

Abstract
Background
The pathological features of Parkinson disease (PD) include motor deficits, glial cell activation, and neuroinflammation. The neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), has an oxidation product, 1-methyl-4-phenylpyridinium ion (MPP+). This study aimed to investigate the effects of 2-aminoquinoline on motor deficits in a mouse model of MPTP-induced PD and cultured mouse astrocytes treated with MPP+, to determine the effects on astrocyte proliferation and apoptosis.
Material/Methods
Motor deficits in the mouse model of MPTP-induced PD were investigated using the climbing time, suspension time, and swim time tests. Cultured mouse astrocytes were treated with MPP+, and mice with MPTP-induced PD were treated with increasing doses of 2-aminoquinoline. The MTT assay was used to measure astrocyte viability. Astrocyte apoptosis was assessed by confocal fluorescence microscopy using Annexin-V and fluorescein isothiocyanate (FITC) staining. Western blot measured the levels of Bax, p-JNK, Bcl-2, and caspase-3.
Results
In the mouse model of MPTP-induced PD, motor deficit tests showed that 2-aminoquinoline reduced the impaired motor function during the climbing time, the suspension time, and the swim time tests in a dose-dependent manner. Pre-treatment with 2-aminoquinoline significantly reduced the proliferation and apoptosis of astrocytes induced by MPP+ in vitro, in a dose-dependent manner (P<0.05). The levels of p-JNK and cleaved caspase-3 levels were significantly reduced in astrocytes treated with MPP+ following pre-treatment with 2-aminoquinoline, which also reversed the increase in the Bax/Bcl-2 ratio.
Conclusions
In the mouse model of MPTP-induced PD, 2-aminoquinoline reduced motor deficiencies, inhibited MPP+ activated astrocyte apoptosis, and regulated the Bax/Bcl-2 ratio by targeting p-JNK.
MeSH Keywords: Anti-Inflammatory Agents, Astrocytes, Neuroglia
Background
Parkinson disease (PD) is characterized by the reduction in the number of neurons in the substantia nigra and the formation of Lewy bodies [1]. PD is the second most common form of neurodegenerative disorder, after Alzheimer’s disease [1]. The basal ganglia are most severely affected during PD, leading to disturbance of balance, the development of a tremor, and bradykinesia [2]. Previous studies have shown that the development of PD is associated with neuroinflammation, oxidative stress, and dysfunction of mitochondria [3–9]. Although the cause of PD remains unknown, oxidative stress, inflammation, aging, genetic, and environmental factors are believed to be involved in the development of PD [10–12]. Inflammatory damage to neurons is followed by neuritic beading associated with activated microglia, and the formation of apoptotic and necrotic cells associated with glutamate, oxidative radicals, and cytotoxic compounds [13].
Animal models of PD have been developed using specific compounds that cause neuronal damage. The neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), has an oxidation product, 1-methyl-4-phenylpyridinium ion (MPP+). In animal models, biochemical and cellular changes that are similar to PD have been developed by the administration of MPTP [14]. The neurotoxicity of MPTP results from the effects of MPP+, which is produced by monoamine oxidase-B during MPTP oxidation [15]. Cell death due to MPTP results from the targeting of the electron transport chain in the neuronal mitochondria, inflammation, and the generation of reactive oxygen species (ROS) [14]. The uptake of MPP+ by neuronal cells leads to toxic changes that include altered calcium ion exchange, oxidative free radical formation, and the activation of nitric oxide synthase (NOS) [16]. Astrocyte dysfunction and reduced proliferation of astrocytes are critical to the survival of neurons and have significance in neurodegenerative diseases [17]. Previously astrocyte cell death was believed to be mainly due to necrosis when studied in animal models of spinal and brain injury [18,19]. However, recently published studies have shown that astrocyte apoptosis is a major component of the pathogenesis of neurodegenerative disorders, including PD [18,19].
Therefore, this study aimed to investigate the effects of 2-aminoquinoline (Figure 1) on motor deficits in a mouse model of MPTP-induced PD and cultured mouse astrocytes treated with MPP+, to determine the effects on astrocyte proliferation and apoptosis.
Figure 1.
The structure of 2-aminoquinoline.
Material and Methods
The mouse model of Parkinson disease (PD)
Sixty mice aged 7 weeks and weighing between 21–26 gm were obtained from Beijing Animal Institute, China. The mouse model of Parkinson Disease (PD) was developed using 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) treatment. The mice were housed in plastic cages individually in controlled conditions at 23±2°C and given standard chow and freshwater. The mice were divided into six groups of ten mice: the negative control (NC) group; the MPTP and 0.5 mg/kg of 2-aminoquinoline, the 2AQ+MPTP (low dose) group; the MPTP and 1 mg/kg of 2-aminoquinoline, the 2AQ+MPTP (medium dose) group; and the MPTP and 2 mg/kg of 2-aminoquinoline, the 2AQ+MPTP (high dose) group.
PD in the mouse model was induced by intraperitoneal injection of 10 μl of 30 mg/kg of MPTP solution over five days. The 2-aminoquinoline was dissolved in normal saline and injected as a single dose, 40 minutes before injecting the MPTP. Motor deficits in the mice were measured after 8 h of MPTP injection one day before the development of the PD model, and on the first, fourth, seventh, and tenth day of the PD model. The experimental protocols were performed in accordance with the guidelines from the National Institutes of Health (NIH, Bethesda, MD, USA). Ethical approval for this study was given by the Animal Care and Use Committee, Changjiang University, Hubei Province, China.
Measurement of the climbing time
A pole test was used to measure climbing time for mice in the model of MPTP-induced PD treated with 2-aminoquinoline using previous methodology [20]. Briefly, a wooden pole of 50 cm length and 1 cm girth was fixed vertically, and a ball of 2.5 cm diameter was placed on top. Digital counters recorded the time taken by each mouse to come down from the top of the ball. When the mouse climbed back before climbing down from the ball or stopped, then the experiment was repeated. The test was performed daily in triplicate for every mouse.
Measurement of the suspension time for mice in the model of MPTP-induced PD treated with 2-aminoquinoline was made using the traction test [21]. A wire of around 30 cm in length was fixed horizontally on a platform, and mice were suspended from it. The time spent by the mice while hanging on the wire was recorded and scored as follows: 0, for 0–4 seconds; 1, for 5–9 seconds; 2 for 10–14 seconds; 3, for 15–19 seconds; 4, for 20–24 seconds; and 5, for 25–29 seconds.
Measurement of swimming time
The swimming test for mice in the model of MPTP-induced PD after 2-aminoquinoline treatment was conducted in a pool of 20×30×20 cm in dimension, as previously described [22]. The temperature in the pool was maintained at 28°C and mice were allowed to swim for 1 min. A successful swimming score was 3.0; swimming for a maximum time, scored 2.5; mice that floated for more than 30 sec scored 2.0; mice that swam only occasionally scored 1.5; and mice that floated only scored 0.
Cell culture of mouse astrocytes
The cerebral cortex was excised from the rats, and the tissue was minced in Dulbecco’s modified Eagle’s medium (DMEM) mixed with DNase (30 μg/ml) and bovine serum albumin (BSA) (0.25%). The digested tissue was treated with a 0.3% solution of trypsin for 40 min at 37°C, followed by filtration of the suspension using a 70 μm mesh nylon filter, The suspension was centrifuged and the cell pellets were placed in DMEM/F12 containing 10% FBS and antibiotics. The cells were transferred into culture flasks for incubation under standard conditions. When the cells reached confluence, the flasks were shaken gently to remove the microglial cells. The separated astrocytes were washed three times in PBS for 5 min, trypsinized, and then cultured in DMEM/F12. The mixture of FBS (12%), with insulin and L-glutamine, were combined with the medium for the culture of the astrocytes until 90% confluence was reached. The PD model of astrocytes was obtained by treating the cells with a 4 mM/L solution of MPP+. Astrocytes were treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline, 2 h before MPP+ treatment.
MTT cell viability assay
The astrocytes were plated into 96-well plates at 2×105 cells per well to measure cell proliferation. Astrocytes were treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline or normal saline (control), and after 2 h, MPP+ was added. Incubation of the astrocytes with 2-aminoquinoline for 24 h was followed by adding a 5 mg/ml solution of MTT (20 μl) into the wells. After 4 hours of incubation with MTT, the astrocytes were rinsed three times in PBS. The medium-free astrocytes were treated with dimethyl sulfoxide (DMSO) (150 μl) and then shaken in the microtiter plate using shakers. Optical density (OD) measurement at 457 nm were made to indirectly calculate the astrocyte viability using the Spectra MAX M5 microplate spectrophotometer (Thermofisher Scientific, Waltham, MA, USA).
Fluorescence-activated cell sorting (FACS) using Annexin-V and fluorescein isothiocyanate (FITC)
Apoptosis suppression in PD astrocytes by 2-aminoquinoline was confirmed using a commercially available Annexin V-FITC kit. Astrocytes were treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline or normal saline (control), and after 2 h, MPP+ was added. At 24 h of incubation with 2-aminoquinoline, astrocytes were harvested and then rinsed 3-times in PBS. Centrifugation of the astrocytes at 12,000×g for 10 min was followed by dyeing with Annexin V-FITC (5 μL) and PI (5 μL) stains. Apoptosis in astrocytes was examined using a fluorescence-activated cell sorter (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blot
Astrocytes were treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline or normal saline (control) for 24 h. The MPP+ was added to the plates at 2 h of 2-aminoquinoline addition for developing the PD model. The lysis of astrocytes was done in RIPA buffer [sodium chloride (150 Mm), NP-40 (1%), sodium deoxycholate (0.5%), SDS (0.2%), Tris-hydrochloride (50 mM at pH 8.0), EDTA (10 mM) and PMSF (1 mM) (Sigma-Aldrich, St. Louis, MO, USA). The lysate was centrifuged for 20 min at 12,000×g at 4°C. The harvested supernatant was used for protein concentration measurement using the Lowry method. The protein samples (equal amounts) were resolved after loading onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and then transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked at room temperature for 1.2 h using TBS + Tween-20, followed by overnight incubation with primary antibodies at 4°C. The PBS washed membranes were treated for 1 h with horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature. Detection of the immunostained signals was made using the enhanced chemiluminescence (ECL) system (Amersham, Piscataway, NJ, USA). The primary antibodies used were to GAPDH, p-JNK, caspase-3, Bax, and Bcl-2 (1: 1,000) (Santa Cruz Biotechnology, Inc. Dallas, TX, USA).
Statistical analysis
Data were expressed are the mean±standard deviation (SD) or standard error (SE) of values from studies performed in triplicate. One-way analysis of variance (ANOVA) and Student’s t-test were used to determine the differences between groups. A P-value <0.05 was considered to be statistically significant
Results
Treatment with 2-aminoquinoline improved the climbing time test scores in the mouse model of MPTP-induced Parkinson disease (PD)
The climbing time test for mice in the model of MPTP-induced PD treated with 2-aminoquinoline or without treatment was recorded using the pole test one day before the development of the PD model, on the first, fourth, seventh, and tenth day of the PD model (Figure 2). The climbing time was almost same for the normal control (NC) group, the MPTP group, the low dose group, the medium dose group, the high dose group, and the 2AQ group, one day before and on the day of creation of the PD model. The climbing time for the MPTP group of mice was significantly longer compared with the 2AQ and NC groups on day 4, day 7, and day 10 of the PD model. MPTP treatment increased the climbing time, which was significantly reduced by 2-aminoquinoline treatment in a dose-dependent manner in the mouse model of MPTP-induced PD.
Figure 2.

Treatment with 2-aminoquinoline improved the climbing time test scores in the mouse model of MPTP-induced Parkinson disease (PD). The mice were pre-treated with a low dose (0.5 mg/kg), a medium dose (1.0 mg/kg), and a high dose (2.0 mg/kg) of 2-aminoquinoline one day before the development of the mouse PD model, and on the first, fourth, seventh, and tenth day of PD development. The climbing time was measured using the pole test 8 h before (one day before) and 8 h after treatment with MPTP. * P<0.05, and ** P<0.01 vs. the NC group.
Treatment with 2-aminoquinoline improved the suspension time test scores in the mouse model of MPTP-induced Parkinson disease (PD)
The suspension times in the mouse model of MPTP-induced PD treated with 2-aminoquinoline or without treatment were recorded using a traction test one day before the development of the PD model, and on the first, fourth, seventh, and tenth day of the PD model (Figure 3). The difference in suspension score was not significant one day before and on the day of the development of the PD model between the NC, MPTP, the low dose (0.5 mg/kg), the medium dose (1.0 mg/kg), and the high dose (2.0 mg/kg), and 2AQ groups. However, the data obtained on days 4, 7, and 10 of the mouse model of MPTP-induced PD mouse model showed a significantly (P<0.05) lower suspension score for the MPTP group (P<0.05). The suspension score was significantly increased in the mouse model of MPTP-induced PD treated with low dose (0.5 mg/kg), medium dose (1.0 mg/kg), and high dose (2.0 mg/kg) 2-aminoquinoline. MPTP induced a significant reduction in the suspension score in the mouse model of PD that was prevented by treatment with the maximum or high dose of 2-aminoquinoline.
Figure 3.

Treatment with 2-aminoquinoline improved the suspension time test scores in the mouse model of MPTP-induced Parkinson disease (PD). The mice were pre-treated with a low dose (0.5 mg/kg), a medium dose (1.0 mg/kg), and a high dose (2.0 mg/kg) of 2-aminoquinoline one day before the development of the mouse PD model, and on the first, fourth, seventh, and tenth day of PD development. The suspension time was measured using a traction test 8 h before (one day before) and 8 h after treatment with MPTP. * P<0.05, and ** P<0.01 vs. the NC group.
Treatment with 2-aminoquinoline improved the swimming time test scores in the mouse model of MPTP-induced Parkinson disease (PD)
Measurement of the swimming scores for the mice in the model of MPTP-induced PD pre-treated with 2-aminoquinoline or without pre-treatment was performed using the swimming test one day before the development of PD, and on the first, fourth, seventh, and tenth day of the PD model (Figure 4). The swimming scores of the mice in the model of MPTP-induced PD showed a significant reduction on day 4, 7, and 10 of PD compared with the 2AQ and NC group. Treatment of the mice in the PD model with 2-aminoquinoline significantly increased the swimming scores in a dose-dependent manner. The swimming score in the mouse model of MPTP-induced PD was increased to the level of the NC group on treatment with high dose 2-aminoquinoline.
Figure 4.

Treatment with 2-aminoquinoline improved the swimming time test scores in the mouse model of MPTP-induced Parkinson disease (PD). The mice were pre-treated with a low dose (0.5 mg/kg), a medium dose (1.0 mg/kg), and a high dose (2.0 mg/kg) of 2-aminoquinoline one day before the development of the mouse PD model, and on the first, fourth, seventh, and tenth day of PD development. The swimming time test score was calculated 8 h before (one day before) and 8 h after MPTP treatment * P<0.05, and ** P<0.01 vs. the NC group.
MPP+-mediated cell viability was reduced by 2-aminoquinoline in vitro
In the in vitro astrocyte model of PD prepared by culture with MPP+, the effect of 2-aminoquinoline on cell proliferation was measured by the MTT assay (Figure 5). MPP+ treatment significantly reduced the proliferation of astrocytes (P<0.05) compared with the untreated astrocytes, as shown by the MTT data (Figure 5A). Pre-treatment of astrocytes with 2-aminoquinoline reversed MPP+-induced reduction in cell proliferation in a dose-dependent manner. There was a significant reduction in MPP+-induced proliferation by 1 μM of 2-aminoquinoline (P<0.05). Pre-treatment of MPP+-treated astrocytes with 8 μM of 2-aminoquinoline increased cell proliferation near to the level of the NC. The data from the MTT assay were obtained by inverted microscopy (Figure 5B).
Figure 5.

The effect of 2-aminoquinoline on MPP+ mediated reduction of astrocyte proliferation. The astrocytes were pre-treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline, 2 h before MPP+ treatment. Astrocyte proliferation was measured using (A) the MTT assay and (B) inverted microscopy at 48 h following 2-aminoquinoline treatment. * P<0.05, ** P<0.02 and *** P<0.01 vs. the MPP+ astrocytes.
Treatment with 2-aminoquinoline suppressed apoptosis in MPP+-treated astrocytes
MPP+ treatment significantly increased apoptosis in cultured astrocytes when compared with the control (Figure 6). Increased astrocyte apoptosis by MPP+ exposure was reduced following 2-aminoquinoline pre-treatment. Although pre-treatment with 0.5 and 1 μM of 2-aminoquinoline slightly reversed MPP+ mediated apoptotic induction in astrocytes, which was maximal at a dose of 8 μM.
Figure 6.

The effects of 2-aminoquinoline on apoptosis of astrocytes in vitro. The astrocytes were pre-treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline, 2 h before MPP+ treatment. The apoptotic changes in the astrocytes were monitored by flow cytometry at 48 h of 2-aminoquinoline pre-treatment. * P<0.05 and ** P<0.01 vs. the MPP+ astrocytes.
Treatment with 2-aminoquinoline reduced MPP+-induced overexpression of p-JNK in astrocytes
In astrocytes, the p-JNK levels were significantly increased on MPP+ exposure when compared with the cultured control cells (Figure 7). Pre-treatment with 2-aminoquinoline of MPP+ exposed astrocytes significantly reduced the p-JNK level in a dose-dependent manner. There was a significant reduction of p-JNK from 1 μM following pre-treatment with 8 μM of 2-aminoquinoline.
Figure 7.

The effects of 2-aminoquinoline on p-JNK in astrocytes in vitro. The astrocytes were pre-treated with 0.5, 1, 2, 4, and 8 μM of 2-aminoquinoline, 2 h before MPP+ treatment. The expression of p-JNK was assessed by Western blot at 48 h following pre-treatment with 2-aminoquinoline. * P<0.05 and ** P<0.01 vs. the MPP+ astrocytes.
Treatment with 2-aminoquinoline maintained the Bax/Bcl-2 ratio in MPP+-treated astrocytes
Treatment of astrocytes with MPP+ significantly reduced the level of Bcl-2 and significantly enhanced Bax and PARP expression when compared with the NC (Figure 8). Therefore, the Bax/Bcl-2 ratio in the astrocytes was significantly increased by MPP+ exposure. However, pre-treatment of the astrocytes with 2-aminoquinoline reduced the MPP+ induced upregulation of Bcl-2 and the suppression of Bax expression. Therefore, 2-aminoquinoline regulated the Bax/Bcl-2 ratio in PD astrocytes.
Figure 8.
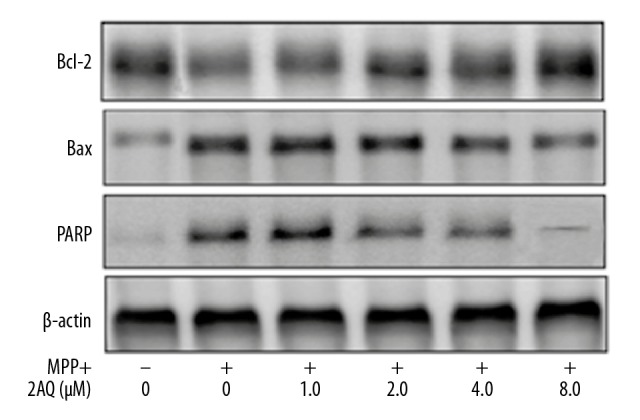
Regulation of the Bax/Bcl-2 ratio and the level of PARP by 2-aminoquinoline on apoptosis in vitro. The astrocytes were incubated with 0, 1, 2, 4, and 8 μM of 2-aminoquinoline before treatment with MPP+. The protein levels were assessed by Western blot at 48 h following 2-aminoquinoline pre-treatment.
Treatment with 2-aminoquinoline reduced caspase-3 cleavage in MPP+-treated astrocytes
Treatment of astrocytes with MPP+ resulted in a significant increase in the immunoreactivity for cleaved caspase-3 (Figure 9). However, pre-treatment of astrocytes with 2-aminoquinoline reduced cleaved caspase-3 immunoreactivity in a dose-dependent manner. The MPP+ mediated increase in the level of cleaved caspase-3 in the astrocytes was significantly reduced by 2-aminoquinoline at a dose of 1 μM. The stimulatory effect of MPP+ on the upregulation of cleaved caspase-3 was reduced by pre-treatment with 8 μM of 2-aminoquinoline.
Figure 9.

Treatment with 2-aminoquinoline reduced caspase-3 cleavage in MPP+-treated astrocytes. The astrocytes after pre-treatment with 0, 1, 2, 4, and 8 μM of 2-aminoquinoline were treated with MPP+ 2 h before. Cleaved caspase-3 was assessed by Western blot at 48 h after 2-aminoquinoline pre-treatment. * P<0.05 and ** P<0.02 vs. the MPP+ astrocytes.
Discussion
The neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), has an oxidation product, 1-methyl-4-phenylpyridinium ion (MPP+). The aims of the present study were to investigate the effects of 2-aminoquinoline on motor deficits in a mouse model of MPTP-induced Parkinson disease (PD) and cultured mouse astrocytes treated with MPP+, to determine the effects on astrocyte proliferation and apoptosis. The findings showed that 2-aminoquinoline reduced PD-associated movement disorders in the mice and inhibited MPP+-mediated astrocyte apoptosis by targeting JNK pathway activation.
The establishment of the PD rat model by the administration of MPTP was supported by the data from previous studies [20–22]. MPTP treatment of the mice in the PD model resulted in significant movement disorders, including an increased climbing time and reduced swimming time, and suspension scores, which were supported by the findings from previous studies [20–22]. The movement deficiencies induced in the mouse model of MPTP-induced PD were significantly reduced by pre-treatment with 2-aminoquinoline in a dose-dependent manner.
Previous studies on neurodegenerative diseases have shown that apoptosis most commonly affects glia and astrocytes [23,24]. Therefore, reduction or inhibition of apoptosis in astrocytes may be an approach to control or treat neurodegenerative disease. In the present study, treatment with 2-aminoquinoline reduced astrocyte apoptosis induced by MPP+ in the mouse PD model. Regulation of Bax, which is a pro-apoptotic molecule, has a major role in the elimination of cells by apoptosis [25], and injury in the CNS [26]. The findings from the present study showed that MPP+ treatment resulted in apoptotic changes in astrocytes by increasing the level of Bax and suppressing Bcl-2 expression. The activation of apoptosis in astrocytes by MPP+ treatment was reduced by 2-aminoquinoline pre-treatment by lowering the Bax/Bcl-2 ratio.
Neuronal stress, including targeting nerve growth factor (NGF), excitotoxicity, and the production of reactive oxygen species (ROS) are associated with JNK signaling activation and cell apoptosis [27,28]. In the mouse model of MPTP-induced PD, the level of p-JNK has previously been shown to be increased in the cell death signaling pathway [29,30]. The activated form of JNK has a key role in apoptosis by the involvement of Bax, Bcl-2, and Bcl-xl, and correlated with p53 [31]. Previous studies have shown that treatment with MPTP is associated with the activation of tumor-associated protein p53 [32,33]. The association between p53 and the JNK pathway induces Bax protein, which results in Bax-dependent apoptosis [33].
The involvement of the Bcl-2 protein family has also been associated with the death of cells induced by MPP+ [34]. The Bax/Bcl-2 ratio in a mouse model of MPTP-induced PD was previously shown to be increased, resulting in the activation of apoptosis [35]. Previous studies have highlighted the role of caspases in programmed cell death, or apoptosis [36,37]. In 2012, Zhang et al. showed that treatment of SH-SY5Y neuroblastoma cells with astragaloside IV prevented MPP+-induced apoptosis associated with the inhibition of Bax-associated pathways [38]. The current study showed a significant increase in the level of p-JNK in astrocytes following MPP+ treatment, which was consistent with the findings from previous studies. However, the findings from this study showed that pre-treatment with 2-aminoquinoline significantly reduced the level of p-JNK in MPP+-treated astrocytes and reduced the effects of impaired motor function in a mouse model of MPTP-induced PD.
Conclusions
This study aimed to investigate the effects of 2-aminoquinoline on motor deficits in a mouse model of MPTP-induced Parkinson disease (PD) and cultured mouse astrocytes treated with MPP+, to determine the effects on astrocyte proliferation and apoptosis. In the mouse model of MPTP-induced PD, 2-aminoquinoline reduced motor deficiencies, inhibited MPP+ activated astrocyte apoptosis, and regulated the Bax/Bcl-2 ratio by targeting p-JNK in a dose-dependent manner. These preliminary findings support the need for further in vivo studies to determine the potential therapeutic role of 2-aminoquinoline in PD.
Footnotes
Conflict of interest
None.
Source of support: Departmental sources
References
- 1.Kidd PM. Parkinson’s disease as multifactorial oxidative neurodegeneration: Implications for integrative management. Altern Med Rev. 2000;5:502–29. [PubMed] [Google Scholar]
- 2.Franco-Iborra S, Vila M, Perier C. The Parkinson disease mitochondrial hypothesis: Where are we at? Neuroscientist. 2016;22:266–77. doi: 10.1177/1073858415574600. [DOI] [PubMed] [Google Scholar]
- 3.Camilleri A, Vassallo N. The centrality of mitochondria in the pathogenesis and treatment of Parkinson’s disease. CNS Neurosci Ther. 2014;20:591–602. doi: 10.1111/cns.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience. 2015;302:47–58. doi: 10.1016/j.neuroscience.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Xie A, Gao J, Xu L, Meng D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed Res Int. 2014;2014 doi: 10.1155/2014/648740. 648780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neil AK, Kimberly HW, Jane BA, et al. High-intensity exercise acutely increases substantia nigra and prefrontal brain activity in Parkinson’s disease. Med Sci Monit. 2017;23:6064–71. doi: 10.12659/MSM.906179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maosheng Y, Yuxin Y, Shixing W, et al. Role of paeonol in an astrocyte model of Parkinson’s disease. Med Sci Monit. 2017;23:4740–48. doi: 10.12659/MSM.906716. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Lagai Y, Zuneng L. Differences in ABCA1 R219K polymorphisms and serum indexes in Alzheimer and Parkinson diseases in northern China. Med Sci Monit. 2017;23:4591–600. doi: 10.12659/MSM.903636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Likun Y, Changfeng G, Jie Z, et al. Increased levels of pro-inflammatory and anti-Inflammatory cellular responses in Parkinson’s disease patients: Search for a disease indicator. Med Sci Monit. 2017;23:2972–78. doi: 10.12659/MSM.904240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J. 2003;17:1954–56. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi H, Wakabayashi K. Controversy: is Parkinson’s disease a single disease entity? Yes. Parkinsonism Relat Disord. 2005;11(Suppl 1):S31–37. doi: 10.1016/j.parkreldis.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi H, Mizuno T, Zhang G, et al. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem. 2005;280:10444–54. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- 13.Schulz JB, Falkenburger BH. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2004;318:135–47. doi: 10.1007/s00441-004-0954-y. [DOI] [PubMed] [Google Scholar]
- 14.Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem. 1993;61:1191–206. doi: 10.1111/j.1471-4159.1993.tb13610.x. [DOI] [PubMed] [Google Scholar]
- 15.Obata T. Nitric oxide and MPP+-induced hydroxyl radical generation. J Neural Transm. 2006;113:1131–44. doi: 10.1007/s00702-005-0415-0. [DOI] [PubMed] [Google Scholar]
- 16.Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi K, Hayashi M, Nakano H, et al. Apoptosis of astrocytes with enhanced lysosomal activity and oligodendrocytes in white matter lesions in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2002;28:238–51. doi: 10.1046/j.1365-2990.2002.00390.x. [DOI] [PubMed] [Google Scholar]
- 18.Szydlowska K, Zawadzka M, Kaminska B. Neuroprotectant FK506 inhibits glutamate-induced apoptosis of astrocytes in vitro and in vivo. J Neurochem. 2006;99:965–75. doi: 10.1111/j.1471-4159.2006.04136.x. [DOI] [PubMed] [Google Scholar]
- 19.Ogawa N, Hirose Y, Ohara S, et al. A simple quantitative hradykinesia test in MPIP treated mice. Res Commun Chem Pathol Pharmacol. 1985;50:435–41. [PubMed] [Google Scholar]
- 20.Kubara H, Higuchi Y, Tadokoro S. Effects of central depressants on rota-rod and action performances in mice. Jpn J Pharmacol. 1977;27:117–26. doi: 10.1254/jjp.27.117. [DOI] [PubMed] [Google Scholar]
- 21.Donnan GA, Willjs GL, Kaczmarczyk SJ, Rowe P. Motor function in the l, methyl-4-phenyl-l,2,3,6-tetrahydropyridine treated mouse. J Neurol Sci. 1987;77:185–91. doi: 10.1016/0022-510x(87)90121-3. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch EC, Breidert T, Rousselet E, et al. The role of glial reaction and inflammation in Parkinson’s disease. Ann NY Acad Sci. 2003;991:214–28. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 23.Halliday GM, Stevens CH. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov Disord. 2011;26:6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- 24.Bar-Peled O, Knudson M, Korsmeyer SJ, Rothstein JD. Motor neuron degeneration is attenuated in Bax-deficient neurons in vitro. J Neurosci Res. 1999;55:542–56. doi: 10.1002/(SICI)1097-4547(19990301)55:5<542::AID-JNR2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Nesic-Taylor O, Cittelly D, Ye Z, et al. Exogenous Bcl-xL fusion protein spares neurons after spinal cord injury. J Neurosci Res. 2005;79:628–37. doi: 10.1002/jnr.20400. [DOI] [PubMed] [Google Scholar]
- 26.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 27.Dickens M, Rogers JS, Cavanagh J, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–96. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 28.Saporito MS, Thomas BA, Scott RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem. 2000;75:1200–8. doi: 10.1046/j.1471-4159.2000.0751200.x. [DOI] [PubMed] [Google Scholar]
- 29.Xia XG, Harding T, Weller M, et al. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci USA. 2001;98:10433–38. doi: 10.1073/pnas.181182298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lotharius J, Falsig J, van Beek J, et al. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25:6329–42. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Findley HW, Gu L, Yeager AM, Zhou M. Expression and regulation of Bcl-2, Bcl-xl, and Bax correlate with p53 status and sensitivity to apoptosis in childhood acute lymphoblastic leukemia. Blood. 1997;89:2986–93. [PubMed] [Google Scholar]
- 32.Mandir AS, Przedborski S, Jackson Lewis V, et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA. 1999;96:5774–79. doi: 10.1073/pnas.96.10.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Malley KL, Liu J, Lotharius J, Holtz W. Targeted expression of BCL-2 attenuates MPP+ but not 6-OHDA induced cell death in dopaminergic neurons. Neurobiol Dis. 2003;14:43–51. doi: 10.1016/s0969-9961(03)00013-5. [DOI] [PubMed] [Google Scholar]
- 34.Blum D, Torch S, Lambeng N, et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol. 2001;65:135–72. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 35.Salvesen GS. Caspases: Opening the boxes and interpreting the arrows. Cell Death Differ. 2002;9:3–5. doi: 10.1038/sj.cdd.4400963. [DOI] [PubMed] [Google Scholar]
- 36.Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: Mutations within caspase genes. J Med Genet. 2009;46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]
- 37.Zhang ZG, Wu L, Wang JL, et al. Astragaloside IV prevents MPP+-induced SH-SY5Y cell death via the inhibition of Bax-mediated pathways and ROS production. Mol Cell Biochem. 2012;364:209–16. doi: 10.1007/s11010-011-1219-1. [DOI] [PubMed] [Google Scholar]