

ABSTRACT
The study of cancer cell invasion in 3D environments in vitro has revealed a variety of invasive modes, including amoeboid migration, characterized by primarily round cells that invade in a protease- and adhesion-independent manner. Here, we delineate a contractility-dependent migratory mode of primarily round breast cancer cells that is associated with extensive integrin-mediated extracellular matrix (ECM) reorganization that occurs at membrane blebs, with bleb necks sites of integrin clustering and integrin-dependent ECM alignment. We show that the spatiotemporal distribution of blebs and their utilization for ECM reorganization is mediated by functional β1 integrin receptors and other components of focal adhesions. Taken together, the work presented here characterizes a migratory mode of primarily round cancer cells in complex 3D environments and reveals a fundamentally new function for membrane blebs in cancer cell invasion.
KEY WORDS: Membrane bleb, Integrin, ECM reorganization, Cancer, Cell migration
Summary: This work demonstrates a migratory mode of primarily round cancer cells in complex 3D environments and reveals a fundamentally new function for membrane blebs in cancer cell invasion.
INTRODUCTION
Aberrant migratory and invasive behavior of cancer cells underlies metastatic dissemination, and limited understanding of such behavior represents a critical bottleneck in the effort to reduce cancer morbidity and mortality. Multiple studies have demonstrated that invading cancer cells can employ a variety of migratory modes and exhibit migratory plasticity, which allows for efficient motility through complex and evolving environments. Two migratory modes commonly associated with cancer invasion are termed the mesenchymal and amoeboid migratory modes (Clark and Vignjevic, 2015). Mesenchymal migration of cancer cells is well described: highly elongated cells exhibit filopodia and lamellipodia at the leading edge and display strong integrin adhesion to and reorganization of surrounding extracellular matrix (ECM) (Friedl and Wolf, 2010; Gardel et al., 2010). This migratory mode is reportedly heavily dependent on the Rac1 signaling pathway as well as on the activity of proteases that effect local degradation of the surrounding ECM. When either proteolytic activity is blocked or Rac1 signaling is suppressed and RhoA signaling is induced, cancer cells switch from mesenchymal to amoeboid migration (Wolf et al., 2003; Sahai and Marshall, 2003; Sanz-Moreno et al., 2008; Bergert et al., 2012; Petrie and Yamada, 2012). The amoeboid migratory mode is so named for the morphological similarities between these cells and amoeba, with cells displaying primarily round morphology that is modulated when they squeeze through small pores in the environment, requiring strong actomyosin contractility but not requiring ECM adhesion, reorganization or proteolytic degradation (Yamada and Sixt, 2019). Amoeboid migration is thus considered path finding in contrast to mesenchymal migration, which is path generating (Wolf and Friedl, 2011; Friedl and Alexander, 2011).
Although cancer cell migration has typically been categorized as mesenchymal or amoeboid, additional and hybrid or mixed modes of migration have been noted (Lehmann et al., 2017; Diz-Muñoz et al., 2010; Orgaz et al., 2014; Petrie et al., 2017; Lorentzen et al., 2011). Moreover, although round cancer cells have typically demonstrated migration consistent with the amoeboid mode as described above, it is unclear whether observed round cell cancer migration constitutes a single mode mechanistically. Indeed, although migratory modes are commonly classified by the predominant type of protrusions that the cells demonstrate, migrating round cells have demonstrated a variety of protrusions and subcellular distributions thereof, depending on both cell type and environmental context (Welch, 2015). In particular, round cells have alternately demonstrated actin-enriched regions, large hemispherical blebs or highly dynamic, small membrane blebs (Wyckoff et al., 2006; Lorentzen et al., 2011; Liu et al., 2015; Ruprecht et al., 2015; Petrie and Yamada, 2012). It is currently unclear whether such observed migratory modes are distinct and whether blebs have specific and critical functions in such invasion. Further clarification of round cancer cell migratory strategies is also demanded by the fact that intravital imaging studies confirm the importance of round invading cancer cells in vivo (Sanz-Moreno et al., 2011; Gligorijevic et al., 2014). Indeed, intravital imaging has revealed enrichment of round cells at the invasive fronts of breast cancers (Giampieri et al., 2009; Pinner and Sahai, 2008a) and both primary and metastatic melanomas (Cantelli et al., 2015; Orgaz et al., 2014; Sanz-Moreno et al., 2011; Tozluoğlu et al., 2013; Herraiz et al., 2016).
RESULTS
To investigate modes of round cancer cell motility in three-dimensional (3D) environments and the potential role of blebs in such migratory modes, we studied migration of MDA-MB-231 and MDA-MB-468 cells in biochemically and biomechanically distinct 3D matrices of either fibrillar (collagen I) or fibrillar/non-fibrillar [collagen I/basement (BME)] composite architecture. Previous work in our laboratory revealed that, of a set of breast cancer cell lines, these two triple-negative cell lines showed the highest invasion efficiency in 3D collagen matrices (Ziperstein et al., 2015). Although these cell lines belong to different aggregate morphology subclasses as classified by Kenny et al. (Kenny et al., 2007), here we observed that both cell lines display a range of cell morphologies and cell protrusions, including round bleb-bearing cells (Fig. 1A), and show comparable degrees of invasiveness in 3D environments (Fig. S1a,b). These cell lines thus represent a suitable set for dissecting the role of blebs in cancer cell invasion.
Fig. 1.

Invasive breast cancer cells of different morphological subclasses show membrane blebs in 3D environments. (A) Representative confocal fluorescence maximum projections of phalloidin-stained MDA-MB-468 and MDA-MB-231 cells embedded in 1 mg/ml 3D collagen I gels (Coll). From left to right: MDA-MB-468 cell displaying both membrane blebs and actin polymerization-driven protrusions, MDA-MB-468 cell depicting exclusively membrane blebs, MDA-MB-231 cell with membrane blebs and actin polymerization-driven protrusions, MDA-MB-231 cell displaying predominantly membrane blebs. In all cases, arrows point to blebs and arrowheads point to actin-driven protrusions. Here, as in all figures, cell line names are presented as MB231 and MB468 for MDA-MB-231 and MDA-MB-468 cells, respectively. (B) Histogram of cell circularity of MDA-MB-468 cells in 1 mg/ml collagen I (Coll) and in 1 mg/ml collagen I plus 3 mg/ml BME (Coll-BME) matrices. Differences in distribution of MDA-MB-468 circularity between different matrices were assessed by KS analysis. (C) Percentage of MDA-MB-468 cells bearing at least one protrusion from each class in Coll and Coll-BME environments. Actin polymerization-driven protrusions are summarized under ‘filopodia’. Differences in occurrence of each protrusion type were assessed by t-tests. (D) Histogram of cell circularity of MDA-MB-231 cells in pure collagen and composite matrices; differences in distribution were assessed by KS analysis. (E) Percentage of MDA-MB-231 cells bearing at least one protrusion from each class in Coll and Coll-BME environments, with differences assessed by t-tests. In B-E, data were pooled from three or four statistically equivalent biological replicates. n=149, 103, 117, 122 for MDA-MB-468 Coll, MDA-MB-468 Coll-BME, MDA-MB-231 Coll, and MDA-MB-231 Coll-BME, respectively. All images and quantified data are from cells imaged 24 h after embedding in the matrix. *P<0.05, ***P<0.001; † indicates insignificance. Scale bar: 10 µm.
Cell morphology and protrusions were investigated on cells dispersed in 3D collagen I and composite (collagen I/BME) matrices. Cell morphology was assessed quantitatively through analysis of cell circularity performed on confocal images of cells, with F-actin fluorescently labeled with phalloidin. This analysis showed that MDA-MB-468 cells in both fibrillar and composite fibrillar/non-fibrillar matrices are statistically equivalent in terms of shape and are constitutively round (median circularity, c=0.87 in both matrices; Fig. 1B), whereas the morphology of MDA-MB-231 cells differs between matrices (c=0.26 versus 0.90 in collagen and composite matrices, respectively; Fig. 1D). Although MDA-MB-231 cells in pure collagen I matrices displayed the most elongation among the tested cell lines and conditions, MDA-MB-231 cells in composite matrix were significantly less elongated and comparable to MDA-MB-468 in both matrices.
Cell protrusions were classified as based on either actin polymerization (lamellipodia, filopodia, brushes) or actomyosin contractility (blebs), and the occurrence of such protrusions alone or in combination was determined. This analysis showed that MDA-MB-468 cells in composite matrix exhibited the highest proportion of bleb-bearing cells and lowest proportion of cells with actin-driven protrusions. MDA-MB-231 cells in collagen I exhibited the lowest proportion of bleb-bearing cells, although 38% of such cells did display blebs (Fig. 1C,E). In composite matrix, MDA-MB-231 cells were more likely to have blebs, consistent with the observed shift toward round cell morphology under this ECM condition. Correlative analysis of protrusion type versus cell circularity revealed that the majority of cells with a high circularity index (c≥0.75) displayed blebs across both cell lines and matrix conditions. In contrast, the proportion of bleb-bearing cells among the elongated cells (c≤0.75) was found to be a function of the cell line and matrix condition (Fig. S2a,b). Taken together, the data suggest that cells with round morphology, dictated by either their biological properties or by the specific extracellular matrix conditions, commonly display blebs, often as the predominant cell protrusion type.
We next inspected whether these different morphologies and protrusions occur during active 3D invasion. For invasion studies, multicellular spheroids embedded in 3D collagen I matrices were used because they resemble solid tumors in their pH, oxygen and nutrient gradients, in the distribution of proliferating cells and in their interaction with the surrounding ECM (Kim et al., 2004; Dufau et al., 2012). Moreover, invasive cells can be straightforwardly identified through their position relative to the spheroid core. Both cell lines showed robust invasion in collagen I matrices, with MDA-MB-468 spheroids displaying a consistently round cell morphology and MDA-MB-231 spheroids showing a dynamic mixture of round and elongated cells (Fig. S1c,d shows representative images from Movies 1,2). Such invading cells demonstrated blebs, as can be appreciated from both the transmitted light images and higher resolution images that are introduced and discussed later.
To analyze cell–ECM interactions of round cancer cells during the dynamic process of spheroid invasion, live cell transmitted light microscopy and confocal reflectance microscopy of 3D collagen I-embedded MDA-MB-468 and MDA-MB-231 spheroids and their surrounding matrix were performed during early time points in spheroid invasion. These experiments demonstrated dynamic radial alignment of the collagen matrix at the spheroid front during the onset of invasion, as well as round bleb-bearing cells re-arranging collagen while emigrating from the spheroid into the surrounding matrix (Fig. 2 shows representative images from Movies 3,4). The radial alignment of collagen fibers in front of individual invading cells suggests that round bleb-bearing cells establish cell–ECM interactions that result in ECM reorganization.
Fig. 2.

Representative images from live-cell imaging. (A,B) MDA-MB-468 and (C,D) MDA-MB-231 cells in a multicellular tumor spheroid were imaged invading in a 1 mg/ml collagen I matrix in (left panels) transmitted light and (right panels) confocal reflectance. In the initial images (A,C), a round bleb-bearing cell with apparent collagen attachment (arrow) is shown at t=10 h 35 min (A) and t=4 h 25 min (C). In the subsequent images taken 5 min later (B,D), extensive collagen I rearrangement is apparent in the direct vicinity of the point on the cell with the initial attachment (arrows). Locations of apparent lack of collagen I are caused primarily by out of plane spheroid and invading cells limiting excitation light reaching the imaging plane. Spheroid core is indicated with a white dotted line in initial transmitted light images. Scale bars: 50 μm. Images are taken from Movies 3,4.
To visualize the cell–ECM interactions of spheroids invading collagen I matrices in more detail, confocal fluorescence microscopy was used on fixed samples of spheroids that had invaded for 24 h (Fig. S3). Such experiments were performed not only with MDA-MB-468 and MDA-MB-231 spheroids but also with an organoid prepared from a primary patient-derived breast tumor. Round invading cells and extensive collagen reorganization were apparent in all cases.
The extensive collagen reorganization around round invading cancer cells suggests that these cells use cellular pathways that are distinct from those described in classical amoeboid migration, which is reportedly independent of integrin-mediated adhesion and protease-mediated ECM degradation. To elucidate the molecular pathways underlying the 3D invasion of round bleb-bearing cells, we addressed invasion dependence on β1 integrin (the integrin subunit common to collagen I binding integrin receptors), ECM degradation-mediating enzymes and the small GTPases Rac and Rho. Rac is the key regulator of protrusive actin polymerization, and Rho acts to regulate the actomyosin contractility required for cell squeezing and membrane blebbing (Sanz-Moreno et al., 2008; Lorentzen et al., 2011; Yang et al., 2012). Although Rho-mediated actomyosin contractility is required for both mesenchymal and amoeboid migration, the amoeboid mode is reported to require higher actomyosin contractility levels than the mesenchymal mode (Sadok et al., 2015; Wilkinson et al., 2005).
Spheroids of both cell lines were embedded in collagen I-based matrices and allowed to invade over 24 h under inhibition of the pathways mentioned above. Rac signaling was inhibited through a small molecule that selectively inhibits Rac1 activation through the respective guanine nucleotide exchange factors (Gao et al., 2004). Inhibition of Rho signaling was achieved by small molecule-mediated inhibition of its direct downstream effector, the p160-Rho-associated coiled-coil kinase (ROCK) (Ishizaki et al., 2000). Integrin signaling was inhibited by siRNA-mediated knockdown of β1 integrin. Inhibition of proteases was achieved through an inhibitor cocktail targeting both matrix metalloproteases (MMPs) and serine/cysteine proteases. The mean relative invasive distance (see Materials and Methods and Fig. S1a) of inhibitor-treated samples normalized to the inhibitor solvent control or non-targeting RNA-transfected control group was used to measure 3D invasion efficiency under inhibition (Fig. 3). Although the inhibition of Rac-mediated actin polymerization did not significantly reduce MDA-MB-468 invasion, the inhibition of Rho-dependent actomyosin contractility led to a striking (90%) decrease in invasive distance (Fig. 3A). This indicates that MDA-MB-468 invasion relies strongly on Rho/ROCK-mediated cell contractility-governed mechanisms, among them bleb formation, as reported for classical amoeboid migration.
Fig. 3.

Round bleb-bearing cancer cells invade 3D collagen in a cell contractility- and integrin-dependent manner. (A) MDA-MB-468 spheroid invasion in 1 mg/ml 3D collagen gels under pharmacological or genetic inhibition of Rac, Rho/ROCK, β1 integrin and proteases (MMPs). n=9, 7, 12, 10 and 9, 8, 12 and 10 for Rac, ROCK, β1 integrin and protease inhibition and their controls, respectively. (B) MDA-MB-231 spheroid invasion in 1 mg/ml 3D collagen gels under inhibition of Rac, Rho/ROCK, β1 integrin and proteases. n=5, 6, 13, 5 and 6, 5, 13, and 6 for Rac, ROCK, β1 integrin and protease inhibition and their controls, respectively. (C) MDA-MB-231 spheroid invasion in composite collagen/BME gels under inhibition of Rac, Rho/ROCK, β1 integrin and proteases. n=11, 7, 14, 8 and 11, 8, 13 and 8 for Rac, ROCK, β1 integrin and protease inhibition and their controls, respectively. In all cases, graphs show mean±s.d. invasion relative to solvent or non-targeting siRNA controls for each inhibitor at t=24 h. Differences were assessed via Wilcoxon Rank-Sum tests; *P<0.05, **P<0.01, ***P<0.001.
Another distinction between mesenchymal and amoeboid migration is the requirement for ECM degradation. Inhibition of proteolytic activity reduced MDA-MB-468 spheroid invasion by 31%, demonstrating that the mode of round cell invasion employed utilizes protease-mediated mechanisms although is not entirely dependent on them (Fig. 3A). Finally, we addressed the requirement for integrin-mediated adhesion, because classical amoeboid migration is characterized by no or low dependency on integrin receptors. We found that MDA-MB-468 invasion was reduced by 45% by β1 integrin knockdown and is thus integrin dependent, in contrast to classical integrin-independent amoeboid migration (Fig. 3A).
To address whether this mode of round cancer cell invasion is only observed in cancer cells with constitutively round morphology, we performed the same series of experiments with MDA-MB-231 cells, which can be induced to switch from predominantly elongated to predominantly round morphology in response to specific ECM conditions (Fig. 1). In pure collagen matrix, which supports a mixture of dynamically interconverting elongated and round morphologies, MDA-MB-231 spheroid invasion was similarly mildly, yet significantly, affected by both Rac and Rho inhibition (28% and 21% reduction in invasive distance, respectively; Fig. 3B), consistent with the idea that under conditions allowing for both round and elongated phenotypes, the cells can interchangeably use Rac-driven and Rho/ROCK-driven invasion modes. MDA-MB-231 cells were rendered more sensitive to ROCK inhibition in the composite matrix in which cells shifted toward round morphology (73% reduction; Fig. 3C). The dependence of round cell invasion on integrin-mediated signaling and ECM degradation, as seen in the MDA-MB-468 cells, was confirmed in the ECM-inducible MDA-MB-231 system.
To confirm the importance of β1 integrin activity in MDA-MB-468 and MDA-MB-231 cells, spheroid invasion assays and gel contraction assays were also performed under antibody-mediated inhibition of β1 integrin. The reduction in collagen invasion mediated by inhibition of integrin receptor ligand-binding function was statistically identical to that observed under integrin receptor knockdown in both cell lines (Fig. S4a,b), indicating that invasion efficiency is dependent on ligand-induced outside–in signaling of β1 integrin receptors. Gel contraction assays, which require both efficient ECM adhesion and actomyosin contractility, confirmed efficient force generation by both MDA-MB-231 and MDA-MB-468 cells in collagen I matrices and its dependency on integrin receptors (Fig. S4c). We noted that MDA-MB-468 cells had lower β1 integrin expression than MDA-MB-231 cells (Ziperstein et al., 2015), as confirmed by flow cytometric analysis (Fig. S4d,e). Thus, the β1 integrin dependency of bleb-mediated cell invasion in collagen environments is not specific to cells with particularly high expression levels of these integrins. These findings, together with the observations captured by the images in Fig. 2, suggest that both MDA-MB-468 and MDA-MB-231 cells display a mode of round bleb-bearing cell invasion in 3D environments that differs from classical amoeboid migration in its strong dependence on integrins.
The findings described above suggested that bleb sites represent locations of β1 integrin-mediated interactions with the ECM. To investigate β1 integrin utilization in cells displaying blebs, subcellular integrin distribution and clustering were analyzed in individual cells dispersed in collagen environments, as well as in cells invading from spheroids. To this end, collagen-embedded dispersed MDA-MB-468 cells, dispersed MDA-MB-231 cells and MDA-MB-231 cells prepared as spheroids were co-stained with a total β1 integrin antibody and a general membrane marker, CellMask (Fig. 4; Fig. S5). For this analysis, only individual isolated blebs were selected in order to allow clear visualization of β1 distribution on and around blebs. However, it should be noted that collagen-embedded cells typically display regions of clustered blebs accumulated in one region of the cell in addition to individual blebs. This is apparent upon close inspection of Fig. 4 and is discussed in further detail later. Intensity profiles of both β1 and CellMask were obtained for the perimeter of the cell (see Materials and Methods; Fig. S5d). Membrane sites with intensities higher than the mean intensity plus two standard deviations (s.d.) were considered ‘clusters’. Bleb necks were specifically analyzed for the presence of total β1 integrin clusters and inspected for co-localization with CellMask clusters. This analysis revealed that in all cases the percentage of blebs displaying a total β1 integrin cluster on one or both sides of the bleb neck was markedly and significantly higher than those displaying a CellMask cluster (Fig. 4D). Thus, the potentially increased amount of membrane material at the bleb neck can be ruled out as the sole cause of apparent β1 integrin clustering at these sites. Statistical analysis of cluster data by a t-test between percentages revealed that, for both cell lines and conditions, the measured occurrence of integrin clusters at bleb necks was significantly higher than that expected if their occurrence were random (Fig. 4D). In contrast, the incidence of both integrin and CellMask clusters on the bulk of the cell membrane was found to be as expected, assuming random and normal distribution (Fig. S5e).
Fig. 4.

β1 integrins cluster at bleb necks. (A-C) Representative images of a 1 mg/ml collagen I-embedded MDA-MB-468 cell (A), MDA-MB-231 cell (B) and the edge of an invading MDA-MB-231 spheroid labeled with a β1 integrin antibody (C). Corresponding images of the general membrane marker CellMask are shown in Fig. S5a-c for each of the three images. In the insets, arrows mark a position at the bleb neck showing an integrin cluster. Scale bars: 10 µm. (D) Analysis of fluorescence intensity distribution of β1 integrin receptors and CellMask at bleb necks. Fluorescence values at the bleb neck exceeding the mean+2s.d. fluorescence of the entire cell were considered clusters. Graph shows the occurrence of blebs (%) with one or two β1 integrin or CellMask clusters at the bleb necks. n=125 blebs for MDA-MB-468, n=104 for dispersed MDA-MB-231 and n=140 for MDA-MB-231 spheroids. Data was pooled from three statistically equivalent independent experiments. The occurrence of β1 integrin and CellMask clusters at bleb necks were compared both with each other and with the expected occurrence using a t-test between percentages. Dashed line indicates the expected incidence of bleb clusters assuming a normal distribution of data. All images and quantified data are from cells imaged 6 h after embedding in the matrix. **P<0.01, ***P<0.001.
We also addressed the question of whether the occurrence of β1 integrin clusters at bleb necks correlated with bleb size, indicating potential preferential occurrence of clusters at a certain stage during bleb expansion or retraction. To this end, the β1 fluorescence intensity at the bleb neck was normalized to the mean intensity of the cell and plotted against the width of the corresponding bleb neck and the perimeter of the bleb. No correlation between the size of the bleb and the incidence of β1 integrin clusters at the bleb neck was found (Fig. S2c,d).
Given the fact that β1 integrin clusters have been associated with focal adhesions, not only on 2D substrates but also in 3D collagen I environments (Harunaga and Yamada, 2011; Doyle et al., 2015; Doyle and Yamada, 2016; Kubow et al., 2013; Fraley et al., 2010), the occurrence of a subset of other focal adhesion associated proteins (in particular, talin and vinculin) was investigated using the same approach as that described for β1 integrins (Fig. 5). This and subsequent experiments and quantitative analyses were performed using dispersed MDA-MB-231 cells, because here β1 integrins are readily visualized as a result of their high expression level (in contrast to MDA-MB-468 cells) and lack of congestion (in contrast to spheroid culture). Talin clusters were found to occur at bleb necks in 25% of blebs, well above that expected statistically and similar to the rate at which total β1 integrin clusters were found. Notably, 79% of talin-positive bleb necks were also positive for total β1 clusters. Vinculin was less likely to be found at bleb necks, with 10% of bleb necks showing vinculin clusters. However, the majority (70%) of vinculin cluster-positive bleb necks also displayed total β1 clusters at the same site. Given the occurrence of talin/vinculin and β1 clusters, the expected number of times a co-cluster of those proteins would be expected was calculated, assuming the occurrence of each was independent. The actual occurrence of co-clusters of both talin and vinculin with β1 was significantly higher than the expected occurrence (Fig. 5C). Such observations are consistent with the idea that bleb necks function as sites of mechanotransduction.
Fig. 5.

Proteins involved in mechanotransduction are present at bleb necks. (A,B) Representative images of a 1 mg/ml collagen I-embedded MDA-MB-231 cell at t=6 h co-stained for talin and β1 integrin (talin channel shown) (A) or vinculin and β1 integrin (vinculin channel shown) (B). Scale bars: 10 µm. (C) Analysis of fluorescence intensity distribution of talin and vinculin at bleb necks as well as their co-clustering with β1 integrin at these sites. n=113 blebs for talin/β1, n=101 for vinculin/β1. Data were collected and pooled from two independent experiments. Dashed line indicates the expected incidence of blebs with co-clusters. Differences between measured and expected occurrence of co-clusters were analyzed by a t-test between percentages; **P<0.01, ***P<0.001.
To address in more detail whether bleb necks represent sites of mechanotransductory signaling, we analyzed the distribution of β1 integrins in the closed inactive conformation and the open active form at bleb necks using antibodies specific for these forms (Arjonen et al., 2012). Here, in analyses performed as described for characterizing total β1 integrins at bleb necks, it was found that inactive integrins were detected at 40% of bleb necks whereas active integrins were present in 19% of bleb necks (Fig. 6A-C). Visualization of dispersed cells in collagen networks showed a high degree of collagen accumulation and alignment coinciding with areas of active integrin accumulation in regions with abundant blebs (Fig. 6D) and, in particular, at bleb necks (Fig. 6A; Fig. S6). Pearson's correlation analysis revealed a significantly higher correlation between active integrins and collagen than between inactive integrins and collagen (Fig. 6F). This analysis showed that active β1 integrin coincides with areas of collagen accumulation and also coincides with areas of clustered bleb formation (Fig. 6D). Because of technical constraints, clustered blebs were not analyzed for subcellular protein distribution, which might explain why the percentage of analyzed bleb necks exhibiting clusters of active β1 was relatively low. In addition to finding evidence of active β1 integrin clustering in individual cells engaged with collagen, plentiful active β1 integrin was evident at the front of cells emigrating from spheroids (Fig. S7). These findings strengthen our hypothesis that bleb necks represent sites of integrin-mediated ECM binding and mechanotransduction. Indeed, collagen-embedded MDA-MB-231 cells under siRNA-mediated integrin knockdown did not depict readily observable accumulation or alignment of collagen fibers (Fig. 6E), further indicating that collagen adhesion and re-organization are mediated by integrin receptors. Moreover, β1 depletion induced a statistically significant shift from elongated to round cell morphology and from actin-driven protrusions to blebs in MDA-MB-231 cells, although the overall percentage of bleb-bearing cells remained unchanged (Fig. S4f,g). Interestingly, the subcellular distribution of blebs in a clustered, polarized manner was heavily compromised upon β1 depletion (Fig. 6H,I). These results indicate that collagen alignment in regions with plentiful blebs, as well as cell polarity and the spatial distribution of blebs, depends on functional integrin receptors.
Fig. 6.

Presence of active integrins and integrin-mediated collagen alignment at bleb necks. (A,B) Representative images of 1 mg/ml collagen I-embedded MDA-MB-231 cells stained for active (A) or inactive (B) β1 integrin show clusters at bleb necks. In A, the cell outline is drawn as assessed through the inactive β1 channel (see Materials and Methods; Fig. S5d). (C) Occurrence of active and inactive β1 integrin clusters at bleb necks of collagen I-embedded MDA-MB-231 cells as assessed by fluorescence intensity distribution analysis. Dashed line indicates the statistically expected value assuming a normal distribution of data. Differences between actual and expected occurrences of clusters were analyzed using a t-test between percentages (n=88 blebs). (D) Representative image of MDA-MB-231 cell stained for active (red) and inactive (orange) β1 integrins and embedded in fluorescently labeled collagen I (green) reveals accumulation of active β1 integrin spatially coinciding with collagen fiber alignment in a region with several blebs. The inset shows a magnified image of the active β1 integrin channel (red). The cell outline was drawn as in A from the inactive β1 channel to visualize the cell perimeter and highlights accumulation of active β1 integrin that spatially coincides with collagen fiber alignment in a region with several blebs. (E) Representative image of a phalloidin-stained MDA-MB-231 cell (red) under siRNA-mediated β1 integrin knockdown and embedded in fluorescently labeled collagen I gel (green) shows much less collagen accumulation and alignment. (F) Correlation analysis of stained inactive/active β1 integrins and fluorescently labeled collagen I of collagen-embedded MDA-MB-231 cells. n=10 cells for inactive β1, n=9 cells for active β1. Differences were assessed via Wilcoxon Rank-Sum tests. The data is presented in a box plot depicting the median and second and third quartiles, with whiskers representing data from 5% to 95%. Squares indicate mean values. (G,H) Occurrence of numerous blebs clustered in a polarized manner is dependent on β1 integrins. Representative confocal fluorescence maximum projections of phalloidin-stained MDA-MB-231 cells either untreated (G) or under siRNA-mediated β1 integrin knockdown (H) and embedded in 3D collagen I gels. Arrow marks a site of blebs clustered in a polarized manner. (I) Occurrence of clustered versus single blebs in collagen-embedded bleb-bearing MDA-MB-231 cells under siRNA-mediated β1 integrin knockdown in comparison with untreated cells. n=50 for untreated controls and n=18 for β1 knockdown. Differences were assessed via t-test between percentages. All images and quantified data are from cells imaged 6 h after embedding in the matrix. **P<0.01, ***P<0.001. Scale bars: 10 µm.
Having demonstrated the importance of functional β1 integrins for efficient invasion of round bleb-bearing cells in 3D environments as well as the presence of active integrins and other mechanotransductory proteins at bleb necks, we performed live-cell imaging to address dynamic aspects of integrin–collagen interactions at bleb sites. Although blebs are highly dynamic structures with a lifetime of approximately 1 min (Aoki et al., 2016) and adhesion complexes in 3D have been reported to exist for several minutes (Kubow et al., 2013; Tolde et al., 2012; Legerstee et al., 2019; Harunaga and Yamada, 2011), ECM reorganization can take place over significantly longer periods of time. To interrogate the dynamics of these processes, live-cell imaging of collagen-embedded cells was performed. To minimize attachment of collagen fibers before imaging commenced, videos were recorded immediately after completion of collagen gelation (t=0 min), about 1 h after placing the cells in the collagen solution. This approach allowed visualization of the initial contact between cells and collagen fibers. A representative cell is shown in Fig. 7 and Movies 5,6. In this cell, a collagen fiber becomes attached and aligned at a bleb neck and remains attached after bleb retraction (Fig. 7A, Movie 5). Such imaging also revealed that cell–collagen adhesion loci established in a region with plentiful blebs are maintained through multiple cycles of bleb formation and retraction on an hour scale, with collagen attachments remaining at the base of blebs (Fig. 7B, Movie 6).
Fig. 7.

Blebs are specific sites of collagen fiber adhesion and alignment in cancer cells. (A) Time-lapse images of initial collagen fiber attachment in an MDA-MB-231 cell labeled with CellMask and embedded in a fluorescently labeled 1 mg/ml collagen matrix. White arrows indicate a collagen fiber that is initially out of plane and, concurrent with the appearance of a bleb, becomes attached to the membrane at the bleb neck at t′=200 s (highlighted by a white circle). (B) Time-lapse images over a 40 min time period of the MDA-MB-231 cell shown in A demonstrate repeated blebbing at other sites of cell–collagen adhesion. White bracket highlights the cell region with recurrent bleb formation; white arrow indicates persistent collagen fiber attachment. Images in A were obtained starting at t=36 min after embedding, shown in B; relevant portions of the full movie are presented in Movies 5,6. Scale bars: 10 µm.
In contrast to the initial phase of bleb–collagen attachment (Fig. 7), when blebs were distributed relatively uniformly, cells exposed to the 3D ECM for longer periods of time typically displayed a polarized distribution of blebs, exhibiting regions with highly clustered blebs and extensive collagen re-arrangement on one side of the cell (Fig. 6D, Fig. 8A-C; Fig. S8). Live-cell imaging of cells after exposure to collagen for 6 h was performed to demonstrate how bleb formation becomes increasingly polarized over time and to show that formation of recurrent clustered blebs coincides spatiotemporally with extensive collagen re-organization (Fig. 8, Movie 7). Time course analysis demonstrated that dynamic collagen fiber displacement occurred preferentially at the site of recurrent clustered blebbing but was barely detectable in other cell regions (Fig. 8C,D, Movie 8).
Fig. 8.

Recurrent clustered blebs are sites of integrin-dependent collagen reorganization. Cells were embedded in 1 mg/ml collagen I. All images were taken starting 6 h after embedding. (A) Representative overlay image of CFM projection of a phalloidin-stained MDA-MB-231 cell displaying extensive radial alignment of collagen at sites of clustered blebs. (B) Time-lapse images of a CellMask-labeled MDA-MB-231 cell showing clustered and recurrent blebs and concurrent collagen rearrangement (also shown in Movie 7). (C) Artificially colored representative CFM time-lapse images of a different CellMask-labeled MDA-MB-231 cell show recurrent blebbing on a portion of the cell over a time period of >40 min, a portion of which is also shown in Movie 8. (D) Artificially colored representative collagen I frames from the same live-cell movie (Movie 7) demonstrate collagen fiber displacement at the blebby region, as evidenced by the apparent multicolored clusters of collagen fibers (arrow and top left inset). Images also show areas of negligible collagen rearrangement on the opposite side of the cell, as evidenced by collagen fibers that appear white due to multiple color overlap (arrowhead and top right inset). (E,F) Categorization of involvement of membrane blebs in collagen reorganization as assessed through collagen fiber density (E) and collagen alignment (F) measured in blebby and non-blebby regions of wild-type MDA-MB-231 as well as β1-depleted (beta1 KD) cells and a cell-free gel. The data is presented in a box plot depicting the median and second and third quartiles, with whiskers representing data from 5% to 95%. For collagen density measurements, n=160, 144, 167 and 225 for cell-free, β1 KD, non-blebby and blebby regions, respectively. For angle alignment, n=20, 18, 50 and 50 for cell-free, β1 KD, non-blebby and blebby regions, respectively. Differences were assessed using t-tests; *P<0.05, **P<0.01, ***P<0.001. Scale bars: 10 μm.
To quantify collagen reorganization by bleb-bearing cells, we developed an approach to characterize collagen fiber density and orientation that does not require isolated blebs. This was done by segmenting the immediate cell surroundings and measuring collagen density and dominant angle of orientation per region, each of which was categorized as regions with plentiful blebs (‘blebby’) or lacking blebs (‘non-blebby’) (see Materials and Methods and Fig. S8). To delineate the contribution of β1 integrins, this analysis was also performed on β1 integrin-depleted cells. The analysis revealed that the immediate environment near blebby regions of β1 integrin-positive cells displayed consistently higher collagen fiber density than non-blebby regions (Fig. 8E). In contrast, β1 integrin-depleted bleb-bearing cells showed significantly less collagen accumulation, with collagen density around cells statistically identical to that in non-blebby regions in β1 integrin-positive cells, as shown by Tukey HSD test (Fig. 8E). Moreover, in β1 integrin-positive cells, 73% of blebby regions contained collagen oriented at an angle corresponding to radial alignment whereas the incidence of radial alignment was significantly reduced in β1 integrin-depleted cells, consistent with expectations for random collagen fiber orientation given the data analysis procedure (Fig. 8F). The robust occurrence of collagen alignment in blebby regions, as reported by collagen density and orientation angle, further implicates blebs as sites of ECM adhesion and re-organization, with β1 integrins being the primary receptors for collagen binding, thus revealing a new role for blebs during 3D cancer cell invasion.
DISCUSSION
Our study demonstrates that invasive cancer cells use blebs in integrin-dependent 3D migration associated with extensive ECM reorganization. Thus, round bleb-bearing cancer cells are capable of efficiently invading complex 3D environments by utilizing an ECM receptor-dependent motility mode that is distinct from previously reported bleb-associated motility mechanisms. To the best of our knowledge, this is the first report of blebs being involved in a path-generating mechanism involving ECM adhesion and alignment.
To date, in cancer cell translocation, membrane blebs have been regarded as either without specified function or associated with outward directed forces, as in ‘pushing and squeezing’ amoeboid migration or in ‘chimneying’ friction-based migration (Friedl and Wolf, 2010; Yip et al., 2015; Wyckoff et al., 2006; Malawista et al., 2000; Lorentzen et al., 2011; Petrie et al., 2017). Contribution of adhesion receptor-mediated cell–ECM interactions, which are central to actin polymerization-driven motility, has not been sufficiently characterized in 3D invasion of bleb-bearing cancer cells. The work presented here reveals that, in 3D environments, cells with persistent clustered blebs display accumulations of active β1 coincident with radially aligned collagen fibers (Fig. 6D; Figs S6-S8), indicative of integrin-mediated adhesion and reorganization of the matrix. Quantitative analysis of collagen around bleb-bearing cells revealed a significant increase in both collagen density and alignment in blebby as compared with non-blebby cell regions, implying binding and reorganization of collagen at these sites (Fig. 8E,F). This increase in collagen density and alignment at bleb sites was shown to be integrin-dependent and active integrin-dependent (Fig. 6D,E; Fig. 8E,F), underlining the central role of integrin receptors in the observed ECM reorganization in blebby regions of cancer cells.
The collagen binding and radial alignment at bleb sites was specifically detected at bleb necks, regions where the bleb bulges out from the surrounding plasma membrane (Figs 7,8; Fig. S6), prompting analysis of the organization of integrin-mediated cell–ECM interactions at these sites. Previously, it was postulated that round bleb-bearing cells show diffuse integrin distribution and lack the focalized integrin accumulations indicative of adhesive complexes and force generation, with the exception of a rear-positioned uropod reported in some contexts (Wolf et al., 2003; Lorentzen et al., 2011; Poincloux et al., 2011). Here, we show that a significant proportion of isolated blebs exhibited β1 integrin clusters at one or both sides of the bleb neck (Fig. 4A-D). Similarly, clusters of active β1 integrin, talin and vinculin were all found at bleb necks at significantly higher rates than expected for random occurrence, and nearly all talin- and vinculin-positive clusters at bleb necks were accompanied by β1 integrin clusters (Figs 5,6). These results implicate the bleb neck as a locus of integrin clustering involved in adhesion and mechanotransduction. The occurrence of adhesion-related protein clusters at bleb necks probably underestimates the number of blebs carrying integrin, talin and vinculin clusters because, for technical reasons, the quantitative analysis was limited to individual, well-isolated blebs and excluded regions of clustered blebs that commonly exhibit stronger accumulations of the analyzed proteins and more pronounced collagen alignment.
Interestingly, the persistence of regions with clustered blebs, with blebs repeatedly expanding and retracting in similar locations, spanned longer periods of time than the lifetimes of individual blebs, which range from 30 s to 3 min as observed here and reported by others (Charras et al., 2008; Aoki et al., 2016). Such polarized accumulations of blebs appeared to persist for at least an hour (Fig. 8B,C; Movies 7,8) and were drastically reduced upon integrin knockdown, suggesting a spatiotemporal coordination and positive feedback between cell–ECM adhesion and bleb formation. The hypothesis of a positive feedback loop is further supported by the observation that blebs appeared uniformly distributed on the plasma membrane during initial contact between the cell and the ECM but occurred in an increasingly polarized manner as the adhesion and reorganization of the surrounding ECM advanced (Fig. 8B-D, Movie 8). Such sites of clustered blebs were often accompanied by collagen-devoid regions at the opposite side of the cell (Fig. 6D, Fig. 8A). These features were found not only on isolated cells but also on cells invading from spheroids (Fig. S3g) and indicate that clustered blebs form at the front of migrating cells that leave microtracks at the rear end (Wolf et al., 2007, 2003). Taken together, these results suggest a mechanism by which membrane blebs, each of which is typically short-lived, can contribute to persistent directional migration. These findings stand in contrast to the previously reported path-finding, but not path-generating, function of membrane blebs in cancer cell migration (Wyckoff et al., 2006; Lorentzen et al., 2011; Wolf et al., 2003) and reveal a new, active role for plasma membrane blebs during 3D cancer invasion. This can occur not only in in vitro environments but also at invasive fronts in vivo where the presence of abundant round, bleb-bearing cancer cells has been described (Pinner and Sahai, 2008a,b; Giampieri et al., 2009; Sanz-Moreno et al., 2008, 2011; Orgaz et al., 2014; Cantelli et al., 2015; Herraiz et al., 2016).
Conclusion
The results described here show that round cancer cells can invade 3D environments utilizing a migratory mode that relies on Rho-mediated actomyosin contractility while being integrin-dependent and associated with extensive ECM reorganization. This study reveals that membrane blebs in 3D invading cancer cells have a previously unreported function in ECM-adhesion and alignment through clusters of integrin receptors and other mechanotransductory proteins at bleb necks. Such path-generating, migration-associated blebs were detected in round cancer cells with blebs as the predominant protrusion, as well as in cancer cells of intermediate and elongated morphologies carrying additional types of protrusions, indicating that blebs can both underpin a primary bleb-mediated migratory mode and complement other migratory modes.
MATERIALS AND METHODS
Cell lines and patient-derived tumor tissue
MDA-MB-231 and MDA-MB-468 breast cancer cells were obtained from the American Type Culture Collection (Manassas, VA). Primary breast tumor tissue was provided by Columbia University Medical Center, where it was obtained from a patient undergoing tumor resection. The research described here was declared Not Human Subjects Research by the Columbia Institutional Review Board (IRB Protocol AAAQ8620). The tumor sample used in this study was characterized as an invasive HER2-negative, PR-positive, ER-negative breast carcinoma.
Reagents
Accutase was purchased from MP Biomedicals (Solon, OH). Pepsin-treated (PT) bovine type I collagen was obtained from Advanced BioMatrix (San Diego, CA) as a 5.9-6.1 mg/ml solution. Acid-solubilized (AS) rat tail type I collagen was obtained as 10 mg/ml solution from Corning (Corning, NY). ATTO 647N dye (with N-hydroxysuccimide ester functionality; λex=646 nm, λem=664 nm). Dimethyl sulfoxide (DMSO) and acetic acid (99.7%) were purchased from Sigma Aldrich (St Louis, MO). Growth factor-reduced, Phenol Red-free BME/Matrigel was obtained as an 8.9-10 mg/ml solution from BD Biosciences (San Jose, CA). DMEM solution (10×), NaOH (1 N) and sodium bicarbonate solution (7.5%) were purchased from Sigma Aldrich and sterile filtered before use. Gibco 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (1 M) and CellLight Talin-GFP BacMam 2.0 were obtained from Invitrogen (Carlsbad, CA) and sterile filtered before use. Protease inhibitor cocktail (P1860) and marimastat (BB-2516) were obtained from Millipore Sigma (Billerica, MA). NSC23766 and Y27362 inhibitors were obtained from Selleck Chemicals (Houston, TX). siRNAs, Lipofectamine RNAiMax and Opti-MEM were obtained from Thermo Fisher Scientific (Waltham, MA). Buffered formalin phosphate (10%) and bovine serum albumin (BSA) were obtained from Fisher Scientific (Pittsburgh, PA). Triton-X was obtained from EMD Millipore Chemicals (Billerica, MA). Unlabeled and FITC-labeled anti-integrin β1 antibodies (clone 4B4, staining inactive β1) were obtained from Beckman Coulter (Brea, CA). AlexaFluor488 and AlexaFluor647-conjugated anti-integrin β1 antibodies (clone P5D2, staining total β1), AlexaFluor647-conjugated vinculin antibody (clone EPR8185) and AlexaFluor488-conjugated goat anti-mouse secondary antibody were obtained from Abcam (Cambridge, MA). AlexaFluor647-conjugated anti-active β1 integrin antibody (clone 12G10, staining active β1) was obtained from Santa Cruz (Dallas, TX) and AlexaFluor-conjugated phalloidin and CellMask membrane dye were obtained from Molecular Probes by Thermo Fisher Scientific (Waltham, MA). FluoroBrite media was obtained from Gibco (Grand Island, NY). Rabbit anti-integrin β1 antibody for detection of endogenous β1 integrin on SDS-PAGE was obtained from Cell Signaling (Danvers, MA), rabbit anti-GAPDH was obtained from Abcam (Cambridge, MA) and peroxidase-conjugated anti-rabbit IgG antibody was obtained from Sigma Aldrich (St Louis, MO).
Cell culture
Cell lines were cultured in growth medium consisting of 1× high glucose DMEM containing 10% (v/v) FBS, 1% (v/v) 100× penicillin/streptomycin/amphotericin solution and 1% (v/v) 100× non-essential amino acids solution at 37°C under 5% carbon dioxide. Patient-derived breast cancer cells were isolated from tumor samples and separated from the tumor-associated fibroblasts using a method based on that of Derose et al. (Derose et al., 2013), followed by an additional Accutase digestion step to ensure dissociation of the tissue into a single cell suspension. Isolated cancer cells were cultured as described (Derose et al., 2013) in modified M87 medium consisting of 1× DMEM/F12, 2% (v/v) FBS, 1% (v/v) insulin-transferrin-selenium-ethanolamine 100× supplement solution, 1% (v/v) 100× penicillin-streptomycin-amphotericin solution, 5 ng/ml human recombinant epidermal growth factor (EGF), 0.3 µg/ml hydrocortisone, 0.5 ng/ml cholera toxin, 5 nM 3,3′,5-triiodo-L-thyronine, 0.5 nM β-estradiol, 5 µM (±)-isoproterenol hydrochloride, 50 nM ethanolamine and 50 nM O-phosphorylethanolamine. Patient-derived cancer cells were subcultured when 80-90% confluent and used in passage 2-5.
Generation of multicellular tumor spheroids and patient-derived organoids
Spheroids and patient-derived organoids were formed in appropriate growth medium supplemented with 0.2575 mg/ml BME using a centrifugation method described previously (Ivascu and Kubbies, 2006) employing ultralow attachment 96-well U-bottom Nunclon Sphera plates from Thermo Fisher Scientific (Waltham, MA). Spheroids and organoids were allowed to form for 24 h at 37°C under 5% carbon dioxide.
Pharmacological cell treatments
Inhibition of Rac1 signaling was achieved using NSC23766, a cell-permeable selective inhibitor of Rac1 GDP/GTP exchange. Spheroids embedded in 200 µl collagen gels were overlaid with 50 µl of 200 µM NSC23766 dissolved in growth medium immediately after gelation (t=1 h) for the duration of the invasion experiment (t=24 h), resulting in an effective NSC23766 concentration of 40 µM.
Inhibition of Rho signaling was achieved using Y27632, an inhibitor of ROCK. Spheroids embedded in 200 µl collagen gels were overlaid with 50 µl of 50 µM Y27632 immediately after gelation for the duration of the invasion experiment, resulting in an effective Y27632 concentration of 10 µM.
Inhibition of endogenous proteases was achieved through application of a protease inhibitor cocktail containing E 64, pepstatin, leupeptin, bestatin, aprotinin and marimastat. All inhibitors except marimastat were contained in the P1860 inhibitor cocktail, which was used at 1:200 dilution, and marimastat was supplemented at 20 µM, as described previously (Wolf et al., 2003). The inhibitors were added to the collagen gel directly as well as to the overlaying medium at the same concentration.
For antibody-mediated inhibition of β1 integrin, spheroids were pretreated for 2.5 h with 5 µg/ml of a function-blocking anti-integrin β1 antibody (clone 4B4) prior to embedding into collagen gels and overlaid with growth medium containing 10 µg/ml of the same antibody 1 h after embedding.
siRNA-mediated integrin knockdown
The target sequences used to mediate silencing of β1 integrin expression were (a) 5′-GGAGGAAUGUUACACGGCUtt-3′ (sense) and 5′-AGCCGUGUAACAUUCCUCCag-3′ (antisense) for siRNA #1 and (b) 5′-GCAGUUGGUUUUGCGAUUAtt-3′ (sense) and 5′-UAAUCGCAAAACCAACUGCtg-3′ (antisense) for siRNA #2. The Silencer Select siRNAs and the non-targeting control siRNA (catalog #4390846) were obtained as annealed duplexes from Thermo Fisher Scientific (Waltham, MA). MDA-MB-231 and MDA-MB468 cells were transiently transfected with the β1 integrin-targeting or the control siRNA using Lipofectamine RNAiMax as follows: prior to transfection, complete culture medium was removed and replaced with serum-free Opti-MEM. siRNA and Lipofectamine RNAiMax (1.5 µl per well in a 24-well plate) were diluted in Opti-MEM according to the manufacturer's instructions, added to cells at a final RNA concentration of 10 nM, and incubated for either 6 h (MDA-MB-468) or 16 h (MDA-MB-231). Opti-MEM was replaced with DMEM containing 10% FBS after the initial incubation period. At 72 h post transfection, cells were returned to complete growth medium containing antibiotics. Depletion of β1 integrin was confirmed by SDS-PAGE/western blot (Fig. S4b). Assays were performed 3-5 days post transfection.
Protein extraction and western blotting
Cells grown in 3.5 cm dishes to 60-80% confluency were washed twice with PBS, detached with Accutase, resuspended in growth medium and pelleted by centrifugation for 3 min at 1600 rpm. After the medium was discarded, the pellet was stored at −80°C. Pellets were lysed in RIPA lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM, Na2EDTA, 1 mM EGTA, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mM DTT) with the addition of protease and phosphatase inhibitors. Lysates were cleared by centrifugation, after which the supernatant was isolated. Subsequently, the protein concentrations of the lysates were quantitated using Bradford assays (Bio-Rad, Hercules, CA) and lysates were adjusted to the same protein concentration. Lysates were mixed with 3× sample buffer containing 5% β-mercaptoethanol, heated to 95°C for 5 min, centrifuged for 3 min at 13,000×g and electrophoresed through a sodium dodecyl sulfate (SDS)-10% polyacrylamide gel. Protein gels were transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA). For β1 integrin detection, the anti-integrin antibody (Cell Signaling, Danvers, MA) was used at a 1:500 dilution; for GAPDH detection, the anti-GAPDH antibody was used at a 1:1000 dilution and in both cases a peroxidase-conjugated secondary antibody was used at 1:5000 dilution. Proteins were visualized with an enhanced chemiluminescence detection system (Thermo Scientific Pierce ECL, Thermo Fisher Scientific, Waltham, MA).
Preparation of collagen solutions
Collagen solution (1 mg/ml) was prepared by diluting the high-concentration PT collagen monomer stock solution. Appropriate amounts of collagen stock solution were prepared with 10% (v/v) 10× DMEM, 2.5% (v/v) HEPES buffer, 2.5% (v/v) sodium bicarbonate and distilled deionized H2O. To prevent self-assembly of collagen monomers, all solutions were held and mixed at 4°C. NaOH was added to adjust the pH to 7.4.
To prepare fluorescently labeled collagen matrices, unlabeled PT collagen was mixed with ATTO 647N-labeled AS collagen in 10:1 (w/w) ratio. Following this, all steps for collagen gel preparation were as described above.
The AS collagen monomers were conjugated with ATTO 647N fluorescent dye molecules following a previously reported procedure (Kalia and Raines, 2010; Stephanopoulos and Francis, 2011). In brief, ATTO dye stock in DMSO was prepared and added to a 3 mg/ml AS collagen solution in 0.01 M sodium bicarbonate (pH 9). The conjugation reaction was carried out at 4°C in the dark for 24 h. For purification, the product was dialyzed against 0.02 M acetic acid for 72 h (or until the acetic acid was colorless after purification) at 4°C in the dark using mini-dialysis tubing, changing the acetic acid solution in the tubing every 12 h. The final product was stored at 4°C in the dark at pH 2 until use.
To prepare composite collagen/BME solutions, first 10× DMEM, HEPES buffer and sodium bicarbonate solutions were mixed. Then, the required amount of BME stock solution was added to achieve the desired final concentration of 3 mg/ml. The BME replaced a proportion of the H2O that would be added in the equivalent pure collagen solution. Subsequently, the PT collagen stock solution was added to achieve final concentration of 1 mg/ml. The resulting solution was brought to pH 7.4 by addition of NaOH.
Preparation of cell-embedded gels
To prepare collagen gels loaded with a single spheroid or organoid, 1 mg/ml collagen solution was prepared as described above at 4°C to prevent gelation. Neutralized collagen solution (200 µl) was added to a chamber consisting of a 5 mm glass cylinder glued to a coverslip-bottom cell culture dish. A nylon mesh was placed on the inner circumference of the cylinder to anchor the collagen gel. The spheroid was added to the liquid collagen in 5 µl growth medium. The chamber was then transferred immediately to a 37°C incubator. The collagen gels were overlaid with 50 µl growth medium after 1 h and surrounded by additional liquid to prevent drying during extended incubation periods. To prepare collagen/BME composite matrices loaded with a single spheroid or organoid, the chambers were first pre-coated with 40 µl collagen/BME solution to prevent sedimentation of spheroids to the bottom of the chamber. The coating was gelled for 1 h at 37°C. Following this, the bulk volume of the matrix solution was added to the chamber and a single spheroid or organoid was implanted as described above.
To prepare collagen or collagen/BME gels loaded with dispersed cells, the 1 mg/ml collagen solution was prepared omitting 50% of the water and neutralized at 4°C. The water volume was substituted with ice-cold growth medium containing the desired number of cells. Subsequently 150-200 µl of the cell-loaded collagen or collagen/BME was added to the chamber and gelled at 37°C as described above for spheroid-loaded collagen gels. After 1 h, gels were overlaid with 50 µl growth medium.
In all cell experiments, time points are described considering time zero (t=0 min) to be the time at which the cells or spheroids were placed into the collagen or collagen/BME solution.
Immunocytochemistry and labeling
For immunocytochemical staining, dispersed cells or spheroids were embedded in 80-150 µl collagen gels according to the following procedures.
For staining of total cell surface β1 integrin receptors, gels were incubated with 100 µl of the fluorescently labeled antibody (clone P5D2) diluted 1:200 per 150 µl gel. Samples were incubated with the antibody at 4°C for 16-20 h in the dark. Following incubation, the antibody solution was aspirated, gels were washed 3× with 200 µl PBS for 10 min at RT and subjected to imaging immediately.
For double staining of cell surface total β1 integrin receptors and the plasma membrane, cells were fixed and washed as described above and each gel (150 µl) was incubated with 100 µl of the staining solution containing the fluorescently labeled β1 antibody (clone P5D2) diluted 1:200 (v/v) and the CellMask membrane dye diluted 1:500 in PBS (v/v). Samples were incubated with the staining solution at 4°C for 16-20 h in the dark. Following incubation, samples were treated as described in the procedure for staining total cell surface β1 integrin receptors.
For triple staining of the active and inactive cell surface β1 integrin receptors and the plasma membrane (as used for the analysis shown in Fig. 6C), cells were fixed and washed as described above followed by 15 min quenching with 50 mM NH4Cl at RT, two 10 min washes with PBS and a 60 min incubation with 1% BSA/PBS (w/v) (blocking solution) at RT. Subsequently, the blocking solution was replaced with the staining solution containing fluorescently labeled antibodies against the inactive (clone 4B4, 1:100 dilution) and active (clone 12G10, 1:50 dilution) β1 integrins as well as the CellMask membrane dye (1:500 dilution) diluted in 1% BSA/PBS. Samples were incubated at 4°C for 16-20 h in the dark. Following incubation, samples were treated as described in the procedure for staining total cell surface β1 integrin receptors.
For double staining active and inactive β1 integrin receptors (as shown in Fig. 6E), cells were fixed and washed as described above followed by 15 min quenching with 50 mM NH4Cl at RT, two 10 min washes with PBS and a 60 min incubation with 1% BSA/PBS (blocking solution) at RT. Subsequently, the blocking solution was replaced with staining solution containing the fluorescently labeled antibodies against the inactive (clone 4B4, 1:100 dilution) and active (clone 12G10, 1:50 dilution) β1 integrins diluted in 1% BSA/PBS. Samples were incubated at 4°C for 16-20 h in the dark. Following incubation, samples were treated as described in the procedure for staining total cell surface β1 integrin receptors.
For staining the actin cytoskeleton, after fixation cells were permeabilized with 0.2% TritonX/H2O for 10 min at RT. After removing the permeabilization solution, samples were washed 4× with 200 µl PBS for 10 min and once for 30 min at RT to remove the detergent completely. Subsequently, 100 µl of the fluorescently labeled phalloidin dye diluted 1:200 in PBS was applied per 150 µl gel, and the gels were incubated at 4°C for 16-20 h in the dark. Following incubation, the staining solution was aspirated, gels were washed as described above and subjected to imaging immediately.
For double staining of talin and total β1 integrin receptors, cells were transfected using BacMam 2.0 Talin-GFP prior to seeding in collagen gels. Transfection involved incubating cells with 25 particles of BacMam 2.0 per cell for 16 h, after which cells were embedded into collagen gels as described above. Gels were subsequently fixed with methanol-free 4% formaldehyde for 20 min followed by three 10 min PBS washes. Gels were then washed and stained as described in the procedure for staining total cell surface β1 integrin receptors.
Finally, for double staining of vinculin and total β1 integrin receptors, cells were fixed and washed as described above and each gel (150 µl) was incubated with 100 µl of the staining solution containing fluorescently labeled β1 antibody diluted 1:200 (v/v) and the vinculin antibody diluted 1:200 in PBS (v/v). Samples were incubated with the staining solution at 4°C for 16-20 h in the dark. Following incubation, samples were treated as described in the procedure for staining total cell surface β1 integrin receptors.
Fluorescence-activated flow cytometry
Cells were detached from the cell culture plate with Accutase and resuspended in ice-cold Phenol Red-free HEPES-containing DMEM supplemented with 10% FBS; samples remained on ice for the remainder of the procedure. Subsequently, 2×105 cells were washed by centrifugation for 2 min at 1300 rpm and resuspended in ice-cold 1% FBS/PBS. After another centrifugation step, the cell pellet was carefully resuspended in 50 µl 1% FBS/PBS containing 1 µg/ml unconjugated β1 integrin antibody (mouse monoclonal, clone 4B4). Cells were incubated with primary antibody for 1 h at 4°C and washed twice with 1% FBS/PBS. Subsequently cells were incubated in 50 µl 1% FBS/PBS containing 20 µg/ml fluorescently labeled secondary antibody (goat anti-mouse AlexaFluor488-conjugated IgG) for 1 h at 4°C and washed with PBS. Finally, cells were resuspended in PBS and measurements were taken on a flow cytometer (Becton Dickinson FACSCalibur with BD CellQuest Pro software).
Collagen contraction assay
Collagen solutions of 1 mg/ml PT collagen containing 5×105 cells/ml were prepared, and 500 µl gels were cast onto the 23 mm coverslip bottom of FluoroDishes (diameter 35 mm). Gels were polymerized for 60 min at 37°C, overlaid with 2 ml growth medium and manually released from the glass bottom. Contraction was allowed for 24 h at normal incubation conditions. Gel contraction was documented using digital photography. Images were taken at t=0 h (before gel release) and at t=24 h, and the diameter of the gel was measured at both time points.
Microscopy
Imaging was performed on an inverted confocal laser-scanning microscope (Olympus Fluoview 300, Zeiss LSM 700 or Zeiss LSM 800) in scanning transmittance, confocal reflectance or confocal fluorescence mode with 10× air (NA 0.4), 60× oil (NA 1.42) or 63× oil (NA 1.4) objectives unless stated otherwise. For excitation of FITC/AlexaFluor488 fluorophores and confocal reflectance (used for imaging of unlabeled collagen), a 488 nm laser was used; for excitation of AlexaFluor568, a 543 nm (Olympus) or 555 nm (Zeiss) laser was used; and for excitation of ATTO647N, a 633 nm (Olympus) or 639 nm (Zeiss) laser was used. A photomultiplier tube detector (PMT) was used for detection on the Olympus and Zeiss LSM 700, and an Airyscan system was used for detection on the Zeiss LSM 800.
For live cell imaging of both dispersed cells and spheroid invasion, gels were prepared as described above and further overlaid with FluoroBrite medium containing a carbonate-independent buffer system; the volume was equal to the gel volume 1 h after onset of gelation. Live cell spheroid invasion (Fig. S1) was performed on the Olympus Fluoview 300 with the 60× oil objective in a live cell imaging chamber (Neue Biosciences) with controlled temperature (37°C) and CO2 (5%) during the 12 h imaging session. The movies of dispersed cells shown in Fig. 7A,B and Fig. 8B were obtained using a Nikon Ti Eclipse spinning disk confocal microscope using a 60× water objective (NA 1.2). The microscope was equipped with a stage-top incubator obtained from Tokai Hit USA (Bala Cynwyd, PA), ensuring a controlled temperature and CO2 environment (37°C and 5% CO2) during imaging. The movies of dispersed cells shown in Fig. 8C,D were obtained using the Zeiss LSM 800 with the 63× oil immersion objective at ambient temperature and CO2 concentration during the 1 h imaging session.
Image analysis
Spheroid invasion
For analysis of invasion distance, spheroids were imaged at 10× magnification in scanning transmittance mode at 2 h and 24 h after implantation. For each spheroid, we used these magnified images to determine the invasive distance, defined as the difference between the periphery of the spheroid at t=2 h and a circle that circumscribes 90% of the invasive cells at t=24 h (Fig. S1a).
Cell morphology and protrusions
For visualization of the actin cytoskeleton and analysis of cell morphology and protrusions, confocal fluorescence z-stacks (Σ10-20 µm, step size 1 µm) of fixed cells stained with fluorescently labeled phalloidin were acquired on a Zeiss LSM 700 using the 10× objective. Cell morphology was determined in a semi-automated fashion using ImageJ. First, maximum fluorescence intensity projections were generated from z-stacks, despeckled and thresholded. Following this processing, the Analyze Particle function was used to assess the circularity, c, of each cell, with 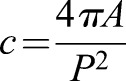 , where A is cell area and P the cell perimeter. Circularity thus ranges from 0 to 1, with c=1 for a circle. Cell protrusion classification was performed by eye using the preprocessed images.
, where A is cell area and P the cell perimeter. Circularity thus ranges from 0 to 1, with c=1 for a circle. Cell protrusion classification was performed by eye using the preprocessed images.
Integrin receptor distribution
Analysis of integrin intensity distribution (total, active and inactive) was performed on individual confocal images of cells with immunolabeled integrin receptors using a custom written ImageJ macro. Single confocal slices displaying individual blebs were selected from z-stacks spanning up to 20 µm. Selections of the membrane perimeter of the entire cell including the bleb(s) of interest were outlined by hand (Fig. S5d). Only blebs that were fully in focus in the slice of interest were included in the outline associated with that particular slice. Images were converted to 8 bit and each selection was fit to a spline curve. The intensity profile along the selection of 2-pixel width was obtained at spaced intervals of 1 pixel. Following this, the bleb neck was defined on the image by visual observation as the sites of greatest membrane bending at both sides of the bleb, and the xy coordinates of the bleb neck were determined. The intensity values of two adjacent pixels most closely corresponding to these xy coordinates were used to calculate the mean intensity value of the fluorescently labeled β1 integrin on either side of the bleb. The intensity profile along the perimeter of the entire cell was used to determine the mean value and standard deviation (s.d.) of the integrin receptors distributed in the bulk of the plasma membrane. Intensity values at bleb necks exceeding the mean+2s.d. were defined as ‘clusters.’ To avoid intensity peaks caused by overlay of membranes in clustered blebs, only individual (rather than clustered) blebs were selected for this analysis.
For analysis of integrin receptor distribution relative to the amount of membrane material, dual-color confocal images of collagen-embedded cells stained with a general membrane dye (CellMask) and immunolabeled integrin receptors were acquired on a Zeiss LSM 800 Airyscan using the 63× objective. Analysis of CellMask clustering was performed as described for β1 integrins. Analysis of talin and vinculin distributions and determination of clusters was also conducted as described above.
Colocalization analysis of active β1 integrin and collagen and inactive β1 integrin and collagen was performed by creating a hand-drawn region of interest (ROI), 20 pixels wide, along the entire cell membrane surface. This ROI was used in the ImageJ plugin Coloc2 (Schneider et al., 2012; Schindelin et al., 2012). Pearson's R values of the non-thresholded images are reported.
Collagen density and alignment
For analysis of bleb-associated collagen density and alignment, confocal images of fluorescently labeled F-actin or β1 integrin and the collagen matrix were acquired on a Zeiss LSM 800 using the 63× objective. Z-stacks spanning 10-15 µm with step size 1 µm were acquired in both channels.
Collagen was characterized using the OrientationJ plugin for Fiji (Püspöki et al., 2016). OrientationJ uses a structure tensor-based approach to measure local orientation of features in an image. Prior to measuring collagen alignment, images were normalized using a histogram-matching algorithm to account for differences in intensities during image acquisition.
To quantify collagen alignment, cell surroundings were segmented into equally sized regions for analysis. Cell segmentation was performed using a custom Fiji macro. Cells were segmented into regions corresponding to 45° slices of a circle. The ROIs started at the edge of the cell membrane and extended 1.5× the cell radius into the surrounding collagen (Fig. S8). For cells with blebby regions, cells were segmented into eight regions of equal size, using OrientationJ to minimize the number of regions containing blebs in only a small part of the region. For each region, a 90° window for collagen alignment was considered; for example, the region corresponding to 45-90° in the circle had an angle cutoff window of 22.5-112.5°. Regions in which the OrientationJ-identified dominant angle fell into this window were considered ‘aligned’. Classification of cell membrane regions as ‘blebby’ or ‘non-blebby’ was performed by eye. The number of aligned regions is expressed as a percentage of the total number of regions of a given type assessed per cell.
Angle alignment is not sufficient to characterize collagen reorganization by the cell because it is insensitive to the amount of collagen present. As another metric of collagen reorganization, collagen density per region was also assessed, with high density indicative of extensive ECM compaction and remodeling by the cell. The energy value output from OrientationJ was used to characterize density. The energy is a sum of the eigenvalues of the structure tensor and provides a general measure of how many collagen fibers are in a given region.
Statistical analysis
All statistical analyses and the software used for these analyses are summarized in Table S1. All statistical tests were conducted at α=0.05. Statistical significance is indicated by *P<0.05, **P<0.01 and ***P<0.001. Lack of statistical significance (P>0.05) is denoted using a dagger (†).
Circularity distributions between biological replicates were compared using Kolmogorov–Smirnov (KS) analysis. Comparison of circularity of the cell lines in various matrices was also conducted using KS analysis. Cell protrusion classifications were compared between replicates using ANOVA tests, whereas the cell protrusion classifications of the cell lines in various matrices were compared using t-tests.
For analysis of spheroid invasion during pharmacological and genetic inhibition experiments, Wilcoxon Rank-Sum tests were used to compare spheroid invasion under inhibition to the appropriate control. For the collagen contraction assays for both cell lines, the percentage contraction of untreated cells versus cells treated with anti-integrin β1 antibody were compared using Wilcoxon Rank-Sum tests.
The occurrence of total β1 integrin versus CellMask clusters on MDA-MB-231 cells was analyzed in a number of ways. First, one outlier bleb was removed from the data set via Grubb's test based on its large perimeter. The average skew and kurtosis were calculated, demonstrating normality of the distribution as well as equal distribution of data between biological replicates via ANOVA analysis. To ensure consistency between biological replicates, the occurrence of total β1 integrin and CellMask cluster-positive blebs from individual experiments were compared using a t-test between percentages. The occurrence of total β1 integrin versus CellMask clusters at bleb necks were similarly compared using a t-test between percentages. Likewise, the occurrence of total β1 integrin and CellMask clusters at bleb necks and the entire cell membrane were compared with statistically expected values using a t-test by percentages. The perimeter of blebs and the width of bleb necks with total β1 integrin clusters were compared with cluster negative blebs using t-tests and KS tests. The occurrence of total β1 integrin clusters in untreated cells versus cells treated with anti-integrin total β1 antibody were compared using t-test between percentages. A t-test between percentages was also used to compare data between biological replicates. Results for the occurrence of clustered versus individual blebs in untreated versus total β1 integrin-depleted cells were compared by t-test between percentages.
All other cluster data (including total β1 integrin in MDA-MB-468 cells and MDA-MB-231 spheroids, active and inactive β1 integrin, talin and vinculin in MDA-MB-231 dispersed cells) were analyzed in a similar manner. The skew and kurtosis of each data set were determined in order to assess normality (Kim, 2013). Statistical consistency between dates was shown using t-tests between percentages. Comparison of dyes/antibodies to one another and of experimental results to statistically expected results were compared using t-tests between percentages.
Comparison of Pearson's R values for active β1 and collagen versus inactive β1 and collagen was carried out using Wilcoxon Rank-Sum tests.
For collagen orientation analysis, the obtained values for collagen energy and orientation angle were subjected to bootstrap resampling to address the normality of the distribution. The mean values of the resampled data were found to be normally distributed and the data was therefore regarded as suitable for parametric statistical analysis. The t-tests were used to compare mean energy values and angle alignment per cell for each region type (blebby, non-blebby, β1 knockdown and cell-free).
Supplementary Material
Acknowledgements
This research used the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.G., L.J.K.; Methodology: A.G., R.C.A., A.J.D., L.J.K.; Software: A.J.D.; Validation: A.G., R.C.A., A.J.D., L.J.K.; Formal analysis: A.G., R.C.A., A.J.D., L.J.K.; Investigation: A.G., R.C.A., A.J.D., O.S.K.; Data curation: A.G., R.C.A., L.J.K.; Writing - original draft: A.G.; Writing - review & editing: A.G., R.C.A., L.J.K.; Visualization: A.G., R.C.A., A.J.D., L.J.K.; Supervision: L.J.K.; Project administration: L.J.K.; Funding acquisition: L.J.K.
Funding
This research was funded in part through the National Institutes of Health/National Cancer Institute Cancer Center Support Grant P30CA013696. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.236778.supplemental
References
- Aoki K., Maeda F., Nagasako T., Mochizuki Y., Uchida S. and Ikenouchi J. (2016). A RhoA and Rnd3 cycle regulates actin reassembly during membrane blebbing. Proc. Natl. Acad. Sci. USA 113, E1863-E1871. 10.1073/pnas.1600968113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjonen A., Alanko J., Veltel S. and Ivaska J. (2012). Distinct recycling of active and inactive β1 integrins. Traffic 13, 610-625. 10.1111/j.1600-0854.2012.01327.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergert M., Chandradoss S. D., Desai R. A. and Paluch E. (2012). Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc. Natl. Acad. Sci. USA 109, 14434-14439. 10.1073/pnas.1207968109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantelli G., Orgaz J. L., Rodriguez-Hernandez I., Karagiannis P., Maiques O., Matias-Guiu X., Nestle F. O., Marti R. M., Karagiannis S. N. and Sanz-Moreno V. (2015). TGF-beta-induced transcription sustains amoeboid melanoma migration and dissemination. Curr. Biol. 25, 2899-2914. 10.1016/j.cub.2015.09.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G. T., Coughlin M., Mitchison T. J. and Mahadevan L. (2008). Life and times of a cellular bleb. Biophys J. 94, 1836-1853. 10.1529/biophysj.107.113605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. G. and Vignjevic D. M. (2015). Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 36, 13-22. 10.1016/j.ceb.2015.06.004 [DOI] [PubMed] [Google Scholar]
- DeRose Y. S., Gligorich K. M., Wang G., Georgelas A., Bowman P., Courdy S. J., Welm A. L. and Welm B. E. (2013). Patient-derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr. Protoc. Pharmacol. 14, 1-43. 10.1002/0471141755.ph1423s60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz-Muñoz A., Krieg M., Bergert M., Ibarlucea-Benitez I., Muller D. J., Paluch E. and Heisenberg C.-P. (2010). Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 8, e1000544 10.1371/journal.pbio.1000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A. D. and Yamada K. M. (2016). Mechanosensing via cell-matrix adhesions in 3D microenvironments. Exp. Cell Res. 343, 60-66. 10.1016/j.yexcr.2015.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A. D., Carvajal N., Jin A., Matsumoto K. and Yamada K. M. (2015). Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 6, 8720 10.1038/ncomms9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufau I., Frongia C., Sicard F., Dedieu L., Cordelier P., Ausseil F., Ducommun B. and Valette A. (2012). Multicellular tumor spheroid model to evaluate spatio-temporal dynamics effect of chemotherapeutics: application to the gemcitabine/CHK1 inhibitor combination in pancreatic cancer. BMC Cancer 12, 15 10.1186/1471-2407-12-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley S. I., Feng Y., Krishnamurthy R., Kim D.-H., Celedon A., Longmore G. D. and Wirtz D. (2010). A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell Biol. 12, 598-604. 10.1038/ncb2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P. and Alexander S. (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992-1009. 10.1016/j.cell.2011.11.016 [DOI] [PubMed] [Google Scholar]
- Friedl P. and Wolf K. (2010). Plasticity of cell migration: a multiscale tuning model. J. Cell Biol. 188, 11-19. 10.1083/jcb.200909003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Dickerson J. B., Guo F., Zheng J. and Zheng Y. (2004). Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 101, 7618-7623. 10.1073/pnas.0307512101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel M. L., Schneider I. C., Aratyn-Schaus Y. and Waterman C. M. (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315-333. 10.1146/annurev.cellbio.011209.122036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampieri S., Manning C., Hooper S., Jones L., Hill C. S. and Sahai E. (2009). Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 11, 1287-1296. 10.1038/ncb1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic B., Bergman A. and Condeelis J. (2014). Multiparametric classification links tumor microenvironments with tumor cell phenotype. PLoS Biol. 12, e1001995 10.1371/journal.pbio.1001995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harunaga J. S. and Yamada K. M. (2011). Cell-matrix adhesions in 3D. Matrix Biol. 30, 363-368. 10.1016/j.matbio.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herraiz C., Calvo F., Pandya P., Cantelli G., Rodriguez-Hernandez I., Orgaz J. L., Kang N., Chu T., Sahai E. and Sanz-Moreno V. (2016). Reactivation of p53 by a cytoskeletal sensor to control the balance between DNA damage and tumor dissemination. J. Natl. Cancer Inst. 108, djv289 10.1093/jnci/djv289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T., Uehata M., Tamechika I., Keel J., Nonomura K., Maekawa M. and Narumiya S. (2000). Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 57, 976-983. [PubMed] [Google Scholar]
- Ivascu A. and Kubbies M. (2006). Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J. Biomol. Screen. 11, 922-932. 10.1177/1087057106292763 [DOI] [PubMed] [Google Scholar]
- Kalia J. and Raines R. T. (2010). Advances in bioconjugation. Curr. Org. Chem. 14, 138-147. 10.2174/138527210790069839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny P. A., Lee G. Y., Myers C. A., Neve R. M., Semeiks J. R., Spellman P. T., Lorenz K., Lee E. H., Barcellos-Hoff M. H., Petersen O. W. et al. (2007). The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 1, 84-96. 10.1016/j.molonc.2007.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-Y. (2013). Statistical notes for clinical researchers: Understanding standard deviations and standard errors. Restor Dent Endod. 38, 263-265. 10.5395/rde.2013.38.4.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. B., Stein R. and O'Hare M. J. (2004). Three-dimensional in vitro tissue culture models of breast cancer-- a review. Breast Cancer Res. Treat. 85, 281-291. 10.1023/B:BREA.0000025418.88785.2b [DOI] [PubMed] [Google Scholar]
- Kubow K. E., Conrad S. K. and Horwitz A. R. (2013). Matrix microarchitecture and myosin II determine adhesion in 3D matrices. Curr. Biol. 23, 1607-1619. 10.1016/j.cub.2013.06.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legerstee K., Geverts B., Slotman J. A. and Houtsmuller A. B. (2019). Dynamics and distribution of paxillin, vinculin, zyxin and VASP depend on focal adhesion location and orientation. Sci. Rep. 9, 10460 10.1038/s41598-019-46905-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann S., Te Boekhorst V., Odenthal J., Bianchi R., van Helvert S., Ikenberg K., Ilina O., Stoma S., Xandry J., Jiang L. et al. (2017). Hypoxia induces a HIF-1-dependent transition from collective-to-amoeboid dissemination in epithelial cancer cells. Curr. Biol. 27, 392-400. 10.1016/j.cub.2016.11.057 [DOI] [PubMed] [Google Scholar]
- Liu Y.-J., Le Berre M., Lautenschlaeger F., Maiuri P., Callan-Jones A., Heuzé M., Takaki T., Voituriez R. and Piel M. (2015). Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659-672. 10.1016/j.cell.2015.01.007 [DOI] [PubMed] [Google Scholar]
- Lorentzen A., Bamber J., Sadok A., Elson-Schwab I. and Marshall C. J. (2011). An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J. Cell Sci. 124, 1256-1267. 10.1242/jcs.074849 [DOI] [PubMed] [Google Scholar]
- Malawista S. E., de Boisfleury Chevance A. and Boxer L. A. (2000). Random locomotion and chemotaxis of human blood polymorphonuclear leukocytes from a patient with leukocyte adhesion deficiency-1: normal displacement in close quarters via chimneying. Cell Motil. Cytoskeleton 46, 183-189. 10.1002/1097-0169(200007)46:3<183::AID-CM3>3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- Orgaz J. L., Pandya P., Dalmeida R., Karagiannis P., Sanchez-Laorden B., Viros A., Albrengues J., Nestle F. O., Ridley A. J., Gaggioli C. et al. (2014). Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat. Commun. 5, 4255 10.1038/ncomms5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie R. J. and Yamada K. M. (2012). At the leading edge of three-dimensional cell migration. J. Cell Sci. 125, 5917-5926. 10.1242/jcs.093732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie R. J., Harlin H. M., Korsak L. I. T. and Yamada K. M. (2017). Activating the nuclear piston mechanism of 3D migration in tumor cells. J. Cell Biol. 216, 93-100. 10.1083/jcb.201605097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinner S. and Sahai E. (2008a). Imaging amoeboid cancer cell motility in vivo. J. Microsc. 231, 441-445. 10.1111/j.1365-2818.2008.02056.x [DOI] [PubMed] [Google Scholar]
- Pinner S. and Sahai E. (2008b). PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat. Cell Biol. 10, 127-137. 10.1038/ncb1675 [DOI] [PubMed] [Google Scholar]
- Poincloux R., Collin O., Lizarraga F., Romao M., Debray M., Piel M. and Chavrier P. (2011). Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA 108, 1943-1948. 10.1073/pnas.1010396108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Püspöki Z., Storath M., Sage D. and Unser M. (2016). Transforms and operators for directional bioimage analysis: a survey. Adv. Anat. Embryol. Cell Biol. 219, 69-93. 10.1007/978-3-319-28549-8_3 [DOI] [PubMed] [Google Scholar]
- Ruprecht V., Wieser S., Callan-Jones A., Smutny M., Morita H., Sako K., Barone V., Ritsch-Marte M., Sixt M., Voituriez R. et al. (2015). Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 160, 673-685. 10.1016/j.cell.2015.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadok A., McCarthy A., Caldwell J., Collins I., Garrett M. D., Yeo M., Hooper S., Sahai E., Kuemper S., Mardakheh F. K. et al. (2015). Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 75, 2272-2284. 10.1158/0008-5472.CAN-14-2156 [DOI] [PubMed] [Google Scholar]
- Sahai E. and Marshall C. J. (2003). Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5, 711-719. 10.1038/ncb1019 [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V., Gadea G., Ahn J., Paterson H., Marra P., Pinner S., Sahai E. and Marshall C. J. (2008). Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510-523. 10.1016/j.cell.2008.09.043 [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V., Gaggioli C., Yeo M., Albrengues J., Wallberg F., Viros A., Hooper S., Mitter R., Féral C. C., Cook M. et al. (2011). ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 20, 229-245. 10.1016/j.ccr.2011.06.018 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S. and Eliceiri K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671-675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanopoulos N. and Francis M. B. (2011). Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 7, 876-884. 10.1038/nchembio.720 [DOI] [PubMed] [Google Scholar]
- Tolde O., Rosel D., Janostiak R., Vesely P. and Brabek J. (2012). Dynamics and morphology of focal adhesions in complex 3D environment. Folia. Biol. 58, 177-184. [DOI] [PubMed] [Google Scholar]
- Tozluoğlu M., Tournier A. L., Jenkins R. P., Hooper S., Bates P. A. and Sahai E. (2013). Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat. Cell Biol. 15, 751-762. 10.1038/ncb2775 [DOI] [PubMed] [Google Scholar]
- Welch M. D. (2015). Cell migration, freshly squeezed. Cell 160, 581-582. 10.1016/j.cell.2015.01.053 [DOI] [PubMed] [Google Scholar]
- Wilkinson S., Paterson H. F. and Marshall C. J. (2005). Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat. Cell Biol. 7, 255-261. 10.1038/ncb1230 [DOI] [PubMed] [Google Scholar]
- Wolf K. and Friedl P. (2011). Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 21, 736-744. 10.1016/j.tcb.2011.09.006 [DOI] [PubMed] [Google Scholar]
- Wolf K., Mazo I., Leung H., Engelke K., von Andrian U. H., Deryugina E. I., Strongin A. Y., Bröcker E. B. and Friedl P. (2003). Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 160, 267-277. 10.1083/jcb.200209006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K., Wu Y. I., Liu Y., Geiger J., Tam E., Overall C., Stack M. S. and Friedl P. (2007). Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9, 893-904. 10.1038/ncb1616 [DOI] [PubMed] [Google Scholar]
- Wyckoff J. B., Pinner S. E., Gschmeissner S., Condeelis J. S. and Sahai E. (2006). ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 16, 1515-1523. 10.1016/j.cub.2006.05.065 [DOI] [PubMed] [Google Scholar]
- Yamada K. M. and Sixt M. (2019). Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 20, 738-752. 10.1038/s41580-019-0172-9 [DOI] [PubMed] [Google Scholar]
- Yang W.-H., Lan H.-Y., Huang C.-H., Tai S.-K., Tzeng C.-H., Kao S.-Y., Wu K.-J., Hung M.-C. and Yang M.-H. (2012). RAC1 activation mediates Twist1-induced cancer cell migration. Nat. Cell Biol. 14, 366-374. 10.1038/ncb2455 [DOI] [PubMed] [Google Scholar]
- Yip A. K., Chiam K.-H. and Matsudaira P. (2015). Traction stress analysis and modeling reveal that amoeboid migration in confined spaces is accompanied by expansive forces and requires the structural integrity of the membrane-cortex interactions. Integr. Biol. 7, 1196-1211. 10.1039/C4IB00245H [DOI] [PubMed] [Google Scholar]
- Ziperstein M. J., Guzman A. and Kaufman L. J. (2015). Breast cancer cell line aggregate morphology does not predict invasive capacity. PLoS ONE 10, e0139523 10.1371/journal.pone.0139523 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.