

SUMMARY
Selective autophagy of organelles is critical for cellular differentiation, homeostasis, and organismal health. Autophagy of the ER (ER-phagy) is implicated in human neuropathy but is poorly understood beyond a few autophagosomal receptors and remodelers. By using an ER-phagy reporter and genome-wide CRISPRi screening, we identified 200 high-confidence human ER-phagy factors. Two pathways were unexpectedly required for ER-phagy. First, reduced mitochondrial metabolism represses ER-phagy, which is opposite of general autophagy and is independent of AMPK. Second, ER-localized UFMylation is required for ER-phagy to repress the unfolded protein response via IRE1a. The UFL1 ligase is brought to the ER surface by DDRGK1 to UFMylate RPN1 and RPL26 and preferentially targets ER sheets for degradation, analogous to PINK1-Parkin regulation during mitophagy. Our data provide insight into the cellular logic of ER-phagy, reveal parallels between organelle autophagies, and provide an entry point to the relatively unexplored process of degrading the ER network.
Graphical Abstract
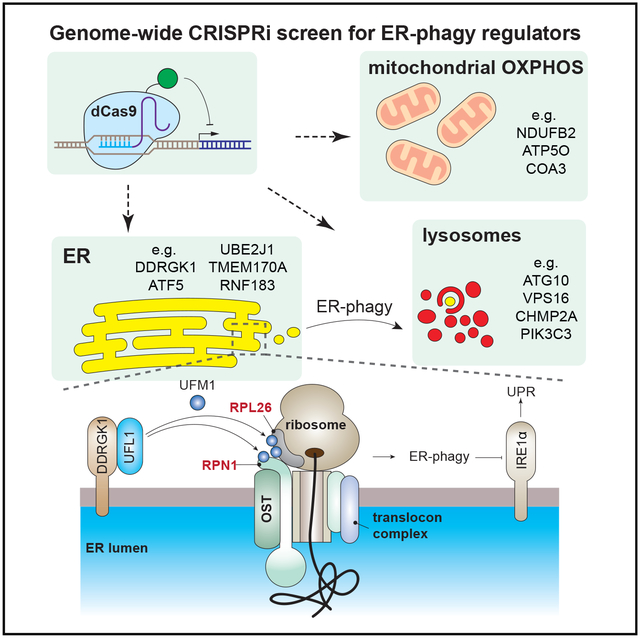
In Brief
An unbiased, genome-wide screen implicates mitochondrial oxidative phosphorylation and ER surface UFMylation as regulators of starvation-induced ER-phagy.
INTRODUCTION
Macroautophagy (herein referred to as autophagy) mediates the delivery of cellular cargo to the lysosome for degradation. Once thought to be a non-specific process, it has become clear that autophagy is complexly regulated and induced by various stresses to remove damaged or excessive cellular components. Targeted removal of entire organelles by autophagy is necessary for cellular homeostasis, and, during selective autophagy of mitochondria (mitophagy), the surface proteins of damaged mitochondria are marked by phosphorylation and ubiquitylation to recruit autophagic machinery (Nguyen et al., 2016; Youle and Narendra, 2011). Dysregulation of selective organelle autophagy negatively impacts cellular fitness and is linked to degenerative diseases, particularly in non-regenerative cell types such as neurons. For example, mutations of key mitophagy genes, such as PINK1 and Parkin, are strongly associated with disorders such as Parkinson’s disease (Deas et al., 2011; Dodson and Guo, 2007; Geisler et al., 2010; Pickrell and Youle, 2015; Pilsl and Winklhofer, 2012).
The endoplasmic reticulum (ER) plays a critical role in numerous cellular functions, such as the the storage of calcium, the biosynthesis of lipids, and the maturation and transport of secretory and membrane proteins (Schwarz and Blower, 2016). The ER is tightly regulated by multiple quality-control mechanisms, such as ER-associated degradation (ERAD) and ER to Lysosome Associated Degradation (ERLAD) (Fregno et al., 2018; Ruggiano et al., 2014). The ER-autophagy (ER-phagy) pathway intersects with the selective autophagy machinery to send portions of the ER for wholesale lysosomal degradation. While ER-phagy has long been observed in yeast (Hamasaki et al., 2005), it has only recently been described in mammalian cells (Khaminets et al., 2015).
During ER-phagy, several ER surface proteins, including FAM134B, RTN3L, TEX264, and ATL3, act as specific receptors through LC3/GABARAP-interacting regions (LIRs/GIMs) to recruit autophagy machinery (An et al., 2019; Chino et al., 2019; Grumati et al., 2017; Khaminets et al., 2015; Chen et al., 2019). ER expansion can also be reversed via ER-phagy that is mediated by the SEC62 and CCPG1 LIR-containing ER-phagy receptors (Fumagalli et al., 2016; Smith et al., 2018). The reticular ER network is remodeled for delivery to the lysosome by Atlastin GTPases that are also involved in basal ER morphology maintenance (Liang et al., 2018; Rismanchi et al., 2008; Wang et al., 2016; Zhao et al., 2016). But beyond the few receptors and remodelers most proximal to autophagosomal function, relatively little is known about the signals that regulate ER-phagy.
We performed a genome-wide CRISPR interference (CRISPRi) reporter-based screen to discover new players in ER-phagy, identifying both activators and inhibitors in a variety of cellular compartments. We deeply interrogated two pathways that positively regulate ER-phagy: (1) mitochondrial oxidative phosphorylation (OXPHOS) and (2) ER-resident UFMylation. While inhibition of OXPHOS reduces cellular energy levels and stimulates general autophagy, genetic or chemical inhibition of OXPHOS instead represses ER-phagy. Surprisingly, OXPHOS-dependent ER-phagy bypasses the canonical energy-sensing AMP-dependent protein kinase (AMPK). We furthermore found that UFMylation, a ubiquitin-like post-translational modification, is required for ER-phagy. The protein DDRGK1 recruits UFMylation machinery to the ER surface in a striking parallel to the mitophagic recruitment of Parkin by PINK1. DDRGK1 is specifically required for the ER-phagy of ER sheets, including ER-phagy mediated by LIR/GIM receptors located on these subdomains. Unbiased proteomics identified Ribophorin 1 (RPN1), an ER-localized quality control factor, as an ER sheet-localized target of DDRGK1-dependent UFMylation. Interfering with UFMylation-dependent ER-phagy leads to the accumulation of misfolded proteins and induces the unfolded protein response (UPR) via IRE1a signaling. Overall, our data provide a detailed map of ER-phagy regulators and highlight how organelle cross-talk and ER-resident factors mediate this emerging process of quality control.
RESULTS
A Genome-wide Flow Cytometry CRISPRi Screen for ER-Phagy
To develop a genome-wide screen for ER-phagy, we employed the previously-developed ER-autophagy tandem reporter (EATR) system (Figure 1A) (Liang et al., 2018). We first exposed HCT116 colon cancer cells stably expressing either the EATR construct or a general autophagy reporter (mCherry-eGFP-LC3B) to several stresses that could induce general autophagy and/or ER stress. Only prolonged amino acid starvation (16 h) using Earl’s buffered saline solution (EBSS) robustly induced both ER-phagy and general autophagy (Figures S1A–S1C). Torin1, which induces general autophagy via inactivation of mTORC1 and mTORC2 complexes, triggered general autophagy but did not induce ER-phagy (Figures S1A–S1C) (Thoreen et al., 2009). Rapamycin, which partially inhibits mTORC1 but spares mTORC2, induced neither general autophagy nor ER-autophagy in HCT116 cells (Figures S1A–S1C) (Jacinto et al., 2004; Thoreen and Sabatini, 2009). Direct activation of the unfolded protein response (UPR) using tunicamycin or thapsigargin also failed to stimulate general autophagy or ER-phagy (Figures S1A–S1C). By quantifying the ratio of eGFP/mCherry reporter fluorescence, we found that EBSS starvation induces ER-phagy in up to 80% of cells, with an average of 8% of the ER present in an acidified compartment at any given time (Figures S1B and S1D).
Figure 1. Unbiased Identification of ER-Phagy Regulators by Genome-wide CRISPRi Screening.
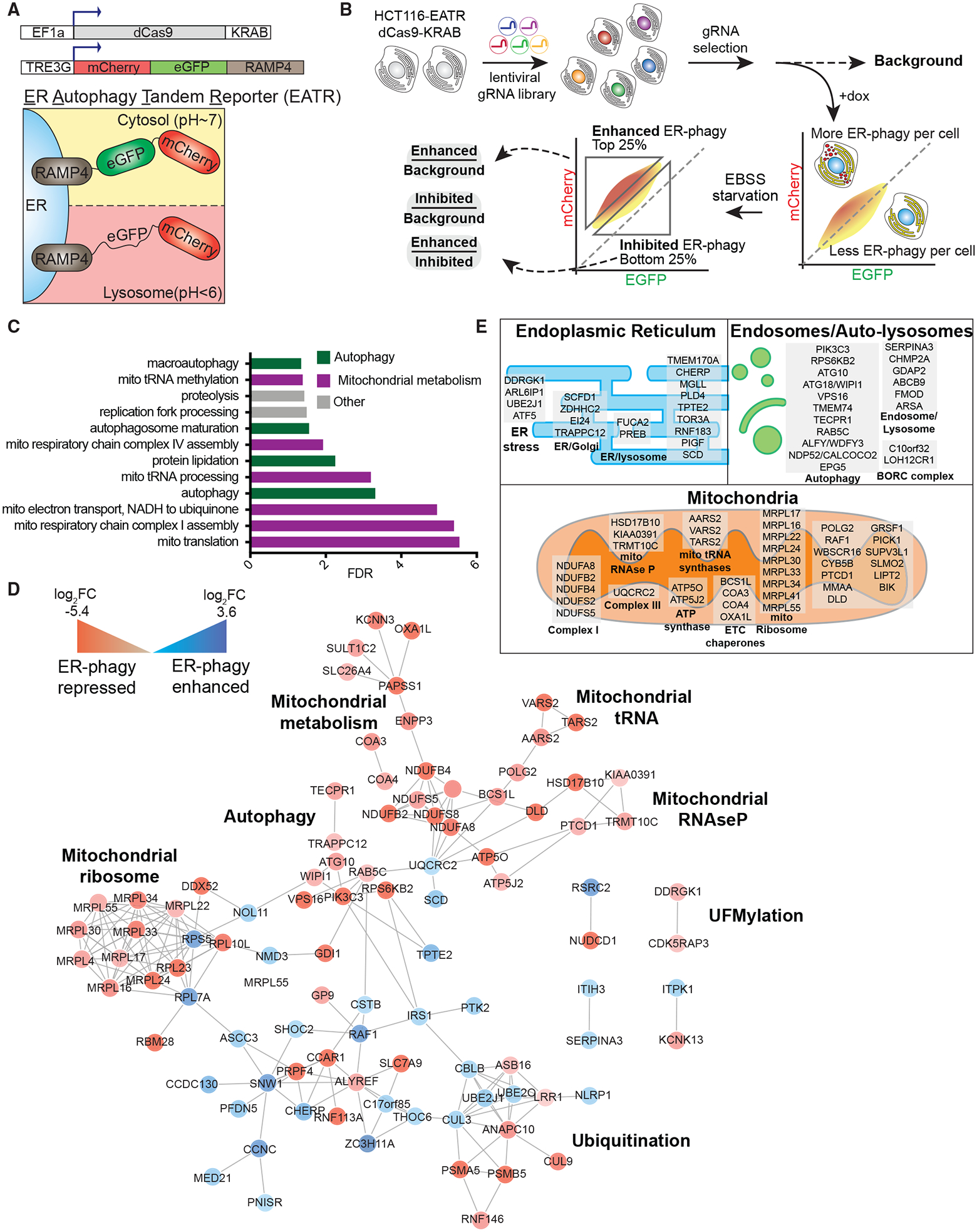
(A) Schematic of the ER Autophagy Tandem Reporter (EATR) and CRISPR inhibition (CRISPRi) system used for screening. HCT116 cells stably express a doxycycline-inducible EATR construct that consists of mCherry and eGFP fused to ER localized RAMP4. Cells also stably express dCas9-KRAB for sgRNA-targeted transcriptional repression.
(B) FACS screening strategy to identify genes whose knockdown enhances or inhibits ER-phagy. HCT116 cells described in (A) are transduced with a genome-wide lentiviral CRISPRi gRNA library. After selection for gRNA expression and removal of essential genes, doxycycline was added to express EATR. Cells were then starved in EBSS for 16 h to induce ER-phagy. The top and bottom quartiles correspond to enhanced and inhibited ER-phagy, respectively. Cells were sorted and processed for next-generation sequencing to identify gRNA representation in each sort bin.
(C) Gene ontology analysis identifies autophagy and mitochondrial metabolism as major signatures of ER-phagy. High-confidence ER-phagy genes were defined as having opposite phenotypes in the enhanced and inhibited sort gates and gene level p < 0.01. Ontologies with Benjamini-Hochberg false discovery rate (FDR) <0.05 are shown.
(D) Genes involved in ER-phagy form a physical interaction network. For clarity, only interactions between two or more high-confidence hits are shown. Red and blue circles represent genes whose knockdown represses and enhances ER-phagy, respectively.
(E) Subcellular classification of high-confidence ER-phagy genes highlights roles in the ER, auto-lysosomes, and mitochondria. See also Figure S1.
Having established amino acid starvation (using EBSS) as a robust ER-phagy stimulus, we coupled EATR-based fluorescence-activated cell sorting (FACS) screening with genome-wide CRISPR transcriptional inhibition (CRISPRi) to identify novel pathways involved in ER-phagy (Figures 1A and 1B) (Gilbert et al., 2014; Horlbeck et al., 2016; Liang et al., 2018). Since autophagy is influenced by the availability of cellular energy, we reasoned that complete knockout of ER-phagy regulators via CRISPR cutting could be detrimental to cells and mask interesting players. Indeed, mTOR is an essential gene and so cannot be queried using CRISPR screens beyond cell survival. The variability in sgRNA efficiencies of CRISPRi leads to different knockdown efficiencies, allowing for allelic series and residual function of essential genes involved in cellular energy regulation (Horlbeck et al., 2016).
As a proof of concept, we first assessed the suitability of EATR for CRISPRi screening by conducting a pilot screen with a custom CRISPRi library targeting known autophagy genes (Table S1). We used EATR-based FACS to isolate the top 25% of cells with the most ER-phagy (“enhanced” sort gate), and the bottom 25% of cells with the least ER-phagy (“inhibited” sort gate) (Figure S1E). This pilot screen successfully identified gRNAs targeting core autophagy genes as required for ER-phagy and correctly assigned their role in promoting ER-phagy such that knockdown of autophagy components was enriched in the inhibited gate (Figure S1F) and depleted in the enhanced sort gate (Figure S1G).
We scaled up to perform an unbiased, genome-wide CRISPRi-v2 screen for ER-phagy regulators using EATR-FACS (Gilbert et al., 2014; Horlbeck et al., 2016). We developed a very stringent list of ER-phagy genes by first performing a cutoff at p < 0.01 and then requiring that true hits have opposite phenotypes in the enhanced and inhibited sort gate. For example, gRNAs that knockdown a bona fide ER-phagy gene should be depleted in a population undergoing more ER-phagy but enriched in one undergoing less ER-phagy. We quantified involvement in ER-phagy by ratiometrically comparing gRNA distributions in the enhanced gate to those in the inhibited gate (Figure 1B), so that a positive log2 fold change indicates a gene whose knockdown increases ER-phagy and a negative log2 fold change indicates a gene whose knockdown inhibits ER-phagy. The resulting high-confidence hit list includes 200 genes, with gene-level log2 fold change phenotypes ranging from 3.62 to −5.37 (Table S2).
As expected, genes involved in multiple stages of general autophagy and membrane trafficking were high-confidence hits in our screen (Figure 1C; Figure S1H). Individual stable knockdown of these factors and testing using EATR, and mCherry-Cleaved ER-phagy Reporter (CCER) western blot assay, and other measures of autophagy verified their requirement for ER-phagy (Figures S1I–S1K). Our stringent criteria for calling a high-confidence hit narrowly exclude some known players in core autophagic pathways (e.g., multiple components of the V-ATPase complex), which have large log2 fold changes but moderate p values. Since our goal was to identify new and bona fide regulators in the relatively unexplored process of ER-phagy, we opted to maintain very strict statistical cutoffs and thus bias toward false negatives rather than introduce false positives. Overall, the presence of many known autophagic components in the screen indicate that the EATR assay and hence the genome-wide screen reports on ER-phagy pathways rather than ERAD or ERLAD, since the latter do not depend on autophagic components (Fregno et al., 2018; Ruggiano et al., 2014).
Consistent with recent reports of functional redundancy between ER-phagy receptors, we found that knockdown of SEC62, TEX264, and FAM134A (paralog of FAM134B) showed consistent but only moderate ER-phagy inhibition upon knockdown (Figure S1H) (Chino et al., 2019). We also examined the performance of individual FAM134B gRNAs by qRT-PCR and found that none of the CRISPRi gRNAs in the genome-wide library successfully knocks down FAM134B (Figure S1L). This highlights a tradeoff in current CRISPRi screening technology, where allelic series enable interrogation of otherwise essential genes but may introduce false negatives. Genetic redundancy and potential underperformance of CRISPRi guide RNAs means that failure to observe a gene in the ER-phagy screen dataset does not exclude potential role in ER-phagy. However, positive membership in the high-confidence set of 200 genes strongly indicates a role in ER-phagy.
As expected, unbiased gene ontology (GO) analysis, shows enrichment of GO related to autophagy (Figure 1C) (Huang et al., 2009). However, multiple aspects of mitochondrial metabolism were unexpectedly prominent. We integrated the high-confidence genetic hits against physical interaction databases to create a putative physical network of ER-phagy (Chatr-Aryamontri et al., 2017; Szklarczyk et al., 2015) (Figure 1D). This network falls into several major classes: autophagic execution, such as ATG10 and WIPI1 (Phillips et al., 2008; Wartosch et al., 2015; Yuan et al., 2013); ubiquitylation, such as the ER-localized UBE2J1 ubiquitin-conjugating enzyme involved in recovery from ER stress (Elangovan et al., 2017); mitochondrial metabolism and OXPHOS genes; and post-translational modification by the ubiquitin-like protein UFM1, including CDK5RAP3 and DDRGK1 (Cai et al., 2015; Wei and Xu, 2016; Wu et al., 2010). Finally, we manually annotated and subdivided all 200 high-confidence hits into those associated with the lysosome/endosome, ER-associated factors, and nuclear-encoded mitochondrial proteins (Table S2; Figure 1E) (Binder et al., 2014).
Mitochondrial Oxidative Phosphorylation Promotes ER-Phagy
We were surprised to find that the largest set of genes required for ER-phagy are involved in OXPHOS (Figure 2A), since cross-talk between ER-phagy and mitochondrial processes has not yet been described. While interference with mitochondrial energy metabolism induces general cytoplasmic autophagy, loss of mitochondrial factors instead repressed ER-phagy. Mitochondrial factors required for ER-phagy are directly involved in multiple aspects of the electron transport chain (ETC) and OXPHOS: complex I (NDUFA8, NDUFB2, NDUFB4, NDUFS2, NDUFS5), complex III (UQCRC2), and the ATP synthase/complex V (ATP5O and ATP5J) (Figure 1E). We also found a large number of factors indirectly required for OXPHOS through either ETC maturation or the synthesis of mitochondrially encoded ETC components (Taanman, 1999): mitochondrial chaperones (BCS1L, COA3, COA4, and OXA1L), mitochondrial ribosome subunits (MRPL17, MRPL16, MRPL22, MRPL24, MRPL30, MRPL33, MRPL34, MRPL41, and MRPL55), mitochondrial tRNAs (AARS2, VARS2, and TARS2), mitochondrial tRNA maturation (PTCD1), and mitochondrial RNase P (KIAA0391, TRMT10C, and HSD17B10). To further interrogate the link between mitochondrial metabolism and ER-phagy, we focused on further investigation of three factors that are involved in different parts of OXPHOS: NDUFB2, NDUFB4, and ATP5O (Figure S2A).
Figure 2. Intact OXPHOS Promotes ER-phagy.
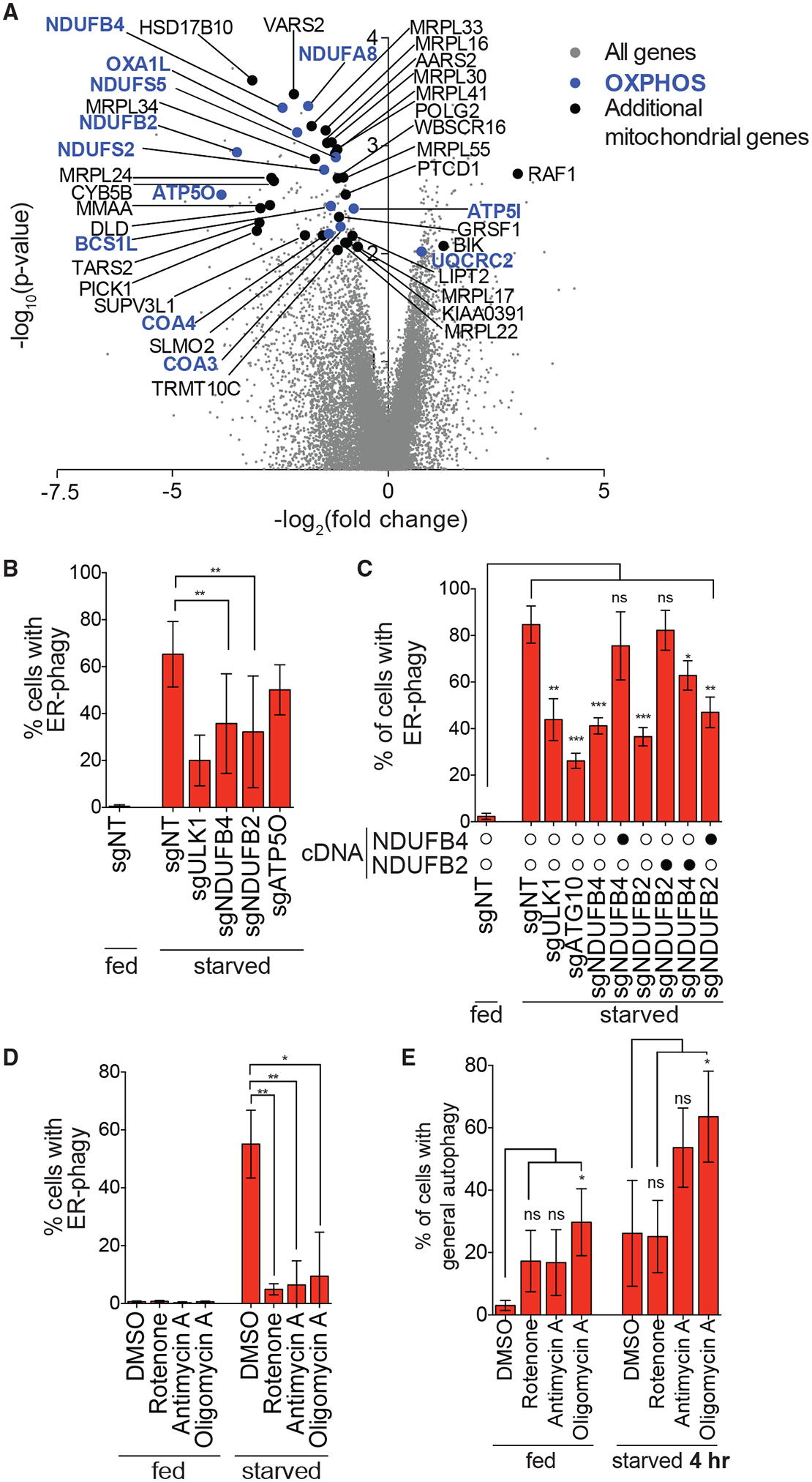
(A) Genes that are components of the OXPHOS pathway were top hits in screen (highlighted in blue). Additional mitochondria-related genes are indicated in black and all other targeting sgRNAs are indicated in gray.
(B) Knockdown of NDUFB4 and NDUFB2 significantly inhibit ER-phagy. HCT116 CRISPRi EATR cells were transduced with sgRNAs targeting ULK1, NDUFB4, NDUFB2, or ATP5O and starved for 16 h before FACS measurement for ER-phagy. Data are presented as mean ± SD of eight biological replicates.
(C) Re-expression of cognate cDNA of each OXPHOS gene rescues ER-phagy. HCT116 CRISPRi EATR cells were transduced with NDUFB4 or NDUFB2 cDNA constructs and then transduced with the indicated sgRNAs. Cells were starved for 16 h before FACS measurement for ER-phagy. Data are presented as mean ± SD of three biological replicates.
(D) Small-molecule inhibitors of the different OXPHOS compartments phenocopy the effect of genetic inhibitions. HCT116 EATR cells were treated with rotenone, antimycin A, or oligomycin A and starved for 16 h before FACS measurement of ER-phagy. Data are presented as mean ± SD of three biological replicates.
(E) General autophagy proceeds in cells where NDUFB2, NDUFB4, or ATP5O are knocked down. HCT116 CRISPRi cells expressing mCherryeGFP-LC3B were transduced with the indicated sgRNAs. Cells were starved for 4 h before FACS measurement for general autophagy. Data represent mean ± SD of four biological replicates.
See also Figures S2 and S3.
Stable knockdown of OXPHOS components quantitatively inhibited starvation-induced ER-phagy in multiple assays in a manner that paralleled knockdown efficiency (Figures 2B and S2B–S2D). However, knockdown of NDUFB2, NDUFB4, or ATP5O did not grossly affect starvation-induced general autophagy (Figure S2E). Knockdown of NDUFB2 destabilized NDUFB4 and vice versa, presumably because both are integral subunits of complex I (Figure S2B). Consistently, re-expressing the cognate OXPHOS cDNA rescued both the cognate protein and the destabilized partner (Figure S2F). Re-expressing the destabilized partner only rescued the abundance of that partner without affecting the knocked-down gene (e.g., NDUFB2 knockdown depletes both NDUFB2 and NDUFB4, and re-expressing NDUFB4 in this background only rescues NDUFB4 levels). Re-expressing the appropriate OXPHOS cDNA rescued ER-phagy, while cross-expressing a non-cognate OXPHOS cDNA had no effect, indicating that each knockdown was specific and on target (Figure 2C; Figures S2F–S2H).
We further explored the necessity of functional OXPHOS for normal ER-phagy using chemical genetics (Figure S2I). Rotenone is a known inhibitor of both general autophagy and complex I (Mader et al., 2012) and reduced both general autophagy and ER-phagy by multiple assays (Figures 2D and 2E; Figures S2J–S2L). By contrast, inhibiting complex III with antimycin A or ATP synthase with oligomycin A reduced ER-phagy but increased general autophagy at both early (4 h) and late time points (16 h) (Figures 2D and 2E; Figures S2J–S2M). Using Cell-Titer Glo to measure ATP levels (Figure S2N) and Seahorse to measure oxygen consumption (Figure S2O), we confirmed that knockdown of the OXPHOS genes reduced cellular energy levels.
We explored several hypotheses to determine how interfering with OXPHOS reduces ER-phagy. First, we found that increased mitophagic flux is not responsible for reduced ER-phagy by titrating away autophagic machinery. OXPHOS conditions that repressed ER-phagy did not grossly alter mitochondrial abundance or membrane potential and starvation actually increased the amount of Mitotracker accumulation (Figure S3A) (Johnson et al., 2014; Xiao et al., 2016). We also found no evidence of increased mitophagy during knockdown of OXPHOS components, either with or without stable overexpression of Parkin (Figures S3B–S3G).
Second, we found that interfering with OXPHOS reduced levels of the autophagic kinase ULK1 during starvation, but this is not causative of reduced ER-phagy. Knockdown of NDUFB2, NDUFB4, and ATP5O did not affect ULK1 under basal conditions, but during starvation reduced levels of ULK1 almost as much as knockdown of ULK1 itself (Figure S3H). Cognate cDNA re-expression in the appropriate stable knockdown background rescued levels of ULK1, and these same conditions also rescued ER-phagy (Figure S3H; Figure 2C). But overexpression of ULK1 itself in the context of OXPHOS knockdown did not rescue ER-phagy, indicating that reduced ULK1 is not limiting (Figure S3I).
Third, we found that reduced ER-phagy upon knockdown of OXPHOS components is independent of 5′ AMP-activated Protein Kinase (AMPK) signaling. This is surprising, since AMPK is a master regulator of general autophagy in response to cellular energy availability (Egan et al., 2011; Kim et al., 2011; Toyama et al., 2016). We generated AMPKɑ CRISPR-Cas9 knockout cells (Figure S3J) and found that they still mount a robust ER-phagy response (Figures S3K and S3L). The ability to perform ER-phagy in AMPKɑ-knockout cells was unaffected by stable re-expression of constitutively active or kinase dead AMPKɑ (Figures S3K and S3L). Overall, while genetically or chemically interfering with mitochondrial OXPHOS potently reduces ER-phagy, the mechanism of this cross-talk is independent of canonical pathways and remains unclear.
DDRGK1-Mediated UFMylation Regulates ER-Specific Autophagy of ER Sheets
The genome-wide screen for ER-phagy regulators yielded several hits that are localized to the ER and/or involved in ER-related processes (Figures 1E and 3A). We focused on one of these factors, DDRGK1/C20orf116/UFBP1, which has emerging roles in ER homeostasis (Leto et al., 2019; Liu et al., 2017; Walczak et al., 2019; Zhu et al., 2019).
Figure 3. DDRGK1 Specifically Regulates ER-phagy.
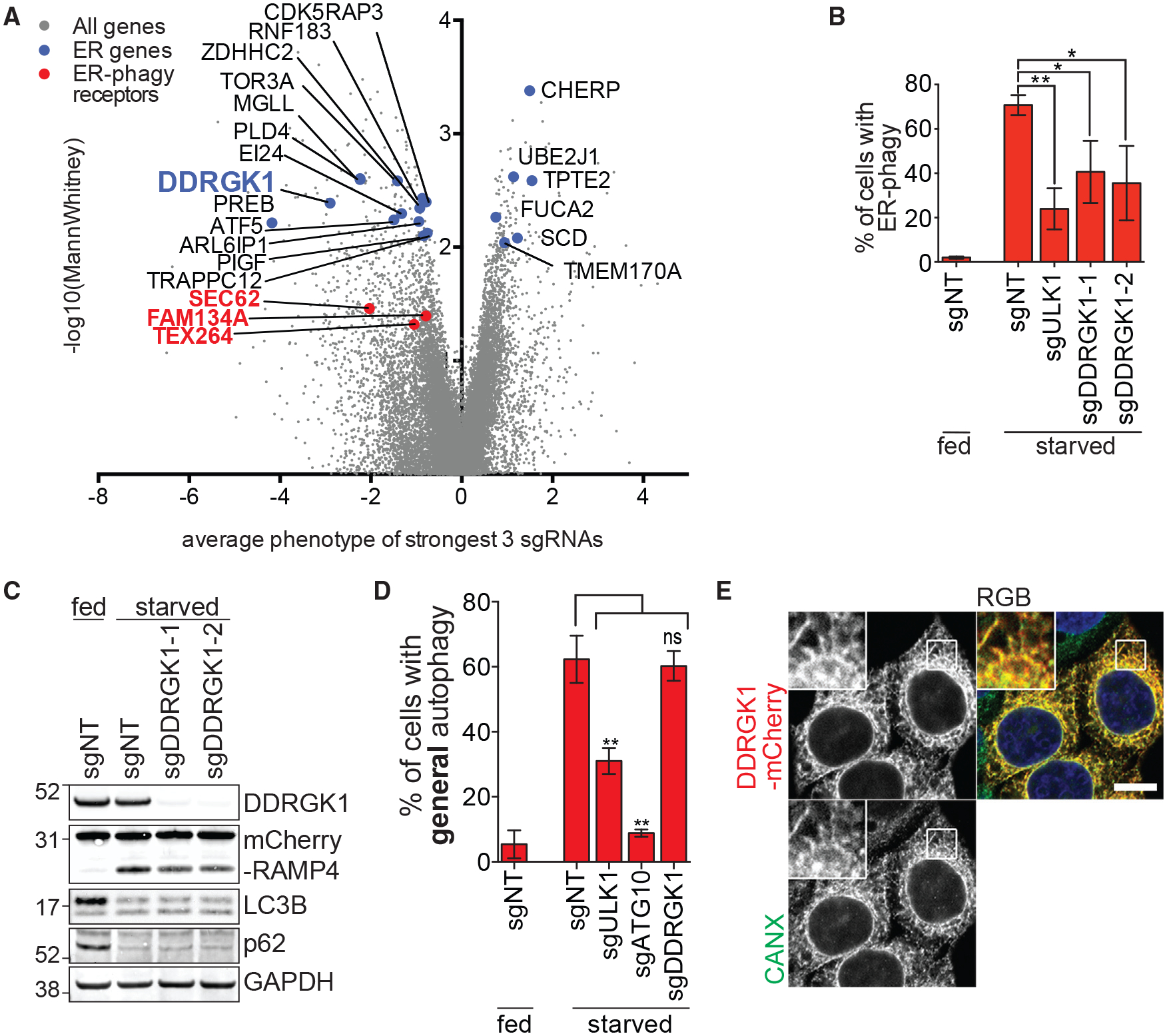
(A) ER-phagy CRISPRi screen identifies genes that are associated with the ER. Previously reported ER-phagy regulators are highlighted in red. DDRGK1 is highlighted in blue.
(B) DDRGK1 depletion results in inhibition of ER-phagy. HCT116 CRISPRi EATR cells were transduced with sgRNAs targeting ULK1 or DDRGK1 and starved for 16 h before FACS measurement for ER-phagy. Data are presented as mean ± SD of three biological replicates.
(C) DDRGK1 specifically inhibits ER-phagy but not general autophagy. HCT116 CRISPRi CCER cells were transduced with sgRNAs targeting DDRGK1 and starved for 16 h. Cells were lysed for western blotting of the indicated proteins.
(D) HCT116 CRISPRi cells stably expressing mCherry-eGFP-LC3B constructs were transduced with sgRNAs targeting either ULK1, ATG10, or DDRGK1. Cells were starved for 16 h before FACS measurement for general autophagy.
(E) DDRGK1 localizes to the ER. HeLa cells were stably transduced with DDRGK1-mCherry construct and immunostained for calnexin (CANX) as an ER marker. Insets represent a 3-fold enlargement of boxed areas. Scale bar represents 10 μm.
See also Figure S4.
Individual, stable knockdown of DDRGK1 resulted in ER-phagy as measured using both EATR and CCER assays (Figures 3B and 3C; Figure S4A) but had no apparent effect on general autophagy as determined using mCherry-eGFP-LC3B assay and the degradation of endogenous p62 and LC3B (Figures 3C and 3D). Immunofluorescence confirmed that an mCherry-tagged DDRGK1 construct co-localized with the ER (Figure 3E).
DDRGK1 is reported to be post-translationally modified by UFMylation, which is in turn required for further UFMylation of other factors (Cai et al., 2015; Liu et al., 2017; Wei and Xu, 2016; Wu et al., 2010). UFMylation involves the sequential activation, conjugation and ligation of UFM1 to a target substrate via an E1 (UBA5), E2 (UFC1), and E3 (UFL1) cascade that mirrors ubiquitin conjugation (Figure 4A) (Daniel and Liebau, 2014; Komatsu et al., 2004). We found that stable knockdown of UFL1 decreases DDRGK1 protein levels in a proteasome-dependent manner and also inhibited ER-phagy (Figures 4B and 4C; Figure S4B), as did knockdown of UFM1 and UBA5 (Figures 4D and S4C). Double knockdown of both UFL1 and DDRGK1 did not further inhibition of ER-phagy as compared to individual depletion of either factor, suggesting that they act in the same pathway to regulate ER-phagy (Figures S4D and S4E). Stable re-expression of UFL1 in UFL1-depleted cells rescued levels of DDRGK1 and restored ER-phagy (Figures 4E and 4F), but overexpression of DDRGK1 in UFL1-depleted cells led to high levels of DDRGK1 without ER-phagy (Figures 4E and 4F). Knockdown of DDRGK1 and UFL1 impeded lysosomal cleavage of both mCherry-RAMP4 and mCherry-KDEL, consistent with UFMylation mediating the autophagic turnover of both ER surface and lumenal proteins (Figures 4C, S4F, and S4G) (Munro and Pelham, 1987).
Figure 4. DDRGK1-Dependent UFMylation Regulates Autophagy of ER Sheets.
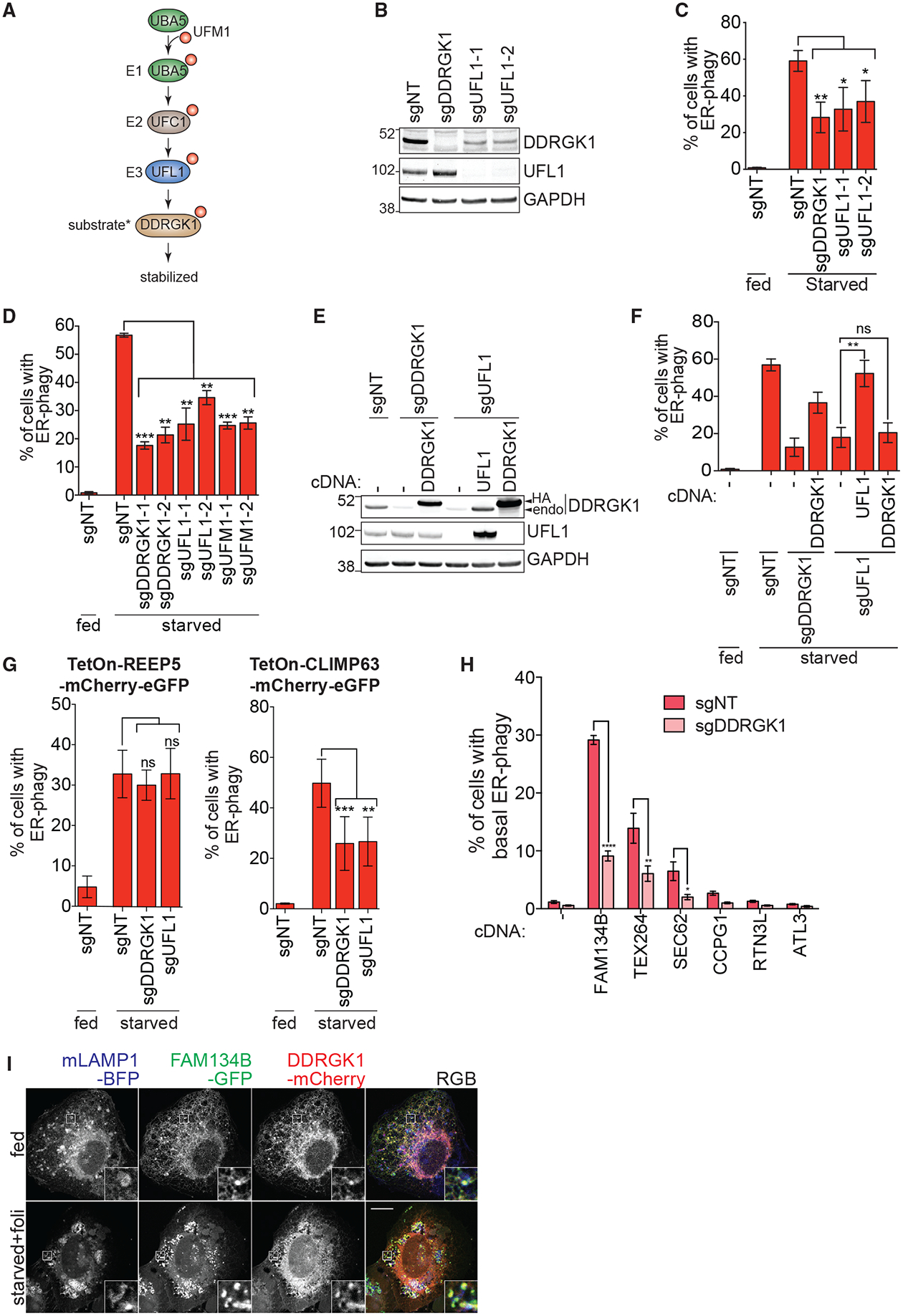
(A) Schematic of the three-step enzymatic reaction of the UFMylation cascade. UBA5 (E1) activate UFM1 and UFC1 acts as an E2 enzyme that interacts with the E3 ligase, UFL1. UFL1 recognizes and transfer UFM1 from UFC1 to its target substrate. Asterisk indicates that DDRGK1 is reported as a substrate of UFMylation in the literature.
(B) UFL1 knockdown reduces DDRGK1 protein levels. HCT116 CRISPRi cells were transduced with the indicated sgRNAs and then harvested to immunoblot for UFL1 protein levels.
(C) UFL1 knockdown phenocopies DDRGK1 knockdown during ER-phagy. The cells generated in (B) were starved for 16 h before FACS measurement for ER-phagy.
(D) UFMylation components are required for ER-phagy. HCT116 CRISPRi EATR cells expressing the indicated sgRNAs were starved for 16 h before FACS measurement for ER-phagy.
(E) UFL1 controls DDRGK1 protein levels. HCT116 CRISPRi cells were transduced with the indicated sgRNAs and further transduced with either DDRGK1-HA or HA-UFL1. Cell lysates were immunoblotted for DDRGK1 and UFL1.
(F) Re-expression of DDRGK1 in UFL1 knockdown cells does not rescue ER-phagy. The cells generated in (E) were starved for 16 h and then subjected to FACS measurement for ER-phagy. Data represent mean ± SD of three biological replicates.
(G) DDRGK1 selectively mediates ER sheets degradation. The RAMP4 in EATR system was replaced with either REEP5 (ER tubule marker) or CLIMP63 (ER sheets). The cells were transduced with the indicated sgRNAs and were starved for 16 h before FACS analysis for ER-phagy progression. Data represent mean ± SD of three biological replicates.
(H) DDRGK1 depletion selectively affect FAM134B, TEX264, and SEC62-mediated ER-phagy. HCT116 CRISPRi EATR cells were stably transduced with the cDNA for the indicated ER-phagy receptors. ER-phagy induced by overexpression of the ER-phagy receptors at basal state was measured by FACS analysis. Data represent mean ± SD of three biological replicates.
(I) DDRGK1 colocalizes with FAM134B and is co-degraded with FAM134B. U2OS cells stably expressing GFP-FAM134B were transiently transfected with mLAMP1-BFP and DDRGK1-mCherry. Cells were then starved for 4 h in the presence of 50 nM folimycin. Scale bar represents 10 μm. Insets represent a 4-fold enlargement of the boxed area.
See also Figure S4.
We made tandem fluorescent reporters for ER sheets (CLIMP63-mCherry-eGFP) and ER tubules (REEP5-mCherryeGFP) and found that knockdown of either DDRGK1 or UFL1 specifically impaired the autophagy of ER sheets (Figures 4G, S4H, and S4I). Different ER-phagy receptors are expressed on ER sheets or ER tubules. We found that overexpression of the sheet-localized ER-phagy receptors FAM134B, TEX264, and SEC62 induced ER-phagy, and this was perturbed by the depletion of DDRGK1 in the presence and absence of starvation (Figures 4H and S4J–S4L) (An et al., 2019; Chino et al., 2019; Fumagalli et al., 2016; Khaminets et al., 2015). Far less ER-phagy was induced by overexpression of the tubule-located ER-phagy receptors CCPG1, RTN3L, and ATL3, and loss of DDRGK1 did not have a statistically significant effect in this context (Figure 4H) (Chen et al., 2019; Grumati et al., 2017; Smith et al., 2018). DDRGK1 co-localizes with FAM134B both on the ER and in the lysosome, suggesting that subdomains of the ER containing DDRGK1 are cargos of FAM134B-mediated ER-phagy (Figure 4I).
DDRGK1 Acts as an ER Surface Adaptor for UFL1 Rather Than a UFMylation Substrate
DDRGK1 is reported to be UFMylated by UFL1 on one or more lysines and thereby stabilized (Figure 5A) (Wu et al., 2010). Using immunoprecipitation of DDRGK1 point mutants, we indeed found higher molecular weight species consistent with lysine post-translational modification of DDRGK1 (Figures 5B and S5A). However, this modification was unaffected by CRISPR-Cas9 knockout of UFL1 or CRISPRi knockdown of UFM1 (Figure 5B and S5A). Furthermore, knockdown of UFM1 had no effect on the abundance of DDRGK1 (Figure S5B), and DDRGK1 still stably interacted with UFL1 even when all 12 conserved lysines were mutated (K-less) (Figure 5B). Taken together, these data indicate that the stability of endogenous DDRGK1 is maintained not by UFMylation but by its interaction with UFL1. Along these lines, we found that DDRGK1’s ability to promote ER-phagy was independent of its major reported site of UFMylation on Lys267 (Figures 5C and S5C). Eleven other lysine mutants also substantially supported ER-phagy, as could a DDRGK1 mutant with all 12 conserved lysines mutated (Figures S5D and S5E). Overall, these data strongly suggest that DDRGK1 is not a target of UFMylation during ER-phagy, as has been reported for DDRGK1’s involved in the UPR and other signaling pathways (Lemaire et al., 2011; Yoo et al., 2014).
Figure 5. DDRGK1 Recruits UFL1 to the ER Surface via the PCI Domain.
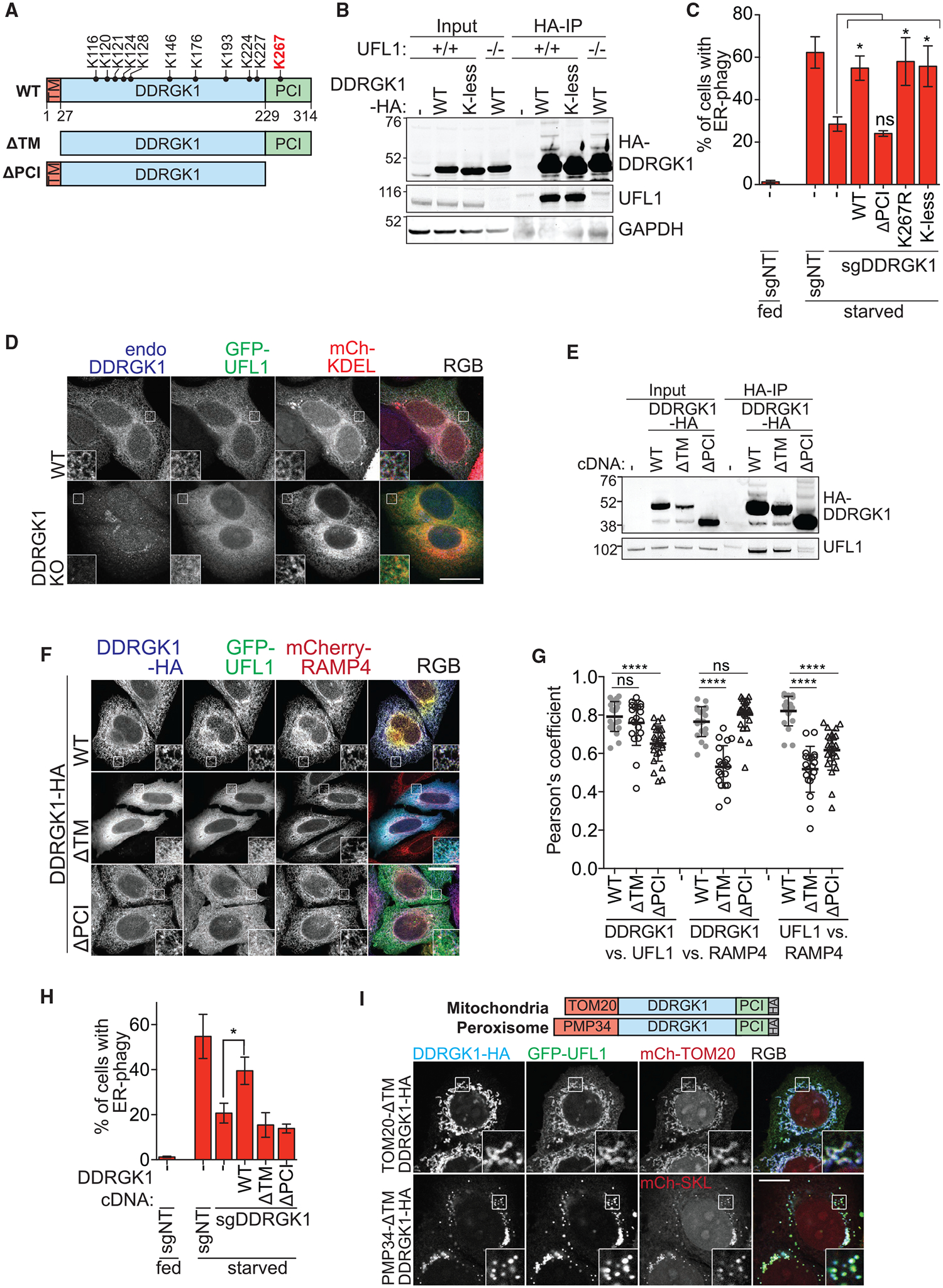
(A) Schematic of DDRGK1 domains and its conserved lysine residues. The reported major lysine residue for UFMylation (K267) is labeled in red. The two truncated forms of DDRGK1 that either lacks the N-terminal transmembrane domain (ΔTM) or the C-terminal proteasome component domain (ΔPCI) are also shown.
(B) Post-translational modification of DDRGK1 occurs on lysine residues. Parental or UFL1 knockout HCT116 cells were transfected with either wild-type (WT) or lysine-less (K-less) DDRGK1-HA constructs. Cells were harvested for HA immunoprecipitation.
(C) DDRGK1’s role during ER-phagy does not require post-translational modification on any lysine residue. HCT116 CRISPRi EATR cells were transduced with DDRGK1 sgRNA and then rescued using the indicated DDRGK1-HA mutant constructs. Cells were starved for 16 h before FACS ER-phagy measurement. Data represent mean ± SD of three biological replicates.
(D) Loss of DDRGK1 relocalizes UFL1 to the cytoplasm. Wild-type or DDRGK1KO HeLa cells were transiently transfected with GFP-UFL1 and mCherry-KDEL for 48 h. Cells were then fixed and immunostained for endogenous DDRGK1. Insets represent a 3-fold enlargement of boxed areas. Scale bar represents 20 μm.
(E) DDRGK1 interacts with UFL1 via its PCI domain. Parental HCT116 cells were stably transfected with the indicated DDRGK1-HA mutant constructs. Cells were then harvested for HA immunoprecipitation.
(F) DDRGK1 recruits UFL1 to the ER. DDRGK1KO HeLa cells were stably transduced with mCherry-RAMP4 (ER marker) and the indicated DDRGK1-HA mutant constructs. Cells were then transiently transfected with GFP-UFL1 for 24 h. Cells were then fixed and immunostained for HA epitope. Representative images are shown. Insets represent a 3-fold enlargement of boxed areas. Scale bar represents 10 μm.
(G) Pearson’s correlation coefficient for (F) was measured between DDRGK1 versus UFL1, DDRGK1 versus RAMP4, and UFL1 versus RAMP4. Data were generated from one biological experiment, and 20–26 cells were analyzed from each condition.
(H) DDRGK1’s role during ER-phagy requires both the SP and PCI domains. HCT116 CRISPRi EATR cells with DDRGK1 knockdown were rescued using the indicated DDRGK1-HA mutant constructs. Cells were then starved for 16 h and ER-phagy was measured by FACS analysis. Data represent mean ± SD of three biological replicates.
(I) DDRGK1’s determines the subcellular localization of UFL1. DDRGK1 knockout HeLa cells were stably transduced with either TOM20MTS-DDRGK1-dSP-HA (MTS, mitochondrial targeting signal) or PMP34-DDRGK1-dSP-HA and transiently transfected with GFP-UFL1 and the respective mCherry-organelle constructs. Insets represent a 3-fold enlargement of boxed areas. Scale bar represents 10 μm. See also Figure S5.
Using immunofluorescence and immunoprecipitation, we found that UFL1’s localization to the ER is dependent on DDRGK1. CRISPR-Cas9 knockout of DDRGK1 abrogated ER localization of UFL1, resulting in diffuse, cytosolic UFL1 (Figure 5D). We re-expressed various cDNA constructs of DDRGK1 in the DDRGK1 knockout background to map functional regions of the protein. Deleting DDRGK1’s N-terminal ER-targeting transmembrane domain (TM) still supported a DDRGK1-UFL1 interaction but led to cytoplasmic localization of both DDRGK1 and UFL1 without ER-phagy (Figures 5E–5H, S6A, and S6B). Removing DDRGK1’s C-terminal PCI domain did not affect normal ER localization of DDRGK1 but abolished its interaction with UFL1 (Figures 5E–5G and S6A). This led to cytoplasmic localization of UFL1 and abolished the cell’s ability to perform ER-phagy (Figures 5E–5G, S5A, and S5B). Re-targeting DDRGK1 to the mitochondria (TOM20-ΔTMDD-HA) or peroxisomes (PMP34-ΔTMDD-HA) was sufficient to re-target GFP-tagged UFL1 to each organelle (Figure 5I). Hence, DDRGK1’s interaction with UFL1 via its PCI domain dictates the subcellular localization of UFL1.
During mitophagy, numerous proteins are ubiquitylated on the mitochondrial surface. But current models of mitophagy suggest that overall ubiquitin load is more important than any one substrate (Chan et al., 2011; Heo et al., 2015; Ordureau et al., 2014). Hence, we asked whether directly recruiting UFL1 to the ER surface could bypass the need for DDRGK1 (Figure S6C). However, expression of UFL1-RAMP4 in DDRGK1-depleted cells was not sufficient to rescue ER-phagy despite robust localization to the ER surface (Figures S6C–S6F). Together, these data indicate that DDRGK1 both recruits UFL1 to the ER and plays a role in activation of its ligase activity or recruitment of substrates during ER-phagy. We find that DDRGK1 is not a UFMylation substrate during ER-phagy and is instead analogous to an ubiquitin substrate adaptor (e.g., an F-box protein) that works with Cullin-RING family proteins.
To identify candidate substrates of DDRGK1/UFL1 dependent UFMylation on the ER, we first knocked down previously reported substrates of UFL1 including CDK5RAP3, SOX9, and ASC1 (Cai et al., 2015; Egunsola et al., 2017; Liu et al., 2017; Wu et al., 2010; Yoo et al., 2014), but none of them affected ER-phagy (Figures S6G and S6H).
We therefore performed immunoprecipitation-tandem mass spectrometry (IP-MS/MS) label-free proteomics to identify new interactors of DDRGK1 and DDRGK1-dependent UFMylation substrates during EBSS starvation. To identify DDRGK1 interactors, non-denaturing IP-MS/MS was performed in DDRGK1 knockout HEK293T clones that either did or did not stably re-express HA-tagged DDRGK1 (Figure 6A). To identify DDRGK1-dependent UFMylated proteins, we counteracted the constitutive deconjugation of UFM1 from substrates by UFSP2 (Walczak et al., 2019) by generating CRISPR-Cas9 UFSP2 knockout clones in both wild-type and DDRGK1 knockout HEK2393T cells (Figure 6B). In the UFSP2 knockout and DDRGK1/UFSP2 double-knockout cells, we transiently expressed UFM1 lacking the C-terminal Cys-Ser residues (HA-UFM1-ΔCS) to override the need for UFSP2 cleavage prior to conjugation (Figure 6B). UFMylated proteins were isolated using a denaturing HA-tag IP during EBSS starvation and folimycin treatment (to prevent lysosomal degradation).
Figure 6. DDRGK1 Mediates UFMylation of ER Surface Proteins.
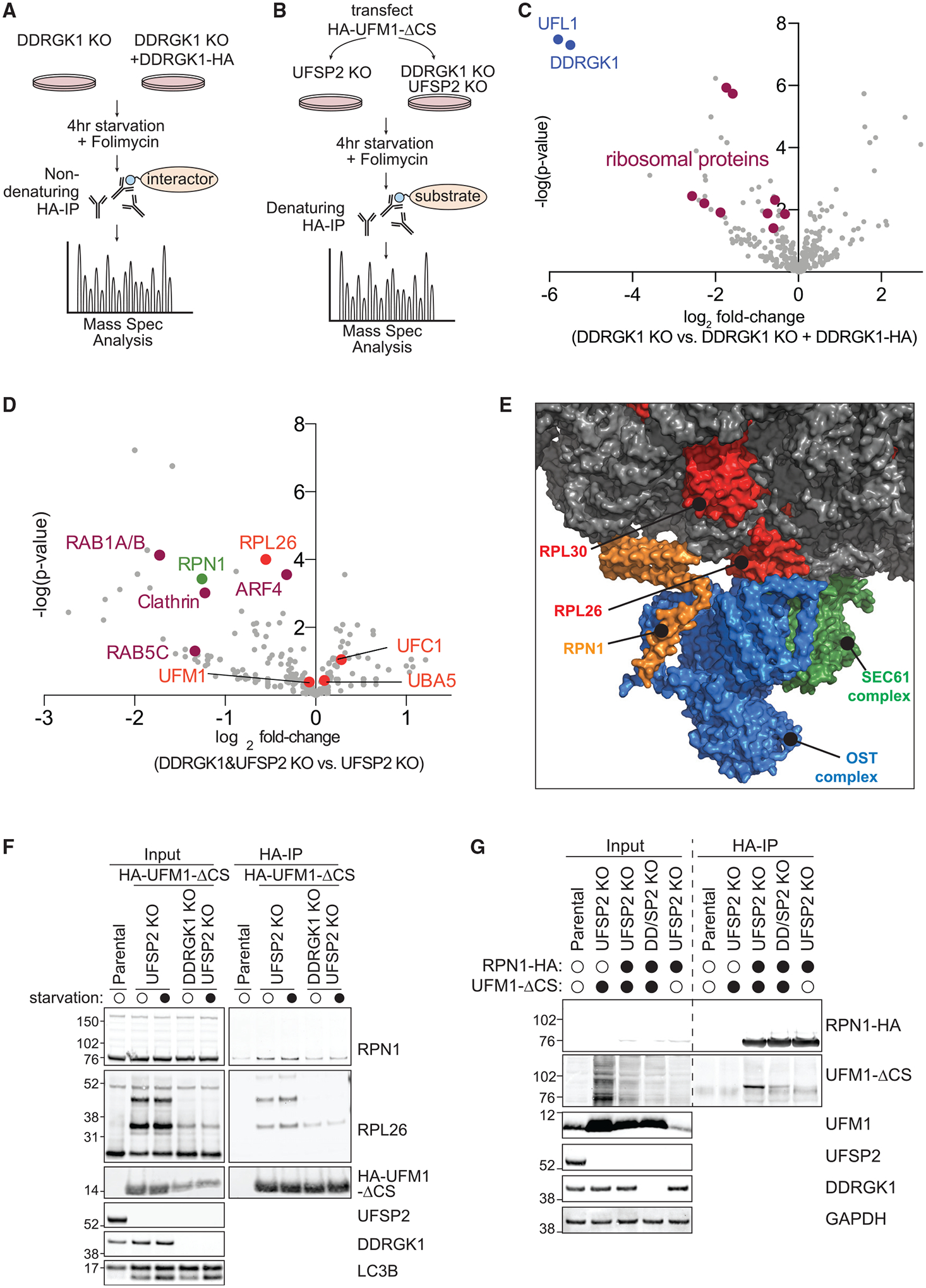
(A) Workflow of mass spectrometry identification of DDRGK1 interactome. DDRGK1KO HEK293T cells ± DDRGK1-HA stable expression were starved for 4 h in the presence of 50 nM folimycin. Cell lysates were harvested for HA immunoprecipitation and mass spectrometry identification of co-immunoprecipitated proteins.
(B) Workflow of mass spectrometry identification of DDRGK1-dependent UFMylation substrates. UFSP2KO or DDRGK1 and UFSP2 double-knockout (KO) HEK293T cells were transfected with HA-UFM1-ΔCS for 48 h. Cells were starved for 4 h in the presence of 50 nM folimycin. The cells were then lysed and denatured prior to HA immunoprecipitation and mass spectrometry identification of UFMylated proteins.
(C) DDRGK1 interacts with several ribosomal subunit proteins. The volcano plot depicts the log2 fold change of the total peptide count of each identified protein between Ctrl (DDRGK1KO) cells and DDRGK1-HA-expressing cells.
(D) Selective enrichment of UFMylated proteins in UFSP2KO cells relative to DDRGK1 and UFSP2 double-KO cells. The volcano plot depicts the log2 fold change of the total peptide count of each identified protein between DDRGK1 and UFSP2 KO versus UFSP2KO cells.
(E) RPN1 is structurally in close proximity with RPL26. Structural model of the ribosome, oligosaccharide transferase (OST), and SEC61 complex generated from Protein Data Bank deposition 6FTG using PyMol (Braunger et al., 2018). RPN1 (orange) is part of the ER-localized OST complex (blue), whereas RPL26 (red) is a component of the large 60S ribosomal subunit (gray). The OST complex and the ribosome are also closely associated with the SEC61 translocon complex (green).
(F) RPL26 and RPN1 are both UFMylated in a DDRGK1-dependent manner. The same experimental setup as in (B) was performed to probe for the indicated proteins. Note that the size shift corresponding to UFMylated RPN1 is not obvious due to the use of MES buffer that better resolves smaller molecular weight proteins, in this case, RPL26.
(G) Reverse immunoprecipitation of RPN1-HA showed DDRGK1-dependent UFMylation of RPN1. The same cell lines as in (B) were transfected with the indicated combinations of RPN1-HA and/or UFM1-ΔCS for 24 h. Cells were then lysed for immunoprecipitation of HA epitope. Samples were resolved using MOPS buffer for better molecular weight separation between unmodified and UFMylated RPN1 proteins.
See also Figure S6.
The top differentially enriched proteins from DDRGK1 immunoprecipitation in cells lacking or expressing DDRGK1 were DDRGK1 itself and UFL1 (Figure 6C; Table S3). We also identified several large ribosomal subunits and ribosome-associated factors, including RPL7A, RPLP0, RPL10A, RPL30, and RPL19. This is consistent with recent reports that the ribosome interacts with and is modified by the UFMylation machinery (Simsek et al., 2017; Walczak et al., 2019).
The top UFMylated proteins in both UFSP2 knockout and DDRGK1/UFSP2 double-knockout cells were UFM1 itself, UBA5, and UFC1; UFL1 and DDRGK1 were not identified (Figure 6D; Table S4). These data confirm that this experiment monitors UFMylation, since UFM1 forms a covalent bond with UBA5 and UFC1 but non-covalent interactions with UFL1 and DDRGK1 (Table S4) (Komatsu et al., 2004; Tatsumi et al., 2010). Comparing UFMylation in UFSP2 knockout cells to DDRGK1/UFSP2 double-knockout cells, we identified RPL26, which is a recently reported substrate of DDRGK1-mediated UFMylation (Figure 6D) (Walczak et al., 2019; Wang et al., 2020). Several factors involved in membrane trafficking between organelles and endosomes were also UFMylated in a DDRGK1-dependent manner, such as RAB1A/B, RAB5C, ARF4, and Clathrin. We further identified Ribophorin1 (RPN1), which was notable for several reasons. First, RPN1 is an ER-resident quality-control factor present on ER sheets that is part of the oligosaccharyltransferase (OST) complex (Figure S6I) (Kelleher et al., 1992). Second, RPN1 contains a significant cytoplasmic domain that could potentially be accessible for UFMylation during ER-phagy (Figure 6E) (Tsao et al., 1992). Third, RPN1 and the rest of the OST are associated with the SEC61/62/63 translocon complex (Yan and Lennarz, 2005). SEC62 has been identified as an ER-phagy receptor, and we found that that SEC62 is epistatic with DDRGK1 during ER-phagy (Figure 5I) (Fumagalli et al., 2016). And fourth, the SEC61/62/63 translocon complex and OST associate with the ribosome during co-translational folding, where they are structurally located immediately adjacent to RPL26 and large ribosomal subunit proteins that we found to interact with DDRGK1 such as RPL30 (Figure 6E) (Braunger et al., 2018). RPN1 and RPL26 are both “common essential” genes, required for the survival of every cell line tested by Dep-Map. The raw guide RNA counts for RPN1 and RPL26 are very low in all populations of the CRISPRi ER-phagy screen, explaining why neither appeared in the high-confidence hit list. Using immunoprecipitation and western blotting, we validated that both RPL26 and RPN1 are UFMylated and this UFMylation is abrogated by knockout of DDRGK1 (Figures 6F and 6G).
DDRGK1-Dependent UFMylation Facilitates ER-Phagy and Represses UPR
IRE1α is an ER stress sensor and was previously reported as a substrate of UFMylation to promote the UPR, but we found no evidence for DDRGK1-mediated UFMylation nor stabilization of IRE1a during ER-phagy by either IP-MS/MS or IP-western blot (Figure 6B; Table S4; Figure S7A) (Liu et al., 2017). Instead, we conversely found that depletion of DDRGK1, UFL1, or UFM1 (HepG2, MCF7 and HeLa cells) resulted in elevated protein levels of IRE1a in multiple cell types (Figures 7A and S7B–S7D). These results suggested that an inability to perform UFMylation-dependent ER-phagy could lead to upregulation of an ER stress response through IRE1α, which senses misfolded proteins in the ER lumen. Indeed, knockdown of UFMylation ER-phagy fac tors led to elevated levels of several other UPR proteins including PERK, BiP, and CANX (Figures 7A and S7B). We also observed an increase in levels of CLIMP63 (ER sheet marker) and REEP5 (ER tubule marker), suggesting possible ER expansion (Figures 7A and S7B) (Schuck et al., 2009). Consistent with this idea, immunofluorescence of DDRGK1 CRISPR-Cas9 knockout HeLa cells showed increased CANX staining (Figures S7D and S7E). Knockdown of DDRGK1 or UFL1 led to modest but consistent transcriptional upregulation of the UPR transcripts PERK and BiP, increased differential splicing of XBP1, and upregulation of CLIMP63 and REEP5 (Figure 7B). As opposed to the other ER stress markers, IRE1α showed higher protein levels upon DDRGK1 knockdown but no change in transcript abundance (Figures 7A and 7B), indicating that IRE1α protein levels could be post-translationally regulated in response to UFM1 signaling. The transcriptional upregulation of multiple UPR transcripts, differential splicing of XBP1, post-translational upregulation of IRE1α, and ER expansion are all consistent with increased ER stress and consequent UPR under conditions where UFMylation-dependent ER-phagy cannot be executed. The upregulation of ER stress markers upon DDRGK1 or UFL1 knockdown are weaker than during acutely toxic tunicamycin treatment, suggesting that the disruption of the UFMylation-mediated ER-phagy represents a chronic, survivable ER stress (Figure 7B).
Figure 7. UFMylation-Mediated ER-phagy Represses IRE1α UPR.
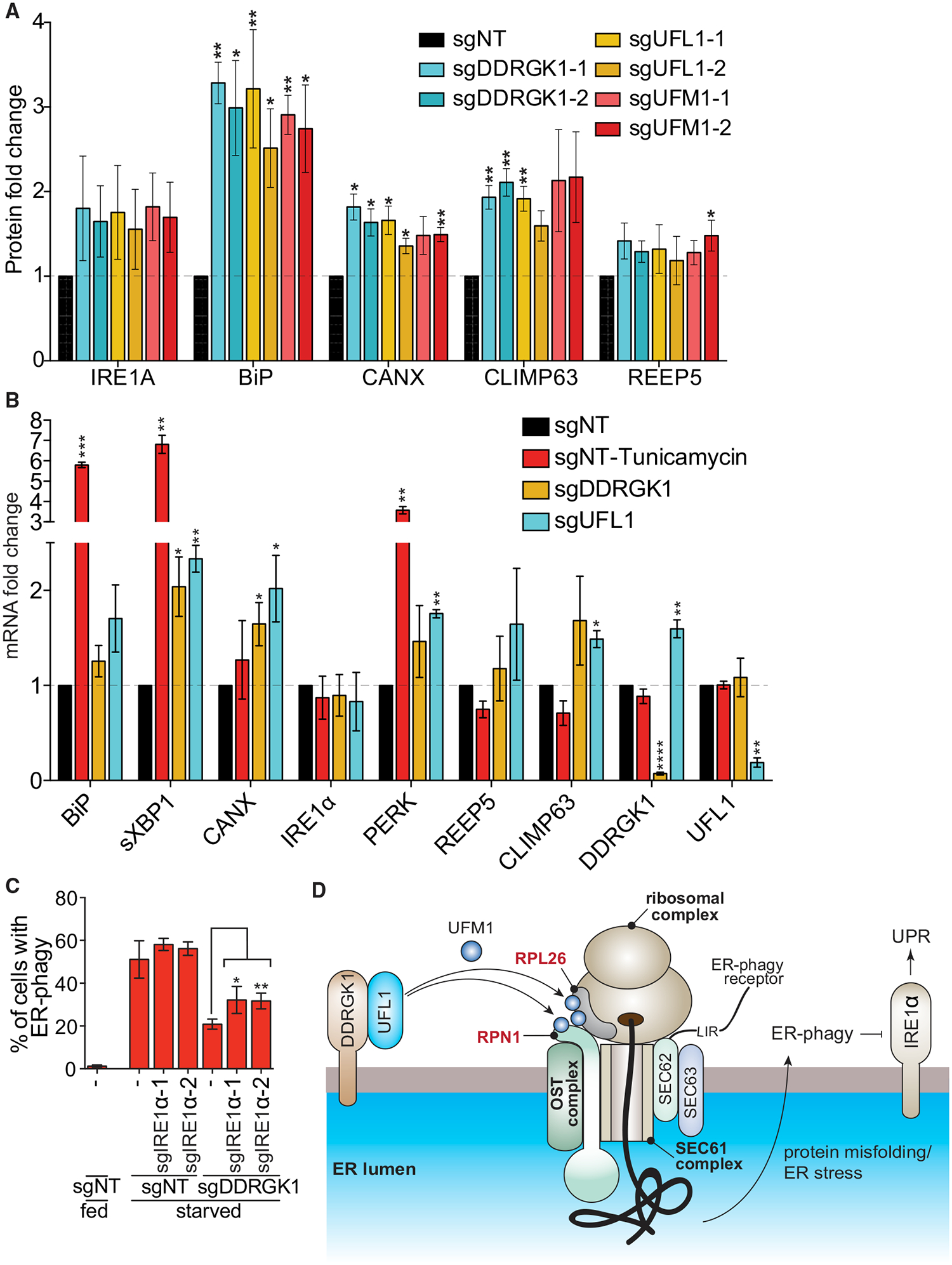
(A) Dysregulation of UFMylation results in upregulation of UPR. HCT116 cells were transduced with the indicated sgRNAs and harvested for western blotting analysis. The graph represents densitometry measurement of the indicated proteins upon sgRNA knockdown. A representative blot is shown in Figure S7B. Data represent mean ± SD of three biological replicates.
(B) Dysregulation of UFMylation transcriptionally upregulates UPR markers except IRE1 α. HCT116 CRISPRi cells were transduced with the indicated sgRNAs. Tunicamyin (0.5 μg/mL; 4 h) was used as a positive control for ER stress. Cells were harvested for qRT-PCR measurement of the indicated ER or UPR genes. Data represent mean ± SD of three biological replicates.
(C) Knockdown of IRE1a partially restores ER-phagy in DDRGK1-depleted cells. HCT116 CRISPRi EATR cells transduced with the indicated sgRNAs and starved for 16 h before FACS measurement for ER-phagy. Data represent mean ± SD of three biological replicates.
(D) Proposed model for the role of UFMylation during ER-phagy. DDRGK1 acts as an ER surface adaptor that recruits UFL1. At least two ER surface proteins that are in close proximity, RPN1 and RPL26, are UFMylated during ER-phagy. Dysregulation of UFMylation inhibits ER-phagy, and this potentially results in accumulation of ER stress and subsequently activates the IRE1 a-mediated unfolded protein response pathway.
See also Figure S7.
IRE1α senses unfolded proteins in the ER lumen and so is a good candidate to mediate stress signals caused by defective ER-phagy. While we did not observe direct UFMylation of IRE1α under either fed or starved conditions (Figure S7A), knockdown of IRE1α in DDRGK1-depleted cells reversed the high levels of UPR markers downstream of IRE1α caused by an inability to execute ER-phagy (Figure S7F). BiP, which is an upstream factor of IRE1α signaling, was upregulated in DDRGK1-depleted cells but was not affected by IRE1α depletion (Figure S7F) (Amin-Wetzel et al., 2017; Oikawa et al., 2009). Knockdown of IRE1α had only a modest reciprocal effect upon ER-phagy, and only somewhat reversed the ER-phagy defect induced by loss of DDRGK1 (Figure 7C). Hence, UFMylation-induced ER-phagy is upstream of IREα signaling.
Overall, our data indicate that DDRGK1-mediated, ER-resident UFMylation is critical for ER-phagy. DDRGK1 recruits UFL1 to promote ER surface UFMylation. We propose that the inability to UFMylate downstream ER substrates such as RPN1 and RPL26 leads to an inability to execute ER-phagy, resulting in the consequent build-up of ER stress, and eventual activation of the unfolded protein response via IRE1α (Figure 7D).
DISCUSSION
Our genome-wide ER-phagy screen provides a rich set of genes and pathways that greatly expands our understanding of ER-phagy and provides many starting points for further investigation. Among these, we identified several aspects of the core autophagy machinery, consistent with studies showing that ER-phagy shares effectors with general autophagy (Grumati et al., 2017; Khaminets et al., 2015; Smith et al., 2018).
So far, nutrient starvation is the only stress known to directly induce ER-phagy. We found that ER stress-inducing compounds do not lead to ER-phagy, but repression of ER-phagy by knockdown of DDRGK1 UFMylation induces ER stress and the UPR. UFMylation has previously been linked to ER stress through unclear mechanisms (DeJesus et al., 2016; Leto et al., 2019; Walczak et al., 2019), and our data suggest that this is connected to the regulation of ER-phagy. How is it that ER stress does not induce ER-phagy but an inability to perform ER-phagy induces ER stress? Under nutrient depletion, protein misfolding may increase in the ER, but these signals are repressed as cells catabolize the protein- and lipid-rich organelle. Blocking ER-phagy could then result in the toxic accumulation of excessive ER and misfolded ER-resident proteins that cannot be sufficiently kept in check by ERAD, thus activating the UPR. Under this model, blocking ER-phagy leads to UPR as a byproduct of ER stress that is no longer relieved by eating portions of the ER. This hypothesis and ordered prioritization of ER stress-relief pathways will require a great deal of investigation but could lead to a molecular rationale for why cells go to the extreme of ER-phagy.
We found extensive interplay between the mitochondria and ER-phagy. The ER and mitochondria are known to crosstalk at membrane contact sites, including transfer and expansion of the lipid bilayer, Ca2+ homeostasis, and mitochondria division (Friedman et al., 2011; Lombardi and Elrod, 2017). Previous studies indicate a unidirectional regulatory role of ER processes toward mitochondrial homeostasis. We found that impairment of mitochondrial OXPHOS represses ER-phagy, demonstrating that mitochondrial metabolism can also inform decisions in the ER. It still remains to be seen whether this communication is directly orchestrated via mitochondria-ER contacts or indirectly as a result of metabolic products. Alternatively, inhibition of OXPHOS could initiate UPR that takes over to repress last-resort ER-phagy. Consistently, mitochondrial dysfunction was reported to trigger the integrated stress response (ISR) which converges with the UPR pathway, further highlighting the complex cross-talk between the two organelles (Guo et al., 2019). Cellular energy levels are regulated by multiple energy sensing mechanisms that have complex roles during general autophagy (Egan et al., 2011; Herzig and Shaw, 2018; Kim et al., 2011), and the interplay between mitochondrial metabolism and ER homeostasis will no doubt involve a rich set of pathways for future investigation.
UFMylation have been implicated in DNA repair, transcriptional regulation, and ribosomal modification, all of which occur in different subcellular compartments (Egunsola et al., 2017; Qin et al., 2019; Walczak et al., 2019; Wang et al., 2019). It has been unclear how the mostly cytoplasmic UFL1 ligase accesses each compartment. We found that DDRGK1 recruits UFL1 to the ER surface for UFMylation-dependent ER-phagy. Post-translational modifications of organelle surfaces are widely involved in organelle autophagy. For example, ubiquitylation of PEX5 serves as a signal for peroxisomal autophagy (Nordgren et al., 2015; Zhang et al., 2015a), and ubiquitylation of multiple mitochondrial substrates promotes mitophagy (Chan et al., 2011; Karbowski and Youle, 2011). Since DDRGK1 recruits UFL1 to the ER surface and their combined ER-resident activity with UFM1 are required for ER-phagy, we speculate that UFMylation of ER surface protein(s) serves as an effector of ER-phagy, similar to PINK1’s recruitment of Parkin to ubiquitylate mitochondrial surface proteins during mitophagy (Chan et al., 2011; Glauser et al., 2011; Karbowski and Youle, 2011; Wang et al., 2011).
We identified RPN1, a subunit of the ER-localized OST complex, as a novel DDRGK1-dependent UFMylation substrate. For several reasons, we propose that RPN1 is part of a UFMylation “hub” that participates in ER-resident quality control. The OST complex associates with ribosomes and the SEC61/62/63 complex for nascent protein translocation and maturation (Braunger et al., 2018; Kelleher et al., 1992; Shibata et al., 2010; Wilson et al., 2005; Yan and Lennarz, 2005). RPL26 is a known UFMylation substrate within the 60S large ribosomal subunit and sits in in close proximity with RPN1 (Figures 6E and 7D) (Walczak et al., 2019). DDRGK1 also physically interacts with multiple components of the large 60S ribosomal subunits that are nearby RPN1.
The downstream parts played by UFMylated RPN1 during ER marking, autophagic engulfment, and degradation remain to be determined. Dysregulation of glycosylation by interfering with the OST complex and inhibition of UFMylation both cause ER stress (Cherepanova et al., 2016; Walczak et al., 2019). The SEC62 ER-stress activated ER-phagy receptor is part of the translocon complex directly adjacent to the OST complex, and we found DDRGK1 to be epistatic to SEC62 during ER-phagy (Fumagalli et al., 2016; Kelleher et al., 1992; Tsao et al., 1992; Walczak et al., 2019; Wilson et al., 2005). Since RPN1 contains an N-terminal cytoplasmic domain and a C-terminal ER lumenal domain, it is possible that RPN1 might relay ER stress signals via ER-surface UFMylation during peptide translocation. Failure to relay and appropriately deal with these signals on the ribosome-rich ER sheets could induce the IRE1α-mediated UPR induced by blockage of DDRGK1-dependent UFMylation. Nascent proteins trapped in the ribosome induces RPL26 UFMylation and lysosomal degradation of the stalled protein and ribosome en bloc (Wang et al., 2020). It is currently unknown whether these UFMylated ribosomes are physically extracted from the ER prior to lysosomal degradation, or if portions of the ER are degraded alongside the UFMylated ribosomes. Importantly, RPN1 and RPL26 might not be the exclusive targets of UFMylation and simultaneous UFMylation of other ER surface proteins could be required to drive ER-phagy in a cooperative manner. Overall, our data unify disparate reports of UFMylation impacting the UPR and provide a mechanistic rationale for how autophagy of ER sheets helps to prevent accumulation of ER stress. This opens up a great deal of further avenues to dissect the subsequent steps by which UFMylation is recognized on the ER surface to mediate ER-phagy.
While defects in ER-phagy have not been explicitly linked to human disease, human mutations in ER-phagy genes such as FAM134B and Atlastins are associated with hereditary neuropathies in OMIM and ClinVar (Abel et al., 2004; Amberger et al., 2015; Kurth et al., 2009). Several ER-phagy genes derived from our screen are also associated with human neurodegenerative phenotypes with previously unclear mechanistic bases, such as Leigh syndrome (mitochondrial OXPHOS, including ETC chaperones) (Lake et al., 2016), spastic paraplegia (ARL6IP1) (Novarino et al., 2014), encephalopathy (TRAPPC12) (Milev et al., 2017), spinocerebellar ataxia and encephalopathy (UBA5) (Daida et al., 2018; Mignon-Ravix et al., 2018), and severe early-onset encephalopathy and progressive microcephaly (UFC1, UFM1) (Nahorski et al., 2018). It is premature to broadly link deficits in ER-phagy to human disease, but the similar phenotypes stemming from mutations in various ER-phagy factors are provocative. Our work lays the foundation for future understanding of ER-phagy and its interplay with the ER stress response, as well as the consequences of ineffective ER-phagy. Further mechanistic dissection of the 200 high-confidence ER-phagy regulators and executors identified here will hopefully shed light on this dramatic process.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
The plasmids generated in this study have been deposited to Addgene (catalog number indicated in Key Resources Table). In the case where the same cDNA with multiple epitope/fluorescence tags are used, only one version is deposited to Addgene. All remaining unique/stable reagents generated in this study are available from the Lead Contact, Jacob Corn (jacob.corn@biol.ethz.ch) with a complete Materials Transfer Agreement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ACC (Acetyl-CoA Carboxylase) | Cell Signaling | Cat#: 3676; RRID: AB_2219397 |
| Actin | Cell Signaling | Cat#: 3700; RRID: AB_2242334 |
| AMPKa | Cell Signaling | Cat#: 5831; RRID: AB_10622186 |
| ATG10 | MBL International | Cat#: M151-3; RRID: AB_1278755 |
| ATP5O | Abcam | Cat#: ab110276; RRID: AB_10887942 |
| BiP | Cell Signaling | Cat#: 3177; RRID: AB_2119845 |
| Calnexin (CANX) | Cell Signaling | Cat#: 2679; RRID: AB_2228381 |
| Calnexin (CANX) | Santa Cruz | Cat#: sc-46669; RRID: AB_626784 |
| CKAP4/ CLIMP63 | Bethyl | Cat#: A302-257A; RRID: AB_1731083 |
| DDRGK1 | ProteinTech | Cat#: 21445-1-AP; RRID: AB_2827383 |
| GAPDH | Cell Signaling | Cat#: #97166; RRID: AB_2756824 |
| GFP | Abcam | Cat#: ab6556; RRID: AB_305564 |
| GFP | Santa Cruz | Cat#: sc-9996; RRID: AB_627695 |
| GST | Cell Signaling | Cat#: 2625; RRID: AB_490796 |
| Ha epitope tag | Cell Signaling | Cat#: 3724; RRID: AB_1549585 |
| IRE1a | Cell Signaling | Cat#: 3294; RRID: AB_823545 |
| LC3B | Novus Biologicals | Cat#: NB100-2220 |
| mCherry | Abcam | Cat#: ab183628; RRID: AB_2650480 |
| MFN1 | Cell Signaling | Cat#: 14739; RRID: AB_2744531 |
| MFN2 | Cell Signaling | Cat#: 11925; RRID: AB_2750893 |
| NDUFB2 | Abcam | Cat#: ab186748; RRID: AB_2827382 |
| NDUFB4 | Abcam | Cat#: ab110243; RRID: AB_10890994 |
| p62 | Santa Cruz | Cat#: sc-28359; RRID: AB_628279 |
| pACC S79 | Cell Signaling | Cat#: 11818; RRID: AB_2687505 |
| PERK | Cell Signaling | Cat#: 3192; RRID: AB_2095847 |
| pRaptor S792 | Cell Signaling | Cat#: 2083; RRID: AB_2249475 |
| pS6K T389 | Cell Signaling | Cat#: 9206; RRID: AB_2285392 |
| pULK1 S555 | Cell Signaling | Cat#: 5869; RRID: AB_10707365 |
| Raptor | Cell Signaling | Cat#: 2280; RRID: AB_561245 |
| REEP5 | ProteinTech | Cat#: 14643-1-AP; RRID: AB_2178440 |
| S6K (p70 S6 Kinase) | Cell Signaling | Cat#: 9202; RRID: AB_331676 |
| TOM20 | Sigma | Cat#: HPA011562; RRID: AB_1080326 |
| UFL1 | Novus Biologicals | Cat#: NBP1-90691; RRID: AB_11040102 |
| UFM1 | Abcam | Cat#: ab109305; RRID: AB_10864675 |
| ULK1 | Cell Signaling | Cat#: #8054; RRID: AB_11178668 |
| UFSP2 (G-11) | Santa Cruz | Cat#: sc-376084; RRID: AB_10989729 |
| RPL26 | Abcam | Cat#: ab59567; RRID: AB_945306 |
| Ribophorin1 (RPN1) | ThermoFisher | Cat#: PA5-27562; RRID: AB_2545038 |
| IRDye 800CW Goat anti-Rabbit IgG H+L | LI-COR | 925-32211; RRID: AB_2651127 |
| IRDye 680 RD Goat anti-Mouse IgG H+L | LI-COR | 926-68070; RRID: AB_10956588 |
| Chemicals, Peptides and Recombinant Proteins | ||
| Torin1 | CST | Cat#: 14379S |
| Rapamycin | Sigma | Cat#: R8781 |
| tunicamycin | Sigma | Cat#: T7765 |
| Thapsigargin | Sigma | Cat#: T9033 |
| CCCP | Sigma | Cat#: C2759 |
| Rotenone | Sigma | Cat#: R8875 |
| Oligomycin A | Sigma | Cat#: 75351 |
| antimycin A | Sigma | Cat#: A8674 |
| Folimycin | Milipore | Cat#: 344085 |
| Epoxomycin | Milipore | Cat#: 324800 |
| EBSS | ThermoFisher | Cat#: 24010043 |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat#: 103015-100 |
| Cell Titer-Glo 2.0 Cell Viability Assay | Promega | Cat#: G9241 |
| Pierce anti-HA magnetic beads | ThermoFisher Scientific | Cat#: 88836 |
| Deposited Data | ||
| CRISPRi screen data (pilot autophagy gene screen and genome-wide screen) | NCBI-SRA | PRJNA599329 |
| Experimental Models: Cell Lines | ||
| HCT116 CRISPRi | Liang et al., 2018 | N/A |
| HCT116 CRISPRi mCherry-eGFP-RAMP4 (EATR) | Liang et al., 2018 | N/A |
| HCT116 CRISPRi mcherry-RAMP4 (CCER) | Liang et al., 2018 | N/A |
| HCT116 CRISPRi REEP5-mCherry-eGFP | This study | N/A |
| HCT116 CRISPRi CLIMP63-mCherry-eGFP | This study | N/A |
| HCT116 mCherry-KDEL | This study | N/A |
| HEK293T UFSP2 KO | This study | N/A |
| HEK293T UFSP2 and DDRGK1 double KO | This study | N/A |
| HCT116 AMPK KO | This study | N/A |
| HeLa DDRGK1 KO | This study | N/A |
| Oligonucleotides | ||
| shNon-targeting sequence- CCTAAGGTTAAGTCGCCCTCG | Liang et al., 2018 | N/A |
| shDDRGK1 sequence - GGCTCTGCTAGTCGGCTTTAT | This study | N/A |
| shUFL1 sequence - GCTTCTTTACTCTGTGCTTGA | Zhang et al., 2015b | N/A |
| Protospacer sequences for all CRISPR KO or CRISPRi experiments | See Table S5 | |
| qPCR primer sequences | See Table S6 | |
| Recombinant DNA | ||
| For more recombinant DNA data, see also Table S6 | This study | N/A |
| pEF1a-dCas9-HA-BFP-KRAB-NLS | Liang et al., 2018 | Addgene 102244 |
| TetOn-mCherry-eGFP-RAMP4 | Liang et al., 2018 | Addgene109014 |
| pLenti-X1-Hygro-mCherry-RAMP4 | Liang et al., 2018 | Addgene 118391 |
| mCherry-mito-7 | Olenych et al., 2007 | Addgene 55102 |
| mCherry-ER-3 | Olenych et al., 2007 | Addgene 55041 |
| mCherry-Peroxisomes-2 | Olenych et al., 2007 | Addgene 54520 |
| pRK5-rLAMP1-BFP | This study | LAMP1 from Rattus norvegicus subcloned from Addgene 55073 |
| pLenti-X1-Neo-NDUFB4 | This study | Addgene 139840 |
| pLenti-X1-Neo-NDUFB2 | This study | Addgene 139841 |
| pLenti-X1-Neo-ATP5O | This study | Addgene139842 |
| pLenti-XI-Neo-GST-Constitutively Active AMPK | Egan et al., 2011 | Addgene139843 |
| pLenti-XI-Neo-GST- AMPK- Kinase Dead (K to R) | This study | Addgene 139844 |
| pBMN-YFP-Parkin | Yamano et al., 2014 | Addgene 59416 |
| pLenti-X1-Neo-DDRGK1-WT-HA | This study | Addgene 139845 |
| pLenti-X1-Neo-DDRGK1-dTM-HA | This study | Addgene 139846 |
| pLenti-X1-Neo-DDRGK1-dPCI-HA | This study | Addgene 139847 |
| pLenti-X1-Neo-DDRGK1-K267R-HA | This study | Addgene139848 |
| pLenti-X1-Neo-DDRGK1-K-less-HA | This study | Addgene 139849, with K116, K120, K121, K124, K128, K146, K176, K193, K224 & K227 mutated to R |
| pLenti-X1-Neo-DDRGK1-K116R-HA | This study | Addgene139850 |
| pLenti-X1-Neo-DDRGK1-K120R-HA | This study | Addgene139851 |
| pLenti-X1-Neo-DDRGK1-K121R-HA | This study | Addgene 139852 |
| pLenti-X1-Neo-DDRGK1-K124R-HA | This study | Addgene139853 |
| pLenti-X1-Neo-DDRGK1-K128R-HA | This study | Addgene139854 |
| pLenti-X1-Neo-DDRGK1-K146R-HA | This study | Addgene139855 |
| pLenti-X1-Neo-DDRGK1-K176R-HA | This study | Addgene139856 |
| pLenti-X1-Neo-DDRGK1-K193R-HA | This study | Addgene139857 |
| pLenti-X1-Neo-DDRGK1-K224R-HA | This study | Addgene139858 |
| pLenti-X1-Neo-DDRGK1-K227R-HA | This study | Addgene139859 |
| pLenti-X1-Neo-HA-UFL1 | This study | Addgene139860 |
| pQCXIN- myc ULK1 wt | Egan et al., 2011 | Addgene 27626 |
| pLenti-X1-Neo-TOM20MTS-DDRGK1-dTM-HA | This study | Addgene 139861 |
| pLenti-X1-Neo-PMP34-DDRGK1-dTM-HA | This study | Addgene 139862 |
| pLenti-X1-Neo-HA-UFL1-RAMP4 | This study | Addgene139863 |
| pRK5-HA-UFM1-dCS | This study | Addgene 139869 |
| prk5-UFM1-dCS-no tag | This study | cDNA of HCT116 cells, NM_016617.4, deleted C-terminal a.a.84–85 |
| TetOn-REEP5-mCherry-eGFP | This study | Addgene 139870 |
| TetOn-CLIMP63-mCherry-eGFP | This study | Addgene139871 |
| pLenti-X1-Neo-HA-FAM134B | This study | subcloned from Addgene 109026 |
| pLenti-X1-Neo-HA-RTN3L | This study | Addgene139864 |
| pLenti-X1-Neo-HA-SEC62 | This study | Addgene139865 |
| pLenti-X1-Neo-HA-ATL3 | This study | subcloned from Addgene 109024 |
| pLenti-X1-Neo-TEX264-HA | This study | Addgene139866 |
| pLenti-X1-Neo-HA-CCPG1 | This study | Addgene139867 |
| pLenti-X1-Neo-mCherry-FAM134B | This study | subcloned from Addgene 109026 |
| pLenti-X1-Neo-mCherry-RTN3L | This study | cDNA of HCT116 cells, NM_001265589.1 |
| pLenti-X1-Neo-mCherry-SEC62 | This study | cDNA of HCT116 cells, NM_003262.4 |
| pLenti-X1-Neo-mCherry-ATL3 | This study | subcloned from Addgene 109024 |
| pLenti-X1-Neo-TEX264-mCherry | This study | cDNA of HCT116 cells, NM_015926.6 |
| pLenti-X1-Neo-mCherry-CCPG1 | This study | cDNA of HCT116 cells, NM_001204450.1 |
| plenti-X1-Neo-RPN1-HA | This study | cDNA of HCT116 cells, NM_002950.4 |
| pLenti-X1-Neo-RPN1-GFP | This study | cDNA of HCT116 cells, NM_002950.4 |
| Software and Algorithms | ||
| Li-Cor’s ImageStudio software V5.2 | Li-Cor | ■ ■ ■ |
| ImageJ | NIH | ■ ■ ■ |
| Adobe Photoshop CS6 | Adobe Systems | ■ ■ ■ |
| Adobe Illustrator CS6 | Adobe Systems | ■ ■ ■ |
| Prism 6 | GraphPad | ■ ■ ■ |
METHOD DETAILS
Design, Production and titering of sgRNA library lentivirus
The genome-wide CRISPRi-V2 library was a gift from the Weismann lab (Addgene catalog #1000000093) and contains 5 sgRNAs per gene. For the pilot autophagy screen, we designed a comprehensive sgRNA library that targets all the reported TSS (10 gRNAs per TSS) of 31 genes that are involved in general autophagy. Overall, a total of 3301 gRNAs were designed (Table S1). The protospacer oligos were annealed and ligated to pCRISPRia vector (Addgene 84832) according to the protocol established by the Weissman lab (https://weissmanlab.ucsf.edu/CRISPR/Pooled_CRISPR_Library_Cloning.pdf) (Horlbeck et al., 2016). In addition, we added in 10% of a custom built non-targeting sgRNA library prior to virus production.
The following paragraph describes the transfection protocol for one 15 cm plate of HEK293T cells. On Day 0, 7.5 million HEK293T cells were seeded in a 15 cm plate in 20 mL of DMEM medium with 10% FBS. The following day HEK293T cells were transfected. In a 15 mL tube, 2.8 mL of Opti-MEM was mixed with 90 μL of Mirus LT1 transfection reagent and incubated at room temperature for 5 minutes. In an eppendorf tube, 12 μg of delta VPR, 3 μg of VSVG, and 15 μg of library plasmid were combined. The plasmids were then added to the Opti-MEM and Mirus mixture and incubated at room temperature for 20 minutes. The media was changed the following day. On Day 3, the virus was harvested using a 0.45 mm syringe filter, aliquoted into 1 mL tubes, and snap frozen. If more than one 15 cm plate of virus was produced for one library, the virus across those plates were pooled and mixed prior to aliquoting into eppendorf tubes. Virus was harvest on Day 4 as well.
Next, the virus was titered to determine the infectivity of the virus in the HCT116 cells. HCT116 were plated in a series of 6 well plates such that each well had cells and there was one 6 well plate per sub-library per time point (i.e., 48 or 72 hour virus harvest). One well on each plate was not transduced with any virus. The virus was titered such that is diluted 2-fold, 4-fold, 8-fold, 16-fold, and 32-fold. Polybrene was used at a concentration of 8 mg/mL. Fresh media were replaced 24hr post transduction. The cells were harvested 48 hours post viral transduction for flow cytometry and the percentage of BFP positive cells was recorded. The optimal virus dilution is defined as dilution-fold that results in less than 20% of BFP positive cells.
CRISPRi screen: cell generation, virus transduction, puro selection, and sort
HCT116 cells stably expressing a dcas9-KRAB and doxycycline-inducible EATR reporter was generated by lentiviral transduction of Addgene constructs 102244 and 109014 (Liang et al., 2018). The library contained seven unique sub-libraries and each sub-library was transduced separately, such that each sgRNA had an average of 500x coverage after transduction (Day 1). Puromycin selection for positively-transduced cells was performed 48 hours post transduction (Day 3). On Day 7, the sub-libraries were pooled proportionally based on the numberof sgRNAs and cells were maintained at 500x coverage. On Day 10, cells were treated with doxycycline (4μg/ml) for 16 hours to induce EATR expression and on Day11, cells were treated with EBSS for 16 hours. Cells were then collected for sorting - cells were gated into the 25% of cells with most ER-phagy and 25% of cells with the least ER-phagy. A background population of cells was collected for downstream NGS analysis of relative enrichment.The entire CRISPRi screen was performed in two biological replicates.
NGS Sample Preparation and screen analysis
Genomic DNA was harvested using the Macherey-Nagel gDNA extraction protocol. The background samples required the XL kit whereas the midi kit was sufficient for sorted cells. After elution, the genomic DNA was treated with SbfI-HI restriction enzyme and incubated overnight at 37°C to liberate the DNA fragment encoding the sgRNA sequences.
Samples were run an agarose gel and the gel piece around the 500 bp size (region containing the sgRNA sequence) was excised. The gel was melted in 55°C water bath and 1/100 by volume of 3 M NaAc (pH 5.2) was added to each tube and then solution was passed through an MN column. Each column was washed twice with NT3 buffer. The column was incubated for 5 minutes in 20 μL of heated elution buffer (98°C) and then spun. The elution step was repeated so that the final elution volume was 40 μL.
A standard PCR protocol was used with Phusion High Fidelity Enzyme and 3% DMSO final concentration. The forward primer contained a TruSeq Index that would be subsequently used during NGS analysis. Before proceeding with a full scale PCR of the samples, a test PCR for each sample was run to determine the proper number of cycles (21, 23, or 25 cycles). The cycle number was identified individually for each sample that allowed a visible band on a TBE gel after staining with ethidium bromide, but not an oversaturated PCR product that could compromise the representation of gRNAs within the sample.
After the optimal cycle number was determine, a total of twelve 100 μL PCRs were done with 3 μL of template per reaction (from the abovementioned elution). The forward primer contained a TruSeq Index that would be subsequently used during NGS analysis. After completion of the PCR, the twelve reactions were pooled together and mixed. 300 μL of the pooled PCR was taken for subsequent PCR clean-up.
195 μL of SPRI beads was added to the pooled PCR and incubated at room temperature for 10 minutes. The samples were attached to a DynaMag for 5 minutes. The supernatant (which has the sample) was transferred to a new tube. 300 μL of SPRI beads were added and incubated for another 10 minutes. The samples were attached to a DynaMag for 5 minutes and the supernatant was discarded (samples attached to the beads). The beads were washed twice with 80% ethanol. After removal of the last supernatant, the beads were spun down, and excess ethanol was removed. The samples were air-dried for 10 minutes and resuspended in 35 μL of water. DNA concentration was quantified using Qubit Fluorometric Quantitation (Thermo Fisher Scientific) and the samples were pooled proportionally to cell number and sequenced on a HiSeq 2500 such that each sgRNA sequence was covered at least 30 times.
Screening data was analyzed using standard protocols in MaGECK and ScreenProcessing (Horlbeck et al., 2016; Li et al., 2014, 2015). MaGECK was used for the pilot autophagy library, while ScreenProcessing was used for the genome-wide library. Briefly, gRNAs were quantified in each pool of cells based by matching reads back to the appropriate library reference, each pool was normalized by total number of reads, and gRNA distributions were compared to the background. Non-targeting gRNAs were explicitly used in each software package. MaGECK and ScreenProcessing integrate multiple gRNAs into gene-level phenotypes (e.g., log2-fold-change) and p values using different approaches (Horlbeck et al., 2016; Li et al., 2014, 2015).
sgRNA plasmid cloning procedures for individual plasmids
The sgRNA sequences for genome-wide screening were based on the Weissman CRISPRi-v2 library and contained 5 sgRNAs per gene. The sgRNA sequences for autophagy-related genes used for the pilot-test run were custom-designed to target all reported transcription start site (TSS) of each gene and contained 10 sgRNAs per TSS. sgRNA plasmids were cloned by annealing and ligating sgRNA-containing short oligos to the CRISPRi-v2 vector (addgene 84832) via the previously described protocol (Horlbeck et al., 2016). Brriefly, the forward and reverse primers of each sgRNA were annealed by pre-incubation at 37°C for 30min in the presence of T4 polynucleotide kinase (PNK; NEB) followed by incubation at 95°C for 5 min and then ramp down to 25°C at 5°C /min. The annealed sgRNA inserts were then ligated to CRISPRia-v2 plasmid (digested using BstXI and BlpI) using Quick ligation kit (NEB). Knockdown efficiency of each guide was measured either by western blot or qRT-PCR. All sgRNA constructs used in this study are detailed in Table S3.
shRNA plasmid cloning for DDRGK1 and UFL1
Short hairpin RNA (shRNA) was used to knockdown DDRGK1 in cell lines that do not express dCas9-KRAB constructs. Non-targeting (5′-CCTAAGGTTAAGTCGCCCTCG-3′), DDRGK1-targeting (5′-GGCTCTGCTAGTCGGCTTTAT-3′) and UFL1-targeting (5′-GCTTCTTTACTCTGTGCTTGA-3′) shRNAs were cloned into pLKO.1 puro construct (Addgene #8453) according to protocol described in Addgene (https://www.addgene.org/protocols/plko/?gclid=Cj0KCQiAm5viBRD4ARIsADGUT25ZCGNPeQSFvLqSwvg2tHDkCc9zOZsLdaUffZzNTRYzI_YOlKFVQdUaAqbfEALw_wcB). Briefly, the shRNAs were cloned by standard annealing (same as sgRNA annealing protocol described in the previous section) and ligated to pLKO.1 construct (digested with AgeI-HF and EcoRI-HF).
cDNA plasmid cloning procedures
Unless stated otherwise, all ORFs described in this article were obtained from PCR amplification of HCT116 cDNA. The ORFs were cloned into pLenti-XI destination vector with neomycin resistance. Briefly, an original pLenti-X1-Neo-eGFP-LC3B vector was first digested with restriction enzymes BamHI and XbaI to remove the eGFP-LC3B insert. Then, Gibson Assembly was used to insert the gene-of-interest and the desired epitope or fluorescent tag into the pLenti-X1 vector (Gibson et al., 2009). All overexpression constructs used in this study are detailed in Table S4.
Cell culture
Cells were cultured at 37°C with 5% CO2 in a humidified atmosphere. All cells were cultured in DMEM-GlutaMAX medium supplemented with 10% FBS, 0.1 mM non-essential amino acids (GIBCO), 1 mM sodium pyruvate (GIBCO), 100 U/mL penicillin (GIBCO), and 100 g/mL streptomycin (GIBCO). Cell lines were obtained from the Berkeley Cell Culture Facility and were verified mycoplasma free with MycoAlert Mycoplasma Detection Kit (Lonza).
Cell Treatments
ER-phagy was induced with media starvation using EBSS with calcium, magnesium, and phenol red (Invitrogen e10043). For EATR and CCER assays, cells were plated 48 hours prior to EBSS treatment. EATR expression is induced using 4 μg/ml doxycycline 24hr prior to starvation. Unless otherwise stated, starvation treatment was carried out for 16 hours. Cells in fed conditions indicate incubation in complete DMEM described above.
For all experiments except the Seahorse assay, rotenone was used at a final concentration of 3 μM, antimycin A was used at a concentration of 0.5 μM, and oligomycin A was used at a concentration of 3 μM. Cells were treated with these drugs in two phases for a total of 40 hours. First, cells were treated for 24 hours with complete DMEM, then immediately treated again for 16 hours in EBSS media or complete DMEM. Unless stated otherwise, epoxomicin and folimycin treatments were co-administered with EBSS starvation at 100 nM final concentration.
Lentiviral packaging and transduction
Lentiviral packaging was performed in HEK293T cells using either TransIT-LT1 Transfection Reagent (Mirus) or Lipofectamine 3000 (ThermoFisher Scientific) according to the manufacturer’s protocol. Briefly, delta-VPR, VSVG, and the construct of interest were transfected at the ratio of 4:1:5. Lentiviral supernatant was harvested at 48hr post-transfection and HEK293T cells were replenished with fresh media for another harvest at 72hr post-transfection.
Knockout Cell Line Generation
AMPK knockout cell lines were generated using Cas9 RNPs and nucleofection as detailed previously (Lingeman et al., 2017). The sgRNA protospacer sequences were validated and used previously by the Shaw lab (Toyama et al., 2016). The protospacer sequences are as follow: AMPKα1-sgRNA1- GGCTGTCGCCATCTTTCTCC; AMPKα1-sgRNA2- GAAGATCGGCCACTACATTC; AMPKα2-sgRNA1- TCAGCCATCTTCGGCGCGCG; AMPKα2-sgRNA2- GAAGATCGGACACTACGTGC. After nucleofection, HCT116 cells were serial diluted into 96 well plates such that there was on average of 0.7 cells/well. AMPK KO clones were screened by western blotting. DDRGK1, UFL1 and UFSP2 knockout cell lines were using pSpCas9(BB)-2A-GFP (PX458) (Addgene #48138) plasmids carrying sgRNAs that targets the respective regions close to the transcription start site. For DDRGK1 and UFL1, two guides were used simultaneously to remove the transcription start sites. The knockout of UFSP2 was performed using only one guide based on reported sequence (Walczak et al., 2019). The protospacer sequences are as follow: DDRGK1-sgRNA1- ATGAGATCCCGGCCT CAGGG; DDRGK1-sgRNA2- TAGGAGATGCCGCTGCACCA; UFL1-sgRNA1- CTGACTCGCAGTAGACGCGG; UFL1-sgRNA2-GCCTAATT TGGGCTCCACAA; UFSP2-sgRNA1- AATAAGAGGAGGCCTTGATT. GFP-positive cells were single-cell sorted 48hr post transfection and knockout clones were screened by western blotting and selected clones were further validated by Sanger sequencing.
Flow Cytometry Analysis of EATR cells
Flow cytometry of EATR assay was performed using an Attune NxT Flow Cytometer and subsequent analysis was performed using FlowJo 10.1 (Liang et al., 2018). All EATR experiments were performed using live cells to prevent reversal of eGFP quenching post-fixation. The intensities for both eGFP and mCherry of the EATR cells at fed condition were used as references to define the gate for zero ER-phagy events. Following stimulation, ER-phagy detection is based on the shift of cell population into the ER-phagy gate. On average, 5 to 10,000 cells were analyzed per condition and all statistical analyses were performed using data from at least three biological replicates.
Quantitative real-time PCR (qRT-PCR)
RNA extraction was performed using Directzol RNA miniprep kit (Zymo Research) according to manufacturer’s instruction. 1μg of RNA per sample were used for reverse transcription using SuperScript III First-Strand Synthesis System (Invitrogen) according to manufacturer’s instruction. qRT-PCR reaction was set up using Fast SYBR Green Mastermix (Applied Biosystems) and run in triplicates using StepOne Plus Real-Time PCR system (Applied Biosystems). A complete list of all primers used are compiled in Table S6.
Western blotting
To prepare samples for western blot, cells were lysed in Radioimmunoprecipitation assay (RIPA) buffer (0.5M Tris-HCl, pH 7.4, 1.5M NaCl, 2.5% deoxycholic acid, 10% NP-40, 10mM EDTA), supplemented with Halt Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail (both ThermoFisher). Cells were lysed on ice for 10 minutes and spun at 14000 rpm for 10 minutes to remove insoluble debris. Protein concentrations were quantified by Bradford assay. Lysates were normalized based on protein concentration and NuPage LDS Sample Buffer (4x) supplemented with B-mercaptoethanol (5% v/v) was added (Invitrogen). Samples were boiled at 98°C for 5 minutes.
Between 20–40mg of samples were run on NuPAGE Bis-Tris 4%–12% gels in NuPage MES SDS Buffer (Invitrogen) for 40 minutes at 200 V and transferred to 0.4-μm nitrocellulose membranes using a semi-dry transfer system (Bio-Rad Catalog #1704150) at 1.3 A and 25 V for 15 minutes. After transfer, membranes were blocked with 5% (w/v) milk in Tris buffered saline containing 0.1% Tween 20 (TBS-T) for 30 minutes, and subsequently washed with TBS-T three times. Primary antibodies were diluted at the appropriate concentration in 5% BSA (w/v) in TBS-T. The membrane was incubated in primary antibody for either 1–2 hours at room temperature or overnight at 4°C. The membrane was washed with TBS-T three times for five minutes each. The blots were incubated for 30 minutes in the milk solution with a 1:10,000 dilution of Li-Cor near-infrared fluorescence secondary antibodies. The blots were scanned using Li-Cor’s Near-InfraRed fluorescence Odyssey CLx Imaging System, and densitometry quantifications were done using Li-Cor’s ImageStudio software complementary of Odyssey.
Immunoprecipitation
Unless specified otherwise, all immunoprecipitation experiments were performed in HCT116 cells stably-expressing the different HA-tagged UFMylation protein constructs using the Perice Anti-HA Magnetic beads Kits according to manufacturer’s protocol. Briefly, cells were harvested and lysed using the IP-lysis buffer supplemented with Halt Protease Inhibitor Cocktail (ThermoFisher). Equal amount of lysates (~5mg) for each condition were mixed with pre-washed 50μl of HA-magnetic bead slurry. Immunoprecipitation was performed at 4’C for 2hr. The beads were then washed twice using ‘high-salt’ IP lysis buffer (IP lysis buffer supplemented with 500mM NaCl), with 5min incubation on a rotor. The final wash was performed using regular IP lysis buffer. Immunoprecipitated proteins were eluted by boiling the samples at 98’C in 1x NuPAGE LDS Sample Buffer (ThermoFisher) for 5min supplemented with NU-PAGE sample reducing agent.
Denaturing Immunoprecipitation and tandem-mass spectrometry analysis (IP-MS/MS)
HEK293T cells with either DDRGK1 knockout or DDRGK1 and UFSP2 double knockout were transiently transfected with pRK5-HA-UFM1-dCS for 72hr. Cells were then starved for 4hr in the presence of folimycin (50nM) and lysed using 1X RIPA lysis buffer supplemented with mammalian protease inhibitor for 10min on ice. Insoluble debris and nuclear fractions were removed by centrifugation. The samples were then denatured using final 2% SDS and boiled for 5min at 95°C. After denaturation, the samples were further diluted at 1:20 in 1X RIPA lysis buffer to dilute the SDS concentration to 0.1% prior to immunoprecipitation using anti-HA magnetic beads (Pierce, #88837). Immunoprecipitation was performed at room temperature for 1hr with constant rotation followed by two washes in high-salt RIPA buffer containing 500mM NaCl and a final wash in 1x RIPA buffer.
The immunoprecipitation samples were then processed by the Proteomics group of Functional Genomics Center Zurich (FGCZ) for on-bead tryptic digestion and mass spectrometry analysis. Briefly, beads were washed twice with 50ul of digestion buffer (10 mM Tris/2 mM CaCl2, pH 8.2). Then, 45μl of digestion buffer and 5ml trypsin (100 ng/ul in 10 mM HCl) were added and samples were microwaved to assist digestion (30 min at 60°C). The supernatant was collected and peptides were extracted from beads with 150ul of 0.1%TFA/50% acetonitrile. Supernatants were then combined and dried, dissolved in 20μl 0.1% formic acid and further diluted 1:10 in 0.1% formic acid and transferred to autosampler vials for LC/MS/MS. Database searches were performed using the Mascot search engine against SwissProt (all species and only human).
Immunofluorescence
Immunofluorescence was conducted as previously described (Liang et al., 2018). Briefly, cells were fixed in 4% (wt/vol) paraformaldehyde for 15 min followed by permeabilization using 0.1% Triton X-100 in PBS for 10 min. Cells were then blocked in 1% BSA in PBS for 20 min. Primary antibodies were incubated for 1hr at room temperature, followed by three PBS washes for 5min each. Alexa Fluor 488/568/660-conjugated goat anti-mouse or anti-rabbit IgG secondary antibodies were incubated for 30 min at room temperature, followed by three PBS washes for 5min each. Coverslips were mounted onto glass slides using ProLong Gold Antifade reagent with or without DAPI addition for nucleus visualization. Images were taken using either Zeiss LSM 710 Axio Observer (in Berkeley) or Leica TCS SP8 confocal microscope (in ETH Zurich) with 63x objective lens and post-processed in Adobe Photoshop for specific inset enlargement and RGB channel separation. Colocalization analysis in Figure 6J was determined by Pearson’s Correlation coefficient using ImageJ with colocalization plugin from McMaster Biophotonics Facility (MBF). The frequency scatterplot in Figure S6A was generated using the same plugin.
MitoTracker
The MitoTracker assay was performed according the manufacturer’s protocol (ThermoFisher Catalog #M7512). Cells were plated 48 hours before starvation. Cell starvation and drug concentrations was performed according to protocols described above. The MitoTracker Red CMXRos was dissolved in DMSO for a stock concentration of 1 mM. MitoTracker was added to samples such that the concentration in each well was 50 nM. The cells were incubated for 30 minutes, washed with media, and then fixed in 4% formaldehyde. The cells were stained with calnexin according to the immunofluorescence protocol. For flow cytometry measurement, the experiment was performed the same way as mentioned above but the cells were trypsinized after incubation with MitoTracker dye to measure the fluorescence intensity of the staining.
ATP Assay
The assay was performed according to the manufacturer’s protocol for the CellTiter-Glo 2.0 Cell Viability Assay (Promega; Cat #: G9241). Briefly, the cells treated with starvation were starved for 25 hours. The cells treated with rotenone or antimycin A were used as positive controls and cells were treated for 1 hour. Cells were harvested, washed with PBS, and counted and normalized. The cells were spun down again and resuspended such that there were 25,000 cells per 50 μL of PBS. 50 μL of PBS was added to each well in an opaque-walled 96-well plate. Each sample was done in technical triplicate. Wells with PBS, but no cells, were used as a blank control. 50 μL of CellTiter-Glo 2.0 reagent was added to each well. The plate was placed on an orbital shake for 2 minutes, followed by a 10 minute bench-top incubation to stabilize the signal. Sample luminescence was determined by the SpectraMax M2 Microplate Reader (Molecular Devices).
Mitochondrial Respiration Measurements
Mitochondrial activity was determined using the Seahorse Flux Analyzer XF24 (Agilent Technology) and the Seahorse XF Cell Mito Stress Test Kit (Agilent; Cat. #:103015–100) according to the manufacturer’s protocol. Briefly, 4 X 104 HCT1 16 cells were seeded on XF24-well cell culture microplates. After 24hr, growth medium was exchanged with XF assay base medium supplemented with 1 mM sodium pyruvate (Invitrogen; Cat. #11360–070), 2mM L-glutamine (Invitrogen; Cat. #25030–081), and 10mM D-glucose (pH 7.4) (Invitrogen; Cat. #:D16–500). The microplates were incubated at 37°C without CO2 for 1hr prior to the assay. Samples were mixed for 3 min, time delayed for 2 min, and measured for 3 min. Oligomycin (1 μM), FCCP (1 μM), and rotenone / antimycin (0.5 μM) were sequentially injected at the indicated time points. OCR data were normalized by protein concentration and the average values were taken for each experiment. Seven replicates were performed for each cell line. The mean ± SEM was determined and statistical significance was evaluated using the Student’s t test with a P value < 0.05.
Statistical analysis
All analysis was performed using data from at least three independent biological replicates (exact number of replicates are stated in the figure legend). Unless stated otherwise, all statistical analyses were performed in PRISM6 software using paired Student’s t test. P values are indicated as follow: * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001. The distribution of the data was assumed to be normal, but this was not formally tested.
DATA AND CODE AVAILABILITY
All sequencing datasets generated as part of this study are publicly available in NCBI-SRA under Bioproject PRJNA599329. Original western blot data for all main and supplemental figures in this paper is available at Mendeley Data (https://doi.org/10.17632/ztzfkww2jx.2).
Supplementary Material
(A) Trial of different ER stress or autophagy stimuli to induce ER-phagy. HCT116 EATR cells were treated with Earl’s buffered saline solution (EBSS – amino acid starvation) or the indicated drugs (Thapsigargin: 1μM, Tunicamycin: 0.5μg/ml, Rapamycin: 1μM, Torin1: 1μM) for 16hr. Cells were then harvested for western blotting to check for UPR and autophagy response.
(B) Only amino acid starvation (using EBSS) inhibits ER-phagy. The same cells and treatment conditions in Figure S1A were also subjected to FACS measurement of ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01).
(C) EBSS and Torin1 treatments induce general autophagy. HCT116 GR-LC3B cells were treated with the same conditions in (A and B) and were subjected to measurement of general autophagy by FACS. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (***p < 0.001).
(D) FACS measurement of eGFP and mCherry fluorescence at individual cell level shows about 8% of ER-phagy per cell upon starvation. A representative plot for the eGFP/mCherry fluorescence intensity ratio of each cell was plotted for HCT116 EATR cells to represent the amount of ER-phagy upregulation after 16hr of starvation. A total of 10,000 cells from each condition was shown. Values on top of each condition represents the mean eGFP/mCherry intensity ratio and the associated standard error. P value indicates two-tailed paired t test (****p < 0.0001).
(E) Gating strategy for ER-phagy screen. HCT116 CRISPRi EATR cells transduced with the CRISPRi library of sgRNAs were starved for 16hr and subjected for FACS measurement for ER-phagy. Based on the EATR assay, the top and bottom 25% of cells correspond to sgRNA knockdown that results in ‘enhanced’ and ‘inhibited’ ER-phagy, respectively and were processed for next generation sequencing of sgRNA barcode.
(F) Multiple autophagy genes (highlighted in blue) were enriched in the ‘inhibited’ ER-phagy sort gate. The normalized sgRNA count from the ‘Inhibited’ ER-phagy sort gate was plotted against the normalized sgRNA count of the background sample. The labeled genes are enriched in the ‘Inhibited’ ER-phagy gate upon knockdown and indicate active transcription start sites (TSS) that are being used in HCT116 cells.
(G) Autophagy genes (highlighted in blue) that are depleted in the ‘enhanced’ ER-phagy sort gate corresponds to the genes that are enriched in the ‘inhibited’ sort gate of (F).
(H) ER-phagy receptor, general autophagy and membrane trafficking genes are hits in the ER-phagy screen. Those genes that are significantly enriched in either the ‘inhibited’ or ‘enriched’ gate are indicated in blue. Volcano plot describes data from the genome-wide CRISPRi screen. All negative control sgRNAs are indicated in gray and targeted sgRNAs are indicated in black. Data represent two biological replicates. log2 fold change and Mann-Whitney P value were calculated as described in (Horlbeck et al., 2016).
(I) Knockdown of genes involved in general autophagy also inhibit ER-phagy. HCT116 CRISPRi EATR cells were transduced with sgRNAs targeting the indicated candidate genes reported from the CRISPRi genome wide screen. Cells were starved for 16hr and subjected to FACS measurement for ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05, **p < 0.01; ****p < 0.0001).
(J) CCER assay complements the EATR data in Figure S1I. HCT116 CRISPRi CCER cells were also transduced with the same sgRNAs as Figure S1I. Cells were then starved for 16hr and harvested for western blot analysis for the levels of two general autophagy markers, p62 and LC3B.
(K) All sgRNAs efficiently knockdown the respective general autophagy genes. The same cell lines in (J) were harvested for qRT-PCR analysis to measure the knockdown efficiency of individual sgRNAs.
(L) The FAM134B sgRNAs used in this CRISPRi screen do not effectively knock down FAM134B. HCT116-CRISPRi EATR cells stably expressing FAM134B sgRNAs were harvested for qRT-PCR to determine the knockdown efficiency.
(A) Schematic of the oxidative phosphorylation (OXPHOS) pathway. The genes implicated in ER-phagy regulation are annotated in red in their respective electron-transport chain compartment.
(B) Cells from Figure 2B were harvested to assess the knockdown efficiency of each sgRNA by western blotting.
(C) Knockdown of NDUFB4, NDUFB2 and ATP5O shows ER-phagy inhibition using the CCER assay. HCT116 CRISPRi CCER cells were transduced with the indicated sgRNAs and subjected to 16hr starvation. Cells were then harvested for western blot analysis.
(D) The cleaved and full length mCherry-RAMP4 band intensities in (C) were measured and normalized to starved sgNT sample. Data represents mean ± SD of three biological replicates. P value represents two-tailed paired t test (**p < 0.001).
(E) Knockdown of NDUFB4, NDUFB2, and ATP5O does not affect general autophagy. HCT116 CRISPRi cells expressing mCherry-eGFP-LC3B were transduced with sgRNAs targeting either ULK1, ATG10, NDUFB4, NDUFB2, or ATP5O. Cells were starved for 16hr before FACS measurement for general autophagy. Data represents mean ± SD of four biological replicates. P value represents two-tailed paired t test (*p < 0.05).
(F) ULK1 protein levels corresponds to the disruption of the OXPHOS complex. Cells from the same experiment as Figure 2C were harvested for western blot analysis to verify the protein levels of ULK1, ATG10, NDUFB4 and NDUFB2.
(G) Re-expression of ATP5O restores ULK1 protein levels. HCT116 CRISPRi EATR cells transduced with sgATP5O were rescued with ATP5O cDNA and harvested for western blot analysis to assess the protein levels of ULK1 and ATP5O.
(H) Re-expression of ATP5O rescues ER-phagy. The same cells in (G) were starved for 16hr before FACS measurement for ER-phagy. Data presented as mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01; ***, p < 0.001).
(I) Schematic of experimental setup for chemical genetic inhibition of electron transport chain function in ER-phagy. HCT116 CRISPRi cells were treated with each inhibitor for 24 hours in complete media, then maintained in inhibitor for another 16 hours in fed or EBSS starvation media. Cells were then harvested for FACS to measure autophagy.
(J) OXPHOS inhibitors disrupt ER-phagy as measured by CCER assay. HCT116 CRISPRi CCER cells were treated with rotenone, antimycin A, and oligomycin A and starved for 16hr. Cells were lysed for western blotting to measure mCherry-RAMP4 cleavage.
(K) Densitometry measurement of the ratio between the cleaved and full length mCherry-RAMP4 bands in (J). Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05).
(L) OXPHOS inhibitors disrupt ER-phagy as measured by flow cytometry. HCT116 EATR cells were treated with small molecule inhibitors of rotenone, antimycin A, or oligomycin A, and starved for 16 hours before FACS measurement of ER-phagy. Data presented as mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05, ****p < 0.0001).
(M) General autophagy is unaffected by antimycin A and oligomycin A treatment in starvation conditions. HCT116 cells were treated with the indicated small molecule inhibitors of antimycin A, or oligomycin A for 24hr and then were subsequently starved for 16hr with treatment of small molecule inhibitors. Cells were lysed for western blot analysis to measure protein levels of LC3B.
(N) NDUFB2, NDUFB4 and ATP5O depletions affect cellular ATP levels. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O and starved for 16hr or treated with rotenone or oligomycin A. Cells were collected for ATP Glo luminescence assay. Data represents mean ± SD of three biological replicates.
(O) Oxygen consumption is reduced in NDUFB4, NDUFB2, and ATP5O knockdown cells. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB2 or ATP5O and the Seahorse Flux Analyzer was conducted to determine the oxygen consumption rate.
(A) Disruption of the mitochondrial OXPHOS components does not affect the mitochondrial membrane potential. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O or treated with indicated small molecule inhibitors. Cells were subsequently treated with MitoTracker, then fixed and immunostained for calnexin (CANX). Representative images are shown. Scale bar represents 10μm.
(B) Quantitative measurement of MitoTracker staining by flow cytometry. Cells from the same experiment in (A) were stained with MitoTracker and then trypsinized from flow cytometry measurement of the staining intensity. Data presented as mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05, **p < 0.01).
(C) Disruption of the OXPHOS pathway does not induce mitophagy based on the protein levels of MFN2. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O and starved for 16hr or treated with CCCP. Cells were harvested for western blot analysis and immunoprobed for the indicated proteins. A representative blot is shown.
(D) Densitometry measurement of the ratio between MFN2/actin from (C). Data represents mean ± SD of two biological replicates.
(E) Starvation does not induce mitophagy in Parkin-overexpressing cells. HCT116 CRISPRi were transduced with YFP-Parkin construct and treated with starvation or CCCP (10 uM) for 16hr. Cells were harvested for western blot analysis and immunoprobed for the indicated Parkin substrates to verify mitophagy in CCCP-treated cells.
(F) OXPHOS disruption does not trigger mitophagy in Parkin-overexpressing cells. HCT116 CRISPRi-YFP-Parkin cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O and starved for 16hr or treated with CCCP. Cells were harvested for western blot analysis and immunoprobed for the indicated proteins. A representative western blot is shown.
(G) Densitometry measurement of the ratio between MFN2/actin from (E). Data represents mean ± SD of three biological replicates.
(H) Changes in ULK1 protein levels during starvation correspond to the protein levels of NDUFB4, NDUFB2, and ATP5O. HCT116 CRISPRi EATR cells were transduced with NDUFB4, NDUFB2, or ATP5O cDNA constructs, and then transduced with sgRNAs targeting ULK1, NDUFB4, NDUFB2, or ATP5O. Cells were lysed for western blotting and immunoprobed for the indicated proteins.
(I) ULK1 overexpression does not rescue ER-phagy. HCT116 CRISPRi EATR cells with or without ULK1 overexpression were transduced with sgRNAs targeting ULK1, NDUFB4, NDUFB2, or ATP5O and starved for 16hr before FACS measurement for ER-phagy. Data presented as mean ± SD of four biological replicates. (*p < 0.05, ***p < 0.001).
(J) Western blot validation of AMPKa knockout and cDNA re-expression. Parental represents HCT116 CRISPRi EATR cells. AMPK KO (knockout) cells were generated using CRISPR. GST-AMPKa cDNAs were re-expressed in the AMPK KO cells. The catalytically active (GST-AMPKa-CA) form is a truncation of full length AMPKa, resulting in the smaller size. GST-AMPKa-KD (kinase dead) has a K47R point mutation in the CA construct.
(K) Phosphorylation status of downstream AMPKɑ targets were analyzed to verify the catalytically active (CA) and kinase dead (KD) AMPKɑ constructs. AMPKɑ knockout HCT116 EATR clone from (I) were starved for 16 hours and samples were lysed for western blotting and immunoprobed for known target proteins of AMPK phosphorylation.
(L) Catalytically active (CA) AMPKɑ does not inhibit ER-phagy. AMPKɑ knockout HCT116 EATR cells stably expressing catalytically active (CA) AMPKɑ, or kinase dead (KD) AMPKɑ were starved for 16hr before FACS measurement of ER-phagy. Data presented as mean ± SD of three biological replicates.
(A) Densitometry measurement of the ratio between the cleaved and full length mCherry-RAMP4 in Figure 3C. Data represents mean ± SD of five biological replicates. P value indicates two-tailed paired t test (**p < 0.01, ***p < 0.001).
(B) UFL1 protein expression prevents proteasomal degradation of DDRGK1. HCT116 CRISPRi cells were transduced with sgRNA targeting UFL1. Cells were treated with Epoxomicin (100nM) or Folimycin (100nM) for 6hr and then harvested for western blot analysis. p53 and LC3B were used as positive controls for proteasomal and lysosomal inhibitions, respectively.
(C) UBA5 knockdown inhibits ER-phagy. HCT116 CRISPRi EATR cells were transduced with the indicated sgRNA and then starved for 16hr before FACS analysis for ER-phagy. Data represents mean ± SD of three biological replicates. Statistical significance was determined based on two-tailed paired t test. (**p < 0.01; ****p < 0.0001).
(D) UFL1 and DDRGK1 act in series during ER-phagy. HCT116 CRISPRi EATR cells were transduced with the indicated sgRNAs and then starved for 16hr before FACS analysis for ER-phagy. Data represents mean ± SD of three biological replicates. Statistical significance was determined based on two-tailed paired t test.
(E) The same cell lines in (D) were harvested for western blotting to verify the knockdown of targeted genes.
(F) DDRGK1 knockdown prevents ER lumen protein degradation. HCT116 cells stably expressing mCherry-KDEL were transduced with shRNA against either DDRGK1 or UFL1. Cells were then starved for 16hr and harvested for western blot analysis to assess mCherry-KDEL cleavage. A representative blot is shown.
(G) Densitometry analysis of mCherry-KDEL cleavage from (F). Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05; **p < 0.01).
(H) The stable cell lines from Figure 4G were harvested for western blot to verify the knockdown of DDRGK1 and UFL1.
(I) Representative FACS scatterplots from Figure 4G for the measurement of eGFP and mCherry fluorescence using either TetOn-REEP5-mCherry-eGFP or TetOn-CLIMP63-mCherry-eGFP.
(J) Knockdown of DDRGK1 impedes FAM134B-mediated ER-phagy. HCT116 CRISPRi EATR cells stably expressing HA-FAM134B were transduced with sgDDRGK1. Cells were starved for 16hr and subjected for ER-phagy measurement by FACS analysis. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (***p < 0.001; ****p < 0.0001).
(K) HCT116 CRISPRi CCER cells stably expressing HA-FAM134B were transduced with sgDDRGK1. Cells were starved for 16hr and then harvested for western blot analysis of ER protein levels.
(L) Densitometry analysis of mCherry-RAMP4 cleavage from (K). Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05).
(A) Knockdown of UFM1 does not prevent the appearance of higher molecular weight species of DDRGK1. HCT116 CRISPRi cells stably expressing DDRGK1-HA construct were transduced with sgRNA targeting UFM1. Cells were then harvested for HA-immunoprecipitation and western blotted for the indicated proteins.
(B) Knockdown of UFL1 but not UFM1 reduces DDRGK1 protein levels. HCT116 CRISPRi EATR cells transeduced with the indicated sgRNAs were harvested for western blotting to assess the protein levels of DDRGK1 upon knockdown of the targeted genes.
(C) The cDNA of DDRGK1-HA mutant variants in Figure 5C were stably expressed in HCT116 cells to verify their respective protein sizes in DDRGK1 knockdown cells.
(D) All individual Lysine mutant constructs of DDRGK1 are able to rescue ER-phagy in DDRGK1 knockdown cells. HCT116 CRISPRi EATR cells with sgDDRGK1 were transduced with the indicated DDRGK1-HA mutant constructs. Cells were then starved for 16hr before FACS measurement of ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test. (*p < 0.05, **p < 0.01).
(E) The cell lines generated for (D) were harvested for western blotting to verify the expression of the indicated DDRGK1-HA mutant constructs.
RPN1 Is an ER Surface UFMylation Substrate
(A) Additional colocalization data analysis of Figure 6F for DDRGK1, UFL1 and the ER marker, RAMP4 based on frequency of colocalization. Conditions with good or near perfect colocalization have good correlation between the X- and Y-axes. Representative frequency scatterplot from each condition is shown.
(B) The cell lines generated for Figure 5H were harvested for western blotting to verify the expression of the indicated DDRGK1-HA mutant constructs.
(C) Schematic for the different DDRGK1 and UFL1 mutant constructs. UFL1is tethered to the ER surface by synthetic fusion with RAMP4 at the C-terminal of UFL1.
(D) Western blot validation of the expression levels of each mutant constructs of DDRGK1 and UFL1 shown in (C). Dotted line represents cropped images from the same blots but with irrelevant lanes removed.
(E) Immunofluorescence analysis to confirm that the RAMP4-fused constructs localize to the ER surface. Scale bar represents 10μm. Insets represent three-fold enlargement of the boxed areas.
(F) None of the synthetic fusion of UFL1 or DDRGK1 to the ER surface rescued ER-phagy in sgDDRGK1 cells. HCT116 cells stably expressing sgDDRGK1 were transduced with the indicated cDNAs. Cells were starved for 16hr before FACS measurement for ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01).
(G) None of the previously reported targets of UFMylation are involved in ER-phagy regulation. HCT116 CRISPRi EATR cells were transduced with sgRNAs targeting CDK5RAP3, Sox9 or ASC1 and starved for 16hr before FACS measurement of ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01).
(H) The same cell lines used in (G) were harvested for qRT-PCR to assess the knockdown efficiency of each sgRNA.
(I) RPN1 localizes to the ER. U2OS cells were transfected with cDNAs for RPN1-GFP and mCherry-KDEL for 48hr. Scale bar represents 10mm. Inset represents 4x enlargement of the boxed area.
(A) IRE1a is not a target of DDRGK1-mediated UFMylation. HCT116 CRISPRi cells stably expressing HA-UFM1 were transduced with sgRNA targeting DDRGK1. Cells were starved for 16hr before being lysed in non-denaturing condition for HA-immunoprecipitation of UFM1 and western blot analysis of its interaction with IRE1α and DDRGK1.
(B) The same experiment shown in Figure S5B were further probed for the indicated ER or UPR markers. The densitometry measurements for IRE1a, BiP, CANX, CLIMP63 and REEP5 from three biological replicates are shown in Figure 7A.
(C) DDRGK1 depletion results in the upregulation of UPR markers in various cell lines. HepG2 and MCF7 cells were transduced with shRNAs targeting DDRGK1. Cells were then harvested for western blot analysis of DDRGK1 knockdown efficiency and UPR response.
(D) HeLa cells with CRISPR/Cas9-KO of DDRGK1 were harvested for western blot analysis of IRE1α protein levels.
(E) Loss of DDRGK1 results in increased ER content. The same HeLa cell lines generated in (D) were fixed and stained for an ER marker (CANX). Scale bar represents 20mm.
(F) IRE1α depletion prevents UPR response in DDRGK1 knockdown cells. The same cell lines in Figure 7C were harvested for western blotting to measure the protein levels of the indicated UPR genes.
Highlights.
Genome-wide CRISPRi screen identifies 200 high-confidence ER-phagy regulators
Disruption of mitochondrial OXPHOS system inhibits ER-phagy
ER-resident UFMylation mediates autophagy of the ER sheets
ACKNOWLEDGMENTS
J.E.C. and J.R.L. were supported by the National Institutes of Health (DP2-HL-141006), the NOMIS Foundation, the Lotte and Adolf Hotz-Sprenger Stiftung, the Li Ka Shing Foundation, and the Heritage Medical Research Institute. This research was also supported by a grant from the National Institute of Health (R01GM112948 to J.A.O.). J.A.O. is a Chan Zuckerberg Biohub investigator. E.L. was supported by the National Science Foundation. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, which is supported by NIH Instrumentation grant S10 OD018174. Confocal live-and fixed-cell imaging experiments were conducted at the Cancer Research Laboratory-Molecular Imaging Center Berkeley (supported by the Gordon and Betty Moore Foundation) and the Scientific Center for Optical and Electron Microscopy facility at ETH Zurich. Mass spectrometry experiments were performed by the Functional Genomics Center Zurich (ETH Zurich/University of Zurich).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.02.017.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abel A, Fonknechten N, Hofer A, Dürr A, Cruaud C, Voit T, Weissen-bach J, Brice A, Klimpe S, Auburger G, and Hazan J (2004). Early onset autosomal dominant spastic paraplegia caused by novel mutations in SPG3A. Neurogenetics 5, 239–243. [DOI] [PubMed] [Google Scholar]
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, and Hamosh A (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin-Wetzel N, Saunders RA, Kamphuis MJ, Rato C, Preissler S, Harding HP, and Ron D (2017). A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 171, 1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Ordureau A, Paulo JA, Shoemaker CJ, Denic V, and Harper JW (2019). TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell 74, 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O’Donoghue SI, Schneider R, and Jensen LJ (2014). COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database (Oxford) 2014, bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunger K, Pfeffer S, Shrimal S, Gilmore R, Berninghausen O, Mandon EC, Becker T, Förster F, and Beckmann R (2018). Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Pi W, Sivaprakasam S, Zhu X, Zhang M, Chen J, Makala L, Lu C, Wu J, Teng Y, et al. (2015). UFBP1, a Key Component of the Ufm1 Conju gation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLoS Genet. 11, e1005643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RLJ, Hess S, and Chan DC (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet 20, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, O’Donnell L, Oster S, Theesfeld C, Sellam A, et al. (2017). The Bio-GRID interaction database: 2017 update. Nucleic Acids Res. 45 (D1), D369–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Xiao Y, Chai P, Zheng P, Teng J, and Chen J (2019). ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol 29, 846–855. [DOI] [PubMed] [Google Scholar]
- Cherepanova N, Shrimal S, and Gilmore R (2016). N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol 41, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chino H, Hatta T, Natsume T, and Mizushima N (2019). Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 74, 909–921. [DOI] [PubMed] [Google Scholar]
- Daida A, Hamano S-I, Ikemoto S, Matsuura R, Nakashima M, Matsumoto N, and Kato M (2018). Biallelic loss-of-function UBA5 mutations in a patient with intractable West syndrome and profound failure to thrive. Epileptic Disord. 20, 313–318. [DOI] [PubMed] [Google Scholar]
- Daniel J, and Liebau E (2014). The ufm1 cascade. Cells 3, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, Wood NW, and Plun-Favreau H (2011). Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim. Biophys. Acta 1813, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus R, Moretti F, McAllister G, Wang Z, Bergman P, Liu S, Frias E, Alford J, Reece-Hoyes JS, Lindeman A, et al. (2016). Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. eLife 5 Published online June 28, 2016. 10.7554/eLife.17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson MW, and Guo M (2007). Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr. Opin. Neurobiol 17, 331–337. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egunsola AT, Bae Y, Jiang M-M, Liu DS, Chen-Evenson Y, Bertin T, Chen S, Lu JT, Nevarez L, Magal N, et al. (2017). Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia. J. Clin. Invest 127, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elangovan M, Chong HK, Park JH, Yeo EJ, and Yoo YJ (2017). The role of ubiquitin-conjugating enzyme Ube2j1 phosphorylation and its degradation by proteasome during endoplasmic stress recovery. J. Cell Commun. Signal 11, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Soldà T, Galli C, D’Antuono R, Morone D, Danieli A, et al. (2018). ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 37, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Soldà T, et al. (2016). Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol 18, 1173–1184. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, and Springer W (2010). The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6, 871–878. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauser L, Sonnay S, Stafa K, and Moore DJ (2011). Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem 118, 636–645. [DOI] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Hölper S, Mari M, Harwardt MI, Yan R, Müller S, Reggiori F, Heilemann M, and Dikic I (2017). Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 6 Published online June 15, 2017. 10.7554/eLife.25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin Y-HT, Wiita AP, Xu K, Correia MA, and Kampmann M (2019). Mitochondrial dysfunction is signaled to the integrated stress response by OMA1, DELE1 and HRI. bioRxiv. 10.1101/715896. [DOI] [Google Scholar]
- Hamasaki M, Noda T, Baba M, and Ohsumi Y (2005). Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic 6, 56–65. [DOI] [PubMed] [Google Scholar]
- Heo J-M, Ordureau A, Paulo JA, Rinehart J, and Harper JW (2015). The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 60, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, and Shaw RJ (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5 Published online September 23, 2016. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, and Hall MN (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol 6, 1122–1128. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Vidoni S, Durigon R, Pearce SF, Rorbach J, He J, Brea-Calvo G, Minczuk M, Reyes A, Holt IJ, and Spinazzola A (2014). Amino acid starvation has opposite effects on mitochondrial and cytosolic protein synthesis. PLoS ONE 9, e93597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, and Youle RJ (2011). Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol 23, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher DJ, Kreibich G, and Gilmore R (1992). Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell 69, 55–65. [DOI] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan K-L (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, and Tanaka K (2004). A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 23, 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, Baets J, Senderek J, Topaloglu H, Farrell SA, et al. (2009). Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet 41, 1179–1181. [DOI] [PubMed] [Google Scholar]
- Lake NJ, Compton AG, Rahman S, and Thorburn DR (2016). Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol 79, 190–203. [DOI] [PubMed] [Google Scholar]
- Lemaire K, Moura RF, Granvik M, Igoillo-Esteve M, Hohmeier HE, Hendrickx N, Newgard CB, Waelkens E, Cnop M, and Schuit F (2011). Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS ONE 6, e18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto DE, Morgens DW, Zhang L, Walczak CP, Elias JE, Bassik MC, and Kopito RR (2019). Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell 73, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Köster J, Xu H, Chen C-H, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Lingeman E, Ahmed S, and Corn JE (2018). Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol 217, 3354–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingeman E, Jeans C, and Corn JE (2017). Production of purified casrnps for efficacious genome editing. Curr. Protoc. Mol. Biol 120, 31.10.1–31.10.19. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang Y, Song L, Zeng L, Yi W, Liu T, Chen H, Wang M, Ju Z, and Cong Y-S (2017). A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1a stability. Nat. Commun 8, 14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi AA, and Elrod JW (2017). Mediating ER-mitochondrial cross-talk. Science 358, 591–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader BJ, Pivtoraiko VN, Flippo HM, Klocke BJ, Roth KA, Mangieri LR, and Shacka JJ (2012). Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem. Neurosci 3, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignon-Ravix C, Milh M, Kaiser CS, Daniel J, Riccardi F, Cacciagli P, Nagara M, Busa T, Liebau E, and Villard L (2018). Abnormal function of the UBA5 protein in a case of early developmental and epileptic encephalopathy with suppression-burst. Hum. Mutat 39, 934–938. [DOI] [PubMed] [Google Scholar]
- Milev MP, Grout ME, Saint-Dic D, Cheng Y-HH, Glass IA, Hale CJ, Hanna DS, Dorschner MO, Prematilake K, Shaag A, et al. (2017). Mutations in TRAPPC12 manifest in progressive childhood encephalopathy and golgi dysfunction. Am. J. Hum. Genet 101, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, and Pelham HR (1987). A C-terminal signal prevents secretion of luminal ER proteins. Cell 48, 899–907. [DOI] [PubMed] [Google Scholar]
- Nahorski MS, Maddirevula S, Ishimura R, Alsahli S, Brady AF, Bege-mann A, Mizushima T, Guzmán-Vega FJ, Obata M, Ichimura Y, et al. (2018). Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 141, 1934–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, and Lazarou M (2016). Deciphering the molecular signals of pink1/parkin mitophagy. Trends Cell Biol. 26, 733–744. [DOI] [PubMed] [Google Scholar]
- Nordgren M, Francisco T, Lismont C, Hennebel L, Brees C, Wang B, Van Veldhoven PP, Azevedo JE, and Fransen M (2015). Export-deficient monoubiquitinated PEX5 triggers peroxisome removal in SV40 large T antigen-transformed mouse embryonic fibroblasts. Autophagy 11, 1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, et al. (2014). Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Kohno K, and Iwawaki T (2009). Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res 315, 2496–2504. [DOI] [PubMed] [Google Scholar]
- Olenych SG, Claxton NS, Ottenberg GK, and Davidson MW (2007). The fluorescent protein color palette. Curr. Protoc. Cell Biol Chapter 21, Unit 21.5. [DOI] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo J-M, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AR, Suttangkakul A, and Vierstra RD (2008). The ATG12-conjugating enzyme ATG10 Is essential for autophagic vesicle formation in Arabidopsis thaliana. Genetics 178, 1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, and Youle RJ (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsl A, and Winklhofer KF (2012). Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathol. 123, 173–188. [DOI] [PubMed] [Google Scholar]
- Qin B, Yu J, Nowsheen S, Wang M, Tu X, Liu T, Li H, Wang L, and Lou Z (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun 10, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismanchi N, Soderblom C, Stadler J, Zhu P-P, and Blackstone C (2008). Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet 17, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, and Carvalho P (2014). Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol 204, 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Prinz WA, Thorn KS, Voss C, and Walter P (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol 187, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, and Blower MD (2016). The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci 73, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, and Rapoport TA (2010). Mechanisms determining the morphology of the peripheral ER. Cell 143, 774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, Chang HY, and Barna M (2017). The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 169, 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, and Wilkinson S (2018). CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 44, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taanman J-W (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123. [DOI] [PubMed] [Google Scholar]
- Tatsumi K, Sou YS, Tada N, Nakamura E, Iemura S, Natsume T, Kang SH, Chung CH, Kasahara M, Kominami E, et al. (2010). A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem 285, 5417–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, and Sabatini DM (2009). Rapamycin inhibits mTORC1, but not completely. Autophagy 5, 725–726. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, and Gray NS (2009). An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem 284, 8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al. (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao YS, Ivessa NE, Adesnik M, Sabatini DD, and Kreibich G (1992). Carboxy terminally truncated forms of ribophorin I are degraded in pre-Golgi compartments by a calcium-dependent process. J. Cell Biol 116, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak CP, Leto DE, Zhang L, Riepe C, Muller RY, DaRosa PA, Ingolia NT, Elias JE, and Kopito RR (2019). Ribosomal protein RPL26 is the principal target of UFMylation. Proc. Natl. Acad. Sci. USA 116, 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, and Schwarz TL (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tukachinsky H, Romano FB, and Rapoport TA (2016). Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. eLife 5 Published online September 13, 2016. 10.7554/eLife.18605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gong Y, Peng B, Shi R, Fan D, Zhao H, Zhu M, Zhang H, Lou Z, Zhou J, et al. (2019). MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 47, 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xu Y, Rogers H, Saidi L, Noguchi CT, Li H, Yewdell JW, Guydosh NR, and Ye Y (2020). UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 30, 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartosch L, Günesdogan U, Graham SC, and Luzio JP (2015). Recruitment of VPS33A to HOPS by VPS16 Is Required for Lysosome Fusion with Endosomes and Autophagosomes. Traffic 16, 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, and Xu X (2016). Ufmylation: A unique & fashionable modification for life. Genomics Proteomics Bioinformatics 14, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CM, Kraft C, Duggan C, Ismail N, Crawshaw SG, and High S (2005). Ribophorin I associates with a subset of membrane proteins after their integration at the sec61 translocon. J. Biol. Chem 280, 4195–4206. [DOI] [PubMed] [Google Scholar]
- Wu J, Lei G, Mei M, Tang Y, and Li H (2010). A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. J. Biol. Chem 285, 15126–15136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Deng X, Zhou W, and Tan E-K (2016). Flow Cytometry-Based Assessment of Mitophagy Using MitoTracker. Front. Cell. Neurosci 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, and Youle RJ (2014). Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3 Published online February 25, 2014 10.7554/eLife.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan A, and Lennarz WJ (2005). Two oligosaccharyl transferase complexes exist in yeast and associate with two different translocons. Glycobiology 15, 1407–1415. [DOI] [PubMed] [Google Scholar]
- Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW, Lee SW, Ka SH, Sou Y-S, Komatsu M, Tanaka K, et al. (2014). Modification of ASC1 by UFM1 is crucial for ERα transactivation and breast cancer development. Mol. Cell 56, 261–274. [DOI] [PubMed] [Google Scholar]
- Youle RJ, and Narendra DP (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H-X, Russell RC, and Guan K-L (2013). Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 9, 1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait-Mulder J, Lee J-H, Paull TT, et al. (2015a). ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol 17, 1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S, Gurav A, Pi W, Makala L, Wu J, et al. (2015b). RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 22, 1922–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Zhu P-P, Renvoisé B, Maldonado-Báez L, Park SH, and Blackstone C (2016). Mammalian knock out cells reveal prominent roles for atlastin GTPases in ER network morphology. Exp. Cell Res 349, 32–44. [DOI] [PubMed] [Google Scholar]
- Zhu H, Bhatt B, Sivaprakasam S, Cai Y, Liu S, Kodeboyina SK, Patel N, Savage NM, Sharma A, Kaufman RJ, et al. (2019). Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nat. Commun 10, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Trial of different ER stress or autophagy stimuli to induce ER-phagy. HCT116 EATR cells were treated with Earl’s buffered saline solution (EBSS – amino acid starvation) or the indicated drugs (Thapsigargin: 1μM, Tunicamycin: 0.5μg/ml, Rapamycin: 1μM, Torin1: 1μM) for 16hr. Cells were then harvested for western blotting to check for UPR and autophagy response.
(B) Only amino acid starvation (using EBSS) inhibits ER-phagy. The same cells and treatment conditions in Figure S1A were also subjected to FACS measurement of ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01).
(C) EBSS and Torin1 treatments induce general autophagy. HCT116 GR-LC3B cells were treated with the same conditions in (A and B) and were subjected to measurement of general autophagy by FACS. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (***p < 0.001).
(D) FACS measurement of eGFP and mCherry fluorescence at individual cell level shows about 8% of ER-phagy per cell upon starvation. A representative plot for the eGFP/mCherry fluorescence intensity ratio of each cell was plotted for HCT116 EATR cells to represent the amount of ER-phagy upregulation after 16hr of starvation. A total of 10,000 cells from each condition was shown. Values on top of each condition represents the mean eGFP/mCherry intensity ratio and the associated standard error. P value indicates two-tailed paired t test (****p < 0.0001).
(E) Gating strategy for ER-phagy screen. HCT116 CRISPRi EATR cells transduced with the CRISPRi library of sgRNAs were starved for 16hr and subjected for FACS measurement for ER-phagy. Based on the EATR assay, the top and bottom 25% of cells correspond to sgRNA knockdown that results in ‘enhanced’ and ‘inhibited’ ER-phagy, respectively and were processed for next generation sequencing of sgRNA barcode.
(F) Multiple autophagy genes (highlighted in blue) were enriched in the ‘inhibited’ ER-phagy sort gate. The normalized sgRNA count from the ‘Inhibited’ ER-phagy sort gate was plotted against the normalized sgRNA count of the background sample. The labeled genes are enriched in the ‘Inhibited’ ER-phagy gate upon knockdown and indicate active transcription start sites (TSS) that are being used in HCT116 cells.
(G) Autophagy genes (highlighted in blue) that are depleted in the ‘enhanced’ ER-phagy sort gate corresponds to the genes that are enriched in the ‘inhibited’ sort gate of (F).
(H) ER-phagy receptor, general autophagy and membrane trafficking genes are hits in the ER-phagy screen. Those genes that are significantly enriched in either the ‘inhibited’ or ‘enriched’ gate are indicated in blue. Volcano plot describes data from the genome-wide CRISPRi screen. All negative control sgRNAs are indicated in gray and targeted sgRNAs are indicated in black. Data represent two biological replicates. log2 fold change and Mann-Whitney P value were calculated as described in (Horlbeck et al., 2016).
(I) Knockdown of genes involved in general autophagy also inhibit ER-phagy. HCT116 CRISPRi EATR cells were transduced with sgRNAs targeting the indicated candidate genes reported from the CRISPRi genome wide screen. Cells were starved for 16hr and subjected to FACS measurement for ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05, **p < 0.01; ****p < 0.0001).
(J) CCER assay complements the EATR data in Figure S1I. HCT116 CRISPRi CCER cells were also transduced with the same sgRNAs as Figure S1I. Cells were then starved for 16hr and harvested for western blot analysis for the levels of two general autophagy markers, p62 and LC3B.
(K) All sgRNAs efficiently knockdown the respective general autophagy genes. The same cell lines in (J) were harvested for qRT-PCR analysis to measure the knockdown efficiency of individual sgRNAs.
(L) The FAM134B sgRNAs used in this CRISPRi screen do not effectively knock down FAM134B. HCT116-CRISPRi EATR cells stably expressing FAM134B sgRNAs were harvested for qRT-PCR to determine the knockdown efficiency.
(A) Schematic of the oxidative phosphorylation (OXPHOS) pathway. The genes implicated in ER-phagy regulation are annotated in red in their respective electron-transport chain compartment.
(B) Cells from Figure 2B were harvested to assess the knockdown efficiency of each sgRNA by western blotting.
(C) Knockdown of NDUFB4, NDUFB2 and ATP5O shows ER-phagy inhibition using the CCER assay. HCT116 CRISPRi CCER cells were transduced with the indicated sgRNAs and subjected to 16hr starvation. Cells were then harvested for western blot analysis.
(D) The cleaved and full length mCherry-RAMP4 band intensities in (C) were measured and normalized to starved sgNT sample. Data represents mean ± SD of three biological replicates. P value represents two-tailed paired t test (**p < 0.001).
(E) Knockdown of NDUFB4, NDUFB2, and ATP5O does not affect general autophagy. HCT116 CRISPRi cells expressing mCherry-eGFP-LC3B were transduced with sgRNAs targeting either ULK1, ATG10, NDUFB4, NDUFB2, or ATP5O. Cells were starved for 16hr before FACS measurement for general autophagy. Data represents mean ± SD of four biological replicates. P value represents two-tailed paired t test (*p < 0.05).
(F) ULK1 protein levels corresponds to the disruption of the OXPHOS complex. Cells from the same experiment as Figure 2C were harvested for western blot analysis to verify the protein levels of ULK1, ATG10, NDUFB4 and NDUFB2.
(G) Re-expression of ATP5O restores ULK1 protein levels. HCT116 CRISPRi EATR cells transduced with sgATP5O were rescued with ATP5O cDNA and harvested for western blot analysis to assess the protein levels of ULK1 and ATP5O.
(H) Re-expression of ATP5O rescues ER-phagy. The same cells in (G) were starved for 16hr before FACS measurement for ER-phagy. Data presented as mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01; ***, p < 0.001).
(I) Schematic of experimental setup for chemical genetic inhibition of electron transport chain function in ER-phagy. HCT116 CRISPRi cells were treated with each inhibitor for 24 hours in complete media, then maintained in inhibitor for another 16 hours in fed or EBSS starvation media. Cells were then harvested for FACS to measure autophagy.
(J) OXPHOS inhibitors disrupt ER-phagy as measured by CCER assay. HCT116 CRISPRi CCER cells were treated with rotenone, antimycin A, and oligomycin A and starved for 16hr. Cells were lysed for western blotting to measure mCherry-RAMP4 cleavage.
(K) Densitometry measurement of the ratio between the cleaved and full length mCherry-RAMP4 bands in (J). Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05).
(L) OXPHOS inhibitors disrupt ER-phagy as measured by flow cytometry. HCT116 EATR cells were treated with small molecule inhibitors of rotenone, antimycin A, or oligomycin A, and starved for 16 hours before FACS measurement of ER-phagy. Data presented as mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05, ****p < 0.0001).
(M) General autophagy is unaffected by antimycin A and oligomycin A treatment in starvation conditions. HCT116 cells were treated with the indicated small molecule inhibitors of antimycin A, or oligomycin A for 24hr and then were subsequently starved for 16hr with treatment of small molecule inhibitors. Cells were lysed for western blot analysis to measure protein levels of LC3B.
(N) NDUFB2, NDUFB4 and ATP5O depletions affect cellular ATP levels. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O and starved for 16hr or treated with rotenone or oligomycin A. Cells were collected for ATP Glo luminescence assay. Data represents mean ± SD of three biological replicates.
(O) Oxygen consumption is reduced in NDUFB4, NDUFB2, and ATP5O knockdown cells. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB2 or ATP5O and the Seahorse Flux Analyzer was conducted to determine the oxygen consumption rate.
(A) Disruption of the mitochondrial OXPHOS components does not affect the mitochondrial membrane potential. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O or treated with indicated small molecule inhibitors. Cells were subsequently treated with MitoTracker, then fixed and immunostained for calnexin (CANX). Representative images are shown. Scale bar represents 10μm.
(B) Quantitative measurement of MitoTracker staining by flow cytometry. Cells from the same experiment in (A) were stained with MitoTracker and then trypsinized from flow cytometry measurement of the staining intensity. Data presented as mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05, **p < 0.01).
(C) Disruption of the OXPHOS pathway does not induce mitophagy based on the protein levels of MFN2. HCT116 CRISPRi cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O and starved for 16hr or treated with CCCP. Cells were harvested for western blot analysis and immunoprobed for the indicated proteins. A representative blot is shown.
(D) Densitometry measurement of the ratio between MFN2/actin from (C). Data represents mean ± SD of two biological replicates.
(E) Starvation does not induce mitophagy in Parkin-overexpressing cells. HCT116 CRISPRi were transduced with YFP-Parkin construct and treated with starvation or CCCP (10 uM) for 16hr. Cells were harvested for western blot analysis and immunoprobed for the indicated Parkin substrates to verify mitophagy in CCCP-treated cells.
(F) OXPHOS disruption does not trigger mitophagy in Parkin-overexpressing cells. HCT116 CRISPRi-YFP-Parkin cells were transduced with sgRNAs targeting either NDUFB4, NDUFB2, or ATP5O and starved for 16hr or treated with CCCP. Cells were harvested for western blot analysis and immunoprobed for the indicated proteins. A representative western blot is shown.
(G) Densitometry measurement of the ratio between MFN2/actin from (E). Data represents mean ± SD of three biological replicates.
(H) Changes in ULK1 protein levels during starvation correspond to the protein levels of NDUFB4, NDUFB2, and ATP5O. HCT116 CRISPRi EATR cells were transduced with NDUFB4, NDUFB2, or ATP5O cDNA constructs, and then transduced with sgRNAs targeting ULK1, NDUFB4, NDUFB2, or ATP5O. Cells were lysed for western blotting and immunoprobed for the indicated proteins.
(I) ULK1 overexpression does not rescue ER-phagy. HCT116 CRISPRi EATR cells with or without ULK1 overexpression were transduced with sgRNAs targeting ULK1, NDUFB4, NDUFB2, or ATP5O and starved for 16hr before FACS measurement for ER-phagy. Data presented as mean ± SD of four biological replicates. (*p < 0.05, ***p < 0.001).
(J) Western blot validation of AMPKa knockout and cDNA re-expression. Parental represents HCT116 CRISPRi EATR cells. AMPK KO (knockout) cells were generated using CRISPR. GST-AMPKa cDNAs were re-expressed in the AMPK KO cells. The catalytically active (GST-AMPKa-CA) form is a truncation of full length AMPKa, resulting in the smaller size. GST-AMPKa-KD (kinase dead) has a K47R point mutation in the CA construct.
(K) Phosphorylation status of downstream AMPKɑ targets were analyzed to verify the catalytically active (CA) and kinase dead (KD) AMPKɑ constructs. AMPKɑ knockout HCT116 EATR clone from (I) were starved for 16 hours and samples were lysed for western blotting and immunoprobed for known target proteins of AMPK phosphorylation.
(L) Catalytically active (CA) AMPKɑ does not inhibit ER-phagy. AMPKɑ knockout HCT116 EATR cells stably expressing catalytically active (CA) AMPKɑ, or kinase dead (KD) AMPKɑ were starved for 16hr before FACS measurement of ER-phagy. Data presented as mean ± SD of three biological replicates.
(A) Densitometry measurement of the ratio between the cleaved and full length mCherry-RAMP4 in Figure 3C. Data represents mean ± SD of five biological replicates. P value indicates two-tailed paired t test (**p < 0.01, ***p < 0.001).
(B) UFL1 protein expression prevents proteasomal degradation of DDRGK1. HCT116 CRISPRi cells were transduced with sgRNA targeting UFL1. Cells were treated with Epoxomicin (100nM) or Folimycin (100nM) for 6hr and then harvested for western blot analysis. p53 and LC3B were used as positive controls for proteasomal and lysosomal inhibitions, respectively.
(C) UBA5 knockdown inhibits ER-phagy. HCT116 CRISPRi EATR cells were transduced with the indicated sgRNA and then starved for 16hr before FACS analysis for ER-phagy. Data represents mean ± SD of three biological replicates. Statistical significance was determined based on two-tailed paired t test. (**p < 0.01; ****p < 0.0001).
(D) UFL1 and DDRGK1 act in series during ER-phagy. HCT116 CRISPRi EATR cells were transduced with the indicated sgRNAs and then starved for 16hr before FACS analysis for ER-phagy. Data represents mean ± SD of three biological replicates. Statistical significance was determined based on two-tailed paired t test.
(E) The same cell lines in (D) were harvested for western blotting to verify the knockdown of targeted genes.
(F) DDRGK1 knockdown prevents ER lumen protein degradation. HCT116 cells stably expressing mCherry-KDEL were transduced with shRNA against either DDRGK1 or UFL1. Cells were then starved for 16hr and harvested for western blot analysis to assess mCherry-KDEL cleavage. A representative blot is shown.
(G) Densitometry analysis of mCherry-KDEL cleavage from (F). Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05; **p < 0.01).
(H) The stable cell lines from Figure 4G were harvested for western blot to verify the knockdown of DDRGK1 and UFL1.
(I) Representative FACS scatterplots from Figure 4G for the measurement of eGFP and mCherry fluorescence using either TetOn-REEP5-mCherry-eGFP or TetOn-CLIMP63-mCherry-eGFP.
(J) Knockdown of DDRGK1 impedes FAM134B-mediated ER-phagy. HCT116 CRISPRi EATR cells stably expressing HA-FAM134B were transduced with sgDDRGK1. Cells were starved for 16hr and subjected for ER-phagy measurement by FACS analysis. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (***p < 0.001; ****p < 0.0001).
(K) HCT116 CRISPRi CCER cells stably expressing HA-FAM134B were transduced with sgDDRGK1. Cells were starved for 16hr and then harvested for western blot analysis of ER protein levels.
(L) Densitometry analysis of mCherry-RAMP4 cleavage from (K). Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (*p < 0.05).
(A) Knockdown of UFM1 does not prevent the appearance of higher molecular weight species of DDRGK1. HCT116 CRISPRi cells stably expressing DDRGK1-HA construct were transduced with sgRNA targeting UFM1. Cells were then harvested for HA-immunoprecipitation and western blotted for the indicated proteins.
(B) Knockdown of UFL1 but not UFM1 reduces DDRGK1 protein levels. HCT116 CRISPRi EATR cells transeduced with the indicated sgRNAs were harvested for western blotting to assess the protein levels of DDRGK1 upon knockdown of the targeted genes.
(C) The cDNA of DDRGK1-HA mutant variants in Figure 5C were stably expressed in HCT116 cells to verify their respective protein sizes in DDRGK1 knockdown cells.
(D) All individual Lysine mutant constructs of DDRGK1 are able to rescue ER-phagy in DDRGK1 knockdown cells. HCT116 CRISPRi EATR cells with sgDDRGK1 were transduced with the indicated DDRGK1-HA mutant constructs. Cells were then starved for 16hr before FACS measurement of ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test. (*p < 0.05, **p < 0.01).
(E) The cell lines generated for (D) were harvested for western blotting to verify the expression of the indicated DDRGK1-HA mutant constructs.
RPN1 Is an ER Surface UFMylation Substrate
(A) Additional colocalization data analysis of Figure 6F for DDRGK1, UFL1 and the ER marker, RAMP4 based on frequency of colocalization. Conditions with good or near perfect colocalization have good correlation between the X- and Y-axes. Representative frequency scatterplot from each condition is shown.
(B) The cell lines generated for Figure 5H were harvested for western blotting to verify the expression of the indicated DDRGK1-HA mutant constructs.
(C) Schematic for the different DDRGK1 and UFL1 mutant constructs. UFL1is tethered to the ER surface by synthetic fusion with RAMP4 at the C-terminal of UFL1.
(D) Western blot validation of the expression levels of each mutant constructs of DDRGK1 and UFL1 shown in (C). Dotted line represents cropped images from the same blots but with irrelevant lanes removed.
(E) Immunofluorescence analysis to confirm that the RAMP4-fused constructs localize to the ER surface. Scale bar represents 10μm. Insets represent three-fold enlargement of the boxed areas.
(F) None of the synthetic fusion of UFL1 or DDRGK1 to the ER surface rescued ER-phagy in sgDDRGK1 cells. HCT116 cells stably expressing sgDDRGK1 were transduced with the indicated cDNAs. Cells were starved for 16hr before FACS measurement for ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01).
(G) None of the previously reported targets of UFMylation are involved in ER-phagy regulation. HCT116 CRISPRi EATR cells were transduced with sgRNAs targeting CDK5RAP3, Sox9 or ASC1 and starved for 16hr before FACS measurement of ER-phagy. Data represents mean ± SD of three biological replicates. P value indicates two-tailed paired t test (**p < 0.01).
(H) The same cell lines used in (G) were harvested for qRT-PCR to assess the knockdown efficiency of each sgRNA.
(I) RPN1 localizes to the ER. U2OS cells were transfected with cDNAs for RPN1-GFP and mCherry-KDEL for 48hr. Scale bar represents 10mm. Inset represents 4x enlargement of the boxed area.
(A) IRE1a is not a target of DDRGK1-mediated UFMylation. HCT116 CRISPRi cells stably expressing HA-UFM1 were transduced with sgRNA targeting DDRGK1. Cells were starved for 16hr before being lysed in non-denaturing condition for HA-immunoprecipitation of UFM1 and western blot analysis of its interaction with IRE1α and DDRGK1.
(B) The same experiment shown in Figure S5B were further probed for the indicated ER or UPR markers. The densitometry measurements for IRE1a, BiP, CANX, CLIMP63 and REEP5 from three biological replicates are shown in Figure 7A.
(C) DDRGK1 depletion results in the upregulation of UPR markers in various cell lines. HepG2 and MCF7 cells were transduced with shRNAs targeting DDRGK1. Cells were then harvested for western blot analysis of DDRGK1 knockdown efficiency and UPR response.
(D) HeLa cells with CRISPR/Cas9-KO of DDRGK1 were harvested for western blot analysis of IRE1α protein levels.
(E) Loss of DDRGK1 results in increased ER content. The same HeLa cell lines generated in (D) were fixed and stained for an ER marker (CANX). Scale bar represents 20mm.
(F) IRE1α depletion prevents UPR response in DDRGK1 knockdown cells. The same cell lines in Figure 7C were harvested for western blotting to measure the protein levels of the indicated UPR genes.
Data Availability Statement
All sequencing datasets generated as part of this study are publicly available in NCBI-SRA under Bioproject PRJNA599329. Original western blot data for all main and supplemental figures in this paper is available at Mendeley Data (https://doi.org/10.17632/ztzfkww2jx.2).