

Here, Lu et al. use Drosophila to show that opening of promoters from their closed state in precursor cells requires function of the spermatocyte-specific tMAC complex, localized at the promoters. Their findings provide novel insight into how promoter-proximal sequence elements that recruit and are acted on by cell type-specific chromatin-binding complexes help establish a robust, cell type-specific transcription program for terminal differentiation.
Keywords: Drosophila, spermatogenesis, transcription, tMAC, core promoter elements
Abstract
Cell type-specific transcriptional programs that drive differentiation of specialized cell types are key players in development and tissue regeneration. One of the most dramatic changes in the transcription program in Drosophila occurs with the transition from proliferating spermatogonia to differentiating spermatocytes, with >3000 genes either newly expressed or expressed from new alternative promoters in spermatocytes. Here we show that opening of these promoters from their closed state in precursor cells requires function of the spermatocyte-specific tMAC complex, localized at the promoters. The spermatocyte-specific promoters lack the previously identified canonical core promoter elements except for the Inr. Instead, these promoters are enriched for the binding site for the TALE-class homeodomain transcription factors Achi/Vis and for a motif originally identified under tMAC ChIP-seq peaks. The tMAC motif resembles part of the previously identified 14-bp β2UE1 element critical for spermatocyte-specific expression. Analysis of downstream sequences relative to transcription start site usage suggested that ACA and CNAAATT motifs at specific positions can help promote efficient transcription initiation. Our results reveal how promoter-proximal sequence elements that recruit and are acted upon by cell type-specific chromatin binding complexes help establish a robust, cell type-specific transcription program for terminal differentiation.
Transcriptional regulation plays a central role in producing different cell types from the same genomic content. Throughout embryonic development, cells make and respond to cell fate decisions by turning on new transcription programs required to generate progressively more specialized cell types. Similar events drive differentiation of specialized cells from proliferating precursors in the adult stem cell lineages that maintain and repair many tissues throughout the life span. Understanding how cell type-specific transcription is achieved forms the very basis of understanding differentiation and development in multicellular organisms.
Tissue and stage-specific transcription programs are established by intricate interplay among promoter-proximal and distal DNA elements, and protein complexes that interact with them. Much recent work has focused on the role of stage or tissue-specific transcriptional activators and repressors acting upon distal enhancer elements to control the time and place of expression of developmental genes. However, evidence has emerged that variant forms of core promoter motifs and their recognition factors can play roles in cell type-specific transcription programs in certain tissues (D'Alessio et al. 2009; Goodrich and Tjian 2010; Haberle et al. 2014; Danks et al. 2018).
Several canonical core promoter motifs and promoter types have been identified and extensively studied. In Drosophila, TATA-box and/or downstream promoter element (DPE)-containing promoters tend to initiate transcription from a narrow region (Hoskins et al. 2011; Chen et al. 2014). The TATA box is bound by the TATA-binding protein (TBP), while the DPE is bound by certain TBP-associated factors (TAFs) in the general transcription factor TFIID to help precisely position RNA polymerase II for transcript initiation (Goodrich and Tjian 2010). On the other hand, promoters containing the DNA replication-related element (DRE) and/or other Ohler motifs tend to initiate transcription from a broad region and are thought to be associated with housekeeping genes (Ohler et al. 2002; FitzGerald et al. 2006; Graveley et al. 2011; Chen et al. 2014). Recent work in Drosophila has shown that thousands of enhancers exhibit a distinct preference for one or the other of these promoter types (Zabidi et al. 2015; Rennie et al. 2018), suggesting that sequences near the transcription start site can play key roles in gene-selective transcriptional regulation. However, the extent to which these canonical versus other core promoter motifs contribute to cell type-specific gene regulatory programs in differentiating cells and the molecular mechanisms by which they do so are not understood.
Male germ cell differentiation in Drosophila provides an excellent opportunity to study cell type-specific transcriptional regulation, as more than a thousand genes turn on for the first time in development when male germ cells become spermatocytes. In Drosophila, one germline stem cell normally produces a new stem cell and a gonialblast, which founds a clone of proliferating spermatogonia through four rounds of mitosis. The resulting 16 interconnected germ cells undergo a last round of DNA synthesis; then, as spermatocytes, they enter meiotic prophase (Fig. 1A). During this ∼3-d period the spermatocytes express the vast majority of genes needed for later stages of male germ cell development. A recently developed heat-shock–Bam time course system (Kim et al. 2017) provided a way to obtain large quantities of germ cells at similar stages and greatly empowered study of the temporal events and molecular mechanisms that underlie the dramatic, cell type-specific change in transcriptional regulation that accompanies the transition from spermatogonia to spermatocyte.
Figure 1.

Transcriptional changes during the transition from spermatogonia to spermatocyte in Drosophila. (A) Diagram of Drosophila spermatogenesis with timeline as spermatogonia differentiate into spermatocytes and the corresponding time points after heat shock of hsBam;bam−/− flies. (B) Composition of the tMAC and MuvB core protein complexes. (Dark gray) Shared components; (light gray at the same position of the diagram) paralogs; (white) nonhomologous components. (C,D) RNA-seq (C) and CAGE profiles (D) for all protein-coding genes expressed in testes from bam−/− (enriched for spermatogonia [X-axis]) and hsBam;bam−/− 72 hrPHS (enriched for maturing spermatocytes [Y-axis]). Axes show read counts normalized by DESeq2 rlog transformation. (Left) Down-regulated genes. (Middle) Off-to-on genes. (Right) Genes expressed from alternative promoters, with each gene represented by one data point in RNA-seq, and two data points, corresponding to the alternative promoters, in the CAGE plot. (E–G) RNA-seq and CAGE profiles for example genes from each group. Y-axes show read counts normalized by sequencing depth.
Genetic and biochemical studies have identified two sets of cell type-specific proteins that regulate the spermatocyte transcription program, the tMAC complex (Beall et al. 2007) required for turning on most of the spermatocyte-specific gene expression program (Perezgasga et al. 2004; Doggett et al. 2011) and the testis-specific TAFs (tTAFs) (Hiller et al. 2001; Hiller 2004) required for full levels of expression of many genes in that program (Doggett et al. 2011; Lu and Fuller 2015). tMAC interacts physically with Achi/Vis, two highly similar TGIF-related TALE-class homeodomain proteins encoded by tandemly duplicated genes that are required for transcription in spermatocytes of tMAC-dependent genes (Ayyar et al. 2003; Wang and Mann 2003). In flies null mutant for tMAC components, Achi/Vis, or tTAFs, germ cells arrest as mature spermatocytes (Lin et al. 1996; White-Cooper et al. 1998; Ayyar et al. 2003; Wang and Mann 2003; Perezgasga et al. 2004; Jiang et al. 2007; Doggett et al. 2011). tMAC is a spermatocyte-specific version of the widely expressed and evolutionarily conserved MuvB core complex (Fig. 1B), which binds different protein partners, including members of the E2F, DP, Rb, and Myb protein families, to repress or activate key cell cycle and developmental genes (Sadasivam and DeCaprio 2013; Fischer and Müller 2017). The tMAC complex expressed in Drosophila spermatocytes contains two proteins shared with the MuvB core: p55 Caf1 (RBBP4 in humans) and Mip40 (LIN37) (Fig. 1B, in dark gray). tMAC also contains testis-specific paralogs of three of the other MuvB core components: Aly (paralog of Mip130 [LIN9]), Tomb (paralog of Mip120 [LIN54]), and Wuc (paralog of Lin52 [LIN52]) (Fig. 1B, in light gray). In addition, tMAC includes the testis-specific proteins Topi and Comr (Fig. 1B, in white). Among the known tMAC subunits, Tomb, Topi, and Comr have predicted DNA-binding domains (Beall et al. 2007). Despite the importance of tMAC for turning on expression of most of the genes newly expressed in spermatocytes, the mechanism by which tMAC carries out this function is not known.
To investigate how the cell type-specific gene expression program for spermatocyte differentiation turns on, we used RNA-seq to map transcript levels, CAGE to quantitatively map transcription start site (TSS) usage (Shiraki et al. 2003; Murata et al. 2014), and ATAC-seq to map chromatin accessibility (Buenrostro et al. 2013) as proliferating spermatogonia transition to differentiating spermatocytes. Combining these data, we showed that the promoters that turn on when germ cells become spermatocytes lack most of the canonical core promoter motifs. Instead, these promoters are enriched for the tMAC-ChIP motif and putative Achi/Vis-binding motif, and require tMAC function to become open and accessible once germ cells become spermatocytes. Within the local open region that tMAC creates, a good match to an Inr sequence at position +1 of the transcript and ACA and CNAAATT motifs at specific positions downstream correlated with efficient usage of individual TSS. Our results reveal a robust cell type-specific and gene-selective developmental transcription program orchestrated by promoter-proximal elements and protein complexes that interact with them.
Results
A third of the testis transcriptome uses a new promoter as germ cells differentiate into spermatocytes
To follow changes in gene expression over differentiation, male germ cells were induced to differentiate from spermatogonia to spermatocytes in vivo in a heat shock-Bam time course system (Fig. 1A; Materials and Methods; Kim et al. 2017). Briefly, in males mutant for the key differentiation factor bam, spermatogonia undergo several additional rounds of mitotic proliferation rather than differentiate into spermatocytes (McKearin and Ohlstein 1995), were subjected to a brief pulse of Bam expression under control of a heat-shock promoter. In hs-Bam;bam−/− flies, a brief pulse of Bam expression can be induced by 30 min of heat shock, which relieved the developmental arrest and caused accumulated spermatogonia to initiate differentiation into spermatocytes relatively synchronously. By 24 h after heat shock, the germ cells had completed a final round of mitosis and premeiotic DNA replication. By 48 h post-heat shock (“48 hrPHS”) germ cells had progressed to young spermatocytes, a majority of the genes expressed specifically in spermatocytes had begun to be expressed, and a small number of genes up-regulated early in the spermatocyte stage had reached a high level of expression. By 72 h post-heat shock (“72 hrPHS”) the differentiating germ cells had become mature spermatocytes and the majority of genes up-regulated in spermatocytes had reached high levels of expression. By 96 h after heat shock, germ cells had begun to initiate the first meiotic division. For this work, 48 hrPHS and 72 hrPHS are of particular interest (Fig. 1A, bottom panel). Note that as the samples were whole testes, they contained both germ line and somatic cells. Also, new spermatogonia that lack Bam continue to accumulate during the time course after heat shock, so at later time points the testes contained increasing numbers of spermatogonia as well as differentiating spermatocytes.
RNA-seq and CAGE from testes from bam−/− flies and hs-Bam;bam−/− flies 72 hrPHS detected expression of 9371 protein-coding genes (Fig. 1C,D). Analysis of how transcript levels changed in the time course using these data identified three classes of genes dynamically regulated as spermatogonia differentiated into spermatocytes (see Materials and Methods).
One-thousand-one-hundred-fifty-five “down-regulated genes” were expressed at least twofold less in either the 48 hrPHS sample or the 72 hrPHS sample than in bam−/− testes, based on RNA-seq (Fig. 1C, left panel), and contained CAGE clusters at the same genomic positions in bam−/− testis and in 72 hrPHS testis (Fig. 1D, left panel). One example, PCNA (Fig. 1E), encodes a component of the DNA replication machinery. PCNA protein is abruptly down-regulated in Drosophila male germ cells after completion of premeiotic S phase (Insco et al. 2009). The decrease in expression of the down-regulated genes is likely an underestimate because after heat shock new spermatogonia produced from bam−/− germline stem cells accumulate over time.
One-thousand-eight-hundred-forty-one “off-to-on genes” were expressed at negligible or low levels in bam−/− testes but were up-regulated more than eightfold by 48 hrPHS or >16-fold by 72 hrPHS based on RNA-seq (Fig. 1C, middle panel) and contained CAGE clusters that appeared in 72 hrPHS testes but not in bam−/− testes (Fig. 1D, middle panel). One example is fzo (Fig. 1F), which encodes a mitofusin expressed in spermatocytes in preparation for the fusion of mitochondria to form the mitochondrial derivative in haploid round spermatids (Hales and Fuller 1997).
Strikingly, 1230 “genes with alternative promoters” were expressed in both bam−/− and 72 hrPHS testes (Fig. 1C, right panel) and contained CAGE clusters expressed in bam−/− as well as new CAGE clusters that appeared in 72 hrPHS, but not in the bam−/−. In other words, these genes were expressed from a new promoter once spermatogonia differentiate into spermatocytes (Fig. 1D, right panel, in which each gene has two corresponding CAGE data points). One example is αTub84D, which encodes the major α-Tubulin isoform expressed in male germ cells. The RNA-seq and CAGE data show expression of αTub84D in testes containing spermatocytes from a new promoter ∼350 bp downstream from the promoter used in testes containing spermatogonia but lacking spermatocytes (Fig. 1G). Taking the 1841 “off-to-on” and the 1230 “alternative promoter” genes together, >3000 genes (∼30% of all genes expressed in testes at time points taken) were expressed from a new promoter after spermatogonia differentiate into spermatocytes.
The count of “genes with alternative promoters” is an underestimate because we required the new 72 hrPHS-specific CAGE cluster to be separated by at least 40 bp from the old CAGE cluster used in bam−/− (Materials and Methods). This excluded genes expressed from different but overlapping promoter regions in 72 hrPHS compared with bam−/− testes. For example, cyclin B1, which is known to be expressed from a tMAC-independent promoter in spermatogonia and a tMAC-dependent promoter in spermatocytes (White-Cooper et al. 1998), had a new CAGE cluster in the 72 hrPHS sample that was not detected in the bam−/− sample, was nevertheless not included among the “genes with alternative promoters” (Supplemental Fig. S1).
Promoters expressed with differentiation lack canonical core promoter motifs
Analysis of the distribution of CAGE signals within a promoter can reveal information about how promoter-proximal sequence elements might influence usage of specific TSSs. Traditionally, promoter width has been defined as the region that includes all the TSSs for a promoter, a measure sensitive to both the level of expression and the overall depth of sequencing. For this work, we used a different metric that captures information about the distribution of TSS usage: the “region of efficient transcription initiation” (RETI), defined as the region within which 80% of all transcripts initiated, obtained by trimming 10% of the total CAGE signal off of both sides of the CAGE cluster (Haberle et al. 2014) (see Supplemental Fig. S2 for how RETI compares and correlates with canonical promoter width).
The promoter regions of the “down-regulated genes” were enriched for known core promoter motifs previously associated with canonical narrow versus broad promoters (Fig. 2A). The narrow promoters (RETI width < 11 bp) were enriched for the initiator (Inr), TATA-box, and DPE motifs at the previously reported positions (Fig. 2A above dashed line, Fig. 2G. P-value from CENTRIMO all <1×10−50). The broad promoters (RETI width ≥11 bp) showed enrichment for the Ohler1, DRE (aka Ohler2), and Ohler5 motifs located upstream of the TSS (P-value from CENTRIMO all <1×10−9) (Fig. 2A [below dashed line], H). Also consistent with a previous report (Rach et al. 2011), the narrow promoters tended to have a more expansive accessible region, suggesting less well-defined nucleosome positioning, whereas the broad promoters tended to have more restricted accessible regions flanked by inaccessible regions, suggesting well-defined nucleosome positioning (Fig. 2I). In addition, the promoters showed different local GC content (scored by occurrence of trinucleotides consisting of either G or C in each position), with the region immediately surrounding the narrower promoters tending to be more GC-rich than the region immediately surrounding the broader promoters (Fig. 2A, right two columns).
Figure 2.

Off-to-on genes lack most canonical core promoter motifs. (A–F, left column) CAGE signal for each promoter. (Other columns) Occurrence and position of indicated sequence motif for the different groups of genes. Genes were aligned by center of RETI and sorted by increasing RETI width. (A) Down-regulated genes. Dashed line separates genes with narrow (<11-nt) and broad (≥11-nt) RETI. (B) Off-to-on genes. (C–F) Genes with alternative promoters. (G) Enrichment of TATA, Inr, or DPE in promoters of down-regulated gene with narrow RETI. (H) Enrichment of Ohler motifs in promoters of down-regulated genes with broad RETI. (I) ATAC-seq signal at promoters of down-regulated genes, aligned and sorted as in A.
In contrast, the promoter regions of “off-to-on genes,” which became expressed with differentiation of germ cells into spermatocytes, were not enriched for any of the canonical core promoter motifs except the Inr (Fig. 2B). These promoters contained less G/C trinucleotide than the promoters of the down-regulated genes and were more AT rich.
For the “genes with alternative promoters” that began to express transcripts from a new promoter once germ cells differentiated into spermatocytes, the old promoters used in the bam−/− samples resembled the promoters of the down-regulated genes, having either TATA/DPE or Ohler/DRE motifs, correlating with promoter width (Fig. 2C,E). However, the new alternative promoters that began to express with differentiation into spermatocytes resembled the promoters of the off-to-on genes, lacking all the known core promoter motifs except for the Inr (Fig. 2D,F). This held true whether the newly expressed alternate promoter was located upstream of the old promoter, presumably outside of a region of active transcription in bam−/− testes (Fig. 2D), or located downstream from the old promoter, so within a region actively transcribed in bam−/− testes (Fig. 2F).
tMAC promotes opening of promoter regions in spermatocytes
Analysis of chromatin accessibility by ATAC-seq across the differentiation time course revealed that the promoters of the off-to-on genes were closed with little ATAC-seq signal in bam−/− testis, in which germ cells continue to proliferate as spermatogonia (Fig. 3A, bam−/−). By 48 h after heat shock, as spermatocyte-specific transcripts started to become expressed (Supplemental Fig. S3A), ATAC-seq signal appeared at the promoter regions of the off-to-on genes (Fig. 3A, 48 hrPHS). By 72 h after heat shock, when the spermatocyte specific transcription program was fully active, the ATAC-seq signal became strong and robust (Fig. 3A, 72 hrPHS).
Figure 3.

tMAC binds to and promotes opening of promoters that become expressed in spermatocyte-enriched samples. (A–C, left column) CAGE signal for promoters newly expressed in hsBam;bam−/− 72 hrPHS testes aligned by center of the RETI and sorted by RETI width. (Middle columns) ATAC-seq signal in different conditions. (Right column) tMAC ChIP-seq signal data from (Kim et al. 2017). (Top row) Average of data in panels below. Heat map scale: Read counts were normalized by library size, effectively 10-fold of reads per million (RPM) (see the Supplemental Material). (A) Off-to-on genes. (B,C) Genes with alternative promoters with new spermatocyte-specific promoters upstream of (B) or downstream from (C) the old promoters. (D, left) CAGE signal for genes in B sorted by increasing distance between old and upstream new alternative promoters. (Right) Nucleosome positions for the old, spermatogonial promoters sorted in the same order. The darkest regions indicate most likely nucleosome dyad position.
Opening of chromatin at promoters for most of the off-to-on genes required function of tMAC. In testes from flies mutant for the tMAC component Aly, most of the off-to-on gene promoters remained closed, with little or no ATAC-seq signal. Some of the off-to-on promoters did show ATAC-seq signal in aly−/− testes (Fig. 3A, aly−/−), consistent with the fact that transcription of some of these genes were up-regulated in spermatocytes independently of Aly function (Supplemental Fig. S3B). These Aly-independent genes tended to be up-regulated earlier in the spermatocyte differentiation time course than the bulk of the off-to-on genes, which were Aly-dependent (Supplemental Fig. S3C). Many of the genes up-regulated in spermatocytes independently of Aly function still required action of the tMAC component Topi for their promoter regions to become accessible to ATAC-seq (Fig. 3A; Supplemental Fig. S3A,B, topi−/−).
For the “genes with alternative promoters,” opening of the new promoters that became expressed when spermatogonia differentiated into spermatocytes also required function of tMAC. These promoters failed to open in topi−/− and in most cases in aly−/− mutants, similar to the promoters of the off-to-on genes (Fig. 3B,C). Again, most of the new promoters showed a peak of enrichment by ChIP for the tMAC component Aly, indicating that tMAC acts locally to open the new promoter (Fig. 3B,C; tMAC ChIP).
The tMAC complex appears to act locally at the off-to-on and newly expressed alternative promoters to promote chromatin opening. Plotting ChIP-seq data for the tMAC complex component Aly from wild-type testes (Kim et al. 2017) showed tMAC binding to the vast majority of these promoters, with the peak of enrichment centered on the promoter region (Fig. 3A; Supplemental Fig. S3A,B; tMAC ChIP). The tMAC complex did not appear to bind to all promoters, as there was little enrichment at the promoters of down-regulated genes in the same ChIP-seq data set (Fig. 3A; tMAC ChIP, top panel, orange).
Supporting a role for tMAC in modulating nucleosome position or occupancy to promote promoter opening, in many cases where the new alternative promoter was within 500 bp upstream of the old spermatogonial promoter (Fig. 3D, left panel), nucleosome position calculated from the ATAC-seq signal in bam−/− testes revealed that the new alternative promoter was frequently located within the region of DNA wrapped around the phased −1 nucleosome of the old promoter (Fig. 3D, right panel; Supplemental Fig. S4). These data suggest that expression of the new promoters in spermatocytes might often be accompanied by tMAC-dependent moving or removing of the −1 nucleosome that had flanked the old promoters.
Spermatocyte-specific promoters contain tMAC and Achi/Vis motifs
Searches by MEME-ChIP (Machanick and Bailey 2011) found several motifs enriched in the promoter regions of the off-to-on genes and newly expressed alternative promoters. As the median ATAC-seq peak width for the off-to-on genes was 219 bp (Supplemental Fig. S3D), a slightly wider region of 300 bp centered in the middle of the CAGE cluster from each off-to-on gene was pooled for the motif search. The most enriched was the tMAC-ChIP motif (E-value = 2.5×10−104 from MEME) previously identified as enriched under ChIP-seq peaks for the tMAC component Aly (Kim et al. 2017). The tMAC-ChIP motif was most often found ∼60 bp upstream of the 3′/downstream edge of the RETI (Fig. 4A [middle column], B [bottom panel]), indicating that most transcription initiation events leading to mature mRNA occurred within an average of 60 bp downstream from the tMAC-ChIP-binding site. The distance between the tMAC-ChIP motif and the 5′/upstream edge of the RETI was much more variable (Supplemental Fig. S5A).
Figure 4.

Promoter-proximal tMAC-ChIP and Achi/Vis motifs are enriched at certain positions upstream of the off-to-on and new alternative promoters. (A) CAGE signal and occurrence of tMAC-ChIP and Achi/Vis motifs. In all columns, promoters were aligned by 3′ edge of the RETI and sorted by RETI width. (B, top) Histogram of nucleosome dyad positions for all off-to-on gene promoters, calculated from the ATAC-seq signal from hsBam;bam−/− 72 hrPHS testes. (Blue bars) Estimated span of 147-bp nucleosomal DNA centered around the most likely −1 and +1 dyad positions (peaks in histogram). Space between blue bars indicates the open, nucleosome-free region. (Bottom) Motif enrichment by position from 3′ edge of the RETI for promoters of off-to-on genes calculated by Centrimo (P-value shown below the plot). (Red dotted line) Overlay of tMAC ChIP signal averaged across all off-to-on genes. (C) Model of the role of tMAC and Achi/Vis. They may promote transcription by creating an ∼100-bp region of accessible DNA downstream from tMAC-binding site, and therefore limit the 3′ edge of RETI.
The second most enriched motif was TGTCA (E-value = 1.9 × 10−101 from DREME), previously identified as binding motif for the TALE-class homeodomain proteins Achi and Vis (Noyes et al. 2008). The Achi/Vis motif tended to lie between the tMAC-ChIP motif and the 3′/downstream edge of the RETI, with a broad distribution from −50 bp∼−5 bp from the 3′ edge of the RETI (Fig. 4A [right column], B [bottom panel]). Achi/Vis had previously been shown to physically interact and collaborate with tMAC to drive spermatocyte-specific gene expression (Ayyar et al. 2003; Wang and Mann 2003). The tMAC-ChIP and Achi/Vis motifs were not enriched near the promoters of the down-regulated genes (Fig. 4A)
Together, our findings suggest that tMAC, recruited to specific sites in the genome by DNA sequence motifs, may create a local short stretch of open chromatin in which transcription initiation can occur (Fig. 4C). Nucleosome positioning calculated from the ATAC-seq data from 72 hrPHS testes showed that the average distance from the dyad position of the −1 nucleosome to the dyad position of the +1 nucleosome for the off-to-on gene promoters was only 253 bp. Assuming 147 bp of DNA wrapped around each nucleosome, that leaves an average nucleosome-free region of only ∼100 bp at the off-to-on promoters (Fig. 4B, top panel). This region contains the tMAC and Achi/Vis binding motifs (Fig. 4B), with the majority of transcription initiation occurring within 60 bp downstream from the tMAC-ChIP motif (cf. Fig. 4A, CAGE column with tMAC-ChIP motif column).
Analysis of reporter transgenes for several off-to-on genes confirmed that a small genomic region around the TSS was sufficient to drive expression in spermatocytes. For example, a reporter transgene containing 117 bp of genomic sequence from 91 bp upstream of to 26 bp downstream from the 3′ edge of the RETI of sa, one of the testis TAFs, was sufficient to drive expression in spermatocytes but not in spermatogonia or somatic cells at the tip of the testis (Fig. 5A). Likewise, for the testis TAFs can and nht, 121-bp and 120-bp genomic regions from 68 bp and 104 bp upstream of the 3′ edge of the RETI, respectively, were sufficient to drive expression of GFP in spermatocytes (Fig. 5B,C). Note that all of these constructs ended at the translational start codon and contained the full-length 5′ UTR of the respective gene and may therefore maintain tissue-specific translational regulation.
Figure 5.
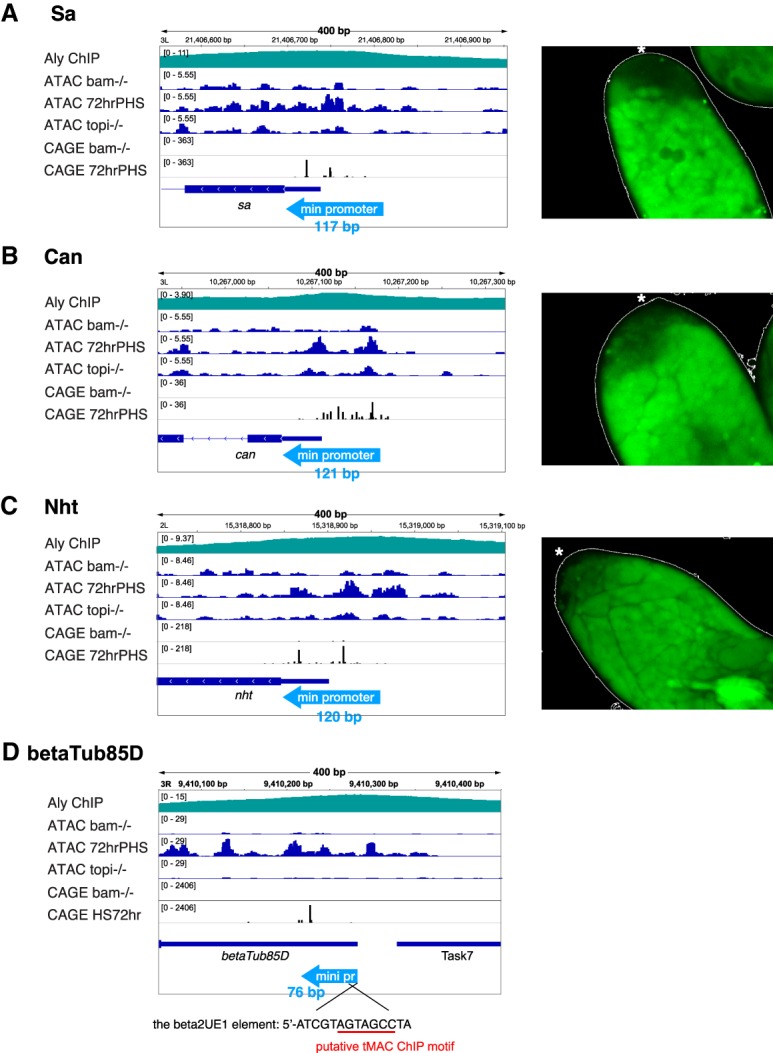
Short stretches of promoter regions are sufficient for spermatocyte-specific expression of tTAFs. (Left) ATAC-seq, CAGE, and tMAC ChIP profile and relative positions of minimal promoter regions (light-blue arrows) tested in the transgene reporters. (Right) Immunofluorescence images of apical ends of testes from flies carrying a transgenes in which the minimal promoter (indicated by light-blue arrows in the left panel) drives expression of GFP. The tip of testis is indicated by asterisks. (A) Sa. (B) Can. (C) Nht. (D) βTub85D, with the 14-bp β2UE1 element required for expression in spermatocytes (Michiels et al. 1989) indicated and the putative tMAC-ChIP motif underlined in red.
The reporter results are consistent with previously published data from multiple genes showing that a short promoter region is sufficient for spermatocyte-specific expression in Drosophila (White-Cooper 2010). Notably among these genes, the βTub85D promoter contains a close match to the tMAC-ChIP motif within the conserved 14-bp β2UE1 element (Fig. 5D) that was shown to be required at a defined position upstream of the TSS for spermatocyte-specific expression. Mutations within the region that resembles the tMAC-ChIP motif abolished expression of reporter transgenes in spermatocytes (Michiels et al. 1989).
Motifs downstream from the TSS correlate with efficient TSS usage
Aligning the promoters of the off-to-on genes by the most highly expressed TSS (the “dominant TSS”) position within each promoter revealed two additional features. First, transcription initiation preferentially occurred at trinucleotides resembling the Inr motif: 36% of the dominant transcript start sites initiated at TCA (the short form of the Inr, with A as +1) (Fig. 6A, TCA column), followed by 15% at TTA, 8% at ACA, 7% at CCA, and 5% at GCA. Second, there was strong enrichment for the trinucleotide ACA with the first A at position +26, +28, or +30 relative to the dominant TSS (defined as +1 following convention) (Fig. 6A [ACA column], B [left panel]).
Figure 6.

Well-positioned promoter proximal downstream motifs ACA and CNAAATT facilitate efficient TSS usage in off-to-on genes. (A) CAGE signal and occurrence of TCA, ACA, and CNAAATT motifs. Promoters in all columns were aligned by the most dominantly used TSS within the CAGE cluster and sorted by RETI width. (B) Histogram of distance from the first base (underlined) in each ACA or CNAAATT to the dominant TSS for all off-to-on genes. Y-axis values were normalized by total number of off-to-on genes. (C) TSS rank composition for TSSs with different combinations of TCA and ACA. Rank 5+ includes the fifth most used and all less frequently used TSSs within a CAGE cluster. (D) Expression level measured by CAGE for dominant TSSs with different combinations of TCA and ACA. P-values were calculated by t-test. (E) TSS rank composition for TSSs with different combinations of TCA and CNAAATT. (F) Expression level measured by CAGE for dominant TSSs with different combinations of TCA, ACA, and CNAAATT. (G) Model of role of ACA and CNAAATT: The region enriched for ACA and/or CNAAATT may promote expression of TSS within a region upstream at the optimal distance, and therefore limit the 5′ edge of RETI. (H) CAGE signals and occurrence of TCA, ACA, and CNAAATT motifs relative to the 5′ edge of the RETI.
The precise positioning of the ACA motif relative to the dominant TSS raised the possibility that ACA may help determine the position where transcription efficiently initiates. This was supported by comparing TSS usage within each promoter as well as by comparing the level of expression of dominant TSSs across different genes.
For TSS within a given promoter, of the 1640 off-to-on genes, 1632 had more than one TSS within the promoter detected by CAGE (more than two CAGE reads at the TSS position). For these promoters, individual TSS can be ranked as the most used (dominant TSS), second most used, third most used, and so on (Fig. 6C,E). Because Inr (TCA) alone made a TSS more highly ranked (Fig. 6C,E, cf. first and third bars), TSSs with or without TCA were analyzed separately. For TSSs not starting at TCA, those with ACA at +26, +28, or +30 tended to be more highly ranked than those without the ACA (Fig. 6C, first and second bars). Likewise, for TSSs starting at TCA, those with ACA positioned at +26, +28, or +30 tended to be more highly ranked than those without the ACA (Fig. 6C, third and fourth bars).
When comparing dominant TSSs across different off-to-on genes, for both TCA-containing and TCA-lacking dominant TSSs, those with ACA at +26, +28, or +30 were more highly expressed than those without ACA (Fig. 6D). The TCA and well positioned ACA motifs both appeared to contribute to TSS usage, with additive effects (Fig. 6D). It is worth noting that as most of the off-to-on genes are under similar upstream regulation and depend on tMAC and Achi/Vis for expression (Perezgasga et al. 2004), it was informative to compare levels of expression of dominant TSSs across different genes to understand the contribution of TSS downstream motifs.
MEME-ChIP analysis of the promoter regions from the off-to-on genes identified a second downstream motif, CNAAATT (E-value = 6.9×1071 from DREME) (Fig. 6A, right column), most enriched in the off-to-on genes between +29 and +60 bp downstream from the dominant TSS (Fig. 6B, right panel). This motif was very similar to what was identified previously as the translational control element (TCE) that was important for testis-specific expression of genes (Schäfer et al. 1990; Katzenberger et al. 2012). The CNAAATT motif appeared to be less prominent in the alternative promoters expressed when germ cells advanced to spermatocytes and was not enriched near the down-regulated promoters (Fig. 6A,H; Supplemental Fig. S5).
Presence of CNAAATT downstream also correlated with more efficient TSS usage, similar to a well-positioned ACA (Fig. 6E,F). In both TCA-containing and TCA-lacking TSSs, those with CNAAATT between +29 and +60 bp downstream tended to be more highly ranked within the CAGE cluster than those without CNAAATT (Fig. 6E). Moreover, comparing dominant TSSs across different off-to-on genes, those with CNAAATT were on average more highly expressed than those without CNAAATT. This was true for dominant TSSs that had one or both of TCA and ACA at +26, +28, or +30 downstream, or for dominant TSS that did not have either TCA or ACA (Fig. 6F). The effects of the TCA, ACA, and CNAAATT motifs appeared to be additive.
Together, the correlation of well-positioned TCA, ACA, and CNAAATT with the level of expression from a given TSS suggests that these motifs may play a role in determining at which potential start sites within the open region created by tMAC transcription can efficiently initiate (Fig. 6G). Consistently, the 5′/upstream edge of the RETI tended to be positioned at a fixed distance upstream of the ACA-dense and CNAAATT-containing regions (Fig. 6H).
The novel motifs in off-to-on genes collaborate in determining a narrow–high subgroup
A distinct population of off-to-on genes were expressed from a narrow RETI (Fig. 7A, genes with width of RETI < 11 bp, same genes as black and gray in Supplemental Fig. S2E, bottom panel; black corresponds to genes in Supplemental Fig. S2C, and gray corresponds to Supplemental Fig. S2A), with the majority highly expressed compared with other off-to-on genes. To understand how this group of promoters correlates with the promoter motifs identified above, the off-to-on genes were divided into four subgroups: high dominant TSS expression from a narrow RETI (“narrow–high,” the group of interest), low expression from a narrow RETI, high expression from a broad RETI, and low expression from a broad RETI (Fig. 7A). The analysis here focused on the expression level of dominant TSS since it was more straightforward to determine relative positions of functional motifs with respect to the dominant TSS than to a cluster of TSSs. However, the grouping based on dominant TSS largely agrees with grouping based on overall promoter expression level measured by CAGE (Supplemental Fig. S6A).
Figure 7.

Off-to-on genes highly expressed from a narrow TSS region often had most or all of the motifs (tMAC-ChIP, Achi/Vis, TCA, ACA, and CNAAATT) at optimal positions. (A) Division of off-to-on genes into four groups based on the width of RETI (X-axis) and level of expression of the dominant TSS in the promoter measured by CAGE (Y-axis). (B) Bar plot showing for every combination of the motifs, the proportion of the four groups. The eight panels correspond to the eight possible combinations of having TCA (at +1), ACA (at +26, 28, or 30), or CNAAATT (between +29 and 60). Within each panel, the four bars correspond to four possible combinations of having the tMAC-ChIP motif (between −70 and −40), and/or the Achi/vis motif (between −50 and −20). (C) Results from a logistic regression model predicting the likelihood of a promoter being narrow–high. (D) Enrichment of indicated motifs from the dominant TSS, calculated by Centrimo. (Top) Broad–high. (Bottom) Narrow–high genes. (E) Model of how upstream and downstream motifs at certain positions may collaborate to create the narrow–high promoters.
A larger proportion of the dominant TSSs in the narrow–high group had match of the motifs identified above (tMAC-ChIP, Achi/Vis, Inr, TCA, ACA, and CNAAATT) at the optimal positions than any of the other groups (Supplemental Fig. S6B). In fact, the more of the identified motifs located at the optimal positions a given dominant TSS had, the more likely the promoter belonged to the narrow-high group (Fig. 7B). To account for the small sample size of genes with each combination of motifs, and to better understand how having the motif combination correlated with promoter type, a logistic regression model was built based on what motifs each dominant TSS had and whether that promoter belonged to the narrow–high group. The model offered two insights. First, all motifs contributed significantly to a gene being narrow–high (P-values for tMAC-ChIP, TCA, and ACA were all <1×10−10, for Achi/Vis was 0.24×10−5, and for CNAAATT was 0.5×10−3). Second, the more motifs at optimal positions a promoter had, the higher the probability that it would be narrow-high. A promoter with all five motifs had a 92% ± 5.5% chance of being narrow–high (Fig. 7C).
Plotting the positions of the best match for each motif relative to dominant TSS for each promoter revealed that the narrow–high group tended to have a longer overall distance from the tMAC-ChIP motif and/or Achi/Vis motif to the ACA and CNAAATT motifs (Fig. 7D). Based on the model proposed earlier, tMAC may create an open stretch of chromatin with defined width that limits the 3′ edge of the RETI (Fig. 7E, top panel), and ACA and CNAAATT may help determine the 5′ edge of the RETI (Fig. 7E, middle panel). Therefore, having tMAC-ChIP motif further away from ACA and CNAAATT motif may limit RETI to only a narrow stretch of promoter region (Fig. 7E, bottom panel).
Discussion
Our results show that the dramatic gene-selective and tissue-specific transcription program that turns on in Drosophila spermatocytes is regulated by specialized promoter-proximal motifs and local action of cell type-specific protein players that act upon them. We identified ∼3000 promoters that require tMAC function to become accessible and initiate active transcription as germ cells transition to the spermatocyte state. Our findings from genome-wide analyses and selected reporter constructs are consistent with published results that regulatory elements sufficient for expression in spermatocytes lie close to the promoter regions in several genes expressed specifically in spermatocytes (White-Cooper 2010).
One of the striking findings from our study is that many genes expressed both in bam−/− mutant testes, in which spermatogonia continue to proliferate, and in 72 hrPHS testes, in which many germ cells have progressed to the spermatocyte state, use an alternate promoter that turns on only in the spermatocyte-containing sample. It is possible that the conditions for productive transcription initiation are so different in spermatocytes compared with spermatogonia that many genes evolved alternative promoters to allow transcription in both cell types. Use of promoters with distinct proximal motifs bound by cell type-specific promoter-interacting factors such as we describe here may contribute to down-regulation of the old program as well as to turning on a new cell type-specific differentiation program. Indeed, the DRE motif, enriched at broad promoters down-regulated when spermatogonia transition to spermatocytes, is bound by the DRE-binding factor protein DREF, which recruits the TBP paralog TRF2 to facilitate transcript initiation (Hochheimer et al. 2002). Because DREF protein is expressed in spermatogonia but down-regulated as spermatocytes mature (Angulo et al. 2019), promoters that depend on the DRE motif and DREF-TRF2 may no longer express efficiently in late spermatocyte stages.
Function of the testis-specific tMAC complex is required for the vast majority of both the off-to-on gene and alternative new spermatocyte-specific promoters to become open and accessible as spermatogonia transition to the spermatocyte state. Where a new alternative spermatocyte-specific promoter is located within the stretch of DNA that wraps around the −1 nucleosome of the old spermatogonial promoter (Fig. 3D), displacement of this −1 nucleosome is likely a prerequisite for the expression of the new promoter. Together with the requirement for tMAC function for opening and expression of the new alternative promoter, this suggests that tMAC may play a role in remodeling nucleosomes. It is not known whether tMAC opens local chromatin by binding DNA that is transiently detached from nucleosomes due to loosening or breathing of nucleosomal arrays or other chromosomal events, and then holding it in an open position, or whether tMAC can bind to DNA wrapped around nucleosomes, similar to a pioneer transcription factor. The tMAC subunit Comr has a winged helix domain (White-Cooper 2010), and certain winged helix domain proteins are able to bind DNA on one side and allow simultaneous histone binding (Zaret and Carroll 2011). Although none of the core components of tMAC have as yet been shown to have nucleosome remodeling activity, genetic analysis in C. elegans suggests that the generally expressed MMB/dREAM components (SynMuvB genes in worms) can interact with and may recruit components of the nucleosome remodeling and histone deacetylase (NuRD) complex (Solari and Ahringer 2000).
It is possible that many of the alternative new promoters arose as a by-product of an ability of tMAC to promote chromatin opening and/or transcription initiation at many sites in the genome. In fact, mechanisms have evolved to keep this propensity under restraint: We recently showed that action of the multiple zinc finger protein Kmg expressed in spermatocytes is required to limit activity of tMAC to its normal target genes. With loss of Kmg function, tMAC binds to many additional sites in the genome that are not previously annotated promoters, with some of these being activated for transcription initiation (Kim et al. 2017).
Our motif and TSS analyses suggest that the off-to-on genes that express from a narrow region of efficient transcription initiation differ from the canonical, previously described narrow promoters (Supplemental Fig. S2). Among the down-regulated genes, most of the promoters with a narrow RETI also had narrow total span of CAGE signals, likely a result of TFIID precisely positioning Pol II for transcription initiation (Nogales et al. 2017). In contrast, most of the off-to-on genes with narrow regions of efficient transcript initiation had a wide total span of CAGE signals (Supplemental Fig. S2D,E). In other words, although these off-to-on promoters had many usable and permissive TSS positions, they were dominantly expressed from just a few TSSs within a narrow region, likely facilitated by the upstream and downstream promoter motifs and Inr at optimal positions.
Traditionally, there has been much focus on the importance of distal enhancer elements rather than promoters in specifying cell type-specific transcription programs. This view, however, may be somewhat biased by the intense analysis of developmental regulatory genes like evenskipped (Fujioka et al. 1999), the cell cycle regulatory phosphatase string/cdc25 (Lehman et al. 1999), or the close-range developmental signaling molecule BMP5 (Guenther et al. 2008, 2015), which are expressed and function in several disparate places in the body. Because the regionally expressed transcriptional activators and repressors that combine to establish positional identity differ in different regions of the body, it stands to reason that a gene that is expressed in different specific places, such as Pitx1 in jaw, pituitary, and pelvis, would need to use different enhancer elements to specify activation in different regions, with the regulatory input from the different enhancers perhaps feeding in to a common generic promoter. The situation and constraints may be quite different for terminal differentiation genes that are only expressed in a single tissue. In this case, as we showed here for differentiation of male germ cells, it may be more possible for the key regulatory sequences that specify cell type-specific transcriptional activation to be built into the core promoter and promoter-proximal regulatory sequences.
Materials and methods
Fly strains and husbandry
Drosophila strains were maintained in standard molasses medium at 22°C. For the bam heat-shock time course, male hs-Bam-HA/CyO; bamΔ86,e/TM3,e,Sb and female ; ;bam1,e/TM6b,e,Hu were crossed in molasses medium at 22°C, grown for 9 d, shifted for 30 min to 37°C for heat shock, and then brought back for 48 h (48 hrPHS) or 72 h (72 hrPHS) to 25°C before collection of hs-Bam-HA/+;bam1/bamΔ86 flies for dissection. With the same cross scheme, +/CyO;bam1/bamΔ86 flies were collected without heat shock as bam−/− mutants (Kim et al. 2017). For tMAC mutants, aly2/aly5p (White-Cooper et al. 2000) and topiZ0707/topiZ2139 (Perezgasga et al. 2004) were used with crosses made at 25°C and grown for 10 d.
RNA-seq library preparation and sequencing
One-hundred pairs of testes from <2-d-old male flies were used per replicate. Library preparation was carried out using the Ribo-Zero rRNA removal kit (Illumina MRZH116) and SMARTer stranded RNA-seq kit (Clonetech 634839). Sequencing was done with NextSeq 500 with nine libraries pooled in each run. Approximately 34 million to 40 million reads were obtained per replicate and each condition had two biological replicates (see the Supplemental Material).
RNA-seq data analysis
Adapters and low-quality bases were trimmed with trimGalore (0.4.1) (Martin 2011), and mapped to Drosophila melanogaster genome build dm6 using STAR (2.5.3b) (Dobin et al. 2013). Reads that fell within gene regions were counted with STAR using Ensembl annotation BDGP6.84. Differential expression analyses were carried out using DEseq2 (Love et al. 2014).
CAGE library preparation and sequencing
Three-hundred pairs of testes from <2-d-old male flies were used per replicate. Total RNA (20∼50 µg) was sent to DNAform (https://www.dnaform.jp/en/products/library/cage) for CAGE library preparation using published protocol nAnTi-CAGE (nonamplifying–nontagging illumina CAGE) (Murata et al. 2014) and sequenced using Illumina HiSeq with 75-bp single-end sequencing. Around 25 million to approximately 30 million reads were obtained per replicate, and each condition had two biological replicates.
CAGE data analysis
Low-quality bases were trimmed using TrimGalore (0.4.4_dev) (Martin 2011) and mapped to Drosophila melanogaster genome build dm6 using STAR (2.5.4b) (Dobin et al. 2013). The most 5′ mapped nucleotide of reads were counted using bedtools coverage (2.27.1) (Quinlan and Hall 2010) and used as input into CAGEr to build CAGE clusters (1.20.0) (see the Supplemental Material; Haberle et al. 2015).
ATAC-seq library preparation and sequencing
ATAC-seq was carried out with a modified version of published protocols (Buenrostro et al. 2015). For each technical replicate, 10∼20 pairs of testes from <1-d-old male flies were dissected and lysed for incubation with transposase from the Nextera kit (Illumina FC-121-1030) for 25 min at 30°C. Libraries from one or two technical replicates done in parallel using testis from the same cross were combined as one biological replicate. Sequencing was done with HiSeq 4000 with 75-bp paired-end reads. Ten million to approximately 30 million reads were obtained for each biological replicate, and each condition had at least two biological replicates.
ATAC-seq data analysis
Adapters and low-quality bases were trimmed using trimGalore (0.4.1) (Martin 2011), mapped to Drosophila melanogaster genome build dm6 using bwa aln (0.7.10) (Li and Durbin 2009). PCR duplicates were removed using Picard tools (1.130). Reproducibility was checked across biological replicates and the replicates were combined to plot heatmaps using DeepTools (3.3.0) (Ramírez et al. 2016) and to calculate nucleosome positions using NucleoATAC (0.3.2) (Schep et al. 2015).
Motif enrichment analysis
Motif discovery was done using MEME-ChIP (Machanick and Bailey 2011) with 150 bp flanking the RETI center on both sides. MEME-ChIP runs MEME for de novo long motif searches (Bailey and Elkan 1994), DREME for de novo short motif searches (Bailey 2011), and CENTRIMO for positional motif enrichment of both known transcription factor-binding sites and motifs found by MEME and DREME (Bailey and MacHanick 2012). Motifs that showed enrichment were further plotted on all groups of promoters using Seqpattern (1.16.0) (Haberle et al. 2014) for visualization. Positions of motifs in promoter regions were calculated using Seqpattern, except for the tMAC-ChIP motif, where motif occurrence was calculated by FIMO (Grant et al. 2011) in the MEME suite with a cutoff of P-value 0.01. In cases where one promoter had multiple matches, only the best match was kept.
Logistic regression model for narrow–high promoters
The logistic regression model was built in R with function glm() with option family = binomial(link = “logit”). The independent variables are for each motif whether the dominant TSS has the motif at the optimal position (0 or 1) and the dependent variable is whether the dominant TSS belonged to the narrow-high group (0 or 1). All positions corresponded to the first nucleotide of the motif except for TCA, where A is +1 by convention.
Data availability
All sequencing data were submitted to GEO GSE145975 and analysis scripts are available at https://github.com/danrlu/Fuller_Lab_paper.
Supplementary Material
Acknowledgments
We thank members of the Fuller laboratory and Joe Lipsick for many constructive discussions throughout this project. We also thank the Stanford Functional Genomics Core Facility for all of the sequencing runs. We thank Joe Lipsick, Joanna Wysocka, Paul Khavari, and members of the Fuller laboratory for critical reading of the manuscript. This work was supported by National Institutes of Health (NIH) F32 5F32HD086986 to D.L., Urology Care Foundation Research Scholar Award Program and AUA Western Section Research Scholar Fund II to H.-S.S., and NIH grant R01GM061986 and funds from the Reed-Hodgson professorship in Human Biology to M.T.F.
Author contributions: D.L. and M.T.F. conceived and designed the study. D.L. carried out ATAC-seq and all data analysis. H.-S.S. carried out the RNA-seq experiments. C.L. carried out the promoter reporter experiments. D.L. and M.T.F. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.335331.119.
References
- Angulo B, Srinivasan S, Bolival BJ, Olivares GH, Spence AC, Fuller MT. 2019. DREF genetically counteracts Mi-2 and Caf1 to regulate adult stem cell maintenance. PLoS Genet 15: e1008187 10.1371/journal.pgen.1008187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar S, Jiang J, Collu A, White-Cooper H, White RAH. 2003. Drosophila TGIF is essential for developmentally regulated transcription in spermatogenesis. Development 130: 2841–2852. 10.1242/dev.00513 [DOI] [PubMed] [Google Scholar]
- Bailey TL. 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659. 10.1093/bioinformatics/btr261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Bailey TL, MacHanick P. 2012. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40: e128 10.1093/nar/gks433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall EL, Lewis PW, Bell M, Rocha M, Jones DL, Botchan MR. 2007. Discovery of tMAC: a Drosophila testis-specific meiotic arrest complex paralogous to Myb–Muv B. Genes Dev 21: 904–919. 10.1101/gad.1516607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 2015: 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Sturgill D, Qu J, Jiang H, Park S., Boley N., Suzuki A. M., Fletcher A. R., Plachetzki D. C., FitzGerald P. C. et al. 2014. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res 24: 1209–1223. 10.1101/gr.159384.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio JA, Wright KJ, Tjian R. 2009. Shifting players and paradigms in cell-specific transcription. Mol Cell 36: 924–931. 10.1016/j.molcel.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danks GB, Navratilova P, Lenhard B, Thompson EM. 2018. Distinct core promoter codes drive transcription initiation at key developmental transitions in a marine chordate. BMC Genomics 19: 164 10.1186/s12864-018-4504-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett K, Jiang J, Aleti G, White-Cooper H. 2011. Wake-up-call, a lin-52 paralogue, and Always early, a lin-9 homologue physically interact, but have opposing functions in regulating testis-specific gene expression. Dev Biol 355: 381–393. 10.1016/j.ydbio.2011.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Müller GA. 2017. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol 52: 638–662. 10.1080/10409238.2017.1360836 [DOI] [PubMed] [Google Scholar]
- FitzGerald PC, Sturgill D, Shyakhtenko A, Oliver B, Vinson C. 2006. Comparative genomics of Drosophila and human core promoters. Genome Biol 7: R53 10.1186/gb-2006-7-7-r53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka M, Emi-Sarker Y, Yusibova GL, Goto T, Jaynes JB. 1999. Analysis of an even-skipped rescue transgene reveals both composite and discrete neuronal and early blastoderm enhancers, and multi-stripe positioning by gap gene repressor gradients. Development 126: 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, Tjian R. 2010. Unexpected roles for core promoter recognition factors in cell-type-specific transcription and gene regulation. Nat Rev Genet 11: 549–558. 10.1038/nrg2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, Van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. 10.1038/nature09715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther C, Pantalena-Filho L, Kingsley DM. 2008. Shaping skeletal growth by modular regulatory elements in the Bmp5 gene. PLoS Genet 4: e1000308 10.1371/journal.pgen.1000308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CA, Wang Z, Li E, Tran MC, Logan CY, Nusse R, Pantalena-Filho L, Yang GP, Kingsley DM. 2015. A distinct regulatory region of the Bmp5 locus activates gene expression following adult bone fracture or soft tissue injury. Bone 77: 31–41. 10.1016/j.bone.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, Li N, Hadzhiev Y, Plessy C, Previti C, Nepal C, Gehrig J, Dong X, Akalin A, Suzuki AM, et al. 2014. Two independent transcription initiation codes overlap on vertebrate core promoters. Nature 507: 381–385. 10.1038/nature12974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, Forrest ARR, Hayashizaki Y, Carninci P, Lenhard B. 2015. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Res 43: e51 10.1093/nar/gkv054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales KG, Fuller MT. 1997. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 90: 121–129. 10.1016/S0092-8674(00)80319-0 [DOI] [PubMed] [Google Scholar]
- Hiller M. 2004. Testis-specific TAF homologs collaborate to control a tissue-specific transcription program. Development 131: 5297–5308. 10.1242/dev.01314 [DOI] [PubMed] [Google Scholar]
- Hiller MA, Lin TY, Wood C, Fuller MT. 2001. Developmental regulation of transcription by a tissue-specific TAF homolog. Genes Dev 15: 1021–1030. 10.1101/gad.869101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochheimer A, Zhou S, Zheng S, Holmes MC, Tjian R. 2002. TRF2 associates with DREF and directs promoter-selective gene expression in Drosophila. Nature 420: 439–445. 10.1038/nature01167 [DOI] [PubMed] [Google Scholar]
- Hoskins RA, Landolin JM, Brown JB, Sandler JE, Takahashi H, Lassmann T, Yu C, Booth BW, Zhang D, Wan KH, et al. 2011. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res 21: 182–192. 10.1101/gr.112466.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insco ML, Leon A, Tam CH, McKearin DM, Fuller MT. 2009. Accumulation of a differentiation regulator specifies transit amplifying division number in an adult stem cell lineage. Proc Natl Acad Sci 106: 22311–22316. 10.1073/pnas.0912454106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Benson E, Bausek N, Doggett K, White-Cooper H. 2007. Tombola, a tesmin/TSO1-family protein, regulates transcriptional activation in the Drosophila male germline and physically interacts with Always early. Development 134: 1549–1559. 10.1242/dev.000521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenberger RJ, Rach EA, Anderson AK, Ohler U, Wassarman DA. 2012. The Drosophila translational control element (TCE) is required for high-level transcription of many genes that are specifically expressed in testes. PLoS One 7: e45009 10.1371/journal.pone.0045009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lu C, Srinivasan S, Awe S, Brehm A, Fuller MT. 2017. Cell fate: blocking promiscuous activation at cryptic promoters directs cell type-specific gene expression. Science 356: 717–721. 10.1126/science.aal3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman DA, Patterson B, Johnston LA, Balzer T, Britton JS, Saint R, Edgar BA. 1999. Cis-regulatory elements of the mitotic regulator, string/Cdc25. Development 126: 1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, Viswanathan S, Wood C, Wilson PG, Wolf N, Fuller MT. 1996. Coordinate developmental control of the meiotic cell cycle and spermatid differentiation in Drosophila males. Development 122: 1331–1341. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Fuller MT. 2015. Recruitment of Mediator complex by cell type and stage-specific factors required for tissue-specific TAF dependent gene activation in an adult stem cell lineage. PLoS Genet 11: e1005701 10.1371/journal.pgen.1005701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL. 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697. 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- McKearin D, Ohlstein B. 1995. A role for the Drosophila Bag-of-marbles protein in the differentiation of cystoblasts from germline stem cells. Development 121: 2937–2947. [DOI] [PubMed] [Google Scholar]
- Michiels F, Gasch A, Kaltschmidt B, Renkawitz-Pohl R. 1989. A 14 bp promoter element directs the testis specificity of the Drosophila β2 tubulin gene. EMBO J 8: 1559–1565. 10.1002/j.1460-2075.1989.tb03540.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Nishiyori-Sueki H, Kojima-Ishiyama M, Carninci P, Hayashizaki Y, Itoh M. 2014. Detecting expressed genes using CAGE. Methods Mol Biol 1164: 67–85. 10.1007/978-1-4939-0805-9_7 [DOI] [PubMed] [Google Scholar]
- Nogales E, Louder RK, He Y. 2017. Structural insights into the eukaryotic transcription initiation machinery. Annu Rev Biophys 46: 59–83. 10.1146/annurev-biophys-070816-033751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA. 2008. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133: 1277–1289. 10.1016/j.cell.2008.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U, Liao G, Niemann H, Rubin GM. 2002. Computational analysis of core promoters in the Drosophila genome. Genome Biol 3: research0087.1 10.1186/gb-2002-3-12-research0087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perezgasga L, Jiang JQ, Bolival B Jr, Hiller M, Benson E, Fuller MT, White-Cooper H. 2004. Regulation of transcription of meiotic cell cycle and terminal differentiation genes by the testis-specific Zn-finger protein matotopetli. Development 131: 1691–1702. 10.1242/dev.01032 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rach EA, Winter DR, Benjamin AM, Corcoran DL, Ni T, Zhu J, Ohler U. 2011. Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet 7: e1001274 10.1371/journal.pgen.1001274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation Web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie S, Dalby M, Lloret-Llinares M, Bakoulis S, Vaagensø CD, Jensen TH, Andersson R. 2018. Transcription start site analysis reveals widespread divergent transcription in D. melanogaster and core promoter-encoded enhancer activities. Nucleic Acids Res 46: 5455–5469. 10.1093/nar/gky244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, DeCaprio JA. 2013. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 13: 585–595. 10.1038/nrc3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer M, Kuhn R, Bosse F, Schäfer U. 1990. A conserved element in the leader mediates post-meiotic translation as well as cytoplasmic polyadenylation of a Drosophila spermatocyte mRNA. EMBO J 9: 4519–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. 2015. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res 25: 1757–1770. 10.1101/gr.192294.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, et al. 2003. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci 100: 15776–15781. 10.1073/pnas.2136655100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solari F, Ahringer J. 2000. NURD-complex genes antagonise Ras-induced vulval development in Caenorhabditis elegans. Curr Biol 10: 223–226. 10.1016/S0960-9822(00)00343-2 [DOI] [PubMed] [Google Scholar]
- Wang Z, Mann RS. 2003. Requirement for two nearly identical TGIF-related homeobox genes in Drosophila spermatogenesis. Development 130: 2853–2865. 10.1242/dev.00510 [DOI] [PubMed] [Google Scholar]
- White-Cooper H. 2010. Molecular mechanisms of gene regulation during Drosophila spermatogenesis. Reproduction 139: 11–21. 10.1530/REP-09-0083 [DOI] [PubMed] [Google Scholar]
- White-Cooper H, Schäfer MA, Alphey LS, Fuller MT. 1998. Transcriptional and post-transcriptional control mechanisms coordinate the onset of spermatid differentiation with meiosis I in Drosophila. Development 125: 125–134. [DOI] [PubMed] [Google Scholar]
- White-Cooper H, Leroy D, MacQueen A, Fuller M. 2000. Transcription of meiotic cell cycle and terminal differentiation genes depends on a conserved chromatin associated protein, whose nuclear localisation is regulated. Development 127: 5463–5473. [DOI] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, Stark A. 2015. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518: 556–559. 10.1038/nature13994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25: 2227–2241. 10.1101/gad.176826.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data were submitted to GEO GSE145975 and analysis scripts are available at https://github.com/danrlu/Fuller_Lab_paper.