

Structured Abstract
Purpose:
Protein kinase C alpha (gene:PRKCA) is a key regulator of cardiac contractility. Two genetic variants have recently been discovered to regulate PRKCA expression in failing human heart tissue (rs9909004 [T→C] and rs9303504 [C→G]). The association of those variants with clinical outcomes in patients with heart failure (HF), and their interaction with HF drug efficacy, is unknown.
Methods:
Patients with HF in a prospective registry starting in 2007 were genotyped by whole genome array (n=951). The primary outcome was all-cause mortality. Cox proportional hazards models adjusted for established clinical risk factors and genomic ancestry tested the independent association of rs9909004 or rs9303504 and the variant interactions with cornerstone HF pharmacotherapies (beta-blockers or angiotensin-converting enzyme inhibitors/angiotensin receptor blockers) in additive genetic models.
Results:
The minor allele of rs9909004, but not rs9303504, was independently associated with decreased risk for all-cause mortality: adjusted HR = 0.81 (95% CI = 0.67 – 0.98) p = 0.032. The variants did not significantly interact with mortality benefit associated with cornerstone HF pharmacotherapies (p > 0.1 for all).
Conclusions:
A recently discovered cardiac-specific regulatory variant for PRKCA (rs9909004) was independently associated with decreased risk for all-cause mortality in patients with HF. The variant did not interact with mortality benefit associated with cornerstone HF pharmacotherapies.
Keywords: heart failure, protein kinase C α (PRKCA), regulatory variants, mortality
Introduction
The protein kinase C (PKC) family plays an integral role in the pathophysiology of heart failure [1]. This family of nine serine/threonine kinases transmits neurohormonal and mechanical signals in the cardiac myocyte [2]. The alpha member of the PKC family (PRKCA) is a critical mediator of cardiomyocyte calcium handling, and thereby regulates cardiac contractility and the development of heart failure [3-5]. Increased expression of PRKCA, which encodes for PRKCA, decreased cardiac contractility in transgenic mice [6]. Alternatively, knockout of PRKCA in mice increased cardiac contractility and protected against dilated cardiomyopathy and heart failure [6]. In humans, PRKCA expression is increased by 70% in failing human hearts [3]. Pharmacologic inhibition of PRKCA enhances cardiac contractility, and affords cardioprotection, in animal models of heart failure [7-9]. The sum of this experimental evidence suggests that decreased PRKCA activity/expression may be protective in heart failure.
Over 200 genetic variants are significantly associated with PRKCA expression in human left ventricle [10]. Until recently, the true causative variant(s) among the over 200 associated was/were unknown. Two independent studies recently discovered two single nucleotide polymorphisms (SNPs) as the likely causative variants: rs9909004 (T→C) [11] and rs9303504 (C→G) [12]. These variants are common (global minor allele frequencies = 42% and 40%, respectively) and in strong linkage disequilibrium (r2 = 55% - 98%, depending on ancestry) [13]. Both SNPs are intronic and located in cardiac specific enhancer regions [14]. In both studies, the minor allele at either locus was significantly associated with decreased cardiac expression of PRKCA. Given the prior experimental evidence suggesting that decreased cardiac expression of PRKCA would be associated with improved heart failure outcomes, the minor alleles of these two SNPs could be protective in patients with heart failure. However, the association of rs9909004 and rs9303504 with clinical outcomes in patients with heart failure is unknown. Therefore, the primary objective of this study was to assess the independent association of PRKCA rs9909004 and rs9303504 with all-cause mortality in patients with heart failure. Based on the prior experimental data, our hypothesis was that the minor alleles of rs9909004 and rs9303504 would be protective.
If rs9909004 and rs9303504 impact heart failure clinical outcomes, it needs to be considered whether those variants are acting by modulating the efficacy of cornerstone heart failure medications. Based on their potential to affect PRKCA expression, rs9909004 and rs9303504 may alter the efficacy of medications targeting elements of PRKCA signaling pathways. The mechanisms of action of three cornerstone heart failure medication classes [15], angiotensin-converting enzyme inhibitors (ACEI), angiotensin II receptor type 1 (AT1) blockers (ARB), and beta-adrenergic receptor blockers, all involve PRKCA. ARB and ACEI either directly or indirectly target AT1, which has been shown to regulate contractility in cardiomyocytes via PRKCA signaling [16-18]. Moreover, PRKCA is known to regulate two proteins (phospholamban and protein phosphatase inhibitor-1) involved in the beta-adrenergic signaling cascade and the associated contractile response [6, 19]. Increased cardiomyocyte PRKCA activity can therefore increase therapeutic response to ACEI, ARB, and beta-blockers. Therefore our second hypothesis is that the minor alleles of rs9909004 and rs9303504, which reduce PRKCA expression, will be associated with decreased response to these cornerstone heart failure therapies. Accordingly, the secondary objectives of this study were to assess the potential for PRKCA rs9909004 or rs9303504 to modify benefit from ACEI/ARB or beta-blockers in heart failure patients (i.e., ACEI/ARB or beta-blocker associated reductions in all-cause mortality).
Methods
Patients
Patients for this study came from a prospective, heart failure registry designed to discover novel ways to predict prognosis and the response to heart failure therapies. The registry is based at Henry Ford Health System in Detroit, MI, USA, and it started in October 2007 and completed enrollment in March 2015. Patients were included in the heart failure registry if they were 18 years of age or older, had health insurance coverage, and met the definition for heart failure as defined by the Framingham Heart Study [20]. Patients included in the analysis had at least one documented left ventricular ejection fraction <50%. Patients were excluded if they required dialysis or supplemental oxygen. Detailed clinical information (e.g., demographics, physical examination results, past medical history, laboratory values, functional status, and medication use) and blood samples were collected at the time of study enrollment. Patient deaths through July 28, 2016 were identified using the Social Security Administration Death Master File, the Michigan State Division of Vital Records, and administrative data from Henry Ford Health System. This study was approved by the institutional review board of the Henry Ford Health System, and all patients gave written informed consent prior to participating.
Calculation of Drug Exposure
Exposure of the patients to beta-blockers and ACEI or ARB was calculated using dose standardization and pharmacy claims data as previously described [21]. Briefly, doses of specific drugs were standardized into dose equivalents by the target dose used in clinical trials of heart failure with reduced ejection fraction (HFrEF) or, if the drug was not tested in a HFrEF clinical trial (e.g., atenolol), by the maximum daily dose. For example, 25 mg of carvedilol per day (i.e., 12.5 mg twice daily) was considered a 0.5 beta-blocker standardized dose equivalent. Drug exposure was then calculated by multiplying the standardized dose equivalent by the quantity of medication dispensed from a pharmacy in a 6-month time block, divided by the total number of days in the 6-month time block. Drug exposure was calculated for each patient for each day of observation, and thus this method accounts for both dose and adherence over a rolling period of time (6 months). For example, if a patient was prescribed 12.5 mg of carvedilol twice daily, and had picked up their prescription from the pharmacy so that there was continuous availability over the previous 6 months, then their calculated average beta-blocker exposure over this time period would be 0.5. We have previously demonstrated that this approach for calculating drug exposure is superior to a single time point, dichotomous classification of drug exposure (e.g., discharge medication status) [22].
Genotyping
Blood samples were collected at enrollment into the heart failure registry and were immediately processed and stored at −70°C. All patients donated blood and were genotyped using the Axiom™ Biobank array (ThermoFisher Scientific). The array includes ~600K polymorphisms: 1) ~300K genome-wide association study (GWAS) markers with minor allele frequencies >1%; 2) ~250K low frequency (<1%) coding variants selected from global exome sequencing projects; and an 3) additional ~50K variants to improve African ancestry coverage (Yoruba in Ibadan, Nigeria [YRI] booster). The two PRKCA SNPs rs9909004 and rs9303504 were not directly genotyped by the array, and imputed genotypes for the SNPs were used. Genome-wide imputation using the 1000 Genomes Project (1KGP) haplotype reference panel was performed using the Michigan Imputation Server with Minimac3 [23]. The accepted imputation score was >0.3. The accuracy of the genotypes for rs9909004 and rs9303504 was checked by performing the chi-square test for Hardy Weinberg equilibrium within each self-reported race group and by comparing the minor allele frequencies in each self-reported race group in this registry to minor allele frequencies reported in the 1KGP.
Statistical Analysis:
Continuous baseline variables were summarized by the mean ± standard deviation and categorical baseline variables were summarized by counts and percentages. The primary outcome was time-to all-cause mortality. Each SNP was shown to have an additive effect on PRKCA cardiac expression [12, 11], and thus they were coded in the additive genetic model for the number of minor alleles (i.e., rs9909004 genotypes TT, TC, and CC were coded as 0, 1, and 2; rs9303504 genotypes CC, CG, and GG were coded as 0, 1, and 2). Each SNP was tested individual in time-dependent Cox proportional hazards regression models adjusted for known clinical risk factors for mortality in patients with heart failure: the Meta-Analysis Global Group in Chronic Heart Failure (MAGGIC) risk score [24], self-reported race, and N-terminal pro b-type natriuretic peptide (NT pro-BNP) level. To overcome the influence of population stratification on genetic effects [25], principal components (PC) analysis was performed by PLINK on the SNP array data after linkage disequilibrium pruning [26]. The top three most significant PCs were included in the association models as covariates. The secondary objective, evaluating a potential interaction of each of the PRKCA SNPs with beta-blocker or ACEI/ARB exposure, was tested by incorporating a multiplicative interaction term between the PRKCA SNPs and the drug exposure variable in the Cox proportional hazards models for all-cause mortality (i.e., number of minor alleles of each SNP*beta-blocker exposure and number of minor alleles of each SNP*ACEI/ARB exposure in separate models). Linkage disequilibrium between rs9909004 and rs9303504 was calculated among African American and white study participants using PLINK [26]. All statistical analyses were performed in SAS version 9.4 (SAS Institute, Cary, NC) unless noted otherwise above; p <0.05 was considered statistically significant for main effects and p <0.1 was considered statistically significant for interaction effects. Given a level of statistical significance of p < 0.05 and our sample size, we estimated 80% power to detect a hazard ratio ≤ 0.83 for each SNP.
Results
This analysis consisted of 951 heart failure patients with a mean duration of follow-up of 1104 days (standard deviation [SD] ±701 days) and a total of 239 deaths (25%). Baseline characteristics of the registry overall and stratified by rs9909904 and rs9303504 genotypes are presented in Tables 1 and 2, respectively. The genotyping results and accuracy check stratified by patients’ self-reported race are displayed in Table 3. The minor allele frequencies in the registry were similar to publicly reported frequencies in 1KGP [13], and the genotypes did not significantly deviate from Hardy Weinberg equilibrium for either SNP. The two SNPs were in strong linkage disequilibrium in both the self-reported African American (r2 = 0.86; D’ = 0.95) and white (r2 = 0.96; D’ = 0.99) patients.
Table 1.
Baseline characteristics of the heart failure genetic registry overall and stratified by rs9909004 genotypes
| rs9909004 genotype | |||||
|---|---|---|---|---|---|
| Baseline characteristics | All patients (n = 951) |
TT (n = 309; 32%) |
CT (n = 452; 48%) |
CC (n = 190; 20%) |
*p-value |
| Female | 341 (35.9%) | 104 (33.7%) | 165 (36.5%) | 72 (37.9%) | 0.584 |
| Self-reported African-American race | 481 (50.6%) | 171 (55.3%) | 231 (51.1%) | 79 (41.6%) | 0.011 |
| Age (years) | 68.3 ± 11.7 | 67.5 ± 11.5 | 68.2 ± 11.5 | 69.7 ± 12.2 | 0.119 |
| Left ventricular ejection fraction (%) | 34.7 ± 10.9 | 34.7 ± 10.9 | 34.3 ± 11.0 | 35.7 ± 10.8 | 0.368 |
| Ischemic etiology | 416 (43.7%) | 123 (39.8%) | 199 (44.0%) | 94 (49.5%) | 0.106 |
| Hypertension | 855 (89.9%) | 283 (91.6%) | 402 (88.9%) | 170 (89.5%) | 0.480 |
| Chronic obstructive pulmonary disease | 220 (23.1%) | 74 (24.0%) | 95 (21.0%) | 51 (26.8%) | 0.256 |
| Chronic kidney disease | 222 (23.3%) | 81 (26.2%) | 106 (23.5%) | 35 (18.4%) | 0.136 |
| Atrial fibrillation | 267 (28.1%) | 94 (30.4%) | 120 (26.6%) | 53 (27.9%) | 0.505 |
| Stroke/transient ischemic attack | 122 (12.8%) | 36 (11.7%) | 65 (14.4%) | 21 (11.1%) | 0.388 |
| Diabetes | 399 (42.0%) | 119 (38.5%) | 198 (43.8%) | 82 (43.2%) | 0.324 |
| Body mass index (kg/m2) | 31.0 ± 7.3 | 31.4 ± 7.5 | 30.8 ± 7.3 | 30.9 ± 7.3 | 0.457 |
| Systolic blood pressure (mmHg) | 129 ± 23 | 129 ± 24 | 129 ± 23 | 129 ± 21 | 0.988 |
| Heart rate (beats per minute) | 71.2 ± 12.9 | 72.4 ± 13.7 | 70.8 ± 12.3 | 70.2 ± 12.8 | 0.131 |
| NT pro-BNP (pmol/L) | 368 ± 384 | 368 ± 389 | 393 ± 404 | 308 ± 316 | 0.038 |
| Serum creatinine (mg/dL) | 1.29 ± 0.93 | 1.26 ± 0.76 | 1.36 ± 1.15 | 1.15 ± 0.46 | 0.050 |
| MAGGIC risk score (w/o beta-blocker) | 18.2 ± 7.3 | 17.9 ± 7.1 | 18.2 ± 7.4 | 18.5 ± 7.1 | 0.645 |
| 1st principal component | 0.0007 ± 0.035 | 0.0017 ± 0.037 | −0.0004 ± 0.037 | 0.0014 ± 0.025 | 0.687 |
| 2nd principal component | 0.0038 ± 0.042 | 0.0053 ± 0.057 | 0.0030 ± 0.031 | 0.0034 ± 0.032 | 0.737 |
| 3rd principal component | −0.0002 ± 0.032 | 0.0015 ± 0.040 | 0.0010 ± 0.026 | −0.0057 ± 0.029 | 0.029 |
| Beta-blocker exposure | 0.27 ± 0.29 | 0.26 ± 0.29 | 0.28 ± 0.30 | 0.27 ± 0.30 | 0.453 |
| Any beta-blocker exposure | 623 (65.5%) | 206 (66.7%) | 291 (64.4%) | 126 (66.3%) | 0.782 |
| Any ACEI/ARB exposure | 580 (61.0%) | 194 (62.8%) | 272 (60.2%) | 114 (60.0%) | 0.733 |
| Length of follow-up (days) | 1104 ± 701 | 1052 ± 712 | 1145 ± 714 | 1092 ± 646 | 0.194 |
| Deaths | 239 (25.1%) | 93 (30.1%) | 99 (21.9%) | 47 (24.7%) | 0.037 |
ACEI: angiotensin converting enzyme inhibitor; ARB: angiotensin receptor blocker; MAGGIC: Meta-Analysis Global Group in Chronic Heart Failure risk score; NT pro-BNP: N-terminal pro b-type natriuretic peptide
p-value is for difference among the three genotypes. Bolded p-values indicate p < 0.05.
Table 2.
Baseline characteristics of the heart failure genetic registry overall and stratified by rs9303504 genotype
| rs9303504 genotype | |||||
|---|---|---|---|---|---|
| Baseline characteristics | All patients (n = 951) |
CC (n = 307; 32%) |
CG (n = 451; 48%) |
GG (n = 193; 20%) |
*p-value |
| Female | 341 (35.9%) | 107 (34.9%) | 161 (35.7%) | 73 (37.8%) | 0.793 |
| Self-reported African-American race | 481 (50.6%) | 169 (55.1%) | 227 (50.3%) | 85 (44.0%) | 0.056 |
| Age (years) | 68.3 ± 11.7 | 67.7 ± 11.6 | 68.4 ± 11.4 | 69.0 ± 12.4 | 0.445 |
| Left ventricular ejection fraction (%) | 34.7 ± 10.9 | 34.9 ± 10.9 | 34.4 ± 11.0 | 35.2 ± 10.8 | 0.691 |
| Ischemic etiology | 416 (43.7%) | 121 (39.4%) | 202 (44.8%) | 93 (48.2%) | 0.130 |
| Hypertension | 855 (89.9%) | 282 (91.9%) | 400 (88.7%) | 173 (89.6%) | 0.362 |
| Chronic obstructive pulmonary disease | 220 (23.1%) | 73 (23.8%) | 95 (21.1%) | 52 (26.9%) | 0.255 |
| Chronic kidney disease | 222 (23.3%) | 79 (25.7%) | 107 (23.7%) | 36 (18.7%) | 0.184 |
| Atrial fibrillation | 267 (28.1%) | 94 (306%) | 119 (26.4%) | 54 (28.0%) | 0.444 |
| Stroke/transient ischemic attack | 122 (12.8%) | 36 (11.7%) | 65 (14.4%) | 21 (10.9%) | 0.368 |
| Diabetes | 399 (42.0%) | 120 (39.1%) | 199 (44.1%) | 80 (41.5%) | 0.381 |
| Body mass index (kg/m2) | 31.0 ± 7.3 | 31.4 ± 7.5 | 30.7 ± 7.3 | 30.9 ± 7.3 | 0.430 |
| Systolic blood pressure (mmHg) | 129 ± 23 | 130 ± 24 | 129 ± 23 | 129 ± 20 | 0.885 |
| Heart rate (beats per minute) | 71.2 ± 12.9 | 72.1 ± 13.8 | 70.8 ± 12.3 | 70.6 ± 12.6 | 0.312 |
| NT pro-BNP (pmol/L) | 368 ± 384 | 370 ± 387 | 389 ± 406 | 315 ± 319 | 0.082 |
| Serum creatinine (mg/dL) | 1.29 ± 0.93 | 1.23 ± 0.59 | 1.38 ± 1.22 | 1.15 ± 0.47 | 0.014 |
| MAGGIC risk score (w/o beta-blocker) | 18.2 ± 7.3 | 17.8 ± 7.2 | 18.3 ± 7.4 | 18.3 ± 7.0 | 0.661 |
| 1st principal component | 0.0007 ± 0.035 | 0.0019 ± 0.038 | -0.0002 ± 0.037 | 0.0003 ± 0.026 | 0.726 |
| 2nd principal component | 0.0038 ± 0.042 | 0.0055 ± 0.058 | 0.0028 ± 0.031 | 0.0034 ± 0.032 | 0.672 |
| 3rd principal component | −0.0002 ± 0.032 | 0.0018 ± 0.040 | 0.0008 ± 0.026 | −0.0057 ± 0.028 | 0.023 |
| Beta-blocker exposure | 0.27 ± 0.29 | 0.26 ± 0.29 | 0.28 ± 0.29 | 0.28 ± 0.31 | 0.663 |
| Any beta-blocker exposure | 623 (65.5%) | 204 (66.5%) | 288 (63.9%) | 131 (67.9%) | 0.565 |
| Any ACEI/ARB exposure | 580 (61.0%) | 191 (62.2%) | 271 (60.1%) | 118 (61.1%) | 0.840 |
| Length of follow-up (days) | 1104 ± 701 | 1056 ± 714 | 1138 ± 721 | 1103 ± 627 | 0.286 |
| Deaths | 239 (25.1%) | 91 (29.6%) | 100 (22.2%) | 48 (24.9%) | 0.066 |
ACEI: angiotensin converting enzyme inhibitor; ARB: angiotensin receptor blocker; MAGGIC: Meta-Analysis Global Group in Chronic Heart Failure risk score; NT pro-BNP: N-terminal pro b-type natriuretic peptide
p-value is for difference among the three genotypes. Bolded p-values indicate p < 0.05.
Table 3.
Genotyping results and accuracy check stratified by patients’ self-reported race
| Self-reported race | ||
| African-American (n = 575) 51% |
White (n = 547) 49% |
|
| Minor allele frequencies | ||
| rs9909004 (C) | ||
| registry | 0.40 | 0.47 |
| 1000 Genomes | 0.35 (African) |
0.47 (European) |
| rs9303504 (G) | ||
| registry | 0.41 | 0.47 |
| 1000 Genomes | 0.37 (African) |
0.47 (European) |
| Hardy Weinberg Equilibrium p-value | ||
| rs9909004 | 0.839 | 0.265 |
| rs9309504 | 0.422 | 0.516 |
| Linkage disequilibrium | ||
| r2 | 0.86 | 0.96 |
| D’ | 0.95 | 0.99 |
In the survival analyses (adjusted for MAGGIC risk score, self-reported race, NT pro-BNP level, and the first three principal components of genomic ancestry), rs9909004 was significantly associated with all-cause mortality in the additive genetic model: rs9909904 adjusted HR = 0.81 (95% CI = 0.67 – 0.98) p = 0.032. rs9303504 was not significantly associated: adjusted HR = 0.83 (95% CI = 0.69 – 1.01) p = 0.059. Kaplan-Meier curves for each individual SNP are displayed in Figures 1 and 2, and the log rank p-values for rs9909004 and rs9303504 were p = 0.019 and p = 0.038, respectively.
Figure 1.
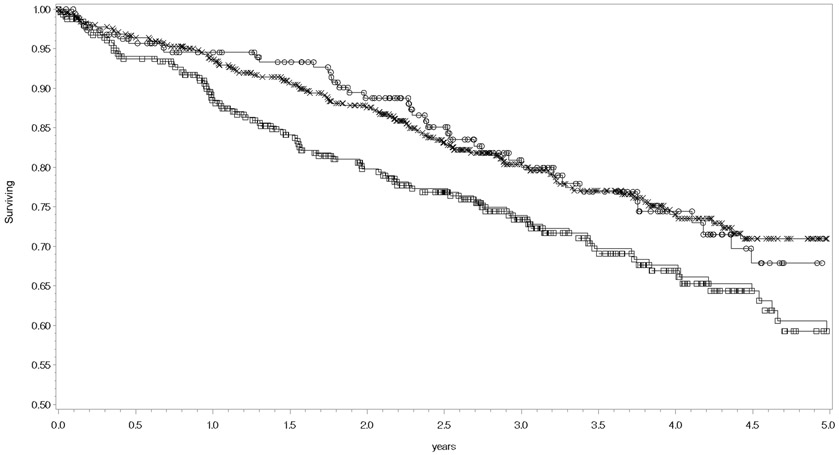
Kaplan-Meier curves for all-cause mortality stratified by rs9909004 genotypes. Censored patients with the T/T genotype are symbolized as squares,, C/T genotype as the letter X, and C/C genotype as circles. The logrank p-value = 0.0189.
Figure 2.
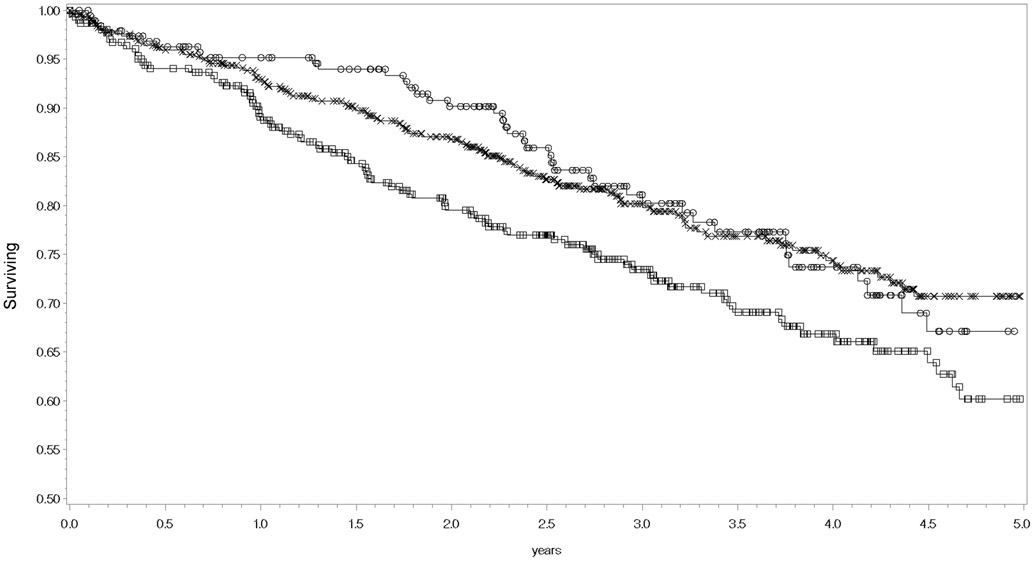
Kaplan-Meier curves for all-cause mortality stratified by rs9303504 genotypes. Censored patients with the C/C genotype are symbolized as squares, C/G as the letter X, and G/G genotype as circles. The logrank p-value = 0.0375.
The secondary analysis of interactions between the PRKCA SNPs and beta-blocker or ACEI/ARB associated benefit for all-cause mortality did not yield statistically significant findings. For an interaction with beta-blockers, the adjusted p-values for rs9909004 and rs9303504 were p = 0.500 (β = 0.23) and p = 0.445 (β = 0.26), respectively. For an interaction with ACEI/ARB, the adjusted p-values for rs9909004 and rs9303504 were p = 0.157 (β = 0.37) and p = 0.301 (β = 0.27), respectively. The associations of beta-blocker exposure and ACEI/ARB exposure with a reduction in all-cause mortality were independent of the total number of minor alleles of the PRKCA SNPs (adjusted HR comparing zero to target beta-blocker exposure in the model including the PRKCA SNPs = 0.47 [0.29 – 0.74] p = 0.001; adjusted HR comparing zero to target ACEI/ARB exposure in the model including the PRKCA SNPs = 0.63 [0.44 – 0.90] p = 0.011). These results suggested that the PRKCA SNPs did not modify the effectiveness of beta-blockers or ACEI/ARB in patients with heart failure.
Discussion
Two previous studies recently discovered two SNPs (rs9909004 and rs9303503) regulating expression of PRKCA in heart tissue [12, 11], but the association of those SNPs with clinical outcomes in patients with heart failure was unknown. Our results from a prospective registry of patients with heart failure showed that rs9909004 was significantly associated with the risk of all-cause mortality, and the association was independent of established clinical risk factors. The association of rs9909004 is in the direction expected from previous experimental evidence: the minor allele, which decreases cardiac PRKCA expression [12, 11], was the protective allele. Mechanistically, rs9909004 is in enhancer regions in 12 different tissue types, DNAse hypersensitivity sites in 14 different tissues, and it also changes the regulatory motif for transcription factor Pou3f1 [14]. These regulatory effects are supported by data from the Genotype-Tissue Expression (GTEx) project, in which the minor allele of rs9909004 is significantly associated with decreased expression of PRKCA in left ventricular and atrial tissues [10]. Based on the additive effects of each SNP on cardiac expression of PRKCA [12, 11], we a priori defined the additive genetic model in our statistical analysis. However, upon inspection of the Kaplan-Meier survival curves, the association of rs9909004 with all-cause mortality appears to more closely follow a dominant genetic model. Thus, future replication studies with clinical outcomes should consider using the dominant genetic model.
Our results are consistent with several previous human ex vivo and animal in vivo studies suggesting that increased cardiac expression of PRKCA could cause adverse effects related to heart failure [1]. Pharmacologic inhibition of PRKCA enhances cardiac contractility and cardioprotection in animal models [6-9], further supporting our findings of a protective association for the minor alleles. A mixed PRKCA/PRKCB inhibitor is currently in phase I/II clinical trials for patients with heart failure (clinicaltrials.gov identifier: NCT02769611), and our study supports inhibiting PRKCA as a potential mechanism for treating heart failure. These data may also indicate a potential opportunity for precision therapy with PRKCA pharmacologic inhibition. Specifically, heart failure patients without the protective minor allele of PRKCA rs9909004 may derive greater benefit from PRKCA inhibition.
Despite our results being consistent with previous experimental studies, our results are not consistent with a clinical association study by Hu et al. [12]. Hu et al. found the reverse association with similar clinical outcomes: the minor alleles of these PRKCA SNPs were associated with adverse left ventricular remodeling, impaired contractile function, and dilated cardiomyopathy. It remains unclear why the findings by Hu et al. are inconsistent with the human ex vivo studies, animal in vivo studies, and our study. Hu et al. suggested that there could be species-specific differences in PRKCA effects, and the timing of PRKCA effects is important (i.e., acute phase of heart failure in animal models versus chronic increased PRKCA exposure in affected humans). The difference between our results and the results by Hu et al. could be influenced by differences in the patient populations and the clinical outcomes studied. For example, all patients in our study had documented heart failure, while Hu et al. excluded patients with heart failure. It is well known that heart failure induces dramatic changes in cardiac gene expression [27], and the expression of PRKCA in normal hearts is known to be much lower when compared to expression in heart failure [3]. The effects of the PRKCA SNPs may also differ depending on the clinical outcome investigated. Specifically, Hu et al. investigated left ventricular remodeling, contractile function, and new onset dilated cardiomyopathy, whereas we investigated all-cause mortality (in patients with established heart failure). Our results regarding mortality are consistent with an aggregate analysis of clinical trials of a PRKCA inhibitor in patients with diabetes [28]. There were fewer deaths in the groups treated with the PRKCA inhibitor than the groups treated with placebo [28].
It is also important to note that the PRKCA association with mortality was not mediated via heart failure medication effectiveness (i.e., it was not a pharmacogenetic effect). Despite the biologic plausibility [16-18], the studied PRKCA SNPs did not interact with beta-blocker or ACEI/ARB exposure in terms of survival benefit. This suggests that the mechanism through which these PRKCA SNPs may affect mortality in patients with heart failure is independent of the effects of these treatments. It could be that PRKCA is acting in the drug pathways, but that the effect is simply proportionate across genotypes. PRKCA plays a myriad of roles within the cardiac myocyte, including the transmission of neurohormonal, mechanical, and electrical signals [29]. So alternatively, the mechanism could be completely outside the beta-blocker and ACEI/ARB pathways, such as via calcium cycling or phosphorylation of ion channels.
Our study has several limitations. The registry enrolled patients from a single site, and thus our results may not be generalizable to broader patient populations. While our cohort is quite diverse in terms of race and stage of heart failure, our findings need to be replicated in an independent heart failure patient population. To our knowledge, a similar dataset consisting of heart failure patients with highly granular clinical and genomic data is not available. Our registry is underpowered to perform a genome-wide association study, or GWAS, which is why we relied on previous experimental data to select these candidate genetic variants. It is possible that rs9303504 is also associated with all-cause mortality in patients with heart failure, but our study was underpowered to detect it. Given the strong linkage disequilibrium between the two PRKCA SNPs, our study was also underpowered to distinguish the independent associations of the two SNPs. We used imputed genotypes for both SNPs; however, these were common alleles and the imputation used a well-described reference sample with good confidence. Finally, since this study was an observational registry, the interactions of the PRKCA SNPs with beta-blocker, ACEI, or ARB efficacy are limited by confounding by indication. We tried to mitigate this using extensive covariate adjustment, and our cohort includes a significant proportion of untreated patients, enhancing power. However, due to this fact we could not truly test causality, which would require a randomized controlled trial.
Conclusions
The minor allele of PRKCA rs9909004, but not rs9303504, was independently associated with decreased risk for all-cause mortality in patients with heart failure. The SNPs did not significantly interact with benefit from beta-blockers or ACEI/ARB; suggesting a mechanism that is independent of adrenergic or angiotensin signaling. Further studies are needed to replicate this association in additional heart failure patient populations. These results support PRKCA as a drug target in patients with heart failure.
Acknowledgments
Funding sources
This research was supported by the National Heart, Lung, and Blood Institute (Luzum L30HL110279-03, Lanfear R01HL103871 and R01HL132154; Williams R01HL118267). Dr. Williams was also supported by the National Institute of Allergy and Infectious Diseases (R01AI079139) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK064695, R01DK113003). Dr. Luzum was also supported by a New Investigator Award from the American Association of Colleges of Pharmacy.
Abbreviations
- ACEI
angiotensin converting enzyme inhibitor
- ARB
angiotensin receptor blocker
- AT1
angiotensin II receptor type 1
- CI
confidence interval
- eQTLs
expression quantitative trait loci
- GWAS
genome-wide association study
- HFrEF
heart failure with reduced ejection fraction
- HR
hazard ratio
- MAGGIC
Meta-Analysis Global Group in Chronic Heart Failure risk score
- NT pro-BNP
N-terminal pro b-type natriuretic peptide
- PKC
protein kinase C family
- PRKCA
alpha member of the protein kinase C family
- PRKCA:
gene for the alpha member of the protein kinase C family
- SNP
single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Disclosures
None.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institution (Henry Ford Health System institutional review board approval number 4562) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
References
- 1.Liu Q, Molkentin JD. Protein kinase Calpha as a heart failure therapeutic target. Journal of Molecular and Cellular Cardiology. 2011;51(4):474–8. doi: 10.1016/j.yjmcc.2010.10.004 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayer AL, Heidkamp MC, Patel N, Porter M, Engman S, Samarel AM. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Molecular and cellular biochemistry. 2003;242(1-2):145–52. [PubMed] [Google Scholar]
- 3.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99(3):384–91. [DOI] [PubMed] [Google Scholar]
- 4.Braz JC, Bueno OF, De Windt LJ, Molkentin JD. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2). The Journal of cell biology. 2002;156(5):905–19. doi: 10.1083/jcb.200108062 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorn GW 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. The Journal of clinical investigation. 2005;115(3):527–37. doi: 10.1172/JCI24178 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nature medicine. 2004;10(3):248–54. doi: 10.1038/nm1000 [doi]. [DOI] [PubMed] [Google Scholar]
- 7.Ladage D, Tilemann L, Ishikawa K, Correll RN, Kawase Y, Houser SR et al. Inhibition of PKCalpha/beta with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circulation research. 2011;109(12):1396–400. doi: 10.1161/CIRCRESAHA.111.255687 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle AJ, Kelly DJ, Zhang Y, Cox AJ, Gow RM, Way K et al. Inhibition of protein kinase C reduces left ventricular fibrosis and dysfunction following myocardial infarction. Journal of Molecular and Cellular Cardiology. 2005;39(2):213–21. doi:S0022-2828(05)00097-0 [pii]. [DOI] [PubMed] [Google Scholar]
- 9.Liu Q, Chen X, Macdonnell SM, Kranias EG, Lorenz JN, Leitges M et al. Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circulation research. 2009;105(2):194–200. doi: 10.1161/CIRCRESAHA.109.195313 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45(6):580–5. doi: 10.1038/ng.2653 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Zhang L, Binkley PF, Sadee W, Wang D. Regulatory Variants Modulate Protein Kinase C alpha (PRKCA) Gene Expression in Human Heart. Pharmaceutical research. 2017;34(8):1648–57. doi: 10.1007/s11095-017-2102-x [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu R, Morley MP, Brandimarto J, Tucker NR, Parsons VA, Zhao SD et al. Genetic Reduction in Left Ventricular Protein Kinase C-alpha and Adverse Ventricular Remodeling in Human Subjects. CirculationGenomic and precision medicine. 2018;11(3):e001901. doi: 10.1161/CIRCGEN.117.001901 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632; 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40(Database issue):D930–4. doi: 10.1093/nar/gkr917 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Writing Committee M, Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–327. doi: 10.1161/CIR.0b013e31829e8776 [doi]. [DOI] [PubMed] [Google Scholar]
- 16.Zou Y, Komuro I, Yamazaki T, Aikawa R, Kudoh S, Shiojima I et al. Protein kinase C, but not tyrosine kinases or Ras, plays a critical role in angiotensin II-induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. The Journal of biological chemistry. 1996;271(52):33592–7. [DOI] [PubMed] [Google Scholar]
- 17.Malhotra A, Kang BP, Cheung S, Opawumi D, Meggs LG. Angiotensin II promotes glucose-induced activation of cardiac protein kinase C isozymes and phosphorylation of troponin I. Diabetes. 2001;50(8):1918–26. [DOI] [PubMed] [Google Scholar]
- 18.Liang W, Oudit GY, Patel MM, Shah AM, Woodgett JR, Tsushima RG et al. Role of phosphoinositide 3-kinase {alpha}, protein kinase C, and L-type Ca2+ channels in mediating the complex actions of angiotensin II on mouse cardiac contractility. Hypertension (Dallas, Tex: 1979). 2010;56(3):422–9. doi: 10.1161/HYPERTENSIONAHA.109.149344 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C et al. Type 1 phosphatase, a negative regulator of cardiac function. Molecular and cellular biology. 2002;22(12):4124–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. The New England journal of medicine. 1971;285(26):1441–6. doi: 10.1056/NEJM197112232852601 [doi]. [DOI] [PubMed] [Google Scholar]
- 21.Lanfear DE, Hrobowski TN, Peterson EL, Wells KE, Swadia TV, Spertus JA et al. Association of beta-blocker exposure with outcomes in heart failure differs between African American and white patients. CirculationHeart failure. 2012;5(2):202–8. doi: 10.1161/CIRCHEARTFAILURE.111.965780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanfear D, Peterson E, Wells K, Williams LK. Discharge Medication Status Compares Poorly with Claims-Based Outpatient Medication Exposure Estimates. Circulation: Cardiovascular Quality and Outcomes. 2015;4(Suppl 2):AP234–AP. [Google Scholar]
- 23.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A et al. Next-generation genotype imputation service and methods. Nature genetics. 2016;48(10):1284–7. doi: 10.1038/ng.3656 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pocock SJ, Ariti CA, McMurray JJ, Maggioni A, Kober L, Squire IB et al. Predicting survival in heart failure: a risk score based on 39 372 patients from 30 studies. European heart journal. 2013;34(19):1404–13. doi: 10.1093/eurheartj/ehs337 [doi]. [DOI] [PubMed] [Google Scholar]
- 25.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38(8):904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan FL, Moravec CS, Li J, Apperson-Hansen C, McCarthy PM, Young JB et al. The gene expression fingerprint of human heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(17):11387–92. doi: 10.1073/pnas.162370099 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Javey G, Schwartz SG, Flynn HW, Aiello LP, Sheetz MJ. Ruboxistaurin: Review of Safety and Efficacy in the Treatment of Diabetic Retinopathy. Clinical Medicine Insights: Therapeutics. 2010;2:CMT.S5046. doi: 10.4137/CMT.S5046; 06 10.4137/CMT.S5046. [DOI] [Google Scholar]
- 29.Ferreira JC, Mochly-Rosen D, Boutjdir M. Regulation of cardiac excitability by protein kinase C isozymes. Frontiers in bioscience (Scholar edition). 2012;4:532–46. doi:283 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]