agar plates containing the appropriate antibiotics at the following concentration: spectinomycin, 200 μg/ml; tetracycline, 2 μg/ml (pGG8A). All strains constructed were confirmed by PCR followed by sequencing at Eurofins (Germany).
Competence assays
For spontaneous competence assays, pre‐cultures of R6 (Ottolenghi & Hotchkiss, 1962) and R6Δcbf1::Sp were grown in C+Y (Lacks & Hotchkiss, 1960), pH 8 at 37°C without shaking to an OD620 nm of 0.5. The pre‐cultures were refreshed in the same medium to a starting OD620 nm of 0.05 and grown at 37°C without shaking to an OD620 nm of 0.13. 50 μl aliquots were diluted 1:10 in C+Y pre‐warmed at 30°C and incubated for 30 min at 30°C. 100 ng/ml of a 524‐bp PCR product encompassing the SmR‐rpsL allele (see below) was added, and incubation was continued at 30°C for 60 min, followed by 90 min at 37°C. Samples were serially diluted and plated on blood agar plates containing 150 μg/ml streptomycin to enumerate transformant colony‐forming units (CFU) and on blood agar plates without antibiotics to enumerate total viable CFU. Results were expressed as the percentage of total CFU resistant to streptomycin. The rpsL PCR product used in transformation experiments was amplified using primer pair rpsLF/101‐rpsLR, gDNA from an S. pneumoniae strain carrying a point mutation in the rpsL gene conferring resistance to streptomycin (Muschiol et al, 2017) as the template and Phusion Flash High‐Fidelity PCR Master Mix (Thermo Fisher Scientific) following manufacturer's instructions. The product was purified using the Wizard SV Gel and PCR Clean‐Up System (Promega) and visualized on a 1% agarose gel run in 1× TAE and stained with GelRed (Biotium).
Author contributions
BH‐N, GG, JH, and JV conceived and designed the study. GG, JH, and L‐MH performed experiments. KUF, JH, and SDG analyzed RNA‐seq and MS data. AS and JTV performed MS and analyzed MS data. BH‐N and JV supervised the project. JH and JV wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Youssef El Mouali, Gianluca Matera, and other members of the Vogel laboratory for comments, ideas, and fruitful discussions. We further thank Thorsten Bischler and Lars Barquist for help with the CLIP‐seq analysis and the Core Unit SysMed at the University of Würzburg for RNA‐seq data generation. This work was funded by a DFG Gottfried Wilhelm Leibniz Award to JV (DFG Vo875‐18) and by the IZKF at the University of Würzburg (project Z‐6), as well as by grants from the Swedish Research Council, Knut and Wallenberg Foundation, the Swedish Foundation for Strategic Research (SSF), and Stockholm County Council.
The EMBO Journal (2020) 39: e103852
Contributor Information
Birgitta Henriques‐Normark, Email: Birgitta.Henriques@ki.se.
Jörg Vogel, Email: joerg.vogel@uni-wuerzburg.de.
Data availability
RNA‐seq data: GEO GSE145605 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145605)
MS data: PRIDE PXD015835 and PXD015842 (http://www.ebi.ac.uk/pride/archive/projects/PXD015835 and http://www.ebi.ac.uk/pride/archive/projects/PXD015842)
Code used: Zenodo 250598 and 3475892 (https://doi.org/10.5281/zenodo.250598 and https://doi.org/10.5281/zenodo.3475892)
References
- Acebo P, Martin‐Galiano AJ, Navarro S, Zaballos A, Amblar M (2012) Identification of 88 regulatory small RNAs in the TIGR4 strain of the human pathogen Streptococcus pneumoniae . RNA 18: 530–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian D, Shen K, Zhang X, Shan S‐o (2013) Signal recognition particle: an essential protein‐targeting machine. Annu Rev Biochem 82: 693–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprianto R, Slager J, Holsappel S, Veening J‐W (2018) High‐resolution analysis of the pneumococcal transcriptome under a wide range of infection‐relevant conditions. Nucleic Acids Res 46: 9990–10006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitzke P, Lai Y‐J, Renda AJ, Romeo T (2019) Posttranscription initiation control of gene expression mediated by bacterial RNA‐binding proteins. Annu Rev Microbiol 73: 43–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechhofer DH, Deutscher MP (2019) Bacterial ribonucleases and their roles in RNA metabolism. Crit Rev Biochem Mol Biol 54: 242–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt H, Harnisch C, Rammelt C, Stöhr N, Zirkel A, Dohm JC, Himmelbauer H, Tavanez J‐P, Hüttelmaier S, Wahle E (2012) Maturation of mammalian H/ACA box snoRNAs: PAPD5‐dependent adenylation and PARN‐dependent trimming. RNA 18: 958–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantl S, Brückner R (2014) Small regulatory RNAs from low‐GC Gram‐positive bacteria. RNA Biol 11: 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AC, Gutmann M, Mueller TD, Lühmann T, Meinel L (2018) Bioresponsive release of insulin‐like growth factor‐I from its PEGylated conjugate. J Control Release 279: 17–28 [DOI] [PubMed] [Google Scholar]
- Burakovsky DE, Prokhorova IV, Sergiev PV, Milón P, Sergeeva OV, Bogdanov AA, Rodnina MV, Dontsova OA (2012) Impact of methylations of m2G966/m5C967 in 16S rRNA on bacterial fitness and translation initiation. Nucleic Acids Res 40: 7885–7895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burenina OY, Hoch PG, Damm K, Salas M, Zatsepin TS, Lechner M, Oretskaya TS, Kubareva EA, Hartmann RK (2014) Mechanistic comparison of Bacillus subtilis 6S‐1 and 6S‐2 RNAs–commonalities and differences. RNA 20: 348–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron TA, Matz LM, De Lay NR (2018) Polynucleotide phosphorylase: not merely an RNase but a pivotal post‐transcriptional regulator. PLoS Genet 14: e1007654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron TA, Matz LM, Sinha D, De Lay NR (2019) Polynucleotide phosphorylase promotes the stability and function of Hfq‐binding sRNAs by degrading target mRNA‐derived fragments. Nucleic Acids Res 47: 8821–8837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh AT, Sperger JM, Wassarman KM (2012) Regulation of 6S RNA by pRNA synthesis is required for efficient recovery from stationary phase in E. coli and B. subtilis . Nucleic Acids Res 40: 2234–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker AF, Ingraham KA, Lunsford RD, Bryant AP, Bryant J, Wallis NG, Broskey JP, Pearson SC, Holmes DJ (2000) The bacA gene, which determines bacitracin susceptibility in Streptococcus pneumoniae and Staphylococcus aureus, is also required for virulence. Microbiology 146: 1547–1553 [DOI] [PubMed] [Google Scholar]
- Chihara K, Bischler T, Barquist L, Monzon VA, Noda N, Vogel J, Tsuneda S (2019) Conditional Hfq association with small noncoding RNAs in Pseudomonas aeruginosa revealed through comparative UV cross‐linking immunoprecipitation followed by high‐throughput sequencing. mSystems 4: e00590‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codemo M, Muschiol S, Iovino F, Nannapaneni P, Plant L, Wai SN, Henriques‐Normark B (2018) Immunomodulatory effects of pneumococcal extracellular vesicles on cellular and humoral host defenses. mBio 9: e00559‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran CP, Rieder R, Podkaminski D, Hofmann B, Vogel J (2012) Use of aptamer tagging to identify in vivo protein binding partners of small regulatory RNAs. Methods Mol Biol 905: 177–200 [DOI] [PubMed] [Google Scholar]
- Cossa G, Roeschert I, Prinz F, Baluapuri A, Silveira Vidal R, Schülein‐Völk C, Chang Y‐C, Ade CP, Mastrobuoni G, Girard C et al (2020) Localized inhibition of protein phosphatase 1 by NUAK1 promotes spliceosome activity and reveals a MYC‐sensitive feedback control of transcription. Mol Cell in press. 10.1016/j.molcel.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgranges E, Marzi S, Moreau K, Romby P, Caldelari I (2019) Noncoding RNA. Microbiol Spectr 7: GPP3‐0038‐2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Condon C (2018) RNases and helicases in Gram‐positive bacteria. Microbiol Spectr 6: RWR‐0003‐2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt A, Wu LJ, Errington J, Vollmer W, Veening J‐W (2009) Cellular localization of choline‐utilization proteins in Streptococcus pneumoniae using novel fluorescent reporter systems. Mol Microbiol 74: 395–408 [DOI] [PubMed] [Google Scholar]
- Erickson HP (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol Proced Online 11: 32–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Zeisberg W‐M, Condon C, Ogryzko V, Danchin A, Mechold U (2009) Degradation of nanoRNA is performed by multiple redundant RNases in Bacillus subtilis . Nucleic Acids Res 37: 5114–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Rutherford ST, Papenfort K, Bagert JD, van Kessel JC, Tirrell DA, Wingreen NS, Bassler BL (2015) A qrr noncoding RNA deploys four different regulatory mechanisms to optimize quorum‐sensing dynamics. Cell 160: 228–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstner KU, Vogel J, Sharma CM (2014) READemption‐a tool for the computational analysis of deep‐sequencing‐based transcriptome data. Bioinformatics 30: 3421–3423 [DOI] [PubMed] [Google Scholar]
- Fröhlich KS, Papenfort K, Berger AA, Vogel J (2012) A conserved RpoS‐dependent small RNA controls the synthesis of major porin OmpD. Nucleic Acids Res 40: 3623–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Mejia A, Gámez G, Hammerschmidt S (2018) Streptococcus pneumoniae two‐component regulatory systems: the interplay of the pneumococcus with its environment. Int J Med Microbiol 308: 722–737 [DOI] [PubMed] [Google Scholar]
- Gorski SA, Vogel J, Doudna JA (2017) RNA‐based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Biol 18: 215–228 [DOI] [PubMed] [Google Scholar]
- Halfmann A, Kovács M, Hakenbeck R, Brückner R (2007) Identification of the genes directly controlled by the response regulator CiaR in Streptococcus pneumoniae: five out of 15 promoters drive expression of small non‐coding RNAs. Mol Microbiol 66: 110–126 [DOI] [PubMed] [Google Scholar]
- Han BW, Hung J‐H, Weng Z, Zamore PD, Ameres SL (2011) The 3′‐to‐5′ exoribonuclease Nibbler shapes the 3′ ends of microRNAs bound to Drosophila argonaute 1. Curr Biol 21: 1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häuser R, Pech M, Kijek J, Yamamoto H, Titz B, Naeve F, Tovchigrechko A, Yamamoto K, Szaflarski W, Takeuchi N et al (2012) RsfA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet 8: e1002815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques‐Normark B, Tuomanen EI (2013) The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3: a010215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt LM, Teupser D, Hackermüller J et al (2014) A multi‐split mapping algorithm for circular RNA, splicing, trans‐splicing and fusion detection. Genome Biol 15: R34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist E, Li L, Bischler T, Barquist L, Vogel J (2018) Global maps of ProQ binding in vivo reveal target recognition via RNA structure and stability control at mRNA 3′ ends. Mol Cell 70: 971–982.e6 [DOI] [PubMed] [Google Scholar]
- Holmqvist E, Vogel J (2018) RNA‐binding proteins in bacteria. Nat Rev Microbiol 16: 601–615 [DOI] [PubMed] [Google Scholar]
- Hör J, Gorski SA, Vogel J (2018) Bacterial RNA biology on a genome scale. Mol Cell 70: 785–799 [DOI] [PubMed] [Google Scholar]
- Hör J, Matera G, Vogel J, Gottesman S, Storz G (2020) Trans‐acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica . EcoSal Plus 10.1128/ecosalplus.ESP-0030-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Janga SC, Babu M, Díaz‐Mejía JJ, Butland G, Yang W, Pogoutse O, Guo X, Phanse S, Wong P et al (2009) Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol 7: e96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Schaffitzel C, Bingel‐Erlenmeyer R, Ban N, Korber P, Koning RI, de Geus DC, Plaisier JR, Abrahams JP (2009) Recycling of aborted ribosomal 50S subunit‐nascent chain‐tRNA complexes by the heat shock protein Hsp15. J Mol Biol 386: 1357–1367 [DOI] [PubMed] [Google Scholar]
- Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, Keseler IM, Krummenacker M, Midford PE, Ong Q et al (2019) The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinform 20: 1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavita K, de Mets F, Gottesman S (2018) New aspects of RNA‐based regulation by Hfq and its partner sRNAs. Curr Opin Microbiol 42: 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC (2015) Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol 13: 285–297 [DOI] [PubMed] [Google Scholar]
- Korber P, Stahl JM, Nierhaus KH, Bardwell JC (2000) Hsp15: a ribosome‐associated heat shock protein. EMBO J 19: 741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacks S, Hotchkiss RD (1960) A study of the genetic material determining an enzyme in pneumococcus. Biochim Biophys Acta 39: 508–518 [DOI] [PubMed] [Google Scholar]
- Lalaouna D, Carrier M‐C, Semsey S, Brouard J‐S, Wang J, Wade JT, Massé E (2015) A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol Cell 58: 393–405 [DOI] [PubMed] [Google Scholar]
- Lalaouna D, Baude J, Wu Z, Tomasini A, Chicher J, Marzi S, Vandenesch F, Romby P, Caldelari I, Moreau K (2019) RsaC sRNA modulates the oxidative stress response of Staphylococcus aureus during manganese starvation. Nucleic Acids Res 47: 9871–9887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberger D, Bermudez‐Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF (2009) Evidence for human microRNA‐offset RNAs in small RNA sequencing data. Bioinformatics 25: 2298–2301 [DOI] [PubMed] [Google Scholar]
- Laux A, Sexauer A, Sivaselvarajah D, Kaysen A, Brückner R (2015) Control of competence by related non‐coding csRNAs in Streptococcus pneumoniae R6. Front Genet 6: 246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécrivain A‐L, Le Rhun A, Renault TT, Ahmed‐Begrich R, Hahnke K, Charpentier E (2018) In vivo 3′‐to‐5′ exoribonuclease targetomes of Streptococcus pyogenes . Proc Natl Acad Sci USA 115: 11814–11819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnyak DV, Osipiuk J, Skarina T, Sergiev PV, Bogdanov AA, Edwards A, Savchenko A, Joachimiak A, Dontsova OA (2007) Methyltransferase that modifies guanine 966 of the 16 S rRNA: functional identification and tertiary structure. J Biol Chem 282: 5880–5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y (2011) FastCloning: a highly simplified, purification‐free, sequence‐ and ligation‐independent PCR cloning method. BMC Biotechnol 11: 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zhu L, Lau GW (2016) Disentangling competence for genetic transformation and virulence in Streptococcus pneumoniae . Curr Genet 62: 97–103 [DOI] [PubMed] [Google Scholar]
- Liu N, Abe M, Sabin LR, Hendriks G‐J, Naqvi AS, Yu Z, Cherry S, Bonini NM (2011) The exoribonuclease Nibbler controls 3′ end processing of microRNAs in Drosophila . Curr Biol 21: 1888–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Lloyd SP (1982) Least squares quantization in PCM. IEEE Trans Inf Theory 28: 129–137 [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten LJP, Hinton GE (2008) Visualizing high‐dimensional data using t‐SNE. J Mach Learn Res 9: 2579–2605 [Google Scholar]
- Mann B, van Opijnen T, Wang J, Obert C, Wang Y‐D, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI et al (2012) Control of virulence by small RNAs in Streptococcus pneumoniae . PLoS Pathog 8: e1002788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marasovic M, Zocco M, Halic M (2013) Argonaute and Triman generate dicer‐independent priRNAs and mature siRNAs to initiate heterochromatin formation. Mol Cell 52: 173–183 [DOI] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnetjournal 17: 10 [Google Scholar]
- Melamed S, Adams PP, Zhang A, Zhang H, Storz G (2020) RNA‐RNA interactomes of ProQ and Hfq reveal overlapping and competing roles. Mol Cell 77: 411–425.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang H‐Y, El‐Gebali S, Fraser MI et al (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47: D351–D360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscoso M, Claverys J‐P (2004) Release of DNA into the medium by competent Streptococcus pneumoniae: kinetics, mechanism and stability of the liberated DNA. Mol Microbiol 54: 783–794 [DOI] [PubMed] [Google Scholar]
- Muschiol S, Erlendsson S, Aschtgen M‐S, Oliveira V, Schmieder P, de Lichtenberg C, Teilum K, Boesen T, Akbey U, Henriques‐Normark B (2017) Structure of the competence pilus major pilin ComGC in Streptococcus pneumoniae . J Biol Chem 292: 14134–14146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschiol S, Aschtgen M‐S, Nannapaneni P, Henriques‐Normark B (2019) Gram‐positive type IV pili and competence. Microbiol Spectr 7: PSIB‐0011‐2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria‐Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T et al (2009) Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374: 893–902 [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Storz G (2017) ProQ/FinO‐domain proteins: another ubiquitous family of RNA matchmakers? Mol Microbiol 104: 905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Camilli A (2012) A fine scale phenotype‐genotype virulence map of a bacterial pathogen. Genome Res 22: 2541–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolenghi E, Hotchkiss RD (1962) Release of genetic transforming agent from pneumococcal cultures during growth and disintegration. J Exp Med 116: 491–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oussenko IA, Sanchez R, Bechhofer DH (2002) Bacillus subtilis YhaM, a member of a new family of 3′‐to‐5′ exonucleases in Gram‐positive bacteria. J Bacteriol 184: 6250–6259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oussenko IA, Abe T, Ujiie H, Muto A, Bechhofer DH (2005) Participation of 3′‐to‐5′ exoribonucleases in the turnover of Bacillus subtilis mRNA. J Bacteriol 187: 2758–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A, Bergstrand J, Sender V, Spelmink L, Aschtgen M‐S, Muschiol S, Widengren J, Henriques‐Normark B (2018) Factor H binding proteins protect division septa on encapsulated Streptococcus pneumoniae against complement C3b deposition and amplification. Nat Commun 9: 3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V et al (2011) Scikit‐learn: machine learning in python. J Mach Learn Res 12: 2825–2830 [Google Scholar]
- Peterson SN, Sung CK, Cline R, Desai BV, Snesrud EC, Luo P, Walling J, Li H, Mintz M, Tsegaye G et al (2004) Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol Microbiol 51: 1051–1070 [DOI] [PubMed] [Google Scholar]
- Quereda JJ, Cossart P (2017) Regulating bacterial virulence with RNA. Annu Rev Microbiol 71: 263–280 [DOI] [PubMed] [Google Scholar]
- Rajagopala SV, Sikorski P, Kumar A, Mosca R, Vlasblom J, Arnold R, Franca‐Koh J, Pakala SB, Phanse S, Ceol A et al (2014) The binary protein‐protein interaction landscape of Escherichia coli . Nat Biotechnol 32: 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Redko Y, Condon C (2010) Maturation of 23S rRNA in Bacillus subtilis in the absence of Mini‐III. J Bacteriol 192: 356–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said N, Rieder R, Hurwitz R, Deckert J, Urlaub H, Vogel J (2009) In vivo expression and purification of aptamer‐tagged small RNA regulators. Nucleic Acids Res 37: e133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadori G, Junges R, Morrison DA, Petersen FC (2019) Competence in Streptococcus pneumoniae and close commensal relatives: mechanisms and implications. Front Cell Infect Microbiol 9: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorpfeil A, Kranz M, Kovács M, Kirsch C, Gartmann J, Brunner I, Bittmann S, Brückner R (2013) Target evaluation of the non‐coding csRNAs reveals a link of the two‐component regulatory system CiaRH to competence control in Streptococcus pneumoniae R6. Mol Microbiol 89: 334–349 [DOI] [PubMed] [Google Scholar]
- Sergeeva OV, Prokhorova IV, Ordabaev Y, Tsvetkov PO, Sergiev PV, Bogdanov AA, Makarov AA, Dontsova OA (2012) Properties of small rRNA methyltransferase RsmD: mutational and kinetic study. RNA 18: 1178–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR (2011) Assembly of bacterial ribosomes. Annu Rev Biochem 80: 501–526 [DOI] [PubMed] [Google Scholar]
- Sinha D, Zimmer K, Cameron TA, Rusch DB, Winkler ME, De Lay NR (2019) Redefining the sRNA transcriptome in Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol 201: e00764‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager J, Aprianto R, Veening J‐W (2018) Deep genome annotation of the opportunistic human pathogen Streptococcus pneumoniae D39. Nucleic Acids Res 46: 9971–9989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager J, Aprianto R, Veening J‐W (2019) Refining the pneumococcal competence regulon by RNA‐sequencing. J Bacteriol 201: e00780‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A, Förstner KU, Holmqvist E, Otto A, Günster R, Becher D, Reinhardt R, Vogel J (2016) Grad‐seq guides the discovery of ProQ as a major small RNA‐binding protein. Proc Natl Acad Sci USA 113: 11591–11596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A, Schneider C, Hör J, Vogel J (2017a) Discovery of new RNA classes and global RNA‐binding proteins. Curr Opin Microbiol 39: 152–160 [DOI] [PubMed] [Google Scholar]
- Smirnov A, Wang C, Drewry LL, Vogel J (2017b) Molecular mechanism of mRNA repression in trans by a ProQ‐dependent small RNA. EMBO J 36: 1029–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son A, Park J‐E, Kim VN (2018) PARN and TOE1 constitute a 3′ end maturation module for nuclear non‐coding RNAs. Cell Rep 23: 888–898 [DOI] [PubMed] [Google Scholar]
- Storz G, Papenfort K (2019) Regulating with RNA in bacteria and archaea. Washington, DC: American Society of Microbiology; [Google Scholar]
- Suzuki S, Tanigawa O, Akanuma G, Nanamiya H, Kawamura F, Tagami K, Nomura N, Kawabata T, Sekine Y (2014) Enhanced expression of Bacillus subtilis yaaA can restore both the growth and the sporulation defects caused by mutation of rplB, encoding ribosomal protein L2. Microbiology 160: 1040–1053 [DOI] [PubMed] [Google Scholar]
- Tang W, Tu S, Lee H‐C, Weng Z, Mello CC (2016) The RNase PARN‐1 trims piRNA 3′ ends to promote transcriptome surveillance in C. elegans . Cell 164: 974–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ et al (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae . Science 293: 498–506 [DOI] [PubMed] [Google Scholar]
- Tomasini A, Moreau K, Chicher J, Geissmann T, Vandenesch F, Romby P, Marzi S, Caldelari I (2017) The RNA targetome of Staphylococcus aureus non‐coding RNA RsaA: impact on cell surface properties and defense mechanisms. Nucleic Acids Res 45: 6746–6760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber T, Treiber N, Plessmann U, Harlander S, Daiß J‐L, Eichner N, Lehmann G, Schall K, Urlaub H, Meister G (2017) A compendium of RNA‐binding proteins that regulate MicroRNA biogenesis. Mol Cell 66: 270–284.e13 [DOI] [PubMed] [Google Scholar]
- Treiber T, Treiber N, Meister G (2018) Identification of microRNA precursor‐associated proteins. Methods Mol Biol 1823: 103–114 [DOI] [PubMed] [Google Scholar]
- Trotochaud AE, Wassarman KM (2005) A highly conserved 6S RNA structure is required for regulation of transcription. Nat Struct Mol Biol 12: 313–319 [DOI] [PubMed] [Google Scholar]
- Tseng C‐K, Wang H‐F, Burns AM, Schroeder MR, Gaspari M, Baumann P (2015) Human telomerase RNA processing and quality control. Cell Rep 13: 2232–2243 [DOI] [PubMed] [Google Scholar]
- Wagner EGH, Romby P (2015) Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv Genet 90: 133–208 [DOI] [PubMed] [Google Scholar]
- Warrier I, Ram‐Mohan N, Zhu Z, Hazery A, Echlin H, Rosch J, Meyer MM, van Opijnen T (2018) The transcriptional landscape of Streptococcus pneumoniae TIGR4 reveals a complex operon architecture and abundant riboregulation critical for growth and virulence. PLoS Pathog 14: e1007461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Storz G (2000) 6S RNA regulates E. coli RNA polymerase activity. Cell 101: 613–623 [DOI] [PubMed] [Google Scholar]
- Wassarman KM (2018) 6S RNA, a global regulator of transcription. Microbiol Spectr 6: RWR‐0019‐2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JN, Bergendahl LT, Marsh JA (2016) Operon gene order is optimized for ordered protein complex assembly. Cell Rep 14: 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann AJ, Venturini E, Sellin ME, Förstner KU, Hardt W‐D, Vogel J (2019) The major RNA‐binding protein ProQ impacts virulence gene expression in Salmonella enterica serovar typhimurium. mBio 10: e02504‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton J, Acebo P, Herranz C, Gómez A, Amblar M (2015) Small regulatory RNAs in Streptococcus pneumoniae: discovery and biological functions. Front Genet 6: 126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther AR, Kjos M, Stamsås GA, Håvarstein LS, Straume D (2019) Prevention of EloR/KhpA heterodimerization by introduction of site‐specific amino acid substitutions renders the essential elongasome protein PBP2b redundant in Streptococcus pneumoniae . Sci Rep 9: 3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S, Rajagopala SV, Blazie SM, Parrish JR, Khuri S, Finley RL, Uetz P (2017) The protein interactome of Streptococcus pneumoniae and bacterial meta‐interactomes improve function predictions. mSystems 2: e00019‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J, Chen S (2012) FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7: e52249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Soares de Oliveira S, Colangeli R, Gennaro ML (1997) Binding of a novel host factor to the pT181 plasmid replication enhancer. J Bacteriol 179: 684–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JJ, Perez AJ, Tsui H‐CT, Massidda O, Winkler ME (2017) Absence of the KhpA and KhpB (JAG/EloR) RNA‐binding proteins suppresses the requirement for PBP2b by overproduction of FtsA in Streptococcus pneumoniae D39. Mol Microbiol 106: 793–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
RNA‐seq data: GEO GSE145605 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145605)
MS data: PRIDE PXD015835 and PXD015842 (http://www.ebi.ac.uk/pride/archive/projects/PXD015835 and http://www.ebi.ac.uk/pride/archive/projects/PXD015842)
Code used: Zenodo 250598 and 3475892 (https://doi.org/10.5281/zenodo.250598 and https://doi.org/10.5281/zenodo.3475892)