

Abstract
Motivation: The Expectation Maximization (EM) algorithm, in the form of the Baum–Welch algorithm (for hidden Markov models) or the Inside-Outside algorithm (for stochastic context-free grammars), is a powerful way to estimate the parameters of stochastic grammars for biological sequence analysis. To use this algorithm for multiple-sequence evolutionary modelling, it would be useful to apply the EM algorithm to estimate not only the probability parameters of the stochastic grammar, but also the instantaneous mutation rates of the underlying evolutionary model (to facilitate the development of stochastic grammars based on phylogenetic trees, also known as Statistical Alignment). Recently, we showed how to do this for the point substitution component of the evolutionary process; here, we extend these results to the indel process.
Results: We present an algorithm for maximum-likelihood estimation of insertion and deletion rates from multiple sequence alignments, using EM, under the single-residue indel model owing to Thorne, Kishino and Felsenstein (the ‘TKF91’ model). The algorithm converges extremely rapidly, gives accurate results on simulated data that are an improvement over parsimonious estimates (which are shown to underestimate the true indel rate), and gives plausible results on experimental data (coronavirus envelope domains). Owing to the algorithm's close similarity to the Baum–Welch algorithm for training hidden Markov models, it can be used in an ‘unsupervised’ fashion to estimate rates for unaligned sequences, or estimate several sets of rates for sequences with heterogenous rates.
Availability: Software implementing the algorithm and the benchmark is available under GPL from http://www.biowiki.org/
Contact: ihh@berkeley.edu
1 INTRODUCTION
A cornerstone of stochastic grammar-based profiling of DNA, protein and RNA sequences is the fast and accurate estimation of the probability parameters of the model, given a ‘training set’. This is achieved for hidden Markov models (HMMs) using the Baum–Welch algorithm, and for stochastic context-free grammars (SCFGs) using the Inside-Outside algorithm (Durbin et al., 1998); both are examples of the Expectation Maximization (EM) algorithm (Dempster et al., 1977).
Currently, most HMM- and SCFG-based methods are consensus- or pair-based; they do not use the underlying phylogenetic tree directly in their training [although consensus methods use it indirectly, when over-represented clades are downweighted in the estimation of the consensus profile (Durbin et al., 1998)]. The full power of evolutionary methods to model mutation rates and phylogenetic correlations is therefore unused. This may be because evolutionary models relating multiple sequences, using an arbitary phylogeny, are more complicated than consensus models, which effectively assume a star-shaped phylogeny (with the consensus sequence at the centre of the star). Nonetheless, there has been some progress in implementing stochastic evolutionary models for multiple sequence alignment and profiling (Mitchison and Durbin, 1995; Hein et al., 2000; Hein, 2001; Holmes and Bruno, 2001; Holmes and Rubin, 2002; Holmes, 2003; Miklós et al., 2004). To develop this work further, a natural step is to extend the probabilistic results used for HMMs and SCFGs to the evolutionary domain.
Previous work demonstrated the potential of the EM algorithm as a quick and effective tool for finding maximum-likelihood (ML) rate matrices for point substitution processes (Holmes and Rubin, 2002). As with all EM algorithms, this algorithm proceeds by repeated application of an E-step followed by an M-step. During the E-step, limited information about the inferred evolutionary history is gathered in the form of certain ‘sufficient statistics’, which are then used during the M-step to improve the estimate of the rate parameters. Specifically, these sufficient statistics are (1) the expected composition of the root sequence  , (2) the expected number of times,
, (2) the expected number of times,  , that each mutation of type i → j occurred and (3) the expected length of time,
, that each mutation of type i → j occurred and (3) the expected length of time, 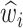 , that each residue was available for substitution. All expectations are taken over the posterior distribution of substitution histories. In the M-step, equilibrium frequencies are set proportional to
, that each residue was available for substitution. All expectations are taken over the posterior distribution of substitution histories. In the M-step, equilibrium frequencies are set proportional to  and substitution rates equal to
and substitution rates equal to  . This process converges extremely rapidly and, assuming the absence of significantly suboptimal local minima in the likelihood function (which in practise is often the case), results in ML substitution rate matrices.
. This process converges extremely rapidly and, assuming the absence of significantly suboptimal local minima in the likelihood function (which in practise is often the case), results in ML substitution rate matrices.
This paper presents an EM algorithm for the estimation of indel rates in the TKF91 model of sequence evolution (Thorne et al., 1991). The main insight is that the analogues of the sufficient statistics (1) 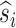 , (2)
, (2)  and (3)
and (3)  for the TKF91 indel process are (1) the expected initial sequence length, (2) the expected number of insertions and deletions and (3) the expected mean sequence length over the evolutionary interval (together with the length of the interval itself, T). We have implemented the algorithm in freely available software. We show that it accurately and quickly recovers the true indel length on simulated data; in contrast, the ‘naive’ or ‘parsimonious’ estimate that associates every observed gap with a single indel event is shown to underestimate the true indel rate by a small amount. Previous ML parameterization approaches have relied on sampling methods, which are significantly slower, albeit less prone to local minima in the likelihood function (Metzler et al., 2001). We report on an application of the algorithm to estimate indel rates in coronavirus proteins.
for the TKF91 indel process are (1) the expected initial sequence length, (2) the expected number of insertions and deletions and (3) the expected mean sequence length over the evolutionary interval (together with the length of the interval itself, T). We have implemented the algorithm in freely available software. We show that it accurately and quickly recovers the true indel length on simulated data; in contrast, the ‘naive’ or ‘parsimonious’ estimate that associates every observed gap with a single indel event is shown to underestimate the true indel rate by a small amount. Previous ML parameterization approaches have relied on sampling methods, which are significantly slower, albeit less prone to local minima in the likelihood function (Metzler et al., 2001). We report on an application of the algorithm to estimate indel rates in coronavirus proteins.
Using methods, such as this rate-based EM algorithm, a number of novel sequence analysis approaches become feasible. For example, it is theoretically possible to do multiple sequence alignment without manually setting scoring parameters; instead, unbiased estimates of these parameters can be obtained quickly, directly and accurately from the data. The algorithms described here can be coupled with a recent ‘structural EM’ algorithm for the estimation of the ML phylogenetic topology (Friedman et al., 2001). Another application is phylogenetic profiling: previous workers, such as Thorne et al. (1996) have shown that it is possible to partition heterogenous multiple alignments using the varying signature of the substitution process, e.g. to predict secondary structure in proteins or transcription factor binding sites in promoters (Kellis et al., 2003). Using methods based on the TKF model, it is now possible to use similar methods to partition the alignment according to the indel rates, and to estimate the relevant heterogenous rate parameters in an unsupervised fashion, from aligned or unaligned sequences, using EM, exactly as one might do with a single-sequence or pair HMM (Durbin et al., 1998). Thus, this algorithm represents a further step in combining the achievements of HMM-based profiling and evolutionary modelling for multiple-sequence comparative genomics.
2 MODELS
We assume some familiarity with continuous-time Markov models for sequence evolution. For a more thorough introduction, the reader is referred to Thorne et al. (1991) or Felsenstein (2003).
Following Thorne et al. (1991), we make the simplifying assumption that the evolutionary model can be separated into independent components: the point substitution process, modelled as a continuous-time finite-state Markov chain at each site of the sequence, and the indel process, modelled as a linear birth–death process with immigration.
The two models share certain features, in that they are both continuous-time Markov models. We begin by introducing some notation for such models, then describe each in detail.
2.1 Continuous-time Markov models
A continuous-time Markov chain is specified by parameters ϑ = {π, R}. Here, the initial distribution is π and the transition rate matrix is R.
The probability of the model being in state j at time t, given that it started in state i at time 0, is Mij(t), where M(t) = eRt is the matrix exponential.
2.2 The point substitution model
The substitution model is a finite-state continuous-time Markov chain. If the substitution model is reversible, then the rate matrix R obeys detailed balance (πiRij = πjRji) and is related to a symmetric matrix S (by  ). The eigenvalues μ(k) and orthonormal eigenvectors v(k) of S (satisfying Sv(k) = μ(k)v(k) and v(k) · v(l) = δkl) can easily be found using standard algorithms (Press et al., 1992), yielding the following result for the entries of M(t)
). The eigenvalues μ(k) and orthonormal eigenvectors v(k) of S (satisfying Sv(k) = μ(k)v(k) and v(k) · v(l) = δkl) can easily be found using standard algorithms (Press et al., 1992), yielding the following result for the entries of M(t)
 |
(1) |
2.3 The TKF91 indel model
In contrast to the substitution model, the state space for the TKF91 indel model is infinite (the sequence length is unbounded). However, much of the theory from continuous-time Markov models still applies.
The TKF91 model describes the evolution of a single sequence under the action of two kinds of mutation event: (1) point substitutions, which act on a single residue only (this process uses the finite-state Markov chain framework described in the previous section); and (2) single-residue indels, which insert or delete a single residue (Thorne et al., 1991). Insertions occur at rate λ per available site; deletions at rate μ.
Consider first the simple example where we ignore the alignment and just look at the sequence length. We then have a linear birth-death process with immigration whose state space is the non-negative integers (Karlin and Taylor, 1975). The rate matrix is sparse: Rij = (i + 1)λ if j = i + 1, Rij = iμ if j = i − 1 and Rij = 0 if |i − j| > 1. Thus Rii = −(i + 1)λ − iμ. The equilibrium distribution over sequence lengths is geometric, πi = gi (1 − g), where g = λ/μ. We assume that the process is always started at equilibrium.
To find the likelihood of an individual alignment, consider starting with some ancestral sequence of length L and allowing it to evolve for some time t. A feature of the TKF91 model is that this process can be simplified by splitting the ancestral sequence into a series of independently evolving ‘links’. For a sequence of length L, there are L + 1 such links, including one immortal link (representing the insertion site at the leftmost end of the sequence) and L mortal links (representing each ancestral residue of the sequence, and the insertion site immediately to its right).
Starting from the initial (ancestral) state, we allow each link to evolve stochastically for some time t, and then examine the new (descendant) state of the link. We may describe this descendant state as follows: first, by the number of newly inserted residues (n); and second (for mortal links) by specifying whether the original ancestral residue survived or was deleted. Let the probability distribution of n be rn(t) for immortal links and pn(t) + qn(t) for mortal links, where pn accounts for the survival of the ancestral residue and qn accounts for its deletion. Then it can be shown (Thorne et al., 1991) that
 |
(2) |
where
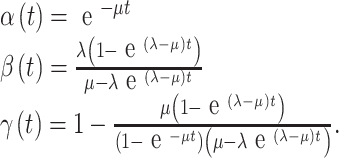 |
(3) |
This probabilistic model for pairwise alignments can be expressed as a pairwise HMM using, e.g. the notation of Durbin et al. (1998) as shown in Figure 1.
Fig. 1.

A Pairwise HMM (Pair HMM) representation of the TKF91 evolutionary model. Transition probabilities are shown beside each transition arrow. The model contains Start (S), End (E), Match (M), Delete (D) and Insert (I) states, plus three null states (closed circles) introduced to simplify the transition probability expressions. (Note that Start and End may also be considered null states.)
The restriction to single-residue indel events leads to the geometric term βn in the above expressions, and is therefore roughly equivalent to using a linear gap penalty to score a sequence alignment. This is widely acknowledged to be unrealistic (Thorne et al., 1992; Hein et al., 2000; Knudsen and Miyamoto, 2003; Miklós et al., 2004). Models that incorporate more realistic length distributions over indel sequences are less tractable than TKF91: such models have been analysed using simulation and combinatoric approximations, but have not yet yielded any algebraic expression for the alignment likelihood (Knudsen and Miyamoto, 2003; Miklós et al., 2004). A tractable alternative is provided by the TKF92 model, which essentially replaces the single residues of TKF91 with artificial, indivisible, multi-residue ‘fragments’. The lengths of these fragments are geometrically distributed. Where TKF91 is a birth-death process on residues, TKF92 is a birth-death process on fragments.
3 ALGORITHMS
We now describe the EM algorithms for estimating model parameters. Again, we start with a general theory for continuous-time Markov models, and then proceed to the specific cases (the point substitution model and the TKF91 indel model).
3.1 The EM algorithm for continuous-time Markov models
Let h represent a ‘history’ of the process: that is, a complete specification of the state of the system at all times. The situation we wish to address is the one where we observe some data O which partially constrains the allowable histories h. For example, we might know what state the process is in at times t = 0 and t = T, but not in between. The state of the process at times 0 < t < T thus constitutes missing information.
The EM algorithm (Dempster et al., 1977) consists of maximizing the following sum over possible histories with respect to ϑ′
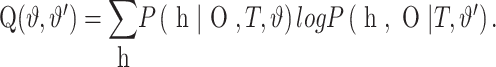 |
(4) |
Previous work (Holmes and Rubin, 2002) showed that, if we break the time interval [0, T] into small intervals Δt, we obtain
 |
(5) |
where  is the expected number of paths that start in state i,
is the expected number of paths that start in state i,  is the expected wait in state i (i.e. the amount of time spent in i) and
is the expected wait in state i (i.e. the amount of time spent in i) and 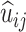 is the expected usage of transition i → j. Note that
is the expected usage of transition i → j. Note that  and
and  .
.
Here, f(ϑ, Δt) is a function that does not depend on the ‘new’ parameters ϑ′. In fact f(ϑ, Δt) ∼ −log (Δt).
Ultimately, we are interested in the infinitesimal limit Δ t → dt. In this limit 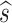 ,
, 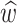 and
and 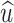 retain their interpretations as the expected initial state occupancy, the expected waiting time and the expected transition usage. The function f is problematic: limΔt → 0f(ϑ, Δ t) = −∞. However, f can effectively be dropped from
retain their interpretations as the expected initial state occupancy, the expected waiting time and the expected transition usage. The function f is problematic: limΔt → 0f(ϑ, Δ t) = −∞. However, f can effectively be dropped from 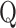 , as it disappears when we differentiate with respect to ϑ′ to maximize the expected log-likelihood of the new parameters.
, as it disappears when we differentiate with respect to ϑ′ to maximize the expected log-likelihood of the new parameters.
We want to maximize  while ensuring that R is a valid rate matrix (i.e. ∑jRij = 0) and π is a normalized probability vector (i.e. ∑i πi = 1).
while ensuring that R is a valid rate matrix (i.e. ∑jRij = 0) and π is a normalized probability vector (i.e. ∑i πi = 1).
Introducing these constraints via Lagrange multipliers  and dropping f, the function to be maximized is
and dropping f, the function to be maximized is
 |
(6) |
3.2 The EM algorithm for the substitution model
Suppose that the observed data O comprises the initial state a and the final state b. Then
 |
(7) |
where
 |
with
 |
The maximum of 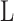 is at
is at 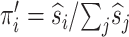 ,
, 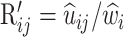 (for j ≠ i) and
(for j ≠ i) and  .
.
3.3 The EM algorithm for the TKF91 model
We substitute into Equation (6) the definitions of R in terms of λ′, μ′, given in Section 2.3.
For the equilibrium distribution π, use independent geometric length parameter g, so that πi = gi (1 − g) (as long as the process stays reversible, we will end up with g = λ/μ)
 |
where 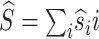 is the expected initial sequence length,
is the expected initial sequence length,  is the expected total time accrued by mortal links, T is the expected time accrued by the immortal link,
is the expected total time accrued by mortal links, T is the expected time accrued by the immortal link,  is the expected number of deletions,
is the expected number of deletions, 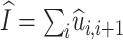 is the expected number of insertions and f′(ϑ) does not depend on ϑ′.
is the expected number of insertions and f′(ϑ) does not depend on ϑ′.
Note the Lagrange multipliers  ,
,  disappear, since our definitions for R and π are already normalized. The maximum of
disappear, since our definitions for R and π are already normalized. The maximum of  is
is 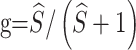 ,
, 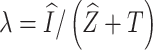 ,
,  .
.
The Supplementary information describes a procedure for calculating  ,
,  ,
,  and
and 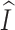 from sequence data.
from sequence data.
4 RESULTS
4.1 Simulated data
We implemented the above-described EM algorithm for estimating indel rates in under 300 lines of C. The implemented code is available free of charge under the GNU Public License from the website www.biowiki.org. [The related EM algorithm for estimating substitution rates was previously implemented in the xrate program, available from the same website (Holmes and Rubin, 2002).]
To test the EM algorithm, we ran extensive numerical simulations, generating random pairwise alignments under the TKF model and comparing the expected insertion counts  and site-times
and site-times  with the actual values of these counts, known from the simulation. We also looked at the following ‘naive’ estimates of these quantities: for I, we simply counted the number of insertions in the simulated alignments, while for Z, we multiplied the lapsed time T by the average of the ancestor and descendant sequence lengths.
with the actual values of these counts, known from the simulation. We also looked at the following ‘naive’ estimates of these quantities: for I, we simply counted the number of insertions in the simulated alignments, while for Z, we multiplied the lapsed time T by the average of the ancestor and descendant sequence lengths.
Figure 2 shows the results of these simulations. There is extremely close agreement between the computed expectations  and the actual values of these statistics. As for the ‘naive’ estimates, we observe that simply counting the number of insertions in the pairwise alignment is a systematically biased underestimate of I, as expected (because some insertions were deleted before being observed). However, perhaps surprisingly, the naive estimate of Z (based on the average of the ancestor and descendant sequence lengths) turned out to be rather good. On closer investigation, we found that the naive Z estimate deteriorates significantly when λ is much less than μ, and also appears to perform worse at larger timescales. In practise, for long sequences (so λ ≃ μ) that are closely related (so T is small), the naive estimator for Z may be appropriate, although it seems likely that I and D will still be underestimates.
and the actual values of these statistics. As for the ‘naive’ estimates, we observe that simply counting the number of insertions in the pairwise alignment is a systematically biased underestimate of I, as expected (because some insertions were deleted before being observed). However, perhaps surprisingly, the naive estimate of Z (based on the average of the ancestor and descendant sequence lengths) turned out to be rather good. On closer investigation, we found that the naive Z estimate deteriorates significantly when λ is much less than μ, and also appears to perform worse at larger timescales. In practise, for long sequences (so λ ≃ μ) that are closely related (so T is small), the naive estimator for Z may be appropriate, although it seems likely that I and D will still be underestimates.
Fig. 2.

Estimated versus actual results for number of insertions (left) and accrued site-time (right) obtained by simulation with λ = 0.0457, μ = 0.0458 (mean sequence length ≃ 50 residues), T ranging from 0.5 to 2, 105 trials per timepoint. The average over each 105 trials is plotted; to give an impression of error on the individual estimates, one in every 5 × 104 individual trials is also plotted. The ‘naive’ estimates, taken without correcting for statistical bias, are also shown. The target values are shown as a dashed straight line.
4.2 Biological data
To test the application of the indel EM algorithm to experimental data, we estimated the indel rates for multiple alignments of Coronavirus protein domains, including domains from the SARS coronavirus (Marra et al., 2003). Lengths of the SARS domains in amino acids are reported below. The first domain is GS1 (629 amino acids) and the second is GS2 (626 amino acids); both are derived from peptide cleavage of the ‘Spike’ surface glycoprotein. The third domain is C16 (277 amino acids), the papain-like peptidase domain from the long RNA polymerase gene product. The analysed proteins were sequenced from viruses responsible for SARS, murine hepatitis, porcine transmissible gastroenteritis, porcine epidemic diarrhoea, equine arteritis and avian infectious bronchitis.
Indel rates were estimated using three different methods: the EM method presented here, a Markov Chain Monte Carlo (MCMC) method and a naive estimate. The MCMC runs included 105 datapoints constituting ∼103 effective independent samples. Confidence intervals for the MCMC-estimated rate parameters are reported as m ± 2s where m is the mean and s the standard deviation of the marginal posterior distribution over the relevant parameter. The EM and naive methods do not return error estimates.
The domain boundaries were first identified by reference to the Pfam database (Bateman et al., 1999). We then estimated a separate phylogenetic tree for each domain, first estimating a pairwise distance matrix using a 20 × 20 amino acid substitution rate matrix previously estimated from PFAM alignments by the EM algorithm (Holmes and Rubin, 2002), then finding an approximate phylogenetic tree using weighted neighbour-joining (Bruno et al., 2000). The three trees thus estimated are shown in Figures 3–5.
Fig. 3.

Phylogenetic trees estimated by the neighbour-joining method applied to pairwise amino acid-level distance matrices for the 5′ GS1 domain from the coronavirus glycosurface protein ‘Spike’. Key: Chicken, AAF69334.1 (murine hepatitis virus); ChickenII, AAF19386.1 (murine hepatitis virus strain II); Cow, NP_150077.1 (bovine coronavirus); SARS, NP_828851.1 (SARS coronavirus); SARSCUHKW1, AAP13567.1 (SARS strain CUHK-W1); SARSUrbani, AAP13441.1 (SARS strain Urbani); Bird, NP_740621.1 (avian infectious bronchitis virus); 229E, NP_073551.1 (human coronavirus strain 229E); PigED, NP_598310.1 (porcine epidemic diarrhea virus); PigTG, NP_058424.1 (porcine transmissible gastroenteritis virus); and Horse, NP_705583.1 (equine arteritis virus).
Fig. 5.

Phylogenetic trees estimated by the neighbour-joining method applied to pairwise amino acid-level distance matrices for the papain-like peptidase domain from coronavirus RNA polymerase. Key: Chicken, AAF69341.1; ChickenII, AAF19383.1; Cow, NP_742129.1; SARS, NP_828860.1; SARSUrbani, AAP13439.1; and SARSCUHKW1, AAP13575.1.
Next, we used the tkfalign program (Holmes and Bruno, 2001) to infer ML alignment paths for the missing ancestral sequences, using that program's default values for the indel rates (λ = 0.049505, μ = 0.050495). We used the alignments containing these inferred ancestral sequences in all subsequent analyses. This strategy is likely to systematically underestimate the true indel rates, since the ML alignment paths will tend to minimize the number of inferred events. Ideally, we should integrate out the ancestral sequences, e.g. by Monte Carlo alignment sampling (Holmes and Bruno, 2001) or multidimensional dynamic programming (Hein, 2001). However, fixing the alignments simplifies the comparison of the parameter estimation methods.
We proceeded to estimate the indel rates, using the three methods described (naive, EM and MCMC). The results of this analysis are shown in Table 1. We note that the Spike protein has elevated indel rates compared with the peptidase protein, and that the first (5′) domain has higher rates than the second (3′) domain. Some caution is, however, needed in interpreting these results, because the indel rate estimates depend strongly on the estimated tree. Since we have estimated independent phylogenies (including branch lengths) for the GS1 and GS2 domains, and since the branch length estimates are dominated by the substitutions in the alignment, what we are actually measuring here is not the absolute indel rate but the ratio of the indel rate to the substitution rate. If substitutions are generally occurring faster in one of the domains (as indeed they are, in the GS1 domain) this may significantly skew the estimate.
Table 1.
Indel rates for coronavirus protein domains
| Domain | Naive | EM | MCMC |
|---|---|---|---|
| λ (Insertion rate) | |||
| GS1 | 0.135352 | 0.145626 | 0.145 ± 0.008 |
| GS2 | 0.0928362 | 0.0971039 | 0.098 ± 0.008 |
| C16 | 0.0341402 | 0.0346793 | 0.035 ± 0.009 |
| μ (Deletion rate) | |||
| GS1 | 0.135459 | 0.145741 | 0.146 ± 0.008 |
| GS2 | 0.0929047 | 0.0971758 | 0.098 ± 0.008 |
| C16 | 0.0342055 | 0.0347457 | 0.035 ± 0.009 |
An absolute comparison of the rates of evolution of the Spike domains can be made by the following procedure. First, estimate a phylogenetic tree for the full alignment of the entire Spike protein (Fig. 6); next, estimate λ and μ independently for the two domains, using the same tree (and branch lengths) for each domain. The results of this test are given in Table 2. Here, it can be seen that the ‘absolute’ rate of indel evolution is, in fact, greater in GS1. Thus, the overall picture is that GS1 is mutating faster than GS2, but substitutions are elevated relative to indels. Given the relative mutation rates, we predict that the GS1 domain has more contact interactions with the host immune system than GS2.
Fig. 6.

Phylogenetic trees estimated by the neighbour-joining method applied to pairwise amino acid-level distance matrices for the entire ‘Spike’ coronavirus glycosurface protein (including both domains GS1 and GS2). For sequence accession numbers, see legend to Figure 3.
Table 2.
Indel rates for coronavirus Spike protein domains
| Domain | Naive | EM | MCMC |
|---|---|---|---|
| λ (Insertion rate) | |||
| GS1 | 0.105146 | 0.110403 | 0.110 ± 0.006 |
| GS2 | 0.0514705 | 0.0527830 | 0.053 ± 0.006 |
| μ (Deletion rate) | |||
| GS1 | 0.105219 | 0.110480 | 0.111 ± 0.006 |
| GS2 | 0.0515080 | 0.0528216 | 0.053 ± 0.006 |
The same phylogenetic tree was used for both domains (Fig. 6). Key to domain names: GS1 is the 5′ domain of the SARS coronavirus Spike surface glycoprotein, while GS2 is the 3′ domain of the same protein.
We note that although there is a clear systematic bias to the naive rate estimates, this bias ranges from 2 to ∼9% and, as such, is usually slightly smaller than the sampling error (as revealed by MCMC). The exception to this is the GS1 domain, for which the bias is slightly larger (but not by much). These results seem to indicate that the naive estimates would probably be acceptable in most practical situations, although in order to avoid the possibility of downstream or compounded errors [e.g. in more elaborate TKF91-derived models containing more parameters (Holmes, 2004)] it would be better to use the unbiased estimators derived here.
5 DISCUSSION
We have shown that the expected initial sequence length 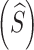 , numbers of mortal insertions
, numbers of mortal insertions 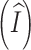 and deletions
and deletions  , and accrued time for mortal
, and accrued time for mortal  and immortal (T) links, along with the expected initial composition
and immortal (T) links, along with the expected initial composition 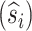 , substitution counts
, substitution counts 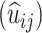 and accrued times
and accrued times  for each residue, constitute ‘sufficient statistics’ for maximizing the expected log-likelihood of a given set of alignments under the TKF91 evolutionary model. This EM algorithm is orders of magnitude faster than brute-force methods of searching the indel/substitution rate space, requiring only an O((T/Δ t)2) overhead for numerical integration, where Δ t is the timestep for the discretized integral. We have implemented such an EM algorithm and tested it by simulation and application to biological data.
for each residue, constitute ‘sufficient statistics’ for maximizing the expected log-likelihood of a given set of alignments under the TKF91 evolutionary model. This EM algorithm is orders of magnitude faster than brute-force methods of searching the indel/substitution rate space, requiring only an O((T/Δ t)2) overhead for numerical integration, where Δ t is the timestep for the discretized integral. We have implemented such an EM algorithm and tested it by simulation and application to biological data.
In the prevalent situation where a multiple alignment is unknown, the calculation of the sufficient statistics by summing over all alignments takes time O(LN), where L is the (geometric mean) sequence length and N is the number of sequences. However, a stochastic version of the EM algorithm, which uses MCMC alignment sampling on local neighbourhoods of the tree, can approximate these statistics efficiently (and improve alignment accuracy) in O(SLK) time, where K is the neighbourhood size (i.e. the number of nodes whose mutual alignment is simultaneously resampled at any one step) and S is the number of sampling steps between stochastic EM updates. Naturally, one can also proceed on the assumption that the best alignment found by an alignment algorithm is the ‘true’ multiple alignment, although such an assumption may lead to systematic biases, such as undercounting indels.
Recent years have seen considerable interest in the derivation of stochastic evolutionary processes for biological sequences and/or structures (Thorne et al., 1992; Knudsen and Miyamoto, 2003; Miklós et al., 2004; Lunter and Hein, 2004; Siepel and Haussler, 2004; Holmes, 2004). Several of these are so closely related to TKF91 that the theory derived here is directly applicable. For example, the TKF92 model (Thorne et al., 1992) (see also Section 2.3) is a birth-death process on sequence fragments, rather than residues, so that the number of matches (a quantity appearing in the expressions for the EM-sufficient statistics) is found by counting the number of aligned fragments rather than the number of aligned residues. Similarly, an evolutionary model for RNA secondary structure has recently been described, together with a recipe for computing the sufficient statistics using the Inside-Outside algorithm (Holmes, 2004). Models that depart more substantially from TKF91, such as ‘long indel’ models (Knudsen and Miyamoto, 2003; Miklós et al., 2004) and context-sensitive substitution models (Lunter and Hein, 2004; Siepel and Haussler, 2004), have generally not been solved completely (i.e. we still lack exact algebraic expressions for the sequence likelihood) and so the TKF91 theory developed here will be less directly applicable. These models may mandate different approaches to rate estimation, such as enumeration of mutation trajectories (Miklós et al., 2004), truncated Taylor expansions (Lunter and Hein, 2004) and variational approximations (Siepel and Haussler, 2004).
The EM algorithm—in the form of the Baum–Welch algorithm—is one cornerstone of the application of profile HMMs in bioinformatics. We hope that the theoretical developments described here may contribute to the efforts to make evolutionary models similarly useful for probabilistic sequence analysis and phylogenomics.
Fig. 4.

Phylogenetic trees estimated by the neighbour-joining method applied to pairwise amino acid-level distance matrices for the 3′ GS2 domain from the coronavirus glycosurface protein ‘Spike’. For sequence accession numbers, see legend to Figure 3.
Supplementary Material
Acknowledgments
The author would like to thank Bob Griffiths, Von Bing Yap and Terry Speed for helpful discussions, and two anonymous reviewers for their suggestions. This work was partially supported by grants from EPSRC (code HAMJW) and MRC (code HAMKA).
REFERENCES
- Bateman, A., et al. 1999. Pfam 3.1: 1313 multiple alignments match the majority of proteins. Nucleic Acids Res. 27260–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno, W.J., et al. 2000. Weighted neighbor joining: a likelihood-based approach to distance-based phylogeny reconstruction. Mol. Biol. Evol. 17189–197 [DOI] [PubMed] [Google Scholar]
- Dempster, A.P., et al. 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B 391–38 [Google Scholar]
- Durbin, R., Eddy, S., Krogh, A., Mitchison, G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids 1998, Cambridge, UK Cambridge University Press
- Felsenstein, J. Inferring Phylogenies 2003. ISBN 0878931775 Sinauer Associates, Inc
- Friedman, N., Ninio, M., Pe'er, I., Pupko, T. 2001. A structural EM algorithm for phylogenetic inference. Proceedings of the Fifth Annual International Conference on Computational Biology , New York Association for Computing Machinery
- Hein, J. 2001. An algorithm for statistical alignment of sequences related by a binary tree. In Altman, R.B., Dunker, A.K., Hunter, L., Lauderdale, K., Klein, T.E. (Eds.). Pacific Symposium on Biocomputing , Singapore World Scientific, pp. , pp. 179–190 [DOI] [PubMed]
- Hein, J., et al. 2000. Statistical alignment: computational properties, homology testing and goodness-of-fit. J. Mol. Biol. 302265–279 [DOI] [PubMed] [Google Scholar]
- Holmes, I. 2003. Using guide trees to construct multiple-sequence evolutionary HMMs. Proceedings of the Eleventh International Conference on Intelligent Systems for Molecular Biology , Menlo Park, CA AAAI Press, pp. 147–157
- Holmes, I. 2004. A probabilistic model for the evolution of RNA structure. BMC Bioinformatics 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, I. and Bruno, W.J. 2001. Evolutionary HMMs: a Bayesian approach to multiple alignment. Bioinformatics 17803–820 [DOI] [PubMed] [Google Scholar]
- Holmes, I. and Rubin, G.M. 2002. An Expectation Maximization algorithm for training hidden substitution models. J. Mol. Biol. 317757–768 [DOI] [PubMed] [Google Scholar]
- Karlin, S. and Taylor, H. A First Course in Stochastic Processes 1975, San Diego, CA Academic Press
- Kellis, M., et al. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423241–254 [DOI] [PubMed] [Google Scholar]
- Knudsen, B. and Miyamoto, M. 2003. Sequence alignments and pair hidden Markov models using evolutionary history. J. Mol. Biol. 333453–460 [DOI] [PubMed] [Google Scholar]
- Lunter, G.A. and Hein, J. 2004. A nucleotide substitution model with nearest-neighbour interactions. Bioinformatics 20Suppl. 1,I216–I223 [DOI] [PubMed] [Google Scholar]
- Marra, M.A., et al. 2003. The genome sequence of the SARS-associated coronavirus. Science 3001399–1404 [DOI] [PubMed] [Google Scholar]
- Metzler, D., et al. 2001. Assessing variability by joint sampling of alignments and mutation rates. J. Mol. Evol. 53660–669 [DOI] [PubMed] [Google Scholar]
- Miklós, I., et al. 2004. A long indel model for evolutionary sequence alignment. Mol. Biol. Evol. 21529–540 [DOI] [PubMed] [Google Scholar]
- Mitchison, G.J. and Durbin, R. 1995. Tree-based maximal likelihood substitution matrices and hidden Markov models. J. Mol. Evol. 411139–1151 [Google Scholar]
- Press, W.H., Teukolsky, S.A., Vetterling, W.T., Flannery, B.P. Numerical Recipes in C 1992, Cambridge, UK Cambridge University Press
- Siepel, A. and Haussler, D. 2004. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 21468–488 [DOI] [PubMed] [Google Scholar]
- Thorne, J.L., et al. 1991. An evolutionary model for maximum likelihood alignment of DNA sequences. J. Mol. Evol. 33114–124 [DOI] [PubMed] [Google Scholar]
- Thorne, J.L., et al. 1992. Inching toward reality: an improved likelihood model of sequence evolution. J. Mol. Evol. 343–16 [DOI] [PubMed] [Google Scholar]
- Thorne, J.L., et al. 1996. Combining protein evolution and secondary structure. Mol. Biol. Evol. 13666–673 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.