

Abstract
The use of replication-competent viruses for the treatment of cancer is an emerging technology that shows significant promise. Among the various different types of viruses currently being developed as oncolytic agents, retroviral replicating vectors (RRVs) possess unique characteristics that allow highly efficient, non-lytic, and tumor-selective gene transfer. By retaining all of the elements necessary for viral replication, RRVs are capable of transmitting genes via exponential in situ amplification. Their replication-competence also provides a powerful means whereby novel and useful RRV variants can be generated using natural selection. Their stringent requirement for cell division in order to achieve productive infection, and their preferential replication in cells with defective innate immunity, confer a considerable degree of natural specificity for tumors. Furthermore, their ability to integrate stably into the genome of cancer cells, without immediate cytolysis, contributes to long-lasting therapeutic efficacy. Thus, RRVs show much promise as therapeutic agents for cancer and are currently being tested in the clinic. Here we describe experimental methods for their production and quantitation, for adaptive evolution and natural selection to develop novel or improved RRV, and for in vitro and in vivo assessment of the therapeutic efficacy of RRVs carrying prodrug activator genes for treatment of cancer.
1. INTRODUCTION
The idea that replication-competent viruses might be used for the treatment of cancer originated more than a century ago, with the first documented case report in 1904 of leukemia remission after influenza infection (Dock, 1904). In 1922, it was reported that vaccinia virus can inhibit rodent tumors (Levaditi and Nicolau, 1922), and during its heyday in the 1950s-1970s, the field of oncolytic virotherapy witnessed numerous clinical trials testing a variety of different viruses administered to patients with advanced cancers (Hammill et al., 2010; Kelly and Russell, 2007). However, in most cases, tumor regression achieved by viral oncolysis was found to be transient, typically followed by immune clearance of the virus, and subsequent tumor recurrence. Faced with such invariably disappointing results, these clinical trials were largely abandoned with the advent of modern chemotherapy and radiation therapy. For many years, the field of oncolytic virotherapy was largely forgotten and has virtually been ignored in recent chronicles recounting the development of modern cancer treatment (Davis, 2007; Mukherjee, 2010; Olson, 1989).
Fast forward to the mid-1980s: advances in molecular biology provided the tools to engineer viruses into efficient gene transfer vectors, spawning the field of gene therapy. However, in clinical trials of gene therapy conducted over the ensuing decade, conventional non-replicating vectors generally showed disappointing levels of gene transfer and inadequate therapeutic effectiveness (Friedmann, 1996; McCormick, 2001; Orkin and Motulsky, 1995). Accordingly, in recent years, there has been renewed interest in the use of tumor-selectively replicating forms of viruses and viral vectors for the treatment of cancer. Various types of replicating viruses, including adenovirus, herpesvirus, reovirus, poliovirus, rhabdovirus, paramyxoviruses, and vaccinia virus, have entered clinical trials (for recent general reviews, see: (Donnelly et al., 2011; Eager and Nemunaitis, 2011). While all other virus species being investigated for this purpose are inherently cytolytic, retroviral replicating vectors (RRVs), based on simple gammaretroviruses such as murine leukemia virus (MLV), possess properties that render them uniquely well suited for use in cancer therapy as a non-lytic tumor-selective gene transfer agent.
As with MLV itself, MLV-derived RRVs have a strict requirement for cell division, at least in part because their nucleocapsids lack nuclear localization signals for active transport across intact nuclear membranes in quiescent cells, and infection is innately restricted to cells that are mitotically active (Lewis and Emerman, 1994; Roe et al., 1993; Seamon et al., 2002). As most normal cells in the body are quiescent, RRV-mediated gene transfer in vivo is largely restricted to the rapidly growing cells of malignant tissue. Furthermore, intrinsic restriction factors activated by innate immunity such as APOBEC-3G, TRIM-5α, tetherin, etc. (Douville and Hiscott, 2010; Jolly, 2011; Oliveira et al., 2010; Takeuchi and Matano, 2008), as well as humoral and cellular adaptive immune responses including neutralizing antibodies and cytotoxic T lymphocytes (Biasi et al., 2011; Hein et al., 1995; Kende et al., 1981), are active against retrovirus infection in normal cells. In contrast, signaling pathways activating innate immunity are frequently mutated or lost in cancer cells (Critchley-Thorne et al., 2009), and the tumor microenvironment is immunosuppressive to adaptive immunity (Flavell et al., 2010; Zitvogel et al., 2006), thereby forming a niche for preferential replication of RRVs in vivo. Since retroviruses permanently integrate their reverse-transcribed cDNA into the cancer cell genome and newly-formed virions bud off from the surface of infected cells without causing cytolysis, RRVs can spread relatively stealthily and achieve widespread gene transfer throughout tumors without harming cells or triggering a strong antiviral immune response that could impair or prevent viral propagation. In contrast, oncolytic viruses kill cells as a natural part of their replication cycle, resulting in piecemeal destruction of the microenvironment shielding them from immune clearance.
The cell-killing function of RRVs lies in the protein encoded by the transgene, and to date most studies evaluating these vectors for cancer therapy have employed prodrug activator (‘suicide’) genes, which generally encode metabolic enzymes that can convert an inactive substrate into an active chemotoxin (Kirn et al., 2002). As noted above, RRVs can stealthily mediate efficient delivery and stable expression of prodrug activator genes throughout the tumor, and subsequently, simultaneous en masse killing of infected cells can be effected at the desired time through administration of the corresponding prodrug. This strategy thus permits a substantially greater amplification of viral replication and spread as compared with other oncolytic viruses, as each cell upon infection becomes a stable virus-producing cell itself, rather than undergoing immediate lysis. Based on highly promising studies conducted by multiple groups in a variety of experimental cancer models (Dalba et al., 2005; Hiraoka et al., 2006; Hiraoka et al., 2007; Hlavaty et al., 2011; Kikuchi et al., 2007a; Kikuchi et al., 2007b; Logg et al., 2001b; Metzl et al., 2006; Solly et al., 2003; Tai et al., 2010; Tai et al., 2005; Wang et al., 2006; Wang et al., 2003), a clinical trial of an RRV encoding the yeast cytosine deaminase (CD) prodrug activator gene for treatment of brain cancer has been initiated and is currently underway in the United States (www.clinicaltrials.gov, NCT01156584).
RRVs are relatively easy to design and construct, due to the small size of their genomes and the breadth of knowledge of MLV biology that already exists, and the production of moderately high titer virus preparations is rapid and simple. The ability of RRV to greatly amplify itself in situ furthermore largely abrogates the need for extensively concentrated or massive doses of virus. These vectors are encoded on a single plasmid harboring a full-length vector provirus. No accessory plasmids are required for virus production as is the case with standard defective vector systems. Initial production of most RRV is driven in transfected cells by the cytomegalovirus (CMV) promoter, which is present within the 5' LTR in place of the MLV U3 sequence. In 293T human embryonic kidney cells, which are used for virus production, expression from the CMV promoter is higher than from the enhancer/promoter of the MLV U3 region. As described in further detail below, upon infection with virus produced by transfection, the CMV sequence is replaced with the wild type MLV enhancer promoter of the U3 region.
Earlier versions of RRVs, developed in the 1980s, contained the transgene sequences within the long terminal repeat (LTR) (Lobel et al., 1985; Reik et al., 1985; Stuhlmann et al., 1989). These earlier vectors, however, exhibited a strong tendency to rapidly delete the inserted transgene sequences upon replication. We therefore developed a different RRV design, in which the transgene is located immediately downstream of the env stop codon (Logg et al., 2001a; Logg et al., 2001b). We and others (Metzl et al., 2006; Paar et al., 2007) have found that this configuration affords much greater genomic stability, and allows replication kinetics comparable to that of wild type MLV.
2. VIRUS PRODUCTION BY TRANSIENT TRANSFECTION
To produce virus, we routinely perform calcium phosphate transfection of 293T human embryonic kidney cells in 10-cm cell culture dishes. The transfection protocol below is for transfection of one 10-cm dish. If more dishes will be transfected or if culture vessels of different size are used, the components can be scaled in proportion to total surface area. A low-endotoxin Maxi-prep kit (e.g., an Invitrogen HiPure or Qiagen Endo-Free kit) is used to generate plasmid preparations of adequate purity for high transfection efficiency. We have found that the E. coli strains DH-5α, DH10B, and derivatives such as NEB-5α (NEB) and TOP10 (Invitrogen) serve as good hosts for propagating RRV plasmids, reliably providing high yields of high-quality plasmid without recombination.
2.1. Required materials
2.1.1. Cell culture
SV40 T antigen-transformed human embryonic kidney (293T) cells: 293T cells may be obtained from the American Type Culture Collection (ATCC; catalog no. CRL-11268).
Culture medium: Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin.
The 293T cells are maintained in the above culture medium at 37°C and 5% CO2 in a humidified incubator, and are split every 3-4 days at a ratio between 1:4 and 1:8. During propagation, cells should not be allowed to reach full confluency at any point as this may reduce transfection efficiency. Frozen aliquots of early passage stocks should be maintained and thawed periodically, as optimal transfection efficiencies are obtained with cells that have been passaged fewer than 40 times.
2.1.2. Reagents and solutions
HEPES-buffered saline (HBS) solution: Prepare 2X HBS stock by adding the following to 400 ml dH2O: 50 ml 1 M HEPES, 28 ml 5 M NaCl and 1.5 ml 0.5 M Na2HPO4. After adjusting the pH to 7.05-7.10 with 5 M and 1 M NaOH, bring the total volume to 500 ml with dH2O.
Calcium chloride solution: Prepare 2.5 M CaCl2 by bringing 3.68 g of cell culture grade reagent (dihydrate form, Sigma cat# C7902) to 10 ml with dH2O.
Sodium butyrate solution: Prepare 0.5 M sodium butyrate (NaB; Sigma cat# B5887) by bringing 551 mg to 10 ml with dH2O.
The HBS, CaCl2 and NaB solutions should all be sterile filtered with 0.2 μM filters, aliquoted and stored at −20°C. The solutions are stable at this temperature for several months.
Poly-L-lysine solution (to enhance cell adhesion to culture dishes): 0.01% poly-L-Lysine solution is obtained from Sigma (cat# P4832). Coating of culture dishes with poly-L-lysine allows the cell monolayer to grow to high density without detaching.
2.2. Transfection procedure
2.2.1. One day before transfection
All procedures should be performed with aseptic technique in a biosafety hood. Coat one 10-cm cell culture dish with poly-L-lysine solution. This is carried out by pipetting 3-4 ml of 0.01% poly-L-lysine solution onto the dish, distributing it over the entire surface and then removing all excess liquid. Allow dish to air dry for 10-15 minutes. If the dish will not be used right away, it can be wrapped in Saran wrap and stored at 4°C.
The day before the cells are to be transfected, seed 2-4 x 106 293T cells on the coated dish in 10 ml growth media. The number of cells plated should be such that, at the time of transfection, cell confluency is on the order of 50-75%. (Note: different sublines of 293T cells, and even the same subline after prolonged culture, may show different growth rates; hence for each batch of cells, various initial plating densities should be checked to determine what level of confluency results in optimal transfection efficiencies.)
2.2.2. The day of transfection
At least two hours before the transfection is performed, replace the medium on the cells with 10 ml of fresh medium.
- Cell transfection is performed using the following procedure:
- Bring 23 μg of the RRV plasmid to 450 μl with dH2O. Sterile filter by spinning in a 0.22 μm Costar Spin-X centrifuge tube filter at 15,000 × g for 2 minutes at room temperature.
- Add 50 μl 2.5 M CaCl2 to the plasmid solution and mix by gently flicking the tube.
- Pipet 500 μl of 2X HBS into a 5-ml polystyrene tube. Add the 500 μl DNA/CaCl2 solution dropwise to the HBS. Briefly vortex on medium speed and let sit at room temperature for 5 minutes.
- Distribute the 1 ml of precipitate drop-wise over the cells.
- Gently rock the dish back-and-forth and side-to-side and return to the incubator.
2.2.3. One day after transfection
Replace the medium with 10 ml of fresh medium containing 10 mM NaB.
6-8 hours later, completely aspirate the NaB-containing medium and gently replace with 5 ml fresh medium containing no NaB.
2.2.4. Two days after transfection
24 hours after the media change, collect the supernatant from cells and filter through a 0.45-μm surfactant-free cellulose acetate syringe filter.
Aliquot volumes of 0.2-1.0 ml directly into sterile microfuge tubes. If the virus will be used within one day, it may be kept at 4°C with negligible loss in titer. If the virus will not be used within one day, place immediately at −80°C for storage. Smaller volumes should be avoided for freezing as they can lead to an accelerated decrease in titer during storage.
2.3. Notes
If the RRV encodes a fluorescent reporter transgene, transfection efficiency can be readily assessed by flow cytometry. With an RRV encoding GFP, the percentage of GFP-positive 293T cells at two days post-transfection is typically 60-80%.
As the limit of efficient packaging capacity for RRVs is on the order of up to 1.5 kb of inserted transgene sequences in addition to the full-length retroviral genome, it is often difficult to incorporate both a marker gene and a therapeutic gene into the same vector. In this case, RRV titers can be determined by molecular methods, as described below.
3. VECTOR COPY NUMBER ASSAY FOR TITER DETERMINATION AND BIODISTRIBUTION STUDIES
In many cases, RRVs carrying therapeutic genes cannot be readily quantitated using reporter assays. An alternative approach is to determine the number of RRV copies stably integrated into cells or tissues of interest. A method of determining copy number employing quantitative PCR (qPCR) analysis of cellular DNA using fluorogenic 5' nuclease ("TaqMan") chemistry is described here. This assay can be used both for determination of biological titer of virus preparations and for evaluating RRV biodistribution in animals.
Advantages of this method include: A) its ability to detect RRV containing any transgene. As the assay specifically detects MLV sequence within the RRV it thus can be used for vectors carrying any insert; B) its measurement of biological titer, which may better correlate with functionality, rather than physical particle titer (as with any biological titer method, different cell lines can give widely different functional titers even when exposed to the same number of physical particles); C) the ability to measure copy number regardless of vector expression levels in transduced cells, which may be influenced by the strength of viral promoter activity in a particular cell type; D) the ability to determine titer without testing of a dilution series. In contrast, methods based on determination of the number of marker-expressing cells require low (<20%) transduction levels to preclude multiple infection of individual cells.
Additionally, this assay can distinguish between and quantitate contaminating vector plasmid versus bona fide reverse-transcribed virus in genomic DNA from transduced cells. This specificity relies on the mechanism whereby the retroviral long terminal repeats (LTR) at each end of the proviral genome are reconstituted during each replication cycle. The LTR consists of the U3, R and U5 regions, and during reverse transcription, the U3 region of the 3' LTR serves as template for reconstitution of the 5' LTR. Within most RRV vector plasmids, however, the 5' LTR contains the CMV promoter in place of U3, where it serves to drive robust initial transcription during virus production in transfected cells. As the start site of transcription is at the 5' border of the R region, the upstream CMV sequence is not included in the viral genomic transcript that is packaged into virions (Fig 1A), and in cells infected with these virions, the MLV U3 in the 3' LTR is copied over to the 5' LTR. Thus, a qPCR primer pair that selectively amplifies the sequence from the CMV promoter to the packaging signal (ψ) will be specific for vector plasmid, whereas a MLV U3-specific primer paired with the ψ primer will detect the reverse-transcribed virus (Fig. 1A). Using genomic DNA from uninfected cells spiked with serial dilutions of vector plasmid DNA containing either the U3 or CMV sequence in the 5' LTR, we found that reactions with oligonuclotides designed according to this strategy are highly specific for their respective targets and that the quantitation is linear over seven orders of magnitude, from 50 to 5 × 108 copies per reaction, with correlation coefficients of >0.99 (not shown).
Figure 1.

qPCR assay for determining provirus copy number and biological titer of RRV. (A) Strategy for detection of virus versus RRV plasmid contaminants. The stepwise changes in the RRV genome that occur through transcription of the plasmid and reverse transcription of the viral RNA are shown. Colored arrows indicate the binding location of primers that specifically amplify either RRV plasmid or virus. The use of a CMV-specific forward primer (red) will detect RRV plasmid in infected cells, either carried over from transfection or from another source, whereas a U3-specific primer (green) will detect only genuine reverse-transcribed RRV genomes. (B) RRV-mediated transmission of GFP expression following infection of cultured cells at low MOI with virus produced by transient transfection. In control cultures, AZT was included in the medium from the time of infection. GFP was detected by flow cytometry at 2, 4, 7, 10 and 13 days post-infection. (C) PCR quantitation of virus versus plasmid copies in genomic DNA from the same cultures as in panel B. The reactions employed a forward primer specific either for MLV U3 or for the CMV promoter. Note that the copy numbers per reaction below 50 are outside of the linear range of the assay. (D) Correlation between titer as determined by copy number determination by qPCR versus analysis of GFP expression by flow cytometry.
3.1. Preparation of template DNA
3.1.1. For vector titer determination
Plate cells of interest in 6-well tissue culture plates. Plate at a density such that 72 hours later the cultures will have just reached or be nearing confluence.
The following day, determine cell numbers in two of the wells using a hemacytometer. Add polybrene to a final concentration of 4 μg/ml to the remaining wells. Add 100 μl of the RRV of interest and gently swirl the plate to mix. For virus samples of very low titer (<104 TU/ml), this volume may be increased to up to half that of the culture medium to ensure that the quantitation is within the linear range of the assay.
24 hours after infection, replace the medium with an equal volume of fresh medium containing 50 μM AZT to block secondary replication cycles.
48 hours after infection, harvest the cells for extraction of genomic DNA as indicated below.
3.1.2. For vector biodistribution studies
At the desired time point following administration of RRV, animals are sacrificed and tissue samples are harvested for isolation of genomic DNA. Tissues may be placed at −80°C for storage if genomic DNA will not be immediately extracted. 10-25 mg of each tissue is processed with a column-based kit as indicated below. The expected yield is typically in the range of 3-30 μg DNA, and yield can vary by tissue.
3.1.3. Genomic DNA extraction
Commercially available kits, such as the PureLink Genomic DNA Mini Kit (Invitrogen) or DNeasy Blood & Tissue Kit with the optional Rnase A treatment (Qiagen), are used according to the manufacturer’s guidelines for genomic DNA extraction. The resulting DNA is high molecular weight (20-50 kb), but not so high as to make pipetting difficult, and is free of contaminating RNA that might confound spectrophotometric quantitation.
The concentration of each purified genomic DNA sample should be determined by spectroscopic measurement of absorbance at 260 nm, and run on a 0.5% agarose gel stained with ethidium bromide to confirm that the size is within the expected range. No low molecular weight smears, which may indicate RNA contamination or DNA degradation, should be present.
For use in qPCR reactions, all genomic DNA samples are normalized to 10 ng/μl in the same Tris-EDTA buffer supplied in the kit for elution.
3.2. qPCR assay for determination of RRV titer and copy number
3.2.1. Materials required
Real-time PCR instrument (e.g., Applied Biosystems 7900HT or Bio-Rad MyiQ2).
qPCR primers at 100 μM: U3 forward primer 5′-AGCCCACAACCCCTCACTC-3′; CMV forward primer 5′-GGTGGGAGGTCTATATAAGCAGAG-3′; reverse primer, 5′-TCTCCCGATCCCGGACGA-3′.
qPCR probe, at 100 μM: 5′-CCCCAAATGAAAGACCCCCGCTGACG-3′. The probe is labeled with a 5′ reporter and a 3′ quencher suitable for the qPCR instrument to be used. Common labels are FAM, HEX or TET at the 5′ end with TAMRA or BHQ1 at the 3′ end.
2X qPCR master mix (e.g., Applied Biosystems Taqman Gene Expression Master Mix or Bio-Rad iQ Super Mix).
Optical 96-well plates.
Optical adhesive film.
Centrifuge with swinging bucket rotor for microtiter plates.
3.2.2. Prepare standard curve
To determine absolute RRV copy numbers in gDNA samples, a standard curve is generated with RRV plasmid containing a wild type 5' LTR. Plasmids pAZE-GFP (Logg et al., 2001a) or pAZ3-GFP (Kimura et al., 2010) can be used for this purpose. The number of molecules in one ng of any given plasmid can be conveniently calculated with the following formula.
One ng of the 12,255-bp pAZE-GFP or 12,136-bp pAZ3-GFP plasmid thus contains approximately 74.8 million or 75.6 million copies, respectively. To derive a standard curve, the plasmid DNA is serially diluted in H2O to produce five separate 10-fold dilutions covering a range of 10 to 100,000 plasmid copies per μl. Each dilution will be amplified in triplicate, using 5 μl of diluted plasmid per reaction. The standard curve reactions are set up as summarized for the experimental samples in Table 1 (below), but the volume of water is reduced by 5 μl per reaction to accommodate the volume of plasmid.
Table 1.
Preparation of reagents for qPCR analysis
| Component | Volume per 30-μl reaction | Final concentration |
|---|---|---|
| 2X qPCR master mix | 15 μl | 1X |
| Forward primer | 0.15 μl | 500 nM |
| Reverse primer | 0.15 μl | 500 nM |
| Probe | 0.06 μl | 200 nM |
| Water | to 25 μl (to 20 μl for std. curve) |
— |
3.2.3. Perform qPCR reactions
Prepare two master mixes in accordance with the table below and the total number of wells to be used. One is for the experimental samples and one is for the standard curve. To allow for loss during pipetting, prepare 10% more than will be required for all wells.
Add 5 μl of template gDNA to each well. For the standard curve wells, use gDNA from uninfected cells.
Add 5 μl of diluted plasmid to the standard curve wells.
Add either 25 or 20 μl master mix to the experimental or standard curve wells, respectively.
Seal the plate with a sheet of optical adhesive film.
Spin the plate at 2000 rpm for 2 minutes in a swinging bucket rotor.
Run the plate in the qPCR instrument under the conditions specified in Table 2.
Table 2.
Time and temperature parameters for qPCR amplification
| Initial denaturation and enzyme activation | 1 cycle | 95°C x 10 min |
| Amplification | 40 cycles | 95°C x 15 sec |
| 60°C x 1 min |
3.2.3. Analysis of results
Using the qPCR instrument's software, obtain threshold cycle (Ct) values for each sample. The Ct is defined as the fractional cycle number at which fluorescence crosses a threshold set above background but sufficiently low to be within the exponential region of the amplification curve.
Export the results to Microsoft Excel.
For the standard curve reactions, plot the log of plasmid copy number versus Ct.
Right-click on a data point on the plot and select "Add trendline."
Under Trendline Options, select "linear" and check "Display equation on chart." The equation will be of the form y = mx+b, where m is the slope and b is the y-intercept.
- For each experimental sample, calculate the log of the copy number per well using the numbers for the y-intercept and slope obtained in the previous step using the following formula:
For the experimental samples, determine the copy number per well by taking the antilog of the log copy number. In Excel, this is calculated with the function = 10^X, where X is log copy number.
Calculate the copy number per cell by dividing the copy number with the number of cell equivalents represented by the 50 ng of gDNA present in the reaction. For normal human or mouse cells, 50 ng corresponds to approximately 7140 or 7460 cells, respectively. The mass of gDNA per cell for other species may be obtained at www.genomesize.com.
-
For calculation of titer (in transducing units per ml) use the following formula:
"Total cells" is the number of cells in the culture at the time of infection. The dilution factor is the ratio of the final culture volume to the volume of virus preparation added.
3.3. Illustrative results
We used this assay to monitor viral spread and to measure the level of plasmid carried over in cells exposed to RRV produced by transient transfection. PC-3 cells were infected at low multiplicity of infection (MOI) with RRV expressing GFP. The reverse transcriptase inhibitor 3′-azido-3′-deoxythymidine (AZT) was included in the medium of some cultures to 50 μM starting from the time of infection to inhibit replication, while others were given no AZT. At 2, 4, 7, 10 and 13 days post-infection, the cells were passaged and aliquots were taken for flow cytometric detection of GFP and for copy number determination by qPCR. Flow cytometry showed that in the absence of AZT, the virus spread with the expected sigmoidal kinetics, while inclusion of AZT had effectively prevented infection (Fig. 1B). Copy number determinations were carried out with either the plasmid- or virus-specific primer/probe set. The results revealed that a substantial amount of plasmid DNA, as detected by the plasmid-specific oligo set, was indeed carried over from transfection, but by day 10 had dropped to undetectable levels (Fig. 1C). The virus-specific set showed that the cultures exposed to virus but not AZT underwent a burst of replication in the first four days after infection, followed by a peak and plateau, while those that contained AZT exhibited vastly lower, relatively constant signals comparable to that obtained with genomic DNA from naïve cells.
Measurement of the titers of a large number of independent RRV preparations using both this copy number assay and flow cytometric detection of GFP revealed a very strong correlation between the two methods (r2 = 0.9537, P<0.0001) (Fig. 1D). Titers as determined by qPCR, however, were roughly two- to four-fold higher than those determined by flow cytometry. This difference may at least in part be explained by the low level of GFP expressed from RRV integrated at some loci and the relatively low detection sensitivity of GFP fluorescence.
4. DEVELOPMENT OF NOVEL RRV USING MOLECULAR EVOLUTION
The ability of RRVs to replicate provides a powerful tool for vector development. Within a spreading virus population, genetic variants with characteristics providing a replicative advantage will eventually predominate. Such variant RRVs can then be cloned and employed as novel vectors. The genetic diversity in a population can either be experimentally introduced, by the construction of libraries of virus mutants, or it can occur naturally during replication. Retroviruses exhibit a high rate of spontaneous mutation, in large part due to errors by reverse transcriptase. The rate of base misincorporation per nucleotide for retroviruses is estimated to be about a millionfold higher than the rate for eukaryotic cells (Preston and Dougherty, 1996).
We and others have taken advantage of the propensity of retroviruses to naturally undergo genetic diversification to evolve novel RRVs with useful properties. The general approach is to passage a poorly replicating, or potentially even replication-defective RRV on susceptible cells, so to allow the random appearance of mutations that facilitate replication. This strategy has been employed in the isolation of more efficiently replicating (Barsov and Hughes, 1996) and less cytotoxic (Barsov et al., 2001) mutants of a Rous sarcoma virus-based RRV containing the env gene of 4070A MLV; to improve the replication efficiency of a doxycycline-dependent human immunodeficiency virus-based RRV (Marzio et al., 2001); and to generate robustly replicating MLV-GALV chimeras (Logg et al., 2007).
An advantage of this evolutionary approach over rational methods of vector development is that knowledge of the mechanism behind any given deficiency in replication is unnecessary. Furthermore, little experimental intervention is required: viral replication itself mediates the selection, and spontaneous mutations provide the genetic diversity. The guidelines provided here for adapting RRVs assume that a poorly- or non-replicating "prototype" RRV, designed to possess some novel property, is in hand.
4.1. Design and construction of prototype RRV for adaptation
The prototype vector might be constructed to contain, for example, a gene or cis-acting element from another viral strain or species, exogenous regulatory elements, a targeting moiety, etc. For the vector to be a viable candidate for molecular evolution, it should contain all of the elements necessary for viral replication, including genes for production of Gag, Pol and Env proteins and sequences for expression, reverse transcription and integration of the viral RNA. In order for the vector to be a viable candidate for molecular evolution, it should be capable of producing at least small amounts of transduction-competent particles upon transfection.
The presence of an easily assayed marker transgene in the RRV will greatly facilitate the titration and monitoring of replication of RRVs during attempts to evolve mutants. GFP is perhaps best suited for this purpose as it can be rapidly quantitated on a cell-by-cell basis by flow cytometry, and the Emerald and EGFP mutants are among the most sensitive fluorescent reporters when detected with a flow cytometer containing the common 488-nm argon laser. We therefore recommend that prototype RRVs be constructed to contain one of these two genes if possible.
4.2. Virus production
Virus is prepared by transfection with the prototype RRV plasmid as detailed in Section 2. As a control, a GFP-encoding RRV that is known to replicate in the cells to be used for infection should be prepared in parallel.
After harvest of virus, the transfected 293T cells are analyzed by flow cytometry to assess transfection efficiency and to confirm that expression from the vector plasmid is occurring.
The preparation should be titered on the cells of interest by flow cytometric detection of GFP, as described previously (Logg et al., 2002). Polybrene (hexadimethrine bromide; Sigma) should be included in the culture medium to 4 μg/ml during titer determinations and all other infections to maximize the available effective titer of prototype virus.
4.3. Molecular evolution and natural selection
To initiate replication, the cells of interest are infected with the prototype and control vectors at two or more MOIs between 0.05 and 0.5. If an MOI as high as 0.5 is not possible due to low titer, the maximum amount of virus supernatant the cells will tolerate (as a fraction of total media volume) should be used. While virus doses at the higher end of this range will provide greater genetic diversity in the initial infection, those at the lower end may be sufficient and will leave a greater number of cells for infection during secondary and later replication cycles.
The kinetics of vector spread is monitored by analyzing the cells by flow cytometry every 2-3 days thereafter for 1-2 weeks or until all of the cells are GFP-positive. Polybrene may also be included in the media throughout the culturing of the infected cells to enhance the effective titer of any virus produced from them and thereby stimulate the generation of genetic diversity. Adaptation is indicated by a change in replication kinetics.
If a poorly replicating prototype RRV exhibits no sign of horizontal infection during the 10-20 days following initial inoculation, the procedure may be repeated with a higher MOI to improve the likelihood that an adaptive mutation will occur during the initial transduction. Alternatively, the use of different cells as hosts may improve the ability of the virus to undergo initial replication and adaptation.
Once evidence of adaptation is observed in cultures infected with the prototype vector, the virus should be passaged through cultures of naive cells at low MOI (0.01 to 0.05) to confirm the improved phenotype and to further purify the adapted vector or vectors. The change in kinetics may not be readily apparent until such infection of naïve cells is performed with the passaged virus.
In preparation for cloning, 2-3 additional passages at low MOI are performed to further refine the population. During each passage the virus should be given enough time to infect >90% of the cells.
4.4. Identification and cloning of mutations
The passaged vector is screened for replication-enabling or -enhancing mutations by cloning and functional testing of PCR fragments amplified from integrated provirus in the cells used in the final virus passage.
PCR primers are designed to flank restriction sites in the prototype RRV plasmid so as to facilitate re-insertion of the amplified sequence into the prototype RRV plasmid for testing and subsequent sequencing.
Genomic DNA is prepared from cell lines infected with the passaged virus according to the procedures described in Section 3.1.
Subregions of the provirus of 2-4 kb are amplified from genomic DNA for this purpose using a very high-fidelity polymerase such as Phusion (NEB) or PfuUltra II Fusion (Agilent).
The amplification products are fractionated by agarose electrophoresis, gel extracted, cut at the selected restriction sites and cloned back into the parental prototype RRV plasmid for testing. The pool of re-cloned RRV plasmids are transformed into a suitable bacterial strain such as DH-5α or DH10B, or a derivative thereof, as recommended above.
As the cells may include a mixture of proviruses containing both adaptive and incidental mutations, or no mutations, several plasmid clones are picked and prepped from bacteria. Miniprep-scale amounts of plasmid are normally sufficient for the screening process.
Virus is prepared by transfection of 293T cells as detailed in Section 2, but scaled down for transfection of single wells of 6-well or 12-well plates with minipreps of each plasmid clone.
Virus preparations are individually harvested, and tested for titer and replication on the same cell type previously used for evolutionary adaptation.
The replication kinetics of each clone is compared to that of the prototype and control RRVs by infection at low MOI followed by monitoring of GFP expression by flow cytometry. Improved kinetics in any of the clones relative to the prototype vector will indicate the presence of adaptive mutations. If no improvement is detected, a different region of the vector should be amplified and tested using the same procedure until one is observed.
After clones exhibiting improved replication are obtained, the entire inserted fragment is sequenced. Since the insert may also contain mutations unrelated to the adaptation, multiple independent clones exhibiting the phenotype should be sequenced. The existence of particular mutations in multiple clones will suggest an adaptive role. If necessary, the importance of these mutations can be confirmed by testing them in isolation after introduction into the prototype vector plasmid.
4.5. Illustrative example
Figure 2 shows the results of infections with an impaired prototype vector, GZAP-GFP, and an efficiently replicating control, AZE-GFP. The two are identical MLV-based RRV, except that the former contains the env gene of GALV and the latter contains the env gene of MLV 4070A. The GALV Env protein utilizes the Pit-1 receptor to mediate viral entry into cells whereas the 4070A Env utilizes Pit-2. While AZE-GFP spread efficiently and without delay, GZAP-GFP exhibited a lag phase of about 9 days before rapid replication commenced (Fig. 2A). Transfer of the supernatants from the infected cultures to naïve cells demonstrated that during the initial passage, GZAP-GFP had acquired the ability to spread with kinetics similar to that of AZE-GFP (Fig. 2B). Cloning and testing of the passaged GZAP-GFP revealed that it had acquired a mutation in the MLV 3' splice site, causing an increase in the level of spliced viral RNA and thereby greatly improving replication.
Figure 2.
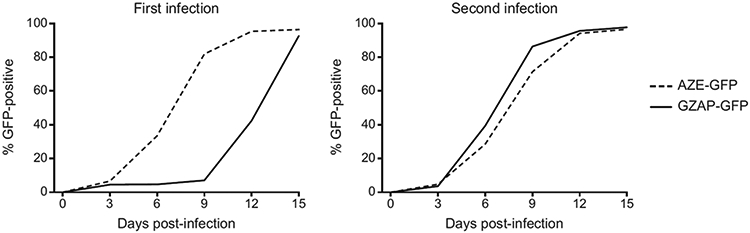
Adaptation of a prototype RRV. A: (left panel) Replication of AZE-GFP, an efficiently replicating amphotropic MLV RRV, and GZAP-GFP, a replication-impaired prototype MLV-GALV hybrid RRV, following initial infection of cultured cells at equal MOI. The virus used for these infections were generated by transient transfections with the corresponding plasmids and replication in infected cells was monitored by flow cytometry. B: (right panel) Infection of fresh cells with virus taken from day 15 of the initial infection, demonstrating greatly improved kinetics for GZAP-GFP.
5. MTS ASSAY OF RRV-MEDIATED CELL KILLING IN VITRO
In this section a procedure is presented for assessing of the capacity of RRVs encoding a prodrug activator (‘suicide’) gene to kill tumor cells in culture upon addition of prodrug. The procedure can be adapted for use in evaluating any prodrug activator gene/prodrug combination. Cytotoxicity is determined using a colorimetric assay that quantitates the reducing potential of cells, which is a measure of their metabolic function. Upon addition to culture medium, the chromogenic substrate MTS (3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium) is reduced by metabolically active cells into a product having a peak absorbance of 490 nm. The absorbance at this wavelength is directly proportional to the number of viable cells present.
In the example provided, U-87 MG malignant glioma cells are infected with an RRV expressing the yeast cytosine deaminase (CD) gene, which converts the non-toxic prodrug 5-Fluorocytosine (5-FC) to the classic chemotherapy drug 5-Fluorouracil (5-FU) (Kievit et al., 1999). Naïve or infected RRV-infected cancer cells are exposed to various concentrations of 5-Fluorocytosine (5-FC) to determine the IC50, the prodrug concentration at which cell viability is reduced by 50%. IC50 values are useful when comparing the potency of different prodrugs, different vectors carrying the same suicide gene, or different cell lines subjected to the same vector/prodrug treatment. An example kill curve from treatment of infected and uninfected U-87 cells exposed to several concentrations of 5-FC is given in Fig. 3. Statistical software was used to fit curves to the collected data points. As illustrated, the IC50 represents the concentration at which the curve reaches the midpoint between the upper and lower asymptotes.
Figure 3.
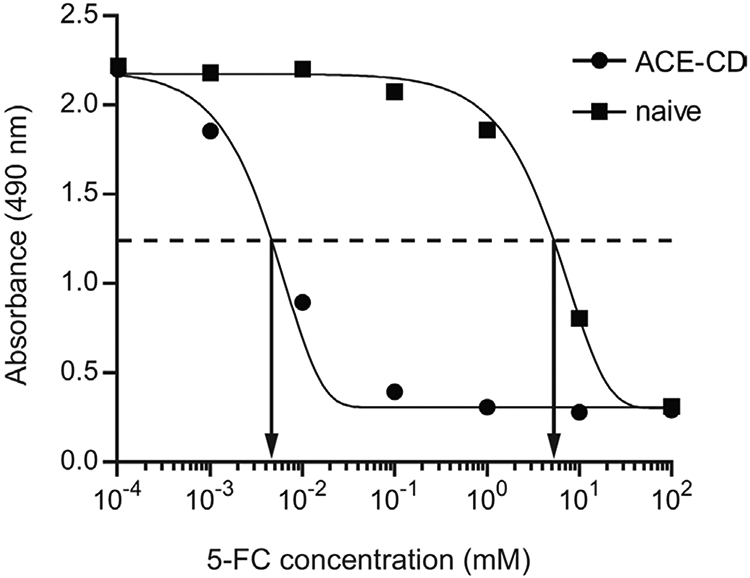
Illustration of IC50 determination by nonlinear regression analysis of MTS assay results. U-87 cells, either naive or infected with ACE-CD, were exposed to a series of 5-FC concentrations and cell viability was subsequently determined by the MTS assay. Sigmoidal dose-response curves were fit to each data set using Prism software. As shown, the IC50 values correspond to the concentration at which the curves cross the midpoint between the baseline and the maximal response, which is indicated by the dashed line.
5.1. Required materials
Flat-bottomed 96-well tissue culture plates.
U-87 human glioma cells (U-87 MG, ATCC catalog no. HTB-14).
Growth media for U-87 cells: DMEM supplemented with 10% fetal bovine serum.
Titered preparation of RRV virus encoding CD (e.g., ACE-CD).
5-Fluorocytosine (Sigma-Aldrich, catalog no. F7129): prepare 10 mg/ml aqueous stock solution, and sterile-filter through a 0.2-micron filter. Alternatively, pre-sterilized stock solutions are also commercially available (e.g., InvivoGen Inc., catalog no. sud-5fc).
MTS reagent (e.g., Promega CellTiter 96 AQueous One Solution Cell Proliferation Assay).
Spectrophotometric plate reader capable of reading absorbance at 490 nm (e.g. Spectra Max 190 Plate Reader).
GraphPad Prism 5 software.
5.2. Assay Procedures
Generate U-87 cells fully infected with ACE-CD: expose the cells to virus at an MOI of 1 and allow the infection to proceed for one week.
Optional: complete infection by can be verified by exposure of the cells to ACE-GFP at an MOI of 1, followed by flow cytometric analysis 3-4 days later. Interference mediated by the first virus should prevent any cells from becoming GFP positive.
Plate the infected cells and naïve control cells in 96-well plates at a density of 2,000 cells/well in growth medium containing 5-FC concentrations of 0, 0.5, 5, 50, 500, 5,000 and 50,000 μM. The final volume in each well should be 200 μl. For determination of background signal generated from the growth medium itself, set up "no-cell" wells containing 200 μl of medium alone. All samples should be in triplicate, and great care should be taken to plate the same number of cells in each well as consistently as possible (e.g., periodically invert and remix cell suspension between plating each row of wells). In order to avoid confounding effects of medium evaporation on assay results (Patel et al., 2005), either use only the inner 32 wells of the plate and fill the outer and any unused wells with 200 ul of sterile water, or use plates specifically designed to eliminate evaporation (e.g., Thermo Scientific Nunc Edge plates).
Incubate the plate at 37°C in a humidified, 5% CO2 atmosphere for 7 days. Note that the number of days necessary to reach a maximal therapeutic index will depend on the vector, suicide gene, prodrug and cells used and will need to be experimentally determined for other systems. Note that it is often difficult to maintain viability of cells in untreated control wells for longer than 8-10 days or so.
Thaw the MTS reagent and add 40 μL to each well. Return the plate to the incubator and leave for 2 hours.
Measure absorbance at 490 nm. If readings are below 1 absorbance unit for wells containing no 5-FC, incubate for another 30-60 minutes and repeat the measurement. Absorbances can be measured up to 4 h after addition of the MTS solution. To obtain the background-corrected absorbances, subtract the average reading from the three no-cell wells from all of the other readings.
- Enter data into GraphPad Prism to plot the kill curve and determine the IC50:
- Create an XY data table with triplicate data values. Enter the seven tested 5-FC concentrations into the X column. Enter the corrected absorbances from the infected cells into column A and those from the naïve cells in column B.
- Log transform the concentrations: click the "Analyze" button and select "Transform" from the list of analyses. In the Transform dialog box, check the "Transform X values using" box, select "X=Log(X)".
- Fit a four-parameter dose-response curve to the transformed data: From the data table, click "Analyze" and select "Nonlinear regression (curve fit)", then from the list of equations, under "Dose-response - Inhibition", choose "log(inhibitor) vs. response - Variable slope (four parameters)", click "OK."
- A new table will appear containing the IC50 values along with other statistics from the regression analysis.
6. IN VIVO GLIOMA MODEL FOR TESTING RRV-MEDIATED GENE THERAPY
Glioblastoma multiforme (GBM) is the most common primary brain tumor in adults, and due to its dismal prognosis and the lack of effective conventional therapeutic options, this disease offers a risk:benefit ratio that justifies its use as a target for testing various experimental treatment strategies, particularly gene therapy (Aghi and Chiocca, 2006; Dent et al., 2008; Rainov and Heidecke, 2011). Furthermore, lessons learned in treating primary brain tumors such as GBM can also be applied to CNS metastasis of systemic cancers, which occur in 10-30% of patients, and similarly carry a dire prognosis (Aragon-Ching and Zujewski, 2007). In fact, much of the preclinical testing which supported the entry of RRV into clinical trials for treatment of patients with recurrent GBM, was performed in subcutaneous and intracranial brain tumor models (Hlavaty et al., 2011; Tai et al., 2005; Wang et al., 2006; Wang et al., 2003).
Subcutaneous models of glioma provide a larger tumor mass and allow examination of a longer time course of replication without rapid lethality, representing a tumor size that (although still small) is closer to what would be encountered clinically. Procedures for establishment of subcutaneous tumors and testing RRV in these models have been previously described in detail (Logg and Kasahara, 2004).
Accordingly, here we will focus on the use of intracranial models of GBM. Intracranial tumor models are, of course, smaller in mass, but are more rapidly fatal, since death is induced through the same mechanisms of increased intracranial pressure and loss of neuroregulation that maintains respiratory function as seen in malignant gliomas in human patients. Intracranial GBM xenografts in immunodeficient hosts can be used to model intratumoral virus replication in human cells in the absence of confounding effects from the immune system, while intracranial syngeneic tumors in immunocompetent hosts can model viral replication and potential antiviral immune responses in the context of the immunosuppressive tumor microenvironment placed within the semiprivileged immunological milieu of the CNS.
In this illustrative example, intracranial tumor models are established by stereotactic intracerebral injection of U-87 MG human glioma cells in athymic nude mice (Tai et al., 2005; Wang et al., 2003). Other intracranial tumor models can also be used, for example Tu-2449 murine gliomas in syngeneic B6C3F1 mice (Hlavaty et al., 2011), or RG2 gliomas in syngeneic Fischer rats (Wang et al., 2006). Subsequently, animals receive intratumoral injections of RRV through the same stereotactic procedure. After allowing virus spread to proceed for 2 – 3 weeks, treatment groups then receive systemic injections of the non-toxic prodrug 5-FC, administered as a single or twice daily IP injection, while control groups receive PBS, for 7 to 8 consecutive days. Subsequently, these 7 – 8 day cycles of prodrug administration are repeated at 2 – 3 week intervals to achieve long-term survival. In previously published studies (Tai et al., 2005), survival could be maintained in the majority of treated animals receiving both vector and prodrug for >135 days, as compared to control groups receiving vector only, prodrug only, or no treatment, which all showed a median survival time on the order of 35 days (p<0.0001).
6.1. Required materials
6.1.1. Animals:
Athymic nude (nu/nu) laboratory mice (Mus musculus), Balb/c- or C57Bl/6-background strain, 6- 9 weeks old.
Note: Animals selected for use in this study should be as uniform in age and weight as possible. Each animal should be identified by ear tag or other standard method of identification. The animals should be housed individually in microisolator cages so they do not disturb each other’s wounds. The room in which the animals are kept should be well ventilated (> 10 air changes per hour) with 100% fresh air (no air recirculation), and maintained with a 12-hour light/12-hour dark cycle, and room temperatures between 18-26°C. All cages, equipment, bedding, and water for immunodeficient animals should be autoclaved prior to use, and rodent chow should be irradiated. All study animals should be acclimatized to their designated housing for 3-7 days prior to the first day of experimentation.
6.1.2. Therapeutic and Analgesic Agents
Ketamine/Xylazine: anesthetic/analgesic combination, 100 mg/kg Ketamine + 10 mg/kg Xylazine via single intraperitoneal (IP) injection, 1 hour prior to surgical procedures.
Bacitricin Zinc-Polymyxin S04 ointment: antibiotic for topical use, to cover wounds, once after surgery and as needed thereafter.
Sulfamethoxazole / Trimethoprim: antibiotic, per os (PO), 1 mg/40 ml in drinking water, for 5 days following intracranial procedures.
Betadine (povidone-iodine): topical use, applied once over the surgical site at the beginning of surgery to achieve sterile field.
Carprofen: Non-steroidal anti-inflammatory drug (NSAID), 5 mg/ml, single subcutaneous (SC) injection daily for 5 days following intracranial procedures.
Buprenex (buprenorphine): analgesic, 0.01 - 0.05 mg/kg SC or IP, once following cranial procedures; also to manage pain if any adverse effects are seen.
Sodium pentobarbital: 100 mg/kg IP, for euthanasia.
Phosphate-buffered saline (PBS) solution: sterile-filtered, given IP as needed for fluid replacement and as control vehicle for prodrug administration.
5-Fluorocytosine (Sigma-Aldrich, catalog no. F7129): 10 mg/ml stock solution, sterile-filtered.
6.1.2. Virus and cells
RRV carrying GFP marker gene (e.g., ACE-GFP)
RRV carrying yeast CD prodrug activator gene (e.g., ACE-CD).
Human embryonic kidney cell line 293T (for virus production) and human glioma cell line U-87 (for tumor establishment). Cell lines are cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin, and maintained in a humidified atmosphere with 5% CO2.
6.1.3. Surgical equipment
Stereotaxic mounting frame with micromanipulator, for mice (e.g., Narishige Instruments catalog no. SR-5M or SR-6M; or Stoelting Co. Lab Standard catalog no. 51600 or 51603).
Cordless surgical drill (e.g., Ideal Micro-Drill with 1 - 1.2 mm burr; or Dremel MiniMite cordless drill with 1/64 inch drill bit).
Electric shaver (e.g., Oster Mini-Clipper).
Electrical heating pad.
Surgical scalpels (#10 blade), forceps, scissors.
Serum separator tube (BDMicrotainer, #365956) for blood collection.
Tissue-Tek Optimal Cutting Temperature (OCT) compound and tissue sample cassettes, for embedding frozen sections.
Nunc cryovials for tissue collection and freezing.
Hamilton syringe, 700-series, 26s-gauge (e.g., catalog no. 20734 or 20779).
Calibrated stereotaxic injector apparatus (e.g., Stoelting Co. Stereotaxic Injector with universal adaptor for Hamilton syringes).
For IP and SC injections: 1 to 1 ml Luer-lock syringe with 20-25 gauge, 5/8 to 1 inch length needle (e.g., Becton-Dickinson BD Eclipse series).
6.2. Study Procedures
6.2.1. Stereotactic injection procedure for intracranial tumor establishment and intratumoral vector administration
Mice are anesthetized with ketamine/xylazine and mounted into a stereotaxic frame with blunt ear bars.
Once properly positioned and immobilized, the fur overlaying the scalp is shaved and betadine is swabbed to prepare the surgical site.
The animal is placed on a heating pad and a scalpel is used under sterile conditions to make a midline incision through the skin. Retraction of the skin and reflection of the fascia at the incision site allows for visualization of the skull.
A small burr hole is drilled in the skull, and a Hamilton syringe containing the tumor cells is positioned at the appropriate intracranial stereotaxic coordinates. The coordinates that we typically employ are AP= 3.0 mm, ML=2.0 mm, DV=3.0 mm (from midline, lambda, and dura, respectively). Exact coordinates for animals at different ages and weights should be determined in a pilot experiment by injecting dye and determining its location.
The tumor cell suspension (2 × 105 U-87 cells in 1-5 μl PBS) is injected into the brain. The injection should be performed very slowly over an interval of 5-10 minutes, using a calibrated injector assembly which allows precise control of injection speed and volume (e.g., screw-type plunger controller gradiated in 0.01mm increments, one complete revolution advances the plunger of the syringe 0.5mm), to minimize injection pressure and equilibrate with tissue pressure. If the plunger is advanced too rapidly, the injected cell suspension will immediately leak out via the route of least resistance, i.e., up the needle track.
Five minutes after completing the injection, the injector assembly is slowly raised out of the skull (again, rapid removal will result in a suction effect that causes the tumor cell suspension to leak out via the injection track) and the skin is closed with suture or surgical staples.
The animals are monitored during anesthesia recovery. Analgesics should be administered once before the end of the procedure, and again every 12-24 hours for up to 3 days. Antibiotics should be administered topically and in the drinking water for 5 days after surgery, and the animals should be monitored on a daily basis and weighed weekly after each procedure.
For intratumoral injection of virus vectors (RRV at titers of up to 106 total transduction units (TU) in 1-5 μl total volume), the stereotactic injection procedure is repeated at the same coordinates. Control animals are injected with PBS instead of vector solution. In the intracranial U-87 model, vector injection is generally performed 5 – 7 days after glioma cell inoculation, in order to allow an adequate time interval for virus spread before control animals succumb to intracranial tumor growth (starting at about 4 weeks post-tumor establishment).
6.2.2. Prodrug administration
After allowing adequate time for viral spread (generally 2-3 weeks post-intratumoral vector injection, which corresponds to 3-4 weeks post-tumor establishment), the prodrug (5-Fluorocytosine, 500 mg/kg) is administered systemically via intraperitoneal (IP) injection. Control animals should receive IP injections of PBS instead of prodrug.
Mice should be manually restrained, but need not be anesthetized for IP injections. The abdominal area is swabbed with Betadine or ethanol, and injection into the abdomen is performed with care taken to avoid the bladder.
Prodrug administration is performed daily for a period of 7 – 8 consecutive days. Depending on the tumor model, if an adequate therapeutic effect is not achieved in this time interval, twice daily IP injections of 5-Fluorocytosine at the same dose may also be performed.
Prodrug administration is stopped after 7 – 8 days and the animals are monitored daily. This 7- to 8-day cycle of prodrug or vehicle administration should be repeated at intervals of every 2 – 3 weeks. Of course, the exact duration and scheduling of these prodrug cycles to achieve the optimum therapeutic effect will depend on the particular tumor model used.
6.2.3. In-Life observations and measurements
Animals should be observed within their cages at least once daily throughout the acclimation and study period.
Starting Day -3 to -5 and continuing throughout the study, each animal should be observed for changes in general appearance and behavior. In particular, after intracranial procedures, if any one or combination of adverse symptoms (bleeding, paresis, hunching, inactivity or not feeding or grooming or weight loss >15% of baseline body weight) is observed and no sign of improvement is shown within a two-day period, the animal should be humanely euthanized.
Body weights should be measured prior to group assignment and weekly thereafter until termination.
6.2.4. Tissue collection
Animals should be fully anesthesized prior to initiation of terminal procedures and tissue collection. Urine is collected prior to blood collection via direct bladder tap, then transferred to a sterile 1.5mL tube and kept refrigerated.
Blood samples are also collected under anesthesia, by cardiac puncture. Half of the blood is transferred into a serum separator tube, and the collected serum is frozen at −80°C.
Euthanasia can be performed by sodium pentobarbital overdose, followed by cervical dislocation. Only sterile single-use instruments, draping, and collection tubes or containers should be used for all dissections and tissue collection, which should be conducted according to a protocol designed to minimize the possibility of cross-contamination by harvesting specimens in a fixed order: e.g., Skin → Axillary lymph node → Lung → Esophagus → Heart → Intestine → Kidney → Liver → Spleen → Ovary → Femur dissection / Bone Marrow flush → Spinal cord → Brain. Representative samples from each organ are harvested using sterile, single-use biopsy punches or scalpels, placed in sterile cryovials, and flash frozen in liquid nitrogen and stored at −80°C until analysis for vector biodistribution by qPCR as described in Section 3. Dissection of the femur to obtain bone marrow is performed from the distal portion of the thigh in order to prevent contamination from intraperitoneal organs. Tissues for immunohistochemistry are preserved by embedding in OCT media and freezing.
6.2.5. Statistical analysis
According to a statistical power analysis, n=14 animals per treatment group would be required to achieve a significant result at p<0.05 using Fisher's exact test, assuming 0% survival of the reference group and 50% survival with the treatment of interest at the same time of followup.
Survival curves are constructed by use of the Kaplan-Meier method and compared by the log-rank test, with P values of <0.05 considered statistically significant.
6.3. Notes
Based on the time course of cell killing observed in MTS in vitro assays over a period of 8 days (above), the majority of cell killing should occur within the first 4 to 5 days after exposure to prodrug, followed by asymptotic reduction in infected cells during the remaining few days. However, as 5-FU produced intracellularly in the infected tumor cells (by 5-FC prodrug conversion) generally acts through interfering with DNA synthesis, it appears that not all the infected cells are dividing within this time interval and hence a small fraction of infected cells remain without being killed.
In vivo, we have observed that further daily administration of prodrug beyond this time interval likely results in eventual complete loss of virus-infected tumor cells, but this is actually associated with a shorter duration of survival (Wang et al., 2003), as these cells seem to act as a reservoir for re-infection of recurrent tumor growth.
Accordingly, we have found that to achieve an optimal therapeutic effect and long-term survival in intracranial glioma models, 7 – 8 day cycles of daily prodrug administration should be interspersed with periods of no treatment for up to 3 to 4 weeks to allow re-infection during tumor recurrence (Tai et al., 2005). Thus, a single injection of RRV carrying a prodrug activator gene can achieve long-lasting therapeutic benefit with repeated cycles of non-toxic prodrug administration, without incurring the systemic adverse side effects associated with conventional chemotherapy.
Figure 4.


A: The concept of Controlled Active Gene Transfer: Vector spreads through tumor but not normal tissue, hence the prodrug is activated into an anti-cancer drug only in the infected tumor cells. In the case of human clinical trials currently being conducted by Tocagen Inc., the RRV contains the cytosine deaminase (CD) gene from yeast. This enzyme converts the prodrug 5-FC, an FDA-approved anti-fungal compound that can be taken as an oral pill, into the anti-cancer drug 5-FU directly within the RRV-infected cancer cells. B: RRV-mediated prodrug activator gene therapy can achieve long-term survival in a U-87 intracranial glioma model. PBS: Control group injected with phosphate-buffered saline instead of vector and prodrug. 5-FC: Control group injected with PBS instead of RRV, followed by systemic prodrug administration. ACE-CD: Control group injected with RRV expressing CD gene, but without receiving prodrug afterward. ACE-CD/5-FC: Treatment group receiving both RRV and prodrug. In the treatment group, only a single injection of vector was performed (green circle), followed by multiple cycles of prodrug (red circle).
ACKNOWLEDGEMENTS
This work was supported in part by Tocagen Inc., NIH grants R01 CA121258 and U01 NS059821 (to NK), and a pilot project grant from the UCLA CURE Digestive Disease Research Center (to CRL). We would like to acknowledge the UCLA Vector Core & Shared Resource facility for their assistance with vector construction and characterization.
REFERENCES
- Aghi M, Chiocca EA, 2006. Gene therapy for glioblastoma. Neurosurg Focus. 20, E18. [PubMed] [Google Scholar]
- Aragon-Ching JB, Zujewski JA, 2007. CNS metastasis: an old problem in a new guise. Clin Cancer Res. 13, 1644–7. [DOI] [PubMed] [Google Scholar]
- Barsov EV, Hughes SH, 1996. Gene transfer into mammalian cells by a Rous sarcoma virus-based retroviral vector with the host range of the amphotropic murine leukemia virus. J Virol. 70, 3922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsov EV, et al. , 2001. Adaptation of chimeric retroviruses in vitro and in vivo: isolation of avian retroviral vectors with extended host range. J Virol. 75, 4973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasi G, et al. , 2011. Immune response to Moloney-murine leukemia virus-induced antigens in bone marrow. Immunol Lett. 138, 79–85. [DOI] [PubMed] [Google Scholar]
- Critchley-Thorne RJ, et al. , 2009. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci U S A. 106, 9010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalba C, et al. , 2005. Beyond oncolytic virotherapy: replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr Gene Ther. 5, 655–67. [DOI] [PubMed] [Google Scholar]
- Davis D, 2007. The Secret History of the War on Cancer. Basic Books, New York, NY. [Google Scholar]
- Dent P, et al. , 2008. Searching for a cure: gene therapy for glioblastoma. Cancer Biol Ther. 7, 1335–40. [DOI] [PubMed] [Google Scholar]
- Dock G, 1904. Influence of complicating diseases upon leukaemia. J Med Sci. 127, 563–592. [Google Scholar]
- Donnelly OG, et al. , 2011. Recent Clinical Experience With Oncolytic Viruses. Curr Pharm Biotechnol. [DOI] [PubMed]
- Douville RN, Hiscott J, 2010. The interface between the innate interferon response and expression of host retroviral restriction factors. Cytokine. 52, 108–15. [DOI] [PubMed] [Google Scholar]
- Eager RM, Nemunaitis J, 2011. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 18, 305–17. [DOI] [PubMed] [Google Scholar]
- Flavell RA, et al. , 2010. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 10, 554–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann T, 1996. The maturation of human gene therapy. Acta Paediatr. 85, 1261–5. [DOI] [PubMed] [Google Scholar]
- Hammill AM, et al. , 2010. Oncolytic virotherapy reaches adolescence. Pediatr Blood Cancer. 55, 1253–63. [DOI] [PubMed] [Google Scholar]
- Hein A, et al. , 1995. Effects of adoptive immune transfers on murine leukemia virus-infection of rats. Virology. 211, 408–17. [DOI] [PubMed] [Google Scholar]
- Hiraoka K, et al. , 2006. Tumor-selective gene expression in a hepatic metastasis model after locoregional delivery of a replication-competent retrovirus vector. Clin Cancer Res. 12, 7108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka K, et al. , 2007. Therapeutic efficacy of replication-competent retrovirus vector-mediated suicide gene therapy in a multifocal colorectal cancer metastasis model. Cancer Res. 67, 5345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavaty J, et al. , 2011. Comparative evaluation of preclinical in vivo models for the assessment of replicating retroviral vectors for the treatment of glioblastoma. J Neurooncol. 102, 59–69. [DOI] [PubMed] [Google Scholar]
- Jolly C, 2011. Cell-to-cell transmission of retroviruses: Innate immunity and interferon-induced restriction factors. Virology. 411, 251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E, Russell SJ, 2007. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 15, 651–9. [DOI] [PubMed] [Google Scholar]
- Kende M, et al. , 1981. Naturally occurring humoral immunity to endogenous xenotropic and amphotropic type-C virus in the mouse. Int J Cancer. 27, 235–42. [DOI] [PubMed] [Google Scholar]
- Kievit E, et al. , 1999. Superiority of yeast over bacterial cytosine deaminase for enzyme/prodrug gene therapy in colon cancer xenografts. Cancer Res. 59, 1417–21. [PubMed] [Google Scholar]
- Kikuchi E, et al. , 2007a. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 14, 279–86. [DOI] [PubMed] [Google Scholar]
- Kikuchi E, et al. , 2007b. Highly efficient gene delivery for bladder cancers by intravesically administered replication-competent retroviral vectors. Clin Cancer Res. 13, 4511–8. [DOI] [PubMed] [Google Scholar]
- Kimura T, et al. , 2010. Optimization of enzyme-substrate pairing for bioluminescence imaging of gene transfer using Renilla and Gaussia luciferases. J Gene Med. 12, 528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn D, et al. , 2002. The emerging fields of suicide gene therapy and virotherapy. Trends Mol Med. 8, S68–73. [DOI] [PubMed] [Google Scholar]
- Levaditi C, Nicolau S, 1922. Affinite du virus herpetique pour les neoplasmes epitheliaux. Comptes Rendus Societe Biologie. 87, 498–500. [Google Scholar]
- Lewis PF, Emerman M, 1994. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol. 68, 510–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobel LI, et al. , 1985. Construction and recovery of viable retroviral genomes carrying a bacterial suppressor transfer RNA gene. Science. 228, 329–32. [DOI] [PubMed] [Google Scholar]
- Logg CR, et al. , 2007. Adaptive evolution of a tagged chimeric gammaretrovirus: identification of novel cis-acting elements that modulate splicing. J Mol Biol. 369, 1214–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logg CR, Kasahara N, 2004. Retrovirus-mediated gene transfer to tumors: utilizing the replicative power of viruses to achieve highly efficient tumor transduction in vivo. Methods Mol Biol. 246, 499–525. [DOI] [PubMed] [Google Scholar]
- Logg CR, et al. , 2002. Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J Virol. 76, 12783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logg CR, et al. , 2001a. Genomic stability of murine leukemia viruses containing insertions at the Env-3' untranslated region boundary. J Virol. 75, 6989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logg CR, et al. , 2001b. A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum Gene Ther. 12, 921–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzio G, et al. , 2001. In vitro evolution of a highly replicating, doxycycline-dependent HIV for applications in vaccine studies. Proc Natl Acad Sci U S A. 98, 6342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick F, 2001. Cancer gene therapy: fringe or cutting edge? Nat Rev Cancer. 1, 130–41. [DOI] [PubMed] [Google Scholar]
- Metzl C, et al. , 2006. Tissue- and tumor-specific targeting of murine leukemia virus-based replication-competent retroviral vectors. J Virol. 80, 7070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, 2010. The Emperor of All Maladies: A Biography of Cancer. Scribner, New York, NY. [Google Scholar]
- Oliveira NM, et al. , 2010. A novel envelope mediated post entry restriction of murine leukaemia virus in human cells is Ref1/TRIM5alpha independent. Retrovirology. 7, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JS, 1989. The History of Cancer: An Annotated Bibliography. Greenwood Press, Westport, CT. [Google Scholar]
- Orkin SH, Motulsky AG, Report and Recommendations of the Panel to Assess the NIH Investment in Research on Gene Therapy. Bethesda, MD, 1995. [Google Scholar]
- Paar M, et al. , 2007. Effects of viral strain, transgene position, and target cell type on replication kinetics, genomic stability, and transgene expression of replication-competent murine leukemia virus-based vectors. J Virol. 81, 6973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MI, et al. , 2005. A Pitfall of the 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2 H-tetrazolium (MTS) assay due to evaporation in wells on the edge of a 96 well plate. Biotechnol Lett. 27, 805–8. [DOI] [PubMed] [Google Scholar]
- Preston BD, Dougherty JP, 1996. Mechanisms of retroviral mutation. Trends Microbiol. 4, 16–21. [DOI] [PubMed] [Google Scholar]
- Rainov NG, Heidecke V, 2011. Clinical Development of Experimental Virus-Mediated Gene Therapy for Malignant Glioma. Anticancer Agents Med Chem. [DOI] [PubMed]
- Reik W, et al. , 1985. Replication-competent Moloney murine leukemia virus carrying a bacterial suppressor tRNA gene: selective cloning of proviral and flanking host sequences. Proc Natl Acad Sci U S A. 82, 1141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe T, et al. , 1993. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 12, 2099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamon JA, et al. , 2002. Inserting a nuclear targeting signal into a replication-competent Moloney murine leukemia virus affects viral export and is not sufficient for cell cycle-independent infection. J Virol. 76, 8475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solly SK, et al. , 2003. Replicative retroviral vectors for cancer gene therapy. Cancer Gene Ther. 10, 30–9. [DOI] [PubMed] [Google Scholar]
- Stuhlmann H, et al. , 1989. Construction and properties of replication-competent murine retroviral vectors encoding methotrexate resistance. Mol Cell Biol. 9, 100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CK, et al. , 2010. Enhanced efficiency of prodrug activation therapy by tumor-selective replicating retrovirus vectors armed with the Escherichia coli purine nucleoside phosphorylase gene. Cancer Gene Ther. 17, 614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CK, et al. , 2005. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 12, 842–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Matano T, 2008. Host factors involved in resistance to retroviral infection. Microbiol Immunol. 52, 318–25. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. , 2006. Use of replication-competent retroviral vectors in an immunocompetent intracranial glioma model. Neurosurg Focus. 20, E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WJ, et al. , 2003. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum Gene Ther. 14, 117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, et al. , 2006. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 6, 715–27. [DOI] [PubMed] [Google Scholar]