

In this study, Mona et al. set out to investigate the role of Ptf1a in specifying a subset of dorsal spinal cord inhibitory neurons in mice in vivo. The authors used CRISPR to target multiple noncoding sequences with putative cis-regulatory activity controlling Ptf1a and demonstrate a requirement for positive transcriptional autoregulatory feedback to attain the levels of PTF1A necessary for generating correctly balanced neuronal circuits.
Keywords: autoregulation, transcriptional control, cell fate specification, neuronal identity, bHLH transcription factor, somatosensory, itch, inhibitory neuron, spinal cord development
Abstract
Peripheral somatosensory input is modulated in the dorsal spinal cord by a network of excitatory and inhibitory interneurons. PTF1A is a transcription factor essential in dorsal neural tube progenitors for specification of these inhibitory neurons. Thus, mechanisms regulating Ptf1a expression are key for generating neuronal circuits underlying somatosensory behaviors. Mutations targeted to distinct cis-regulatory elements for Ptf1a in mice, tested the in vivo contribution of each element individually and in combination. Mutations in an autoregulatory enhancer resulted in reduced levels of PTF1A, and reduced numbers of specific dorsal spinal cord inhibitory neurons, particularly those expressing Pdyn and Gal. Although these mutants survive postnatally, at ∼3–5 wk they elicit a severe scratching phenotype. Behaviorally, the mutants have increased sensitivity to itch, but acute sensitivity to other sensory stimuli such as mechanical or thermal pain is unaffected. We demonstrate a requirement for positive transcriptional autoregulatory feedback to attain the level of the neuronal specification factor PTF1A necessary for generating correctly balanced neuronal circuits.
Cell fate determination during development requires precise spatiotemporal gene expression achieved by tightly orchestrated transcriptional programs. Combinatorial actions of lineage determining transcription factors binding to noncoding regulatory elements are central to this process. Despite increasing evidence of mutations in noncoding regions causing diseases (Qu and Fang 2013; Siggens and Ekwall 2014), understanding the complexity of interactions between enhancers directing tissue specificity and precise levels of gene expression, and the consequences to development when these interactions are disrupted remain largely unexplored. Here we identify distal noncoding sequences that control levels of a developmentally critical transcription factor that function to specify inhibitory neurons in somatosensory neuronal circuitry in the spinal cord.
The developing nervous system provides a unique opportunity to study transcriptional regulation and cell fate decisions due to the diversity of neuronal subtypes necessary to form the neuronal circuitry that controls behavior. During development, exposure of neural progenitor cells to extrinsic morphogen signals sets up combinatorial expression of transcription factors that commit the progenitors to distinct lineages. While there are many different classes of transcription factors such as homeodomain and zinc finger-containing factors, the basic helix–loop–helix (bHLH) factors stand out as essential to both neurogenesis and neuronal subtype specification in the dorsal neural tube (for review, see Lai et al. 2016). Some bHLH transcription factors activate cascades of lineage determining genes, including homeodomain factors, to generate diverse neuronal subtypes (Mizuguchi et al. 2006; Lai et al. 2011; Borromeo et al. 2014). Pancreas transcription factor 1A (PTF1A) is one such factor that, in addition to its requirement in the developing and adult pancreas (Kawaguchi et al. 2002), is transiently expressed in a subset of neural progenitors, and is required for specification of inhibitory neurons in multiple regions of the central nervous system including the dorsal spinal cord and brain stem, Purkinje cells and other inhibitory interneurons in cerebellum, and amacrine and horizontal cells in retina (Kawaguchi et al. 2002; Glasgow et al. 2005; Hoshino et al. 2005; Fujitani et al. 2006; Dullin et al. 2007; Nakhai et al. 2007; Pascual et al. 2007; Yamada et al. 2007; Millen et al. 2014; Iskusnykh et al. 2016).
Through loss- and gain-of-function studies, PTF1A was shown to direct neural progenitors toward an inhibitory, GABAergic neuronal cell fate while repressing gene expression programs for an excitatory, glutamatergic neuronal cell fate. Maintaining the balance between inhibitory and excitatory neurons is critical for functional neural circuits and disruption of this balance leads to perinatal lethality in Ptf1a null mice (Glasgow et al. 2005; Hori et al. 2008). PTF1A activates transcription of genes encoding homeodomain transcription factors such as Pax2, Lhx1, Lhx5, and inhibitory neurotransmitter biosynthesis and transporter proteins such as Gad1, Slc32a1, and Slc6a5 (Borromeo et al. 2014). PTF1A also indirectly suppresses the excitatory neuronal gene expression program by activating transcription of Prdm13, a repressor that silences transcription of genes encoding factors specifying excitatory neurons such as Tlx3 and Lmx1b. Thus, controlling where and when Ptf1a itself is transcribed is central to generating correctly balanced neuronal circuits.
The dorsal spinal interneurons form the first level of information processing for most somatosensory modalities; they receive primary sensory input from the periphery, they modulate the input in local microcircuits, and they send the processed information to the brain or other spinal circuits to generate the appropriate motor response. Molecular studies of Ptf1a knockout mice identified multiple neuronal subpopulations in the dorsal spinal cord that require PTF1A such as GLYT2-, NPY-, PNOC-, PDYN-, GAL-, SST-, and PENK-positive neurons (Bröhl et al. 2008; Huang et al. 2008), neuronal populations shown to process distinct somatosensory modalities such as nociception (pain), pruritic sensation (itch), mechanoreception (touch), and thermosensation (heat or cold) (Koch et al. 2018). Thus, Ptf1a is indispensable for specification of many subtypes of inhibitory neurons involved in somatosensory circuits in the spinal cord.
In an effort to understand how the spatiotemporal expression of Ptf1a is controlled, multiple noncoding genomic regulatory regions have been identified. Spontaneous noncoding mutations in human and mouse have pancreas-specific or cerebellum-specific functions with no reported disruption in the spinal cord (Weedon et al. 2014; Gonc et al. 2015; Gabbay et al. 2017; Evliyaoğlu et al. 2018). However, in enhancer/reporter assays in mouse and chick embryos, two enhancer regions were found sufficient to direct a heterologous reporter gene to a Ptf1a pattern in the developing neural tube. One enhancer is activated by PTF1A itself, and thus is defined as an autoregulatory enhancer (Masui et al. 2008; Meredith et al. 2009). It is sufficient to direct GFP expression in transgenic mice to all known Ptf1a domains in the pancreas and the nervous system. However, given the lack of sustained Ptf1a expression into the mature nervous system, the importance of positive autoregulation for this tissue was not clear. A second enhancer comprises a highly conserved sequence 3′ of Ptf1a that was defined as a dorsal neural tube specific enhancer (Mona et al. 2016). Here we test the requirement for these enhancer sequences for generating the correct spatiotemporal expression of Ptf1a in the mouse neural tube. Findings reveal an unexpected requirement for the autoregulatory enhancer to attain levels of PTF1A required for specifying dorsal spinal cord inhibitory neurons, even when maintenance of expression to retain neuronal subtype identity is not needed. Loss of this feedback regulation results in uncontrolled spontaneous itch and subsequent skin lesions.
Results
Mutating noncoding Ptf1a enhancer regions using CRISPR–Cas9 gene targeting in mice
In previous studies, we identified multiple regulatory regions for Ptf1a that are sufficient to direct expression of a heterologous gene to the Ptf1a expression domain in the neural tube. These include a 2.3-kb autoregulatory (AR) enhancer located 13.4 kb upstream of Ptf1a (Masui et al. 2007, 2008; Meredith et al. 2009) and another 1.2-kb dorsal neural tube (DNT) enhancer located 11 kb downstream from Ptf1a (Fig. 1A, top; Mona et al. 2016). The AR enhancer is directly regulated by the PTF1 transcriptional activator complex, a trimeric complex comprising PTF1A, an E-protein partner such as E47 or HEB (TCF3 or TCF12), and RBPJ (Beres et al. 2006; Hori et al. 2008). This enhancer is functional in all regions where PTF1A is expressed, and it contains two PTF1 binding sites separated by ∼1.2 kb. The PTF1-binding site consists of an E-box recognized by the PTF1A/E-protein heterodimer, and a TC-box recognized by RBPJ. The spacing between these motifs is constrained to one to three DNA helical turns for the PTF1 trimer to bind and activate transcription. The DNT enhancer contains a highly conserved 132-bp region required for its function (Mona et al. 2016). The assays used to demonstrate enhancer activity of a DNA sequence test sufficiency to direct tissue-specific gene expression but do not address their requirement in vivo. Here we test the in vivo requirement for the AR and DNT enhancers in regulating Ptf1a expression, and assess the consequences to neuronal specification and somatosensory-related behaviors.
Figure 1.

Mutations in the Ptf1a-AR enhancer lead to a spontaneous scratch phenotype. (A) Graphical representation of the Ptf1a locus. The previously identified autoregulatory (AR) enhancer with two PTF1 trimer complex binding sites consisting of an E-box and a TC-box, and the dorsal neural tube (DNT)-specific enhancer with the Pd-HD motif are shown. The sgRNA and donor templates used in the CRISPR targeting strategies in three separate experiments (Exp I–III) are depicted. (B) Representative mutant mice with lesions (arrows) due to excessive spontaneous scratching. (C) Timing for the onset of the scratch phenotype in three of the mutant strains. N > 25 in each group. (D) A subset of the mutant strains with and without the scratch phenotype used for analysis in this study. Both PTF1A-binding sites in the AR enhancer must be disrupted for the mice to exhibit the scratch phenotype (in AR2, a new cryptic PTF1 motif is generated). For details on each mutant strain, see Supplemental Figure S1 and Table S1.
Using CRISPR–Cas9, mice were generated with mutations targeted to the Ptf1a enhancers individually and in combination. Experiment I introduced sgRNAs targeting multiple enhancers simultaneously. sgRNA1 and sgRNA2 target the two PTF1-binding motifs in the AR, while sgRNA3 targeted near a paired homeodomain binding motif (Pd-HD) within the DNT (Fig. 1A). Experiments II and III targeted mutations to the AR and DNT enhancers individually. Additionally, in experiment II where the AR enhancer was targeted, donor templates were included to generate discrete mutations in the E-boxes and TC-boxes in the PTF1 motifs.
Resulting founder mice were screened for mutations by PCR and sequencing. Nineteen founder mutant strains were selected and bred to homozygousity (Supplemental Table S1). All 19 of the homozygous enhancer mutant mice survived past weaning. This is in contrast to null alleles of Ptf1a, which are perinatal lethal (Kawaguchi et al. 2002; Glasgow et al. 2005), suggesting the enhancer mutations do not eliminate Ptf1a expression completely, if at all. Notably, at 3–5 wk of age, five of the mutant strains began exhibiting a severe scratch phenotype in 100% of the homozygotes that requires euthanasia due to self-wounding (Fig. 1B,C; Supplemental Table S1). Whereas heterozygotes of these strains are indistinguishable from wild-type littermates, homozygous mutants start scratching at the neck and haunches, or licking/biting the front limbs causing deep lesions (Fig. 1B, arrows). Some enhancer mutant strains were crossed to a Ptf1a-null strain, Ptf1aCRE, to test for complementarity. The scratch phenotype was the same whether animals were homozygous for the enhancer mutant alleles or whether they had one enhancer mutant allele and one null allele, supporting the conclusion that the excessive scratching in the enhancer mutants is due to disruption of the Ptf1a gene locus (Supplemental Table S1).
The AR enhancer but not the DNT enhancer is required to generate the levels of PTF1A needed for normal spinal cord development
We hypothesized that mutation of the DNT enhancer would result in a decrease of Ptf1a in dorsal spinal cord development, resulting in loss of inhibitory neurons, and a possible disruption of somatosensory circuits. On the other hand, the AR enhancer was hypothesized to be most relevant in the acinar pancreas because Ptf1a expression persists throughout adulthood in this tissue, while it is only transiently expressed in the nervous system where no PTF1A is present in mature neurons. To determine which regulatory elements are responsible for the scratching phenotype, we compared the mutated sequences of affected mice relative to those strains with no excessive scratching behavior. Contrary to our expectations, this analysis revealed mutations in the AR enhancer are solely responsible for the phenotype. All five mutant strains exhibiting the scratch phenotype had both PTF1-binding sites in the AR enhancer disrupted. Surprisingly, mutations in the DNT enhancer, an enhancer sufficient to drive reporter gene expression specifically in the dorsal neural tube in transgenic assays, did not display any observable phenotype in vivo, even when 118 bp of a highly conserved region of the enhancer was deleted DNT118 (Fig. 1D; Supplemental Fig. S1; Supplemental Table S1).
Given that we generated multiple mutant alleles in the AR enhancer, we next asked whether one, both, or neither of the PTF1-binding motifs were required. Four of the five enhancer mutants exhibiting excessive scratch (AR-DNT1–3 and AR1) have relatively large deletions that disrupt both PTF1 motifs and encompass the 1.2 kb between them (Fig. 1D). Notably, the mutants that retained at least one intact PTF1 complex binding site did not display any phenotype (AR-DNT5–10 and AR5–6), demonstrating redundancy of these motifs within the AR enhancer (Supplemental Table S1). Two additional mutants support the conclusion that at least one PTF1 site is sufficient to maintain Ptf1a expression at levels needed to avoid the excessive scratching. First, in AR2 mutants, despite a large 1.2-kb deletion between the PTF1 sites, an E-box from one PTF1 site and a TC-box from the other site are brought together such that a new PTF1 trimeric complex binding site is reconfigured. Homozygous mutants in this strain are indistinguishable from wild-type littermates. Second, AR4 have specific mutations that disrupt each PTF1-binding motif but leave the 1.2 kb between the sites intact. These mutants also exhibit excessive scratch behaviors, albeit with a delayed onset relative to the mutants with the intervening 1.2 kb deleted (Fig. 1C). The AR4 mutant phenotype demonstrates the requirement for the PTF1 motifs within the enhancer for function. These results reinforce the importance of the PTF1 trimer as the functional transcription complex rather than individual binding of the PTF1A/E-protein heterodimer to the E-boxes or RBPJ acting through the TC-boxes (Beres et al. 2006; Masui et al. 2007; Hori et al. 2008). For further phenotype analysis, we focused mainly on AR-DNT1, AR1, and AR4 representing mice with the scratch phenotype, and utilized AR2 and DNT118 as controls representing mice lacking the scratch phenotype.
We next asked how Ptf1a expression is disrupted in the Ptf1a enhancer mutants, particularly in the caudal neural tube that gives rise to the spinal cord. Here, PTF1A is spatiotemporally restricted from E10.5 to E12.5 to the dorsal progenitor 4 (dP4) domain (Glasgow et al. 2005). Immunofluorescence for PTF1A at E10.5 shows PTF1A restricted to the dP4 domain, and although the number of cells positive for PTF1A is comparable between AR-DNT1, AR1, AR4, and their wild-type littermates, the level of protein is dramatically reduced. The reduced levels are shown either by quantifying fluorescence intensity of the PTF1A domain or by quantification of the number of cells with a particular fluorescence intensity from immunostaining for PTF1A (Fig. 2A–C; Supplemental Fig. S2). Supporting this result, in situ hybridization (ISH) for Ptf1a in these strains shows reduced levels of Ptf1a mRNA compared with wild-type littermates at E10.5 and E11.5 (Fig. 2D,F; Supplemental Fig. S2). Importantly, this decrease in PTF1A levels was only observed in mutant strains with the scratch phenotype but not in mutants lacking the scratch phenotype (AR2 and DNT118) (Fig. 2A–D). For a more quantitative analysis, we isolated RNA from E11.5 neural tubes for each strain and performed RT-qPCR. AR-DNT1, AR1, and AR4 strains exhibiting the scratch phenotype had less than a third Ptf1a mRNA than that in wild-type littermates (15%, 23%, and 27%, respectively), although AR4 did not reach significance (Fig. 2E). In contrast, AR2 and DNT118 had close to wild-type levels of Ptf1a mRNA (85% and 90%). These results highlight the role of the AR enhancer in driving the high levels of PTF1A required for normal development.
Figure 2.
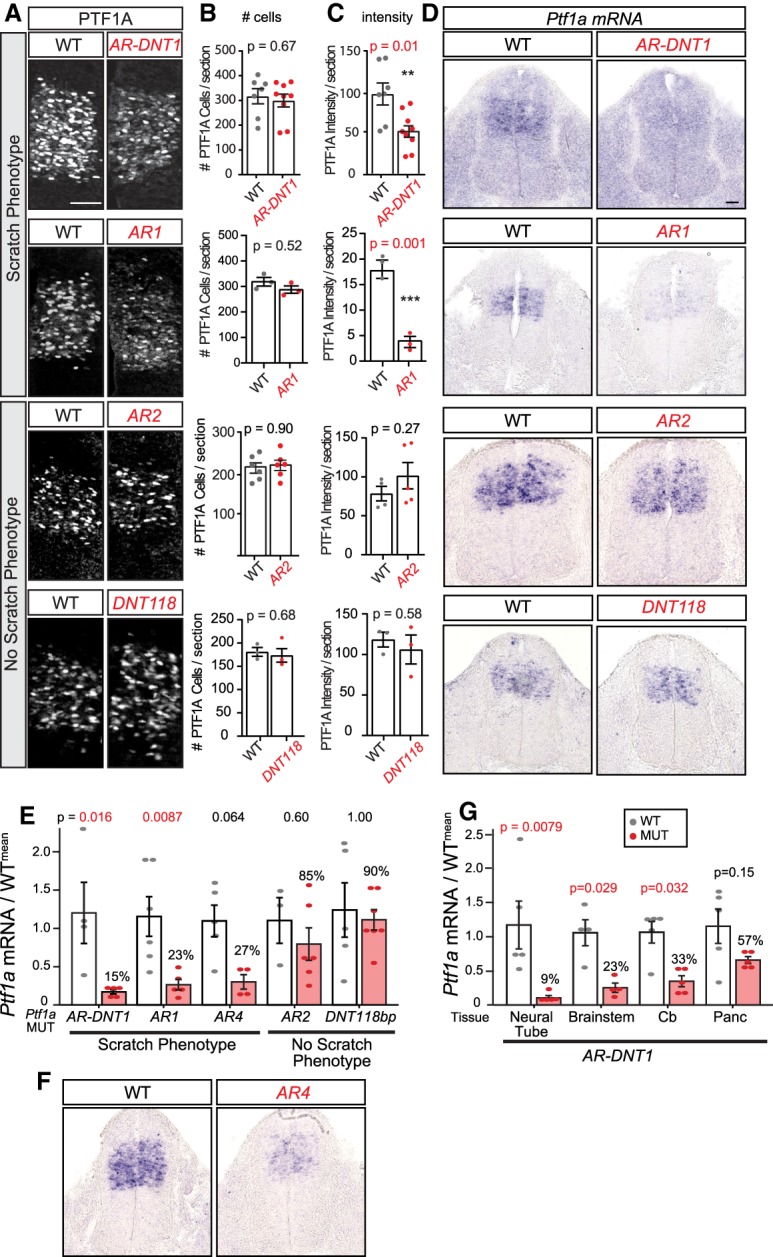
Ptf1a enhancer mutants with a scratch phenotype have lower levels of PTF1A relative to controls. (A) Immunofluorescence shows the PTF1A expression domain in transverse hemisections of the mouse neural tube at E10.5 comparing littermate controls (WT) with Ptf1a homozygous mutants with the scratch phenotype (AR-DNT1 and AR1) and without the scratch phenotype (AR2 and DNT118). (B) The number of PTF1A+ cells per E10.5 section is not different between WT and homozygous mutants. (C) The fluorescence intensity from immunostaining for PTF1A shows reduced levels in AR-DNT1 and AR1 but not AR2 and DNT118 compared with their wild-type littermates. Intensity measurements between strains cannot be compared due to imaging differences on different confocal microscopes in different experiments. (D–F) ISH for Ptf1a mRNA at E10.5, and RT-qPCR analysis of Ptf1a normalized to Gapdh at E11.5 in neural tubes of Ptf1a mutants and littermate controls show reduced levels of Ptf1a mRNA only in mutants with the scratch phenotype. (G) Ptf1a levels are also decreased relative to controls in other Ptf1a expressing tissues at E11.5 as assessed by RT-qPCR. Each data point represents a biological replicate (N), error bars indicate SEM. Student's t-test (B,C) and one-way ANOVA (E,G) were used to determine significant differences relative to WT. P-values are as indicated. (**) P < 0.01; (***) P < 0.001. Scale bar, 50 µm. See also Supplemental Fig. S2.
PTF1A is expressed in multiple regions of the developing nervous system besides the spinal neural tube such as cerebellum, brain stem, and hypothalamus (Kawaguchi et al. 2002; Glasgow et al. 2005; Hoshino et al. 2005; Fujitani et al. 2006; Dullin et al. 2007; Nakhai et al. 2007; Pascual et al. 2007; Yamada et al. 2007; Millen et al. 2014; Iskusnykh et al. 2016). Considering previous studies have shown activity of the AR enhancer across all Ptf1a expression domains (Meredith et al. 2009), we analyzed PTF1A protein and RNA in these regions in AR-DNT1 mutants. Immunofluorescence for PTF1A at E11.5 shows PTF1A is restricted to its normal domain but is reduced in the developing cerebellum and brain stem in AR-DNT1 relative to WT (Supplemental Fig. S2). Validation by RT-qPCR showed that the level of Ptf1a mRNA in AR-DNT1 was 9%, 23%, 33%, and 57% compared with WT littermates in neural tube, cerebellum, brain stem, and pancreas, respectively (Fig. 2G). In the pancreas, 55% of normal levels of Ptf1a is predicted to be sufficient for normal pancreas development based on previous reports (Fukuda et al. 2008). Indeed, we detect no obvious disruption in pancreas in the enhancer mutants (R MacDonald, unpubl.).
Sufficient PTF1A levels are required to specify subsets of inhibitory neurons in the dorsal neural tube during development
PTF1A is required for specification of dI4 PAX2+ neurons during development, evident in Ptf1a-null mice that have a complete loss of PAX2+ cells in the dorsal neural tube. The loss of dorsal PAX2+ neurons in the Ptf1a-null is in contrast to the increase in the neighboring TLX1/3+ and LMX1B+ neurons, reflecting a transfating of progenitors that would have been specified as dI4 in wild type into dI3 and dI5 neurons in the null (Glasgow et al. 2005). To understand how neural specification is affected in the Ptf1a AR enhancer mutant mice, we performed immunostaining for these markers at E10.5 and E14.5. Although AR-DNT1, AR1, and AR4 mutants retain some dorsal PAX2+ cells, they have significantly fewer compared with wild type (Fig. 3A,D; Supplemental Fig. S3). A comparable increase in TLX1/3+ and LMX1B+ cells in the mutants compared with wild-type controls was also seen (Fig. 3B,C,E,F). At E10.5, this increase in dI5 markers is detected spatially in the region normally occupied by dI4 (Fig. 3B,C, red arrowheads). At E14.5, the decrease in number of PAX2+ cells is specific to the Ptf1a lineage cells in the dorsal neural tube, as there is no difference in PAX2+ cells in the ventral domain relative to controls (Fig. 3D). In contrast to these cell identity phenotypes detected in AR-DNT1, AR1, and AR4 mutants, the mutant strains AR2 and DNT118 with no excessive scratch behaviors and no decrease in PTF1A were indistinguishable from wild type (Fig. 3; Supplemental Fig. S3). These results highlight the requirement of the AR enhancer to attain the levels of PTF1A necessary to specify the correct number of PAX2+ neurons in the dorsal spinal cord.
Figure 3.
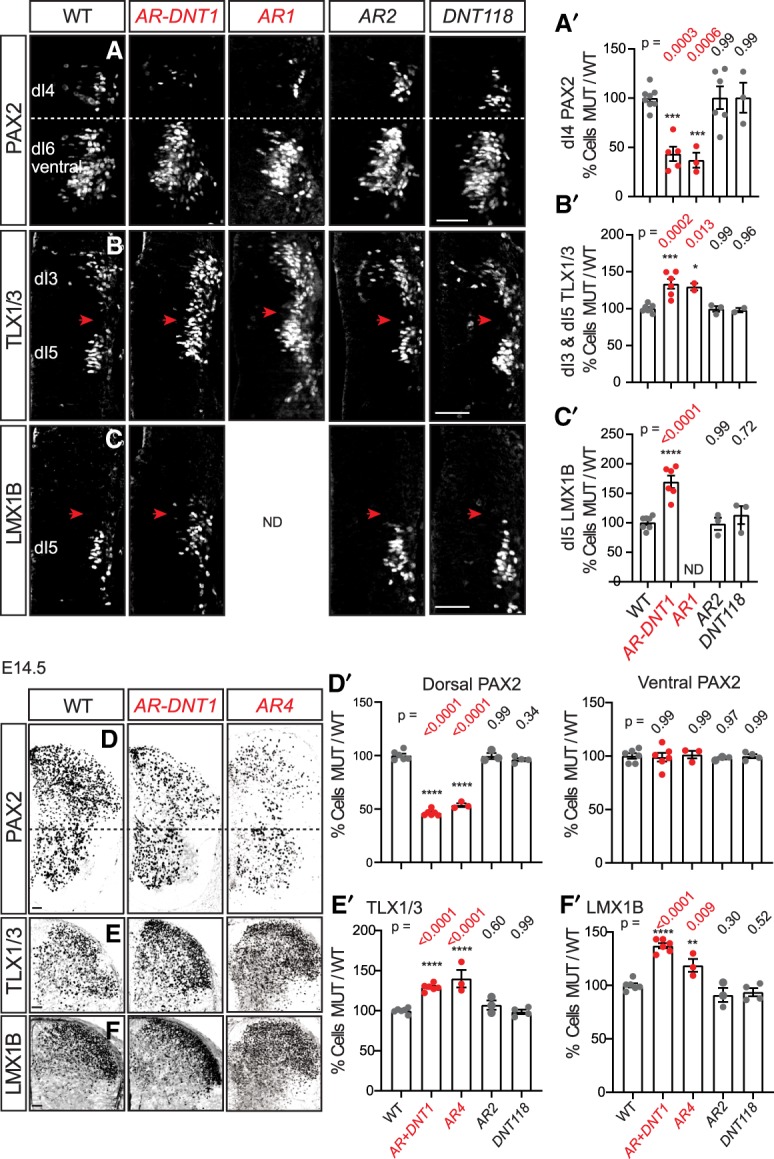
Ptf1a enhancer mutants have a reduced number of PAX2+ and an increase in TLX1/3+ and LMX1B+ neurons. (A–C) Expression of PAX2, TLX1/3, and LMX1B in transverse hemisections of mouse E10.5 neural tube. (A,A′) PAX2+ cells (dI4) are decreased in Ptf1a homozygous mutants with the scratch phenotype (AR-DNT1 and AR1) but not in mutants with no scratch phenotype (AR2 and DNT118) relative to littermate controls. In contrast, the number of TLX1/3+ cells (dI3/5) (B,B′) and LMX1B+ cells (dI5) (C,C′) are increased, occupying the dI4 domain (red arrowheads). (D–F) Expression of PAX2, TLX1/3, and LMX1B in transverse hemisections of mouse E14.5 neural tube. Similar to earlier stages, there is a loss of PAX2+ cells in the dorsal but not ventral spinal cords (D,D′), and there is an increase in TLX1/3+ (E,E′) and LMX1B+ (F,F′) cells in AR-DNT1 and AR4 but not in AR2 and DNT118 relative to littermate controls. Quantification reports the change in number of marker+ cells per section in mutant relative to WT controls. Each data point represents a biological replicate (N) and error bars indicate SEM. One-way ANOVA was used to determine significant differences relative to WT. P-values are as indicated. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. Scale bar, 50 µm. See also Supplemental Fig. S3.
We next asked how the neurotransmitter phenotypes detected by Gad1 and Glyt2 (Slc6a5) defining inhibitory neurons, and Vglut2 (Slc17a6) defining excitatory neurons are affected in postnatal Ptf1a enhancer mutant spinal cords at P30. In embryonic spinal cords, PTF1A is required for Gad1+ and Glyt2+ neurons while repressing generation of Vglut2+ neurons (Glasgow et al. 2005; Huang et al. 2008). Consistent with the decrease in PAX2+ cells, ISH for Gad1 and Glyt2 mRNA shows significant reduction in these inhibitory neuronal populations in P30 AR-DNT1, AR1, and AR4 mutant dorsal spinal cords compared with wild type (Fig. 4A,B). However, there was no significant change detected in VGlut2+ neurons (Fig. 4C). This was unexpected given the increases in TLX1/3 and LMX1B seen during embryogenesis. In contrast to these changes in the dorsal spinal cord, no decrease in Gad1+ neurons was detected in the cerebellum (Supplemental Fig. S4), consistent with the lack of observable ataxic behavior in these mice. Together, sufficient levels of PTF1A must be reached to generate the correct number of inhibitory neurons in the dorsal spinal cord.
Figure 4.
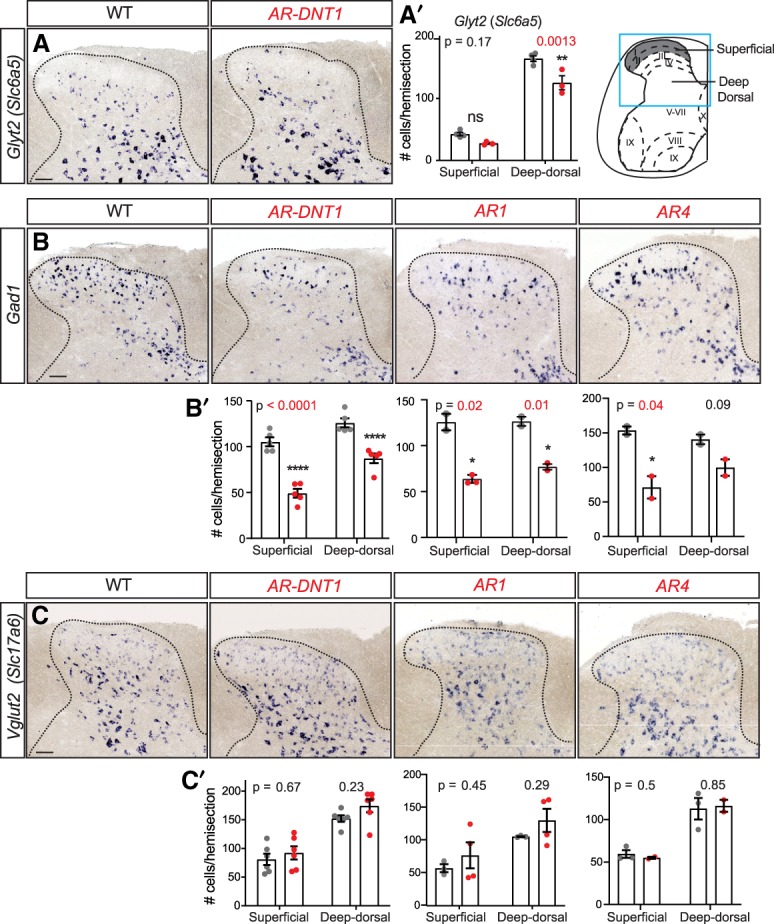
Reduced PTF1A levels affect specification of Gad1 and Glyt2 (Slc6a5) inhibitory neurons. (A–C) ISH shows a decrease in Glyt2 (A) and Gad1 (B), but no significant change in VGlut2 (Slc17a6) (C) neurons in Ptf1a enhancer mutants with the scratch phenotype (AR-DNT1, AR1, and AR4) at P30 compared with WT. (A′–C′) Quantification reports the number of marker+ cells per hemisection from superficial (gray in diagram) or deep dorsal spinal cord regions for the markers shown in A–C, respectively. Each data point represents a biological replicate (N), error bars indicate SEM. Student's t-test was used to determine significant differences relative to WT, P-values are as indicated. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001. Scale bar, 100 µm. See also Supplemental Fig. S4.
Pft1a enhancer mutants have increased sensitivity to chemical pruritogens but have normal acute responses to pain, thermal, and mechanical stimuli
Dorsal spinal interneurons receive somatosensory inputs from the periphery and serve as the first order of processing before the information is relayed to the brain or to motor output. PTF1A is required for specification of PAX2 and Gad1 dorsal inhibitory neurons (Mizuguchi et al. 2001; Glasgow et al. 2005), subsets of which modulate multiple sensory inputs such as pain, itch, touch, and motor control. Given the excessive scratching behavior observed (Fig. 1), it was predicted that the AR enhancer mutants would have increased sensitivity to chemical pruritogens as well. To test this, we investigated the scratch response after chloroquine or histamine intradermal injections in neck regions in postweaned animals prior to the appearance of the spontaneous scratching behavior (Fig. 5A,B). The AR-DNT1 mutants exhibit increased sensitivity to both pruritogens compared with littermate controls, consistent with a disinhibition of itch modulating circuits. Similarly, increased sensitivity to chloroquine was observed in a second mutant strain, AR-DNT2, that exhibited the spontaneous scratch phenotype (Supplemental Fig. S5A).
Figure 5.
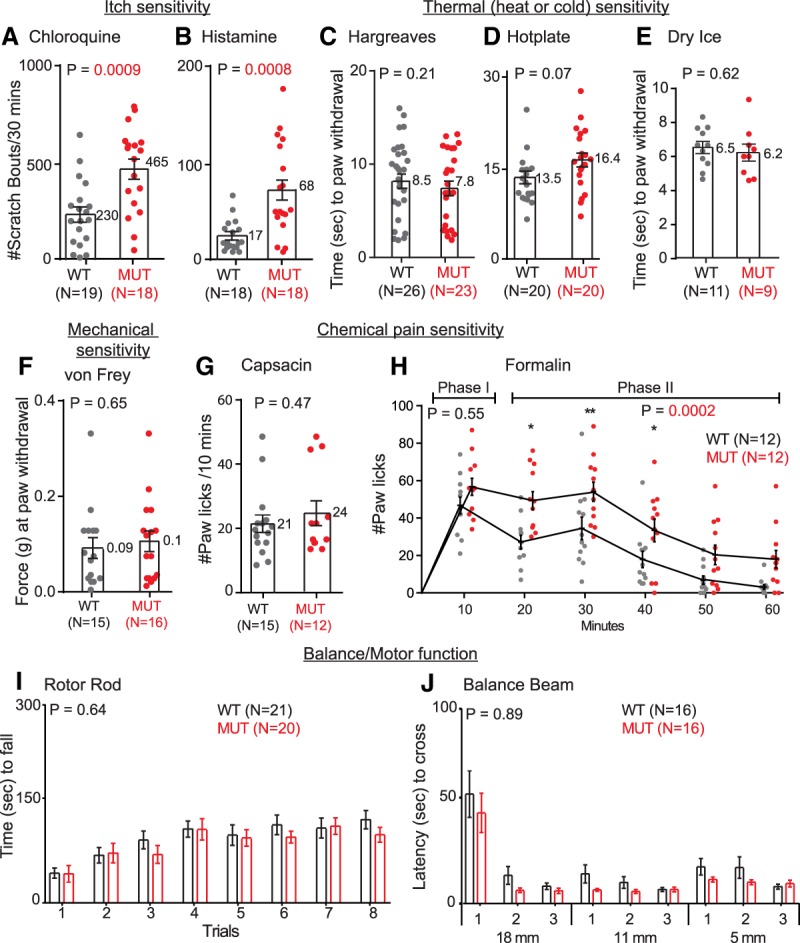
Ptf1a enhancer mutants have increased sensitivity to itch inducing pruritogens. (A,B) Quantification of the number of scratch bouts in 30 min after injection of chloroquine (A) or histamine (B) at the neck reveals increased sensitivity to itch in the AR-DNT1 mutants relative to WT littermates. (C–J) Thermal sensitivity behavior tests including Hargreaves (C), hot plate (D), and cold sensitivity (E), von Frey testing for mechanical sensitivity threshold (F), acute chemical pain, Capsaisin induced (G), the rotor rod (I), and the balance beam (J) showed no significant difference in the AR-DNT1 mutants relative to WT. (H) AR-DNT1 mutants do show increased sensitivity to phase II nocifensive formalin-induced pain but not phase I acute pain. Each data point represents a biological replicate (N); error bars indicate SEM. Student's t-test (A–G) or two-way ANOVA (H–J) was used to determine significant differences relative to WT littermate controls, N-values and P-values are as indicated. See also Supplemental Fig. S5.
To determine whether other somatosensory-related behaviors are altered in the AR enhancer mutants, we examined their responses in a battery of behavior tests. Tests for reflexive responses to thermal (Hargreaves, hot plate and dry ice), and mechanical (von Frey) sensitivities (Fig. 5C–F) and tests of balance and gross motor function (balance beam and rotor rod) (Fig. 5I,J) showed similar responses in AR-DNT1 mutants when compared with WT littermates. Similar results were observed in another mutant strain (AR-DNT2) for thermal (Hargreaves) and mechanical (von Frey) sensitivity (Supplemental Fig. S5). While capsaicin-induced and formalin-induced phase I acute pain (Fig. 5G,H, within 10 min after treatment) were similar among the WT and Ptf1a enhancer mutants, there was a significant difference in formalin-induced phase II nocifensive pain (Fig. 5H, 10–60 min after treatment). No additional disruption in somatosensory behaviors was detected. Thus, although there is a substantial loss in the number of PAX2 and Gad1 inhibitory neurons in the dorsal spinal cords of the Ptf1a enhancer mutants, only behaviors related to increased sensitivity to itch and nocifensive pain were detected.
Sufficient PTF1A levels are required to specify BHLHB5+/PAX2+ neurons in the dorsal neural tube during development
The excessive scratch behavior appearing in 3- to 5-wk-old mice is similar to that reported in mice mutant for the transcription factor BHLHB5 (Bhlhe22) (Ross et al. 2010). BHLHB5 is normally found in both excitatory and inhibitory neurons in superficial dorsal spinal lamina, but the loss of BHLHB5 specifically in the inhibitory PAX2+ lineage was shown to be responsible for the increased sensitivity to itch. Notably, these mice also had increased sensitivity to formalin-induced inflammatory pain, similar to the phenotype observed here for Ptf1a enhancer mutants. As a cellular basis for the excessive scratching in the AR enhancer mutants, we examined P5 cervical spinal cords from AR-DNT1 mutants for PAX2+/BHLHB5+ neurons. This analysis revealed a specific loss of these neurons relative to wild-type controls (Fig. 6A). The reduction in the number of BHLHB5+ neurons can be accounted for by the specific loss of those coexpressing PAX2 (Fig. 6A, arrowheads). Thus, the generation of BHLHB5 inhibitory neurons is dependent on sufficient levels of PTF1A, and the loss of these neurons in AR-DNT1 mutants at least in part explains the excessive scratching and nocifensive pain behavior in the mutants.
Figure 6.

Reduced PTF1A levels alter the balance of inhibitory and excitatory neurons in the dorsal spinal cord. (A,A′) Immunocytochemistry for PAX2 and BHLHB5 on P5 cervical spinal cords from superficial lamina I & II show a reduced number of PAX2/BHLHB5 neurons (yellow arrowheads) in AR-DNT1 mutants when compared with WT. Quantification reports the number of marker+ cells per hemisection (A′). Each data point represents a biological replicate (N), error bars indicate SEM. Student's t-test was used to determine significant differences relative to WT. P-values are as indicated. (*) P < 0.05; (**) P < 0.01. Scale bar, 50 µm. (B–D) UMAP visualization of the snRNA-seq from P25 cervical spinal cords from Ptf1aCre/AR-DNT1;Ai14 and Ptf1aCRE/+;Ai14 controls. (B) Clusters identified by major cell type. (C) Neurons were analyzed separately generating 31 clusters. Annotations are from pairwise Pearson correlation analysis between the data sets generated in this study and that of Sathyamurthy et al. (2018). (DI) dorsal inhibitory; (DE) dorsal excitatory; (VI) ventral inhibitory; (VE) ventral excitatory; (MI) mid inhibitory; (ME) mid excitatory; (VC) ventral cholinergic. (D) illustrates the separation of excitatory and inhibitory neuronal clusters. (E) A comparison of the proportions of control (Ctrl) and MUT nuclei were calculated relative to the total number of nuclei in the respective population and are represented by the size of the circle, and those clusters that are over or under represented in AR-DNT1 mutants relative to Ctrl are shown. Cluster annotations from this study (C), the associated annotation from Sathyamurthy et al. (2018), and marker genes for the cluster are shown. Genes in bold were also used as defining markers in Sathyamurthy et al. (2018). See Supplemental Table S2 for details including the P-values used to determine whether a cluster had significantly different contributions from the different genotypes. See Supplemental Figure S6 for visualization of genes supporting UMAP visualizations in B–D.
Multiple subtypes of spinal interneurons are misspecified in Pft1a enhancer mutants
The Ptf1a enhancer mutants establish the importance of PTF1A levels for specification of neuronal subsets in the dorsal spinal cord. To identify distinct neuronal populations affected by reduced PTF1A levels, we performed single-nucleus RNA-seq (snRNA-seq) on P25 cervical spinal cords from two AR-DNT1 mutants (Ptf1aCre/AR-DNT1;Ai14) and two littermate controls (Ptf1aCre/+;Ai14). Using 10X Genomics, we obtained sequence on 10,889 and 9969 nuclei of each genotype, respectively, that passed quality control criteria, of which 36% of each were identified as neurons (3875 and 3629), and the other nuclei were identified as other major spinal cord classes including astrocytes, oligodendrocytes, and OPCs (Fig. 6B; Supplemental Fig. S6A). We analyzed nuclei from the neurons only and identified 31 discrete clusters (Fig. 6C). Using uniquely expressed genes within the top 50 genes defining each neuronal cluster, we assigned neuronal subtype identities through pair-wise Pearson correlation analysis using data and nomenclature from (Sathyamurthy et al. 2018) who performed snRNA-seq on adult lumbar spinal cords (Fig. 6C; Supplemental Table S2). Similar to this previous study, we found that the inhibitory and excitatory neurons generate largely distinct clusters, particularly in the dorsal spinal cord where neuronal populations are more discretely clustered than the ventral neuronal populations (Fig. 6D; Supplemental Fig. 6B). Further analysis focused on the dorsal spinal cord populations where Ptf1a lineage neurons almost exclusively reside.
To pinpoint neuronal subtypes specifically affected in AR-DNT1 mutants that could underlie the scratch phenotype, we identified clusters that were either overrepresented or underrepresented in the AR-DNT1 mutant cervical spinal cords relative to controls (Fig. 6E; Supplemental Table S2). This analysis revealed a significant underrepresentation of all dorsal inhibitory neuron populations (annotated as DI from Sathyamurthy et al. 2018) in the AR-DNT1 mutants, consistent with the broad reduction of dorsal PAX2+ cells at E14.5, and the reduction of dorsal Gad1 and Glyt2 neurons at P30 described above (Figs. 3, 4). The most dramatic loss, however, was detected in the cluster DI-3* (* indicates the cluster from our study that is similar but not a perfect match with those in Sathyamurthy et al. 2018). This cluster is defined by marker genes Pnoc, Pdyn, and Gal. Notably, Pdyn and Gal are coexpressed in the BHLHB5 inhibitory neuronal lineage involved in inhibiting itch (Kardon et al. 2014). In contrast, two dorsal excitatory neuron clusters (DE in Sathyamurthy et al. 2018) were overrepresented in the AR-DNT1 mutants relative to controls, albeit the differences here were more modest (Fig. 6E; Supplemental Table S2). Grp, encoding a neuropeptide in excitatory neurons that mediate itch (Kardon et al. 2014; Sun et al. 2017) is a defining gene in one of these clusters, DE-1*.
The results from the snRNA-seq were validated by ISH for select genes in the dorsal horn of AR-DNT1 and AR1 mutants (Fig. 7). Consistent with the snRNA-seq, there is a reduction in the number of neurons expressing Pdyn and Gal in the mutant spinal cords relative to wild-type littermates (Fig. 7A,B). The remaining Pdyn and Gal in the mutant spinal cords may be due to the incomplete loss of DI-3*, or in the case of Pdyn, the signal may be from the unaffected excitatory neuronal population DE-15* that also expresses Pdyn. We next examined Grp by ISH to determine whether we could detect an increase in this population in the mutant spinal cords. A statistically significant increase in Grp neurons in AR-DNT1, but not in AR1 was detected (Fig. 7C). Grp is expressed in other excitatory neuronal clusters that do not change in the mutant, DE-2* and DE-3*, with DE-1* only contributing 15% of the Grp+ neurons detected in the snRNA-seq. Additionally, no change was detected in Penk or Nos1, genes encoding neuropeptides found in both inhibitory and excitatory neurons in multiple clusters such as DE-6*, 11*, and 16* for Penk (Fig. 7D,E). We also examined Npy+ by ISH since Npy-expressing inhibitory neurons are known to be within the Ptf1a lineage, and it is present in two inhibitory neuronal clusters underrepresented in the mutants, DI-4*, 9*; however, no loss of these neurons was detected by ISH (Fig. 7F). Taking the ISH and snRNA-seq together, the excessive scratch phenotype in Ptf1a enhancer mutants may be attributed to a particularly robust loss of DI-3* inhibitory neurons marked by Pdyn+ and Gal+, with a possible increase in DE-1* Grp+ neurons.
Figure 7.
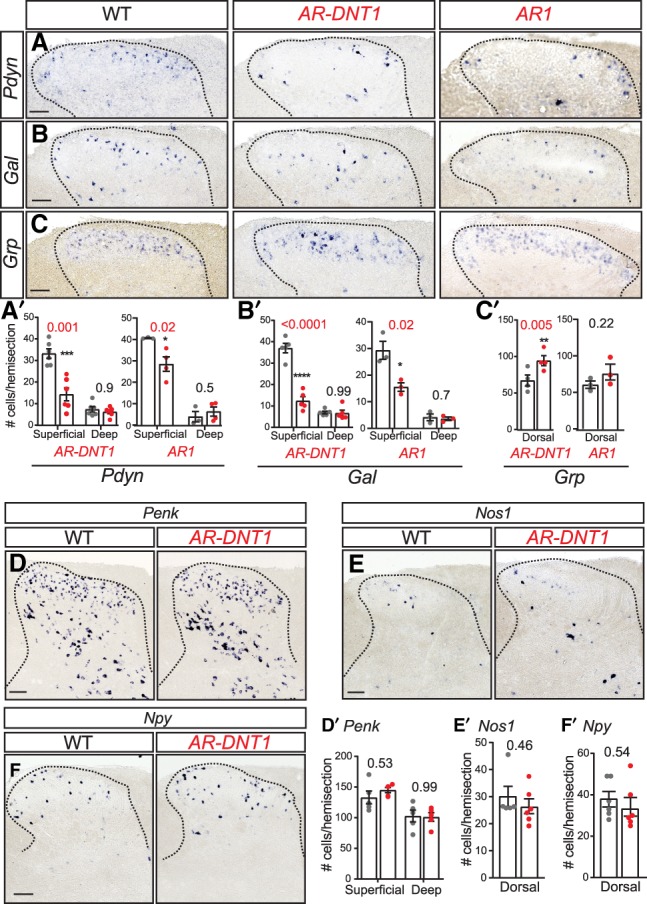
Reduced PTF1A levels affect specification of multiple neuronal subtypes in the dorsal spinal cord. (A–F) ISH for neuronal markers in transverse sections of spinal cord at cervical levels 1–4 in P25 mice. Ptf1a enhancer mutants with the scratch phenotype (AR-DNT1 and AR1) have a reduced number of Pdyn (A) and Gal (B) neurons, and an increase in Grp (C) neurons. Penk (D), Nos1 (E), and Npy (F) neurons were comparable between AR-DNT1 mutants and WT littermates. (A′–F′) Quantification reports the number of marker+ cells per hemisection from the superficial or deep-dorsal spinal cord regions. Each data point represents a biological replicate (N), error bars indicate SEM. Student's t-test was used to determine significant differences relative to WT. P-values are as indicated. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. Scale bar, 100 µm.
Discussion
Functional redundancy of noncoding genomic regulatory regions, particularly for essential developmental regulators, is thought to be important for a robust biological system (Cannavò et al. 2016; Dickel et al. 2018). However, this feature makes it challenging to uncover functions of individual regulatory regions in situ, and has hampered the ability to study noncoding genomic regions. CRISPR technologies are overcoming some of the experimental hurdles (Osterwalder et al. 2018; Torbey et al. 2018). Here we target multiple putative noncoding regulatory elements in situ to gain a deeper understanding of transcriptional regulation of an essential developmental gene. These studies connect mutations in the noncoding genome to subsequent disruption of cell fate decisions with adversive consequences to somatosensory-related behaviors. We found that functionally distinct inhibitory neurons, associated in particular with chemical itch, rely on transcriptional autoregulation of Ptf1a.
Mutations in a Ptf1a noncoding region result in adverse itch sensitivity and an increased nocifensive chemical pain response without affecting other somatosensory behaviors
Most genome-wide association studies (GWAS) have demonstrated that >90% of disease-associated genetic variants are in noncoding regions (Gallagher and Chen-Plotkin 2018). Examples include a cis-regulatory element for Dmrt3 deleted in patients with gait abnormalities and impaired development of the forebrain (Kubota et al. 2018), and single nucleotide polymorphisms (SNPs) in the regulatory region of Scn10a, coding for the NAV1.8 sodium channel, found associated with decreased sensitivity to mechanical pain (Duan et al. 2018). Additionally, mutations in Ptf1a cis-regulatory regions identified in humans cause pancreatic agenesis (Weedon et al. 2014). Our study reveals that loss of PTF1-complex binding sites in the autoregulatory enhancer of Ptf1a leads to disrupted dorsal spinal cord development with increased itch sensitivity and an apparent break in feedback mechanisms that suppress scratching behaviors.
Spinal cord neural circuits comprise complex interactions between excitatory and inhibitory neurons for somatosensory information processing. Ptf1a enhancer mutants have a general decrease in inhibitory neuronal populations in the dorsal spinal cord as revealed in the snRNA-seq and the Gad1 ISH. However, this general decrease in dorsal inhibitory neurons is not accompanied by a comparable increase in excitatory neurons when assessed by snRNA-seq or VGlut2 ISH in P25 spinal cords. The increase in TLX1/3 marking dorsal excitatory neuronal lineages embryonically may be compensated for by P25, or these neurons may be localized to more ventral populations. Indeed, the snRNA-seq did reveal some increase in ventral excitatory neurons in the AR-DNT1 mutants over controls (Supplemental Table S2).
Notably, the developmental specification of the subset of inhibitory neurons marked by BHLHB5, Pdyn and Gal are particularly sensitive to decreased levels of PTF1. These neurons, which receive direct inputs from nociceptive afferents, are required to gate itch transmission, such that deletion of the gene encoding BHLHB5 results in a spontaneous scratch phenotype similar to that shown here (Ross et al. 2010; Kardon et al. 2014). Development of other classes of inhibitory neurons, including NPY+ inhibitory neurons that are involved in mechanical itch (Bourane et al. 2015) appear less affected by decreased PTF1A. The robust disruption in itch circuitry may also be modulated by a modest increase in the number of Grp neurons, excitatory neurons that are specified by TLX3 during development and are known to mediate itch information in the spinal cord (Xu et al. 2008, 2013). Thus, the scratch phenotypes seen in the Ptf1a enhancer mutants could result from the loss of inhibitory neurons, the increase in excitatory neurons, or a combination of these alterations.
Importantly, the loss of inhibitory neurons, as detected by PAX2, Gad1, and Glyt2 in tissue sections, and revealed in the snRNA-seq, is more general than just the loss of itch circuit inhibitory neurons. However, the only other behavioral phenotype detected was an increase in nocifensive pain behavioral responses. Here, the acute response to a chemical pain stimulus was not different between the Ptf1a mutants and wild-type littermates; however, an increase in a sustained response in the mutants, thought to include more complex information processing, was detected. Whether this is due to loss of inhibition in the spinal cord or other higher brain regions was not determined. The lack of other somatosensory phenotypes, particularly mechanical and thermal pain sensitivities, was surprising given the close interactions within itch and pain circuits in the spinal cord. Pdyn inhibitory neurons have been implicated in the gating of both mechanical pain and chemical itch (Duan et al. 2014). The existence of separate subsets of Pdyn-expressing neurons that carry out these distinct roles was proposed. Loss of the AR elements may cause a selective loss of bHLHB5/Pdyn neurons functioning for itch inhibition, whereas the remaining Pdyn neurons could be sufficient to gate mechanical sensitivity. It is also possible that itch circuits have a greater inhibitory drive that is less protected by redundant pathways than other sensory modalities whose signals need to be sustained until sensory stimuli are removed. Nevertheless, mutation of noncoding genomic regions disrupting positive feedback on transcription of the Ptf1a locus leads to specific adversive, somatosensory behavioral phenotypes, likely through preferential disruption in specification of Pdyn/Gal-expressing neurons.
An autoregulatory enhancer is required to achieve levels of PTF1A necessary for specification of dorsal spinal cord inhibitory neurons
Comparison of the different enhancer mutant mice generated in this study reveals that the AR enhancer is necessary to attain high levels of PTF1A in the developing nervous system. Autoregulation, whereby a transcription factor feeds back to activate or repress transcription of its own gene, is a common transcriptional control mechanism utilized by developmentally important transcription factors. In transiently expressed genes, or in genes requiring oscillating levels of expression, negative autoregulation is key, such as that seen with the negative autoregulation of Hes1 (Hirata et al. 2002; Kageyama et al. 2007). In contrast, positive autoregulation is important with transcription factors that turn on in development but need to be maintained into adult stages to lock in cell-fate identities, such as seen with Pou4f1 (Trieu et al. 2003). With these models in mind, it was unexpected to find that positive feedback through the autoregulatory enhancer is key in neural development where Ptf1a is only transiently expressed, whereas there was no phenotype detected in the pancreas where Ptf1a expression is maintained throughout adulthood, and where it is required for acinar cell maintenance (Kawaguchi et al. 2002; Hoang et al. 2016). Here we demonstrate a different role for positive autoregulation: a boost to transcription such that sufficient levels of the factor, in this case PTF1A, are attained to specify particular cell fates. Similarly, transcriptional autoregulation was recently reported in Caenorhabditis elegans where the che-1 zinc-finger transcription factor was shown to be required to amplify che-1 expression during embryonic development to reach a minimal threshold to specify the ASE neuronal fate (Leyva-Díaz and Hobert 2019). In that case however, the positive autoregulation is also important for sustained expression. For the transiently expressed Ptf1a, additional mechanisms such as feedback inhibition by the transcription repressor PRDM13 (Mona et al. 2017) must be in play to interrupt this positive feedback loop to turn off expression.
Redundancy in the regulation of Ptf1a in the dorsal neural tube
Transcriptional regulation is a complex process comprising combinations of transcription factors working through multiple cis-regulatory sequences to obtain the necessary levels of gene expression with defined spatial and temporal characteristics. A recent example of this complexity is seen in the transcriptional regulation of Indian Hedgehog (Ihh) where nine different regulatory regions, each with a tissue-specific function, were identified that direct expression of Ihh in an additive manner (Will et al. 2017). Previously identified enhancers for Ptf1a hint at similar transcriptional regulatory complexity. A mouse strain (Ptf1acbll; Cerebelless) with a large deletion 60 kb 3′ of Ptf1a disrupted Ptf1a expression, and resulted in cerebellar and pancreatic agenesis (Hoshino et al. 2005; Fukuda et al. 2008). Whole-genome sequencing of humans with isolated pancreatic agenesis identified multiple noncoding genomic regions in the Ptf1a locus with apparent pancreas specific functions (Weedon et al. 2014; Gonc et al. 2015; Gabbay et al. 2017; Evliyaoğlu et al. 2018). In these cases, no disruption of somatosensory behaviors was reported, suggesting complex tissue specificity of regulatory regions for Ptf1a.
The DNT enhancer was identified through direct tests of seven genomic regions downstream from the Ptf1a-coding region selected due to extensive sequence conservation across mammals (Mona et al. 2016). Nevertheless, deletion of a 118-bp region within the DNT had no detectable consequence to Ptf1a transcription or to Ptf1a-dependent behaviors. These results suggest Ptf1a expression in the dorsal neural tube is controlled by redundant regulatory regions. This redundancy could be from sequences within the DNT enhancer not targeted here, or regulatory regions located elsewhere in the Ptf1a locus. Further studies will be needed to elucidate the complexity of this DNT enhancer and other inputs into controlling the spatiotemporal characteristics of Ptf1a expression in the developing spinal cord.
Even within the AR enhancer, functional redundancy of cis-regulatory elements was seen. This enhancer contains two PTF1 complex-binding sites ∼1.2 kb apart that can act independently, and the presence of either is sufficient to obtain enough PTF1A to generate the inhibitory neurons needed for correct modulation of the itch circuitry. This functional redundancy supports findings from reporter assays that used targeted mutations of the E-box and TC-box motifs within the enhancer (Masui et al. 2007; Meredith et al. 2009). The allelic series generated for this study adds additional support for redundancy in the AR enhancer. In particular, the AR2 mutant, where a 1.2-kb deletion between the two PTF1 motifs regenerated a single cryptic PTF1 site, had sufficient Ptf1a transcription to support normal itch behaviors. Only alleles that disrupted both PTF1 sites elicited the scratch phenotypes.
Materials and methods
Star methods
Further information and requests for resources and reagents are available on request.
Resources
The following antibodies were used in this study: rabbit anti-PAX2 (1:500; Invitrogen 71-6000), guinea pig anti-TLX1/3 (1:10,000; C. Birchmeier, Max Delbrück Center for Molecular Medicine, Berlin), guinea pig anti-PTF1A (1:10,000; laboratory-generated TX507), guinea pig anti-LMX1B (1:5000; C. Birchmeier, Max Delbrück Center for Molecular Medicine, Berlin), and guinea pig anti BHLHB5 (1:10,000; S. Ross). The chemicals, peptides, and recombinant proteins used were chloroquine diphosphate (Sigma C6628), histamine (Sigma H7125), capsaicin (Sigma M2028), formalin (Sigma HT501128), NBT (Roche 11383213001), and BCIP (Roche 1138322100). We used Adobe Illustrator and Photoshop, Graphpad Prism 5, Microsoft Excel 365, and National Institutes of Health ImageJ. The following sgRNA/oligos/primers were used: sgRNA1 (5′-CACAAGTGGCGACATTCCCA-3′), sgRNA2 (5′-CCGCAGAGCACGCCAGTCCG-3′), sgRNA2.1 (5′-ATAACACATGTGCTGGGGCG-3′), sgRNA3 (5′-GGGTAACCATTGGTTTGATT-3′), donor 1 (5′-ATTTATAATGTTCATTGAGCATTTTGCCTAATTGTGTCTGTCTCTGAAAGGGCAGCGCACCCCTCCCGACCACGAGCGGCACTCGAGGCGACATTCTATTGGAGCGGCCGCGCCGCAGCTGCCGTATTCAGTACGGGCTGCCCGGACGTGTGCTTCTGGCCCCGGCGACCGGGCGCCT-3′), donor 2 (5′-CGGCGGGAGGCCAGGAGGCCCAGCTCAGGGAGGGCCGGGTGGTCCCCACTCCCCACCCCCGCCCCGCCAGCCTCGCCCCAGCAGATATCTTATGATTCAATCGAACCGGCATGCTCTGCGGCTGGAGGCCCGGGTGCCCGGGGCCCCGGAGACGTGCAGGACATAGAGGGGTTTGTACAC-3′), PCR primers for genotyping the Ptf1a locus (WT: 123 bp) (forward 5′-TGAGGAAGATTTCTTCACCGACCAGTCCTC-3′ and reverse 5′-CGGTAGCAGTATTCGTGTAGCTGGTG-3′), PCR primers for genotyping the Ptf1aCre locus (Cre: 250 bp) (forward 5′-ATAGGCTACCTGGCCATGCCC-3′ and reverse 5′-CGGGCTGCAGGAATTCGTCG-3′), PCR primers for the AR-far locus (WT: 259 bp) (forward 5′-CCCGCGAGCGACCATATAAT-3′ and reverse 5′-TGAGCCGCGAGCTATTAGTG-3′), PCR primers for the AR-near locus (WT: 316 bp) (forward 5′-GATTTCCCCGAGCGTCTGAA-3′ and reverse 5′-ACCTGAGCCCTTGACTGGTA-3′), PCR primers for the DNT locus (WT: 308 bp) (forward 5′-GCAGTGATCTCACCATCCCC-3′ and reverse 5′-AACCTGCAGAGCTCGAAAAG-3′), PCR primers for deletions between the AR-far and AR-near loci (i.e., for AR1) (forward 5′-CAACACCGAGTCTTTCAGTTGTATT-3′ and reverse 5′-GTGTACAAACCCCTCTATGTCCTG-3′), sequencing primers for the AR-far locus (5′-CCATATAATTTGATTTGCCAGG-3′), sequencing primers for the AR-near locus (5′-GCGTCTGAACACCCCATTCG-3′), sequencing primers for the DNT locus (5′-CACCATCCCCAATCATTTTTTATAC-3′), mouse Ptf1a qPCR primers (forward 5′-TAGACACGCTGCGCTTGGCCATAGGCTAC-3′ and reverse 5′-ACAAAGACGCGGCCAACCCGATGT-3′), and mouse Gapdh qPCR primers (forward 5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse 5′-TGTAGACCATGTAGTTGAGGTCA-3′).
Mouse strains
The Ptf1a enhancer mutant mouse lines were generated by pronuclear injection of Cas9 mRNA and sgRNA targeting previously identified enhancer regions of Ptf1a (Meredith et al. 2009; Mona et al. 2016) in the Transgenic Technology Center at University of Texas Southwestern. Three experimental strategies were used (Fig. 1A). Experiment I included the combination of sgRNA1 (targeting AR-far), sgRNA2 (targeting AR-near), and sgRNA3 (targeting DNT). Experiment II included sgRNA1 and sgRNA2, and to improve frequency of generating indels in AR-near, sgRNA2.1. In this strategy, two donor templates were also included to facilitate homologous repair. Donor 1 included a mutation in the E-box and TC-box (sequence underlined) to disrupt the PTF1-binding site at the AR-far locus. A XhoI site was introduced for ease of genotyping. Donor 2 included a mutation in the E-box and TC-box (sequence underlined) to disrupt the PTF1-binding site in the AR-near locus. An EcoRV site was introduced for ease of genotyping. Experiment III included only sgRNA3 to generate deletions in the DNT enhancer. The sgRNAs were designed to target specific genomic loci with careful consideration of high on-target score and low off-target risks. Using Cas-OFFinder, no off-target was found when allowed for one mismatch for each sgRNA and only two off-targets (on chr13 and chrX; not near Ptf1a locus on chr2) were found when allowed for two mismatches.
Out of 49 founder mouse strains from experiment I, 53 from experiment II, and 102 from experiment III, 19 independent mouse strains were chosen for breeding to obtain homozygous mutants for further analysis (Supplemental Table S1). To further reduce the risk of off-target effects each mouse strain was outbred with ICR and maintained as heterozygotes before obtaining homozygous mutants for analysis. Genotyping of each allele was PCR-based followed by sequence confirmation. No heterozygous phenotypes were detected. All homozygote mutant strains survived postnatally but five strains exhibited excessive scratching, and in four of these strains subviability was detected (Supplemental Table S1). Due to required euthanasia of homozygous mutant animals at 3–5 wk due to the excessive scratching, animals were carried as heterozygotes and outbred to ICR over multiple generations. To demonstrate complementarity with Ptf1a, a subset of the enhancer mutant mice exhibiting excessive scratching were also bred with Ptf1aCRE mice containing a Ptf1a-null allele (Kawaguchi et al. 2002). In each case, compound heterozygotes with one Ptf1aCRE allele and one enhancer mutation allele exhibited excessive scratching, a phenotype never detected in heterozygous animals. Genotyping for the Ptf1aCRE allele was performed as described previously (Glasgow et al. 2005).
All animal work was approved by the Institutional Animal Care and Use Committee at University of Texas Southwestern. All mouse strains were maintained on a mixed background of ICR and C57Bl/6.
Tissue preparation, immunohistochemistry (IHC), in situ hybridization (ISH), and microscopy
Mouse embryos at embryonic day 10.5 (E10.5), E11.5, or E12.5 were dissected in ice-cold 0.1 M sodium phosphate buffer pH 7.4 and fixed in 4% paraformaldehyde for 1 h, 1.5 h, or 2 h, respectively, at 4°C. For E14.5, spinal cords were dissected out of the embryo prior to fixation for 2 h at 4°C. For postnatal day 5 (P5) spinal cords, the mice were anesthetized on ice, transcardially perfused, first with PBS and then with 4% paraformaldehyde. Dissected vertebral columns were postfixed for 2 h at 4°C. Tissue was washed three times in ice-cold 0.1 M sodium phosphate buffer (pH 7.4) for 15 min and sunk overnight in 30% sucrose in PBS for E10.5, E11.5, and E12.5 and 30% sucrose in water for E14.5 embryos and P5. Tissue was embedded in Tissue-Plus O.C.T. compound (Fisher Healthcare) and cryosectioned at 30 µm focusing on sections from the upper limb level. For IHC, cryosections were blocked with a PBS/1% normal goat or donkey serum/0.2% NP-40 or 0.1% Triton-X for at least 1 h at room temperature and incubated overnight with primary antibody at 4°C. The appropriate secondary antibody (Alexa 488, 568, and/or 657; Invitrogen) was incubated for an hour at room temperature. ISH for Ptf1a on neural tubes was as described previously (Chang et al. 2013).
For ISH on P30 spinal cords, hydraulic extrusion of spinal cords was performed (Richner et al. 2017). The lower cervical (C4–8) segments were embedded in OCT and cryosectioned at 20–30 µm. Slides dried at room temperature were fixed in 4% paraformaldehyde in DEPC-PBS for 20 min at room temperature. Quick wash in DEPC-water at room temperature was followed by acetylation (for 200 mL of 0.1 M RNase-free triethanolamine-HCl at pH 8.0, add 500 µL of acetic anhydride). Peroxide treatment (3% H2O2/methanol) for 30 min was followed by incubation in RIPA buffer for 45–60 min. Slides were postfixed in 4% paraformaldehyde in DEPC-PBS for 20 min at room temperature and prehybridized for 1–4 h at 60°C–70°C followed by hybridization with 1–2 ng/µL of fresh probe overnight. Multiple washes with varying concentrations of saline-sodium citrate were performed with RNAse treatment followed by overnight incubation with antidigoxygenin antibody. NBT and BCIP were used to visualize digoxygenin. Probes used were Gad1, Gal, Grp, and Nos1 (Allen Brain Atlas), and Pdyn, Penk, Npy, GlyT2 (Slc6a5), and Vglut2 (Slc17a6) (Cheng et al. 2004).
Imaging was performed in the Neuroscience Microscopy Facility with a Zeiss LSM 710 or 880 confocal microscope with 5-µm optical slice at 20× zoom or as appropriate. Images were pseudocolored using Adobe Photoshop (Adobe). ISH sections were imaged with a Nanozoomer C9600-12 (Hamamatsu).
Quantification and statistical analysis
Quantification of cell number and intensity was assisted by ImageJ software on three or more sections for each embryo (N = number of embryos, biological replicates). Each data point in the graphs corresponds to a biological replicate averaged over multiple sections. For intensity measurements using ImageJ, either the overall PTF1A (dI4) domain was chosen, or individual cells were selected with a filter size of 5–10 µm. The individual cells with particular intensity were plotted in a histogram using bins for various expression levels. For postnatal spinal cords, IB4 IHC was used to distinguish lamina I + IIi from the rest of the spinal cord and the central canal was used as reference to distinguish dorsal versus ventral. Significant differences between control and mutants in each case were calculated using a two-tailed two-sample unequal variance (homoscedastic) Student t-test, one-way ANOVA or two-way ANOVA, as appropriate. For each genotype, mutants were compared with their WT littermates to ensure direct comparison and account for embryonic staging or technical differences. Error bars indicate SEM.
RT-qPCR analysis
Mouse E11.5 neural tubes caudal to brain stem were dissected in ice-cold PBS and dissociated in Trizol reagent (Invitrogen 15596-026). Total RNA was extracted using the Arcturus PicoPure RNA isolation kit (Applied Biosystems 12204-01), cDNA was synthesized with the SuperScript III first strand synthesis supermix (Invitrogen 11752-050). Real-time RT-qPCR reactions were performed in technical triplicates using the 7500 Fast real-time PCR system (Applied biosystems) with Fast SYBR Green master mix (Applied Biosystem 4385612) for SYBR assay. Each data point in the graphs corresponds to a biological replicate. The RT-PCR primers used are listed above in “Resources.”
Single-nucleus RNA-seq
Lower cervical spinal cord segments were isolated by hydraulic extrusion from two Ptf1aCre/+;Ai14 (Control) and two Ptf1aCre/AR-DNT1;Ai14 P25 (Mut). Nuclei were prepared essentially as described in Jove video (Matson et al. 2018). 10× Genomics Next GEM single-cell 3′ reagent kit v3.1 was used. The Agilent Tapestation 4200 with the DNAHS 5000 tape and a Qubit 4 fluorometer (Thermo Fisher) using the DNA HS assay were used to assess samples at multiple stages before and after library construction. Samples were sequenced on an Illumina NextSeq500 high-output flow cell using V2.5 chemistry. Single-nucleus reads were mapped to annotated pre-mRNA sequences from mouse reference genome (mm10) with sequences from tdTomato and its 3′ UTR from Ai14 added, and raw expression UMI counts were obtained using the 10× Genomics CellRanger (V3.1.0) tool. TdTomato transcripts were not efficiently detected and were not used in the subsequent analysis. snRNA-seq data generated here are available on the GEO database (GSE146238).
Clustering analysis
Downstream analysis of snRNA-seq raw expression data was performed using Seurat R package (v3.1.1). Only nuclei with >200 genes/features expression and <5% of mitochondrial reads were retained. After filtering, we merged the expression counts from the two genotype samples and log normalized the counts. The normalized data was used to identify the anchors using the FindIntegrationAnchors function, which uses the canonical correlation analysis (CCA) method, and integrates data from two objects. Integrated data was scaled and used for PCA analysis using 20 dimensions. We used these principal components and performed UMAP clustering with 0.1 resolution to identify the most abundant clusters in the data.
Cell type annotation
To annotate different cell type clusters in our data, we used signatures defined in Sathyamurthy et al. (2018) to annotate neurons, astrocytes, oligodendrocytes, oligodendrocyte progenitor cells (OPCs), and meningeal cells and annotated the clusters (Fig. 6B; Supplemental Fig. S6A).
Neuron reclustering and annotation
Based on neuronal marker expression, we separated nuclei from C1, C5, C6, C7, C9, C10, and C12 and reclustered them using the UMAP method with 0.8 resolution. Eight small clusters lacked neuronal markers and were removed. UMAP clustering on the remaining neuronal clusters with 0.8 resolution identified 31 clusters (Supplemental Table S2).
Neuron cell type annotation
To assign cluster identities relative to those in (Sathyamurthy et al. 2018), the top 50 marker genes from each cluster were combined, the redundancies removed, and the average gene expression extracted for each cluster. This was done for the Sathyamurthy data as well and a pair-wise Pearson correlation analysis between the data sets was performed. Each cluster from our study was annotated based on the best-fit cluster (that with the highest correlation coefficient) defined in Sathyamurthy et al. (2018).
Enrichment of nuclei count
To identify the enrichment of Ctrl or MUT nuclei in a given annotated cluster, the proportions of Ctrl and MUT nuclei were calculated relative to the total number of nuclei in the respective population. Determination of overrepresentation or underrepresentation used the prop.test R function to calculate the significance of enrichment. The P-values were adjusted for multiple hypothesis testing using the “BH” method.
Behavioral assessment
Unless otherwise noted, 3- to 4-wk-old mice with no observable scratch phenotype and blinded to genotype were used for all behavioral tests. Mice were placed in clear plastic containers (3.5 in × 3.5 in × 5 in) for observation and allowed to acclimate for 30–60 min prior to all behavioral assessments. Significant differences between control and experimental samples in each case were calculated using a two-tailed two-sample unequal variance (homoscedastic) t-test or one-way repeated measures ANOVA in GraphPad. Error bars indicate SEM. Each data point in the graphs represents individual mouse performance in the behavior paradigm tested.
Itch sensitivity
The injection site was shaved a few hours prior to the experiment. Histamine (100 µg/µL) or chloroquine (200 µg/µL) dissolved in PBS was injected intradermally in a total volume of 20 µL into the nape of the neck. The number of scratch bouts was counted over a 30-min observation period. All assays were videotaped, and subsequently scored.
Capsaicin-induced pain sensitivity
To test for acute pain, capsaicin (2.5 µg in 20 µL) was injected subcutaneously into the plantar surface of the hindpaw. All assays were videotaped for 10 min after injection, and subsequently scored by counting the number of paw licks.
Formalin-induced pain sensitivity
Formalin (20 µL of 2.5% in saline) was injected subcutaneously into the plantar surface of the hindpaw. All assays were videotaped for 60 min after injection, and subsequently scored by counting the number of paw licks within each 10 min interval. Phase I (within 0–10 min) measures the acute pain response while phase II (10–60 min) defines the nocifensive/inflammatory response (Dubuisson and Dennis 1977).
Cold sensitivity
For cold sensitivity, fresh dry ice within a syringe was used (Brenner et al. 2012). Mice were tested by exposing the plantar surface of the hind paw to the tip of the dry ice pellet past the end of the syringe by pressing it to the glass underneath the hind paw. The withdrawal latency was measured with a stopwatch, with 20 sec as the cutoff time to avoid tissue damage. Each paw was tested three times at an interval of 10 min and the mean time to response was calculated for each biological replicate.
von Frey hairs
Sensitivity to mechanical pain was assayed by the von Frey hairs simplified up-down method (Bonin et al. 2014). Mice were acclimated in acrylic cylinders on a wire mesh for 2–3 h on the day of testing. Graded filaments from 0.4 to 15 g were applied for ∼3 sec to the plantar hindpaw with at least 5 min between each application. Toe spreading, flinching, or licking was recorded as a response.
Hargreaves
Animals were placed in plastic boxes and the plantar paw surface was exposed to a beam of radiant heat according to the Hargreaves method using the IITC Plantar Analgesia Meter (serial number 10616-336). Paw withdrawal latency was then recorded (beam intensity was adjusted to result in a latency of 8–12 sec for control animals). The heat stimulation was repeated three times at an interval of 10 min for each animal and the mean was calculated. A cutoff time of 20 sec was set to prevent tissue damage.
Hot plate
Mice were placed directly on a hot plate set to 52°C. The response latency to hind paw licking or jumping was recorded over two trials for each mouse. Mice that did not respond at 45 sec were removed from the hot plate to avoid tissue damage.
Balance beam
Mice traversed a 75-cm beam of increasing difficulty (18-mm, 9-mm, and 5-mm widths) toward dark housing on the opposite side. Three trials were performed per beam width before proceeding to the narrower beam. Time to cross and number of foot slips were recorded.
Motor coordination
Mice were trained on the rotor rod at a constant speed of 16 rpm until they could remain on for one minute without falling. On test day, each mouse performed eight trials over 2 d on the rotarod, which was accelerated from 5 rpm to 45 rpm over a period of 5 min. Trials were separated by 30-min intervals for rest, and the time to fall off the apparatus was noted across the trials for each mouse.
Supplementary Material
Acknowledgments
We acknowledge the many hours of helpful discussions with Dr. H. Lai, and critical reading of the manuscript by Dr. H. Lai and Dr. S. Ross. We are grateful for the excellent transgenic mouse services provided by the University of Texas Southwestern Transgenic Core (Dr. R. Hammer, Director), and the access to microscopy in the Neuroscience Microscopy Facility supported by the University of Texas Southwestern Neuroscience Department and the Peter O'Donnell Jr. Brain Institute. We acknowledge the generous gifts from Dr. T. Muller and Dr. C. Birchmeier (TLX1/3 and LMX1B antibodies), as well as Dr. S. Ross (BHLHB5 antibodies). B.E.B. was a University of Texas Southwestern SURF-Stem Cell student supported by the Hamon Center for Regenerative Science and Medicine. This work was supported by the National Institutes of Health R01 HD037932 and R37 HD091856 to J.E.J.
Author contributions: B.M. and J.E.J. conceived the study and performed the methodology. B.M., J.V., T.K.S., R.K.K., and B.E.B. performed the investigation. B.M. and J.E.J. wrote the manuscript. J.E.J. acquired the funding and resources and supervised the study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.332577.119.
References
- Beres TM, Masui T, Swift GH, Shi L, Henke RM, MacDonald RJ. 2006. PTF1 is an organ-specific and Notch-independent basic helix–loop–helix complex containing the mammalian Suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol Cell Biol 26: 117–130. 10.1128/MCB.26.1.117-130.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin RP, Bories C, De Koninck Y. 2014. A simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments. Mol Pain 10: 26 10.1186/1744-8069-10-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borromeo MD, Meredith DM, Castro DS, Chang JC, Tung KC, Guillemot F, Johnson JE. 2014. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development 141: 2803–2812. 10.1242/dev.105866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourane S, Duan B, Koch SC, Dalet A, Britz O, Garcia-Campmany L, Kim E, Cheng L, Ghosh A, Ma Q, et al. 2015. Gate control of mechanical itch by a subpopulation of spinal cord interneurons. Science 350: 550–554. 10.1126/science.aac8653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, Gereau RWT. 2012. A novel behavioral assay for measuring cold sensation in mice. PLoS One 7: e39765 10.1371/journal.pone.0039765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröhl D, Strehle M, Wende H, Hori K, Bormuth I, Nave KA, Müller T, Birchmeier C. 2008. A transcriptional network coordinately determines transmitter and peptidergic fate in the dorsal spinal cord. Dev Biol 322: 381–393. 10.1016/j.ydbio.2008.08.002 [DOI] [PubMed] [Google Scholar]
- Cannavò E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, Ciglar L, Korbel JO, Furlong EE. 2016. Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol 26: 38–51. 10.1016/j.cub.2015.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Meredith DM, Mayer PR, Borromeo MD, Lai HC, Ou YH, Johnson JE. 2013. Prdm13 mediates the balance of inhibitory and excitatory neurons in somatosensory circuits. Dev Cell 25: 182–195. 10.1016/j.devcel.2013.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Arata A, Mizuguchi R, Qian Y, Karunaratne A, Gray PA, Arata S, Shirasawa S, Bouchard M, Luo P, et al. 2004. Tlx3 and Tlx1 are post-mitotic selector genes determining glutamatergic over GABAergic cell fates. Nat Neurosci 7: 510–517. 10.1038/nn1221 [DOI] [PubMed] [Google Scholar]
- Dickel DE, Ypsilanti AR, Pla R, Zhu Y, Barozzi I, Mannion BJ, Khin YS, Fukuda-Yuzawa Y, Plajzer-Frick I, Pickle CS, et al. 2018. Ultraconserved enhancers are required for normal development. Cell 172: 491–499.e15. 10.1016/j.cell.2017.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, et al. 2014. Identification of spinal circuits transmitting and gating mechanical pain. Cell 159: 1417–1432. 10.1016/j.cell.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan G, Sun J, Li N, Zheng H, Guo S, Zhang Y, Wang Q, Ying Y, Zhang M, Huang P, et al. 2018. A variant in the SCN10A enhancer may affect human mechanical pain sensitivity. Mol Pain 14: 1744806918763275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson D, Dennis SG. 1977. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 4: 161–174. 10.1016/0304-3959(77)90130-0 [DOI] [PubMed] [Google Scholar]
- Dullin JP, Locker M, Robach M, Henningfeld KA, Parain K, Afelik S, Pieler T, Perron M. 2007. Ptf1a triggers GABAergic neuronal cell fates in the retina. BMC Dev Biol 7: 110 10.1186/1471-213X-7-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evliyaoğlu O, Ercan O, Ataoğlu E, Zübarioğlu U, Özcabi B, Dağdeviren A, Erdoğan H, De Franco E, Ellard S. 2018. Neonatal diabetes: two cases with isolated pancreas agenesis due to homozygous PTF1A enhancer mutations and one with developmental delay, epilepsy, and neonatal diabetes syndrome due to KCNJ11 mutation. J Clin Res Pediatr Endocrinol 10: 168–174. 10.4274/jcrpe.5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani Y, Fujitani S, Luo H, Qiu F, Burlison J, Long Q, Kawaguchi Y, Edlund H, MacDonald RJ, Furukawa T, et al. 2006. Ptf1a determines horizontal and amacrine cell fates during mouse retinal development. Development 133: 4439–4450. 10.1242/dev.02598 [DOI] [PubMed] [Google Scholar]
- Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, Kawaguchi M, Terao M, Doi R, Wright CV, et al. 2008. Reduction of Ptf1a gene dosage causes pancreatic hypoplasia and diabetes in mice. Diabetes 57: 2421–2431. 10.2337/db07-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbay M, Ellard S, De Franco E, Moisés RS. 2017. Pancreatic agenesis due to compound heterozygosity for a novel enhancer and truncating mutation in the PTF1A gene. J Clin Res Pediatr Endocrinol 9: 274–277. 10.4274/jcrpe.4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MD, Chen-Plotkin AS. 2018. The post-GWAS era: from association to function. Am J Hum Genet 102: 717–730. 10.1016/j.ajhg.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow SM, Henke RM, Macdonald RJ, Wright CV, Johnson JE. 2005. Ptf1a determines GABAergic over glutamatergic neuronal cell fate in the spinal cord dorsal horn. Development 132: 5461–5469. 10.1242/dev.02167 [DOI] [PubMed] [Google Scholar]
- Gonc EN, Ozon A, Alikasifoglu A, Haliloğlu M, Ellard S, Shaw-Smith C, Kandemir N. 2015. Variable phenotype of diabetes mellitus in siblings with a homozygous PTF1A enhancer mutation. Horm Res Paediatr 84: 206–211. 10.1159/000435782 [DOI] [PubMed] [Google Scholar]
- Hirata H, Yoshiura S, Ohtsuka T, Bessho Y, Harada T, Yoshikawa K, Kageyama R. 2002. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 298: 840–843. 10.1126/science.1074560 [DOI] [PubMed] [Google Scholar]
- Hoang CQ, Hale MA, Azevedo-Pouly AC, Elsasser HP, Deering TG, Willet SG, Pan FC, Magnuson MA, Wright CV, Swift GH, et al. 2016. Transcriptional maintenance of pancreatic acinar identity, differentiation, and homeostasis by PTF1A. Mol Cell Biol 36: 3033–3047. 10.1128/MCB.00358-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Cholewa-Waclaw J, Nakada Y, Glasgow SM, Masui T, Henke RM, Wildner H, Martarelli B, Beres TM, Epstein JA, et al. 2008. A nonclassical bHLH Rbpj transcription factor complex is required for specification of GABAergic neurons independent of Notch signaling. Genes Dev 22: 166–178. 10.1101/gad.1628008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino M, Nakamura S, Mori K, Kawauchi T, Terao M, Nishimura YV, Fukuda A, Fuse T, Matsuo N, Sone M, et al. 2005. Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron 47: 201–213. 10.1016/j.neuron.2005.06.007 [DOI] [PubMed] [Google Scholar]
- Huang M, Huang T, Xiang Y, Xie Z, Chen Y, Yan R, Xu J, Cheng L. 2008. Ptf1a, Lbx1 and Pax2 coordinate glycinergic and peptidergic transmitter phenotypes in dorsal spinal inhibitory neurons. Dev Biol 322: 394–405. 10.1016/j.ydbio.2008.06.031 [DOI] [PubMed] [Google Scholar]
- Iskusnykh IY, Steshina EY, Chizhikov VV. 2016. Loss of Ptf1a leads to a widespread cell-fate misspecification in the brainstem, affecting the development of somatosensory and viscerosensory nuclei. J Neurosci 36: 2691–2710. 10.1523/JNEUROSCI.2526-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Kobayashi T. 2007. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development 134: 1243–1251. 10.1242/dev.000786 [DOI] [PubMed] [Google Scholar]
- Kardon AP, Polgar E, Hachisuka J, Snyder LM, Cameron D, Savage S, Cai X, Karnup S, Fan CR, Hemenway GM, et al. 2014. Dynorphin acts as a neuromodulator to inhibit itch in the dorsal horn of the spinal cord. Neuron 82: 573–586. 10.1016/j.neuron.2014.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. 2002. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet 32: 128–134. 10.1038/ng959 [DOI] [PubMed] [Google Scholar]
- Koch SC, Acton D, Goulding M. 2018. Spinal circuits for touch, pain, and itch. Annu Rev Physiol 80: 189–217. 10.1146/annurev-physiol-022516-034303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota N, Yokoyama T, Hoshi N, Suyama M. 2018. Identification of a candidate enhancer for DMRT3 involved in spastic cerebral palsy pathogenesis. Biochem Biophys Res Commun 496: 133–139. 10.1016/j.bbrc.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Lai HC, Klisch TJ, Roberts R, Zoghbi HY, Johnson JE. 2011. In vivo neuronal subtype-specific targets of Atoh1 (Math1) in dorsal spinal cord. J Neurosci 31: 10859–10871. 10.1523/JNEUROSCI.0445-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HC, Seal RP, Johnson JE. 2016. Making sense out of spinal cord somatosensory development. Development 143: 3434–3448. 10.1242/dev.139592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyva-Díaz E, Hobert O. 2019. Transcription factor autoregulation is required for acquisition and maintenance of neuronal identity. Development 146: dev177378 10.1242/dev.177378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui T, Long Q, Beres TM, Magnuson MA, MacDonald RJ. 2007. Early pancreatic development requires the vertebrate Suppressor of Hairless (RBPJ) in the PTF1 bHLH complex. Genes Dev 21: 2629–2643. 10.1101/gad.1575207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui T, Swift GH, Hale MA, Meredith DM, Johnson JE, Macdonald RJ. 2008. Transcriptional autoregulation controls pancreatic Ptf1a expression during development and adulthood. Mol Cell Biol 28: 5458–5468. 10.1128/MCB.00549-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson KJE, Sathyamurthy A, Johnson KR, Kelly MC, Kelley MW, Levine AJ. 2018. Isolation of adult spinal cord nuclei for massively parallel single-nucleus RNA sequencing. J Vis Exp 10.3791/58413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith DM, Masui T, Swift GH, MacDonald RJ, Johnson JE. 2009. Multiple transcriptional mechanisms control Ptf1a levels during neural development including autoregulation by the PTF1-J complex. J Neurosci 29: 11139–11148. 10.1523/JNEUROSCI.2303-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millen KJ, Steshina EY, Iskusnykh IY, Chizhikov VV. 2014. Transformation of the cerebellum into more ventral brainstem fates causes cerebellar agenesis in the absence of Ptf1a function. Proc Natl Acad Sci 111: E1777–E1786. 10.1073/pnas.1315024111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi R, Sugimori M, Takebayashi H, Kosako H, Nagao M, Yoshida S, Nabeshima Y, Shimamura K, Nakafuku M. 2001. Combinatorial roles of olig2 and neurogenin2 in the coordinated induction of pan-neuronal and subtype-specific properties of motoneurons. Neuron 31: 757–771. 10.1016/S0896-6273(01)00413-5 [DOI] [PubMed] [Google Scholar]
- Mizuguchi R, Kriks S, Cordes R, Gossler A, Ma Q, Goulding M. 2006. Ascl1 and Gsh1/2 control inhibitory and excitatory cell fate in spinal sensory interneurons. Nat Neurosci 9: 770–778. 10.1038/nn1706 [DOI] [PubMed] [Google Scholar]
- Mona B, Avila JM, Meredith DM, Kollipara RK, Johnson JE. 2016. Regulating the dorsal neural tube expression of Ptf1a through a distal 3′ enhancer. Dev Biol 418: 216–225. 10.1016/j.ydbio.2016.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mona B, Uruena A, Kollipara RK, Ma Z, Borromeo MD, Chang JC, Johnson JE. 2017. Repression by PRDM13 is critical for generating precision in neuronal identity. Elife 6: e25787 10.7554/eLife.25787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhai H, Sel S, Favor J, Mendoza-Torres L, Paulsen F, Duncker GI, Schmid RM. 2007. Ptf1a is essential for the differentiation of GABAergic and glycinergic amacrine cells and horizontal cells in the mouse retina. Development 134: 1151–1160. 10.1242/dev.02781 [DOI] [PubMed] [Google Scholar]
- Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. 2018. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554: 239–243. 10.1038/nature25461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Abasolo I, Mingorance-Le Meur A, Martinez A, Del Rio JA, Wright CV, Real FX, Soriano E. 2007. Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc Natl Acad Sci 104: 5193–5198. 10.1073/pnas.0605699104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H, Fang X. 2013. A brief review on the human encyclopedia of DNA elements (ENCODE) project. Genomics Proteomics Bioinformatics 11: 135–141. 10.1016/j.gpb.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner M, Jager SB, Siupka P, Vaegter CB. 2017. Hydraulic extrusion of the spinal cord and isolation of dorsal root ganglia in rodents. J Vis Exp 10.3791/55226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, Hu L, Mok SI, Shah A, Savner EM, et al. 2010. Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron 65: 886–898. 10.1016/j.neuron.2010.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, Levine AJ. 2018. Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep 22: 2216–2225. 10.1016/j.celrep.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggens L, Ekwall K. 2014. Epigenetics, chromatin and genome organization: recent advances from the ENCODE project. J Intern Med 276: 201–214. 10.1111/joim.12231 [DOI] [PubMed] [Google Scholar]
- Sun S, Xu Q, Guo C, Guan Y, Liu Q, Dong X. 2017. Leaky gate model: intensity-dependent coding of pain and itch in the spinal cord. Neuron 93: 840–853.e5. 10.1016/j.neuron.2017.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbey P, Thierion E, Collombet S, de Cian A, Desmarquet-Trin-Dinh C, Dura M, Concordet JP, Charnay P, Gilardi-Hebenstreit P. 2018. Cooperation, cis-interactions, versatility and evolutionary plasticity of multiple cis-acting elements underlie krox20 hindbrain regulation. PLoS Genet 14: e1007581 10.1371/journal.pgen.1007581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu M, Ma A, Eng SR, Fedtsova N, Turner EE. 2003. Direct autoregulation and gene dosage compensation by POU-domain transcription factor Brn3a. Development 130: 111–121. 10.1242/dev.00194 [DOI] [PubMed] [Google Scholar]
- Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, Rodríguez-Segui SA, Shaw-Smith C, Cho CH, Lango Allen H, et al. 2014. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet 46: 61–64. 10.1038/ng.2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will AJ, Cova G, Osterwalder M, Chan WL, Wittler L, Brieske N, Heinrich V, de Villartay JP, Vingron M, Klopocki E, et al. 2017. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat Genet 49: 1539–1545. 10.1038/ng.3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lopes C, Qian Y, Liu Y, Cheng L, Goulding M, Turner EE, Lima D, Ma Q. 2008. Tlx1 and Tlx3 coordinate specification of dorsal horn pain-modulatory peptidergic neurons. J Neurosci 28: 4037–4046. 10.1523/JNEUROSCI.4126-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lopes C, Wende H, Guo Z, Cheng L, Birchmeier C, Ma Q. 2013. Ontogeny of excitatory spinal neurons processing distinct somatic sensory modalities. J Neurosci 33: 14738–14748. 10.1523/JNEUROSCI.5512-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Terao M, Terashima T, Fujiyama T, Kawaguchi Y, Nabeshima Y, Hoshino M. 2007. Origin of climbing fiber neurons and their developmental dependence on Ptf1a. J Neurosci 27: 10924–10934. 10.1523/JNEUROSCI.1423-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.