

Abstract
Azobenzene is one of the most studied light‐controlled molecular switches and it has been incorporated in a large variety of supramolecular systems to control their structural and functional properties. Given the peculiar isomeric distribution at the photoexcited state (PSS), azobenzene derivatives have been used as photoactive framework to build metastable supramolecular systems that are out of the thermodynamic equilibrium. This could be achieved exploiting the peculiar E/Z photoisomerization process that can lead to isomeric ratios that are unreachable in thermal equilibrium conditions. The challenge in the field is to find molecular architectures that, under given external circumstances, lead to a given isomeric ratio in a reversible and predictable manner, ensuring an ultimate control of the configurational distribution and system composition. By reviewing early and recent works in the field, this review aims at describing photoswitchable systems that, containing an azobenzene dye, display a controlled configurational equilibrium by means of a molecular recognition event. Specifically, examples include programmed photoactive molecular architectures binding cations, anions and H‐bonded neutral guests. In these systems the non‐covalent molecular recognition adds onto the thermal and light stimuli, equipping the supramolecular architecture with an additional external trigger to select the desired configuration composition.
Keywords: supramolecular chemistry, out-of-the equilibrium, azobenzenes, molecular recognition, functional systems
In control! Gaining control of the distribution and related properties of a given molecular population is a hot topic in systems chemistry. This can be achieved in a multi‐stimuli system in which the out‐of‐equilibrium population of a photoexcited photochromic switch is finely tuned by a second supramolecular interaction such as a reversibly guest binding process. This concept is discussed taking examples from the large family of supramolecular architectures incorporating an azobenzene moiety.

1. Introduction
Azobenzene is one of the most famous light‐controlled molecular switches and since the discovery of its photoinduced isomerization,1 it has been integrated in an incredible number of different functional chemical systems. Actually, it is difficult to think of a supramolecular system and function that has not be controlled or regulated by an azobenzene moiety.2, 3, 4 Noticeable applications range from classical supramolecular devices (recognition,5, 6 self‐assembly,7 transport, sensing, catalysis,8 etc.) to photoresponsive architectures,9, 10, 11, 12, 13 passing through responsive materials14, 15, 16, 17, 18, 19, 20 and polymer science,21, 22, 23 energy‐related technologies,24, 25 molecular machines,26, 27, 28 and biology.29, 30, 31, 32, 33, 34 The preference for azobenzene with respect to other molecular switches relays on its easy preparation and functionalization, on the neat photophysical behavior that normally allows fast E/Z isomerization and selective irradiation of the two isomers, and on the large geometrical rearrangement that accompanies the isomerization process.35 The majority of these studies focused on the influence of the E/Z‐photoisomerization of azobenzene on the supramolecular structure aiming to control their supramolecular functions. However, it was evident since the seminal work of Shinkai 36 in the eighties that the integration of an azobenzene moiety in a supramolecular system strongly affected its isomerization process, influencing the kinetics of the photo and thermal processes and consequently the thermodynamic and photodynamic equilibria. This aspect was somehow overlocked in the past, but in the recent years it has increasingly attracted interest, because the possibility of controlling the configurational equilibria of a molecule is important in the field of “system chemistry”, where the control of the distribution of a given molecular population is relevant.37, 38, 39, 40, 41, 42 In this context, the possibility to fine tune the population distribution by means of a second stimulus that, being different from light, could rely on a chemical perturbation such as that created by a supramolecular interaction with a given guest could add new tools and perspectives to the field (Scheme 1).43, 44, 45, 46
Scheme 1.

General concept of a dual‐stimuli configurational selection process of an azobenzene molecular switch. A) Energy level diagram showing the dual‐stimuli model building on a fast light‐induced switching between the two azobenzene states and the chemical perturbation of the product distributions caused by a molecular recognition event; B) schematic representation of the equilibria involving an azobenzene architecture undergoing molecular recognition through non‐covalent interactions with a given chemical guest (red sphere).
In the classical case of a thermodynamic equilibrium, the ratio between two equilibrating isomers, e.g. that of the E/Z isomers of an olefin, is governed by the Gibbs free energy difference between the two states (ΔG Θ), which is reflected in the equilibrium constant (K Δ eq). For a molecular system in a solution of a given solvent, the only adjustable parameter to vary the E/Z ratio is the temperature and, in any case, it is not possible to achieve higher concentrations of the less stable isomer. Another approach would rely on the non‐covalent binding of isomers to guests to tune the energy levels of the two complexes and alter their equilibrium concentration without temperature adjustment. However, it is difficult to realize a complete inversion of the stability so that higher concentration of the less stable isomers could be reached. In this scenario, it would be much easier to start from a metastable state in which the higher energy state is more populated and find some artefact able to regulate the E/Z ratio from 0 to 1. This is actually possible in the case of a photo‐triggerable switch, because after irradiation the molecule in its excited state can relax through a different energy surface toward both isomers and a photodynamic equilibrium is reached.47 In photodynamic equilibria, the high‐energy metastable state can be effectively populated reaching a E/Z composition that depends on the ratio of the photochemical forward and backward reactions, which in turn are regulated by the molar absorption coefficients ϵ and reaction quantum yields Φ at the given excitation wavelength λ exc. Therefore at a given λ exc, the composition of the photostationary state (PSS) is dictated by the equilibrium constant K λ eq=ϵ λ E Φ E→Z/ϵ λ Z Φ Z→E. In the absence of light, and in the presence of a system featuring a sufficiently high energy barrier that separates the two states, the re‐equilibration to the thermodynamic stable state is slow and the E/Z composition can be maintained for a long time. This is normally the case of the azobenzene switch, which reverts thermally to the thermodynamic stable state with half‐life typically in the order of hours or days.2
To gain control of the E/Z composition of the irradiated mixture several approaches are in principle possible. The first one is the “covalent” modification of the azobenzene core by adding substituents that modify the photophysical properties of the molecule affecting the rate of the photochemical forward and backward reactions.35 Although in this way it is possible to alter the isomeric composition of the PSS and its temporal stability, a fine tuning is non achievable unless an “almost continuum” range of modified azobenzene could be devised for this scope. A more convenient approach appears the use of a second non‐covalent reversible interaction that can be used as a second stimulus to regulate the composition of the PSS (Scheme 1). This can be, for example, the formation of a reversible supramolecular complex between an external guest with a binding site built in close proximity with the azobenzene moiety (Scheme 1). If the formation of the complex modifies the composition of the PSS influencing the K λ eq, by changing the concentration of added guest, one could tune the amount of supramolecular complex formed and in principle a full spectra of PSS compositions can be spanned. Alternatively, if the formation of the complex alters the thermal stability of the Z isomer, accelerating its isomerization to the thermodynamically stable E configuration, it would be possible to subtract from the PSS composition only the fraction of Z complexed to the guest and again this fraction could be finely controlled by adjusting the concentration of guest. Clearly the slow thermal recovery of the E isomer is always present, but this slow drift toward the thermodynamic equilibrium could be corrected by further, time‐sparse “refreshing” photoactivation steps.
In this review we will focus on the concept of supramolecular configurational selection limiting the field to those systems incorporating an azobenzene moiety and a recognition motif. As a second stimulus, besides that of the photoexcitation used to reach the metastable state, we will consider those chemical perturbations deriving from non‐covalent interactions established in supramolecular complexes with cationic, anionic and H‐bonded neutral guests. In the following sections, selected examples of the different molecular systems and developed approaches will be reviewed with a particular focus on the control of the composition of the equilibrating mixture of isomers by a dual‐stimuli approach.
2. Configurational Selection Following Cations Complexation
2.1. Alkali Metals Ions
As mentioned above, one of the first examples of supramolecular configurational selection in azobenzene‐based switches was reported by Shinkai and co‐workers back in the eighties.48 In their report, the authors focused on the photoregulation of the binding affinity of crown and azacrown ethers towards metal cations using a switchable azobenzene subunit. However, it was immediately evident that the binding of the cation strongly influenced the thermodynamic and kinetic of the isomerization reaction, suggesting that the reaction could be controlled thermodynamically. To our opinion, the most famous receptor of this family is “butterfly crown ethers” 1 shown in Figure 1 and reported in 1981.36
Figure 1.

a) Butterfly crown ether reported by Shinkai and b) binding mode for Na+ (left) and K+ (right) cations.36
In the structure of receptor 1, two 15‐crown‐5 ethers are linked to the two phenyl rings of an azobenzene moiety. The photoinduced E/Z isomerization strongly affects the geometrical relationship between the two crown ethers, which are far apart in the E configuration and come closer in the Z isomer. In this configuration the crown ethers cooperate in the binding of the cations, behaving like a tweezer. Molecule E‐1 is readily isomerized to Z‐1 when irradiated with a high‐pressure Hg lamp reaching a photostationary state (PSS) with a 52/48 Z/E composition. The thermal back reaction is relatively slow in ODCB/n‐butyl alcohol with a first order rate constant k t=1.85×10−3 s−1. The presence of a cation strongly modifies the isomerization kinetics and the composition of the PSS. The effect is strictly correlated with the size of the metal cation with the PSS composition progressively shifted toward isomer Z‐1 and the thermal recovery slowed down descending the group, from Na+ to Cs+. The maximum effect is observed for Rb+ for which a 98/2 Z/E composition and k t value of 0.24×10−3 s−1 (ΔΔG #=1.23 kcal mol−1) were measured. In analogy to the slowing down of the thermal recovery, the shift in the PSS composition could be also attributed to a retardation of the reverse reaction in the photoisomerization process. These effects are ascribed to the complexation preferences of the 15‐crown‐5 ether that forms complexes with 1 : 2 and 1 : 1 stoichiometries with Na+ and K+, respectively. While with Na+ cation there is not a large difference in the binding affinity between the E and Z isomers, with larger cations (e.g., K+) a 1 : 1 sandwich‐type complex is strongly favored by the Z‐isomer (Figure 1b). The “tying” of two crown ethers by one alkali metal cation stabilizes the complex formed by the Z isomer with respect to that formed by the E configuration, effectively shifting the equilibrium toward Z‐1. The practical consequence is that Z‐1 is 42 times more effective than E‐1 in extracting K+ from an aqueous solution to an organic phase. Also, isomer Z‐1 is able to transport the cation across a bulk chloroform membrane playing with the photogeneration of the Z isomer to increase extraction from the source phase and to the relaxation to the E configuration for the release in the receiving phase.48 Interestingly, when increasing the concentration of larger metal ions (Rb+ or Cs+) from 0 to 4 equivalents, the percentage of the Z isomer at the PSS can be finely tuned from about 50 % to almost 100 %, demonstrating a supramolecular control of the equilibrium mixture composition. Following this approach, several receptors incorporating crown ethers and azobenzene switches have been engineered for photoregulated cation recognition. Figure 2 depicts the most significant examples.49, 50, 51, 52 In the case of molecule 2, the E→Z isomerization induces a significant expansion of the ligand cavity favoring the binding of large cations, in particular that of K+ cations, when compared to the E isomer.48, 49 A retardation of the thermal Z→E reaction is observed and the K+ cation, which best fits the size of the macrocycle, exerts the most efficient inhibition, suggesting a strong contribution of the complex stabilization in the retardation effect observed. However, this interpretation may be too simplistic because a similar rate decrease is observed also with the ammonium ion, which shows higher affinity for E‐2. Thus, it is postulated that with the ammonium ion other factors affecting the stability of the transition state may play a role. On the other hand, the E form of 3, which is the dipyridine E analog of molecule 2, shows a higher affinity for transition metal ions (e. g. Cu2+ and Pb2+) than that observed for the Z one. This corresponds to an acceleration of the thermal Z→E isomerization observed only for the metal ions that bind into the crown ether ring.50 This behavior was confirmed when macrocycles 4 a–c, behaving as “all‐or‐nothing” receptors, revealed to bind alkali cations only in the Z configuration.51 By photoisomerization of E‐4, a PSS state containing 70–80 % of the Z isomer was reached. The thermal back conversion is rather slow (one day at 30 °C in the dark), allowing the full characterization of the binding and extraction ability of the Z isomers. As expected, the recognition pattern of Z‐4 a–c is related to the size of the cavity of the crown‐ether moiety, with Z‐4 a, Z‐4 b and Z‐4 c depicting higher affinity toward Na+, K+ and Rb+ cations, respectively. Interestingly, the coordination of the metal ions slows down the rate of the thermal Z→E isomerization showing a trend, which can be positively correlated to the binding affinity and extractability of the cations. In this case, however, no effect on the PSS composition was observed, suggesting that the binding of the cations does not influence the rate of the photoinduced reisomerization processes, as in this case the complexes showed a low thermodynamic stability. These results, together with other observations obtained using related crown‐ether/azobenzene derivatives,53, 54 confirmed the idea for which the binding of metal cations to a crown ether could influence the chemical Z/E equilibrium of the azobenzene moiety, protecting the relevant isomer as a consequence of the additional energy required to disrupt the crown‐metal interaction.
Figure 2.

Examples of photo‐controlled crown ethers reported by Shinkai and co‐workers.49, 50, 51, 52
A related receptor (5) to those shown in Figure 2, incorporating an azobenzene switch, was reported by Tamaoki and Oka in 2010 (Figure 3).55 In the crystal structure, molecule E‐5 binds an Ag+ cation (E‐5@Ag+) with an unusual coordination environment comprising a nitrogen atom of the azobenzene moiety, an η2‐naphthalene and two oxygen atoms of a triflate counterion. Photoirradiation of E‐ 5 and of E‐5@Ag+ lead to the formation of the Z isomers. While E‐ 5 is converted almost quantitatively into the Z isomers, the PSS composition in the case of the silver complex show the presence of only 77 % of the Z isomer. The complexation of the metal ion accelerates the rate of the thermal Z→E isomerization. Data from NMR studies indicated that the photoisomerization of E‐5@Ag+ into Z‐5@Ag+ leads to the cleavage of the π‐cation interaction and to the weakening of the complex, and vice versa. Therefore, the most stable complex is favored, and complexation of the metal ion gives different compositions of the PSS, following a perturbation of the reaction kinetics.
Figure 3.

Isomerization reaction in the Ag+ receptor described by Tamaoki and Oka.55
Other examples in which the coordination of metal cation influences the composition of PSS are the calix[4]arene based receptors illustrated in Figure 4. Calix[4]arene 6 was reported by the Vicens’ group and studied in the extraction of alkali metal ions from water to an organic phase.56, 57 Molecule E‐6 readily photoisomerizes, reaching a PSS characterized by the presence of only 18 % of the Z isomer. However, the Z isomer is more efficient in extracting the metal ions, leading to stronger complexes than those formed by E‐6. As a consequence, the presence of the metal ions increases the percentage of the Z isomer at the PSS reaching, in the case of Cs+, a concentration value of 71 %.
Figure 4.

Calix[4]arene‐based receptors incorporating an azobenzene moiety.56, 58
At the same time, the back thermal relaxation of the Z isomer is strongly suppressed by the metal ions and no change in PSS composition was observed after 100 h, while the same Z→E process in the absence of the metal ions was completed after only 80 minutes. These observations suggested that the PSS composition depends on the apparent formation constants of the complexes, and on the concentration of the relevant cation. Therefore, by properly choosing the cation and its concentration it is possible to fine tune the PSS composition of receptor 6. In a parallel avenue, receptor 7 was reported by Tuntulani and coworkers and shows an interestingly behavior in the presence of Na+ and K+ cations.58 At the PSS state, the Z form is favored by the complexation with Na+, whereas the amount of E isomer is increased in the presence of K+ cations. Although these effects are small, the authors showed that it could have been possible to drive out of the equilibrium the photostationary state in opposite directions by selecting the relevant cation.
A system working with a triply stimuli has been reported by the group of Shinkai, in which light/pH/cation effectors could be used to control the equilibrium composition (Figure 5).59 The ability of this ligand to bind metal ions changes as a function of the photoisomerization outcome of the azobenzene group. The pH‐dependent competitive intramolecular interaction of the amine/ammonium tail with the crown ether occurring in the Z form triggers the binding ability of the cavity. Building on the hypothesis for which the ammonium derivative interacts with stronger associations than the amino analogue, strong competition with the metal complexation is expected under acidic conditions with the Z isomer. Accordingly, the cation affinity of the protonated 8 (8 .H+) is markedly reduced by photoisomerization to the Z isomer, and the rate of the thermal isomerization (i.e., from Z‐8 .H+ to E‐8 .H+) of 8 .H+ is slower than that observed for the free‐amino analogue (Z‐8→E‐8). At last, the rate of the Z‐8 .H+ to E‐8 .H+ process is accelerated upon addition of K+ ions.
Figure 5.

Triply stimuli responsive crown ether reported by Shinkai.59
2.2. Transition metals ions
Beside the works describing the complexation with alkaline cations, a large number of studies also demonstrated that transition metals such as palladium, zinc, iron, nickel, copper, iridium, etc may strongly perturb the photoisomerization equilibrium of the azobenzene switch.60, 61, 62 The transition metal ion can directly bind to the nitrogen atoms of the N=N bond or to a dedicated functional group connected to the azobenzene moiety, and it may influence the switching process with different mechanisms. For instance, these processes could involve direct electronic communication or energy transfer from the excited azobenzene group to the lateral complexes. Moreover, playing with the oxidation state of the metal or with the insertion of a redox active moiety such as Fc, the spectrum of possible interactions and mechanisms of action could be further broadened.63 Given the large body of literature on this topic, in this review we will limit our discussion only to those examples that are pertinent to the focus of this review. The interested reader is addressed to the comprehensive reviews on the topic.60, 61, 62
In a first example, the group of Nishihara tackled the problem of the interplay between light and red‐ox reaction on metal centers for the control of the PSS composition of an azobenzene‐based molecular switch.64 The structure is composed of an o‐dimethyl‐2,2’‐bipyridine ligand moiety functionalized with a azobenzene appendage (9 in Figure 6). In this case, due to the two ortho‐methyl substituents ligand 9 prefers to form tetrahedral bis‐chelate complexes with Cu+ with respect to square planar complexes with Cu2+. As a consequence, oxidation of the metal center from Cu+ to Cu2+ in the presence of 2,2’‐bipyridine (bpy) auxiliary ligand gives rise to a facile ligand exchange, so that the most abundant species in solution are [Cu(9)2]+ and [Cu(bpy)2]2+ at the Cu+ and Cu2+ states, respectively, in agreement with the well‐known coordination aptitudes of both oxidation states (Figure 6b). When starting from the Cu+ and Cu2+ complexes under irradiation at 365 nm, PSS states enriched respectively with either the E or Z isomers were obtained. Considering that free 9 is released from the Cu2+ complex during the E→Z isomerization, the E/Z photoisomerization behavior of the azobenzene moiety can be synchronized with the ligand exchange dynamics in the coordination to the copper metal ions. Moreover, the PSS composition can be reversibly controlled by red‐ox switching of the metal ion. Starting from a mixture of [Cu(9)2]+ and 2 equivalents of bpy in CH2Cl2 under irradiation at 365 nm, a PSS containing 32 % of the Z isomer was found. After chemical oxidation of Cu+ to Cu2+ with [Fe(η5‐C5H4Cl)2]PF6 followed by re‐irradiation, the Z isomer concentration increased to 70 %. The PSS composition was brought back to the initial value by reduction of the metal ion (Cu2+ to Cu+) with Co(η5‐C5H4−COCH3)2 and further irradiation. Therefore, the isomerization behavior revealed to be controllable with UV light and the ligand's binding/release reaction, which is reversibly performed with the Cu2+/Cu+ redox cycle (Figure 6c). Further elaboration of this rex‐od controlled ligand exchange reaction coupled with the photoisomerization of azobenzene led to even more complex systems, in which the light excitation causes reciprocal Cu+ translocation between two coordination environments, resulting in the modification of the redox potential of Cu+ ions. In this system, light is transformed into an electrode potential change and a positive/negative current response, similar to the process of natural visual transduction.65
Figure 6.

Light and redox control of the PSS composition in copper complexes.64
In a collaborative report from Yamamura, Nabeshima and co‐workers, the use of transition metal ions to fully lock the isomerism of azobenzene made possible the reversible selection of one configuration of the azobenzene switch.66 Thus, azobenzene‐linked bis‐terpyridine ligand E‐10 was designed (Figure 7). When E‐10 is irradiated with 365 nm light in CHCl3 gave a PSS characterized by 82 : 18 Z/E composition after 10 seconds. Isomer E‐10 formed with Fe2+ ion a stable dimeric (E‐10)2 ⋅ Fe2+ 2 complex, which hardly isomerize to Z‐10 and could be purified by silica chromatography. On the other hand, the complexation of Z‐10 with Fe2+ to give Z‐10 ⋅ Fe2+ (70 % yield) could be performed from the E isomer under irradiation with UV light, giving the mononuclear complex Z‐10 ⋅ Fe2+. Complex Z‐10 ⋅ Fe2+ is remarkably stable because of the rigid macrocyclic structure, and the thermal isomerization of the azobenzene is essentially hampered. Indeed, the first‐order rate constant for the Z→E isomerization at 70 °C was determined to be 1.17×10−6 s−1 (t 1/2=164 h), which is more than 2000 times slower than that measured for free Z‐10 (k t=2.38×10−3 s−1 at 70 °C, t 1/2=4.85 min). Moreover, complex Z‐10 ⋅ Fe2+ is also extraordinarily stable against photoisomerization and irradiation under visible light, i.e. irradiation (436 nm) for 5 h did not lead to any change in the 1H‐NMR and UV/Vis spectra. Remarkably, the isomerization reaction was reversible by demetalation reaction with H2O2, leading to free Z‐10, the latter thermally reverting into the E isomer. In contrast to Z‐10 ⋅ Fe2+, (E‐10)2 ⋅ Fe2+ 2 isomerized by irradiation at 365 nm lead to a mixture of complexes containing (E‐10)(Z‐10) ⋅ Fe2+ 2 and (Z‐10)2 ⋅ Fe2+ 2 with a 83 : 17 Z/E ratio at the PSS. Building on these observations, the authors have shown that the formation of metal complex Z‐10 ⋅ Fe2+ fully locked the Z configuration, suppressing both the thermic and the photochemical processes. Interestingly, changing the metal ion to Co3+ resulted in the locking of the E isomer, suppressing the photochemical isomerization probably due to excitation quenching by energy or electron transfer from the azo moiety to the metal complexes. Other examples, of suppression of E→Z isomerization of azobenzene by coordination‐driven macrocyclization were reported by the same authors67 and by Yam and co‐workers.68
Figure 7.

Structure of the E isomer of bis‐terpyridine‐azobenzene ligand 10 along with the isomerization reactions involving the Fe2+‐complexes of ligands E‐10 and Z‐10. Adapted with permission from reference [66]. Copyright 2014 John Wiley and Sons.
3. Configurational Selection Following Anion Complexation
A complementary approach to gain control of the E/Z ratio at the equilibrium relays on the incorporation of binding sites for anions. Exploiting the reversible complexation of anions, one could imagine using the binding event as allosteric stimulus to modify either the thermodynamic and/or the kinetic of the E/Z isomerization process. Despite the large body of research on supramolecular recognition of anions,69, 70 the examples of photoregulated anion receptors are scarcer with respect to those designed for cations71 and often incorporate different switches than those deriving from azobenzenes.72, 73, 74, 75, 76
The group of Jiang has reported77 an anion receptor containing an azobenzene unit that, bridging two phenyl‐1,2,3‐triazole moieties, can allow the binding of anions using as H‐bond donors the acidic C−H functional groups78 (Figure 8). Upon photoisomerization of the azobenzene switch, receptor 11 changes its configuration, passing from an extended E isomer to a folded Z‐architecture, structurally suited for the binding of an anion. Accordingly, higher affinities for binding anions is expected for the Z‐isomer. 1H‐NMR titration in d6‐acetone confirmed this hypothesis, showing that the Z‐isomer is able to bind several anions (Cl−, Br−, I−, NO3 − and HSO4 −) with enhanced affinity with respect to E‐11. Strong binding affinities were obtained with Cl− ion, which binds 4.1 times stronger to Z‐11 with respect to E‐11 isomer. Alternating UV and visible light irradiation in the presence of Cl− demonstrated that the switching ability of the receptor is maintained in the presence of anions, thus ensuring a controllable modulation of the binding affinity for anions. The effect of the anion binding on the PSS composition and on the switching rates has not been investigated in details.
Figure 8.

Photoswitchable anion receptor reported by the Jiang and co‐workers.77
In a parallel research endeavor, the group of Flood has reported aryl‐triazole foldameric receptor 12 shown in Figure 9.79 Being functionalized with two azobenzene termini, the receptor can adopt three different states. In the more thermodynamically stable E/E state, the receptor assumes a helical conformation forming a Cl− binding pocket. Subsequent photoisomerization of the E/E configuration to E/Z‐ 12 and Z/Z‐12 isomers decreases the stability of the foldamer, as it destabilizes π‐π interactions, leading to coiled architectures. The destabilization of the helical architecture disrupts the preorganization of the receptor pocket, weakening the binding affinity for the Cl− anion. Indeed, CD, UV/Vis and 1H‐NMR spectroscopic investigations showed a nine‐fold difference in the relative Cl− anion affinity between the E/E and Z‐dominated states. The different affinities of the folded and coiled architectures were exploited to regulate the concentration of Cl− anion in solution. By alternating irradiation of UV and Vis light the photoisomerization of the azobenze switches the conformation of the receptor from helical to random‐coil, modulating the concentration of the highly diffusive Cl− ions in solution, as signaled by the alternating decreasing and increasing of the electrical conductivity of the chloride salt solution. Elaborating the structure of receptor 12 by addition of residues able to stabilize the folded helical conformation, the group of Flood demonstrated that is possible to enhance the difference in the binding affinity for Cl− ions between the E/E and Z‐dominated states up to 84 times.80 Further, they also showed that double‐to‐single helix switching using anions of different size as allosteric regulators could be achieved on structurally related photo‐foldamer.81 In analogy with the butterfly‐like cation receptor of Shinkai, the authors postulated that the formation of the foldamer‐chloride complexes affects the relative stability of the isomers at the PSS. Overall, these systems are examples of conformational selection controlled by the cooperation of light and a reversible binding of anions to an allosteric site.
Figure 9.

Photoswitchable aryl‐triazole foldamer reported by Flood and co‐workers (top) and proposed cycle of photodriven binding and release of Cl− ions (bottom). Adapted with permission from reference [79]. Copyright 2010 American Chemical Society.
A different approach has been undertaken by the group of Jeong, who designed tweezer‐like anion receptors bearing two urea binding sites spaced by an azobenzene moiety (13, Figure 10).82 Irradiation with UV light of the thermally stable E isomer (98 %) induces the formation of the corresponding Z isomer as major component of the PSS (90–96 %), as shown by 1H‐NMR investigations in DMSO‐d6. In the Z form, four convergent NH protons can participate in the H‐bonding with the Cl− ion, while this is not possible with the extended E configuration. As a consequence, molecule Z‐13 showed larger binding constants for the anion than the E isomer by approximately one order of magnitude. Receptor 13 was subsequently tested in the transport of Cl− ions across a phospholipid membrane. Notably, only isomer Z‐13 showed meaningful transport ion activity. However, when the membrane loaded with E‐13 was irradiated with UV‐light, the Z‐ 13‐Z isomer is formed and the transport process activated. A similar approach was reported by the group of Wang (Figure 10), who prepared receptor 14.83 Compound 14 was tested in the binding of di‐carboxylate anions and the Z isomer showed high association constants, in particular for adipate. However, no quantitative investigations about the influence of anion binding on the photoisomerization process of azobenzenes shown in Figure 10 was reported.
Figure 10.

Tweezer‐like receptors reported by the groups of Jeong 82 (above) and Wang 83 (below).
In 2014 the group of Jurczak reported the synthesis of receptor 15 (Figure 11), featuring an azobenzene moiety bearing either one (15 a) or two (15 b) urea recognition sites in the para position.84 Building on the idea for which it would be desirable to trigger the switching using a different stimulus than light and heat,85 in this work, the scientists focused their studies on gaining kinetic control of the thermal Z→E isomerization through the use of chemical triggers. Capitalizing on the experimental observations for which a retardation effect of the Z→E isomerization can be achieved introducing an electron withdrawing group on the azobenzene rings,86 the authors hypothesized that a non‐covalent binding of an anion to the π‐conjugated urea would have an opposite outcome, namely a rate enhancement of the thermal Z→E reaction. Photoisomerization studies showed that compound 15 can be readily switched between the E and Z forms using UV (368 nm) and blue light (410 nm) for several cycles without photodegradation. The thermal Z→E reaction occurred with a t 1/2 of 13.1 and 1.8 h for 15 a and 15 b, respectively. Addition of different anions (F−, Br−, RCOO−, HSO4 −, H2PO4 −) as tetra‐n‐butylammonium (TBA) salts strongly accelerated the thermal Z→E isomerization, with the highest enhancement observed for derivative Z‐15 a with the acetate anion. A reduction in the t 1/2 of about 200 times was recorded in the presence of 10 equivalents of acetate anion, which is known to display the strongest association with the urea binding site amongst all the tested anions. Interestingly, the retardation effect is dependent on the concentration of the anion and it can be reverted or restored by addition of either TFA or TEA. An accurate kinetic analysis paralleled by 1H‐NMR titrations allowed to determine the rate constants of the thermal Z→E isomerization in the fully bound complexes between 15 a,b and benzoate. Interestingly, Z‐15 a binds benzoate 4.5 times stronger than Z‐15 b (first association constant) while no difference in the affinity for the anion was observed between Z‐ and E‐15 a. In a subsequent work by the same authors,87 these concepts were applied to the chiral recognition of α‐amino acid carboxylates using symmetrical receptors 16 a, b, exposing D‐carbohydrate termini as chiral selectors. The E and Z isomers of 16 display different binding modes (i.e., 1 : 2 and 1 : 1, respectively) and different affinities (E>Z) towards the phenylalanine and tryptophan anions, while the enantioselective discrimination of the two amino acids was moderate. Interestingly, the rate of the thermal Z→E isomerization was found to depend on the binding strength of Z‐16 with the given anion. In general, the more strongly carboxylate enantiomer binds, the stronger is the acceleration of the Z→E isomerization.
Figure 11.

Urea‐based photoswitchable receptors reported by the group of Jurczak.84, 87
In 2017 the group of Jurczak reported a new anion receptor incorporating an azobenzene switch (17, Figure 12).88 The receptor bears four amides connected in meta positions to an azobenzene moiety. As opposed to the cases of compounds 15 and 16, in molecule 17 there is not direct π‐conjugation between the binding sites and the N=N bond. Compound 17 showed a complex anion coordination chemistry influenced by the configuration of the azobenzene group. In general, the Z‐isomer, in which the four amide groups cooperate in the binding of the anion, showed higher association constants with respect to the E‐isomer. Computational studies on the complexes of Z ‐17 with H2PO4 − and acetate anions showed all the four NH amide protons interacting with the acetate, while only three H‐bonds were detected for the complexation with H2PO4 −. Interestingly, an excess of Cl− anion did not influence the rate of the thermal isomerization of Z ‐17, whereas in the case of H2PO4 − a small rate retardation was observed. These results supports the idea that the anion induced destabilization of the Z isomer observed in compounds 15 and 16 was due to an increased electron density on the azo‐bond as a consequence of the donation from the bound anion via the conjugated nitrogen of the urea moiety. As in molecule 17 the binding sites are not in π‐conjugation with the N=N bond, the formation of the complex stabilizes the cleft‐type structure of the Z isomer, retarding its thermal relaxation. These binding‐induced behaviors are in full agreements with the observations by Shinkay and co‐workers with the butterfly‐like crown ether receptors for cations.36
Figure 12.

Photoswitchable anion receptor reported by the group of Jurczak.88
A further example of photomodulated anion receptor was reported by the group of Kohnke (Figure 13).89 Receptor 18 is constituted by two calix[4]pyrroles linked via an azobenzene unit. Building on the idea that the E/Z configurational switching should affect the ability of the two calixpyrrole components to cooperate towards the binding of ditopic anions, the association ability of host 18 was probed with different bis‐carboxylate anions. 1H‐NMR titration in DMSO‐d6 showed that Z‐18 strongly binds dicarboxylates such as succinate and glutarate anions, whereas the E isomer displayed weaker affinity. The highest affinity constant was measured for succinate (KZ=5.3×105 M−1) with a KZ/KE ratio of 82. On the other hands, when dicarboxylates featuring long spacing units are used, e.g. adipate, the strongest complexes were formed with the E‐18 (e.g., KE=8.6×104 M−1 and KE/KZ ratio of 3.9 for adipate). A detailed kinetic analysis allowed to define the effect of the dianion complexation on the isomerization process. In the case of the UV‐triggered (λ=365 nm) E→Z photoisomerization, a retardation effect positively correlated with the strength of the association constant of the complex was observed. This suggested that the N=N isomerization became more difficult when isomer E‐18 is complexed to the bis‐anion. The effect on the thermal Z→E reaction was complex, showing a bi‐exponential kinetic behavior not correlated with the strength of the complex. Indeed, binding of succinate to Z‐18 slowed down the thermal isomerization while all the other anions accelerate the process. This behavior was interpreted with the occurrence of two competitive phenomena: i) de‐acceleration of the Z→E isomerization due to the bridging of the calix units and ii) acceleration of the same reaction by electron donation effects from the bound substrate to the N=N bond. The first effect predominates with succinate anion, while the second was prevalent with the other anions. In a structurally‐related dimeric resorcin[4]arene cavitand linked by a azobenzene spacer, Rebek and co‐workers90 found a direct correlation between the strength of the association in a family of bisadamantane guests with different lengths and both the composition of the PSS and the rate of thermal Z→E isomerization. That is, with long guests that bind more favorably to the E isomer the amount of the Z at the PSS is reduced and vice versa for shorter guests that fit better within the Z isomer, enhancing its stability. Taken all together, these findings highlight the complex interpretation that one needs to consider when evaluating the effects of a guest binding on the isomerization of azobenzenes.
Figure 13.
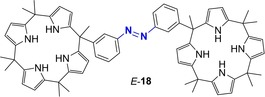
Bis‐calix[4]pyrrole receptor reported by the group of Kohnke.89
4. Configurational Selection Following H‐Bonding Complexes Formation
A H‐bond is another classical reversible supramolecular interaction that can be exploited for the regulation of the photochemical equilibrium of an azobenzene switch. It is well‐known that intramolecular H‐bonding interactions involving the nitrogen atom of the N=N group may strongly influence the switching equilibrium of the azobenzene moiety. For example, in molecules 19 a–b, the intramolecular H‐bonds involving the amine group a H‐bond donor and the azo group and the pyridine nitrogen as the acceptor suppress the E→Z photoisomerization and strongly accelerates the thermal recovery process (Figure 14).91, 92 The isomerization of azobenzene occurs via a concerted inversion mechanism where both aryl rings must adopt a collinear arrangement prior to inversion. Theoretical calculation and experimental studies show that the H‐bond in molecules 19 a–b forces the molecule to adopt a rigid planar structure, preventing both aryl rings to rotate and introducing a substantial barrier to the concerted E→Z inversion mechanism. This rigid conformation also makes these compounds more emissive than classical azobenzenes at low temperatures. Disruption of the intramolecular H‐bonding interaction by methylation of the amine nitrogen atom restores the photoisomerization process and quenches the radiative emission. In analogy, intramolecular H‐bonds between the hydroxyl group and the azo‐nitrogen atom in 20 causes rapid thermal Z→E isomerization although the photochemistry of this class of compounds is complicated by the tautomeric equilibrium involving the corresponding phenylhydrazone.93
Figure 14.

Examples of intramolecular H‐bonded azobenzene architectures.91, 92, 93
The photoisomerization of the azobenzenes has also been exploited to regulate intramolecular hydrogen‐bonding interaction in photoswitchable catalysts.94 For example, compound 21 was conceived and prepared as a catalyst for the ring opening polymerization (ROP) of rac‐lactide (Figure 15).95 In the Z isomer, the NH proton of the guanidinium moiety establishes intramolecular H‐bonds with the acyl group of the azobenzene. After photoisomerization to the E isomer, the H‐bond is broken releasing the gaunidinium in an active catalytic form. Although the reversible light‐induced switchability was demonstrated, compound 21 revealed to be inactive in the catalysis of the polymerization of lactide. Therefore, a variant of 21 was proposed, namely compound 22 (Figure 15).96 In this case, the nitro substituent in the Z isomer engages in intramolecular H‐bonds with the thiourea moiety. The photoisomerization generates the active form of the catalyst in which the thiourea functional group acts as a H‐bond donor that, cooperating with an H‐bond acceptor (pentamethyldiethylenetriamine (PMDETA)) as co‐catalyst in the ROP of lactic acid, triggers the polymerization to produce poly(lactic acid). Similar results were reported for a structurally related photoswitchable thiourea.97 In all cases, however, the effect of the intramolecular H‐bonding on the isomerization of the azobenzene has not been investigated in detail.
Figure 15.
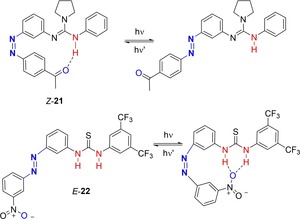
Photoswitchable catalysts in which the off‐state is characterized by intramolecular H‐bonding.95, 96
An interesting example of locking and unlocking of the E isomer of an azobenzene switch has been reported by Hecht and co‐workers, who exploited H‐bonding interactions between ammonium cations and crown ethers (Figure 16).98 The architecture is the photochromic axle 23, carrying two terminal secondary ammonium‐binding sites linked to an azobenzene core. Stilbene analogue 24 was also prepared for comparison purpose. Axle E‐23 binds very strongly to the ditopic host 25, the latter featuring two dibenzo‐24‐crown‐8 linked to an anthracene‐spacer. The complex features elevate effective molarity and a highly positive chelate cooperativity. On the contrary, no appreciable association between the host 25 and the Z isomers of axle 23 and of the stilbene analogue 24 was observed. In the absence of host 25, E‐23 showed the typical isomerization processes of azobenzene reaching, upon irradiation with UV light, a PSS containing about 90 % of the Z isomer. This isomer could be thermally re‐converted to the E form with a half‐life of 127 hours at room temperature. However, in the presence of one equivalent of host 25, the E→Z photo isomerization is essentially suppressed, while the reverse process can be still reached. Indeed, irradiation at 436 nm of a PSS mixture (containing 90 % Z‐23 and 10 % E‐23) in the presence of 25 showed clean Z→E photoisomerization. Unexpectedly, the presence of host 25 instead of accelerating the thermal Z→E reaction by selective stabilization of complex E‐23@25, slowed down the process. Interestingly, a clean E→Z photo isomerization was observed when irradiating E‐23 in the presence of an excess of monovalent host 26. This forms complex E‐23@26 2, suggesting that the complexation of the ammonium group to the crown ether was not responsible for the observed inhibition effect. Moreover, the E→Z photoisomerization of E‐23 could be reversibly restored by dethreading the axel upon deprotonation. Successive re‐locking could be achieved by reprotonation with acid. This gating effect was attributed to the reversible formation of the pseudo‐rotaxane architecture, that locks the conformation of the E isomer.
Figure 16.

Isomerization of divalent azobenzene and stilbene axles 23 and 24, divalent host 25, monovalent host 26 and pseudo[2]rotaxanes E‐23@25. Adapted with permission from reference [98]. Copyright 2015 The Royal Society of Chemistry.
The conjugation of the large geometrical rearrangement that accompanies the E/Z isomerization in azobenzene with the principle of shape complementarity has been used for the light‐controlled recognition of guests and to direct the auto‐association preferences in H‐bonded complexes. One example of the first approach is the receptor reported by S. Goswami and co‐workers in which two acylamino pyridines were attached in the para‐position of the phenyl rings of an azobenzene.99 In the Z isomer the two acylamino pyridines cooperates in the binding of di‐carboxylic acids through H‐bonds shifting the isomerization equilibrium toward the Z isomer, slowing down its thermal Z→E conversion. More recently Hunter and co‐workers reported ditopic receptor 27 (Figure 17a).100 The E/E form of 27 adopts an extended conformation in which the two carboxylate moieties are separated by a considerable distance. Therefore, binding of guanidium in DMSO may occur only with one of the two carboxylate receptors, with an association constant of 2.2×103 M−1. Irradiation at 345 nm led to a complex mixture of E/E, E/Z and Z/Z isomers in a 1 : 1 : 1 ratio. 1H‐NMR titration allowed to measure the association constants, which revealed to be 1.5×103 M−1 and 2.0×104 M−1 for the E/Z and Z/Z isomers, respectively. The increase of one order of magnitude of the association constant value for the Z/Z complex, with respect to the other isomers, was explained considering the folding of receptor 27 into a cleft‐type structure, where the two carboxylates cooperate in the binding of the substrate. The system developed by Sleiman and co‐workers of Figure 17b, describes how the reversible isomerization of the azobenzene moiety switches the self‐assembly preference of a monomer from a cyclic tetramer in the Z‐form to a linear oligomer in the E‐form.101 This is the consequence of the change of the angle between the two carboxylic acids in p‐azodibenzoic acid 28, which change from a linear arrangement in the E isomer to an about 90° angle in the Z one. However, in these cases no information on the effect of these recognition phenomena on the equilibrium of the azobenzene isomerization was given.
Figure 17.

Hydrogen‐bonded self‐assembled architectures triggered by the isomerization of an azobenzene.100, 101
In a conceptually related approach, Ghadiri and co‐workers reported the exchange between inter‐ and intramolecularly assembled cylindrical structures using peptide derivative 29 shown in Figure 18.102 Peptide 29 is made of two cyclic octapeptides with alternating D‐ and L‐α‐amino acids, that are bridged by an azobenzene moiety. 1H‐NMR studies showed that in nonpolar organic solvents, E‐29 existed as a mixture of dimers and higher oligomers, formed as a result of intermolecular hydrogen‐bonding interactions between the cyclic peptides. Irradiation with UV light generated isomer Z‐29 and promoted the conversion from intermolecular assemblies into the single intramolecularly hydrogen‐bonded species (Figure 18).
Figure 18.

Light‐induced folding and unfolding of peptide 29.102
Remarkably, the isomerization process was quantitative, and the Z isomer was the only species present at the PSS. The inverse transition to the E isomer could be achieved by thermal stimulus or irradiation with visible light (85 : 15 E/Z ratios at the PSS). Notably, the thermally induced switching was substantially retarded (about 7.5 times at 293 K) with respect to a reference compound. This reflects the high activation energy required to break the intramolecular H‐bonds. The reversible switching between inter‐ and intramolecular H‐bonds was also demonstrated in thin films of peptide 29, formed either at the air/water interface or on mica and quartz glass. Light control of intramolecular H‐bonds has been also recently demonstrated by Ballester and co‐workers, who prepared a dimeric capsule formed by a tetraureacalix[4]arene with four appended terminal azobenzene groups.103 In CD2Cl2 solution, the thermally equilibrated all‐E‐tetraurea dimerized quantitatively forming a capsule held together by an array of H‐bonds and encapsulating one Me4P+ cation. Light‐induced isomerization of all‐E encapsulation complex produced a mixture of isomeric Z‐enriched assemblies, featuring either reduced cavity size or in noncapsular dimers as a consequence of the disruption of the H‐bonding pattern. Being these Z‐enriched dimers not suitable for the encapsulation of the cation, the structures are partially released to the bulk solution.
A different approach to the modification of the affinity in a complementary H‐bonded complex has been reported by Rotello and co‐workers.104 The authors prepared a DAD (donor‐acceptor‐donor) diaminotriazine H‐bonding receptor linked in close proximity to an azobenzene moiety (30) and studied the recognition of a complementary ADA (acceptor‐donor‐acceptor) naphthalenediimide substrate (Figure 19). UV/Vis studies on host 30 demonstrated the rapid and efficient interconversion of the azobenzene unit between the E and Z configurations and subsequent reversion to E‐30. No noticeable changes in the UV/Vis spectrum of 30 (in both forms) in the presence of the guest were observed. 1H‐NMR studies of the photoswitching in the presence of the guest showed efficient reversible isomerization. Interestingly, E‐30 binds a naphthalenediimide derivative about 16 times stronger than the Z isomer. This was explained considering the loss of the favorable aromatic stacking interactions upon isomerization, possibly accentuated by unfavorable dipolar interactions between the azo group and the naphthalenediimide guest. Further electrochemical reduction of the naphthalenediimide to the mono and di‐anionic species allowed the creation of a single host‐guest dyad that possesses six states featuring different binding affinities.
Figure 19.

Photoswitchable host‐guest complexes synthesized by Rotello and co‐workers.104
The group of Wisner has studied a photoswitchable self‐complementary H‐bonded system composed of an azoheteroaromatic backbone.105 As shown in Figure 20a, E‐31 formed stable dimers through six H‐bonding interactions, also involving a nitrogen atom of the azo group. Irradiation with UV light produces the less stable Z isomer (max 32 % at PSS in CD3CN) in which a reduced number of potentially interacting sites led to the formation of a weaker dimer. The analysis of the system was complicated by the relatively fast thermal Z→E conversion, which was completed in about 30 min. The fast change in the concentration of Z‐31 did not allow to use standard 1H‐NMR experiments to measure the dimerization constant of the Z isomer. This problem was solved by developing a new mathematical and experimental approach that allowed the determination of all the relevant affinity constants involved, comprising those related to formation of the complexese of the E and the Z isomers. The obtained association constants ranged, in toluene‐d8 at 298 K, from 590 M−1 to 1200 M−1 for the E/E dimer and from 18 M−1 to 46 M−1 for the Z/Z dimer, with those for the E/Z mix dimer being between these two values.
Figure 20.

H‐bonding receptors developed by Wisner and co‐workers.105, 106
In a second study, the same group studied the formation of H‐bonding complexes between di‐pyridylazobenzene ligands of the type shown in Figure 20b and derivatives of 2‐aminoindolo.106 Molecule E‐32 features an AAA‐type H‐bonding array formed by the pyridine and azo nitrogen atoms, which is complementary to the DD set of the indole. Upon E→Z isomerization, the H‐bonded array is lost and a higher affinity of the E isomer for the guest with respect to the Z configuration is anticipated. E‐32 is readily isomerized to Z‐32 by irradiation at 360 nm reaching a PSS containing 70 % of Z‐32. The Z isomer was isolated in the pure form by column chromatography. However, the thermal back reaction allows the full formation of the E isomer in one h. Using analytical tools developed in the group, it was calculated that E‐32 isomer binds the indole substrate about 30 times stronger than Z‐32. Moreover, alternating irradiation at 350 nm and 530 nm, the complexation and decomplexation from the E‐isomer was cycled. Unfortunately, the effect of the binding on the kinetic of the isomerization process was not analyzed in detail.
In 2017, our groups reported a detailed study on the E/Z isomerization process of the uracil‐azobenzene derivative 33 in which the nucleobase is conjugated to a phenyldiazene tail and on the effect of the formation of a triply H‐bonded complexes with a complementary 2,6‐diacetylamino‐4‐pyridine ligand (DAP, Figure 21)107 Thanks to an accurate UV/Vis and 1H‐NMR analysis all the relevant thermodynamic and kinetics parameters were determined.
Figure 21.

Isomerization reaction of the uracil‐azobenzene derivative 33 in the free form and in the H‐bonded complex with DAP. Adapted with permission from reference [107]. Copyright 2017 American Chemical Society.
When irradiated at 360 nm, E‐33 rapidly isomerized to the Z isomer that can be quickly reconverted back to the E configuration by irradiation at 442 nm. The molecular system depicted excellent fatigue resistance, and remarkable photostability when cycled several times and nearly quantitate two‐way isomerization. On the other hand, the thermal recovery of the E isomer in the dark was slow and it was characterized by a k est=2.5×10−5 s−1. In toluene‐d8, E‐33 formed an H‐bonded complex with NAP with an association constant KE=6710±800 M−1 which is lowered to KZ=4040±355 M−1 for the Z isomer. The formation of the complex influenced also the rate of thermal Z→E conversion which was about four times faster than that occurring for Z‐33 alone. This behavior was interpreted on the basis of DFT calculations, suggesting that the formation of the triply H‐bonded complex weakened and elongated the azo bond as a consequence of the increase of its πg* antibonding character, thus reducing the N=N torsional barrier. We proposed that the binding induced acceleration of the Z→E conversion could be exploited in the configuration control of the isomeric mixture of 33. Indeed, photogeneration of Z‐33 in the presence of a given concentration of DAP will form complex Z‐33@DAP, which quickly isomerizes to the E isomer. Therefore, a Z/E mixture with an isomeric composition determined by the amount of Z‐33@DAP complex will be produced, with the system slowly drifting toward the thermodynamic equilibrium composition due to the slower background thermal isomerization of free Z‐33. Clearly, the possibility of producing a Z/E mixture with a defined isomeric composition and stable in time depends on the ratio of the rate constants of the thermal recovery i) in the free 33 and ii) in the DAP complex that, in the present case, is only 4, thus not allowing a complete separation between the two kinetic processes. Building on thermodynamic and the dynamic properties of the reversible H‐bonding complexes, in this paper we have demonstrated that is possible to select and control a given configuration fraction using a second stimulus the concentration of a guest.
5. Conclusions
Azobenzenes have a long and successful history and are now consolidated and powerful tools in the hands of supramolecular chemists. The use of light as an external stimulus to vary geometrical and chemical‐physical parameters offers indeed a clean tool to gain control over a large number of different systems and functions. Moreover, the shortcut through the highly energetic excited states of the photochemical switches allows to escape from the landscape of the ground state energy profile and its thermodynamic pitfall. In this way, it is possible to reach metastable states in which the isomeric ratio is far from that of the thermic equilibrium, adding a new controllable parameter to govern the system, and trigger its population distribution. A further step in the field is the fine tuning of the isomeric ratio in a reversible and predictable manner to ensure an ultimate control of the configurational equilibria and of the system composition. This review has explored this concept re‐visiting old and new photoswitchable systems containing an azobenzene dye. The review of selected examples demonstrates that it is indeed possible to gain control of the azobenzene configurational equilibrium exploiting the reversible binding of different guests to selected recognition sites. We have limited our attention to classical guests such as cations, anions and H‐bonded neutral molecules. Nevertheless, a complex and rich range of behaviors that span from the complete freezing of the configurational equilibrium in one of the two states to a fine continuous tuning of the E/Z isomeric ratio has emerged from the literature. In these systems a molecular recognition is added to the light stimulus, allowing a more sophisticated control of the whole composition of the properties of a chemical system.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Davide Bonifazi was born in Guastalla (Italy) in 1975. After obtaining the “Laurea” in “Industrial Chemistry” from the University of Parma working with Prof. Enrico Dalcanale, he joined the group of Prof. François Diederich as PhD student at the ETH Zürich (2000–2004). He was awarded the Silver Medallion of the ETH for his doctoral dissertation (2005). After a one‐year postdoctoral fellowship with Prof. Maurizio Prato at University of Trieste, he joined the same University as a research associate first, and then as a part‐time Professor (2012–2015). In 2006, he joined the University of Namur (BE) as Junior Professor (2006–2011) and as Professor of Organic Chemistry (2012–2015). In 2016 he moved to the School of Chemistry at Cardiff University (UK) as Chair Professor of Organic Supramolecular Chemistry until his current appointment (2020) as Chair Professor of Organic Chemistry at the Institute of Organic Chemistry at the University of Vienna. His activities are focused on the creation of functional organic molecules in interdisciplinary projects through targeted organic synthesis, self‐assembly, and self‐organization of architectures in solution and on surfaces, physical‐organic studies, material‐ and bio‐based design.

Biographical Information
Paolo Tecilla obtained his PhD in Chemistry from the University of Padova (Italy) with Prof. Umberto Tonellato in 1989. After post‐doctoral work with Prof. A. D. Hamilton at the University of Pittsburgh, he got a Lecturer position at the University of Padova. In 1998 he moved to the University of Trieste where he is now full Professor of Organic Chemistry. His scientific interests are in the field of supramolecular chemistry with a background in physical organic chemistry and focus on metallo‐catalysts of hydrolytic reactions, on fluorescence chemosensors, and synthetic ionophores.

Acknowledgements
P.T and D.B. gratefully acknowledge the EU through the MSCA‐RISE (project: INFUSION) and MSCA‐ITN (project PHOTOTRAIN) funding schemes for the financial support.
P. Tecilla, D. Bonifazi, ChemistryOpen 2020, 9, 529.
Dedicated to Prof. J. M. Lehn on the occasion of his 80th birthday
Contributor Information
Prof. Paolo Tecilla, Email: ptecilla@units.it.
Prof. Davide Bonifazi, Email: bonifazid@cardiff.ac.uk.
References
- 1. Hartley G. S., Nature 1937, 140, 281–282. [Google Scholar]
- 2. Bléger D., Hecht S., Angew. Chem. Int. Ed. 2015, 54, 11338–11349. [DOI] [PubMed] [Google Scholar]
- 3. Baroncini M., Ragazzon G., Silvi S., Venturi M., Credi A., Pure Appl. Chem. 2015, 87, 537–545. [Google Scholar]
- 4. Klajn R., Pure Appl. Chem. 2010, 82, 2247–2279. [Google Scholar]
- 5. Isaacs L., Acc. Chem. Res. 2014, 47, 2052–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Díaz-Moscoso A., Ballester P., Chem. Commun. 2017, 53, 4635–4652. [DOI] [PubMed] [Google Scholar]
- 7. Yao X., Li T., Wang J., Ma X., Tian H., Adv. Opt. Mater. 2016, 4, 1322–1349. [Google Scholar]
- 8. Dorel R., Feringa B. L., Chem. Commun. 2019, 55, 6477–6486. [DOI] [PubMed] [Google Scholar]
- 9. Dattler D., Fuks G., Heiser J., Moulin E., Perrot A., Yao X., Giuseppone N., Chem. Rev. 2020, 120, 310–433. [DOI] [PubMed] [Google Scholar]
- 10. Kanj A. B., Müller K., Heinke L., Macromol. Rapid Commun. 2018, 39, 1700239. [DOI] [PubMed] [Google Scholar]
- 11. Galanti A., Diez-Cabanes V., Santoro J., Valášek M., Minoia A., Mayor M., Cornil J., Samorì P., J. Am. Chem. Soc. 2018, 140, 16062–16070. [DOI] [PubMed] [Google Scholar]
- 12. Galanti A., Santoro J., Mannancherry R., Duez Q., Diez-Cabanes V., Valášek M., De Winter J., Cornil J., Gerbaux P., Mayor M., Samorì P., J. Am. Chem. Soc. 2019, 141, 9273–9283. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Zeitouny J., Aurisicchio C., Bonifazi D., De Zorzi R., Geremia S., Bonini M., Palma C.-A., Samorì P., Listorti A., Belbakra A., Armaroli N., J. Mater. Chem. 2009, 19, 4715–4724; [Google Scholar]
- 13b. Zeitouny J., Belbakra A., Llanes-Pallas A., Barbieri A., Armaroli N., Bonifazi D., Chem. Commun. 2011, 47, 451–453. [DOI] [PubMed] [Google Scholar]
- 14. Xu W. C., Sun S., Wu S., Angew. Chem. Int. Ed. 2019, 58, 9712–9740. [DOI] [PubMed] [Google Scholar]
- 15. Nie H., Self J. L., Kuenstler A. S., Hayward R. C., Read de Alaniz J., Adv. Opt. Mater. 2019, 7, 1900224. [Google Scholar]
- 16. Liaros N., Couris S., Maggini L., De Leo F., Cattaruzza F., Aurisicchio C., Bonifazi D., ChemPhysChem 2013, 14, 2961–2972. [DOI] [PubMed] [Google Scholar]
- 17. Kundu P. K., Klajn R., ACS Nano 2014, 8, 11913–11916. [DOI] [PubMed] [Google Scholar]
- 18. Das S., Ranjan P., Maiti P. S., Singh G., Leitus G., Klajn R., Adv. Mater. 2013, 25, 422–426. [DOI] [PubMed] [Google Scholar]
- 19. Döbbelin M., Ciesielski A., Haar S., Osella S., Bruna M., Minoia A., Grisanti L., Mosciatti T., Richard F., Prasetyanto E. A., De Cola L., Palermo V., Mazzaro R., Morandi V., Lazzaroni R., Ferrari A. C., Beljonne D., Samorì P., Nat. Commun. 2016, 7, 11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baroncini M., d′Agostino S., Bergamini G., Ceroni P., Comotti A., Sozzani P., Bassanetti I., Grepioni F., Hernandez T. M., Silvi S., Venturi M., Credi A., Nat. Chem. 2015, 7, 634–640. [DOI] [PubMed] [Google Scholar]
- 21. Pianowski Z. L., Chem. Eur. J. 2019, 25, 5128–5144. [DOI] [PubMed] [Google Scholar]
- 22. Stoychev G., Kirillova A., Ionov L., Adv. Opt. Mater. 2019, 7, 1900067. [Google Scholar]
- 23. Pang X., -an Lv J., Zhu C., Qin L., Yu Y., Adv. Mater. 2019, 31, 1904224. [DOI] [PubMed] [Google Scholar]
- 24. Dong L., Feng Y., Wang L., Feng W., Chem. Soc. Rev. 2018, 47, 7339–7368. [DOI] [PubMed] [Google Scholar]
- 25. Sun C.-L., Wang C., Boulatov R., ChemPhotoChem 2019, 3, 268–283. [Google Scholar]
- 26. Abendroth J. M., Bushuyev O. S., Weiss P. S., Barrett C. J., ACS Nano 2015, 9, 7746–7768. [DOI] [PubMed] [Google Scholar]
- 27. Lancia F., Ryabchun A., Katsonis N., Nat. Rev. Chem. 2019, 3, 536–551. [DOI] [PubMed] [Google Scholar]
- 28. Baroncini M., Silvi S., Credi A., Chem. Rev. 2020, 120, 200–268. [DOI] [PubMed] [Google Scholar]
- 29. Zhu M., Zhou H., Org. Biomol. Chem. 2018, 16, 8434–8445. [DOI] [PubMed] [Google Scholar]
- 30. Dong M., Babalhavaeji A., Samanta S., Beharry A. A., Woolley G. A., Acc. Chem. Res. 2015, 48, 2662–2670. [DOI] [PubMed] [Google Scholar]
- 31. Albert L., Vázquez O., Chem. Commun. 2019, 55, 10192–10213. [DOI] [PubMed] [Google Scholar]
- 32. Broichhagen J., Frank J. A., Trauner D., Acc. Chem. Res. 2015, 48, 1947–1960. [DOI] [PubMed] [Google Scholar]
- 33. Mart R. J., Allemann R. K., Chem. Commun. 2016, 52, 12262–12277. [DOI] [PubMed] [Google Scholar]
- 34. Cabré G., Garrido-Charles A., Moreno M., Bosch M., Porta-de-la-Riva M., Krieg M., Gascón-Moya M., Camarero N., Gelabert R., Lluch J. M., Busqué F., Hernando J., Gorostiza P., Alibés R., Nat. Commun. 2019, 10, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bandarab H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825. [DOI] [PubMed] [Google Scholar]
- 36. Shinkai S., Nakaji T., Ogawa T., Shigematsu K., Manabe O., J. Am. Chem. Soc. 1981, 103, 111–115. [Google Scholar]
- 37. Ashkenasy G., Hermans T. M., Otto S., Taylor A., Chem. Soc. Rev. 2017, 46, 2543–2554. [DOI] [PubMed] [Google Scholar]
- 38. Kindermann M., Stahl I., Reimold M., Pankau W. M., von Kiedrowski G., Angew. Chem. Int. Ed. 2005, 44, 6750–6755. [DOI] [PubMed] [Google Scholar]
- 39. Ludlow R. F., Otto S., Chem. Soc. Rev. 2008, 37, 101–108. [DOI] [PubMed] [Google Scholar]
- 40. Lehn J.-M., Chem. Soc. Rev. 2007, 36, 151–160. [DOI] [PubMed] [Google Scholar]
- 41. -M Lehn J., Chem. Eur. J. 1999, 5, 2455–2463. [Google Scholar]
- 42. Vantomme G., Jiang S., Lehn J.-M., J. Am. Chem. Soc. 2014, 136, 9509–9518. [DOI] [PubMed] [Google Scholar]
- 43. Lehn J.-M., Top. Curr. Chem. 2012, 322, 1–32. [DOI] [PubMed] [Google Scholar]
- 44. Lehn J.-M., Angew. Chem. Int. Ed. 2013, 52, 2836–2850. [DOI] [PubMed] [Google Scholar]
- 45. Lehn J.-M., Angew. Chem. Int. Ed. 2015, 54, 3276–3289. [DOI] [PubMed] [Google Scholar]
- 46. Baggi G., Casimiro L., Baroncini M., Silvi S., Credi A., Loeb S. J., Chem. Sci. 2019, 10, 5104–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kathan M., Hecht S., Chem. Soc. Rev. 2017, 46, 5536–5550. [DOI] [PubMed] [Google Scholar]
- 48. Shinkai S., Shigamatsu K., Sato M., Manabe O., J. Chem. Soc. Perkin Trans. 1 1982, 2735–2739. [Google Scholar]
- 49. Shinkai S., Nakaji T., Nishida Y., Ogawa T., Manabe O., J. Am. Chem. Soc. 1980, 102, 5860–5865. [Google Scholar]
- 50. Hammon H. L., Bhattacharjee S. K., Shinkai S., Honda Y., J. Am. Chem. Soc. 1984, 106, 262–263. [Google Scholar]
- 51. Shinkai S., Kouno T., Kusano Y., Manabe O., J. Chem. Soc. Perkin Trans. 1 1982, 2741–2747. [DOI] [PubMed] [Google Scholar]
- 52. Shinkai S., Minami T., Kusano Y., Manabe O., J. Am. Chem. Soc. 1983, 105, 1851–1856. [Google Scholar]
- 53. Shinkai S., Miyazaki K., Manabe O., J. Chem. Soc. Perkin Trans. 1 1987, 449–456. [Google Scholar]
- 54. Akabori S., Miura Y., Yotsumoto N., Uchida K., Kitano M., Habata Y., J. Chem. Soc. Perkin Trans. 1 1995, 2589–2594. [Google Scholar]
- 55. Oka Y., Tamaoki N., Inorg. Chem. 2010, 49, 4765–4767. [DOI] [PubMed] [Google Scholar]
- 56. Saadioui M., Reynier N., Dozol J.-F., Asfari Z., Vicens J., J. Inclusion Phenom. 1997, 29, 153–165. [Google Scholar]
- 57. Reynier N., Dozol J. F., Saadioui M., Asfari Z., Vicens J., Tetrahedron Lett. 1998, 39, 6461–6464. [Google Scholar]
- 58. Pipoosananakaton B., Sukwattanasinitt M., Jaiboon N., Chaichit N., Tuntulani T., Bull. Korean Chem. Soc. 2000, 21, 867–874. [Google Scholar]
- 59. Shinkai S., Ishihara M., Ueda K., Manabe O., J. Chem. Soc. Chem. Commun. 1984, 727–729. [Google Scholar]
- 60. Tylkowski B., Trojanowska A., Marturano V., Nowak M., Marciniak L., Giamberini M., Ambrogi V., Cerruti P., Coord. Chem. Rev. 2017, 351, 205–217. [Google Scholar]
- 61. Bianchi A., Delgado-Pinar E., García-Espãna E., Giorgi C., Pina F., Coord. Chem. Rev. 2014, 260, 156–215. [Google Scholar]
- 62. Moreno J., Grubert L., Schwarz J., Bléger D., Hecht S., Chem. Eur. J. 2017, 23, 14090–14095. [DOI] [PubMed] [Google Scholar]
- 63. Xia X., Yu H., Wang L., ul-Abdin Z., RSC Adv. 2016, 6, 105296–105316. [Google Scholar]
- 64. Kume S., Kurihara M., Nishihara H., Inorg. Chem. 2003, 42, 2194–2196. [DOI] [PubMed] [Google Scholar]
- 65. Kume S., Murata M., Ozeki T., Nishihara H., J. Am. Chem. Soc. 2005, 127, 490–491. [DOI] [PubMed] [Google Scholar]
- 66. Yamamura M., Yamakawa K., Okazaki Y., Nabeshima T., Chem. Eur. J. 2014, 20, 16258–16265. [DOI] [PubMed] [Google Scholar]
- 67. Yamamura M., Okazaki Y., Nabeshima T., Chem. Commun. 2012, 48, 5724–5726. [DOI] [PubMed] [Google Scholar]
- 68. Tang H.-S., Zhu N., Yam W.-W. V., Organometallics 2007, 26, 22–25. [Google Scholar]
- 69. Gale P. A., Howe E. N. W., Wu X., Spooner M. J., Coord. Chem. Rev. 2018, 375, 333–372. [Google Scholar]
- 70. Chen L., Berry S. N., Wu X., Howe E. N. W., Gale P. A., Chem 2020, 6, 61–141. [Google Scholar]
- 71. Lee S., Flood A. H., J. Phys. Org. Chem. 2013, 26, 79–86. [Google Scholar]
- 72. Shimasaki T., Kato S. I., Ideta K., Goto K., Shinmyozu T., J. Org. Chem. 2007, 72, 1073–1087. [DOI] [PubMed] [Google Scholar]
- 73. Li Z. Y., Zhang C., Ren Y. L., Yin J., Liu S. H., Org. Lett. 2011, 13, 6022–6025. [DOI] [PubMed] [Google Scholar]
- 74. Wezenberg S. J., Vlatkovic M., Kistemaker J. C. M., Feringa B. L., J. Am. Chem. Soc. 2014, 136, 16784–16787. [DOI] [PubMed] [Google Scholar]
- 75. Wezenberg S. J., Feringa B. L., Org. Lett. 2017, 19, 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vlatković M., Feringa B. L., Wezenberg S. J., Angew. Chem. Int. Ed. 2016, 55, 1001–1004. [DOI] [PubMed] [Google Scholar]
- 77. Wang Y., Bie F., Jiang H., Org. Lett. 2010, 12, 3630–3633. [DOI] [PubMed] [Google Scholar]
- 78. Hua Y., Flood A. H., Chem. Soc. Rev. 2010, 39, 1262–1271. [DOI] [PubMed] [Google Scholar]
- 79. Hua Y., Flood A. H., J. Am. Chem. Soc. 2010, 132, 12838–12840. [DOI] [PubMed] [Google Scholar]
- 80. Lee S., Hua Y. R., Flood A., J. Org. Chem. 2014, 79, 8383–8396. [DOI] [PubMed] [Google Scholar]
- 81. Parks F. C., Liu Y., Debnath S., Stutsman S. R., Raghavachari K., Flood A. H., J. Am. Chem. Soc. 2018, 140, 17711–17723. [DOI] [PubMed] [Google Scholar]
- 82. Choi Y. R., Kim G. C., Jeon H. G., Park J., Namkung W., Jeong K. S., Chem. Commun. 2014, 50, 15305–15308. [DOI] [PubMed] [Google Scholar]
- 83. Yuan Y. X., Wang L., Han Y. F., Li F.-F., Wang H.-B., Tetrahedron Lett. 2016, 57, 878–882. [Google Scholar]
- 84. Dabrowa K., Niedbala P., Jurczak J., Chem. Commun. 2014, 50, 15748–15751. [DOI] [PubMed] [Google Scholar]
- 85. Garcia-Amorós J., Díaz-Lobo M., Nonell S., Velasco D., Angew. Chem. Int. Ed. 2012, 51, 12820–12823. [DOI] [PubMed] [Google Scholar]
- 86. Bléger D., Schwarz J., Brouwer A. M., Hecht S., J. Am. Chem. Soc. 2012, 134, 20597–20600. [DOI] [PubMed] [Google Scholar]
- 87. Dabrowa K., Niedbala P., Jurczak J., J. Org. Chem. 2016, 81, 3576–3584. [DOI] [PubMed] [Google Scholar]
- 88. Dąbrowa K., Jurczak J., Org. Lett. 2017, 19, 1378–1381. [DOI] [PubMed] [Google Scholar]
- 89. Cafeo G., Kohnke F. H., Mezzatesta G., Profumo A., Rosano C., Villari A., White A. J. P., Chem. Eur. J. 2015, 21, 5323–5327. [DOI] [PubMed] [Google Scholar]
- 90. Busseron E., Lux J., Degardin M., Rebek J., Chem. Commun. 2013, 49, 4842–4844. [DOI] [PubMed] [Google Scholar]
- 91. Bandara H. M. D., Friss T. R., Enriquez M. M., Isley W., Incarvito C., Frank H. A., Gascon J., Burdette S. C., J. Org. Chem. 2010, 75, 4817–4827. [DOI] [PubMed] [Google Scholar]
- 92. Bandara H. M. D., Basa P. N., Yan J., Camire C. E., MacDonald J. C., Jackson R. K., Burdette S. C., Eur. J. Org. Chem. 2013, 4794–4803. [Google Scholar]
- 93. Emond M., Le Saux T., Maurin S., Baudin J.-B., Plasson R., Jullien L., Chem. Eur. J. 2010, 16, 8822–8831. [DOI] [PubMed] [Google Scholar]
- 94. Blanco V., Leigh D. A., Marcos V., Chem. Soc. Rev. 2015, 44, 5341–5370. [DOI] [PubMed] [Google Scholar]
- 95. Viehmann P., Hecht S., Beilstein J. Org. Chem. 2012, 8, 1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dai Z., Cui Y., Chen C., Wu J., Chem. Commun. 2016, 52, 8826–8829. [DOI] [PubMed] [Google Scholar]
- 97. Osorio-Planes L., Rodríguez-Esrich C., Pericàs M. A., Org. Lett. 2014, 16, 1704–1707. [DOI] [PubMed] [Google Scholar]
- 98. Lohse M., Nowosinski K., Traulsen N. L., Achazi A. J., von Krbek L. K. S., Paulus B., Schalley C. A., Hecht S., Chem. Commun. 2015, 51, 9777–9780. [DOI] [PubMed] [Google Scholar]
- 99. Goswami S., Ghosh K., Halder M., Tetrahedron Lett. 1999, 40, 1735–1738. [Google Scholar]
- 100. Hunter C. A., Togrul M., Tomas S., Chem. Commun. 2004, 108–109. [DOI] [PubMed] [Google Scholar]
- 101. Rakotondradany F., Whitehead M. A., Lebuis A. M., Sleiman H. F., Chem. Eur. J. 2003, 9, 4771–4780. [DOI] [PubMed] [Google Scholar]
- 102. Vollmer M. S., Clark T. D., Steinem C., Ghadiri M. R., Angew. Chem. Int. Ed. 1999, 38, 1598–1601. [DOI] [PubMed] [Google Scholar]
- 103. Arroyave F. A., Ballester P., J. Org. Chem. 2015, 80, 10866–10873. [DOI] [PubMed] [Google Scholar]
- 104. Goodman A., Breinlinger E., Ober M., Rotello V. M., J. Am. Chem. Soc. 2001, 123, 6213–6214. [DOI] [PubMed] [Google Scholar]
- 105. Mendez I. J. L., Pleizier J. S., Wang H.-B., Wisner J. A., J. Phys. Org. Chem. 2018, 31, e3805. [Google Scholar]
- 106.J. S. Pleizier, “Photo-Isomerizable Complementary H-bond Arrays” (2018). Electronic Thesis and Dissertation Repository. 5983. https://ir.lib.uwo.ca/etd/5983.
- 107. Vulcano R., Pengo P., Velari S., Wouters J., De Vita A., Tecilla P., Bonifazi D., J. Am. Chem. Soc. 2017, 139, 18271–18280. [DOI] [PubMed] [Google Scholar]