

Abstract
Scientists have long wondered how maternal diabetes, malnutrition and placental dysfunction impair fetal nephrogenesis. Anew study discovered a link between prenatal metabolic stress and nephron deficit via dysregulation of DNA methylation — an epigenetic mechanism that is essential for the renewal and differentiation of nephron progenitors.
Human nephrogenesis begins around week 5–8 of gestation and ends by week 34. Interestingly, the final number of nephrons at birth (the so-called nephron endowment) varies widely in humans (by approximately tenfold)1. Epidemiological studies have demonstrated that low nephron number at birth is associated with the development of chronic kidney disease and hypertension in later life2. Prenatal stressors, such as prematurity, maternal diabetes and exposure to nephrotoxins and infections, have been postulated to rewire the fetal nephrogenesis programme leading to renal hypoplasia, but how such adverse prenatal events reprogramme fetal nephrogenesis is unknown. New findings from Wanner et al.3 show that DNA methylation is a key mechanism that links the intrauterine environment with fetal nephrogenesis.
Using two rodent models of gestational stress (induced by hyperglycaemia and placental blood flow insufficiency — known causes of intrauterine growth restriction (IUGR)), Wanner et al.3 show that global DNA hypomethylation is associated with renal hypoplasia. They further show that conditional disruption of Dnmt1 in nephron progenitor cells (NPCs) impairs their renewal and differentiation. Dnmt1 encodes the maintenance DNA methyltransferase DNMT1, which faithfully methylates cytosine residues on daughter DNA strands in accordance with the methylation pattern of the parental DNA strands. Surprisingly, however, deletion of the de novo DNA methyltransferases, Dnmt3a and Dnmt3b — which unlike Dnmt1 can establish new methylation patterns — had no effect on NPC homeostasis. A strength of this study is the use of sophisticated quantitative 3D imaging reconstruction techniques to characterize the nephron progenitor deficits in the mutant kidneys. Specifically, the investigators used in vivo cell labelling with the nucleoside analogue 5-ethynyl-2’-deoxyuridine (EdU) and co-staining of the whole organ using NPC and other compartment-specific markers followed by optical projection tomography and confocal microscopy. The imaging phase was followed by quantitative analysis with modelling and integration of the morphogenetic events at a cell and tissue level in 3D space and across developmental time. The total number of NPC niches and NPC per niche can be quantified fairly accurately using these techniques. The researchers also used gene expression profiling to reveal that renal hypoplasia occurs in these models as a result of at least three mechanisms: a modest downregulation of a subset of genes that are important for NPC proliferation; downregulation of the WT1-WNT4 signalling axis, which is necessary for NPC differentiation; and perhaps the most novel finding of this study, derepression of the stress interferon-p53-p21 pathway as well as endogenous retroviral elements and non-lineage germline genes. Dnmt1 seems to uniquely affect self-renewing NPCs, as deletion of this methyltransferase in terminally differentiated podocytes did not affect glomerular development.
Despite their similar phenotypes, the magnitude of DNA hypomethylation in embryonic kidneys of mice with gestational stress reported by Wanner at al.3 is relatively modest in comparison with the almost complete DNA hypomethylation seen in NPCs of Dnmt1-deficient mice. This difference in hypomethylation raises the question of which specific intrarenal compartments (for example, cap mesenchyme, stroma or ureteric bud) are most affected by gestational stress. Moreover, since gene expression profiles of the nephrogenic niche in the gestational stress models were not reported, we do not know if prenatal metabolic stress and/or IUGR are accompanied by aberrant activation of the interferon-p53 pathway and retroviral elements, as observed in Dnmt1-deficient mice. Thus, while IUGR and Dnmt1 deficiency might both manifest renal hypoplasia, additional studies are needed to confirm that downstream pathways are similarly affected in the two models.
Studies from the past decade have identified a link between folate metabolism and IUGR that involves DNA methylation (reviewed elsewhere4). Folate, a coenzyme of one-carbon metabolism, has a direct role in the transfer of methyl groups in DNA methylation. Reduced folate intake or altered folate metabolism as can occur in IUGR might therefore directly affect fetal organ DNA methylation. This raises the question of whether the global reduction in DNA methylation in the stressed prenatal kidney, as observed by Wanner et al., may be partly related to altered one-carbon metabolism. Although this hypothesis has been challenged by studies of rats with gestational folate deficiency5, the bulk of evidence suggests that perinatal folate consumption is linked to DNA methylation and thus transcriptional regulation of developmental genes6. Thus, a simple strategy involving gestational supplementation of methyl donors, for example, in the form of folate or methionine, might be effective in restoring DNA methylation and gene expression in the NPCs affected by gestational stress.
Upregulation of the tumour suppressor and guardian of the genome, p53, in Dnmt1-mutant kidneys is reminiscent of the accumulation of p53 that occurs in developing kidneys of animals with IUGR induced by low protein intake during gestation7. Following genotoxic and metabolic stress, the p53 protein can be stabilized through post-translational modifications such as acetylation and phosphorylation. It should be noted that the embryo is very sensitive to even modest variations in p53 activity. We have shown8 that aberrant p53 protein stabilization in NPCs due to impaired degradation, resulting from conditional deletion of the p53 ubiquitin ligase MDM2, is sufficient to disrupt NPC renewal and differentiation — defects that were rescued by concomitant p53 gene deletion. These findings beg the question of whether restoration of normal p53 function by pharmacological inhibition might rescue NPC homeostasis in the context of IUGR.
The study by Wanner et al.3 reports derepression of retroviral elements in Dnmt1-mutant NPCs. Although this is a new finding in kidney cells, suppression of retroelements and germline genes by DNMTs and histone methyltransferases has been well characterized in embryonic stem cells and somatic cells9. Conversely, derepression of retroelements and germline genes along with DNA hypomethylation have been observed in cancer. Thus, derepression of these retroelements and genes not only disrupts organogenesis but might also predispose developing cells to neoplastic transformation.
The study by Wanner et al.3 therefore reports for the first time that DNA methylation is a key mechanism for the control of nephron progenitor renewal and differentiation and provides a mechanistic view of how DNA methylation might affect the gene expression landscape in the developing nephron. Further studies are needed to directly link IUGR in humans and in experimental models with altered DNA methylation of developmental pathways, as well as to identify preventive and therapeutic interventions that target metabolic and epigenetic dysregulation in IUGR (FIG. 1).
Fig. 1|. The link between gestational stress, epigenetic mechanisms and congenital nephron deficit.
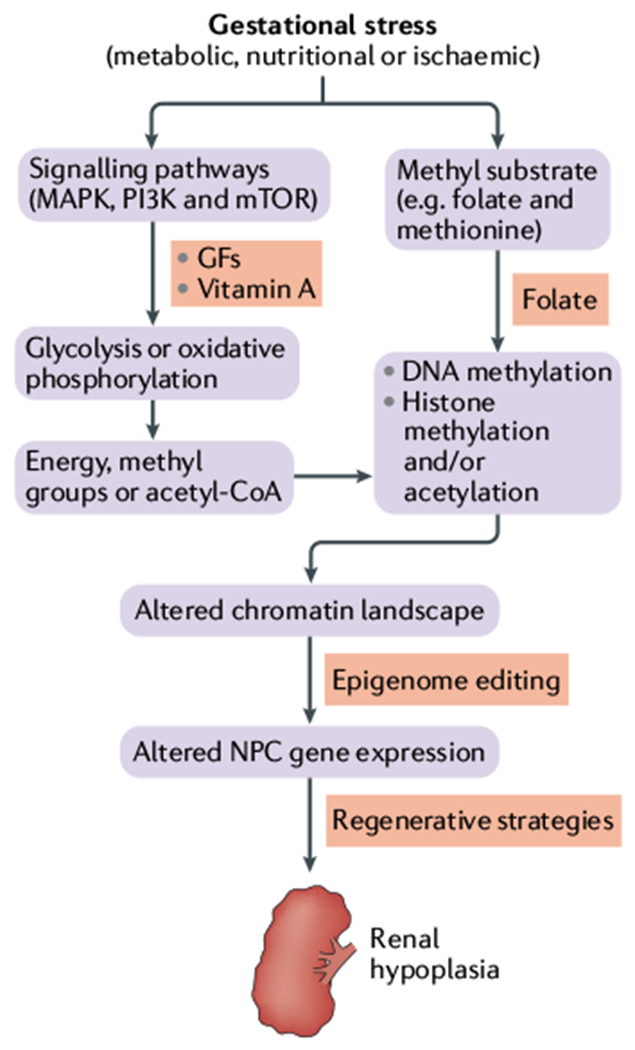
The renewal of nephron progenitor cells (NPCs), and thus maintenance of the nephrogenic niche, depends on activation of growth and proliferation pathways, such as those involving the mitogen-activated protein kinase (MAPK), phosphoinositide 3- kinase (PI3K) and mechanistic target of rapamycin (mTOR), and their downstream energy-producing pathways. A high level of glycolysis relative to oxidative phosphorylation favours NPC self-renewal. Glycolysis also provides acetyl-CoA and methyl group substrates for DNA and histone modifications. Intrauterine growth restriction (IUGR) can limit the supply of these essential substrates from the mother to the fetus, altering the chromatin landscape and leading to renal hypoplasia. Interventions to enhance availability of these substrates include supplementation with growth factors (GFs) to stimulate glycolysis and folate supplementation. Delivery of targeted epigenome editors might also modulate the accessibility of dynamic developmental enhancers that are affected by IUGR.
Acknowledgements
The author is funded by grant RO1 DK 114050 from the US National Institutes of Health.
Footnotes
Competing interests
The author declares no competing interests.
References
- 1.Hughson M et al. Glomerular number and size in autopsy kidneys: the relationship to birth weight. Kidney Int 63,2113–2122 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Keller G et al. Nephron number in patients with primary hypertension. N. Engl. J. Med 348, 101–108 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Wanner N et al. DNA methyltransferase 1 controls nephron progenitor cell renewal and differentiation. J. Am. Soc. Nephrol 30, 63–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKay JA, Williams EA & Mathers JC Folate and DNA methylation during in utero development and aging. Biochem. Soc. Trans 32, 1006–1007 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Maloney CA, Hay SM & Rees WD Folate deficiency during pregnancy impacts on methyl metabolism without affecting global DNA methylation in the rat fetus. Br. J. Nutr 97, 1090–1098 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Kim KC, Friso S & Choi SW DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. J. Nutr. Biochem 20,917–926 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pham TD et al. Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full-term IUGR rat kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol 285, R962–R970 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Hilliard SA, Yao X & El-Dahr SS Mdm2 is required for maintenance of the nephrogenic niche. Dev. Biol 387, 1–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karimi ΜM et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]