

Abstract
Aims: To develop a specific and highly sensitive loop‐mediated isothermal amplification (LAMP) technique for the rapid detection of canine parvovirus (CPV) DNA directly in suspected faecal samples of dogs by employing a simple method of template preparation.
Methods and Results: LAMP reaction was developed by designing two sets of outer and inner primers, which target a total of six distinct regions on VP2 gene of CPV. The template DNA was prepared by a simple boiling and chilling method. Of the 140 faecal samples screened by the developed LAMP and the conventional PCR assays, 104 samples (74·28%) were found positive by LAMP, whereas 81 samples (57·85%) were found positive by PCR. The specificity of the LAMP assay was tested by cross‐examination of common pathogens of dogs and further confirmed by sequencing. The detection limit of the LAMP was 0·0001 TCID50 ml−1, whereas the detection limit of the PCR was 1000 TCID50 ml−1.
Conclusions: The developed LAMP assay detects CPV DNA in faecal specimens directly within an hour by following a simple and rapid boiling and chilling method of template preparation. The result also shows that the developed LAMP assay is specific and highly sensitive in detecting CPV.
Significance and Impact of the Study: The result indicates the potential usefulness of LAMP which is a simple, rapid, specific, highly sensitive and cost‐effective field‐based method for direct detection of CPV from the suspected faecal samples of dogs.
Keywords: canine parvovirus, loop‐mediated isothermal amplification, polymerase chain reaction, primers, VP2 gene
Introduction
Canine parvovirus (CPV) is a small non‐enveloped ssDNA virus having a genome of approximately 5000 bases that encodes two structural (VP1 and VP2) and two non‐structural (NS1 and NS2) proteins (Cotmore and Tattershall 1987).
CPV‐2 is the most significant viral cause of canine enteritis responsible for neonatal death in pups (Parrish 1999). It emerged as a potentially fatal and highly contagious viral disease in 1978 (Appel et al. 1979). Few years after its emergence, CPV‐2 was completely replaced by two antigenic variants designated as CPV‐2a and CPV‐2b which is now distributed worldwide (Parrish et al. 1988). In 2001, a novel CPV mutant with an amino acid substitution at position 426 (Asp → Glu), called CPV‐2c, emerged in Italy (Buonavoglia et al. 2001). Currently, CPV‐2c is broadly distributed and co‐exists with CPV‐2a and CPV‐2b types in Europe, Australia, North and South American countries (Decaro et al. 2007; Spibey et al. 2008). Similarly, CPV types 2a/2b, having mutation at residue 297 (Ser→Ala) and designated as New CPV‐2a/2b, have been reported from various countries including India in recent times (Ohshima et al. 2008; Mohanraj et al. 2010). In Puducherry, southern India, 16 CPV‐2a variants (297 Ser →Ala) termed as ‘new CPV‐2a’ were detected among vaccinated and unvaccinated dog population (Mohanraj et al. 2010). Recently, 24 new CPV‐2a and two new CPV‐2b strains were detected among vaccinated and unvaccinated dog populations from 5 different states/union territories in South India (Vivek 2011).
Several detection methods have been developed to detect proteins and nucleic acids of CPV, and many of these tests are effective and accurate in detecting the virus infection in laboratory, but they require the use of expensive equipments and are often laborious and time‐consuming. Early and rapid diagnosis is necessary so that the infected dogs can be isolated to prevent the spread of the disease and also to administer supportive treatment to reduce morbidity and mortality. Therefore, a better detection method would be one that is not only speedy, accurate and sensitive, but also simple and economical for its practical field‐based applications.
Several PCR‐based detection methods for CPV in faecal samples including PCR–restriction fragment length polymorphism (Sakulwira et al. 2001), nested PCR (Hirasawa et al. 1994) and real‐time PCR (Decaro et al. 2005) have been applied for laboratory diagnosis because of their sensitivity and specificity. However, these assays require 2–4 h, special equipments and technical expertise. Therefore, a novel nucleic acid amplification method, termed loop‐mediated isothermal amplification (LAMP), which amplifies specific DNA sequences under isothermal conditions within 60 min, was developed as a simple, rapid, specific and cost‐effective alternative (Notomi et al. 2000). Specific amplification can be detected by observing directly the turbidity (Mori et al. 2001), the fluorescence after adding DNA‐binding dyes like propidium iodide (Hill et al. 2008) or by the presence of ladder‐like multiple bands on 2·0% agarose gel (Parida et al. 2006).
LAMP technique can be used as a potent field diagnostic test for the detection of CPV as it is less sensitive than PCR to inhibitory substances present in biological samples (faeces). This method has been applied successfully for the detection of porcine parvovirus (Chen et al. 2009), goose parvovirus (JinLong et al. 2010), canine distemper virus (Cho and Park 2005), Leptospira spp. (Lin et al. 2009), Haemophilus influenzae (Torigoe et al. 2006), E. coli (Hill et al. 2008), Dirofilaria immitis (Aonuma et al. 2009), Babesia canis (Muller et al. 2010) and many other pathogens. Detection of CPV by LAMP having a detection limit of 0·1 TCID50 ml−1 was reported by Cho et al. (2006), where the extraction kit using DNAzol was used for template DNA preparation from faecal samples.
The purpose of the study hereby reported was to develop and evaluate a LAMP technique, for direct detection of CPV DNA in faecal samples of suspected dogs without using any DNA extraction kit/reagents so as to make it economical, rapid and a simple field‐based technique. Two sets of highly specific primers were designed and used for the detection of CPV by LAMP, and the results were compared with those from a conventional PCR assay.
Materials and methods
CPV strains
CPV‐2 vaccine strain (Strain C154; Intervet, India Pvt Ltd, Pune, India), new CPV‐2b vaccine strain (Fort Dodge Animal Health Inc., KS, USA) and new CPV‐2a and new CPV‐2b strains (GenBank Accession nos. GU139554.1 and JN008393.1, respectively) maintained in the Department of Veterinary Microbiology, Rajiv Gandhi College of Veterinary and Animal Sciences, Puducherry, India, were utilized to develop the LAMP technique.
Designing of LAMP primers
The highly conserved region of the VP2 gene of the CPV (GenBank Accession no. NC_001539.1) was selected as the LAMP target for designing four specific LAMP primers. LAMP primers were designed using the Primer Explorer version 4.0 http://loopamp.eiken.co.jp/e/: forward inner primer (FIP), backward inner primer (BIP), forward outer primer (F3) and backward outer primer (B3). The location and the sequences of the primers are shown in Fig. 1 and Table 1, respectively.
Figure 1.
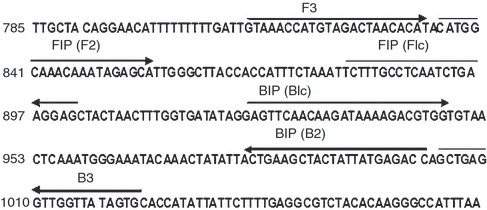
Location of primers in VP2 gene of canine parvovirus (GenBank Accession no. NC_001539.1) The forward inner primer and backward inner primer inner primers contain two distinct sequences (F1c + F2 and B1c + B2, respectively).
Table 1.
Sequence of primers
| Primers* | 5′pos | 3′pos | Sequence |
|---|---|---|---|
| F3 | 812 | 834 | 5′ GTAAACCATGTAGACTAACACAT 3′ |
| B3 | 1003 | 1022 | 5′ GCACTATAACCAACCTCAGC 3′ |
| FIP | GCTCCTTCAGATTGAGGCAAAGA‐CATGGCAAACAAATAGAGCA | ||
| BIP | GAGTTCAACAAGATAAAAGACGTGG‐GGTCTCATAATAGTAGCTTCAGT |
*FIP, Forward inner primer. BIP, backward inner primer.
Extraction of DNA from vaccine/standard strains
DNA was extracted from CPV strains by a boiling and chilling method. Approximately 100 μl of supernatant was taken from the reconstituted cell culture fluid, boiled at 96°C for 10 min, followed by an immediate chilling in crushed ice for 5 min and centrifuged at 12 000 g for 10 min. The collected supernatant was stored at −20°C until further use.
Optimization of LAMP reaction
The template DNA prepared from CPV‐2 (Strain C154; Intervet) was used to optimize the LAMP reaction. LAMP technique was performed as described by Notomi et al. (2000). LAMP was performed in 25 μl total reaction volume containing 2 μl of extracted DNA (template), 1 μl (40 pmol) each of FIP and BIP, 1 μl (5 pmol) each of F3 and B3, 2·5 μl of a 10× ThermoPol reaction buffer, 3·5 μl of 1·4 mmol l−1 deoxynucleotide triphosphates, 1·5 μl of 8 mmol l−1 MgSO4, 4 μl of 0·8 mol l−1 betaine, 1 μl of 8 U Bst DNA polymerase large fragment and 6·5 μl of sterile triple distilled water. Except betaine (Sigma‐Aldrich, St Louis, MO), all the other reagents were obtained from New England Biolabs (Ipswich, MA, USA) The following temperatures were tested to determine the optimum reaction temperature for the LAMP reaction: 62, 63, 64 and 65°C, with a termination temperature of 80°C for 2 min. After confirming the optimum reaction temperature, reaction times (15, 30, 45 and 60 min) were tested to determine the most favourable reaction time. Amplified products were analysed on 2% agarose gel electrophoresis. Varying concentrations of MgSO4 like 5, 6 and 8 mmol l−1 were also tried for optimization of LAMP. The positivity of LAMP reaction was detected initially by the development of turbidity and the addition of 2 μl of propidium iodide (1 mg ml−1; Sigma‐Aldrich) to LAMP products followed by the presence of bands in ladder‐like pattern on 2% agarose gel electrophoresis.
Comparison of DNA extraction by DNA isolation kit with a boiling and chilling method
DNA was extracted from 0·25 g of healthy dog faeces spiked with CPV‐2 vaccine strain (strain C154; Intervet, 106·5 TCID50) by employing Ultraclean® Faecal DNA isolation kit (Mo Bio laboratories, Inc., Carlsbad, CA) according to manufacturer’s instructions and also by the rapid boiling and chilling method described earlier. Templates that are diluted (serial tenfold) (10−1–10−7), prepared by both the methods, were subjected to LAMP assay.
Specificity and sensitivity of LAMP assay
The specificity of the designed LAMP primers was determined by cross‐examination of templates extracted from canine adenovirus 1 (Indian Immunologicals Ltd), canine coronavirus (Pfizer Animal Health, New York, USA) and canine distemper virus (Intervet) vaccines and Leptospira icterohemorrhagiae (strain RGA; National Leptospirosis Reference Centre, Regional Medical Research Centre, Andaman and Nicobar Islands, India). The specificity of the LAMP assay was further confirmed by bidirectional custom sequencing of LAMP products by employing outer primers (F3 and B3). The sequences were analysed and identified using Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) from the GenBank database. The sensitivity of the LAMP assay was determined by a tenfold serial dilution of the CPV‐2 strain (strain C154; Intervet, 106·5 TCID50) in faecal emulsions obtained from a healthy dog to resemble the field conditions. The template DNA from each dilution was prepared in duplicate by applying the boiling and chilling method and subjected to LAMP and conventional PCR assays (Buonavoglia et al. 2001).
Collection and processing of faecal samples for LAMP and conventional PCR
A total of 140 faecal samples were collected from CPV‐suspected dogs from eight different states/union territories in India during 2010–2011. Faecal samples were directly collected from rectum using sterile swabs, immersed in 1·5 ml of sterile PBS, stored at 4°C and transported as early as possible to the laboratory under refrigeration. The emulsions were centrifuged at 6000 g for 15 min at 4°C, and the supernatant was collected and stored at −40°C until further use. The details of the faecal samples collected are presented in Table 2.
Table 2.
Details of collected samples
| Sl. no. | State/Union Territory | Place | Clinical samples |
|---|---|---|---|
| 1 | Union Territory of Pondicherry | Puducherry | 26 |
| 2 | Tamil Nadu | Chennai | 9 |
| Coimbatore | 6 | ||
| Tirupur | 5 | ||
| Erode | 8 | ||
| Salem | 3 | ||
| Chengalpattu | 3 | ||
| 3 | Kerela | Palakkad | 11 |
| Thrissur | 4 | ||
| Trivandrum | 5 | ||
| 4 | Andhra Pradesh | Hyderabad | 6 |
| Tirupati | 10 | ||
| Kakinada | 9 | ||
| 5 | Karnataka | Bangalore | 12 |
| 6 | Uttar Pradesh | Bareilly | 8 |
| 7 | Maharashtra | Mumbai | 3 |
| 8 | Goa | Panaji | 12 |
| Total | 140 |
Template DNA was prepared from all the 140 faecal samples by the boiling and chilling method as detailed above. Extracted DNA was subjected to LAMP assay as described earlier.
Conventional PCR assay
All the 140 faecal samples of dogs suspected for CPV were also subjected to conventional PCR by using H primers for amplifying a 630‐bp fragment of the VP2 gene encoding capsid protein (Buonavoglia et al. 2001). The template DNA was prepared by the similar boiling and chilling method as in LAMP assay, and the supernatant was diluted 1 : 10 in sterile distilled water to reduce residual inhibitors of Taq DNA polymerase activity (Decaro et al. 2006).
Statistical analysis
The differences in CPV detection rates by LAMP and PCR assays were determined by McNemar test. This was calculated as 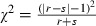 , d.f. = 1 (r and s are the numbers of discordant pairs) (Riffenburgh 2006).
, d.f. = 1 (r and s are the numbers of discordant pairs) (Riffenburgh 2006).
Results
Optimization of LAMP reaction was carried out using different time–temperature combinations and MgSO4 concentrations. The amplicons that were formed at 62, 63, 64, and 65°C at different time intervals were analysed on 2% agarose gel electrophoresis. The clearest bands were detected after incubation at 63°C for 60 min. Among various concentrations of MgSO4 tried, ladder pattern was the clearest at 8 mmol l−1 concentration. The comparative analysis of DNA extraction carried out by the DNA isolation kit and the boiling and chilling methods revealed that there is no difference as the templates got amplified in all the seven dilutions (10−1–10−7).
The LAMP products were initially screened by observing turbidity (Fig. 2a) and by adding 2 μl of propidium iodide (1 mg ml−1), a DNA‐binding dye to each reaction tube under ambient and UV light. A change of colour from reddish orange to pink under ambient light (Fig. 2b) and fluorescence under UV light by propidium iodide indicated a positive reaction (Fig. 2c). All the LAMP products were also subjected to 2% agarose gel electrophoresis for confirmation. Of the 140 samples screened by LAMP, 104 samples (74·28%) were positive yielding bands in ladder‐like pattern upon 2% agarose gel electrophoresis. All the 140 samples were also screened by conventional PCR using H primers, and 81 samples (57·85%) were found as positive yielding 630‐bp products (Fig. 3a,b). McNemar test was performed to compare the sensitivities of both the assays, and the P value obtained was <0·0001 which suggests that the proportion of positive results is significantly different for the two assays (Table 3).
Figure 2.
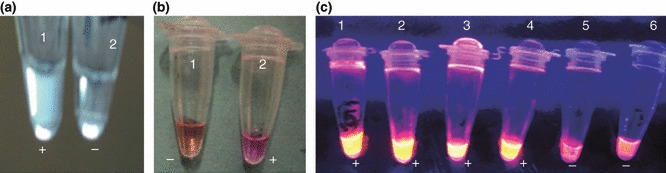
Visual detection of loop‐mediated isothermal amplification (LAMP) products by (a) observing white turbidity, tube 1 shows turbidity which is considered as positive. (b) Addition of propidium iodide in the LAMP reaction tube and observed under ambient light, positive reaction (tube 2) shows a change of colour to pink, which can be differentiated from reddish orange colour of a negative reaction (tube 1) (c) Addition of propidium iodide in the LAMP reaction tube and viewed under UV for fluorescence (tube 1‐positive control, tube 2 to 4‐CPV positive clinical samples, tube 5‐negative clinical sample, tube 6‐negative control).
Figure 3.
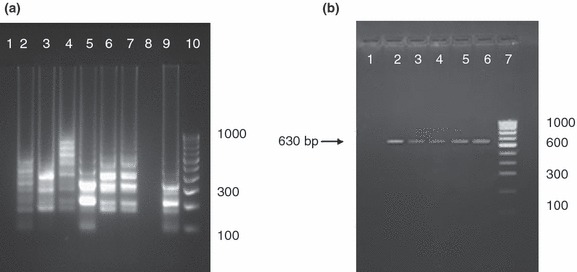
(a) Screening of canine parvovirus (CPV)‐suspected faecal samples by agarose gel electrophoresis of loop‐mediated isothermal amplification products; Lane 1, negative control (faecal sample from healthy dog); Lanes 2–7, positive samples; Lane 8, negative sample; Lane 9, positive control (CPV‐2); and Lane 10, 100‐bp DNA ladder (b) Screening of CPV‐suspected faecal samples by agarose gel electrophoresis analysis of PCR products showing amplification of specific region of VP2 gene (630 bp) using H primers; Lane 1, negative control (faecal samples from healthy dog); Lanes 2–5, positive samples; Lane 6, positive control (CPV‐2); and Lane 7, 100‐bp DNA ladder.
Table 3.
Comparison of loop‐mediated isothermal amplification (LAMP) and PCR for the detection of canine parvovirus from suspected faecal samples of dogs
| PCR | LAMP | Total | |
|---|---|---|---|
| + | − | ||
| + | 81 | 0 | 81 |
| − | 23 | 36 | 59 |
| Total | 104 | 36 | 140 |
The specificity of the LAMP assay was confirmed using template DNA prepared from canine adenovirus 1, canine coronavirus, canine distemper virus and Leptospira icterohemorrhagiae. No amplification products were detected in the above template DNA, whereas only CPV‐2, new CPV 2a and new CPV 2b strains showed positive reactions (Fig. 4). The specificity of the amplified products was further confirmed by bidirectional custom sequencing of the LAMP products by employing outer primers and the sequences of the amplified products perfectly aligned with nucleotide sequences of CPV available in GenBank database (Supporting information Table S1). The sensitivity of LAMP and PCR assays were determined and compared by subjecting the templates prepared from serially diluted (tenfold) CPV‐2 strain (106·5 TCID50 ml−1) in faecal emulsions. The detection limits of LAMP and PCR assays were 0·0001 TCID50 ml−1 and 1000 TCID50 ml−1, respectively (Fig. 5a,b).
Figure 4.
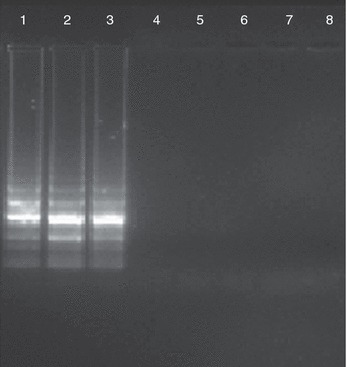
Specificity of loop‐mediated isothermal amplification assay. Lane 1, canine parvovirus (CPV)‐2; Lane 2, new CPV‐2a; Lane 3, new CPV‐2b; Lane 4, Canine coronavirus; Lane 5, Canine adenovirus 1; Lane 6, Leptospira icterohemorrhagiae; Lane 7, Canine distemper virus; and Lane 8, Negative control. No amplification was seen in Lanes 4–7.
Figure 5.
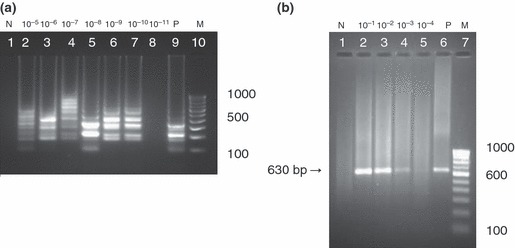
(a) Sensitivity of loop‐mediated isothermal amplification assay. Lane 1, negative control; Lane 2–8 serial dilutions (10−5–10−11); Lane 9, positive control; Lane 10, 100‐bp DNA ladder, Detection limit up to 10−10 dilution (i.e.) 0·0001 TCID50 ml−1 (b) Sensitivity of PCR assay using H primers. Lane 1, negative control; Lane 2–5 serial dilutions (10−1–10−4); Lane 6, positive control; Lane 7, 100‐bp DNA ladder, Detection limit up to 10−3 dilution (i.e.) 1000 TCID50 ml−1.
Discussion
CPV is one of the most dreadful diseases in young pups. Early detection and isolation of the affected animals is one of the most logical strategies to reduce the risk of further transmission. The LAMP assay, developed in this study for direct detection of CPV DNA from suspected faecal samples, could identify CPV within one hour of DNA extraction. The boiling and chilling method for template preparation was as sensitive as the kit method. The boiling and chilling method for template preparation adopted in this study was simple, cost‐effective, less labour oriented and less time‐consuming when compared to kit method. The boiling and chilling method of DNA extraction is easy to perform and can be readily adopted in clinical laboratories for field diagnosis of CPV infection by LAMP assay. Nemoto et al. (2011) used a similar technique for direct detection of equine herpes virus‐1 DNA in nasal swabs of horses. In the present study, of the 140 clinical samples tested, 104 (74·28%) and 81 samples (57·85%) were detected positive for CPV by LAMP and PCR assays, respectively. In an earlier study, Cho et al. (2006) reported that of the 50 CPV‐suspected faecal samples, 37 (74·0%) were positive by PCR, whereas 40 of 50 faecal samples (80·0%) were found positive by LAMP. Chen et al. (2009) compared the detection rates of porcine parvovirus by PCR (93·6%) and LAMP (97·6%) from 125 clinical samples and reported higher detection rate in LAMP. Decaro et al. (2005) developed a rapid and sensitive real‐time PCR assay for detecting and quantifying CPV‐2 DNA in the faeces of dogs with diarrhoea. However, use of real‐time PCR assay is limited because of cost of instrumentation, cost of enzyme mixes sold by the machine manufacturers and the cost of reagents especially fluorogenic probe that is more expensive than the regular primers (Curry et al. 2002). JinLong et al. (2010) analysed LAMP and fluorescent quantitative real‐time PCR (FQ‐PCR) assay for goose parvovirus infected gosling tissues. Twenty one of the 30 samples were tested positive, while nine were negative, based on FQ‐PCR and LAMP.
In the present study, significant difference was found between LAMP and conventional PCR assays for direct detection of CPV DNA in faecal samples. Bst polymerase used in LAMP is reported to be more tolerant than Taq polymerase to inhibitors present in biological materials like faecal samples (Monteiro et al. 1997; Bakheit et al. 2008). As a simple and cost‐effective boiling and chilling method of template preparation was adopted in this study, it would have probably resulted in inactivation of Taq polymerase by faecal inhibitors in certain cases leading to problems related to sensitivity of PCR assay. Moreover, the size of the amplicon was 630 bp, and a longer amplicon size would have resulted in false negative result in few cases as it has been demonstrated by Tilley (2004) during PCR amplification of wheat sequences from DNA. LAMP was also found to detect more numbers of positive cases than PCR for the identification of toxoplasma DNA (Krasteva et al. 2009), taenid DNA (Nkouawa et al. 2010) and equine rotavirus in faecal specimens (Nemoto et al. 2010) which corroborates with the findings of the present study.
The specificity of the LAMP assay developed in this study was confirmed by the presence of multiple DNA bands in ladder‐like pattern with CPV‐2, new CPV‐2b and new CPV‐2a strains and the absence of the amplification products with the other commonly occurring pathogens of dogs like canine adenovirus 1, canine coronavirus, canine distemper virus and Leptospira icterohemorrhagiae.
The detection limit of the developed LAMP assay was 0·0001 TCID50 ml−1 as the virus diluted up to 10−10 revealed the amplified products. On the other hand, the conventional PCR assay could detect only up to 1000 TCID50 ml−1 of virus (up to 10−3 dilution). Therefore, the developed LAMP assay was found to be more sensitive in comparison with conventional PCR assay. Similar higher sensitivity of LAMP over PCR was also reported by Krasteva et al. (2009) and Nemoto et al. (2010), whereas Cho et al. (2006) reported the detection limit of LAMP as 0·1 TCID50 ml−1 from CPV‐suspected faecal samples. There are several reports of CPV infection among vaccinated and unvaccinated dogs from different parts of India (Chinchkar et al. 2006; Panneer et al. 2008; Mohanraj et al. 2010). Therefore, the development of a simple, rapid, sensitive, cost‐effective and specific diagnostic test like loop‐mediated isothermal amplification (LAMP) technique, for direct detection of CPV in faecal specimens without the elaborate steps involved in template DNA preparation, would be a real boon to veterinary clinicians in resource‐limited veterinary clinics and laboratories for effective control of CPV infections in canines.
Supporting information
Figure S1 (a) Screening of CPV suspected feacal samples by agarose gel electrophoresis of LAMP products; Lane 1, 100 bp DNA ladder, Lanes 2,3,5 and 8, Positive samples; Lane 4,6 and 7, Negative sample; Lane 9, Positive control (CPV‐2) and Lane 10, Negative control (faecal sample from a healthy dog).
Table S1 Nucleotide sequence and sequence analysis of VP2 gene of Canine parvovirus (LAMP product)
Supporting info item
Acknowledgements
The grant (BT/PR14677/ADV/57/111/2010 dt. 20‐5‐2011 of DBT, Govt. of India) provided by DBT, Govt. of India for conducting the study is sincerely acknowledged. The facilities provided by Dean, RAGACOVAS are also highly acknowledged. The authors declare that there is no potential source of conflict of interest.
References
- Aonuma, H. , Yoshimura, A. , Perera, N. , Shinzawa, N. , Bando, H. , Oshiro, S. , Nelson, B. and Fukumoto, S. (2009) Loop‐mediated isothermal amplification applied to filarial parasites detection in the mosquito vectors‐Dirofilaria immitis as a study model. Parasit Vectors 2, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel, M.J. , Scott, F.W. and Carmichael, L.E. (1979) Isolation and immunization studies of a canine parvo‐like virus from dogs with haemorrhagic enteritis. Vet Rec 105, 156–159. [DOI] [PubMed] [Google Scholar]
- Bakheit, M.A. , Torra, D. , Palomino, L.A. , Thekisoe, O.M. , Mbati, P.A. , Ongerth, J. and Karanis, P. (2008) Sensitive and specific detection of Cryptosporidium species in PCR‐negative samples by loop‐mediated isothermal DNA amplification and confirmation of generated LAMP products by sequencing. Vet Parasitol 158, 11–22. [DOI] [PubMed] [Google Scholar]
- Buonavoglia, C. , Martella, V. , Pratelli, A. , Tempesta, M. , Cavalli, A. , Buonavoglia, D. , Bozzo, G. , Elia, G. et al. (2001) Evidence for evolution of canine parvovirus type 2 in Italy. J Gen Virol 82, 3021–3025. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Zhang, J. , Yang, S. , Ma, L. , Ma, Y. , Liu, X. , Cai, X. , Zhang, Y. et al. (2009) Rapid detection of Porcine Parvovirus DNA by sensitive loop‐mediated isothermal amplification. J Virol Methods 158, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchkar, S.R. , Mohana Subramanian, B. , Hanumantha Rao, N. , Rangarajan, P.N. , Thiagarajan, D. and Srinivasan, V.A. (2006) Analysis of VP2 gene sequences of canine parvovirus isolates in India. Arch Virol 151, 1881–1887. [DOI] [PubMed] [Google Scholar]
- Cho, H.S. and Park, N.Y. (2005) Detection of Canine distemper virus in blood samples by reverse transcription loop‐mediated isothermal amplification. J Vet Med 52, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, H.S. , Kang, J.I. and Park, N.Y. (2006) Detection of canine parvovirus in faecal samples using loop‐mediated isothermal amplification. J Vet Diagn Invest 18, 81–84. [DOI] [PubMed] [Google Scholar]
- Cotmore, S.F. and Tattershall, P. (1987) The autonomously replicating parvoviruses of vertebrates. Adv Virus Res 33, 91–174. [DOI] [PubMed] [Google Scholar]
- Curry, J.D. , McHale, C. and Smith, M.T. (2002) Factors influencing real‐time RT‐PCR results: application of real‐time RT‐PCR for the detection of leukemia translocations. Mol Biol Today 3, 79–84. [Google Scholar]
- Decaro, N. , Elia, G. , Martella, V. , Desario, C. , Campolo, M. , Trani, L.D. , Tarsitano, E. , Tempesta, M. et al. (2005) A real‐time PCR assay for rapid detection and quantitation of canine parvovirus type 2 in the feces of dogs. Vet Microbiol 105, 19–28. [DOI] [PubMed] [Google Scholar]
- Decaro, N. , Martella, V. , Desario, C. , Bellacico, A.L. , Camero, M. , Manna, L. , d’Aloja, D. and Buonavoglia, C. (2006) First detection of canine parvovirus type 2c in pups with haemorrhagic enteritis in Spain. J Vet Med B 53, 468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro, N. , Desario, C. , Elia, G. , Campolo, M. , Lorusso, A. , Mari, U. , Martella, V. and Buonavoglia, C. (2007) Occurrence of severe gastroenteritis in pups after canine parvovirus vaccine administration: a clinical and laboratory diagnostic dilemma. Vaccine 25, 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, J. , Beriwal, S. , Chandra, I. , Paul, V.K. , Kapil, A. , Singh, T. and Wadowsky, R.M. (2008) Loop‐mediated isothermal amplification assay for rapid detection of common strains of Escherichia coli . J Clin Microbiol 46, 2800–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa, T. , Kaneshige, T. and Mikazuki, K. (1994) Sensitive detection of canine parvovirus DNA by the nested polymerase chain reaction. Vet Microbiol 41, 35–45. [DOI] [PubMed] [Google Scholar]
- JinLong, Y. , Rui, Y. , AnChun, C. , MingShu, W. , LiZhi, F. , SongQuan, Y. , SuHui, Z. , Liu, Y. et al. (2010) A simple and rapid method for detection of Goose Parvovirus in the field by loop‐mediated isothermal amplification. Virology J 7, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasteva, D. , Toubiana, M. , Hartati, S. , Kusumawati, A. , Dubremetz, J.F. and Widada, S.J. (2009) Development of loop‐mediated isothermal amplification (LAMP) as a diagnostic tool of toxoplasmosis. Vet Parasitol 162, 327–331. [DOI] [PubMed] [Google Scholar]
- Lin, X. , Chen, Y. , Lu, Y. , Yan, J. and Yan, J. (2009) Application of loop‐mediated isothermal amplification for the detection of pathogenic leptospira. Diagn Microbiol Infect Dis 63, 237–242. [DOI] [PubMed] [Google Scholar]
- Mohanraj, J. , Mukhopadhyay, H.K. , Thanislass, J. , Antony, P.X. and Pillai, R.M. (2010) Isolation, molecular characterization and phylogenetic analysis of canine parvovirus. Infect Genet Evol 10, 1237–1241. [DOI] [PubMed] [Google Scholar]
- Monteiro, L. , Bonnemaison, D. , Vekris, A. , Petry, K.G. , Bonnet, J. , Vidal, R. , Cabrita, J. and Megraud, F. (1997) Complex polysaccharides as PCR inhibitors in feces: Helicobacter pylori model. J Clin Microbiol 35, 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, Y. , Nagamine, K. , Tomita, N. and Notomi, T. (2001) Detection of loop‐mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun 289, 150–154. [DOI] [PubMed] [Google Scholar]
- Muller, H. , Aysul, N. , Liu, Z. , Salih, D.A. , Karagenc, T. , Beyer, D. , Kullmann, B. , Ahmed, J.S. et al. (2010) Development of a loop‐mediated isothermal amplification (LAMP) assay for rapid diagnosis of Babesia canis . Transbound Emerg Dis 57, 63–65. [DOI] [PubMed] [Google Scholar]
- Nemoto, M. , Imagawa, H. , Tsujimura, K. , Yamanaka, T. , Kondo, T. and Matsumura, T. (2010) Detection of equine rotavirus by reverse transcription loop‐mediated isothermal amplification (RT‐LAMP). J Vet Med Sci 72, 823–826. [DOI] [PubMed] [Google Scholar]
- Nemoto, M. , Ohta, M. , Tsujimura, K. , Bannai, H. , Yamanaka, T. , Kondo, T. and Matsumura, T. (2011) Direct detection of equine herpesvirus type 1 DNA in nasal swabs by loop‐mediated isothermal amplification (LAMP). J Vet Med Sci 73, 1225–1227. [DOI] [PubMed] [Google Scholar]
- Nkouawa, A. , Sako, Y. , Li, T. , Chen, X. , Wandra, T. , Swastika, K. , Nakao, M. , Yanagida, T. et al. (2010) Evaluation of a loop‐mediated isothermal amplification method using fecal specimens for differential detection of Taenia species from humans. J Clin Microbiol 48, 3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi, T. , Okayama, H. , Masubuchi, H. , Yonekawa, T. , Watanabe, K. , Amino, N. and Hase, T. (2000) Loop‐mediated isothermal amplification of DNA. Nucleic Acids Res 28, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima, T. , Hisaka, M. , Kawakami, K. , Kishi, M. , Tohya, Y. and Mochizuki, M. (2008) Chronological analysis of canine parvovirus type 2 isolates in Japan. J Vet Med Sci 70, 769–775. [DOI] [PubMed] [Google Scholar]
- Panneer, D. , Mukhopadhyay, H.K. , Antony, P.X. and Pillai, R.M. (2008) Comparison of diagnostic tests and antigenic typing of canine parvovirus. Indian J Virol 19, 7–11. [Google Scholar]
- Parida, M.M. , Santhosh, S.R. , Dash, P.K.N. , Tripathi, K. , Saxena, P. , Sahni, A.K. and Ambuj, S. (2006) Development and evaluation of reverse transcription loop‐mediated isothermal amplification assay for rapid and real‐time detection of Japanese Encephalitis virus. J Clin Microbiol 44, 4172–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, C.R. (1999) Host range relationships and the evolution of canine parvovirus. Vet Microbiol 69, 29–40. [DOI] [PubMed] [Google Scholar]
- Parrish, C.R. , Aquadro, C.F. and Carmichael, L.E. (1988) Canine host range and a specific epitope map along with variant sequences in the capsid protein gene of canine parvovirus and related feline, mink, and raccoon parvoviruses. Virology 166, 293–307. [DOI] [PubMed] [Google Scholar]
- Riffenburgh, R.H. (2006) Statistics in Medicine. pp. 274–280. Burlington, MA, USA: Elsevier Academic Press. [Google Scholar]
- Sakulwira, K. , Oraveerakulb, K. and Poovorawanc, Y. (2001) Detection and genotyping of canine parvovirus in enteritic dogs by PCR and RFLP. Sci Asia 27, 143–147. [Google Scholar]
- Spibey, N. , Greenwood, N.M. , Sutton, D. , Chalmer, W.S.K. and Tarpey, I. (2008) Canine parvovirus type 2 vaccine protects against virulent challenge with type 2c virus. Vet Microbiol 128, 48–55. [DOI] [PubMed] [Google Scholar]
- Tilley, M. (2004) PCR amplification of wheat sequences from DNA extracted during milling and baking. Cereal Chem 81, 44–47. [Google Scholar]
- Torigoe, H. , Yamashita, Y. , Seki, M. , Sugaya, A. and Maeno, M. (2006) Detection of Haemophilus influenzae by loop‐mediated isothermal amplification of the outer membrane protein P6 gene. Jpn J Infect Dis 60, 55–58. [PubMed] [Google Scholar]
- Vivek, V.M. (2011) Molecular epidemiology of canine parvovirus in Southern India. MVSc thesis submitted to Pondicherry University, Puducherry.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (a) Screening of CPV suspected feacal samples by agarose gel electrophoresis of LAMP products; Lane 1, 100 bp DNA ladder, Lanes 2,3,5 and 8, Positive samples; Lane 4,6 and 7, Negative sample; Lane 9, Positive control (CPV‐2) and Lane 10, Negative control (faecal sample from a healthy dog).
Table S1 Nucleotide sequence and sequence analysis of VP2 gene of Canine parvovirus (LAMP product)
Supporting info item