

Abstract
BACKGROUND & AIMS:
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive malignancy that invades surrounding structures and metastasizes rapidly. Although inflammation is associated with tumor formation and progression, little is known about the mechanisms of this connection. We investigate the effects of interleukin (IL) 22 in the development of pancreatic tumors in mice.
METHODS:
We performed studies with Pdx1-Cre;LSL-KrasG12D;Trp53+/−;Rosa26EYFP/+ (PKCY) mice, which develop pancreatic tumors, and PKCY mice with disruption of IL22 (PKCY IL22−/−mice). Pancreata were collected at different stages of tumor development and analyzed by immunohistochemistry, immunoblotting, real-time polymerase chain reaction, and flow cytometry. Some mice were given cerulean to induce pancreatitis. Pancreatic cancer cell lines (PD2560) were orthotopically injected into C57BL/6 mice or Il22−/−mice, and tumor development was monitored. Pancreatic cells were injected into the tail veins of mice, and lung metastases were quantified. Acini were collected from C57BL/6 mice and resected human pancreata and were cultured. Cell lines and acini cultures were incubated with IL22 and pharmacologic inhibitors, and protein levels were knocked down with small hairpin RNAs. We performed immunohisto-chemical analyses of 26 PDACs and 5 nonneoplastic pancreas specimens.
RESULTS:
We observed increased expression of IL22 and the IL22 receptor (IL22R) in the pancreas compared with other tissues in mice; IL22 increased with pancreatitis and tumorigenesis. Flow cytometry indicated that the IL22 was produced primarily by T-helper 22 cells. PKCY IL22−/−mice did not develop precancerous lesions or pancreatic tumors. The addition of IL22 to cultured acinar cells increased their expression of markers of ductal metaplasia; these effects of IL22 were prevented with inhibitors of Janus kinase signaling to signal transducer and activator of transcription (STAT) (ruxolitinib) or MEK (trametinib) and with STAT3 knockdown. Pancreatic cells injected into IL22−/− mice formed smaller tumors than those injected into C57BL/6. Incubation of IL22R-expressing PDAC cells with IL22 promoted spheroid formation and invasive activity, resulting in increased expression of stem-associated transcription factors (GATA4, SOX2, SOX17, and NANOG), and increased markers of the epithelial-mesenchymal transition (CDH1, SNAI2, TWIST1, and beta catenin); ruxolitinib blocked these effects. Human PDAC tissues had higher levels of IL22, phosphorylated STAT3, and markers of the epithelial-mesenchymal transition than nonneoplastic tissues. An increased level of STAT3 in IL22R-positive cells was associated with shorter survival times of patients.
CONCLUSIONS:
We found levels of IL22 to be increased during pancreatitis and pancreatic tumor development and to be required for tumor development and progression in mice. IL22 promotes acinar to ductal metaplasia, stem cell features, and increased expression of markers of the epithelial-mesenchymal transition; inhibitors of STAT3 block these effects. Increased expression of IL22 by PDACs is associated with reduced survival times.
Keywords: Immune Response, Transcriptional Regulation, EMT, Cancer Stem Cell
Graphical Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains among the most lethal cancers, with an expected 5-year survival of <10%.1 Although much is known about the genetic mutations contributing to the formation of pre-malignant lesions (pancreatic intraepithelial neoplasia [PanIN]), the precise events that lead to initiation, invasion, and dissemination are poorly understood. A hallmark of PDAC progression is the establishment and accumulation of a fibroinflammatory stroma, which functions to protect tumors from immune-mediated destruction2 and supports the maintenance of transformed cells.3,4 Epithelial immune cell cross-talk within this microenvironment results in the polarization of immune cells and enhancement of epithelial malignant signaling.5,6
Inflammation is crucial for tumor initiation, as evidenced by the up to 50-fold increased risk of PDAC in patients with chronic and familial pancreatitis.7–9 Further supporting the important role of inflammation in tumorigenesis is the finding that induction of pancreatitis in murine models expressing a pancreas-specific oncogenic form of KRAS leads to the earlier development and spread of tumors and, in some instances, is required for tumor initiation.7,10,11 Abrogation of inflammation with steroids in these models results in a dramatic decrease in tumor formation and dissemination.11 Although the precise mediators of inflammation-induced tumorigenesis remain unknown, growing evidence supports an important role for innate inflammation in PDAC development.4,12
Among potential candidate cytokines contributing to PDAC initiation and spread is interleukin (IL) 22, which is primarily secreted by a subset of helper T cells (Th22) and type 3 innate lymphoid cells (ILCs) and has emerged as an important cytokine in host defense, tissue repair, and tumor formation in the gastrointestinal tract.13,14 Unlike most cytokines, its receptor (IL22R) is found only on nonimmune cells, positioning IL22 as an ideal mediator of epithelial immune cell cross-talk.15,16 Further evidence of its potential importance in PDAC is the high expression of IL22R on pancreatic acinar cells12,17 and data showing that IL22 is critical to organ repair after acute pancreatitis.18,19 Recent data have also shown that elevated IL22 and IL22R in surgically resected PDAC specimens are associated with poor disease-specific patient survival.20,21 Based on its important role in pancreatic inflammation and prior association with tumor formation elsewhere in the gastrointestinal tract, we sought to determine the impact of IL22 on initiation, progression, and metastasis using murine models of PDAC and resected surgical specimens.
Methods
Cell Lines
Previously established mouse KPC derived cell lines (PD2560,11 PKCY,11 MT3,22 and 7940B23) were grown at 37°C and 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum or Dulbecco’s modified Eagle medium supplemented with 10% FBS. Capan2 cells were grown in 15% FBS McCoy’s 5a modified medium and BxPC3, CF-PAC, Panc 10.05, Jurkat, and HepG2 cells were grown in 15% FBS supplemented EMEM, RPMI 1640, Iscove’s modified Dulbecco’s medium or Dulbecco’s modified Eagle medium.
Animal Studies
All animal protocols were approved by the University of Michigan University Committee on the Use and Care of Animals. Six- to 10-week-old mice (both sexes), housed in specific pathogen-free facilities at the University of Michigan, were used in orthotopic and intravenous inoculations. Mice were fed regular LabDiet, housed in micro-isolated cages with corn cob/Pure-o’Cel bedding with enrichment. For experiments with transgenic mice, sibling littermates were used as control. Orthotopic tumors were generated by injecting 5 × 104 syngeneic MT3 cells in 50% Matrigel (Corning Life Sciences, Corning, NY) in RPMI directly into the pancreas of C57BL/6 mice. Mice were killed per the animal protocol at the endpoint or when predetermined signs were present. Details of reagents are described in Supplementary Table 1. The metastatic lung model was established by direct intravenous tail vein injection of 5 × 105 syngeneic MT3 cells. Mice were monitored and subsequently killed 21 days after inoculation, and tumors were quantified by intratracheal India ink instillation.
Sphere Formation Assay
A total of 2000 single cells per well were plated in ultra-low-attachment plates in X-VIVO medium with or without respective treatment or supplements and incubated in 37°C and 5% CO2. Spheres were imaged up to 2 months every 3 to 5 days. The numbers reflect spheres >250 μm in size per field of view to avoid the inclusion of nonspecific cell clumping.
Acinar and Tumor Derived 3-Dimensional Organoid Cultures
Whole acini were isolated from mouse or human pancreatic samples obtained from mice or human surgical resections. Resected pancreata were washed 3 times in cold Hank’s balanced salt solution (HBSS) supplemented with 5% FBS. Tissue was minced into pieces smaller than 1 mm in diameter. Tissue was washed 3 more times in 5% FBS-supplemented HBSS; then the pellet was resuspended in HBSS with 1 mg/mL collagenase and incubated for 15 minutes at 37°C with rotation. Digestion was stopped in ice cold HBSS supplemented with 5% FBS, and cells were washed 2 more times with 5% HBSS. The acinar suspension was successively filtered through 500-μm and 100-mm meshes, collected, and centrifuged in 10 mL of 30% FBS/HBSS. The pellet was resuspended in medium supplemented with 5% Matrigel and cold Waymouth media for mouse or supplemented PTOM media24 for human samples. A total of 1.5 × 104 acinar cells were plated on Matrigel-coated 96-well plates and incubated at 37°C and 5% CO2.
Invasion Assay
Cells were grown under specified treatment conditions for or 10 days. Culture media was replaced with serum-free media 24 hours before Transwell seeding of 5 × 104 tumor cells in serum-free media (plus experimental cytokine) and 10% FBS media in the receiving well. After 48 hours, invading cells were fixed and stained with the DIFF Stain Kit. Non-invading cells were removed from the upper surface of the membrane with a cotton-tipped swab. The cell number in each membrane cutout was recorded.
Immunohistochemistry and Multiplex Fluorescent Immunohistochemistry
Immunohistochemistry (IHC) and multiplex fluorescent immunohistochemistry (mfIHC) was performed as previously described.25 Briefly, after antigen retrieval and blocking, slides were incubated overnight with their respective primary antibodies. After incubation with a peroxidase-biotinylated secondary antibody, ABC chromogenic reaction (for IHC) or TSA amplification (mfIHC) was performed, followed by counter-staining with hematoxylin or 4’,6-diamidino-2-phenylindole (DAPI). Further details are presented in the supplementary methods.
Immunofluorescence
Cell culture immunofluorescence was performed as previously described.11 In brief, tumor cells were grown on rat tail collagen-treated slides under respective conditions and fixed. Cells were permeabilized and incubated with primary antibodies overnight at 4°C in a moist chamber. After PBS washes, samples were incubated with secondary antibodies diluted in blocking buffer and protected from light for 60 minutes at room temperature. Slides were stained with DAPI before mounting with Prolong Gold (Invitrogen, Waltham, MA).
Immunoblotting
Single-cell suspension pellets or tissue fragments were resuspended in cell extraction buffer supplemented with protease inhibitors (Halt or phenylmethylsulfonyl fluoride). Lysates were loaded in 8%, 10%, or 12% polyacrylamide gel electrophoresis gels and run for 2 hours. Proteins were transferred overnight to polyvinylidene difluoride membranes and after 1 hour of blocking with 5% milk in 0.5% Tween-20 in Tris-buffered saline) and probed with respective primary antibodies (listed in Supplementary Table 1). Membranes were washed and probed with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. Finally, membranes were imaged by autoradiography with entero-chromaffin-like enhanced chemoluminescence or Femto substrates (Pierce, Thermo-Fisher Scientific, Waltham, MA).
Flow Cytometry
Single-cell suspension was prepared from fresh tissue samples and stained with respective antibodies. Intracellular cytokine staining was performed after 4 hours of phorbol myristate acetate/ionomycin activation at 37°C with transport block (monensin, Brefeldin A). After a 30-minute surface staining at room temperature, cells were Fix-permeabilized (BD Pharmingen, Franklin Lakes, NJ) and stained for intracellular markers for 1 hour at 4 °C. After wash, data were analyzed using an LSRII cytometer (BD Biosciences, Franklin Lakes, NJ). All antibodies used for staining are listed in Supplementary Table 1.
RNA Isolation and Quantitative Reverse-Transcription Polymerase Chain Reaction
Tissue or cells were harvested in Trizol (Invitrogen), and RNA was extracted using manufacturers’ protocols. Complementary DNA was synthesized with a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Real-time polymerase chain reaction (PCR) was performed with FastStart SYBR Green Master (Rox) (Roche, Basel, Switzerland) in a StepOnePlus or QuantStudio 3 Real-Time PCR System (Applied Biosystems, Waltham, MA). Primer sequences are described in Supplementary Table 1.
Statistical Analysis
Data are shown as mean ± standard error of the mean or mean ± standard deviation. Statistical analysis was performed using 2-sample t test, or 1-way or 2-way analysis of variance (ANOVA). For all tests, P < .05 was considered significant.
Results
Interleukin 22 and Interleukin 22 Receptor Are Expressed in Human and Murine Pancreatic Ductal Adenocarcinoma
To study the role of IL22 in PDAC, we used a previously established transgenic mouse model, Pdx1-Cre;LSL-KrasG12D;Trp53+/−;Rosa26EYFP/+ (PKCY), with pancreas-specific oncogenic KRAS, a yellow fluorescent protein (YFP) reporter and monoallelic deletion of the tumor suppressor p53, which faithfully mimics progression of the human disease.10,11,26 To characterize IL22R expression in all stages of pancreatic tumorigenesis, tissue was collected from PKCY mice at 6, 16, and 20 weeks of age, representing normal pancreas, PanIN, and invasive cancer, respectively, and from surgically resected human pancreatic specimens. IHC analysis showed robust IL22R expression predominately on epithelial cells in both healthy and diseased pancreata, with some expression on intercalating stromal cells (Figure 1A). mfIHC confirmed the lack of IL22R expression on immune cells (Figure 1B). Established murine tumors were digested to single cells and epithelial cells (EpCAM+), fibroblasts (PDGFRa+), and T cells (CD3+) sorted. As expected, IL22R messenger RNA (mRNA) was not detected in T cells, whereas epithelial cells and fibroblasts had high and intermediate expression levels, respectively (Figure 1C). Immunoblotting showed IL22R expression on cultured human PDAC cell lines (Panc 10.05, Capan 2, CF-PAC, BxPC3) and those derived from tumor-bearing KPC mice (MT3, PKCY, PD2560) but not on immortalized immune Jurkat cells (Supplementary Figure 1A). All cell lines possessing IL22R responded to IL22 stimulation as measured by phosphorylation of signal transducer and activator of transcript (Stat) 3, the pathway involved in canonical signaling (Supplementary Figure 1B). Immuno-blot for IL22 showed elevated protein levels in the spleen and tumors of PKCY mice compared with normal pancreata and muscle (Figure 1D and Supplementary Figure 1C). Antibody specificity was confirmed with activated splenocytes from IL22−/− mice (Supplementary Figure 1D). Although mRNA expression was greatest in PDAC, IL22 expression in the nontransformed pancreas and normal lung was higher than liver and muscle (Figure 1E). Similar results were shown in human tumors, although IL22 expression was more variable when compared with pooled noncancer pancreas specimens (Figure 1F). To establish IL22 concentration during and after acute pancreatitis, mice were subjected to 7 repeated injections of cerulean hourly, and tissue was collected for analysis during acute inflammation and after recovery. IL22 increased in the acute phase of injury (24 hours) and remained elevated 1 week after pancreatitis, when acini had largely returned to normal (Supplementary Figure 1E).
Figure 1.

Human and murine PDAC express high levels of IL22 and its cognate receptor, IL22R. (A) Representative IHC shows diffuse IL22R expression in nontransformed, PanIN, and invasive PDAC samples from mouse (top row) and human (bottom row) pancreas tissue (n = 5; scale bars, = 20 μm). (B) Representative fluorescent multiplex staining of a human PDAC sample labeled with anti-IL22R (green), anti-CD45 (magenta), and DAPI (blue) (n = 5; scale bars, 20 μm). (C) IL22R mRNA expression in fluorescence-activated cell sorted epithelial cells (EC) (EpCAM+), fibroblasts (Fb) (PDGFRa+), and T cells (CD3+). (D) Immunoblotting confirms elevated IL22 presence in murine PDAC compared with normal pancreas tissue. (E) Results were confirmed by measurement of IL22 mRNA transcripts in WT and cancerous murine pancreata. (F) IL22 mRNA expression in human PDAC compared with pooled noncancerous pancreata (n = 5). (G) qRT-PCR analysis of sorted EC, Fb, myeloid (CD11b+), and T cells confirms elevated IL22 mRNA expression in T cells. (H, I) Flow cytometry of CD45+ gated single cells from murine PDAC shows the primary source of IL22 to be CD3+CD90+ lymphocytes, which were significantly more abundant in cancerous pancreata (n = 4 per group, *P < .05). (J) Fluorescent multiplex imaging identified CD45+CD4+IL22+ cells (white arrow) in spatial proximity to PanINs (n = 3; scale bars, 40 μm). DAPI, 4′,6-diamidino-2-phenylindole.
To identify the source of IL22 in normal and malignant pancreata, single cells were separated from established murine pancreatic tumors. IL22 mRNA was measured in sorted epithelial cells (EpCAM+), fibroblasts (PDGFRa+), and immune cells (CD3+ or CD11b+), and its expression was detected only in the last of these (Figure 1G). To further characterize the cellular source, we used flow cytometry. After gating for immune cells (Supplementary Figure 1F), traditional T lymphocytes (CD45+, CD3+, CD90+) and ILCs (CD45+, CD3−, CD90+) were identified (Figure 1H). Although slightly more IL22-producing ILCs existed in tumors compared with normal pancreata, a majority of cytokines came from traditional CD4+ T lymphocytes, and none was present in CD3−CD90− immune cells or T-cell receptor γd+ T cells (Figure 1I and Supplementary Figure 1G-J). Overall, a significantly greater number of IL22-producing lymphocytes was present in tumors compared with non-tumor-bearing control pancreata. The spatial proximity of IL22-producing cells to early-stage pancreatic lesions (PanIN) was established by using IL22 mRNA fluorescent probes and surface markers stained by mfIHC (Figure 1J). These data show robust expression of IL22R on pancreatic epithelial cells and fibroblasts throughout the neoplastic spectrum and increased IL22 expression in the PDAC tumor microenvironment and show the source to be T lymphocytes.
Interleukin 22 Is Needed for Acinar to Ductal Metaplasia
The first step in the cascade from normal pancreatic cells to invasive malignancy is a process whereby acinar cells de-differentiate to a more embryologic ductal state in a process known as acinar-to-ductal metaplasia (ADM). To functionally characterize the role of IL22 in ADM and, therefore, PDAC initiation, we generated IL22−/−PKCY mice and compared their tumor initiation and progression to PKCY mice of a similar mixed background. At 20 weeks, IHC of pancreata showed the absence of PanINs and invasive lesions in the IL22−/−PKCY compared with PKCY mice (Figure 2A and B). Staining for acinar-associated amylase and the duct marker CK19 confirmed the absence of ADM in IL22−/−PKCY mice and comparable appearance to wild-type mice (Supplementary Figure 2A). To confirm that this did not represent merely a delay in tumor formation, IL22−/−PKCY mice were aged for 104 weeks and IHC confirmed lack of neoplastic transformation (n = 5) (Supplementary Figure 2B). We observed decreased expression of pStat3 in IL22-deficient PKCY mice (Supplementary Figure 2C). Although phosphorylated extracellular signal-regulated kinase (pERK) was increased in IL22−/−PKCY compared with IL22−/− wild-type (WT) mice, levels appeared decreased compared with cytokine-proficient PKCY mice (Figure 2C). Flow cytometry showed that pERK expression in EpCAM+ cells before malignant transformation (pancreata harvested at 8 weeks and confirmed to be histologically normal) was lower in IL22−/−PKCY mice compared with PKCY mice (Figure 2D). Whole pancreata were harvested from WT and PKCY mice and activated by incubation in serum-free media for 15 minutes in the presence of IL22, showing that, although pERK was constitutively activated in PKCY mice, IL22 was able to enhance activation in pancreatic tissue from WT mice similar to the classic mitogen-activated protein kinase (MAPK) agonist epidermal growth factor (Supplementary Figure 2D). To test whether IL22 was capable of enhancing ADM, acinar cultures were established from WT mice, which readily undergo ERK-mediated ADM after treatment with the epidermal growth factor receptor ligand transforming growth factor α.27 After IL22 incubation for 48 hours, enhanced development of duct formation was seen (Figure 2E), which was abrogated by pharmacologic inhibition of Janus kinase (JAK)/STAT (ruxolitinib, 2.5 μmol/L) and MEK (trametinib, 0.1 μmol/L) (Figure 2F). PD2560 cells treated with IL22 (20 ng/mL) for 24 hours had up-regulation of the ductal cell-related transcription factor SOX9, with suppression of the acinar markers PTF1A and NR5A2 (Figure 2G). Disruption of canonical IL22 signaling via shStat3 (Supplementary Figure 2E) resulted in suppression of the acinar-related transcription factor PTF1A and enhancement of the ductal markers SOX9 and BMI1 (Figure 2H). Culture with IL22 in the presence or absence of ruxolitinib and trametinib for 15 minutes confirmed the lack of interdependent signaling of pStat3 and pERK (Supplementary Figure 2F) and that JAK/STAT inhibition reversed the previously observed increase in SOX9 expression, whereas MEK inhibition had no effect (Supplementary Figure 2G). The findings that IL22−/−PKCY mice fail to get even the earliest-stage lesions and that IL22 is capable of inducing ADM formation in acinar cell cultures suggest that IL22 may be required for PDAC initiation.
Figure 2.

IL22 promotes PDAC initiation through Stat3-mediated induction of ADM. (A) Representative H&E of murine pancreata from WT, PKCY, and PKCY/IL22−/− mice (top row scale bars, 400 μm; bottom row scale bars, 40 mm). (B) Incidence of PanlNs and invasive cancer in WT, PKCY, and PkCy/IL22−/− mice (*P < .05). (C) IHC staining showed lower intensity of pERK in PKCY/IL22−/− compared with PKCY mice (n = 5) (scale bars, 40 μm). (D) Intensity of pERK before tumor formation by flow cytometric analysis of EpCAM+ cells isolated from the pancreas (representative fluorescence-activated cell sorting data shown, n = 5; *P < .05, **P < .05). (E, F) IL22 (20 ng/mL) treatment of acinar cultures for 48 hours induced acinar-to-ductal metaplasia, which was abrogated by inhibition of JAK/Stat (ruxolitinib, 2.5 mmol/L) and MEK (trametinib, 0.1 mmol/L) (*P < .05, **P < .05, ***P < .05) (scale bars, 400 μm). (G, H) qRT-PCR showed that 24 hours of IL22 treatment enhanced the expression of the ductal marker SOX9, which was dependent on Stat3 (*P < .05, **P < .05, ***P < .05). MFI, mean fluorescence intensity; Nml, normal; sh, short hairpin; UT.
Interleukin 22 Promotes Tumor Establishment by Supporting a Pancreatic Progenitor Cell State
To determine the role of IL22 in the establishment of tumors, MT3 PDAC cells were orthotopically injected into the pancreata of syngeneic WT and IL22−/− mice. Examination at 14 days showed that tumors were significantly smaller in IL22−/− mice (Figure 3A and Supplementary Figure 3A). IHC on explanted tumors confirmed decreased pStat3 expression in tumors from IL22−/− mice compared with WT mice (Figure 3B). Based on known associations between IL22, tumor establishment, and stemness reported in colon cancer,13 its role in the induction of pancreatic progenitor-associated pathways in PDAC was further studied. Incubation of IL22R-expressing PDAC cells with IL22 resulted in enhanced spheroid formation in both murine (PD2560) and human (Capan 2) cell lines (Figure 3C and D and Supplementary Figure 3B). Disruption of the signaling cascade by Stat3 knockdown resulted in abrogation of IL22-mediated sphere formation, suggesting that JAK/STAT signaling was responsible for the observed progenitor like properties (Figure 3E and Supplementary Figure 3C). IL6, another potent activator of Stat3, has previously been implicated PDAC tumorigenesis.28,29 To confirm that Stat3 activation was indeed driven by IL22 rather than IL6, PD2560 cells were incubated with IL6 or IL22 in the presence or absence of IL6 receptor (IL6R) blockade or IL22BP, a naturally occurring neutralizing protein of IL22. Although both cytokines were able to activate Stat3 (Supplementary Figure 3D), IL22 induced significantly greater phosphorylation and was uniquely capable of inducing sphere formation (Figure 3F). Neutralization of IL22 abrogated sphere formation, whereas IL6R blockade had no effect on IL22-mediated activity. To further investigate this, we used the murine PDAC cell line 7940B, which possesses IL22R but lacks a functional IL6R (Supplementary Figure 3E and F). The absence of IL6R had no effect on IL22-induced sphere formation (Supplementary Figure 3G) in this cell line. To better understand the precise progenitor pathways involved, quantitative reverse-transcription (qRT) PCR was performed on PD2560 cells cultured with and without IL22, showing significant increase in GATA4, SOX2, SOX17, and NANOG (Figure 3G).
Figure 3.

IL22 enhances progenitor cell-like properties of PDAC. (A) Orthotopic injection of syngeneic MT3 cells in IL22−/− mice resulted in impaired tumor growth compared with WT mice (n = 6, *P < .05). (B) IHC confirmed decreased pStat3 expression in orthotopic tumors of IL22−/− mice (**P < .05) (top row scale bars, 200μm; bottom row scale bars, 100 μm). (C, D) Sphere assay showed increased mean sphere formation in PD2560 cells treated with varying doses of IL22 for 14 days (n = 5; *P < .05, **P < .05) (scale bars = 100 mm). (E) This was abrogated by Stat3 knockdown (n = 3; *P < .05, **P < .05). (F) Sphere formation in PD2560cells incubated for 21 days with IL6 or IL22 in the absence or presence of IL6R inhibitor or IL22BP, a protein that binds and neutralizes IL22 (*P < .05, **P < .05, ***P = not significant). (G) IL22 (24-hour incubation) resulted in increased mRNA expression of several stemness-related genes by qRT-PCR (P < 0.5 relative to untreated for all genes). (H) Murine PDAC organoids cultured in IL22 for 7 days resulted in increased mean surface area (n = 40 per condition; *P < .05, **P < .05). (I) Human organoids, established from freshly resected surgical specimens, were cultured in IL22 (20 ng/mL) for 7 days (scale bars, 100 mm), resulting in increased (J) mean surface area and (K) number (n = 40 per condition; *P < .05). (L) This was abrogated by the addition of a JAK/STAT (ruxolitinib), but not AKT, inhibitor (n = 40 per condition; * P < .05, **P < .05, ***P = not significant).
Recently, tissue-derived organoids have emerged as an important tool for studying pancreatic tumorigenesis.30 Established from murine or human pancreata, organoids recapitulate all stages of tumor formation and provide a new tool for studying stem cell capabilities in the gastrointestinal tract, specifically in the context of IL22.31 Establishment of murine organoids in media containing increasing concentrations of IL22 resulted in larger mean organoid surface area, confirming its ability to support pancreatic tumor growth (Figure 3H and Supplementary Figure 3H). The culture of human organoids derived from surgically resected PDAC specimens in IL22-containing media resulted in greater size and number of structures supporting the role of IL22 in PDAC establishment and tumorigenesis (Figure 3I-K). The addition of the inhibitor to JAK/STAT, but not AKT (unrelated to traditional IL22 signaling), negated the growth advantage conferred by IL22 (Figure 3L).
IL22 Enhances Pancreatic Ductal Adenocarcinoma Invasion and Metastasis by Up-regulation of Epithelial-Mesenchymal Transition
A hallmark of malignant progression is the cell’s ability to undergo epithelial-mesenchymal transition (EMT) and gain invasive capabilities.32 In vitro, 2-dimensional culture of the murine PD2560 cell line with IL22 resulted in morphologic changes of PDAC cells from a traditional cuboidal architecture to a spindle-like morphology, with long, narrow projections similar to fibroblasts (Supplementary Figure 4A). High-dose IL22 resulted in decreased CDH1 expression, but increasing levels of CDH2, vimentin, and the transcription factor ZEB1, previously shown to be integral to EMT induction in PDAC33 (Figure 4A). Incubation in lower doses of cytokine resulted in a mixed expression pattern with intermediate levels of epithelial- and mesenchymal-related proteins, suggesting that IL22’s control of cell identity may vary with dose. qRT-PCR confirmed increased ZEB1 expression and the EMT transcription factors SNAI1, TWIST1, and β-catenin (Figure 4B). To determine if IL22 signaling was directly linked to transcription of EMT factors, chromatin immunoprecipitation was performed and found to significantly increase the binding of pStat3 to the TWIST1 promoter after cytokine stimulation (Figure 4C).
Figure 4.

IL22 enhances the expression levels of EMT-related transcription factors. (A) Treatment of PDAC cells with high-dose IL22 results in increased ZEB1, vimentin, and CDH2 and decreased CDH1. (B) After 24-hour incubation in IL22, mRNAof EMT-related transcription factors was detected by qRT-PCR (P < .5 relative to untreated for all genes). (C) Chromatin immunoprecipitation assay identified the association of Stat3 with the EMT transcription factor TWIST1 upon IL22 activation (n = 3; *P < .05). (D) Treatment with a JAK/STAT inhibitor abrogated IL22-mediated Stat3 phosphorylation by immunoblot. (E) Immunofluorescence showed increased protein expression of vimentin (red) and decreased CDH1 (green) after 48 hours of culture with IL22.This was blocked by inhibition of JAK/STAT (n = 5; scale bars, 20 μm). DAPI, 4′,6-diamidino-2-phenylindole.
To further establish the impact of IL22 on EMT, cells were treated with cytokine in the presence or absence of ruxolitinib (Figure 4D). Although IL22 resulted in significant down-regulation of CDH1 and up-regulation of vimentin, this was abrogated by the inhibition of JAK/STAT (Figure 4E). Immunofluorescence also confirmed that ZEB1 was induced by IL22, which was nullified by ruxolitinib (Figure 5A). To confirm dependence on canonical IL22 signaling, genetic knockdown of Stat3 in PD2560 cells was used, resulting in reversal of the previously observed increase in EMT transcription factors (Figure 5B).
Figure 5.

IL22 enhances invasion and metastasis by up-regulation of EMT. (A) A 48-hour treatment with IL22 (100 ng/mL) results in increased expression of ZEB1, which is abrogated by blockade of JAK/STAT. (B) Expression of mRNA for key EMT-related transcription factors, as measured by qRT-PCR, in Stat3-silenced PD2560 cells after 24-hour treatment with IL22 (100 ng/mL) or transforming growth factor β (5 ng/mL) (*P < .05). (C) Matrigel invasion assay of PD2560 cells showed that IL22 increased invasion at 48 hours (n = 3; *P < .05, **P < .05). (D) This was abrogated by JAK/STAT blockade (n = 3; *P = not significant, **P = not significant). (E, F) Increased peritoneal metastases were seen in WT compared with IL22−/− mice inoculated with SQ tumors (n = 4 mice per cohort, results of 3 independent experiments; *P < .05). (G, H) Tail vein injection of 105 PDAC cells into WT or IL22−/− mice showed increased tumor formation in the former. Pretreatment of cells with IL22 (20 ng/mL) for 24 hours resulted in enhanced tumor formation in IL22−/− mice (n = 3; *P < .05, **P < .05). DAPI, 4′,6-diamidino-2-phenylindole; Veh, vehicle.
To determine whether the observed increase in EMT resulted in a more malignant phenotype, cells with low- and high-dose IL22 treatment were subjected to Matrigel invasion assays. After 48 hours of culture in IL22, there was a 2- and 4.5-fold increase in invasion with to untreated cells for low- and high-dose cultures, respectively (Figure 5C and Supplementary Figure 5A and B). Blockade of Stat3 phosphorylation impaired the observed IL22-mediated increase in invasion, confirming the importance of canonical signaling in promoting a more malignant phenotype (Figure 5D).
To determine if IL22 contributed to metastasis in vivo, syngeneic subcutaneous tumors were established in WT and IL22−/− mice for 14 days. After mice were killed, examination of the peritoneal cavity showed ulcerated tumors and multiple metastases in the WT mice, with few seen in the IL22−/− mice that ultimately developed established subcutaneous tumors (Figure 5E and F). H&E staining confirmed that the peritoneal implants were consistent with metastatic PDAC (Supplementary Figure 5C). These findings support the ability of IL22 to both enhance invasion and establish metastases. To further investigate the role of IL22 in the establishment of metastasis and distinguish increased disease spread from the effects of invasion alone, we injected syngeneic murine MT3 tumor cells into the tail vein of WT or IL22−/− mice. At 21 days, the lungs showed a 3.5-fold increase in lung weight and tumor number in WT mice compared with those deficient in IL22 (Figure 5G and Supplementary Figure 5D). To confirm the role of IL22 in these findings, MT3 cells were pretreated for 24 hours with IL22 before tail vein injection, which partially abrogated the protective effects seen in the IL22−/− mice (Figure 5H). Thus, these data show that IL22 enhances EMT in PDAC cells and, consequently, tumor invasion and metastasis.
Interleukin 22 Expression Is Associated With Epithelial-Mesenchymal Transition and Reduced Disease-Specific Survival in Human Pancreatic Ductal Adenocarcinoma
To determine the impact of IL22 in human PDAC, patient data sets from The Cancer Genome Atlas (TCGA) were queried for cytokine expression and separated into those with detectable (n = 11) and undetectable (n = 154) IL22 mRNA transcripts. Tumors with IL22 present had decreased expression of cellular adhesion markers including EpCAM and CDH1 (Figure 6A). Pathway analysis showed down-regulation of processes involved in cell polarity and adhesion, classically associated with activation of EMT (Table 1).
Figure 6.

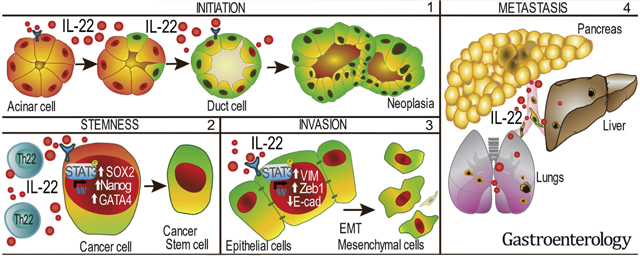
IL22 signaling is associated with EMT in human PDAC, resulting in poor disease-specific survival. (A) Volcano plot (false discovery rate vs fold change) of genes associated with IL22 expression in TCGA patients with PDAC (n = 11 vs 154 for patients with and without IL22 expression, respectively). (B) Representative multiplex fluorescent image of a human PDAC sample (yellow, pancytokeratin; green, pStat3; red, ZEB1; blue, DAPI); arrows indicate cells that lost CK expression and are double-positive for pStat3 and ZEB1 (scale bars, 15 μm). (C) Representative multiplex fluorescent imaging of a human PDAC sample from a TMA (white, CD45; yellow, IL22R1; light blue, pancytokeratin; red, pStat3; magenta, ZEB1; blue, DAPI; n = 25) (scale bars, 100 μm). (D) PDAC samples show an increased percentage of IL22-activated cells (IL22R1+pStat3+ZEB1+) relative to normal pancreas (*P < .05). (E) Kaplan-Meier analysis shows worse disease-specific survival in high (n = 8) compared with low (n = 17) IL22-activated CK− cells (9.4 months vs 17.5 months, respectively; P < .05). (F) IL22 released from Th22 cells promotes ADM, maintenance of a progenitor cell-like state, and EMT which contribute to disease progression and impaired survival.
Table 1.
Pathway Analysis of Genes Associated With IL22 Signaling
| DEG cluster | Number of genes |
Fold change |
Adjusted P value |
|---|---|---|---|
| Plasma membrane region | 417 | 0.178962218 | 5.87 × 10−31 |
| Apical part of cell | 318 | 0.177193222 | 7.24 × 10−25 |
| Apical plasma membrane | 239 | 0.151303452 | 1.01 × 10−23 |
| Apical junction complex | 118 | 0.091033752 | 2.79 × 10−23 |
| Basolateral plasma membrane | 180 | 0.138350864 | 1.05 × 10−20 |
| Tight junction | 102 | 0.101993824 | 4.29 × 10−18 |
| Occluding junction | 102 | 0.101993824 | 4.29 × 10−18 |
| Skin development | 341 | 0.265455055 | 2.10 × 10−14 |
| Cell-cell junction | 345 | 0.299671184 | 2.23 × 10−12 |
NOTE. DEG cluster analysis in TCGA samples with (n = 11) and without (n = 154) IL22 expression shows down-regulation of the pathways associated with cell polarity and contact (cluster 1 vs cluster 2, P < .05).
To determine if IL22 signaling and subsequent Stat3-mediated promotion of EMT may explain the previously reported impaired survival,20,21 surgically resected specimens were stained for pan-cytokeratin (CK), pStat3, and ZEB1. Two distinct cell subsets were identified: (1) pStat3+CK+ZEB1− cells, which tended to be located in duct-like structures within the tumor, and (2) pStat3+CK−ZEB1+ cells, located in close proximity to the former but not clearly attached to other cells (Figure 6B and Supplementary Figure 6A). Because cells undergoing EMT gain ZEB1 and lose cytokeratin expression, we hypothesized that the latter cells may represent IL22 activation of epithelial cells with subsequent induction of a mesenchymal phenotype and invasion. Alternatively, these could represent pStat3-activated fibroblasts. To further study this, 26 stage-matched PDACs and 5 non-neoplastic pancreas specimens were stained as described earlier, with the addition of IL22R and CD45 to confirm IL22 responsiveness and exclude intercalating immune cells (Figure 6C). Cells that were CD45−IL22R+pStat3+ represent IL22 activated nonimmune cells and, of that subset, those that lost CK expression but gained ZEB1 represented cells that have undergone EMT. PDAC specimens had a 4-fold increase in CK−IL22R+pStat3+ZEB1+ cells compared with normal pancreata, suggesting that the previously described increased tumor resident IL22 may be inducing pStat3-dependent EMT in PDAC (Figure 6D and Supplementary Figure 6B-D). To determine if this finding may explain the worse survival experienced in those with elevated IL22 present in the tumor microenvironment, patients with a high abundance of CK−IL22R+pStat3+ZEB1+ (upper quartile: n = 8) were compared with the remaining group (n = 17). Patients with greater IL22 and Stat3 activation of EMT had significantly worse disease-specific survival at 9.4 months compared with 17.5 months in the remainder (P = .044) (Figure 6E). No difference was seen when considering IL22-activated cells that had yet to undergo EMT, defined by retained presence of CK (Supplementary Figure 6E).
Discussion
A complicated cross-talk exists between immune and epithelial cells in the pancreas, with both exerting effects that can favor tumor formation and establishment. The important role of inflammation in pancreatic tumor initiation is well recognized because patients with familial and chronic pancreatitis, characterized by persistent accumulation of innate and adaptive immune cells, have a 50-fold increased risk of developing malignancy,34 and immune-suppression decreases PDAC development in murine models of disease.11 Other supportive data include the finding that, although pancreas-specific constitutive activation of the KRAS oncogene can slowly result in malignant transformation, the process is rapidly accelerated and, in some cases, dependent on induction of pancreatitis.10,26,35 The precise mediators that orchestrate the malignant potential of inflammation and the mechanisms by which they act are poorly understood.
Our work suggests that IL22, a signal that is used by the immune system to promote repair and renewal of pancreatic acinar cells during injury,18 may have deleterious effects on KRAS-mutated cells, leading to cancer formation. Unlike most cytokines, IL22 signals to epithelial cells unidirectionally because its receptor is absent on immune cells16 but is highly expressed on pancreatic acinar cells.17 It has been previously recognized as essential for tissue recovery after pancreatitis and hepatitis36 and mediates cellular regeneration after injury in the colon.37
One of the crucial first components of response to injury and repair in the pancreas is a transient transdifferentiation of acinar cells to an embryologic ductal state, allowing for decreased burden of protein production and improved proliferation. With increased regenerative capacity and reduced production demands, these duct-like cells are poised to replete necrotic acini, restoring exocrine function upon re-differentiation. Failure of re-differentiation back to acinar cells is believed to represent the first stage in the neoplastic cascade toward the formation of PanIN and, eventually, invasive cancer.38 Evidence of the importance of IL22 in ductal differentiation comes from data that IL22−/−/PKCY mice failed to acquire ADM lesions and subsequent PanINs or invasive PDAC. Because it has been established that KRAS signaling alone in transgenic mice is insufficient to force malignant transformation, we hypothesize that IL22 may provide a secondary signal needed for ADM formation and cancer initiation. This was supported by recent data by DEG, differentially expressed gene. Halbrook et al, which confirmed the dependence of ADM formation in pancreatic injury on inflammation-mediated enhancement of MAPK signaling and suggested that persistent ERK phosphorylation prevents re-differentiation of duct cells back to their acinar state.27 We confirmed that the absence of IL22 resulted in decreased global pERK signaling in KRAS mutant pancreata and that, in acini activated ex vivo with IL22, ERK was phosphorylated and ducts readily formed, a phenomenon that was abrogated by blockade of canonical IL22 signaling (Stat3). This represents important evidence linking components of innate inflammation to ADM and suggests that IL22 is an important mediator of inflammation-mediated carcinogenesis in the pancreas (Figure 6F).
Prior data exist showing that innate inflammation affects tumor progression in PDAC. McAllister et al4 found that IL17-producing immune cells present in the microenvironment accelerated the progression of PanIN and that, although cytokine neutralization had no effect on the development of ADM, it did abrogate conversion to early neoplasia. A similar study genetically deleted IL6 from KPC mice and found that, like IL17, cytokine depletion slowed progression of disease but had little effect on ADM.29 Although these prior reports have linked innate inflammation to progression in PDAC,4 they have stopped short of attributing cytokine signaling to formation of ADM and cancer initiation, a key in linking inflammation to carcinogenesis. Our data confirm that IL22, which is elevated during pancreatitis, contributes to and is required for ADM formation, providing important evidence of epithelialimmune cross-talk and inflammation-induced pancreatic cancer initiation.
In line with its impact on cellular plasticity, another physiologic property associated with IL22 that may have deleterious consequences is its impact on stemness. A prior report by Kryczek et al13 showed that IL22 signaling in colon cancer resulted in Stat3-mediated up-regulation of stem cell genes and impaired cancer-specific survival.13 Its role in tumor formation elsewhere in the gastrointestinal tract has also been reported, including in the liver, where transgenic expression of IL22 resulted in increased sensitivity to diethylnitrosamine-induced liver cancer.39 Similar to colon and liver cancers, our data suggest that IL22 promotes PDAC establishment in a Stat3-dependent fashion by up-regulating the genes involved in stem cell biology. Abrogation of signaling by blockade of JAK-STAT signaling resulted in reversal of the progenitor cell phenotype and abrogated tumor formation. These findings are supported by a recent report that described increased stem-like properties in IL22RA1hl cells that were sorted from murine PDACs.12
The ability of IL22 to enhance progenitor pathways likely stems from its physiologic role in promoting tissue repair during damage or infection.16 The pancreas is a site of constant injury because digestive enzymes often breach protective barriers, leading to subacute or acute pancreatitis. The capacity to repair and regenerate pancreatic parenchyma is essential in preventing autodigestion and maintenance of organ integrity, and the role of IL22 in this process is well established.18,19 Mounting evidence also suggests that the pancreas is home to a multitude of commensal microbes, many of which are capable of activating innate inflammation, which may include enhanced production of IL22.40 Although IL22’s ability to restore tissue integrity and promote progenitor cell features is beneficial during times of injury, evidence that persistent overexpression results in malignant transformation in colonic epithelium suggests that it may have yet another important role in pancreatitis-mediated PDAC formation.14,41
Although these data suggest a role for IL22 in early neoplasia, its more likely influence in driving the impaired survival experienced by patients with high IL22/IL22R20,21 are through its impact on progression in the form of invasion and metastasis. Our data suggest that IL22 drives the expression of the genes involved in EMT by direct binding of Stat3 to the promoter for TWIST, enhancing expression of mesenchymal proteins, and that inhibition of IL22-mediated STAT signaling reduces invasion and metastases in vivo. This was confirmed in human PDAC by using analysis of TCGA data, where expression of IL22 signaling was associated with down-regulation of the genes associated with cell-to-cell contact and polarity, a hallmark of EMT. Although the triggers for IL22-mediated EMT are unknown, it stands to reason that signals intended to induce metaplasia for the purposes of physiologic tissue repair could lead to untoward consequences when delivered to a genetically transformed cell. To better understand the relationship between IL22-mediated EMT and survival, we identified IL22-activated cells infiltrating the stroma that expressed pStat3 and Zeb1 and had lost epithelial expression of cytokeratin. The heightened presence of these cells was associated with worse overall survival, providing an important potential link between elevated IL22/IL22R expression in resected pancreatic specimens and worse disease-specific outcomes. It is also possible that these cells represent IL22-activated fibroblasts, because we showed that PDAC-associated fibroblasts express cognate IL22R, and previous reports have characterized their role in the induction of IL22-mediated pancreatic fibrosis.42 The driving factors that lead to increased IL22 signaling in the pancreas and links to known risk factors are unknown and warrant further investigation.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
We investigated the effects of inflammation and interleukin 22 (IL22) in development of pancreatic tumors in mice.
NEW FINDINGS
Levels of IL22 were increased during pancreatitis and pancreatic tumor development; this increase was required for tumor development and metastasis in mice. IL22 promotes acinar to ductal metaplasia, stem cell features, and increased expression of markers of the epithelial-mesenchymal transition; inhibitors of STAT3 blocked these effects. Increased expression of IL22 by PDACs is associated with reduced survival times.
LIMITATIONS
This study was performed in mice and human tissue samples
IMPACT
Strategies to block IL22 in patients with chronic pancreatitis might prevent tumor development or progression.
Acknowledgments
Funding
This work funded by the National Institutes of Health (K08CA201581 to Timothy L. Frankel, R01CA151588 to Mirna Perusina Lanfranca, R01CA198074 to Mirna Perusina Lanfranca, and U01 CA224145 to Mirna Perusina Lanfranca and Howard Crawford), the American Cancer Society (P30CA46592 to Mirna Perusina Lanfranca), and the Rogel Cancer Center (T32CA009672-21 and R01CA152470 to Weiping Zou).
Abbreviations used in this paper:
- ADM
acinar-to-ductal metaplasia
- CK
cytokeratin
- FBS
fetal bovine serum
- EMT
epithelial-mesenchymal transition
- HBSS
Hank’s balanced salt solution
- IHC
immunohistochemistry
- IL
interleukin
- IL6R
interleukin 6 receptor
- IL22R
interleukin 22 receptor
- ILC
innate lymphatic cell
- JAK
Janus kinase
- MAPK
mitogen-activated protein kinase
- mfIHC
multiplex fluorescent immunohistochemistry
- mRNA
messenger RNA
- PanIN
pancreatic intraepithelial neoplasia
- PCR
polymerase chain reaction
- PDAC
pancreatic ductal adenocarcinoma
- pERK
phosphorylated extracellular signal-regulated kinase
- PKCY
Pdx1-Cre;LSL-KrasG12D;Trp53+/−;Rosa26EYFP/+
- STAT
signal transducer and activator of transcription
- TCGA
The Cancer Genome Atlas
- Th
T helper
- WT
wild type
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2019.12.010.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Hidalgo M, Cascinu S, Kleeff J, et al. Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology 2015;15:8–18. [DOI] [PubMed] [Google Scholar]
- 2.Waghray M, Yalamanchili M, di Magliano MP, et al. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroenterol 2013;29:537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vonderheide RH. Tumor-promoting inflammatory networks in pancreatic neoplasia: another reason to loathe Kras. Cancer Cell 2014;25:553–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McAllister F, Bailey JM, Alsina J, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014;25:621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waghray M, Yalamanchili M, Dziubinski M, et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Discov 2016;6:886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cobo I, Martinelli P, Flandez M, et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 2018;554(7693):533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitcomb DC. Inflammation and cancer. V. Chronic pancreatitis and pancreatic cancer. Am J Physiol Gastrointest Liver Physiol 2004;287:G315–G319. [DOI] [PubMed] [Google Scholar]
- 8.Farrow B, Sugiyama Y, Chen A, et al. Inflammatory mechanisms contributing to pancreatic cancer development. Ann Surg 2004;239:763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993; 328:1433–1437. [DOI] [PubMed] [Google Scholar]
- 10.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007;11:291–302. [DOI] [PubMed] [Google Scholar]
- 11.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He W, Wu J, Shi J, et al. IL-22RA1/STAT3 signaling promotes stemness and tumorigenicity in pancreatic cancer. Cancer Res 2018;78:3293–3305. [DOI] [PubMed] [Google Scholar]
- 13.Kryczek I, Lin Y, Nagarsheth N, et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014;40:772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber S, Gagliani N, Zenewicz LA, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012; 491(7423):259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim C, Savan R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev 2014;25:257–271. [DOI] [PubMed] [Google Scholar]
- 16.Perusina Lanfranca M, Lin Y, Fang J, et al. Biological and pathological activities of interleukin-22. J Mol Med (Berl) 2016;94:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aggarwal S, Xie MH, Maruoka M, et al. Acinar cells of the pancreas are a target of interleukin-22. J Interferon Cytokine Res 2001;21:1047–1053. [DOI] [PubMed] [Google Scholar]
- 18.Feng D, Park O, Radaeva S, et al. Interleukin-22 ameliorates cerulein-induced pancreatitis in mice by inhibiting the autophagic pathway. Int J Biol Sci 2012; 8:249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue J, Nguyen DT, Habtezion A. Aryl hydrocarbon receptor regulates pancreatic IL-22 production and protects mice from acute pancreatitis. Gastroenterology 2012;143:1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wen Z, Liao Q, Zhao J, et al. High expression of interleukin-22 and its receptor predicts poor prognosis in pancreatic ductal adenocarcinoma. Ann Surg Oncol 2014;21:125–132. [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Tang Y, Guo S, et al. Increased intratumoral interleukin 22 levels and frequencies of interleukin 22-producing CD4+ T cells correlate with pancreatic cancer progression. Pancreas 2014;43:470–477. [DOI] [PubMed] [Google Scholar]
- 22.Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015; 160:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long KB, Gladney WL, Tooker GM, et al. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov 2016;6:400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L, Holtzinger A, Jagan I, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med 2015;21:1364–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazarus J, Maj T, Smith JJ, et al. Spatial and phenotypic immune profiling of metastatic colon cancer. JCI Insight 2018;3(22):e121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–450. [DOI] [PubMed] [Google Scholar]
- 27.Halbrook CJ, Wen HJ, Ruggeri JM, et al. Mitogen-activated protein kinase kinase activity maintains acinar-to-ductal metaplasia and is required for organ regeneration in pancreatitis. Cell Mol Gastroenterol Hepatol 2017;3:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Yan W, Collins MA, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res 2013;73:6359–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011;19:456–469. [DOI] [PubMed] [Google Scholar]
- 30.Boj SF, Hwang CI, Baker LA, et al. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol Cell Oncol 2016;3:e1014757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindemans CA, Calafiore M, Mertelsmann AM, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015;528(7583):560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol 2018;13:395–412. [DOI] [PubMed] [Google Scholar]
- 33.Dangi-Garimella S, Krantz SB, Shields MA, et al. Epithelial-mesenchymal transition and pancreatic cancer progression In: Grippo PJ, Munshi HG, eds. Pancreatic cancer and tumor microenvironment. Trivandrum, India: Transworld Research Network, 2012. x–xx. [PubMed] [Google Scholar]
- 34.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–446. [DOI] [PubMed] [Google Scholar]
- 35.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cobleigh MA, Robek MD. Protective and pathological properties of IL-22 in liver disease: implications for viral hepatitis. Am J Pathol 2013;182:21–28. [DOI] [PubMed] [Google Scholar]
- 37.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 2011; 12:383–390. [DOI] [PubMed] [Google Scholar]
- 38.Reichert M, Blume K, Kleger A, et al. Developmental pathways direct pancreatic cancer initiation from its cellular origin. Stem Cells Int 2016;2016:9298535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park O, Wang H, Weng H, et al. In vivo consequences of liver-specific interleukin-22 expression in mice: implications for human liver disease progression. Hepatology 2011;54:252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pushalkar S, Hundeyin M, Daley D, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirchberger S, Royston DJ, Boulard O, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 2013; 210:917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue J, Zhao Q, Sharma V, et al. Aryl hydrocarbon receptor ligands in cigarette smoke induce production of interleukin-22 to promote pancreatic fibrosis in models of chronic pancreatitis. Gastroenterology 2016;151:1206–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.