

Abstract
Balanced proliferation and differentiation of neural progenitor cells (NPCs) are critical for brain development, but how the process is regulated and what components of the cell division machinery is involved are not well understood. Here we report that SEPT7, a cell division regulator originally identified in Saccharomyces cerevisiae, interacts with KIF20A in the intercellular bridge of dividing NPCs and plays an essential role in maintaining the proliferative state of NPCs during cortical development. Knockdown of SEPT7 in NPCs results in displacement of KIF20A from the midbody and early neuronal differentiation. NPC-specific inducible knockout of Sept7 causes early cell cycle exit, precocious neuronal differentiation, and ventriculomegaly in the cortex, but surprisingly does not lead to noticeable cytokinesis defect. Our data uncover an interaction of SEPT7 and KIF20A during NPC divisions and demonstrate a crucial role of SEPT7 in cell fate determination. In addition, this study presents a functional approach for identifying additional cell fate regulators of the mammalian brain.
Keywords: cerebral cortical development, KIF20A, neural progenitor cells, proliferation versus differentiation, SEPT7
Introduction
Septins are a family of GTP-binding proteins that are conserved in eukaryotes from yeast to human. They assemble into nonpolar, rod-shaped hetero-oligomeric complexes that can further morph into filament-, ring-, or gauze-like polymeric structures and are thus often regarded as a distinct component of the cytoskeleton (Weirich et al. 2008; McMurray and Thorner 2009; Saarikangas and Barral 2011; Mostowy and Cossart 2012). Septins were originally discovered in mutagenesis screens for key cell division genes in budding yeast Saccharomyces cerevisiae (Hartwell 1971), which led to identification of four cell division cycle genes (cdc3, cdc10, cdc11, and cdc12) displaying mutational defects in cytokinesis. In S. cerevisiae, septins form filaments encircle the neck between mother and the emerging bud during cell division (Kim et al. 1991). The septin filaments were found to be involved in a mechanism responsible for setting up a membrane diffusion barrier across the mother-bud neck and to facilitate segregation of mother and bud proteins (Shcheprova et al. 2008). In higher eukaryotic cells, septins were reported to be also associated with functions in cytokinesis, facilitating chromosomal alignment (Spiliotis et al. 2005) or participating in formation of the cleavage furrow/midbody or regulating abscission (Estey et al. 2010; Kim et al. 2011; El Amine et al. 2013; Estey et al. 2013; Renshaw et al. 2014; Ribet et al. 2017). However, genetic knockout studies in mice showed that the cytokinesis function of mammalian septins appeared to be dependent on cellular context (Menon et al. 2014; Menon and Gaestel 2015). In addition, further studies have reported that septins are not limited to a function in cytokinesis, but are involved in a diverse range of cellular processes including phagocytosis (Huang et al. 2008; Mostowy et al. 2010; Lobato-Marquez et al. 2018), ciliogenesis (Hu et al. 2010; Kim et al. 2010), and dendrite morphogenesis (Tada et al. 2007; Xie et al. 2007; Cho et al. 2011; Hu et al. 2012; Ageta-Ishihara et al. 2013; Yadav et al. 2017). In all these processes, while the precise mechanistic roles of septins are not fully understood, the close associations of septin complexes with membranes, actins, and microtubules suggest that septins may act by providing scaffold structures for spatial organization and regulation of the signaling and/or function of cellular membrane or cytoskeletal microdomains.
During cerebral cortex development, neural progenitor cells (NPCs) initiate the formation of a functional cortical structure through consecutive cycles of NPC expansions followed by sequential steps of neurogenesis and gliogenesis (Rakic 2009; Lui et al. 2011; Greig et al. 2013; Taverna et al. 2014; Lodato and Arlotta 2015). Proper progression of these distinct developmental processes relies on a delicate control of the balance between proliferation and differentiation in NPCs. Many upstream signaling pathways regulating this aspect of neural stem/progenitor cell biology have been identified and extensively studied. These include growth factor pathways such as bFGF and EGF, and Wnt, Notch, or Sonic Hedgehog pathways. While the specific transduction cascade of each pathway is molecularly distinct, signaling commonly initiates from activation of a membrane receptor followed by activation of pathway-specific transcription factors, leading to up-regulation of certain cell fate determinant genes, which in turn regulate the decision of NPCs to adopt either a proliferative or a differentiative mode of division. However, identification of the key cell fate regulatory factors of cell division modes has lagged behind. Data obtained from studies of the Drosophila nervous systems have implicated cellular polarity-related factors as strong candidates suited for cell fate determining functions (Homem and Knoblich 2012; Homem et al. 2015). Of particular importance was mitotic spindle orientation (or cell division plane), which was thought to control symmetric or asymmetric mode of cell divisions through the cleavage plane either retaining or bypassing key fate determinants along the apical-basal membrane domains. In addition, asymmetrically distributed cellular organelles or structures were also implicated for a role in specifying asymmetric cell division. These thoughts have provided a framework for identifying cell fate regulators in the mammalian brain (Delaunay et al. 2017). However, specific molecules that carry out the cell fate functions associated with the mechanisms of mitotic spindle orientations or asymmetrically distributed organelles are not yet clear. So far, key cell fate regulators crucial for controlling proliferative versus differentiative cell divisions in the mammalian brain remain largely to be uncovered.
We have previously shown that regulator of G protein signaling (RGS) motif-mediated Ephrin-B reverse signaling pathway and the Gα signaling pathway work together to regulate the balance between proliferation and differentiation during mammalian cortical neurogenesis (Qiu et al. 2008; Murai et al. 2010; Qiu et al. 2010). The Ephrin-B/RGS pathway is essential for the maintenance of NPCs (Qiu et al. 2008; Qiu et al. 2010), while the Gα signaling pathway works to initiate neuronal differentiation (Murai et al. 2010). Subsequent investigation into additional RGS-interacting factors led to the finding that mitotic kinesin KIF20A functions with the Ephrin-B/RGS pathway to maintain the proliferative state of NPCs (Geng et al. 2018). A particularly intriguing observation from the study was that KIF20A, RGS3, Ephrin-B1, and Gα subunit, all of which participate in regulation of proliferation versus differentiation, are concentrated into the intercellular bridge (ICB) of dividing cortical NPCs during the progression of cytokinesis (Geng et al. 2018). This raised an interesting possibility that cell fate specification in mammalian NPCs occurs within the ICB during the late phase of cytokinesis in association with cell abscission. From a practical point of view, this suggested that some key cell fate regulators should reside in the ICB during the division of NPCs. In this study, we sought to identify additional cell fate regulators by virtue of their potential interaction with KIF20A as well as their presence within the ICB. This led to the identification of an interaction between SEPT7 and KIF20A. Further functional analyses via knockdown and knockout approaches revealed that inactivation of SEPT7 in cortical NPCs causes early cell cycle exit and precocious neuronal differentiation but surprisingly does not lead to obvious defects of cytokinesis, implicating a role of SEPT7 in cell fate determination during cortical neurogenesis.
Materials and Methods
Plasmids and Antibodies
Plasmids of Flag-KIF20A, GFP-SEPT7, and KIF20A deletions, all under the control of CAG promoter, were used for co-IP experiments. Plasmids of GFP-SEPT7wt, GFP-SEPT7G59V, or GFP-SEPT7E202A, all under the control of CMV promoter, were used in utero electroporation (IUE)-based rescue experiments. CAG-Cre-myc-2A-GFP (Addgene ID #116879) and Hiv7CMV-Cre-myc-2A-GFP (Addgene ID #117148) were used in IUE-based and lentivirus-based experiments, respectively. Primary antibodies included Rabbit anti-Flag (Sigma F7425; 1:2000), Mouse anti-GFP (Roche; 1:200), Rabbit anti-SEPT7 (H-120) (Santa Cruz; 1:100), Goat anti-KIF20A(L-13) (Santa Cruz; 1:100), Rabbit anti-MKLP1(N-19) (Santa Cruz; 1:100), Mouse anti-BrdU (Sigma B2531; 1:100), Mouse anti-Ki67(BD Biosciences; 1:25), and Rabbit antiactivated caspase 3 (BD Biosciences; 1:200). Additional antibodies and reagents included Rabbit anti-PAX6 (Covance; 1:500), Chicken anti-TBR2 (Abcam; 1:500), Rabbit anti-SOX5(H-90) (Santa Cruz; 1:50), Rabbit anti-NeuN (Millipore ABN78; 1:200), ConA (Molecular Probes; 1:100), Click-iT EdU Alexa Fluor 594 image kit (ThermoFisher Scientific), Duolink PLA probe antigoat minus, and Duolink PLA probe antirabbit plus (Sigma; 1:5). Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (Cy3, Cy5, and Cy2 AffiniPure conjugated; 1:200).
Yeast Two-Hybrid Screening
The two-hybrid screening was performed by ProteinLinks (Pasadena, CA). cDNA encoding KIF20A was cloned into the bait vector pCWX200, which was subsequently transformed into yeast Y304. The library screening was done by mating Y304 (MATa) with a prey strain (MATα) that expressed a mouse brain cDNA library. About 10 million independent mouse brain cDNA clones were screened on the galactose selective medium lacking leucine, histidine, tryptophan, and uracil. All 50 preliminary clones that could grow in the selective media were picked and replicated onto the four selective plates for examining expression of URA3 and LacZ reporters. To further validate the interaction of TetR-KIF20A with the putative interacting clones, plasmids encoding the putative interactors were isolated from yeast. Each interacting plasmid was retransformed into the prey yeast strain and its interaction with TetR-KIF20A and a control bait was tested. A total of seven interacting clones, which activated both reporters but did not interact with the control bait, were isolated and sequenced. Two of the seven clones encoded the cDNA sequence of SEPT7.
Coimmunoprecipitation and Western Blot
Plasmids were transfected into HEK293 cells using the calcium phosphate method. Transfected cells were sonicated in cold lysis buffer (10-mM Tris-HCl, pH 7.5, 150-mM NaCl, 1%NP40, 1-mM EDTA, 1-mM DTT and protease inhibitors). Either Flag or GFP antibody was used to bind the target protein at final concentration 1 μg/mL, and then magnetic protein G beads (20398, Pierce) were used to precipitate the antibody-protein complex. For endogenous protein co-IP, 12–15 E15.5 brains (including cortices and ganglionic eminences) were pooled in 500 μL of lysis buffer (10-mM Tris-HCl, pH 7.5, 150-mM NaCl, 1%NP40, 1-mM EDTA, 1-mM DTT and 1× protease inhibitor cocktail), triturated using a pipette with a blue tip, and sonicated for 30 s on ice. After incubation for 4 h on a rotation wheel at 4 °C, the lysate was centrifuged at 13 000 g for 15 min at 4 °C. The cleared lysate was diluted with 3 volumes of dilution buffer (10-mM Tris–HCl, pH 7.5, 150-mM NaCl, 0.2% NP40, 1-mM EDTA, 1-mM DTT, 1× protease inhibitor cocktail). One half of the diluted lysate was mixed with 5 μg of anti-KIF20A antibody and the other half was mixed with 5 μg of control IgG, and then incubated with magnetic protein G beads. The beads containing antibody-protein complex were washed and boiled in 2xSDS buffer. Denatured proteins were resolved by SDS-PAGE and transferred to a PVDF membrane for western blot detection by HRP-conjugated antibody with a chemiluminescent substrate (Pierce).
PLA Assay
PLA assay was done according to the manufacturer’s instructions. Briefly, dissociated mouse cortical cells from the E14 brains were cultured for overnight. Cells were fixed with 4% PFA, blocked with Duolink blocking solution at 37 °C for one hour, and incubated with rabbit anti-SEPT7 and goat anti-KIF20A at 4 °C for overnight. After washing the cells, Duolink PLA anti-Goat minus and anti-Rabbit plus probes were applied and incubated at 37 °C in a preheated humidity chamber for one hour. Cells were washed and Duolink PLA ligase and reagents were applied and incubated at 37 °C for 30 min. Cells were washed and Dual-link amplification reagents were applied and incubated at 37 °C for 100 min. After wash, F488 Donkey anti-Goat secondary antibody was applied and incubated at room temperature for one hour. After the final wash, cells were mounted for imaging PLA signal (Red) and KIF20A signal (green). For controls, PLA assay was performed with only one primary antibody present.
Immunohistochemistry
Brain sections were blocked for 2 h at room temperature in blocking solution (1xPBS, 0.5% Triton X-100, 10% serum of different species choice, 0.2% sodium azide), followed by incubation with primary antibody in blocking solution at 4 °C for overnight. The sections were rinsed three times with blocking solution followed by another three times rinse with 0.5% Triton X-100 in PBS. The sections were then incubated with secondary antibody in blocking solution without sodium azide at 4 °C for overnight. After rinsing three times with 0.5% Triton X-100 in PBS followed by twice with PBS, the sections were mounted with Fluoromount-G (SouthernBiotech) for imaging with a confocal microscope.
shRNA Design and Screening
Potential shRNA sequences were selected using a web-based design tool (https://rnaidesigner.thermofisher.com/rnaiexpress/design.do). shRNAs were expressed under the control of a mouse U6 promoter in pNUTS vector which additionally contains an ubiquitin promoter-EGFP expression cassette. Candidate shRNAs were first screened with targets cloned in psi-CHECK vector in transfected HEK293 cells, using a dual luciferase reporter assay (Promega, E1910). Candidate shRNAs were further verified by qPCR of shRNA-infected mouse cells (Neuro2a) containing the endogenous Sept7 transcript. The sequences of 19mer shRNAs (the loop of shRNA is underlined) used in this study were as below:
Scrambled shRNA-.
5′-CGGCTGAAACAAGAGTTGG-TTCAAGAGA-CCAACTCTTGTTTCAGCCG-3′;
shSEPT7-3UTR-.
5′-CTTGAATTGTCTAGGATAT-TTCAAGAGA-ATATCCTAGACAATTCAAG-3′;
shSEPT7-CDS-.
5′-GCTGTGGTAGGTAGTAATA-TTCAAGAGA-TATTACTACCTACCACAGC-3′.
In Utero Electroporation and Phenotype Analysis
Mice were group housed and maintained in the temperature range and environmental conditions recommended by AAALAC. Animal procedures were approved by the Institutional Animal Care and Use Committee of Beckman Research Institute of the City of Hope and were carried out in accordance with NIH guideline and the Guide for the Care and Use of Laboratory Animals. In utero electroporation was performed on E13.5 embryos, and the transfected brains were dissected out two or three days later at E15.5 or E16.5. Different plasmids (e.g., CREmyc-2A-GFP and control GFP) were pair-injected into separate horns of embryos to perform littermate analyses. Consecutive coronal sections (12 μm) of an injected brain were collected and detected by direct visualization of GFP expressed from the shRNAs or over-expression plasmid. The center sections along the anterior-posterior axis of the fluorescent region of individual brains (six or more injected brains for each DNA plasmid combination) were used for quantification with Image-Pro Premier (Media Cybernetics).
Live Cell Imaging
Dissociated cells were prepared using the E13.5 Dcx-mRFP transgenic mouse cortices by trituration in Hanks’ balanced salt solution (HBSS) (Mediatech). Cells were resuspended in Neurobasal Plus medium (Thermofisher Scientific, Cat:A3582901), supplemented with penicillin/streptomycin (Gibco), B27 Plus Supplement (Thermofisher Scientific, Cat:A3582801), N2 Supplement (Thermofisher Scientific, Cat:17502048), and 10-ng/ml bFGF2 (PeproTech, Cat: AF-100-18B), and were plated down into Poly-L-ornithine (Sigma, Cat: P4597) and Laminin (Sigma, Cat: L2020)-coated Hi-Q4 Quadruple well glass bottom plate (MZI00040, Ibidi) at 5 × 104 cells per well. Cells were infected with Lentivirus of shSEPT7-3UTR or scrambled shRNA for 24 h. Live imaging of the infected cells (GFP positive) was then taken with BioStation IM-Q (Nikon) microscopic machine at 15 min intervals for 48–72 h. Time lapse images were processed by BioStation IM (Version 2.22) and Image-Pro Premier (64-bit) software.
Analyses of Sept7 Inducible Knockout Mice
For BrdU and EdU birth dating, pregnant female mice were first administered with tamoxifen in corn oil (80-mg/kg weight body) by intraperitoneal injection at E9.5 and E10.5, and then were labeled with BrdU (100 mg/kg) and EdU (100 mg/kg) sequentially at E12.5 and E15.5, respectively. The labeled pups were delivered by C-section at E19.5 for brain analysis. Cryosections of E19.5 brains (16 μm) were analyzed for immunostaining on BrdU, EdU, and early born neuron marker SOX5.
For fluorescence activated cell sorting (FACS)-based cell cycle analysis, pieces of the E15.5 cortices were gently dissociated in cold HBSS (Mediatech) with 5-mM EDTA. After centrifugation with 300 g at 4 °C for 10 min, cells were washed twice with cold HBSS buffer. Subsequently, large cell/aggregates were removed with a cell strainer (40 μm). After fixation with 70% ethanol drop by drop for overnight at 4 °C, cells were washed in cold PBS twice. For propidium iodide (PI) staining, cells were resuspended in 0.5 mL staining buffer (PBS + 10 μg/mL PI + 0.1% Triton X-100 + 0.2 mg/mL DNAse-free RNAse A) and were incubated at 37 °C for 30 min. Cells were filtered before FACS analysis. Measurements were done using a CyAn ADP instrument (DakoCytomation) in combination with summit software.
Image Acquisition and Processing
Fluorescent images were taken with Zeiss Observer II or confocal microscope of Zeiss LSM 700 or Zeiss LSM 880.
Results
SEPT7 Interacts With KIF20A in the ICB during Cell Division
To search for KIF20A interactors, a mouse brain yeast two-hybrid cDNA library was screened using full-length KIF20A as bait. The screen yielded 50 candidate clones from over 10 million colonies, among which seven clones were confirmed for two-hybrid interaction with KIF20A in the subsequent validation experiments (Supplementary Fig. 1). Two of the seven positive clones encode mouse SEPT7. Because septins have been implicated with a role in cell division control and SEPT7 was seen to be present in the ICB of dividing cells (Estey et al. 2013; Menon and Gaestel 2015), we chose SEPT7 for further characterization. To confirm interaction between SEPT7 and KIF20A observed from two-hybrid screen, constructs encoding GFP-tagged SEPT7 and Flag-tagged KIF20A were cotransfected into HEK293 cells. The two proteins were able to coimmunoprecipitate (co-IP) from the transfected cells in reciprocal experiments (Fig. 1A,B). Further co-IP of GFP-SEPT7 with deletion mutants of KIF20A showed that the KIF20A domain responsible for interaction was located within the coiled-coil region (Fig. 1C).
Figure 1.
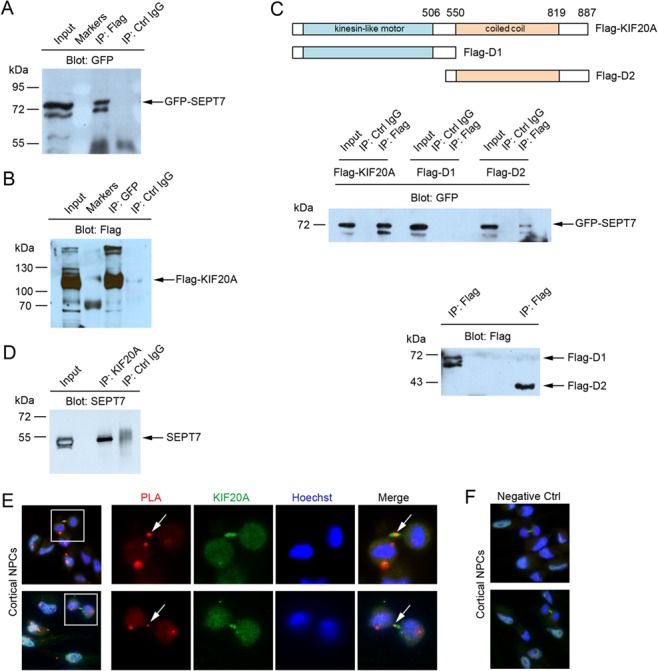
SEPT7 interacts with KIF20A. (A and B) Reciprocal coimmunoprecipitation (co-IP) of GFP-SEPT7 with Flag-KIF20A. Expression plasmids were cotransfected into Hek293 cells. Coexpressed proteins in cell lysates were precipitated for KIF20A (anti-Flag) or SEPT7 (anti-GFP) followed by western blot for the other tagged protein. (C) Two deletions of KIF20A were made and fused with Flag tag (Flag-D1 and Flag-D2). Expression plasmids of Flag-KIF20A or Flag-deletions were cotransfected with GFP-SEPT7 into Hek293 cells. In the top panel, coexpressed proteins were precipitated for KIF20A or deletions (anti-Flag) followed by western blot for GFP-SEPT7 (anti-GFP). In the bottom control panel, the above anti-Flag co-IPs were blotted by anti-Flag antibody to confirm IP of the Flag-tagged deletions. (D) Co-IP of endogenous SEPT7 and KIF20A proteins. Cell lysates were made from the E15.5 mouse brains and proteins were immunoprecipitated with anti-KIF20A antibody followed by western blot for SEPT7. (E) In dissociated dividing cortical NPCs, PLA using both SEPT7 and KIF20A antibodies could detect specific PLA events in discrete areas. Arrows in high magnification images indicate the PLA event detected in the ICB of a dividing NPC at telophase. (F) No PLA events were detected in cultured NPCs when only one antibody (KIF20A antibody, green) was present.
In the developing cerebral cortex, SEPT7 could be seen expressed in cells located across the radial domain (Supplementary Fig. 2), suggesting possible functions in both NPCs and progeny. To examine interaction between endogenous KIF20A and SEPT7, we thus made cell lysate from the E15.5 embryonic brains and performed co-IP with KIF20A antibody. The immunoprecipitation could bring down a protein band reactive to SEPT7 antibody (Fig. 1D), suggesting that the two endogenous proteins were able to interact. To further examine potential interaction between the endogenous proteins, we next performed proximity ligation assay (PLA) on dissociated embryonic cortical NPCs. When both SEPT7 and KIF20A antibodies were present, specific PLA events could be detected at the ICB of telophase NPCs (Fig. 1E). As a control, when only one of the two antibodies was present, no PLA signal was detected (Fig. 1F). These results thus collectively indicated that SEPT7 and KIF20A interact in the ICB of dividing NPCs during cytokinesis.
Knockdown of SEPT7 in Cortical NPCs Causes Early Neuronal Differentiation
The observed interaction of SEPT7 with cell fate regulator KIF20A in the ICB of dividing NPCs suggested that SEPT7 may similarly function to regulate proliferation versus differentiation in NPCs. To examine the possible function of SEPT7, we generated plasmid-based shRNAs against both the coding sequence (CDS) and the 3′-untranslated region (3UTR) of SEPT7 transcript (Supplementary Fig. 3). shRNA plasmids were delivered into the mouse cortex at E13.5 by in utero electroporation (IUE), and the transfected brains were analyzed at E15.5. In brains electroporated with shSept7-CDS or shSept7-3UTR, more transfected cells could be seen to have translocated across the radial cortical domains from the ventricular zone (VZ) into the intermediate zone (IZ) and cortical plate (CP) (Bystron et al. 2008) compared with cells expressing a scrambled control shRNA (Fig. 2A), reflecting the phenotype of early neuronal differentiation and subsequent outward migration by the daughter neurons from the VZ to IZ and CP (Qiu et al. 2008; Murai et al. 2010). Consistent with the idea of activated neuronal differentiation (rather than abnormal NPC displacement), shSept7-expressing cells that migrated out earlier into the CP were positive for neuronal marker NeuN (Fig, 2B).
Figure 2.
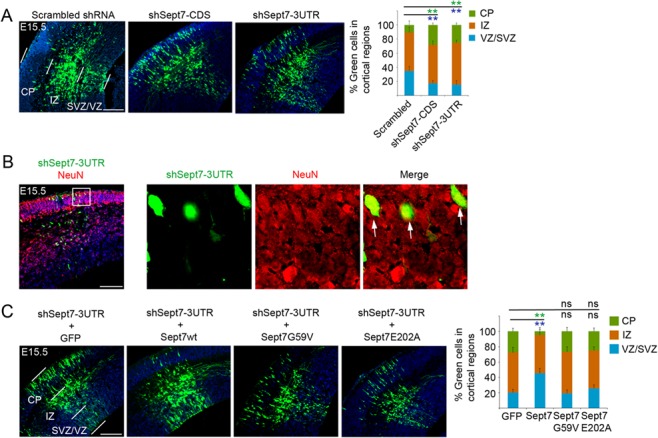
Knockdown of SEPT7 causes neuronal differentiation of NPCs. (A) shRNA-expressing plasmids were introduced into the cortex by IUE at E13.5 and the brains were analyzed at E15.5. The shRNA vector carried an ubiquitin promoter-GFP expression cassette for visualization of transfected cells. Distributions of transfected cells in different radial regions of the cortex were scored (6–9 electroporated brains for each plasmid were used for quantification). ** (in green and blue font) indicated P < 0.01 (Student’s t-test) with respect to the CP and VZ/SVZ distributions compared with control shRNA. Scale bar represents 100 μm. Error bars represent standard deviation (SD). SVZ, subventricular zone. (B) Two days after IUE, shSept7-expressing cells (GFP+ cells) in the CP were positive for NeuN, indicating that these cells were neuronal progeny. Arrows indicated examples of GFP+NeuN+ cells. Scale bar represents 100 μm. (C) shRNA-expressing plasmid and expression plasmid for GFP or different Sept7 proteins were cointroduced into the cortex by IUE at E13.5 and the brains were analyzed at E15.5. Distributions of transfected cells in different radial regions of the cortex were scored. Wild-type Sept7 protein could rescue the inhibition by shSept7-3UTR, but the two mutant Sept7 proteins did not. ** (in green and blue font) indicated P < 0.01 (Student’s t-test) with respect to the CP and VZ/SVZ distributions compared with the control. ns, not significant (P > 0.05). Scale bar represents 100 μm.
The Function of SEPT7 in NPCs is Dependent on the Ability of SEPT7 in Dimerization
The biological functions of septins are believed to rely on their ability to dimerize and assemble into filamentous polymers which further evolve into higher order structures. We therefore examined whether mutant SEPT7 proteins defective of homodimerization could rescue the effect caused by SEPT7 knockdown in NPCs. SEPT7G59V and SEPT7E202A, two SEPT7 mutants with point mutation of conserved residues important for the GTPase domain-mediated dimerization (Abbey et al. 2016), were tested. Our results showed that while wild-type SEPT7 could reverse the effect of knockdown by shSept7-3UTR, neither of the two mutant SEPT7 proteins could (Fig. 2C), suggesting that formation of SEPT7 oligomers or higher order structures of SEPT7 complex may be important for its function in NPC proliferation and differentiation control.
Knockdown of SEPT7 in Cortical NPCs Promotes Differentiative Divisions
To further examine the cellular effect of SEPT7 knockdown on NPCs, we performed live cell imaging on cortical progenitor cells in active divisions. Control shRNA or shSept7-3UTR was introduced into cortical cells derived from the Dcx-mRFP transgenic reporter mice (Wang et al. 2007), so that emerging neuronal fate of daughter cells could be monitored with mRFP expression. Our results showed that knockdown of SEPT7 expression caused more dividing progenitor cells to adopt differentiative divisions (Fig. 3; Supplementary movies).
Figure 3.
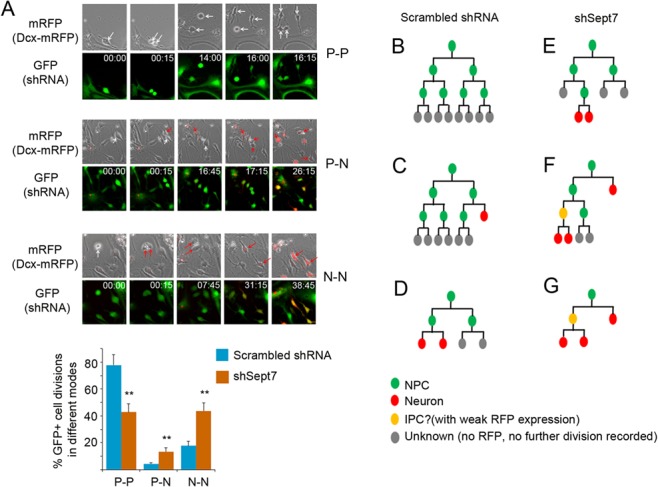
Knockdown of SEPT7 in NPCs promotes differentiative cell divisions. (A) Dissociated cells from the E13.5 cortices of the Dcx-mRFP transgenic reporter mice were infected with lentiviruses for expression of shSept7-3UTR or scrambled control shRNA. shRNA-expressing (GFP positive) progenitor cells were imaged in live culture at intervals of 15 min for 48–72 h. Phase contrast and fluorescent images at indicated time were shown. Appearance of RFP expression in some daughter cells reflected the emerging neuronal fate in these nascent cells. Representative images of dividing cells in different division modes are shown (images are snap shots from supplementary movies). White arrows indicate mother and daughter progenitor cells. Red arrows indicate daughter cells taking a neuronal fate. Progenitor-progenitor (P-P), progenitor-neuron (P-N), or terminal neuron-neuron (N-N) division modes were scored. **P < 0.01 (Student’s t-test). Error bars represent SD. (B–G) Some representative lineage trees reconstructed from NPCs expressing scrambled control (B–D) shRNA or (E–G) shSept7-3UTR. IPC, intermediate progenitor cell.
Electroporation-Mediated Somatic Knockout of Sept7 Causes Displacement of KIF20A from the Midbody and Neuronal Differentiation
We next used IUE-mediated somatic knockout of the Sept7 gene to further examine the function of SEPT7 in cell fate regulation. For this purpose, an expression plasmid of Cre-2A-GFP was electroporated into the cortices of the conditional Sept7 knockout mice carrying floxed alleles (Sept7fl/fl) at E13.5. The transfected brains were then collected at E16.5 for analyses. Our previous data showed that IUE-based expression of Cre-2A-GFP could lead to Cre-mediated recombination in the majority of transfected cortical cells (Geng et al. 2018). In the Sept7fl/fl cortices, Cre expression caused more cortical cells to translocate from the VZ into the IZ and CP than cells expressing the control GFP alone (Fig. 4A), reflecting early differentiation and subsequent outward migration of the transfected cells. Consistent with the idea of activated neuronal differentiation due to Sept7 knockout, Cre expressing cells in the Sept7fl/fl cortices that migrated out earlier into the CP were positive for neuronal marker NeuN (Fig. 4B). We next further examined subcellular localization of KIF20A in Sept7 knockout cells. For this purpose, control GFP or Cre-2A-GFP was introduced into dissociated cortical NPCs using lentivirus, and cells were then cultured for two days. Among the infected cells, significantly fewer dividing NPCs expressing Cre-2A-GFP showed midbody-localized KIF20A protein (Fig. 4C), suggesting that SEPT7 is important for normal organization of KIF20A in the ICB.
Figure 4.
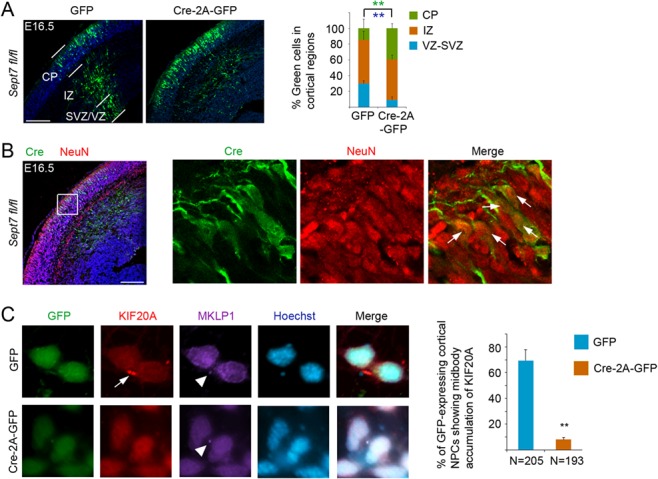
IUE-mediated knockout of SEPT7 causes neuronal differentiation of NPCs. (A) Cre expressing or control GFP-expressing plasmid was introduced into the cortex of Sept7fl/fl embryos by IUE at E13.5, and the brains were analyzed at E16.5. Distributions of transfected cells in different radial regions of the cortex were scored. Scale bar represents 100 μm. ** (in green and blue font) indicated P < 0.01 (Student’s t-test) with respect to the CP and VZ/SVZ distributions compared with control GFP. Error bars represent SD. (B) In the cortices of Sept7fl/fl conditional knockout mice, Cre expressing cells (GFP+ cells) that have migrated into the CP were positive for NeuN, suggesting early differentiation into neurons due to Sept7 knockout. Arrows indicated examples of GFP+NeuN+ cells. Scale bar represents 100 μm. (C) Dissociated cells from the E13.5 cortices of Sept7fl/fl mice were infected with lentiviruses for expression of GFP or Cre-2A-GFP. After culturing for two days post infection, cells were fixed and costained for KIF20A and KIF23/MKLP1. Percentage of telophase NPCs showing midbody (with noticeable MKLP1 staining) accumulation of KIF20A was scored. **P < 0.01 (Student’s t-test). Error bars represent SD.
NPC-Specific Knockout of Sept7 Leads to Cortical Neurogenesis Defect without Causing Obvious Abnormality in Cytokinesis
We next used NPC-specific inducible deletion (via Nestin-CreERT2 transgene allele) of the floxed Sept7 gene to further examine the function of SEPT7 in cortical neurogenesis. Tamoxifen was administered to pregnant mice at gestation days of E9.5 and E10.5 consecutively. Littermates of homozygous brains (Nestin-CreERT2; Sept7fl/fl) and wild-type brains (Sept7fl/fl) were collected at E12.5 or E15.5 for analysis. At E15.5, mutant brains displayed a reduction in overall size and cortical thickness with an enlarged ventricle compared with the wild-type littermate brains (Fig. 5A,B). Examination of NPC and neuron populations showed an apparent reduction (thinning of the marker-labeled radial regions) of radial glial cells (RGC; PAX6+) and intermediate progenitor cells (IPC; TBR2+), as well as neurons (βIII-Tubulin+) in the mutant brains (Nestin-CreERT2; Sept7fl/fl) (Fig. 5C), although a precise quantitative comparison between the mutant and wild-type brains was hard to achieve due to the dilation of mutant ventricles. Because SEPT7 has been implicated in cell division control, we asked whether the loss of NPCs and neurons observed in the Sept7 mutants was due to a possible cytokinesis defect. Using fluorescently labeled Concanavalin A (ConA) to help demarcate membrane peripherals of individual cells and nucleic acid stain propidium iodide (PI) to mark nuclei, we first examined whether there might be an increase of bi- or multinucleated cells in Sept7 mutant brains, an indication of defect in cytokinesis. We found no obvious signs of binucleated cells in the proliferating zone of the mutant brains either at the NPC expansion stage (E12.5) or peak neurogenesis stage (E15.5) (Fig. 5D). We next used FACS-based analysis to more quantitatively assess cell cycle dynamics in the mutant brains of the Sept7 inducible knockout mice. Tamoxifen was administered to pregnant mice at gestation days of E9.5 and E10.5 consecutively, and cortical cells were prepared from littermate brains of homozygous mutants (Nestin-CreERT2; Sept7fl/fl) and wild-types (Sept7fl/fl) collected at E15.5, fixed and stained with PI. FACS-based analysis showed that there was no difference in DNA content between the mutant (Nestin-CreERT2; Sept7fl/fl) and wild-type (Sept7fl/fl) cortical cells at different phases of cell cycle (Fig. 5E), indicating that loss-of-function (LOF) of SEPT7 did not cause noticeable changes in cytokinesis of NPC divisions, or a possible cytokinesis defect was largely compensated by other factors. We also examined the cell apoptosis status by staining of activated caspase 3. In both the wild-type and Sept7 knockout cortices, the overall apoptosis levels were low at the two neurogenesis stages examined (Supplementary Fig. 4). Apoptotic cell numbers in the mutant cortices showed an increase at E12.5 but were comparable to wild-type littermates at E15.5 (Supplementary Fig. 4). These results collectively suggested that a possible defect in cytokinesis or activated cell death was not a major cause of the overall growth retardation observed in the Sept7 mutant brains.
Figure 5.
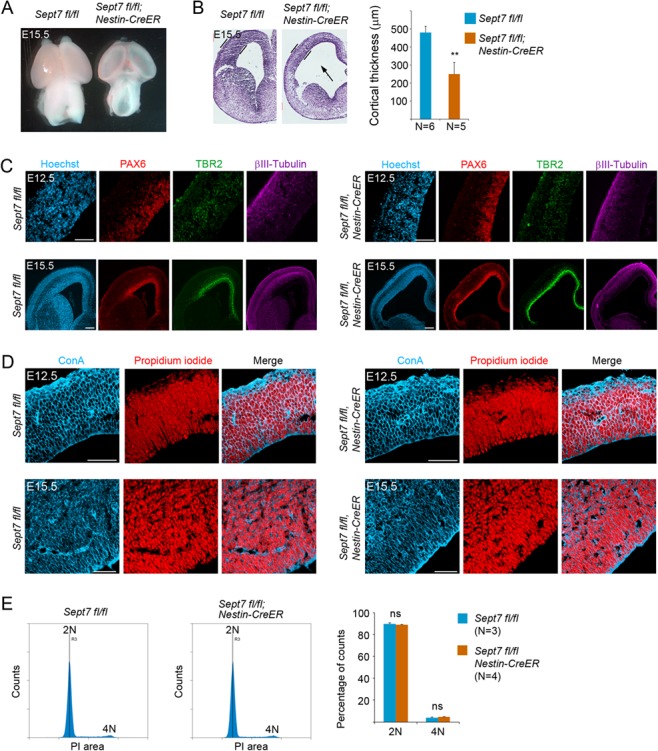
Inducible knockout of Sept7 in NPCs causes growth retardation of the brain without affecting cytokinesis. (A) Tamoxifen was administered to pregnant mother at E9.5 and E10.5 consecutively, and embryonic brains were collected at E15.5 for analyses. At this stage of development, Nestin-CreERT2; Sept7fl/fl homozygous mutant brains showed smaller size and more transparent cortical hemispheres than the control littermate brains. (B) Nissl staining of brain sections revealed thinner cortices of the Nestin-CreERT2; Sept7fl/fl homozygous mutant brains at E15.5. The arrow indicates enlarged ventricle in the mutant brains. **P < 0.01 (Student’s t-test). (C) Cellular marker staining revealed an overall reduction in the thickness of cell ribbons representing radial glial cells (PAX6+), intermediate progenitor cells (TBR2+), and neurons (βIII-Tubulin+) in the cortices of Sept7 inducible knockout mice compared with the wild-type littermate brains. Scale bars represent 100 μm. (D) No obvious bi- or multinucleated cells were seen in the wild-type or Sept7 inducible knockout mutant cortices at E12.5 or E15.5. ConA (blue) and PI (red) were used for visualizing cell peripherals and nuclei, respectively. Scale bars represent 100 μm. (E) Cell cycle analysis of wild-type and mutant cortical cells. Tamoxifen was administered to Sept7 inducible knockout mice at E9.5 and E10.5 consecutively. Littermates of Nestin-CreERT2; Sept7fl/fl and control Sept7fl/fl embryos were collected at E15.5. Cortical cells were examined for their DNA contents by FACS analysis. 2 N, cells in G1/S phase; 4 N, cells in G2/M phase or binucleated cells; ns, not significant (P > 0.05).
NPC-Specific Knockout of Sept7 Causes Early Cell Cycle Exit and Precocious Neuronal Differentiation
We next examined whether cell cycle exit and reentry of cortical NPCs were altered in the Sept7 mutant brains. In this experiment, tamoxifen was administered to pregnant mice at gestation days of E9.5 and E10.5 consecutively followed by injection of 5-Ethynyl-2′-deoxyuridine (EdU) at E14.5. The brains of littermate embryos were collected for analyses 24 h later. Among the three ribbons of EdU+ cells which reflected temporal waves of cell cycle exit/differentiation (Geng et al. 2018), noticeably more cells were present in the ribbons of R1 and R2 than in R3 of the mutant cortices (Nestin-CreERT2; Sept7fl/fl) when compared with the wild-type littermate cortices (Sept7fl/fl) (Fig. 6A). Accumulations of EdU+ cells into the R1 and R2 ribbons of the mutant cortices is an indication that more labeled cells have differentiated than those in the wild-type cortices at this stage. Cell cycle exit index analysis showed that there was a higher portion of EdU+Ki67− cells (cells exited the cell cycle) in the EdU+ population of the mutant cortices than that of the wild-type littermate cortices (Fig. 6A), indicating that there was early cell cycle exit in mutant NPCs. Further examination of cell marker expression showed that EdU+Ki67− cells that had migrated towards the CP were positive for neuronal marker NeuN (Fig. 6B). These results suggested that LOF of SEPT7 in NPCs led to cell cycle exit and early differentiation into neurons.
Figure 6.
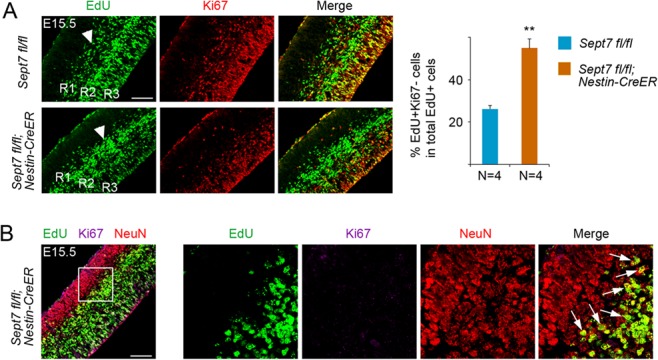
Inducible knockout of Sept7 in NPCs causes early cell cycle exit and precocious neuronal differentiation. (A) Tamoxifen was administered to Sept7 inducible knockout mice at E9.5 and E10.5 consecutively. EdU was labeled at E14.5 and embryonic brains were collected 24 h afterwards. R1, R2, and R3 indicated the EdU-labeled cell ribbons located progressively from superficial regions to the apical surface. EdU+Ki67− cells were progenitor cells that had left the cell cycle. More EdU+Ki67− cells in the total population of EdU+ cells were present in the cortices of the homozygous mutant cortices. Scale bar represents 100 μm. **P < 0.01 (Student’s t-test). Error bars represent SD. (B) EdU+Ki67− cells that had migrated into the CP in the Nestin-CreERT2; Sept7fl/fl homozygous mutant cortices were positive for NeuN, indicating that these cells had become neurons. Arrows indicated examples of EdU+Ki67−NeuN+ cells. Scale bar represents 100 μm.
We further examined cell cycle exit and reentry in temporal progression of early-born and late-born neuronal productions during cortical morphogenesis. Tamoxifen was administered to pregnant mice at gestation days of E9.5 and E10.5 consecutively, and the littermate brains were collected for analyses around birth (E19.5) by Cesarean section delivery of pups. At E19.5, the mutant brains (Nestin-CreERT2; Sept7fl/fl) appeared to show elongated cortical hemispheres, compared with their control wild-type littermate brains (Sept7fl/fl) (Fig. 7A). Sections of these brain samples revealed that mutant brains had thinner cortices and wider ventricles than wild-type brains (Fig 7B), a phenotype similarly seen in the Kif20a knockouts (Geng et al. 2018). Staining of SOX5 (early-born neuron marker) and CUX1 (late-born neuron marker) showed that cortical lamination was not compromised in the mutant brains (Fig. 7C). This was also supported by sequential 5-Bromo-2′-deoxyuridine (BrdU) and EdU labeling of early-born and late-born neurons (Fig. 7D). Costaining of BrdU and EdU showed that BrdU+EdU− cells (early-born neurons) and BrdU+EdU+ cells (late-born neurons) in both mutant and wild-type brains were located in respective deep and upper cortical radial domain (Fig. 7D). Furthermore, within the population of BrdU-labeled cells, there were relatively more BrdU+EdU− cells in the deep layer region of the mutant brains than in the wild-type brains (Fig. 7D,E), suggesting that more NPCs in the mutant brains had left cell cycle and become differentiated after the initial labeling of BrdU. Consistent with this idea, many of these BrdU+EdU− postmitotic cells were positive for early-born neuron marker SOX5 (Fig. 7F), indicating precocious neuronal differentiation by these BrdU+ mutant NPCs. These data collectively suggested that LOF of SEPT7 could tip the balance between proliferation and differentiation in NPCs toward differentiation, leading to an early depletion of NPCs and resulting in a severe impairment in neuronal production and morphogenesis of the cortex.
Figure 7.
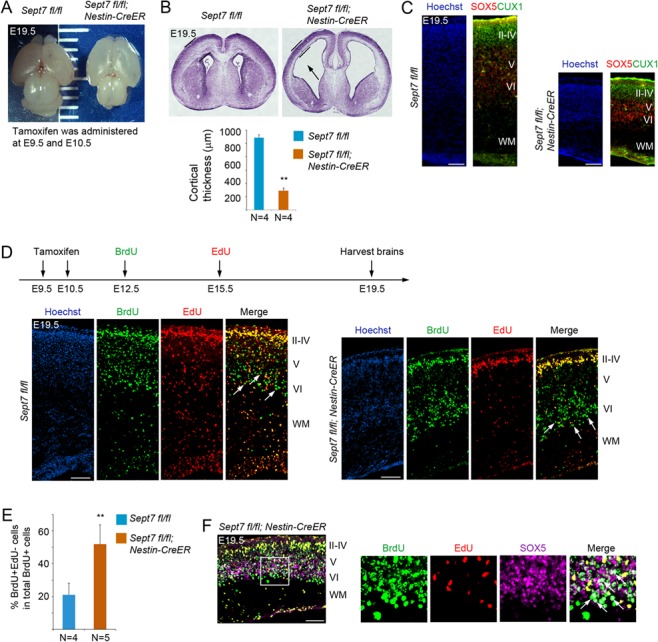
Inducible knockout of Sept7 in NPCs causes ventriculomegaly and defect in neuronal production. (A) Tamoxifen was administered to Sept7 inducible knockout mice at E9.5 and E10.5 consecutively. Pups were delivered by C-section at E19.5. Inducible knockout of Sept7 yielded brains with elongated cortical hemispheres. (B) Sections of Nestin-CreERT2; Sept7fl/fl homozygous mutant brains revealed thinner cortices and dilated ventricles (black arrow). **P < 0.01 (Student’s t-test). Error bars represent SD. (C) Cellular marker staining showed that early-born neurons (SOX5+) and late-born neurons (CUX1+) were correctly located in the respective deep and upper layers in the cortices of Sept7 knockout mice, suggesting that LOF of SEPT7 did not affect cortical lamination. Scale bars represent 100 μm. (D) Tamoxifen was administered to Sept7 inducible knockout mice at E9.5 and E10.5 consecutively. BrdU and EdU were sequentially administered at E12.5 and E15.5, respectively. In Sept7 knockout mice, BrdU+EdU− cells (early-born neurons) and BrdU+EdU+ cells (late-born neurons) were properly located in the respective deep and upper layers. Relatively more BrdU+EdU− cells within the BrdU+ cell population could be seen in the deep layers of Sept7 mutant brains compared with wild-type littermates, reflecting early cell cycle exit due to LOF of SEPT7. Arrows indicate examples of BrdU+EdU− cells. Scale bars represent 100 μm. (E) Quantification of panel D. **P < 0.01 (Student’s t-test). Error bars represent SD. (F) Many BrdU+EdU− cells in the deep layers (boxed area) of Sept7 mutant cortices were positive for SOX5, indicating that these cells have exited cell cycle and differentiated into neurons (early-born neurons). Arrows indicated examples of BrdU+EdU−SOX5+ cells. Scale bar represents 100 μm.
Discussion
Septins were discovered more than 45 years ago in mutants defective in cytokinesis in budding yeast S. cerevisiae. So far, the roles of septins in cell divisions have been best characterized in S. cerevisiae and implicated in processes of bud-site selection, mitotic spindle positioning, polarized growth, and cytokinesis. In budding yeast, septins concentrate at the bud neck during mitosis and form hourglass-shaped collar-like structures separating mother and bud membranes. The septin structures are involved in setting up diffusion barriers at the bud neck restricting protein diffusions between the mother and bud membranes, thereby establishing distinct mother and bud membrane compartments (Shcheprova et al. 2008). In C. elegans and Drosophila melanogaster, septins were reported to be involved in furrow ingression during cytokinesis as well as in cell migration or axon guidance. In mammalian cells, initial experiments suggested that LOF of septins was linked to defect in chromosome reorganization or delay in cytokinesis (Kinoshita et al. 1997; Spiliotis et al. 2005). However, genetic knockout studies of SEPT7 revealed that SEPT7 was indispensable for cytokinesis in some cells (e.g., fibroblasts), but expendable in other cells (e.g., hematopoietic cells) (Menon et al. 2014; Menon and Gaestel 2015). Since SEPT7 is the pivotal subunit constituting the septin oligomeric structures, these genetic data indicated that mitotic septins may have additional functions not directly linked to cytokinesis per se. Here, prompted by the observed interaction between SEPT7 and KIF20A, we hypothesized that similar to KIF20A, SEPT7 might also function in cell fate determination during the divisions of NPCs. Using IUE-mediated Cre expression in the mouse brains carrying the floxed Sept7 gene, we found that somatic knockout of Sept7 in cortical NPCs caused early neuronal differentiation, evident by the noticeable shift of transfected cells translocating from the VZ into the IZ and CP in the cortex (Fig. 3A). The fact that these SEPT7 knockout cells could complete cell divisions to generate new neurons which subsequently migrated normally outwards into the IZ and CP was an indication that LOF of SEPT7 did not compromise cytokinesis per se but cause a shift in NPCs from proliferative to differentiative divisions. Further NPC-specific inducible knockout of Sept7 in the cortex confirmed that LOF of SEPT7 did not cause noticeable defect in cytokinesis, suggested by the lack of obvious appearance of binucleated cells in the mutant cortices and by the comparable cell cycle dynamics (FACS profiles) between the mutant and wild-type littermate cortical cells. We saw a transient increase of apoptosis at the early stage of cortical neurogenesis (E12.5), but this was apparently rescued perhaps by some compensatory effects at a later stage (E15.5). While the possible mechanism was not clear, given that SEPT7 was known to play a role in cell divisions and neurons, this early increase of cell death might have occurred in either progenitors or neurons and contributed to the overall loss of progenitor cells and neurons in the Sept7 knockout brains. Additional data of cell cycle exit and reentry analyses obtained from both E15.5 and E19.5 brains of the inducible knockout mice further showed that LOF of SEPT7 resulted in a shift in NPCs from proliferation to differentiation, which appeared to be the major contributing factor to the early depletion of progenitor cells. Collectively, our data revealed a crucial role of SEPT7 in controlling the balance between proliferative and differentiative divisions of NPCs. Whether this function of SEPT7 is shared by other stem/progenitor cell systems will be an interesting question to address in future studies.
How SEPT7 acts to regulate cell fate specification in NPCs is not clear, but it might be involved in regulating the membrane or microtubule organization of KIF20A, RGS3, and other associated cargos/complexes. Based on the budding yeast models (Weirich et al. 2008; McMurray and Thorner 2009; Mostowy and Cossart 2012; Spiliotis and Gladfelter 2012; Fung et al. 2014; Marquardt et al. 2018), it is conceivable that SEPT7 (or more precisely an oligomeric septin complex containing SEPT7) might provide scaffolds of membrane microdomains to recruit and organize proteins or establish barriers to restrict diffusion of proteins across cellular membranes. Such a membrane modeling/remodeling function of SEPT7 oligomeric complex within the ICB of dividing NPCs might be critical for membrane organization of KIF20A-RGS3 associated cargos/complexes via SEPT7-KIF20A interaction. We speculate that during the course of cytokinesis, particularly at the stage of cell abscission, the function of SEPT7 oligomeric complex might have an influence on segregation or distribution of KIF20A-RGS3 associated proliferative fate-promoting regulators, resulting in either symmetric or asymmetric inheritance of the cell fate regulators in the two nascent daughter cells. When SEPT7 is inactivated, complexes of these cell fate determinants might not be appropriately organized on the membranes of ICB to ensure proper passage into daughter cells, thereby leading them to take a differentiative fate. During cortical neurogenesis, RGCs produce most, if not all, projection neurons through IPCs (Liu et al. 2016), which are committed to becoming neurons. Thus, while it is not clear whether the KIF20A-SEPT7 interaction works in both RGC divisions and IPC divisions, we suspect that cell fate regulatory function of KIF20A and SEPT7 may occur in the RGC divisions. How KIF20A and SEPT7 are distributed in ICB during and after cell division will be essential information for better understanding the precise mechanisms of their functions in the cell fate determination process. Live cell imaging of fluorescently tagged KIF20A or SEPT7 during NPC divisions will be helpful in this regard; however, due to cytotoxicity induced by exogenously expressed KIF20A or SEPT7, we were unable to image transfected NPCs. Future experiment will rely on knock-in of endogenously expressed fluorescent reporter-fused proteins in cortical NPCs. Septins are biochemically very intriguing proteins and they can transform from a linear oligomeric complex into various different structures, which might favor or even dictate different curvature or rigidity of cellular membranes (Cannon et al. 2017; Kang and Lew 2017; Spiliotis 2018). While the specific function of SEPT7 in cell fate regulation is speculative, it is conceivable that the transitions of a SEPT7 complex between oligomeric form and different high order structures might be quite important in cell fate determination. In this regard, it is interesting to note that two SEPT7 mutant proteins defective of forming dimers (Abbey et al. 2016) could not rescue the knockdown effect caused by a SEPT7-targeting shRNA (Fig. 2C), indicating that compositions of septin oligomers or distinct higher order structures associated with SEPT7 complex are likely crucial for cell fate determination. A deeper understanding of the relationships between the biochemical properties of SEPT7 and the various forms of septin structures in the function of membrane organization would provide insight into the molecular mechanisms controlling the balance between proliferation and differentiation in NPCs.
In the past two decades, search for cell fate determinants (regulators that can have an impact on the mode of cell division in stem/progenitor cells) has focused primarily on cellular polarity-related factors, which were implicated for a role in regulation of cell division mode from studies of invertebrate nervous system models. However, genetic studies of mouse homologs of multiple polarity-related genes could not substantiate a similar function in the mouse brains, suggesting that preexisting polarity in NPCs may not be required for fate determination between two nascent daughter cells. Our findings on the essential roles of SEPT7 (this study) and KIF20A (Geng et al. 2018) in maintaining the proliferative divisions of NPCs collectively suggest that key regulators of cell division mode likely reside in the ICB, and their membrane organization within the ICB and stochastic distribution into nascent daughter cells during cell abscission could have a major impact on the fate of the daughter cells. Based on this thought, we suggest the following approach for facilitating continued search for additional cell fate regulators that work during the course of cell divisions. The approach includes three components. First, the proteins of interest would ideally have a presence in the ICB during NPC divisions. Those proteins associated with the midbody structure of the ICB would be particularly good candidates for future studies. Second, IUE-mediated cortical expression of Cre enzyme (vs. control GFP) in homozygous mouse embryos carrying the floxed candidate gene alleles should cause an upshift of cell distribution from the VZ into IZ and CP, as we have observed in the cases of Sept7fl/fl (Fig. 4A) and Kif20afl/fl mice (Geng et al. 2018). This IUE-based assay can provide a quick and reliable initial assessment of the postulated role of the target gene in proliferation versus differentiation. If the results are positive, then finally, NPC-specific inducible deletion of the candidate gene can be employed to further validate the role. We anticipate this approach be straightforward to perform, since many floxed lines of mutant mice have become available from knockout mice projects carried out by research consortiums or commercial entities. More importantly, this approach focuses on functional/phenotypic characterization of candidate genes, which can avoid the limitation of defining the mode of divisions by relying mainly on symmetric or asymmetric distribution of polarity markers in mitotic NPCs but without direct knowledge of the identity of progeny cells. In summary, this study identified an interaction between SEPT7 and KIF20A in the ICB of dividing NPCs and demonstrated an essential role of SEPT7 in the maintenance of NPCs during development. We anticipate that future identifications of additional ICB proteins with a cell fate function would help piece together a picture of how cell fate determination (proliferation versus differentiation) is regulated during mammalian brain development.
Supplementary Material
Author Contributions
R.Q., A.G., and J.L. conducted the experiments. C.W.X. (previously at ProteinLinks, Inc.) performed yeast two-hybrid screen with KIF20A as a bait. M.B.M. and M.G. provided conditional Sept7 knockout mice, expression plasmids of mutant SEPT7 proteins, and insightful discussions on mechanism and function of SEPT7. R.Q., A.G., and Q.L. analyzed data and wrote the manuscript.
Funding
National Institutes of Health (grant NS096130 to Q.L.); National Cancer Institute (award P30CA033572).
Notes
We thank Jeremy LaDou and animal center staff for assistance with animal breeding and care; Lucy Brown and Jeremy Stark and their staff for helping with cell sorting; Brian Armstrong and staff for the use of imaging facility; and Dr Ryoichiro Kageyama for providing Nestin-CreERT2 mice. Conflict of Interest: None declared.
References
- Abbey M, Hakim C, Anand R, Lafera J, Schambach A, Kispert A, Taft MH, Kaever V, Kotlyarov A, Gaestel M, et al. 2016. GTPase domain driven dimerization of SEPT7 is dispensable for the critical role of septins in fibroblast cytokinesis. Sci Rep. 6:20007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageta-Ishihara N, Miyata T, Ohshima C, Watanabe M, Sato Y, Hamamura Y, Higashiyama T, Mazitschek R, Bito H, Kinoshita M. 2013. Septins promote dendrite and axon development by negatively regulating microtubule stability via HDAC6-mediated deacetylation. Nat Commun. 4:2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystron I, Blakemore C, Rakic P. 2008. Development of the human cerebral cortex: boulder committee revisited. Nat Rev Neurosci. 9:110–122. [DOI] [PubMed] [Google Scholar]
- Cannon KS, Woods BL, Gladfelter AS. 2017. The unsolved problem of how cells sense micron-scale curvature. Trends Biochem Sci. 42:961–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SJ, Lee H, Dutta S, Song J, Walikonis R, Moon IS. 2011. Septin 6 regulates the cytoarchitecture of neurons through localization at dendritic branch points and bases of protrusions. Mol Cells. 32:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay D, Kawaguchi A, Dehay C, Matsuzaki F. 2017. Division modes and physical asymmetry in cerebral cortex progenitors. Curr Opin Neurobiol. 42:75–83. [DOI] [PubMed] [Google Scholar]
- El Amine N, Kechad A, Jananji S, Hickson GR. 2013. Opposing actions of septins and sticky on anillin promote the transition from contractile to midbody ring. J Cell Biol. 203:487–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estey MP, Di Ciano-Oliveira C, Froese CD, Bejide MT, Trimble WS. 2010. Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. J Cell Biol. 191:741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estey MP, Di Ciano-Oliveira C, Froese CD, Fung KY, Steels JD, Litchfield DW, Trimble WS. 2013. Mitotic regulation of SEPT9 protein by cyclin-dependent kinase 1 (Cdk1) and Pin1 protein is important for the completion of cytokinesis. J Biol Chem. 288:30075–30086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung KY, Dai L, Trimble WS. 2014. Cell and molecular biology of septins. Int Rev Cell Mol Biol. 310:289–339. [DOI] [PubMed] [Google Scholar]
- Geng A, Qiu R, Murai K, Liu J, Wu X, Zhang H, Farhoodi H, Duong N, Jiang M, Yee JK, et al. 2018. KIF20A/MKLP2 regulates the division modes of neural progenitor cells during cortical development. Nat Commun. 9:2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. 2013. Molecular logic of neocortical projection neuron specification, development and diversity. Nature Reviews. 14:755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH. 1971. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 69:265–276. [DOI] [PubMed] [Google Scholar]
- Homem CC, Knoblich JA. 2012. Drosophila neuroblasts: a model for stem cell biology. Development. 139:4297–4310. [DOI] [PubMed] [Google Scholar]
- Homem CC, Repic M, Knoblich JA. 2015. Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci. 16:647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Bai X, Bowen JR, Dolat L, Korobova F, Yu W, Baas PW, Svitkina T, Gallo G, Spiliotis ET. 2012. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Curr Biol. 22:1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. 2010. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YW, Yan M, Collins RF, Diciccio JE, Grinstein S, Trimble WS. 2008. Mammalian septins are required for phagosome formation. Mol Biol Cell. 19:1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Lew DJ. 2017. How do cells know what shape they are? Curr Genet. 63:75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. 1991. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3 gene product and the timing of events at the budding site. J Cell Biol. 112:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Froese CD, Estey MP, Trimble WS. 2011. SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. J Cell Biol. 195:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, et al. 2010. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 329:1337–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Kumar S, Mizoguchi A, Ide C, Kinoshita A, Haraguchi T, Hiraoka Y, Noda M. 1997. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev. 11:1535–1547. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu X, Zhang H, Qiu R, Yoshikawa K, Lu Q. 2016. Prospective separation and transcriptome analyses of cortical projection neurons and interneurons based on lineage tracing by Tbr2 (Eomes)-GFP/dcx-mRFP reporters. Dev Neurobiol. 76:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato-Marquez D, Krokowski S, Sirianni A, Larrouy-Maumus G, Mostowy S. 2018. A requirement for septins and the autophagy receptor p62 in the proliferation of intracellular Shigella. Cytoskeleton. 76(1):163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, Arlotta P. 2015. Generating neuronal diversity in the mammalian cerebral cortex. Annu Rev Cell Dev Biol. 31:699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, Kriegstein AR. 2011. Development and evolution of the human neocortex. Cell. 146:18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt J, Chen X, Bi E. 2018. Architecture, remodeling, and functions of the septin cytoskeleton. Cytoskeleton. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. 2009. Septins: molecular partitioning and the generation of cellular asymmetry. Cell Div. 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon MB, Gaestel M. 2015. Sep(t) arate or not - how some cells take septin-independent routes through cytokinesis. J Cell Sci. 128:1877–1886. [DOI] [PubMed] [Google Scholar]
- Menon MB, Sawada A, Chaturvedi A, Mishra P, Schuster-Gossler K, Galla M, Schambach A, Gossler A, Forster R, Heuser M, et al. 2014. Genetic deletion of SEPT7 reveals a cell type-specific role of septins in microtubule destabilization for the completion of cytokinesis. PLoS Genet. 10:e1004558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy S, Bonazzi M, Hamon MA, Tham TN, Mallet A, Lelek M, Gouin E, Demangel C, Brosch R, Zimmer C, et al. 2010. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe. 8:433–444. [DOI] [PubMed] [Google Scholar]
- Mostowy S, Cossart P. 2012. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. 13:183–194. [DOI] [PubMed] [Google Scholar]
- Murai K, Qiu R, Zhang H, Wang J, Wu C, Neubig RR, Lu Q. 2010. Galpha subunit coordinates with ephrin-B to balance self-renewal and differentiation in neural progenitor cells. Stem Cells. 28:1581–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu R, Wang J, Tsark W, Lu Q. 2010. Essential role of PDZ-RGS3 in the maintenance of neural progenitor cells. Stem Cells. 28:1602–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu R, Wang X, Davy A, Wu C, Murai K, Zhang H, Flanagan JG, Soriano P, Lu Q. 2008. Regulation of neural progenitor cell state by ephrin-B. J Cell Biol. 181:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. 2009. Evolution of the neocortex: a perspective from developmental biology. Nature Reviews. 10:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw MJ, Liu J, Lavoie BD, Wilde A. 2014. Anillin-dependent organization of septin filaments promotes intercellular bridge elongation and Chmp4B targeting to the abscission site. Open Biol. 4:130190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribet D, Boscaini S, Cauvin C, Siguier M, Mostowy S, Echard A, Cossart P. 2017. SUMOylation of human septins is critical for septin filament bundling and cytokinesis. J Cell Biol. 216:4041–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikangas J, Barral Y. 2011. The emerging functions of septins in metazoans. EMBO Rep. 12:1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheprova Z, Baldi S, Frei SB, Gonnet G, Barral Y. 2008. A mechanism for asymmetric segregation of age during yeast budding. Nature. 454:728–734. [DOI] [PubMed] [Google Scholar]
- Spiliotis ET. 2018. Spatial effects - site-specific regulation of actin and microtubule organization by septin GTPases. J Cell Sci. 131(1). pii: jcs207555. doi: 10.1242/jcs.207555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotis ET, Gladfelter AS. 2012. Spatial guidance of cell asymmetry: septin GTPases show the way. Traffic. 13:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotis ET, Kinoshita M, Nelson WJ. 2005. A mitotic septin scaffold required for mammalian chromosome congression and segregation. Science. 307:1781–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Simonetta A, Batterton M, Kinoshita M, Edbauer D, Sheng M. 2007. Role of Septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr Biol. 17:1752–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E, Gotz M, Huttner WB. 2014. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 30:465–502. [DOI] [PubMed] [Google Scholar]
- Wang X, Qiu R, Tsark W, Lu Q. 2007. Rapid promoter analysis in developing mouse brain and genetic labeling of young neurons by doublecortin-DsRed-express. J Neurosci Res. 85:3567–3573. [DOI] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Barral Y. 2008. The septin family of GTPases: architecture and dynamics. Nat Rev Mol Cell Biol. 9:478–489. [DOI] [PubMed] [Google Scholar]
- Xie Y, Vessey JP, Konecna A, Dahm R, Macchi P, Kiebler MA. 2007. The GTP-binding protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr Biol. 17:1746–1751. [DOI] [PubMed] [Google Scholar]
- Yadav S, Oses-Prieto JA, Peters CJ, Zhou J, Pleasure SJ, Burlingame AL, Jan LY, Jan YN. 2017. TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through Septin7 phosphorylation. Neuron. 93:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.