

Abstract
Cross-coupling reactions enable rapid, convergent synthesis of diverse molecules and provide the foundation for modern chemical synthesis. The most widely used methods employ sp2-hybridized coupling partners, such as aryl halides or related pre-functionalized substrates. Here, we demonstrate copper-catalysed oxidative cross coupling of benzylic C–H bonds with alcohols to afford benzyl ethers, enabled by a redox-buffering strategy that maintains the activity of the copper catalyst throughout the reaction. The reactions employ the C–H substrate as the limiting reagent and exhibit broad scope with respect to both coupling partners. This approach to direct site-selective functionalization of C(sp3)–H bonds provides the basis for efficient three-dimensional diversification of organic molecules and should find widespread utility in organic synthesis, particularly for medicinal chemistry applications.
Graphical Abstract
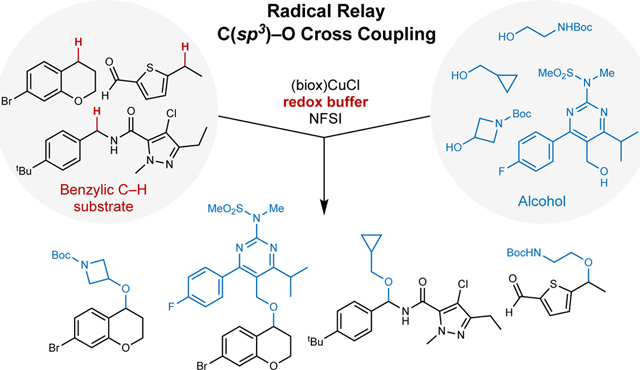
Medicinal chemistry efforts in the pharmaceutical industry rely on efficient synthetic methods to prepare molecules with diverse chemical structures and compositions. Coupling methods that unite molecular fragments from two large pools of substrates, such as amide coupling and palladium-catalysed cross coupling, are among the most important and widely used reaction classes in this domain1,2. The prevalent use of sp2-hybridized coupling partners (i.e., aryl, vinyl, acyl electrophiles), however, constrains the topological diversity of molecules that may be accessed and, in many cases, leads to molecules with less desirable physicochemical and other pharmaceutical properties. These limitations have contributed to a growing demand for cross- coupling methods involving sp3-hybridized carbon atoms to access molecules with more three-dimensional character3,4. C(sp3)–H bonds adjacent to aromatic and heteroaromatic rings are ubiquitous in key pharmacophores, and methods for selective cross coupling of benzylic C–H bonds and other versatile substrate partners (e.g., arylboronic acids, amines, alcohols, Fig. 1a) could have a transformative influence on drug discovery. Such reactions would present a wealth of opportunities for elaboration of simple building blocks and pharmaceutical intermediates, as well as late-stage functionalization of drug molecules5. The comparatively low bond strength of benzylic C–H bonds makes them intrinsically reactive and provides a potential basis for high site selectivity in complex molecules bearing many other C–H bonds. Benzylic sites are also notorious metabolic hot spots in pharmaceuticals, and their selective substitution has important pharmacological implications6.
Fig. 1. Cross-coupling reactions of benzylic C–H bonds and alcohols via a radical relay pathway.
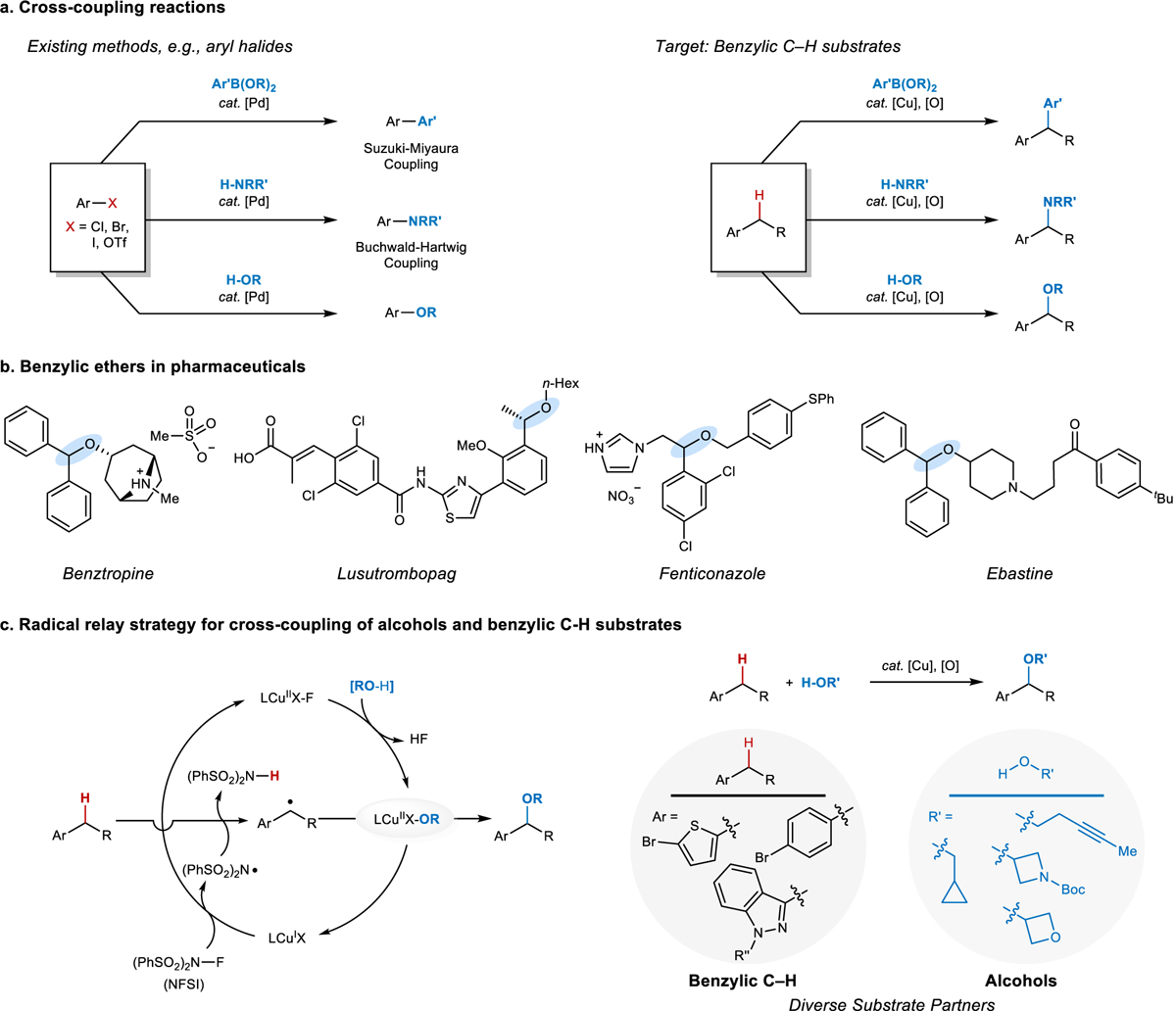
a, Conceptual similarity between traditional cross-coupling reactions of aryl halides and the targeted benzylic C–H functionalization reactions. b, Important examples of existing drug molecules containing benzylic ether moieties. c, Proposed radical relay mechanism for benzylic C–H etherification enabling the coupling of two diverse pools of substrates.
In recent years, a number of methods have been developed for intermolecular functionalization of C(sp3)–H bonds that show good site-selectivity, even in the absence of a directing group. Some of the most effective are those that replace the hydrogen atom of a C–H bond with a small fragment, for example, oxygenation7, amination8,9–10, carbene insertion11, halogenation12,13, and various pseudohalogenation reactions14,15,16–17. Complementary advances have been made in methods for site-selective functionalization of low-cost feedstock molecules, such as alkylarenes18,19, tetrahydrofuran20,21, or simple hydrocarbons22,23–24 that use excess C–H substrate relative to the oxidant and/or functionalization reagent. Collectively, these precedents do not incorporate the characteristics typically associated with “cross coupling” reactions. The most effective cross-coupling methods, such as the Suzuki-Miyaura25 and Buchwald-Hartwig26,27 reactions, have a number of common traits: (a) the most valuable coupling partner is used as the limiting reagent, in ideal cases approaching a 1:1 stoichiometry of the two coupling partners, (b) both coupling partners draw from a diverse pool of readily (ideally commercially) available reagents, and (c) the reactions exhibit broad tolerance of the steric, electronic and functional-group properties of both coupling partners.
Benzyl ethers are prominent motifs in pharmaceuticals and bioactive molecules (Fig. 1b). Alcohols represent an abundant class of building blocks that are widely used as partners in other coupling reactions, including classical methods, such as the Williamson ether synthesis28, in addition to modern catalytic methods26–29. Precedents for the direct oxidative coupling of benzylic C–H bonds and alcohols, however, are confined to electron-rich arenes30–31–32 such as those capable of undergoing hydride transfer to DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone). C–H cleavage via hydrogen-atom transfer (HAT) should be much less sensitive to electronic effects relative to pathways initiated by electron or hydride transfer33, which directly generate cationic intermediates. Thus, we postulated that a “radical relay” strategy14 could provide the basis for selective, broad-scope cross coupling of benzylic C–H bonds and alcohols, using the C–H substrate as the limiting reagent (Fig. 1c). This reaction could be initiated by CuI-mediated activation of an oxidant, such as N-fluorobenzenesulfonimide (NFSI), which generates an N-centered radical capable of promoting HAT from the benzylic C–H bond. The resulting CuII species is then available to mediate coupling of the benzylic radical with the alcohol coupling partner (Fig. 1c). Here, we show that radical-relay cross coupling of benzylic C–H bonds with alcohols is made possible by in situ reductive activation of the catalyst. The latter concept not only provides the basis for successful reactivity in the present reactions, but also sets the stage for development of other benzylic C–H cross-coupling methods.
Results
Identifying the redox buffer effect.
Recent examples of radical relay cyanation14, arylation34, and related functionalization of benzylic C–H bonds35,36–37 provided a starting point for this investigation. We anticipated that reaction conditions similar to these precedents could lead to effective benzylic etherification (Fig. 2a). Attempted coupling of 4-ethylbiphenyl and methanol, however, led to negligible yield of the benzylic ether 4 with little conversion of the substrate or NFSI (Fig. 2a). The good product yields observed from analogous cyanation and arylation reactions indicate that the coupling partner can have a major influence on the reaction outcome. We postulated that the coupling partner could influence the reactive form of the Cu catalyst. Stoichiometric experiments probing the reaction of CuII with the different coupling partners revealed that TMSCN and ArB(OH)2 induce rapid reduction of CuII to CuI, resulting in the formation of cyanogen38 and biaryl39. In contrast, MeOH does not reduce CuII under these conditions (Fig. 2b).
Fig. 2. Cu-catalysed benzylic C–H functionalization with NFSI as the oxidant.
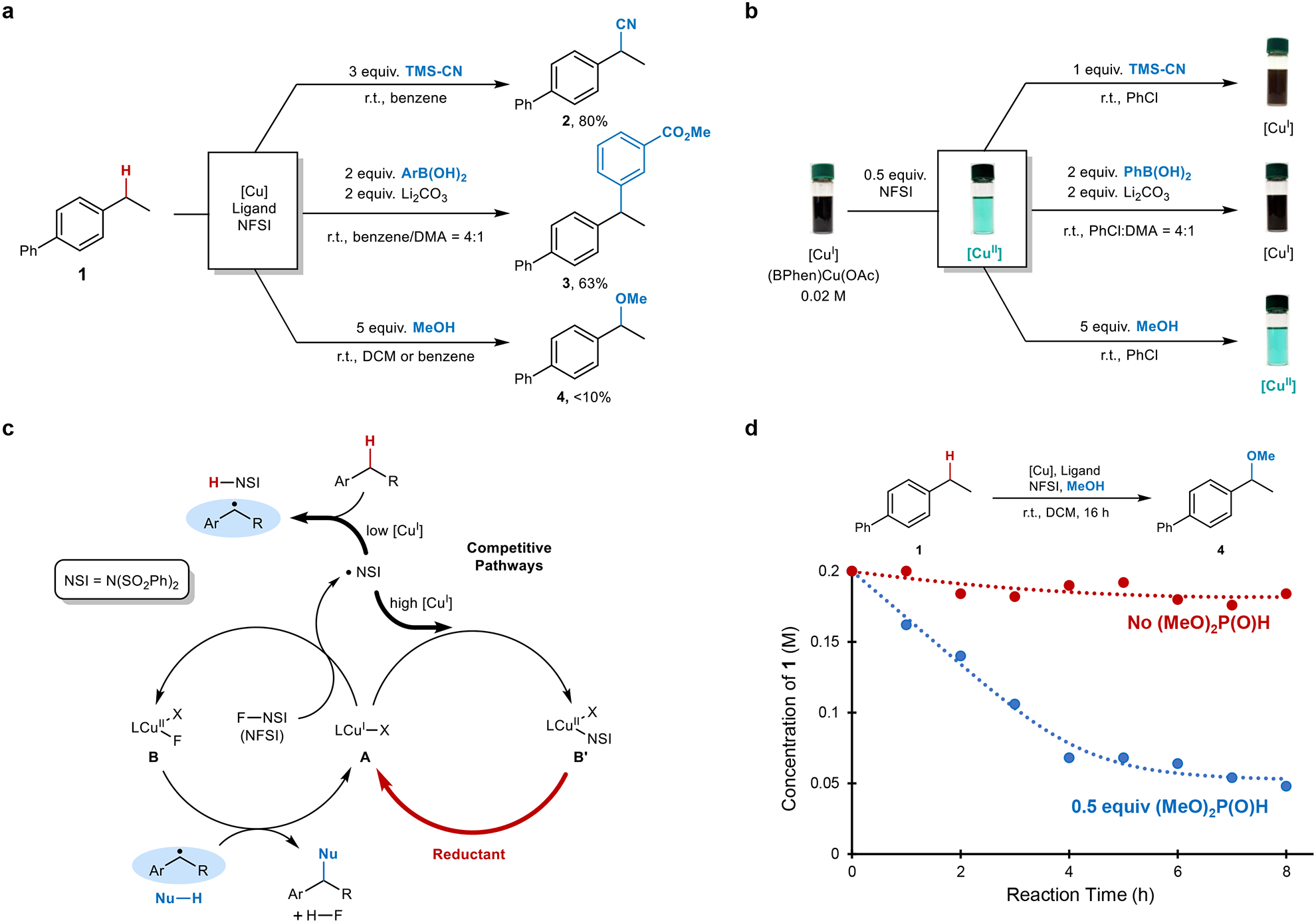
a, Cu-catalysed benzylic C–H functionalization reactions14,34. b, Changes in the Cu redox state between +1 (brown) and +2 (blue-green) upon addition of NFSI to a solution of the CuI catalyst precursor, followed by addition of cross-coupling partners. c, Modified radical relay mechanism (cf. Fig. 1c) to account for quenching of the •NSI by CuI and regeneration of CuI by a reducing substrate or additive. d, Reaction time course for benzylic etherification conducted in the absence (red) and presence of 0.5 equiv of dimethylphosphite (blue). Reaction conditions: 4-ethylbiphenyl (0.2 mmol), NFSI (0.4 mmol), MeOH (1.0 mmol), CuCl (0.02 mmol), 2,2’-bioxazoline (0.02 mmol), DCM (1 mL), room temperature.
These observations indicate that the mechanism in Fig. 1c is overly simplified and needs to be modified to explain successful reaction with certain coupling partners, but not with others. The modified mechanism in Fig. 2c retains reaction of CuI (A) with NFSI to initiate catalysis. This step generates CuII (B) and a nitrogen-centered radical, •NSI (Fig 2c, left cycle). The latter species can either undergo a productive reaction with the benzylic C–H bond, or it can react with a second equivalent of CuI, quenching the radical and forming a second CuII species (B’; Fig 2c, right cycle). Experimental observations suggest that NFSI rapidly oxidizes all of the CuI to CuII when the reaction is initiated, but that certain coupling partners, such as TMSCN or ArB(OH)2, are capable of reducing CuII to regenerate CuI during the course of the reaction (right cycle, red arrow). The CuI generated in this manner will react with NFSI to generate •NSI in the absence of a large pool of CuI, thereby supporting productive HAT from the benzylic substrate. MeOH does not readily reduce CuII, and the Cu catalyst will accumulate as a CuII species, such as B or B’. The inability of CuII to react with NFSI under such conditions will cause the reaction to stall. These mechanistic considerations suggested that a reductant could be identified as a “redox buffer”, leading to controlled regeneration of CuI during the reaction. To test this hypothesis, several reductants were investigated as additives in the etherification reaction with MeOH, including phosphites, silanes, hydrazines, and sodium ascorbate (see Supplementary Table 1 and 2 for details). Promising reactivity was observed with dimethylphosphite [(MeO)2P(O)H], and a representative time course of the reaction in Fig. 2d illustrates the effect of this additive. In the absence of phosphite, the reaction proceeds to <10% conversion of 4-ethylbiphenyl, while inclusion of 0.5 equiv of (MeO)2P(O)H leads to high conversion within 5 h at room temperature, generating the benzyl methyl ether in 52% yield (unoptimized). The latter reaction mixture exhibits a blue-green color (cf. Fig. 2b), implicating a CuII catalyst resting state; however, the results are consistent with the ability of phosphite to serve as a redox buffer (Fig. 2c).40 This hypothesis was probed further with a series of UV-visible and EPR experiments, in which reduction of CuII to CuI by dimethyl phosphite was clearly indicated (see Supplementary Fig. 2 and 3 for details).
Reaction development.
These preliminary results provided the basis for further reaction optimization, and the oxidative coupling of ethylbenzene and methanol was tested with different solvents, ancillary ligands, Cu sources, and reaction temperatures (see the Supplementary Tables 3 and 4 for details). A number of monodentate and bidentate ligands were evaluated, and the unsubstituted 2,2’-bioxazoline (biox) ligand led to the best product yields. Use of chiral ligand derivatives did not lead to enantioselectivity under these conditions (see below for further discussion)14,41. Inclusion of hexafluoroisopropanol (HFIP) as a co-solvent with dichloromethane (DCM:HFIP = 4:1) led to higher yields and significantly increased the reaction rate, allowing the reaction to proceed at lower temperature (40 °C). The activating effect of HFIP suggests that it may enhance the reactivity of the •NSI radical, for example, by hydrogen-bonding to the sulfonyl groups42. Under these conditions, reactions of ethylbenzene and 4-ethylbiphenyl, which are electronically similar, generated the 1-methoxyethylbenzene and 4-(1-methoxyethyl)biphenyl in 80% and 88% yield, respectively (Fig. 3). Use of ethylarenes bearing electron-donating versus electron-withdrawing substituents exhibited variable results, with yields ranging from 10–67% (Fig. 3, red bars); however, the modular nature of the reaction conditions enabled straightforward optimization of these yields by applying intuitive principles. For example, the electron-rich 4-methoxyethylbenzene is more reactive under the standard conditions and undergoes full conversion with the generation of considerable unidentified side products. Use of milder conditions, including removing the co-solvent HFIP (which enhances reactivity) and lowering the temperature from 40 °C to room temperature, led to formation of the desired product in 80% yield. Substrates bearing electron-withdrawing substituents are somewhat less reactive, as evident from incomplete conversion of the starting material under the original conditions. In these cases, the product yield was improved by raising the reaction temperature to 50 °C and/or increasing the Cu catalyst loading to 20 mol %. After applying these variations, the product yields ranged from 66–88%. Only the 4-cyano derivative, which is very electron deficient, retained a low yield (10%) after attempted optimization.
Fig. 3. Electronic effects and site selectivity observed in the oxidative coupling of ethylarenes and methanol.
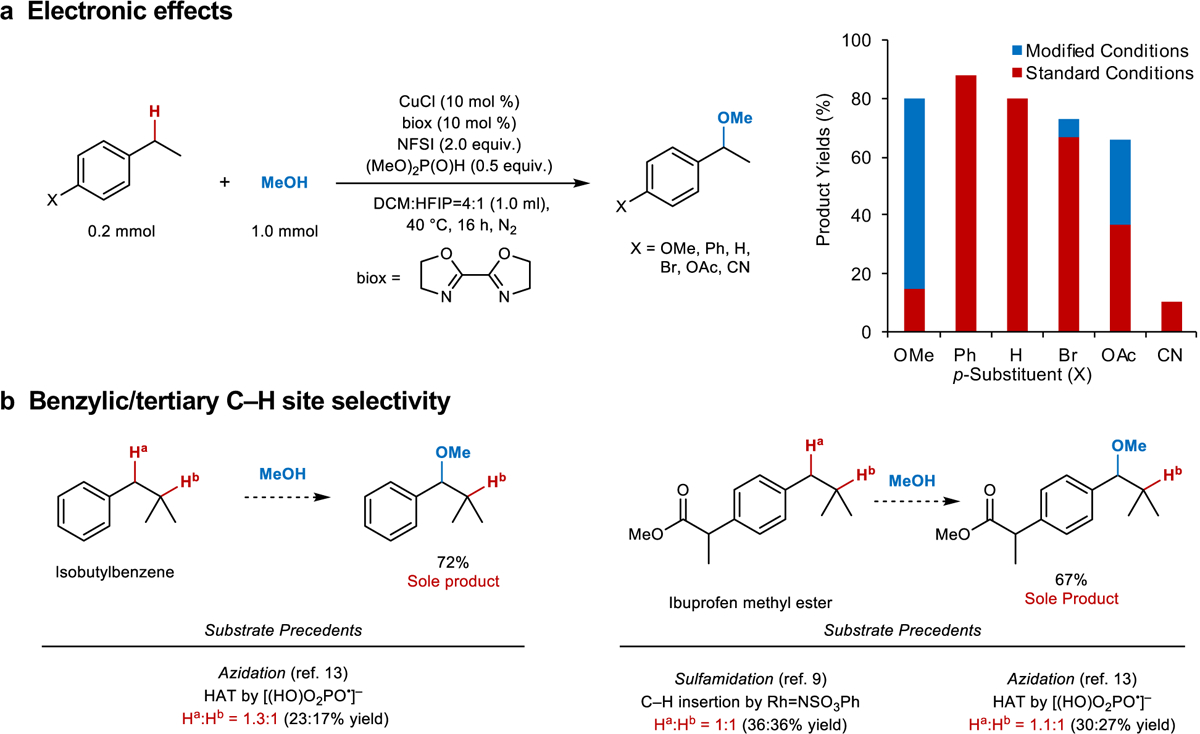
a, Results observed from the reaction under standard (red) and individually optimized (blue) conditions (1H NMR yields with CH2Br2 as the internal standard. Modified conditions: X = OMe: 20 mol % Cu/biox in DCM at r.t.; X = Br: 5 mol % Cu/biox; X = OAc: 20 mol % Cu/biox at 50 °C. b, Analysis of benzylic versus tertiary site selectivity observed in etherification of isobutylbenzene and ibuprofen methyl ester (see Fig. 4 for reaction conditions).
The good results obtained here with electronically differentiated substrates may be rationalized by the HAT C–H activation mechanism. Previous reports of benzylic etherification initiated by hydride or single-electron transfer directly generate cationic intermediates and are typically only effective for electron-rich substrates30–31,32. Loss of a neutral hydrogen atom is much less susceptible to electronic effects33.
The broad tolerance of arene electronic properties is complemented by nearly exclusive site selectivity for benzylic over tertiary C–H bonds. Isobutylbenzene and ibuprofen methyl ester have been used previously to probe selectivity for benzylic versus tertiary C–H activation in photoredox-based azidation13 and nitrene insertion9,43 reactions. Approximately 1:1 product ratios were observed in the reported reactions with these substrates (Fig. 3b). Recognizing that the selectivity depends on the mechanism and reagent involved in the C–H cleavage step, we tested these substrates in the present oxidative coupling conditions with methanol. Exclusive reaction at the benzylic position was observed with both substrates (Fig. 3b), affording 72% and 67% yield of the two methyl ethers, respectively. Complementary experiments with toluene, ethylbenzene, and cumene reveal preferential reactivity at secondary benzylic positions (see Supplementary Fig. 1 for details).
Computational analysis of the proposed mechanism.
The catalytic mechanism proposed in Fig. 2 was analysed by density functional theory (DFT) methods to probe the energetics of individual reaction steps, with a particular focus on the competing pathways involving the •NSI and benzylic radical intermediates, and to gain further insights into C–O bond formation (Fig. 4). The following computation methods were employed in this effort: M06-L/basis-II/SMD(ε = 10.6)//B3LYP-D3(BJ)/basis-I/SMD(ε = 10.6) level of theory (basis-I = 6–31G(d,p) for non-metals and SDD basis and pseudopotential for Cu; basis-II = def2-TZVP for non-metals, def2-TZVP basis and SDD pseudopotential for Cu (See Supplementary Information, section X for details).
Fig. 4. Calculated reaction pathways and energy landscape for (biox)CuI/NFSI-mediated methoxylation of ethylbenzene.
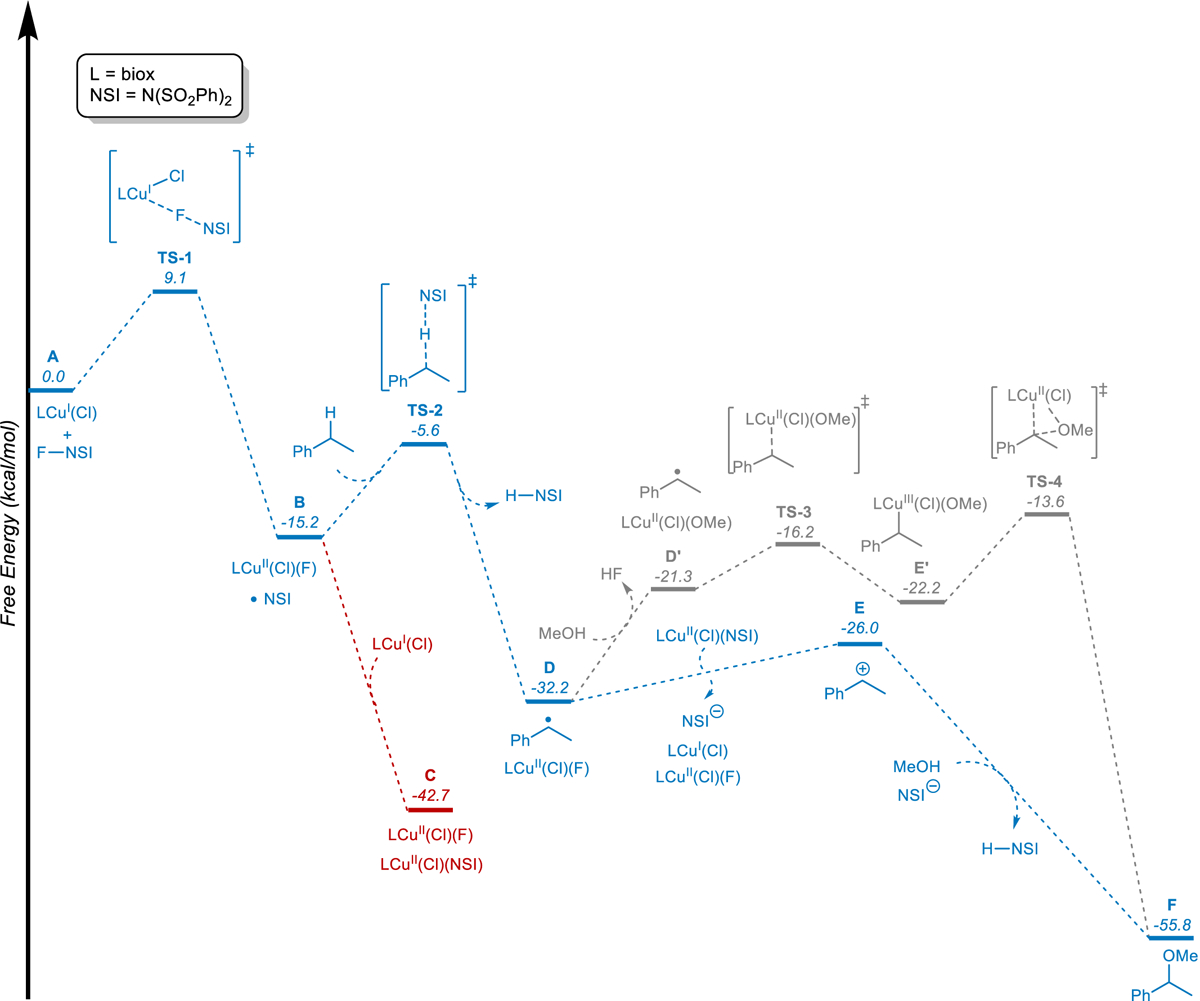
(Gibbs free energies at 313.15 K; computed at M06-L/basis-II/SMD(ε = 10.6)//B3LYP-D3(BJ)/basis-I/SMD(ε = 10.6) level of theory).
The reaction of NFSI with (biox)CuI(Cl) is computed to be highly favorable (ΔG° = –15.2 kcal/mol), generating •NSI and (biox)CuII(Cl)(F)44. Subsequent reaction of •NSI with a second equivalent of CuI is even more favorable (ΔG° = –27.5 kcal/mol), generating (biox)CuII(Cl)(NSI) (Fig. 4, red pathway). This sequence is consistent with the experimental observations in Fig. 2b which show rapid formation of CuII species upon addition of NFSI to solutions of CuI. We then evaluated the energetics of •NSI reactivity with the benzylic C–H bond of ethylbenzene. This HAT reaction, which forms a benzylic radical and H–NSI is also strongly favored (ΔG° = –17.0 kcal/mol) and exhibits an activation free energy (ΔG‡; cf. TS-2) of +9.6 kcal/mol.
Two possible pathways were considered for product formation. The first features benzylic radical addition to CuII and C–O bond formation via reductive elimination from an organocopper(III) intermediate (Fig. 4, grey pathway), while the second features a radical-polar crossover pathway45,46 in which C–O bond formation involves reaction of the alcohol with a benzylic cation (Fig. 4, blue pathway). The former pathway requires incorporation of a methoxide ligand into the CuII coordination sphere, and the calculations indicate that substitution of fluoride is favored over chloride. The resulting process, which affords (biox)CuII(Cl)(OMe) and HF, is endergonic (ΔG° = +10.9 kcal/mol). Addition of the benzylic radical to the CuII species proceeds with a small kinetic barrier (TS-3, ΔG‡ = +5.1 kcal/mol) to form the benzylcopper(III) species E’ in a nearly ergoneutral process (ΔG° = −0.9 kcal/mol). Subsequent C–O reductive elimination yields the methoxylated product via TS-4, which represents the highest energy species along this pathway (+18.6 kcal/mol relative to D). The alternative pathway for C–O bond formation involves one-electron oxidation of the benzylic radical by (biox)CuII(Cl)(NSI) to afford a benzylic cation. This electron-transfer step is only moderately uphill (E, ΔG° = +6.2 kcal/mol), and the resulting cation can undergo a highly favorable reaction with methanol to produce the methoxylated product (F, ΔG° = –29.8 kcal/mol).
The organocopper(III) pathway aligns with the pathway proposed for Cu/NFSI-mediated cyanation of benzylic C–H bonds, which proceeds with high enantioselectivity14. The computational results in Fig. 4, however, favor the radical-polar crossover pathway for the etherification reaction, and this conclusion is further supported by several additional observations. The methoxylation reaction generates racemic products, even when chiral biox ligands are used. Deuterium kinetic isotope effect experiments conducted with PhEt and PhEt-d10 reveal the presence of a small, but significant, primary KIE (competition experiment: KIE = 2.1; independent rate measurement: KIE = 1.7; see Supplementary Figs. 5 and 6). These results are consistent with the radical-polar crossover pathway, which shows that the HAT step has the highest barrier. In contrast, C–O reductive elimination is calculated to be the rate-limiting step in the organocopper(III) pathway, and a negligible KIE is expected for this step.
Synthetic scope and utility.
Benzylic methoxylation is a valuable transformation in medicinal chemistry, particularly in late-stage functionalization applications, because the introduction of small molecular fragments can significantly influence the activity and pharmacological properties of pharmaceuticals4,5. For example, these groups can modulate the physicochemical characteristics and conformational dynamics of the molecule, introduce hydrogen bond donors/acceptors that can lead to enhanced ligand-target binding interactions, and block reactive sites to slow metabolism and excretion. The methoxy group is an appealing fragment because it has minimal impact on the mass or lipophilicity (i.e., LogP) of the molecule and introduces a potential hydrogen bond acceptor site5.
Examination of the substrate scope for benzylic methoxylation began with a number of small molecules and pharmaceutical building blocks as coupling partners (Fig. 5). Longer alkyl chains, including those bearing primary alkyl halide substituents are tolerated by the reaction conditions (10-12, 14), and good reactivity was also observed with a phenylacetic ester derivative (13). Tetralin (14), a substructure present in numerous drugs such as sertraline, treprostinil and rotigotine, underwent effective methoxylation at room temperature. Benzhydryl ethers, including benztropine and ebastine (cf. Fig. 1a) represent an important class of antihistamines47. Methyl ethers were obtained in good yield with a series of benzhydryls (15–19) via oxidative cross coupling with methanol. Substrates included the benzhydryl fragment present in dapagliflozin, an approved drug for treatment of type 2 diabetes (cf. 19). This promising reactivity was extended to C–H bonds adjacent to medicinally relevant sulfur-, oxygen- and nitrogen-containing heterocycles (20–29). Noteworthy features among these examples include tolerance of (hetero)aryl bromides (20, 23, 24) and a formyl group (22), which are versatile functional groups that permit further elaboration of the products. The thiophenylarylmethane core in 24 is a key fragment in canagliflozin, another commercial drug for type 2 diabetes. Chromans and azoles, which represent important pharmacophores48 undergo effective coupling with methanol (25–29). These studies also showed that certain functional groups, such as amines and carboxylic acids required modification (e.g., via acetylation or methyl ester formation, cf. 27 and 28, respectively) to attain the desired reactivity. Other substrates, such as those with pyridine and indole heterocycles, led to lower yields or failed to afford the desired product (see summary provided in Supplementary Table 12).
Fig. 5. Assessment of different benzylic C–H substrates in oxidative cross-coupling reactions with methanol.
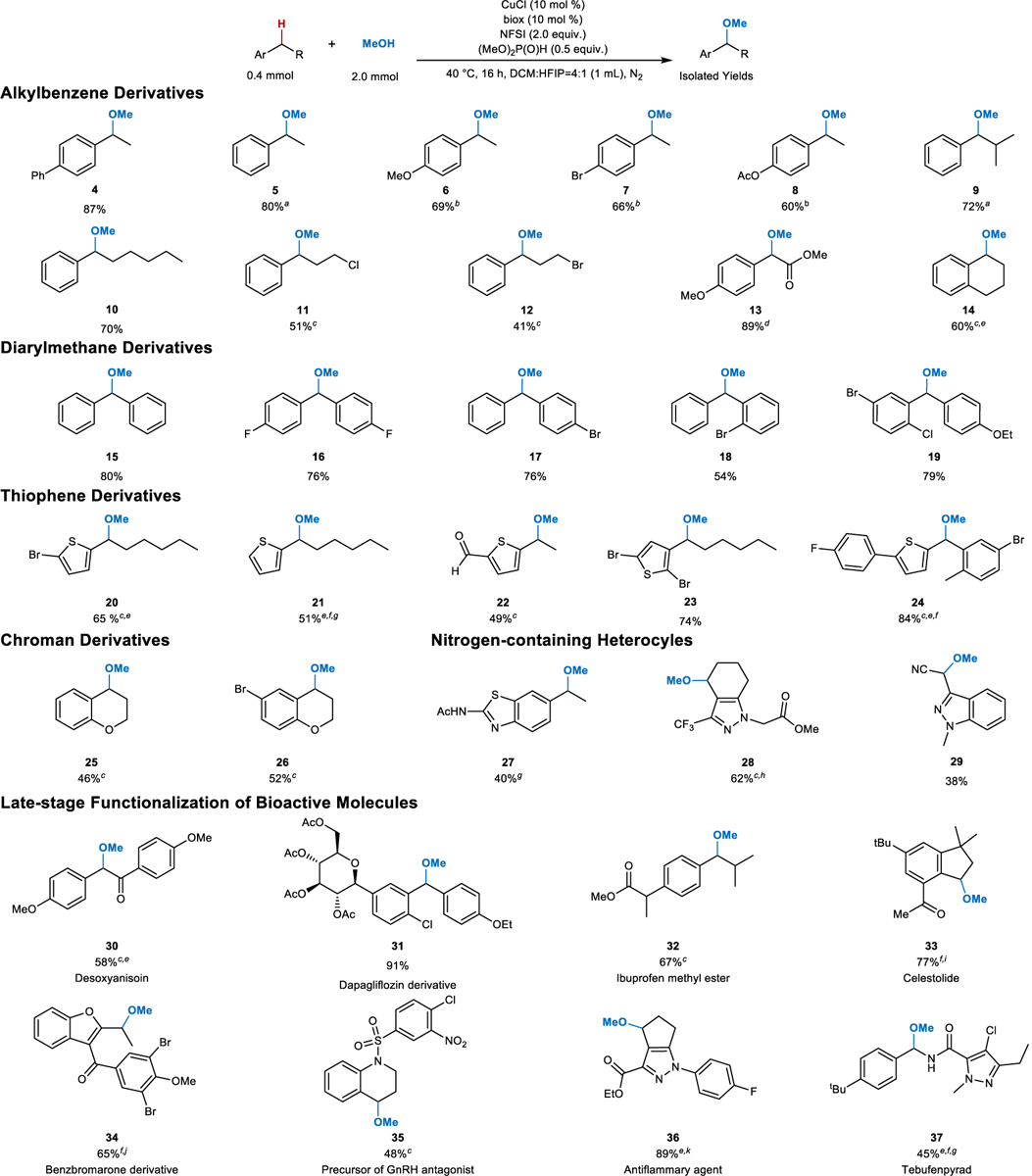
Isolated yields unless otherwise noted. a 1H NMR yield; isolated yield unavailable due to compound volatility. b See Fig. 3a for optimized conditions. c 15 mol % Cu/biox. d Reaction yield at 4 h. e At room temperature. f DCM as the solvent. g 20 mol % Cu/biox. h Only one regioisomer was observed. i At 30 °C. j 30 mol % Cu/biox. k Two regioisomers were observed with a ratio of 9:1.
The methoxylation reaction also proceeded effectively in the late-stage functionalization of a number of pharmaceuticals and related bioactive molecules, including the immunosuppressant desoxyanisoin (30)49; the natural product celestolide (33); a precursor to a GnRH antagonist (35)50; a cyclopentapyrazolyl anti-inflammatory and anti-allergy agent (36)51; and the insecticide, tebufenpyrad (37). Each of these underwent effective coupling with methanol and exhibited excellent selectivity for reaction at the benzylic position. For example, no products were obtained from methoxylation of the aliphatic C–H bond next to the nitrogen atom in 35 or adjacent to the alkoxy oxygen atoms in 30. Good selectivity was also observed between the two similar cyclopentyl C–H positions in 36. The 9:1 ratio favoring the product shown over the alternate regioisomer probably arises from higher reactivity at the more electron-rich site. Free -OH groups, such as those present in a sugar fragment of dapagliflozin (anti-diabetic), a carboxylic acid of ibuprofen (anti-inflammatory), and a phenol in benzbromarone (xanthine oxidase inhibitor52) interfere with the etherification reaction, but successful reactivity proceeds when these groups are suitably protected (31, 32, 34).
The potential utility of this method for medicinal chemistry library synthesis is especially evident from assessment of the C–H cross-coupling reaction with diverse alcohols. The thiophene-containing fragment of canagliflozin was selected as a representative, moderately complex core structure for these studies (Fig. 6a). Initial tests employing 2-chloroethanol as the coupling partner led to two products, the desired 2-chloroethyl ether in addition to the methyl ether product in 64% and 18% yields, respectively. The latter product derives from participation of the methoxy group from the (MeO)2P(O)H reductant in the reaction. Reevaluation of other phosphites showed that this side product formation could be nearly eliminated (<2%) by using diisopropyl phosphite (see Supplementary Table 7 for details). This insight was then implemented in reactions with numerous alcohol coupling partners. Several 2-substituted ethanol derivatives, including those bearing chloro, methoxy, BocNH (Boc = tert-butyloxycarbonyl), alkynyl, vinyl, naphthyl, and benzyl ether substituents (38–44), were effective in the reaction, in most cases affording product in good-to- excellent yields. Only the alkene-containing substrate 42 led to a relatively low yield, possibly reflecting competitive reaction with the allylic C–H bonds. The presence of benzylic C–H bonds in the alcohols 43 and 44 did not interfere with successful reactivity. Both afforded the desired product in good yield. Expanding on this compatibility, a series of ortho-, meta-, and para-substituted benzyl alcohols with different electronic and steric properties proved to be excellent coupling partners in these reactions (45–49, 73–91% yields), with only small amounts of benzaldehyde by-product observed.
Fig. 6. Assessment of different alcohols and C–H/alcohol coupling partners in benzylic C–H etherification reactions. a, Benzylic C–H etherification of a canagliflozin precursor with various alcohols. b, Cross coupling of medicinally relevant benzylic C–H substrates and alcohols.
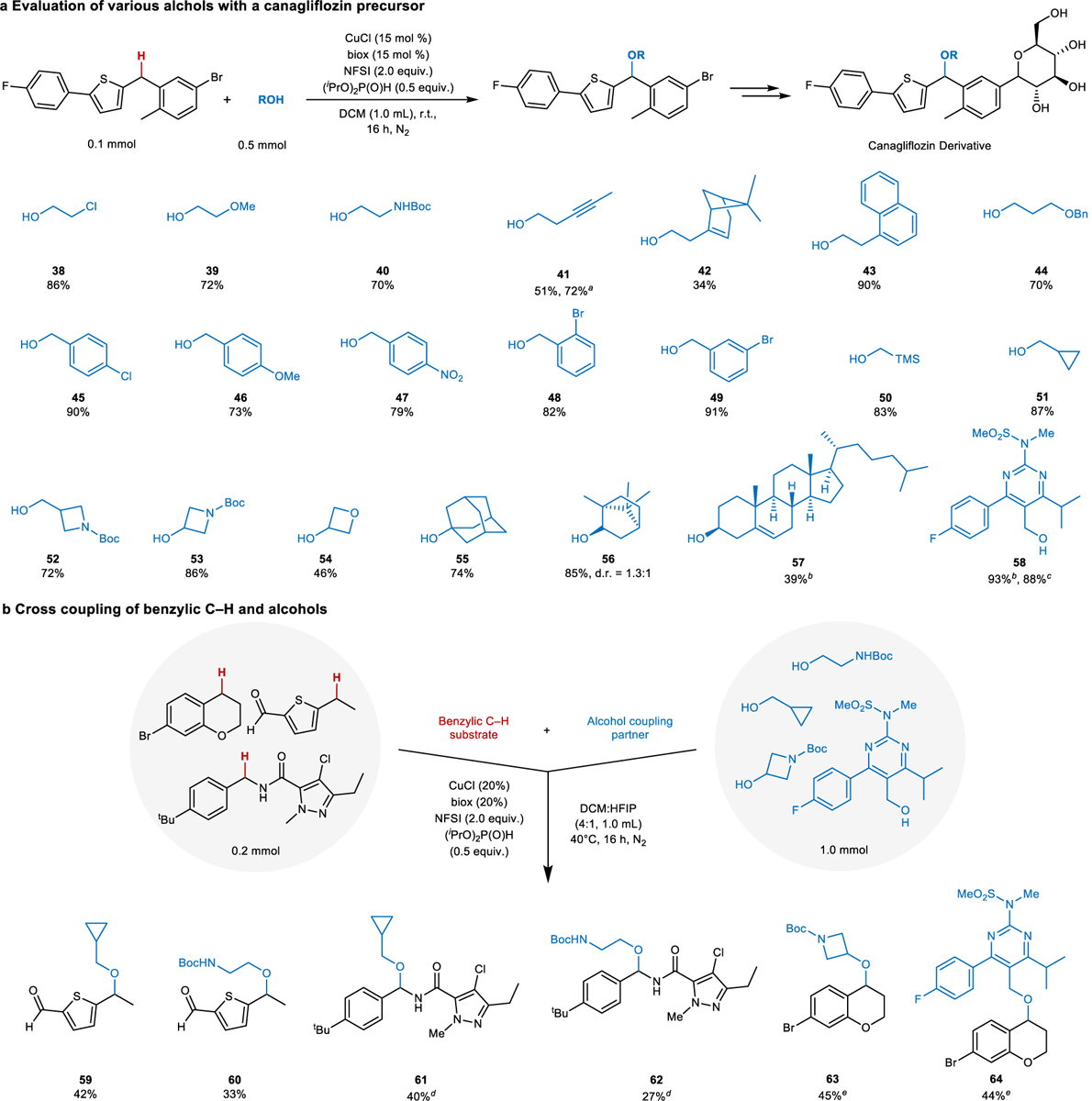
Isolated yields are reported. a 1H NMR yield with 30 mol % Cu/biox. b Conducted with 3.0 equiv. alcohol. c Conducted with 1.1 equiv. alcohol. d 50 °C. e r.t. in DCM.
Other aliphatic alcohols were also successful, including trimethylsilylmethanol, cyclopropylmethanol (50, 51), and several oxetane and azetidine analogs (52–54). Small groups such as these are increasingly featured in preclinical and clinical drug candidates53,54, and the cross coupling of benzylic C–H bonds and alcohols provides a compelling strategy to introduce these units. The effectiveness of adamantanol and (–)-borneol (55, 56) showed that sterically hindered alcohols can serve as effective coupling partners. The method also proved effective with complex alcohols, including cholesterol (57) and a pyrimidinylmethanol precursor to rosuvastatin (58). The latter reaction was demonstrated on >1 g scale (91% yield, 1.3 g) and was also successful with only 1.1 equiv of 58 (88% yield). In a final assessment of the method, six substrate pairs were selected from three representative heterocyclic substrates containing benzylic C–H bonds and four alcohol coupling partners. Moderate to good yields of benzylic ethers were obtained in each of these cross-coupling examples (Fig. 6b).
Conclusions
Collectively, these results demonstrate a new class of highly selective, non-directed C–H cross coupling reactions that create opportunities for efficient synthesis of novel molecules and diversification of chemical structures, ranging from simple aromatic and heteroaromatic building blocks to complex pharmacophores and existing drug molecules. Prominent features of these reactions include good product yields, the ability to use the benzylic substrate as the limiting reagent, high benzylic site selectivity, and access to a broad substrate scope with respect to both reaction partners. Mechanistic insights set the stage for these results by revealing that traditional reaction conditions lead to accumulation of the catalyst in an inactive CuII state, and the key breakthrough arose from identification of dialkylphosphites as effective in situ reductants that convert CuII into catalytically active CuI during the course of the reaction. Further mechanistic studies support a catalytic pathway involving radical-polar crossover initiated by HAT from the benzylic C–H site. This pathway is noteworthy because HAT exhibits a weak dependence on substrate electronic properties that allows for broad substrate scope, and the subsequent trapping of the benzylic cation by alcohols is similarly promiscuous, allowing for broad scope among alcohol coupling partners. Overall, it is likely that the “redox buffering” strategy will not be unique to this reaction class and allow for the discovery and development of other radical relay C–H cross-coupling methods with widespread impact and utility in medicinal chemistry and organic synthesis.
Methods.
General Procedure (I) for Methoxylation of Benzylic C–H Substrates (pressure tube)
Copper(I) chloride (2.0 mg, 0.020 mmol, 10 mol%), 4,4’,5,5’-tetrahydro-2,2’-bioxazole (2.8 mg, 0.020 mmol, 10 mol%), NFSI (126.1 mg, 0.40 mmol, 2.0 equiv.) and benzylic substrate (if solid, 0.20 mmol, 1.0 equiv.) were added to a pressure tube under air, and then the tube was moved to a glove box. Solvent (1.0 mL), benzylic substrate (if liquid, 0.20 mmol, 1.0 equiv.), methanol (42 μL, 1.0 mmol, 5.0 equiv.) and dimethyl phosphonate (9.5 μL, 0.10 mmol, 0.5 equiv.) were added to the tube. The tube was sealed in the glove box and taken out to a hot plate. The sealed tube was heated at 40 °C with stirring for 16 h. When the reaction finished, the mixture was cooled down to room temperature, poured into water and extracted with CHCl3 (10 mL × 3). The organic layers were combined and washed sequentially with saturated sodium bicarbonate and brine, then dried with Na2SO4 and filtered. The mixture was evaporated under vacuum and the crude mixture was purified by automated flash chromatography (silica gel, eluted by pentane:ethyl acetate = 20:1 to 4:1).
General Procedure (II) for Methoxylation of Benzylic C–H Substrates (glass vial)
Copper(I) chloride (2.0 mg, 0.020 mmol, 10 mol%), 4,4’,5,5’-tetrahydro-2,2’-bioxazole (2.8 mg, 0.020 mmol, 10 mol%), NFSI (126.1 mg, 0.40 mmol, 2.0 equiv.) and benzylic substrate (if solid, 0.20 mmol, 1.0 equiv.) were added under air to a 4 mL vial containing a magnetic stir bar. Then the vial was capped with a pierceable Teflon cap. A needle was pierced through the cap to facilitate exchange of the vial headspace with the atmosphere. The vial was moved into a glove box, through three vacuum-nitrogen-backfill cycles. The needle was removed, and the vial was taken out of the glove box (now sealed under an inert gas). Solvent (1.0 mL), benzylic substrate (if liquid, 0.20 mmol, 1.0 equiv.), methanol (42 μL, 1.0 mmol, 5.0 equiv.) and dimethyl phosphonate (9.5 μL, 0.10 mmol, 0.5 equiv.) were added into the vial via injection through the cap. The sealed vial was heated at 40 °C and stirred for 16 h. When the reaction finished, the mixture was cooled down to room temperature and triethylamine (140 μL, 1.0 mmol, 5.0 equiv.) was added to quench any unreacted NFSI. Then the mixture was evaporated under vacuum and the crude mixture was purified by automated flash chromatography (silica gel, eluted by pentane:ethyl acetate = 20:1 to 4:1).
Supplementary Material
Acknowledgements.
We thank Bing Li (Merck & Co., Inc., Kenilworth, NJ, USA) for technical assistance. This work was supported by the NIH (R01 GM126832, to S.S.S. and F32 GM129909, to J.A.B.); Jiangsu Province (BK20161307 and “333” Talents Project, to H.H.) and Huaiyin Normal University (JSKC18014, to H.H.); Merck & Co., Inc., Kenilworth, NJ, USA (S.W.K.; travel funds to S.-J.C.); and M.M. acknowledges a doctoral dissertation fellowship from the University of Minnesota. Spectroscopic instrumentation was supported by a gift from Paul. J. Bender, the NSF (CHE-1048642), and the NIH (1S10 OD020022-1).
Footnotes
Data Availability. The authors declare that all of the data supporting the findings of this study are available within the paper and its supplementary information file.
Supplementary Information is available in the online version of the paper.
Competing Interests.
The authors declare no competing interests.
References
- 1.Brown DG & Boström J Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone?: miniperspective. J. Med. Chem 59, 4443–4458 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Boström J, Brown DG, Young RJ & Keserü GM Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Disc. 17, 709–727 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Lovering F, Bikker J & Humblet C Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Blakemore DC et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Cernak T, Dykstra KD, Tyagarajan S, Vachal P & Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Thompson TN Optimization of metabolic stability as a goal of modern drug design. Med. Res. Rev 21, 412–449 (2001). [DOI] [PubMed] [Google Scholar]
- 7.White MC, Zhao J Aliphatic C–H oxidations for late-stage functionalization. J. Am. Chem. Soc 140, 13988–14009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muniz K & Bosnidou AE Intermolecular radical C(sp3)–H amination under iodine catalysis. Angew. Chem. Int. Ed 58, 7485–7489 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Chiappini N, Mack J & Du Bois J Intermolecular C(sp3)−H amination of complex molecules. Angew. Chem. Int. Ed 57, 4956–4959 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Clark JR, Feng K, Sookezian A & White MC Manganese-catalysed benzylic C(sp3)–H amination for late-stage functionalization. Nat. Chem 10, 583–591 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies HML & Morton D Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev 40, 1857 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Liu W, Groves JT Manganese catalyzed C–H halogenation. Acc. Chem. Res 48, 1727–1735 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Margrey KA, Czaplyski WL, Nicewicz DA & Alexanian EJ A general strategy for aliphatic C–H functionalization enabled by organic photoredox catalysis. J. Am. Chem. Soc 140, 4213–4217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W et al. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma A & Hartwig JF Metal-catalysed azidation of tertiary C–H bonds suitable for late-stage functionalization. Nature 517, 600–604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X & Groves JT Taming azide radicals for catalytic C–H azidation. ACS Catal. 6, 751–759 (2016). [Google Scholar]
- 17.Czaplyski WL, Na CG & Alexanian EJ C–H xanthylation: A synthetic platform for alkane functionalization. J. Am. Chem. Soc 138, 13854–13857 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasilopoulos A, Zultanski SL & Stahl SS Feedstocks to pharmacophores: Cu-catalyzed oxidative arylation of inexpensive alkylarenes enabling direct access to diarylalkanes. J. Am. Chem. Soc 139, 7705–7708 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Zheng Z, Xu X, Mao J & Walsh PJ One-pot aminobenzylation of aldehydes with toluenes. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shield BJ, Doyle AG Direct C(sp3)–H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc 138, 12719–12722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heitz DR, Tellis JC & Molander GA Photochemical nickel-catalyzed C–H arylation: synthetic scope and mechanistic investigations. J. Am. Chem. Soc 138, 12715–12718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry IB et al. Direct arylation of strong aliphatic C–H bonds. Nature 560, 70–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu D et al. Nickel-catalyzed selective oxidative radical cross-coupling: an effective strategy for inert Csp3 –H functionalization. Org. Lett 17, 998–1001 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Tran BL, Li B, Driess M & Hartwig JF Copper-catalyzed intermolecular amidation and imidation of unactivated alkanes. J. Am. Chem. Soc 136, 2555–2563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyaura N & Suzuki A Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev 95, 2457–2483 (1995). [Google Scholar]
- 26.Ruiz-Castillo P & Buchwald SL Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev 116, 12564–12649 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartwig JF Transition metal catalyzed synthesis of arylamines and aryl ethers from aryl halides and triflates: scope and mechanism. Angew. Chem. Int. Ed 37, 2046–2067 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Williamson A Theory of ætherification. Philos. Mag 37, 350–356 (1850). [Google Scholar]
- 29.Enthaler S & Company A Palladium-catalysed hydroxylation and alkoxylation. Chem. Soc. Rev 40, 4912–4924 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Chapman LM, Beck JC, Wu L & Reisman SE Enantioselective total synthesis of (+)-psiguadial B. J. Am. Chem. Soc 138, 9803–9806 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Maloney DJ, Chen S & Hecht SM Stereoselective synthesis of the atropisomers of myristinin B/C. Org. Lett 8, 1925–1927 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Lee BJ, Deglopper KS & Yoon TP Site-Selective Alkoxylation of Benzylic C–H Bonds via Photoredox Catalysis. Angew. Chem. Int. Ed (2019). doi: 10.1002/anie.201910602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rafiee M, Wang F, Hruszkewycz DP & Stahl SS N‑Hydroxyphthalimide-mediated electrochemical iodination of methylarenes and comparison to electron-transfer-initiated C−H Functionalization. J. Am. Chem. Soc 140, 22–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, Chen P & Liu G Copper-catalyzed arylation of benzylic C–H bonds with alkylarenes as the limiting reagents. J. Am. Chem. Soc 139, 7709–7712 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Ni Z et al. Highly Regioselective copper-catalyzed benzylic C-H amination by N-Fluorosulfonimide. Angew. Chem. Int. Ed 51, 1244–1247 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Yang H et al. Silver-promoted oxidative benzylic C−H trifluoromethoxylation. Angew. Chem. Int. Ed 57, 13266–13270 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Xiao H et al. Copper-catalyzed late-stage benzylic C(sp3)–H trifluoromethylation. Chem 5, 940–949 (2019).. [Google Scholar]
- 38.Janz GJ Cyanogen. Inorg. Synth 5, 43–48 (1957). [Google Scholar]
- 39.Demir AS, Reis Ö & Emrullahoglu M Role of copper species in the oxidative dimerization of arylboronic acids: synthesis of symmetrical biaryls. J. Org. Chem 68, 10130–10134 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Hong Y, Yang D, Luo Y, Pao C-W, Lee J-F & Lei A Direct observation of reduction of Cu(II) to Cu(I) by P–H compounds using XAS and EPR spectroscopy. Organometallics 35, 1426–1429 (2016). [Google Scholar]
- 41.Zhang W, Wu L, Chen P & Liu G Enantioselective arylation of benzylic C-H bonds via copper-catalyzed radical relay. Angew. Chem. Int. Ed 58, 6425–6429 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Colomer I, Chamberlain AER, Donohoe TJ Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem 1, 0088 (2017). [Google Scholar]
- 43.Bess EN et al. Analyzing site selectivity in Rh2(esp)2-catalyzed intermolecular C-H amination reactions. J. Am. Chem. Soc 136, 5783–5789 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haines BE et al. Cu-Catalyzed aromatic C–H imidation with N-fluorobenzenesulfonimide: mechanistic details and predictive models. Chem. Sci 8, 988–1001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy JA The Radical-Polar Crossover Reaction In Radicals in Organic Synthesis (eds. Renaud P & Sibi MP) 298–315 (Wiley, 2001). [Google Scholar]
- 46.Banerjee S, Sathyamoorthi S, Du Bois J & Zare RN Mechanistic analysis of a copper-catalyzed C–H oxidative cyclization of carboxylic acids. Chem. Sci 8, 7003–7008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loew ER, MacMillan R & Kaiser ME The anti-histamine properties of benadryl, β-dimethylaminoethyl benzhydryl ether hydrochloride. J. Pharmacol. Exp. Ther 86, 229 (1946). [PubMed] [Google Scholar]
- 48.Du L et al. Molecular hybridization, synthesis, and biological evaluation of novel chroman IKr and IKs dual blockers. Bioorg. Med. Chem. Lett 19, 1477–1480 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Rhodes JR Immunopotentiatory agents and physiologically acceptable salts thereof. US Patent US5508310A (1996). [Google Scholar]
- 50.Fushimi N, Yonekubo S, Ohno K, Miyagi T Nitrogen-containing fused ring derivatives, pharmaceutical compositions containing them and their pharmaceutical use. JP Patent 5308342B2 (2008). [Google Scholar]
- 51.Co Upjohn. Cyclopentapyrazole and tetrahydroindazole compounds. WO Patent Appl. 8607357 (1986). [Google Scholar]
- 52.Schepers G Benzbromarone therapy in hyperuricaemia; comparison with allopurinol and probenecid. J. Int. Med. Res 9, 511–515 (1981). [DOI] [PubMed] [Google Scholar]
- 53.Talele TT The ‘cyclopropyl fragment’ is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem 59, 8712–8756 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Wuitschik G et al. Oxetanes in drug discovery: structural and synthetic insights. J. Med. Chem 53, 3227–3246 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.