

Abstract
Bile acids modulate several gastrointestinal functions including electrolyte secretion and absorption, gastric emptying, and small intestinal and colonic motility. High concentrations of bile acids lead to diarrhea and are implicated in the development of esophageal, gastric and colonic cancer. Alterations in bile acid homeostasis are also implicated in the pathophysiology of irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD). Our understanding of the mechanisms underlying these effects of bile acids on gut functions has been greatly enhanced by the discovery of bile acid receptors, including the nuclear receptors: farnesoid X receptor (FXR), vitamin D receptor (VDR), pregnane X receptor (PXR), and constitutive androstane receptor (CAR); and the G protein-coupled receptors: Takeda G protein-coupled receptor (TGR5), sphingosine-1-phosphate receptor 2 (S1PR2), and muscarinic acetylcholine receptor M3 (M3R).. For example, various studies provided evidence demonstrating the anti-inflammatory effects FXR and TGR5 activation in models of intestinal inflammation. In addition, TGR5 activation in enteric neurons was recently shown to increase colonic motility, which may lead to bile acid-induced diarrhea. Interestingly, TGR5 induces the secretion of glucagon-like peptide-1 (GLP-1) from L-cells to enhance insulin secretion and modulate glucose metabolism. Because of the importance of these receptors, agonists of TGR5 and intestine-specific FXR agonists are currently being tested as an option for the treatment of diabetes mellitus and primary bile acid diarrhea, respectively. This review summarizes current knowledge of the functional roles of bile acid receptors in the gastrointestinal tract.
1. Introduction
Bile acids are synthesized in the liver from cholesterol, secreted into the intestine, and are reabsorbed in the ileum. Reabsorbed bile acids circulate back to the liver and are resecreted to establish their enterohepatic circulation.1,2 In the intestine, the entry of bile acids into enterocytes of the distal ileum stimulates the expression and secretion of fibroblast growth factor FGF19 (FGF15 in rodents) that functions as a hormone affecting several biological processes in the liver, including the suppression of bile acid synthesis (Figure 1). Bile acids in the intestine emulsify dietary lipids and cholesterol to facilitate their absorption. However, extensive research during the last two decades has provided interesting evidence showing that bile acids exert multiple important physiological and pathophysiological roles beside their function as detergents. Since the gut is a large organ that represents the longer limb of the enterohepatic circulation of bile acids, the effects of bile acids on physiological functions of the gut have been extensively studied. The intestine is equipped with elaborate bile acid-sensing mechanisms that allow elegant coordination of different intestinal functions and control the crosstalk between the gut and other organs in the body. Seminal work by several investigators addressed the roles of bile acids as signaling molecules that trigger cellular signaling pathways by activating specific receptors to modulate biological processes. Bile acids have been shown to activate both nuclear receptors and plasma membrane associated receptors expressed in different cell types to elicit the physiological effects of bile acids. In light of the complex nature of the gastrointestinal tract and the intricate network of signaling induced by bile acids, the influence of bile acids on intestinal functions deserves special attention. Many recent advances have been made with respect to our understating of the effects of bile acids on gastrointestinal tract physiology. In this review, we will focus on summarizing the current knowledge about the roles of bile acid receptors in modulating gut functions (Table 1).
Figure 1. Bile acid signaling in the intestine.
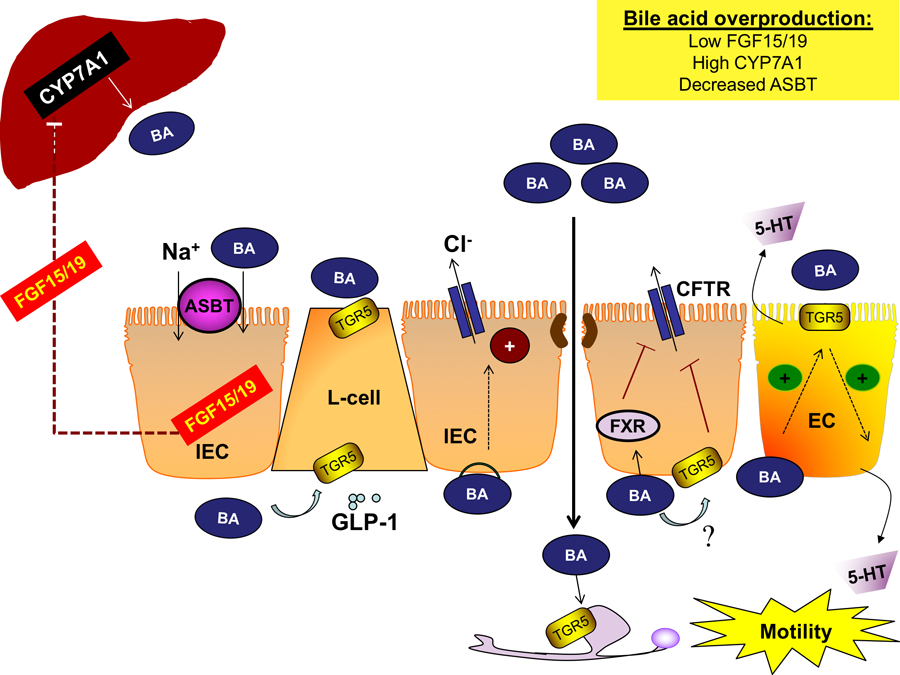
Bile acids are synthesized in the liver by the rate-limiting enzyme CYP7A1 and secreted into the intestine. Bile acids are reabsorbed by IECs in the distal ileum via ASBT where they activate FXR, inducing the expression and secretion of FGF15/19 to inhibit hepatic bile acid synthesis. Luminal and basolateral bile acids activate TGR5 in enteroendocrine L-cells, resulting in the release of the incretin GLP-1, promoting glucose tolerance. Overproduction of bile acids can promote diarrhea by several distinct mechanisms. In the colon, bile acids disrupt barrier integrity allowing bile acids to reach the basolateral membrane of epithelial cells and induce chloride secretion, though the receptor(s) underlying this phenomenon is unknown. Interestingly, colonic FXR and TGR5 activation inhibits the secretion of chloride and other electrolytes, which may be a compensatory mechanism. Excessive bile acids also promote gut motility. Colonic bile acids activate TGR5 on ECs, promoting 5-HT release and motility. Bile acids activate neuronal TGR5, which either stimulates or inhibits motility, depending on the type of neuron and region of the GI tract. Overall, overproduction of bile acids induces colonic secretion and enhances motility, producing a diarrheal phenotype. Abbreviations: 5-HT, 5-hydroxytryptamine; ASBT, apical sodium-dependent bile acid transporter; BA, bile acid; CFTR, cystic fibrosis transmembrane conductance regulator; CYP7A1, cytochrome P450 7A1; EC, enterochromaffin cells; FGF15/19, fibroblast growth factor 15/19; FXR, farnesoid X receptor; GI, gastrointestinal; GLP-1, glucagon-like peptide-1; IEC, intestinal epithelial cell; TGR5, Takega G protein-coupled receptor 5
Table 1.
Functional roles of bile acid receptors in the gut.
| Section | Receptor | Major functions in the intestine | Consequences of loss of function | Selected references |
|---|---|---|---|---|
| Nuclear receptors | ||||
| 2.1 | Farnesoid X receptor (FXR) | • Bile acid and cholesterol homeostasis via ileal FGF15/19 • Suppression of mucosal immune response • Maintenance of intestinal barrier |
• Increased susceptibility to intestinal inflammation • Bacterial overgrowth • Increased bile acid production, bile acid diarrhea • Promoting colorectal tumorigenesis |
3,6,7 |
| 2.2 | Vitamin D receptor (VDR) | • Detoxification • Modulation of bile acid biosynthesis via FGF15/19 |
3,30 | |
| 2.2 | Pregnane X receptor (PXR) | • Detoxification | 3,36 | |
| 2.2 | Constitutive androstane receptor (CAR) | • Detoxification | 3,36 | |
| Membrane receptors | ||||
| 3.1 | Takeda G protein-coupled receptor (TGR5; GPBAR1) | • Delayed gastric emptying • Increased colonic motility • Anti-inflammatory response • Induction of incretin secretion and modulation of glucose homeostasis via GLP-1 |
• Disturbed motility that varies between intestinal regions • Reduced frequency of defecation • Low stool water content • Disturbed glucose homeostasis? |
37,54,77 |
| 3.2 | Muscarinic acetylcholine receptor M3 (M3R) | • Cell proliferation | 87 | |
| 3.3 | Sphingosine-1-phosphate receptor 2 (S1PR2) | • Hepatic lipid homeostasis • Cell proliferation? |
90,94 |
2. Nuclear receptors
One of the mechanisms by which bile acids regulate intestinal physiology is altering gene expression via binding to and activating nuclear receptors.3 These are a group of ligand-activated transcription factors that are involved in regulating a variety of biological processes. At least four nuclear receptors are known to be activated by bile acids: farnesoid X receptor (FXR, NR1H4), vitamin D receptor (VDR, NR1I1), pregnane X receptor (PXR, NR1I2), and constitutive androstane receptor (CAR, NR1I3).3 We will summarize in the following section what is currently known about the roles of bile acid nuclear receptors in influencing gut functions and their relevance to the development of gastrointestinal diseases.
2.1. Farnesoid X-Receptor (FXR).
FXR was the first identified receptor for bile acids and its activation meditates the canonical pathways triggered by bile acids.4–8 FXR is expressed in different types of cells and tissues including the liver and small intestine and has been shown to play crucial roles in the maintenance of bile acid and cholesterol homeostasis as well as glucose and lipid metabolism.9 Bile acids are the endogenous ligands for FXR with different potency in the following order: chenodeoxycholic acid > deoxycholic acid > lithocholic acid > cholic acid (CDCA > DCA > LCA > CA).9 There are two genes for FXR: FXRα and FXRβ. The function of FXRβ is not fully understood in mice and it is considered to be a pseudo gene in humans. Thus, the name FXR in this review refers to FXRα. Intestinal FXR was shown to be crucial for controlling bile acid biosynthesis in the liver. Indeed, tissue-specific knock out in mice provided compelling evidence demonstrating that intestinal FXR, via the induction of FGF15/19, is more important than liver FXR in mediating the negative feedback of bile acids on their synthesis in the liver. Beside its importance in controlling hepatic bile acid synthesis, intestinal FXR is relevant to a number of important intestinal functions and is considered as a target for the treatment of a number of gastrointestinal disorders.9 We will discuss in the following sections the roles of FXR in modulating intestinal functions and its potential roles in the development of intestinal diseases.
2.1.1. FXR and mucosal immune response.
Effects of bile acids on immune cells were known from the early studies of Calmus et al, which showed that incubation of monocytes with CDCA inhibited LPS-stimulated secretion of cytokines including TNFα.10 Since CDCA is a potent activator of FXR, these findings suggested the involvement of FXR in the observed anti-inflammatory effects of bile acids. In this regard, FXR was found to be expressed in human peripheral blood mononuclear cells (PBMCs) along with other nuclear receptors.11 These studies showed FXR mRNA and protein expression in CD4+ T cells, CD8+ T cells, and CD14 monocytes subpopulations, suggesting a modulatory role for this nuclear receptor in immune responses. Consistently, the synthetic FXR agonist INT-747 (obeticholic acid) decreased TNFα secretion from activated human PBMCs and CD14+ monocytes. Also, INT-747 was able to suppress the secretion of TNFα from mononuclear cells isolated from the lamina propria of IBD patients. It was observed that the FXR agonist not only inhibited the secretion of cytokines from active mononuclear immune cells but also decreased the expression of proinflammatory cytokines in differentiated intestinal epithelial cells.12 Activation of FXR was also shown to inhibit cellular signaling pathways known to be triggered during inflammation via interactions with AP-1 and STAT3 transcription factors. In addition, FXR inhibits the expression of genes that are known targets for the major modulators of immune responses, namely, NF-κB and AP-1 transcription factors.13–15 Collectively, studies of FXR and the immune response provided strong evidence for the involvement of FXR in the regulation of innate and adaptive immunity, as well as inhibition of the pro-inflammatory response, suggesting its potential role in the pathophysiology of chronic inflammatory diseases of the intestine.
2.1.2. FXR and intestinal inflammation.
Inflammatory bowel diseases (IBD), which consist of Crohn’s disease (CD) and ulcerative colitis (US), are chronic inflammatory conditions of the gut. While the development of intestinal inflammation is multifactorial, there is recent experimental evidence to suggest that increased levels of luminal bile acids may play a role in the progression of these disorders.16,17 FXR expression was shown to be reduced in patients with CD and in mouse models of colitis.18 Since FXR was shown to influence immune function, recent studies have focused on its involvement in IBD. Furthermore, the studies of Gadaleta et al showed that administration of INT-747 protected mice from dextran sulfate sodium (DSS) and 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis.12 On the other hand, induction of inflammation by DSS and TNBS in FXR knockout mice resulted in more severe inflammation and increased levels of proinflammatory cytokines, which was accompanied by a greater loss in body weight as compared to wildtype mice.18 Also, administration of an FXR agonist failed to attenuate DSS- or TNBS-induced inflammation in FXR knockout mice.18 These findings strongly suggest that a decrease in FXR expression contributes to the development of intestinal inflammation and indicate that exploitation of the anti-inflammatory effects of FXR should be considered for the treatment of IBD.
2.1.3. FXR, gut bacteria, and intestinal permeability.
In addition to the role of bile acids in modulating immune response, it has become evident that blocking the secretion of bile and decreasing bile acid concentrations in the colonic lumen results in bacterial overgrowth and compromised epithelial barrier function, leading to enhanced bacterial translocation across colonic epithelia.19,20 Studies showed that administration of CDCA to rats with obstructive jaundice decreased bacterial overgrowth and translocation, providing supportive evidence for the protective roles of bile acids and suggesting the involvement of FXR in this process.19,21 The first evidence directly supporting the anti-bacterial effects for FXR was established by the studies of Inagaki et al.20 The authors showed that in mice subjected to bile duct ligation, administration of the FXR agonist GW4064 attenuated bacterial overgrowth and translocation. Further, they showed that FXR deficient mice exhibited enhanced bacterial overgrowth accompanied by increased translocation of bacteria to mesenteric lymph nodes. Activating FXR by GW4064 resulted in a significant increase in the expression of genes in epithelial cells that are known to have antibacterial effects, including angiogenin, carbonic anhydrase 12, inducible nitric oxide, and IL-18.19,20 Similar results were shown in recent studies using the INT-747, which further demonstrated that experimentally-induced obstructive jaundice in rats is associated with increased epithelial permeability along with elevated expression of claudin-2.22 The authors showed that obeticholic acid normalized intestinal permeability and increased the expression of tight junction proteins claudin-1 and occludin. The conclusion from these studies and many others has established pivotal roles of FXR in the protection against mucosal damage by inducing antibacterial effects and maintaining epithelial integrity.
2.1.4. FXR and bile acid-induced diarrhea.
Increased levels of luminal bile acids in the colon induces chloride secretion and impairs fluid absorption, leading to bile acid-induced diarrhea (BAD).23 BAD often presents secondary to ileal resection or ileal disease such as CD.23 However, there is also a prevalent type of primary (idiopathic) BAD that is common in patients with functional bowel disorders. Recent studies suggested that primary BAD is associated with overproduction of bile acids from the liver due to a decrease in ileal FGF19/15 secretion and attenuated feedback inhibition.24 Since activation of intestinal FXR stimulates the secretion of FGF19, it is reasonable to propose that FXR agonists are useful for the treatment of bile acid diarrhea. Recent studies indeed have shown promising results demonstrating that treatment with obeticholic acid was well-tolerated in patients with BAD and caused a decrease in hepatic synthesis of bile acids along with improved symptoms of diarrhea.25 It should also be noted that the studies of Mroz et el showed that FXR agonists exhibited antisecretory effects by reducing the expression of the chloride channel CFTR (cystic fibrosis transmembrane conductance regulator) and inhibiting the activity of Na/K ATPase transporter on the basolateral membrane of epithelial cells.26 Thus, the influence of FXR activation on epithelial transport processes may also contribute to the antidiarrheal effects of FXR agonists (Figure 1).
2.1.5. FXR and colorectal cancer.
Multiple lines of evidence suggest a role for FXR in the development of colorectal cancer. The studies of Bailey et al showed that FXR expression is decreased in precancerous lesions and is absent in the majority of samples from stages I to IV colonic adenocarcinoma patients.27 The authors showed that this silencing of FXR was the result of FXR promoter hypermethylation and KRAS signaling. The findings implicate FXR in the suppression of genes involved in the epithelial-to-mesenchymal transition and other oncogenic signaling pathways.27 Similar findings were also reported showing a decrease in FXR in colonic polyps and a remarkable reduction in its expression in colon adenocarcinoma.28 In other studies, the loss of FXR was associated with an increase in the number and the size of intestinal epithelial tumors in mice treated with azoxymethane (AOM).29 Also, the lack of FXR was shown to be associated with an increased tumor progression and early mortality in the ApcMin/+ and in chronic colitis mouse models.29 The authors demonstrated an activation of Wnt signaling by infiltrating neutrophils and elevation in the levels of TNFα secreted from macrophages when FXR was absent. Importantly, the administration of cholestyramine, a bile acid-binding resin, did not affect the promotion of tumorigenicity in FXR−/− mice, clearly demonstrating that the loss of FXR and not the increase in bile acids is responsible for the high susceptibility to tumorigenesis. These findings provided novel insights into the potential role of FXR as a target for the treatment of colon cancer.
2.2. VDR, PXR, and CAR.
In addition to FXR, it has also been shown that bile acids bind to the vitamin D receptor (VDR), the pregnane X receptor (PXR), and the constitutive androstane receptor (CAR). In contrast to FXR, bile acids are not the main ligands for these receptors. VDR is activated be Calcitriol (1,25-dihydroxyvitamin D3) and by the oncogenic secondary bile acid lithocolic acid (LCA), but not by other bile acids.30,31 The activation of VDR resulted in the induction of cytochrome CYP3A4 that is responsible for detoxifying LCA.30 Also, the activation of VDR resulted in a suppression of CYP7A1 expression in the liver, decreasing hepatic bile acid synthesis.32 Interestingly, IP injection of vitamin D3 resulted in induced expression of ileal FGF15 in mice, suggesting that FGF15 is also a target of VDR.33 Furthermore, VDR is also known to increase the expression of the ileal bile acid transporter ASBT.34 Collectively, activation of VDR may be involved in increasing bile acid absorption, suppressing hepatic bile acid synthesis, and promoting the expression of enzymes involved in the detoxification of LCA.
PXR is primarily activated by xenobiotics as well as drugs such as rifampicin and is responsible for induction of phase I and phase II metabolism of many compounds including bile acids. LCA is the main bile acid ligand for PXR, and PXR activation induces the expression of detoxifying enzymes (including CYP3A family enzymes), as well as enzymes involved in sulphation and conjugation of bile acids.3,9 Indeed, activation of PXR has been suggested as a useful approach to attenuate the cytotoxic effects of bile acids during cholestasis.3,9 Furthermore, activating PXR in colonic cell line was shown to induce the expression of the FGF19, suggesting that PXR also contributes to the suppression of bile acid synthesis in the liver.35 CAR has also been shown to bind LCA and cooperate with PXR in stimulating the pathways involved in LCA detoxification in the liver.36
3. G protein-coupled receptors
Beside their roles as ligands for the nuclear receptors, a broad spectrum of bile acid effects throughout the body are now known to be mediated by direct activation of G protein-coupled receptors at the plasma membrane. Three major GPCRs are known to interact with bile acids to modulate gastrointestinal (GI) functions: the Takeda G protein-coupled receptor 5 (TGR5; also known as the G protein-coupled bile acid receptor 1, GPBAR1),37 the M3R muscarinic receptor,38 and the sphingosine-1-phosphate receptor 2 (S1PR2).39 Although little is known about the roles of M3R and SIPR2 in modulating gastrointestinal functions, extensive work has demonstrated the importance of TGR5 in controlling a wide range of GI physiology.40 We will focus in this section on reviewing the current knowledge regarding the roles of these receptors in modulating gut function, with a focus on TGR5.
3.1. Takeda G-protein coupled receptor 5 (TGR5)
A major breakthrough came with the discovery of a plasma membrane-associated G-protein coupled receptor mediating non-genomic effects of bile acids through Gαs.40 This receptor is widely referred to as the Takeda G-protein coupled receptor 5 (TGR5) and has been implicated in a wide range physiological functions in the body such as maintaining energy homeostasis and stimulating insulin secretion.40 Bile acids are the endogenous ligands for TGR5 and the potency of different types of bile acids are in the order of LCA > DCA > CDCA > CA.9 In the following sections, we will attempt to summarize the results of a large body of literature related to the involvement of TGR5 in modulating gastrointestinal functions.
3.1.1. Role of TGR5 in modulating intestinal electrolyte transport and gut motility
Excessive bile acids in the lumen of the small intestine and colon has long been known to cause diarrhea.41–43 Earlier studies in humans showed that perfusion of the colon with bile acids stimulated water and electrolyte secretion and inhibited absorption.41,44 In particular, bile acid-dependent induction of chloride secretion across colonic epithelia was shown using different experimental models.45,46 One noteworthy aspect of this phenomenon is the sidedness of bile acid effects on chloride secretion. Interestingly, chloride secretion from colonic epithelial monolayers was shown to be significantly induced when cells were exposed to bile acids from the basolateral membrane, while higher concentrations of bile acids at the luminal membrane of colonic epithelial monolayers were needed to stimulate chloride secretion.47 The observed effect on chloride secretion from the luminal membrane has been attributed to compromised epithelial tight junctions that allow bile acids to reach the basolateral membrane, where they subsequently stimulate chloride secretion (Figure 1).47 The sidedness of bile acid effects on chloride secretion indicated a mechanism by which bile acids may activate a membrane receptor exclusively expressed on the basolateral membrane of epithelial cells. In this regard, the expression of TGR5 was recently shown in T84 cells and in epithelial cells of the rat colon.47,48 However, potent activators of TGR5 including LCA and ciprofloxacin failed to induce chloride secretion in T84 cells.49 Also, the activation of TGR5 by the synthetic agonist INT-777 rather decreased basal secretion in rat colon and significantly attenuated Ca2+-dependent induced chloride secretion.48 These observations strongly indicated that TGR5 is not involved in bile acid induced chloride secretion.
Bile acids are also well known to modulate intestinal and colonic motility, but until recently the underlying mechanisms were poorly understood. Bile acids inhibit gastric emptying and reduce small intestinal transit time but stimulate colonic peristalsis and increase colonic transit time.50,51 With respect to gastric emptying, TGR5 was found to be expressed in gastric smooth muscle, where its activation triggers a signaling via Epac and PKA-mediated pathway to inhibit RhoA and cause gastric muscle relaxation.52 besides gastric smooth muscle, TGR5 was also shown to be expressed in nitrergic inhibitory neurons of the myenteric plexuses in the small intestine, colon, and stomach, as well as in the cholinergic neurons of the submucosal plexuses in the small intestinal and colon.53 TGR5 expression was also detected in the intrinsic primary afferent neurons (IPAN) that represent the sensory limb of the enteric neurogenic reflexes of the gut.53 Studies in transgenic mice provided important insights into the roles of TGR5 in modulating gut motility. In this regard, the lack of TGR5 in knockout mice resulted in a constipated phenotype with a decline in colonic transit time, a reduction in the frequency of defecation, and a decrease in water content in the stool.54 On the other hand, TGR5 overexpression in transgenic mice caused a faster colonic transit time as well as an increased frequency of defecation. Alemi et al further showed that the induction of colonic peristalsis by bile acids was inhibited by antagonists to 5-hydroxytroptamine (5-HT) and calcitonin gene-related peptide (CGRP), suggesting the involvement of 5-HT and CGRP in mediating the effects of TGR5 receptor on motility (Figure 1).54 It is interesting to note that TGR5 was recently shown to be expressed in enterochromaffin cells (ECs) of the colon but not in the small intestine, suggesting that bile acids may directly induce 5-HT secretion only in the colon.55 The secreted 5-HT from EC may also contribute to bile acid effects on motility.
Overall, TGR5 appears to play a key role in coordinating the responses of different intestinal cell types to bile acids. For instance, activation of TGR5 on L-cells may be involved in mediating the ileal brake phenomenon, resulting in a decrease in small intestinal motility and slowing of gastric emptying. Also, TGR5 on ECs stimulates the secretion of 5-HT to enhance peristalsis in the colon. The presence of TGR5 in IPAN sensory neurons triggers the enteric reflex, promoting peristalsis mediated by CGRP. Peristalsis occurs by orchestrated contraction and relaxation of circular and longitudinal smooth muscles of the GI tract. The direct activation of TGR5 on inhibitory neurons leads to inhibition of longitudinal muscle contractility, consistent with the induction of peristalsis. It is intriguing to note that activation of TGR5 seems to mediate the laxative effects of bile acids mainly by altering gut motility patterns and not via induced secretion. Indeed, the activation of TGR5 on cholinergic secretomotor neurons in the submucosal plexuses was shown to inhibit basal as well as agonist-stimulated electrolyte secretion from the colonic epithelia.56
With regard to diarrhea, it appears that the roles of bile acid receptors are complex and may be summarized as follows: diarrhea is multifactorial and may result from increased colonic motility, increased electrolyte secretion, decrease electrolyte absorption, or a combination of these factors.43 With respect to TGR5, the activation of this receptor on enteric neurons causes an increase in colonic motility. However, TGR5 on cholinergic neurons in submucosal plexuses inhibits electrolyte secretion. Also, TGR5 activation results in a decrease in agonist-induced chloride secretion in epithelial cells. The inhibition of secretion by TGR5 could be construed as compensatory to stimulated colonic motility. Therefore, TGR5 expressed on enteric neurons and ECs, but not the TGR5 expressed on epithelial cells, may explain bile acid-induced diarrhea. However, bile acids directly induce chloride secretion when added to the basolateral membrane of epithelial cell and the receptor(s) involved remain to be identified (Figure 1). Finally, it should be noted that the effects of TGR5 on electrolyte absorption have not been fully delineated. We have previously shown that that bile acids decrease chloride uptake in epithelial cells.44 However, the role of TGR5 or other receptors in mediating bile acid-induced inhibition of chloride uptake has not been thoroughly investigated.
Not only has TGR5 been shown to modulate gastrointestinal motility, it has also been demonstrated that alterations in TGR5 expression may also be involved in the pathophysiology of several disease states. In this regard, an increase in TGR5 expression in the gastric myenteric plexus was implicated in the pathogenesis of delayed gastric emptying in mice.57 These studies showed that depleting the bile acid pool using cholestyramine reversed the effects of high fat diet on TGR5 expression and gastric emptying. These data suggest that the high levels of serum bile associated with high fat diets may explain the increase in TGR5 expression. Along with studies in mice, recent investigations in humans showed that a functionally relevant SNP in the TGR5 gene (rs11554825) increases susceptibility to fast colonic transit time and increased levels of fecal bile acids in carriers of this allele.58 This evidence reinforces the essential role of TGR5 in gut motility and supports the notion that alterations in TGR5 expression may be involved in the development of disorders such as delayed gastric emptying and irritable bowel syndrome. It is clear from these studies that TGR5 is an attractive therapeutic target for the management of functional gastrointestinal and motility disorders.
3.1.2. Emerging role of TGR5 in intestinal inflammation.
The expression of TGR5 has been established in immune cells including monocytes and macrophages.37,59 In macrophages activated by LPS, TRG5 agonists suppressed the production of TNFα through the mTOR pathway.60 These studies also showed that TGR5 deficiency in macrophages was associated with an increase in cytokine secretion and macrophage migration.60 Such findings represent strong evidence for the important role of TGR5 in chronic inflammatory conditions. Incubation of human peripheral blood monocytes with a specific TGR5 agonist (benzyl 2-keto-6-methyl-4-(2-thienyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate) but not an FXR agonist (fexaramine) induced differentiation into dendritic cells with an IL-12 hypo-producing phenotype.61 IL-12 produced by dendritic cells promotes the immune response mediated by type 1 helper T cells, contributing to the pathogenesis of chronic inflammatory diseases such as IBD.
In this regard, Cipriani et al showed TGR5 agonists (ciprofloxacin and oleanolic acid) mitigated intestinal inflammation induced by TNBS in mice.62 Consistent with the anti-inflammatory effects of TGR5 agonists, the authors showed that TGR5 knockout mice are more susceptible to TBNS-induced colitis. Similarly, TGR5 agonists were also shown to reduce the inflammatory index in the DSS model of induced colitis and other models of intestinal and gastric epithelial injury, such as that induced by acetylsalicylic acid.63 It is also notable that the lack of TGR5 resulted in altered epithelial tight junctions associated with increased intestinal permeability, which may contribute to the increased susceptibility to experimentally-induced intestinal inflammation.62
One important observation is that TGR5 expression was increased in the colon of mice with TNBS-induced colitis and in patients with Crohn’s disease.62 Furthermore, it was shown that the majority of CD14+ cells isolated from intestinal lamina propria of inflamed intestine in mice treated with TNBS and from patients with Crohn’s are also positive for TGR5. The fact that TGR5 exhibits anti-inflammatory effects and that its expression is decreased during the course of inflammatory disease highlights the potential for targeting TGR5 in the treatment of intestinal inflammation. Indeed, a recent study by Biagioli et al demonstrated that a putative small molecule activator of TGR5, BAR501, was able to significantly decrease the disease activity index in TNBS-induced colitis by about 70%.64 This decrease in the inflammatory response was associated with a shift in the macrophages of the lamina propria from the classically activated macrophages (CD11b+, CCR7+, F4/F80−) to alternatively activated phenotype (CD11b+, CCR7−, F4/F80+). The treatment with TGR5 agonist resulted in a decrease in the levels of pro-inflammatory cytokines (TNFα, IL1b, IL-6 and CCL2), along with an increase in the levels of anti-inflammatory cytokines (IL10 and TGFβ). Interestingly, TGR5-mediated shift in M1/M2 phenotype of intestinal macrophages was dependent on IL-10, as the anti-inflammatory effects of BAR501 were lost in IL-10 knockout mice.64
3.1.3. TGR5 and gut hormone secretion: beneficial role in metabolic disorders
The link between bile acids and GLP-1 secretion was initially established by Katsura et al, who demonstrated an increase in secretion of this incretin from the mouse STC-1 enteroendocrine cell line in response to incubation with bile acids (Figure 1).65 The increase was blocked by inhibitors of adenylate cyclase (MDL12330A),65 strongly suggesting the involvement of TGR5 receptor. These results were later confirmed using the specific TGR5 synthetic agonist INT-777 and were reproduced in various experimental models.40,66 Similar observations were also made in humans. Rectal administration of taurocholic acid in male human subjects showed an increase in both GLP-1 and PYY peptides.67 Indeed, activation of TGR5 receptor on L-cells also triggers the secretion of peptide YY (PYY) along with GLP-1 via a cAMP-dependent mechanism and the involvement of Epac/PLC-mu/Ca2+ pathway.68 These studies also showed that H2S, which may be generated by gut bacteria, blocks TGR5-mediated secretion of GLP-1 and PYY via the inhibition of PLCε/Ca2+ pathway.68 These findings have significant implications showing that bacterial products may influence the induction of GLP-1 secretion by bile acids.
Because TGR5 activation induces the secretion of the incretin hormone GLP-1, it is reasonable to expect that TGR5 may be intimately involved in the control of glucose homeostasis. Indeed, the beneficial effects of TGR5 activation in the control of hyperglycemia and hyperinsulinemia has been shown in mouse models of diet-induced obesity as well as in human patients. These studies showed that treatment of obese mice with the bile acid sequestrant colesevelam reduced hepatic glucose production by blocking hepatic glycogenolysis.69 Using both TGR5 knockout mice and GLP-1 antagonists, these studies further showed that the effects of colesevelam on hepatic glucose production were mediated by the TGR5-GLP-1 axis. Bile acid sequestrants such as colesevelam are resins that block bile acid absorption, retaining them in the intestinal and colonic lumen. The authors of this study posited that luminal colesevelam-bile acid complexes remain capable of activating TGR5. This hypothesis was proven using non-polarized HEK 293 cells as an in-vitro model. Also, injecting colesevelam-bile acid complexes via the rectum was able to stimulate GLP-1 secretion. However, the idea that bile acid-sequestrant complexes activate TGR5 was later challenged by at least two studies. In the first study, Brighton et al showed that adding bile acids to the basolateral side of the intestinal epithelia was more effective in inducing GLP-1 secretion from L-cells than the luminally added bile acids.70 They also showed that the luminal effects were abolished in the presence of an ASBT inhibitor. In the second study, GLP-1 secretion was assessed in metformin-treated patients after cholecystokinin (CCK) infusion to induce the release of bile acids. The results showed that the secretion of bile acids into the gut lumen by CCK-mediated gallbladder contraction caused an increase in GLP-1 secretion. Importantly, this effect was blocked by oral supplementation of bile acid sequestrant sevelamer.71 Furthermore, administration of CDCA to diabetic patients was able to increase GLP-1 level when given alone but not when it was given with colesevalam.72 Taken together, these findings strongly suggest that the luminal uptake of bile acids via ASBT is essential for the delivery of bile acids to the basolateral side of L-cells to activate TGR5 and secrete GLP-1 (Figure 1). However, this conclusion is in conflict with the earlier proposition of luminal bile acids activating TGR5-GLP1 even if they are complexed with sequestrants. The discrepancy between these findings requires further critical investigation.
In relation to obesity, strong evidence suggests that alterations in bile acid homeostasis and TGR5-mediated increase in GLP-1 levels drive the beneficial effects of Roux-en-Y gastric bypass surgery on weight loss and improved insulin sensitivity, independent of restricted caloric intake.73,74 There is also convincing evidence for the role of TGR5 in the beneficial response to other types of bariatric surgeries such as vertical sleeve gastrectomy.75
In light of these numerous studies, TGR5 receptor agonists (synthetic or naturally produced by gut bacteria) are now considered as therapeutics for the treatment of diabetes mellitus due to their ability to induce GLP-1 secretion.74,76 However, TGR5 agonists my cause systemic side effects such as increased gallbladder filling, as TGR5 is also expressed in the gallbladder.77 Thus, recent studies have focused on the development of poorly absorbed agonists to avoid systemic effects.77 Moreover, the combination of TGR5 agonists with inhibitors of the dipeptidyl peptidase-4 (DPP-4) potentiated the glucose lowering effects, galvanizing the research in this area for developing advanced and more effective interventions for the management of hyperglycemia in diabetic patients.77 For instance, recent studies by Pathan et al using the dual FXR and TGR5 agonist (INT-767) have unraveled novel pathways via which FXR increases TGR5 expression to enhance GLP-1 secretion.78 The same group further showed the important role of altered gut microbial composition in mediating the effects of the intestine-specific FXR agonist (fexaramine) on TGR5 signaling. They demonstrated that antibiotic treatment blunted the effects of fexaramine on GLP-1 secretion, impairing the subsequent improvement in insulin sensitivity and metabolic conditions.79 These studies highlight the increased efficacy of treating obesity and diabetes mellitus by simultaneously activating both FXR and TGR5.
3.1.4. TGR5 and gastrointestinal cancer.
The link between TGR5 signaling and tumorigenesis was first shown in human gastric carcinoma AGS cells.80 These studies of Yasuda et al demonstrated an induction of EGFR phosphorylation in response to incubation with bile acids, an event that was blocked by siRNA-mediated attenuation of TGR5 expression.80 Further, studies by Cao et al showed an increase in TGR5 in the majority of cases of gastric intestinal-type adenocarcinoma.81 Interestingly, the level of TGR5 expression in these studies was also shown to negatively correlate with patient survival. The authors further showed that activation of TGR5 and coupling to Gαq and Gαi-3 proteins enhances the proliferation of gastric adenocarcinoma cells. Studies by Carino et al provided additional evidence for the increased expression of TGR5 in advanced stages of gastric cancer and showed that TGR5 activation increases the expression of markers for epithelial-mesenchymal transition (EMT) in gastric cancer cells.82 These studies suggest the pivotal role for TGR5 in the development of gastric cancer.
The reflux of bile acids back into the stomach and esophagus is also implicated in the development of Barrett’s esophagus and esophageal adenocarcinoma (EAC).83 Strong evidence showed higher levels of TGR5 expression in human tissues of EAC as compared to normal esophageal mucosa and to mucosa from Barrett’s esophagus.84 It was shown that TGR5 expression was associated with worse survival in EAC patients.84 The activation of TGR5 receptor in EAC cells was linked to the increase in inducible nitric oxide synthase, NADPH oxidase NOX5-S, and the production of H2O2.83 The pathway was further delineated, and the evidence suggested that in EAC cells, bile acid-dependent activation of TGR5 caused DNA damage via NOX5-S and CREB transcription factor.85 Interestingly, there was an association between TGR5, VDR, and claudin-2 expression suggesting a potential mechanism that may contribute to the development of EAC.86 It is also interesting to note that increased expression of TGR5 in EAC patients was more prominent in males as compared to females, which may help to explain the increased incidence of EAC in males.84
3.2. Muscarinic receptors.
Earlier studies demonstrated the selective interactions and activation of muscarinic receptors by bile acids in gastric epithelial cells.87 Studies further showed that the activation of muscarinic receptor M3R in colonic epithelial cells caused a transactivation of EGFR and an increase in cell proliferation.88 Interestingly, mice lacking ASBT, in which the colonic levels of bile acids are significantly higher due to the malabsorption, exhibited a significant increase in the number and size of tumors when challenged with the carcinogen azoxymethane (AOM).89 These findings could be explained by the involvement of M3R muscarinic receptor in the induction of proliferation of intestinal epithelial cells and the promotion of tumor progression.
3.3. Sphingosine-1-Phosphate receptors
Sphinogosine-1-phosphate receptor 2 (S1PR2) was recently shown to be activated by bile acids. Several studies showed that conjugated bile acids stimulate ERK1/2 as well as AKT-dependent pathways in hepatocytes in a manner dependent on Gαi to induce the activity of nuclear sphingosine kinase 2 (Sphk2), leading to subsequent increase in nuclear S1P.90 Such an increase in S1P inhibits specific HDACs, resulting in altered gene expression. This novel bile acid-S1PR2-Sphk2 axis has been implicated in the modulation of processes involved in lipid metabolism. Mice deficient in S1PR2 were more susceptible to development of fatty liver when fed with a high fat diet.91 Also, activation of S1PR2 by bile acids regulates hepatic glucose metabolism in a manner similar to that of insulin.92 S1RP2 activation by bile acids was also implicated in the development of cholangiocarcinoma.93
Although much of the research on bile acids and S1PR2 focus on the liver, a recent study showed that S1PR2 is expressed in intestinal epithelial cells and suggested that its activation by conjugated bile acids promotes cell proliferation.94 Nevertheless, the role of S1PR2 in intestinal physiology requires further investigation.
4. Conclusion
Bile acid effects on the gastrointestinal tract have shown regional differences between the different segments of the tract. The advances in our knowledge about the receptors mediating the effects of bile acids has provided clues that have helped to explain the differential effects of bile acids in the segments of the gut. For example, TGR5 mediates the effects of bile acids in the progression of tumors in the upper GI tract (gastric and esophageal), whereas the loss of FXR expression may be involved in the development of colorectal cancer. Also, activation of TGR5 on gastric smooth muscle results in delayed gastric emptying associated with feeding high fat diet, whereas activation of enteric TGR5 in myenteric pluses in the colon may explain the increase in colonic motility and diarrhea associated with increased levels of bile acids in the colon. Despite the extensive work that has been done, additional investigations are still needed to fully understand the complex network of events that underlie the many interactions between bile acids and the gastrointestinal system. The receptors involved in the induction of chloride secretion in intestinal epithelial cells and the mechanisms underlying their activation remains elusive. Also, the exact roles of bile acid receptors in shaping the composition of gut microbiota is in interesting area of investigation. Expanding our knowledge about the functional roles of bile acid receptors in the gut will enhance our understanding of gut related disorders and will unravel potential targets for improved therapeutic interventions to treat diseases such as IBD and gut functional disorders.
Acknowledgement:
The work in the authors’ laboratories is supported by merit review grants from the Department of Veterans Affairs (VA): BX000152 (WAA) and BX002011 (PKD); F30 grant DK117535 (ALT); and R01 grants: DK109709 (WAA), DK54016 (PKD), DK81858 (PKD), DK92441(PKD), DK98170 (RKG), from NIDDK/NIH. WAA is supported by a VA Research Career Scientist Award. PKD is also supported by a VA Senior Research Career Scientist Award.
5. References
- 1.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res 2007;24(10):1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 2.Chiang JYL. Bile acid metabolism and signaling. Compr Physiol 2013;3(3):1191–1212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li T, Chiang JYL. Nuclear receptors in bile acid metabolism. Drug Metab Rev 2013;45(1):145–155. doi: 10.3109/03602532.2012.740048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forman BM, Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995;81(5):687–693. [DOI] [PubMed] [Google Scholar]
- 5.Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol 1995;9(1):72–85. doi: 10.1210/mend.9.1.7760852. [DOI] [PubMed] [Google Scholar]
- 6.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 7.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999;284(5418):1365–1368. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Molecular Cell 1999;3(5):543–553. [DOI] [PubMed] [Google Scholar]
- 9.Schaap FG, Trauner M, Jansen PLM. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11(1):55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 10.Calmus Y, Guechot J, Podevin P, Bonnefis MT, Giboudeau J, Poupon R. Differential effects of chenodeoxycholic and ursodeoxycholic acids on interleukin 1, interleukin 6 and tumor necrosis factor-alpha production by monocytes. Hepatology 1992;16(3):719–723. [DOI] [PubMed] [Google Scholar]
- 11.Schote AB, Turner JD, Schiltz J, Muller CP. Nuclear receptors in human immune cells: expression and correlations. Mol Immunol. 2007;44(6):1436–1445. doi: 10.1016/j.molimm.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 12.Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011;60(4):463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 13.Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev 2005;208(1):126–140. doi: 10.1111/j.0105-2896.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 14.Fiorucci S, Cipriani S, Mencarelli A, Renga B, Distrutti E, Baldelli F. Counter-regulatory role of bile acid activated receptors in immunity and inflammation. Curr Mol Med 2010;10(6):579–595. [DOI] [PubMed] [Google Scholar]
- 15.Ding L, Yang L, Wang Z, Huang W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharmaceutica Sinica B 2015;5(2):135–144. doi: 10.1016/j.apsb.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ananthakrishnan AN, Bernstein CN, Iliopoulos D, et al. Environmental triggers in IBD: a review of progress and evidence. Nat Rev Gastroenterol Hepatol 2018;15(1):39–49. doi: 10.1038/nrgastro.2017.136. [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Cao L, Jiang C, et al. PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nature Communications 2014;5(1):4573. doi: 10.1038/ncomms5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 2009;183(10):6251–6261. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 19.Ding L, Yang L, Wang Z, Huang W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharmaceutica Sinica B 2015;5(2):135–144. doi: 10.1016/j.apsb.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inagaki T, Moschetta A, Lee Y-K, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA 2006;103(10):3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorenzo-Zúñiga V, Bartolí R, Planas R, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 2003;37(3):551–557. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- 22.Verbeke L, Farre R, Verbinnen B, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol 2015;185(2):409–419. doi: 10.1016/j.ajpath.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 23.Keely SJ, Walters JRF. The Farnesoid X Receptor: Good for BAD. Cell Mol Gastroenterol Hepatol 2016;2(6):725–732. doi: 10.1016/j.jcmgh.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walters JRF, Tasleem AM, Omer OS, Brydon WG, Dew T, le Roux CW. A new mechanism for bile acid diarrhea: defective feedback inhibition of bile acid biosynthesis. Clin Gastroenterol Hepatol 2009;7(11):1189–1194. doi: 10.1016/j.cgh.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 25.Walters JRF, Johnston IM, Nolan JD, Vassie C, Pruzanski ME, Shapiro DA. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment Pharmacol Ther 2015;41(1):54–64. doi: 10.1111/apt.12999. [DOI] [PubMed] [Google Scholar]
- 26.Mroz MS, Keating N, Ward JB, et al. Farnesoid X receptor agonists attenuate colonic epithelial secretory function and prevent experimental diarrhoea in vivo. Gut 2014;63(5):808–817. doi: 10.1136/gutjnl-2013-305088. [DOI] [PubMed] [Google Scholar]
- 27.Bailey AM, Zhan L, Maru D, et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am J Physiol Gastrointest Liver Physiol 2014;306(1):G48–G58. doi: 10.1152/ajpgi.00234.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Gottardi A, Touri F, Maurer CA, et al. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig Dis Sci 2004;49(6):982–989. [DOI] [PubMed] [Google Scholar]
- 29.Maran RRM, Thomas A, Roth M, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther 2009;328(2):469–477. doi: 10.1124/jpet.108.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 31.Adachi R, Shulman AI, Yamamoto K, et al. Structural determinants for vitamin D receptor response to endocrine and xenobiotic signals. Mol Endocrinol 2004;18(1):43–52. doi: 10.1210/me.2003-0244. [DOI] [PubMed] [Google Scholar]
- 32.Han S, Li T, Ellis E, Strom S, Chiang JYL. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol Endocrinol 2010;24(6):1151–1164. doi: 10.1210/me.2009-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt DR, Holmstrom SR, Fon Tacer K, Bookout AL, Kliewer SA, Mangelsdorf DJ. Regulation of bile acid synthesis by fat-soluble vitamins A and D. Journal of Biological Chemistry 2010;285(19):14486–14494. doi: 10.1074/jbc.M110.116004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Chen F, Liu S, et al. Transactivation of rat apical sodium-dependent bile acid transporter and increased bile acid transport by 1alpha,25-dihydroxyvitamin D3 via the vitamin D receptor. Mol Pharmacol 2006;69(6):1913–1923. doi: 10.1124/mol.105.020792. [DOI] [PubMed] [Google Scholar]
- 35.Wistuba W, Gnewuch C, Liebisch G, Schmitz G, Langmann T. Lithocholic acid induction of the FGF19 promoter in intestinal cells is mediated by PXR. World J Gastroenterol 2007;13(31):4230–4235. doi: 10.3748/wjg.v13.i31.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner M, Halilbasic E, Marschall H-U, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005;42(2):420–430. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- 37.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem 2003;278(11):9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 38.Raufman J-P, Chen Y, Zimniak P, Cheng K. Deoxycholic acid conjugates are muscarinic cholinergic receptor antagonists. Pharmacology 2002;65(4):215–221. doi: 10.1159/000064347. [DOI] [PubMed] [Google Scholar]
- 39.Studer E, Zhou X, Zhao R, et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012;55(1):267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis 2014;46(4):302–312. doi: 10.1016/j.dld.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mekjian HS, Phillips SF, Hofmann AF. Colonic secretion of water and electrolytes induced by bile acids: perfusion studies in man. J Clin Invest 1971;50(8):1569–1577. doi: 10.1172/JCI106644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camilleri M. Bile Acid Diarrhea: Prevalence, Pathogenesis, and Therapy. Gut Liver 2015;9(3). doi: 10.5009/gnl14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camilleri M, Sellin JH, Barrett KE. Pathophysiology, Evaluation, and Management of Chronic Watery Diarrhea. Gastroenterology 2017;152(3):515–532.e2. doi: 10.1053/j.gastro.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alrefai WA, Saksena S, Tyagi S, Gill RK, Ramaswamy K, Dudeja PK. Taurodeoxycholate modulates apical Cl−/OH− exchange activity in Caco2 cells. Dig Dis Sci 2007;52(5):1270–1278. doi: 10.1007/s10620-006-9090-8. [DOI] [PubMed] [Google Scholar]
- 45.Balistreri WF, Heubi JE, Suchy FJ. Bile acid metabolism: relationship of bile acid malabsorption and diarrhea. J Pediatr Gastroenterol Nutr 1983;2(1):105–121. [PubMed] [Google Scholar]
- 46.Bajor A, Gillberg P-G, Abrahamsson H. Bile acids: short and long term effects in the intestine. Scand J Gastroenterol 2010;45(6):645–664. doi: 10.3109/00365521003702734. [DOI] [PubMed] [Google Scholar]
- 47.Ao M, Sarathy J, Domingue J, Alrefai WA, Rao MC. Chenodeoxycholic acid stimulates Cl(−) secretion via cAMP signaling and increases cystic fibrosis transmembrane conductance regulator phosphorylation in T84 cells. Am J Physiol, Cell Physiol 2013;305(4):C447–C456. doi: 10.1152/ajpcell.00416.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward JBJ, Mroz MS, Keely SJ. The bile acid receptor, TGR5, regulates basal and cholinergic-induced secretory responses in rat colon. Neurogastroenterol Motil 2013;25(8):708–711. doi: 10.1111/nmo.12148. [DOI] [PubMed] [Google Scholar]
- 49.Domingue JC, Ao M, Sarathy J, Rao MC. Chenodeoxycholic acid requires activation of EGFR, EPAC, and Ca2+ to stimulate CFTR-dependent Cl− secretion in human colonic T84 cells. Am J Physiol, Cell Physiol 2016;311(5):C777–C792. doi: 10.1152/ajpcell.00168.2016. [DOI] [PubMed] [Google Scholar]
- 50.Armstrong DN, Krenz HK, Modlin IM, Ballantyne GH. Bile salt inhibition of motility in the isolated perfused rabbit terminal ileum. Gut 1993;34(4):483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shiff SJ, Soloway RD, Snape WJ. Mechanism of deoxycholic acid stimulation of the rabbit colon. J Clin Invest 1982;69(4):985–992. doi: 10.1172/JCI110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajagopal S, Kumar DP, Mahavadi S, et al. Activation of G protein-coupled bile acid receptor, TGR5, induces smooth muscle relaxation via both Epac- and PKA-mediated inhibition of RhoA/Rho kinase pathway. Am J Physiol Gastrointest Liver Physiol 2013;304(5):G527–G535. doi: 10.1152/ajpgi.00388.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poole DP, Godfrey C, Cattaruzza F, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil 2010;22(7):814–25–e227–8. doi: 10.1111/j.1365-2982.2010.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alemi F, Poole DP, Chiu J, et al. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 2013;144(1):145–154. doi: 10.1053/j.gastro.2012.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lund ML, Egerod KL, Engelstoft MS, et al. Enterochromaffin 5-HT cells - A major target for GLP-1 and gut microbial metabolites. Mol Metab 2018;11:70–83. doi: 10.1016/j.molmet.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duboc H, Tolstanova G, Yuan P-Q, et al. Reduction of epithelial secretion in male rat distal colonic mucosa by bile acid receptor TGR5 agonist, INT-777: role of submucosal neurons. Neurogastroenterol Motil 2016;28(11):1663–1676. doi: 10.1111/nmo.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou H, Zhou S, Gao J, Zhang G, Lu Y, Owyang C. Upregulation of bile acid receptor TGR5 and nNOS in gastric myenteric plexus is responsible for delayed gastric emptying after chronic high-fat feeding in rats. Am J Physiol Gastrointest Liver Physiol 2015;308(10):G863–G873. doi: 10.1152/ajpgi.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Camilleri M, Vazquez-Roque MI, Carlson P, Burton D, Wong BS, Zinsmeister AR. Association of bile acid receptor TGR5 variation and transit in health and lower functional gastrointestinal disorders. Neurogastroenterol Motil 2011;23(11):995–9–e458. doi: 10.1111/j.1365-2982.2011.01772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoneno K, Hisamatsu T, Shimamura K, et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013;139(1):19–29. doi: 10.1111/imm.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perino A, Pols TWH, Nomura M, Stein S, Pellicciari R, Schoonjans K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J Clin Invest 2014;124(12):5424–5436. doi: 10.1172/JCI76289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ichikawa R, Takayama T, Yoneno K, et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012;136(2):153–162. doi: 10.1111/j.1365-2567.2012.03554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cipriani S, Mencarelli A, Chini MG, et al. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. Ryffel B, ed. PLoS ONE 2011;6(10):e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cipriani S, Mencarelli A, Bruno A, et al. Activation of the bile acid receptor GPBAR1 protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs and aspirin in mice. Br J Pharmacol 2013;168(1):225–237. doi: 10.1111/j.1476-5381.2012.02128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biagioli M, Carino A, Cipriani S, et al. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J Immunol 2017;199(2):718–733. doi: 10.4049/jimmunol.1700183. [DOI] [PubMed] [Google Scholar]
- 65.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 2005;329(1):386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 66.Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 2009;10(3):167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu T, Bound MJ, Standfield SD, et al. Effects of rectal administration of taurocholic acid on glucagon-like peptide-1 and peptide YY secretion in healthy humans. Diabetes Obes Metab 2013;15(5):474–477. doi: 10.1111/dom.12043. [DOI] [PubMed] [Google Scholar]
- 68.Bala V, Rajagopal S, Kumar DP, et al. Release of GLP-1 and PYY in response to the activation of G protein-coupled bile acid receptor TGR5 is mediated by Epac/PLC-ε pathway and modulated by endogenous H2S. Front Physiol 2014;5:420. doi: 10.3389/fphys.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Potthoff MJ, Potts A, He T, et al. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol 2013;304(4):G371–G380. doi: 10.1152/ajpgi.00400.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brighton CA, Rievaj J, Kuhre RE, et al. Bile Acids Trigger GLP-1 Release Predominantly by Accessing Basolaterally Located G Protein-Coupled Bile Acid Receptors. Endocrinology 2015;156(11):3961–3970. doi: 10.1210/en.2015-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brønden A, Albér A, Rohde U, et al. The bile acid-sequestering resin sevelamer eliminates the acute GLP-1 stimulatory effect of endogenously released bile acids in patients with type 2 diabetes. Diabetes Obes Metab 2018;20(2):362–369. doi: 10.1111/dom.13080. [DOI] [PubMed] [Google Scholar]
- 72.Hansen M, Scheltema MJ, Sonne DP, et al. Effect of chenodeoxycholic acid and the bile acid sequestrant colesevelam on glucagon-like peptide-1 secretion. Diabetes Obes Metab 2016;18(6):571–580. doi: 10.1111/dom.12648. [DOI] [PubMed] [Google Scholar]
- 73.Zhai H, Li Z, Peng M, et al. Takeda G Protein-Coupled Receptor 5-Mechanistic Target of Rapamycin Complex 1 Signaling Contributes to the Increment of Glucagon-Like Peptide-1 Production after Roux-en-Y Gastric Bypass. EBioMedicine 2018;32:201–214. doi: 10.1016/j.ebiom.2018.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaska L, Sledzinski T, Chomiczewska A, Dettlaff-Pokora A, Swierczynski J. Improved glucose metabolism following bariatric surgery is associated with increased circulating bile acid concentrations and remodeling of the gut microbiome. World J Gastroenterol 2016;22(39):8698–8719. doi: 10.3748/wjg.v22.i39.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding L, Sousa KM, Jin L, et al. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 2016;64(3):760–773. doi: 10.1002/hep.28689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jafri L, Saleem S, Calderwood D, Gillespie A, Mirza B, Green BD. Naturally-occurring TGR5 agonists modulating glucagon-like peptide-1 biosynthesis and secretion. Peptides 2016;78:51–58. doi: 10.1016/j.peptides.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 77.Ma S-Y, Ning M-M, Zou Q-A, et al. OL3, a novel low-absorbed TGR5 agonist with reduced side effects, lowered blood glucose via dual actions on TGR5 activation and DPP-4 inhibition. Acta Pharmacol Sin 2016;37(10):1359–1369. doi: 10.1038/aps.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pathak P, Liu H, Boehme S, et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. Journal of Biological Chemistry 2017;292(26):11055–11069. doi: 10.1074/jbc.M117.784322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pathak P, Xie C, Nichols RG, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018;89:147. doi: 10.1002/hep.29857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun 2007;354(1):154–159. doi: 10.1016/j.bbrc.2006.12.168. [DOI] [PubMed] [Google Scholar]
- 81.Cao W, Tian W, Hong J, et al. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am J Physiol Gastrointest Liver Physiol 2013;304(4):G322–G327. doi: 10.1152/ajpgi.00263.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carino A, Graziosi L, D’Amore C, et al. The bile acid receptor GPBAR1 (TGR5) is expressed in human gastric cancers and promotes epithelial-mesenchymal transition in gastric cancer cell lines. Oncotarget 2016;7(38):61021–61035. doi: 10.18632/oncotarget.10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hong J, Behar J, Wands J, et al. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut 2010;59(2):170–180. doi: 10.1136/gut.2009.188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pang C, LaLonde A, Godfrey TE, et al. Bile salt receptor TGR5 is highly expressed in esophageal adenocarcinoma and precancerous lesions with significantly worse overall survival and gender differences. Clin Exp Gastroenterol 2017;10:29–37. doi: 10.2147/CEG.S117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li D, Cao W. Bile acid receptor TGR5, NADPH Oxidase NOX5-S and CREB Mediate Bile Acid-Induced DNA Damage In Barrett’s Esophageal Adenocarcinoma Cells. Sci Rep 2016;6(1):31538. doi: 10.1038/srep31538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abu-Farsakh S, Wu T, LaLonde A, Sun J, Zhou Z. High expression of Claudin-2 in esophageal carcinoma and precancerous lesions is significantly associated with the bile salt receptors VDR and TGR5. BMC Gastroenterol 2017;17(1):33. doi: 10.1186/s12876-017-0590-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Raufman J-P, Cheng K, Zimniak P. Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig Dis Sci 2003;48(8):1431–1444. [DOI] [PubMed] [Google Scholar]
- 88.Cheng K, Raufman J-P. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochemical Pharmacology 2005;70(7):1035–1047. doi: 10.1016/j.bcp.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 89.Raufman J-P, Dawson PA, Rao A, et al. Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis 2015;36(10):1193–1200. doi: 10.1093/carcin/bgv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nagahashi M, Takabe K, Liu R, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2015;61(4):1216–1226. doi: 10.1002/hep.27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kwong E, Li Y, Hylemon PB, Zhou H. Bile acids and sphingosine-1-phosphate receptor 2 in hepatic lipid metabolism. Acta Pharmaceutica Sinica B 2015;5(2):151–157. doi: 10.1016/j.apsb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nagahashi M, Yuza K, Hirose Y, et al. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J Lipid Res 2016;57(9):1636–1643. doi: 10.1194/jlr.R069286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Aoki H, Yang J, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017;65(6):2005–2018. doi: 10.1002/hep.29076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen T, Huang Z, Liu R, Yang J, Hylemon PB, Zhou H. Sphingosine-1 phosphate promotes intestinal epithelial cell proliferation via S1PR2. Front Biosci (Landmark Ed) 2017;22:596–608. [DOI] [PubMed] [Google Scholar]